 Li Xi
Li Xi Xinxi Qin
Xinxi Qin Yumin Song
Yumin Song Jincheng Han
Jincheng Han Zhiqiang Li
Zhiqiang Li Jinliang Zhang
Jinliang Zhang- 1Department of Animal Science, College of Biology and Food, Shangqiu Normal University, Shangqiu, China
- 2Henan Engineering Research Center of Development and Application of Green Feed Additives, College of Biology and Food, Shangqiu Normal University, Shangqiu, China
- 3Linyi Agricultural Science and Technology Career Academy, Linyi, China
The structure and composition of gut microbiota correlate with the occurrence and development of host health and disease. Diarrhea can cause alterations in gut microbiota in animals, and the changes in the gut microbial structure and composition may affect the development of diarrhea. However, there is a scarcity of information on the effects of diarrhea on gut fungal composition and structure, particularly in Baer's pochard (Aythya baeri). The current study was performed for high-throughput sequencing of the fungal-specific internal transcribed spacer 1 (ITS-1) to detect the differences of gut mycobiota in healthy and diarrheal Baer's pochard. Results showed that the gut mycobiota not only decreased significantly in diversity but also in structure and composition. Statistical analysis between two groups revealed a significant decrease in the abundance of phylum Rozellomycota, Zoopagomycota, Mortierellomycota, and Kickxellomycota in diarrheal Baer's pochard. At the genus levels, fungal relative abundance changed significantly in 95 genera, with 56 fungal genera, such as Wickerhamomyces, Alternaria, Penicillium, Cystofilobasidium, and Filobasidium, increasing significantly in the gut of the diarrheal Baer's pochard. In conclusion, the current study revealed the discrepancy in the gut fungal diversity and community composition between the healthy and diarrheal Baer's pochard, laying the basis for elucidating the relationship between diarrhea and the gut mycobiota in Baer's pochard.
Introduction
The gut microbiota is a group of microorganisms that maintains the balance of the intestinal microecological system in birds. They participate in the nutrition, metabolism, immunity, and development of the host (1, 2). Bacteria, fungus, and protozoa inhabiting the gut may engage in a commensal, symbiotic, or parasitic relationship, resulting in a stable intestinal environment (3). Gut microbial alterations can cause many diseases, such as diabetes, obesity, and diarrhea (4, 5).
Although the proportion of fungi in gut microbiota is low, it is closely associated with the feeding, immunity, and metabolic health of the host (6–8). Studies on the relationship between gut fungi and host health have confirmed that an imbalance in the gut fungal flora can cause diarrhea (9, 10). The intestinal abundance of Neocallimastigomycota in diarrhea yaks was found to be significantly higher than that of healthy yaks, suggesting that Neocallimastigomycota may be associated with diarrhea (11). Fecal fungi showed significant correlations with irritable bowel syndrome (IBS) symptoms (12). Moreover, bacterial–fungal interactions markedly declined in diarrhea-predominant IBS patients (13). Candida glabrata and Candida krusei were identified from Amazon parrot, dove, macaw, and cockatiel with gastrointestinal disease (14). Recent studies have shown that Candida species is often a conditional pathogen that has the opportunity to play a pathogenic role when the host's gut microecological balance is disrupted (15, 16). Aspergillus fumigatus is a widespread opportunistic pathogenic fungus in the natural environment and can cause severe pulmonary aspergillosis and other diseases. For instance, Aspergillus fumigatus could cause multisystemic damage to the ostrich body, showing diarrhea, vomiting, severe respiratory suppression, and even death (17). Given the importance of the gut fungal community in dysbacteriosis associated with diarrhea and inflammatory bowel disease, it is crucial to learn more about its composition and structure.
Baer's pochard (Aythya baeri) has been listed in the Category I of the State Key Protected Wildlife List in China and the CR/EN Category of the International Union for Conservation of Nature (IUCN) (18). As a wild bird, it is not easy to get timely diagnosis and treatment after diarrhea, which may result in death and threaten the continuity of the species. Currently, most of the studies on gut microecology in birds focus on gut bacterial diversity. High-throughput sequencing is less used to investigate the gut fungal diversity of birds, especially Baer's pochards. 18S rDNA, and ITS-based gene sequencing technologies have been widely used in fungal taxonomic identification (19–21). The in-depth analysis of changes in the structure and composition of gut mycobiota in diseased animals is beneficial to reveal the correlation of disease and flora, identify new pathogens, establish flora beneficial to maintaining the healthy growth of the body, and put forward effective prevention and control measures (22, 23). In this study, high-throughput sequencing technology was used to study the gut fungal composition and structure in the healthy and diarrheal Baer's pochards in order to investigate the effect of diarrhea on the gut fungal diversity of the Baer's pochards. Our study could provide molecular data for further protection of this endangered water bird and lay a scientific theoretical basis for population recovery and the formulation of effective protection policies and measures.
Materials and Methods
Sample Collection
Fresh fecal samples were collected from eight healthy adult Baer's pochards (group C; marked as C1–C8) and eight diarrheal adult Baer's pochards (group D; marked as D1–D8) from the National wetland park (Shangqiu, China). The samples in each group were collected immediately after Baer's pochard defecation to avoid contamination by environmental microorganisms. For further examination, the feces were put in sterile tubes, snap frozen, and stored at −80°C.
Internal Transcribed Spacer Amplification and Sequencing
The feces were processed as previously described (11). The fungal genomic DNA of fecal samples was extracted using Rapid Fungi Genomic DNA Isolation Kit (Sangon Biotech, Shanghai, China) according to the instructions of the manufacturer. The DNA sample was purified and quantified by 0.8% agarose gel electrophoresis and was diluted with sterile water to 1 ng/μl. Using the diluted genome DNA as the template, PCR amplification was performed using fungal ITS gene primers (ITS1F: 5′-CTTGGTCATTTAGAGGAAGTAA-3′ and ITS2R: 5′-GCTGCGTTCTTCATCG ATGC-3′), and Phusion High-Fidelity PCR Master Mix with GC Buffer (Thermo Fisher Scientific, Waltham, USA). The PCR products were analyzed using 2% agarose gel electrophoresis, and the equivalent PCR products were mixed according to the concentration. The sequencing library was prepared by Illumina TruSeq Nano DNA LT Library Prep Kit and then underwent quality inspection and quantitative analysis. The qualified library with concentrations above 2 nmol/L and single peak were combined and performed 2 × 250-bp paired-end sequencing on Illumina HiSeq 2500 platform (Illumina, CA, USA).
Bioinformatics and Statistical Analysis
To ensure the accuracy of the obtained sequences, raw reads were first filtered by Trimmomatic v0.33 software (24), and then the Cutadapt 1.9.1 software (25) was used to obtain high-quality reads without the primer sequence. Subsequently, the above high-quality reads were spliced through the FLASH v1.2.7 software (26) to obtain the clean reads. Finally, the UCHIME v4.2 software (27) was used to identify and remove the chimera sequence in clean reads to obtain effective reads. The effective reads were clustered into operational taxonomic unit (OTU) using VSEARCH algorithm (1.9.6) (28) according to the UNITE ITS database at 97% sequence similarity. The characteristic sequences are taxonomically annotated using a naive Bayesian classifier. Abundance tables at different classification levels were generated using QIIME 2 software (29). Community structure maps at each taxonomic level of the sample were drawn using the R v3.0.3 software (30). Rarefaction curves and rank abundance curves evaluating the sequencing depth and the species richness and evenness contained in the samples were calculated by the R v3.0.3 software. The α and β diversity indexes were calculated using QIIME 2 software. PCoA (31) were illustrated using R v3.0.3 software to characterize the fungal diversity among samples. Line discriminant analysis (LDA) effect size (LEfSe) (32) was used to find biomarkers with statistical differences between groups. T-testing of species abundance data between groups was performed using the Metastats software (33), and species with significant differences were screened based on p-values. A p-value < 0.05 was considered statistically significant.
Results
Sequencing Quality Assessment
A total of 1,280,719 (C = 639,738, D = 640,981) original sequences were obtained in the 16 fecal samples (Table 1). The effective reads [1,266,771 (C = 632,515, D = 634,256)] were obtained after removing the low-quality data, with an average of 79,173 effective reads per sample. The obtained effective reads were clustered using VSEARCH 1.9.6 software with 97% consistency, yielding 1,489 OTUs (C = 1,373, D = 1,225). Notably, there were 264 OTUs in the healthy group that were not detected in the diarrheal group, while 116 OTUs were present only in the diarrheal group (Figure 1A). The healthy samples shared 69 OTUs, while the diarrheal samples shared 62 OTUs (Figures 1B,C). The rarefaction curve was used to judge whether the sequencing data volume reflects the species diversity in the sample. When the sequencing amount reaches 10,000, all of the curves tend to be flat, indicating that the amount of sampling is reasonable, the sequencing depth has been substantially covering all the species in the sample, and can well reflect the fungal community structure and diversity of all the samples (Figures 1D,E). Except for the smooth decline in the rank abundance curves for C3, C4, and D6 showing better abundance and evenness, rapid and steep decline in other samples indicating that these samples had lower fungal diversity and a high proportion of dominant fungi (Figure 1F).
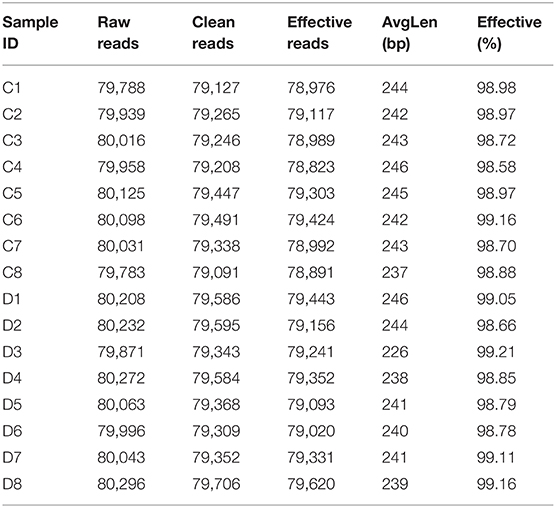
Table 1. The sequencing data processing results of fecal samples.
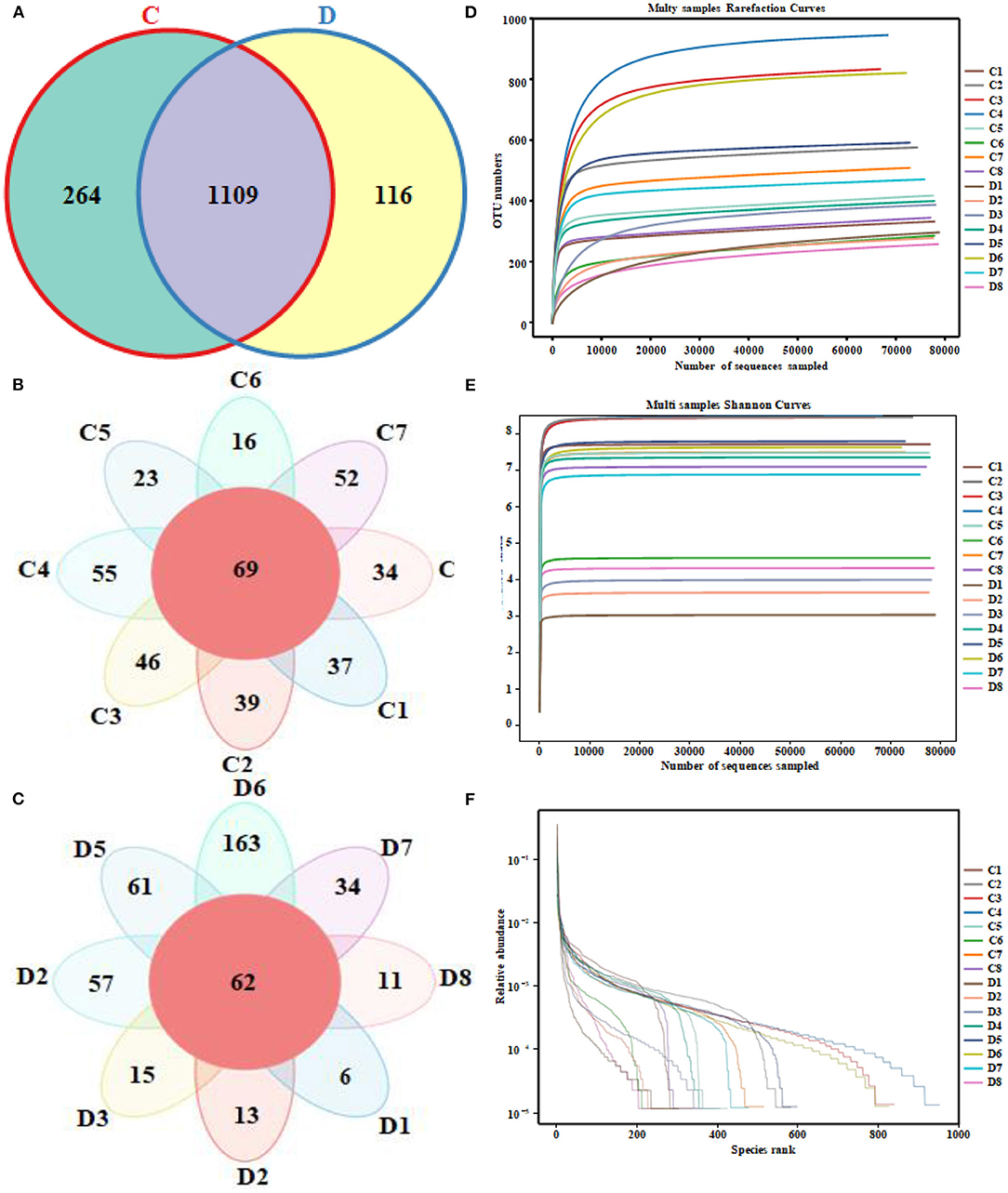
Figure 1. Venn diagrams and sequencing quality evaluation. (A) Venn diagram of operational taxonomic units (OTUs) distribution between group C and group D. (B,C) Flower diagrams of the OTU shared among the samples within the group C and group D, respectively. (D) Rarefaction curve of 16 fecal samples in both groups. (E) Shannon index curve of 16 fecal samples in both groups. (F) Rank abundance curve of 16 fecal samples in both groups.
Effects of Diarrhea on Gut Fungal Diversity
The alpha diversity index of the samples was evaluated using the QIIME2 software. The Good's coverage values for all samples ranged from 99.90 to 99.95%, showing high library coverage. The average Chao1 index was 699.84 and 539.47 in the healthy and diarrheal groups, respectively, with no significant difference (p > 0.05). ACE indices in the healthy and diarrheal group were 982.29 and 586.18 (p < 0.01), respectively. The Shannon index was 7.09 in the healthy group, 5.21 in the diarrheal group, with a significant difference (p < 0.05). The Simpson index also varied significantly (p < 0.05) between the two groups, with 0.97 in the healthy group and 0.85 in the diarrheal group. Alpha diversity index statistics showed that gut fungal diversity in diarrheal Baer's pochard was significantly lower in the healthy group (Figure 2). Principal coordinates analysis (PCoA) further demonstrated beta diversity differences between the two groups. PCoA based on the binary-Jaccard and Bray–Curtis distance revealed that the principal compositions of gut fungal diversity in the control group differed from that in diarrheal groups due to diarrheal effect (Figures 3A,B).
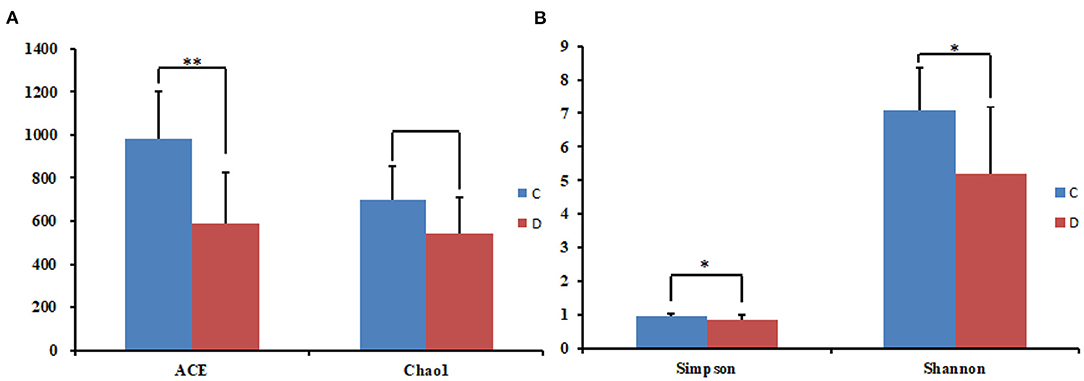
Figure 2. Statistical results of the alpha diversity between the two groups. (A) ACE and Chao1 index; (B) Simpson and Shannon index. *p < 0.05, **p < 0.01.
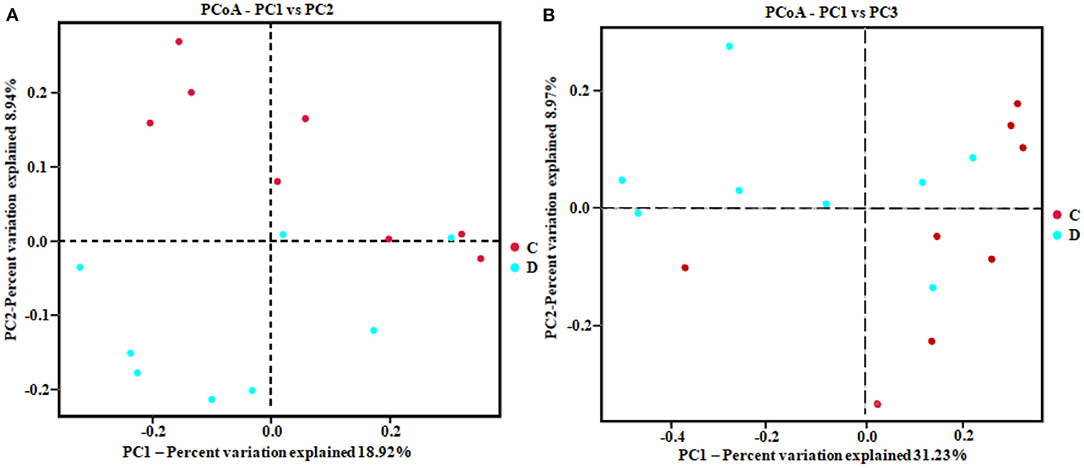
Figure 3. Principal coordinates analysis (PCoA) diagram of heterogeneity between the two groups. Scatterplot from PCoA based on binary-Jaccard (A) and Bray–Curtis dissimilarity (B) in gut fungal community.
Effect of Diarrhea on the Gut Fungal Structure and Composition
The histogram of species distribution showed relative abundances of dominant fungal taxa in all samples. In the current study, 10 phyla and 388 genera were identified from all samples, and the top 10 phyla and genera rank in abundance is presented in Figure 4. At phylum level, Ascomycota (70.26% in group C, 69.47% in group D) and Basidiomycota (18.88% in group C, 25.15% in group D) were the dominant fungal phyla in all samples, accounting for 89.14 and 94.62% of the taxonomic groups identified, respectively (Figure 4A). The other five phyla with lower abundance, including Mortierellomycota (3.14% in group C, 1.35% in group D), Chytridiomycota (0.63% in group C, 0.54% in group D), Rozellomycota (0.77% in group C, 0.23% in group D), Glomeromycota (0.54% in group C, 0.28% in group D), and Mucoromycota (0.27% in group C, 0.09% in group D) accounted for 5.35 and 2.49% of all fungal taxa in two groups, respectively. At the genus level, the relative abundances of Wickerhamomyces, Alternaria, Penicillium, Cystofilobasidium, and Filobasidium were observed higher in the diarrheal group (4.93, 4.67, 2.75, 0.85, and 0.78% in group C vs. 16.68, 8.15, 8.00, 6.83, and 5.53% in group D). Conversely, the relative abundances of Fusarium, Talaromyces, and Mortierella in the diarrheal group decreased (5.15, 3.43, and 2.79% in group C vs. 3.95, 2.72, and 1.30%). The cluster heatmap of species abundance at genus level is shown in Figure 5.
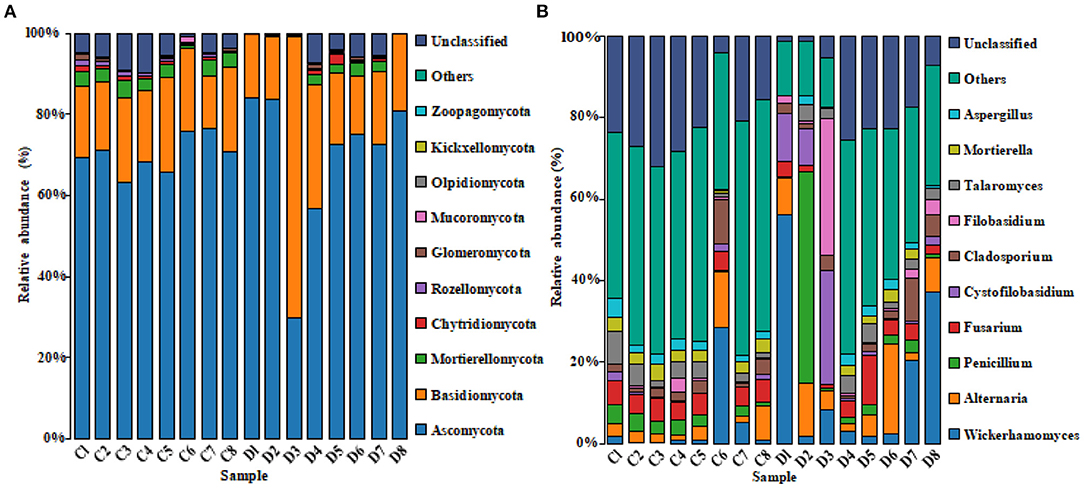
Figure 4. The histogram of the top 10 species distribution at phylum (A) and genus (B) levels.
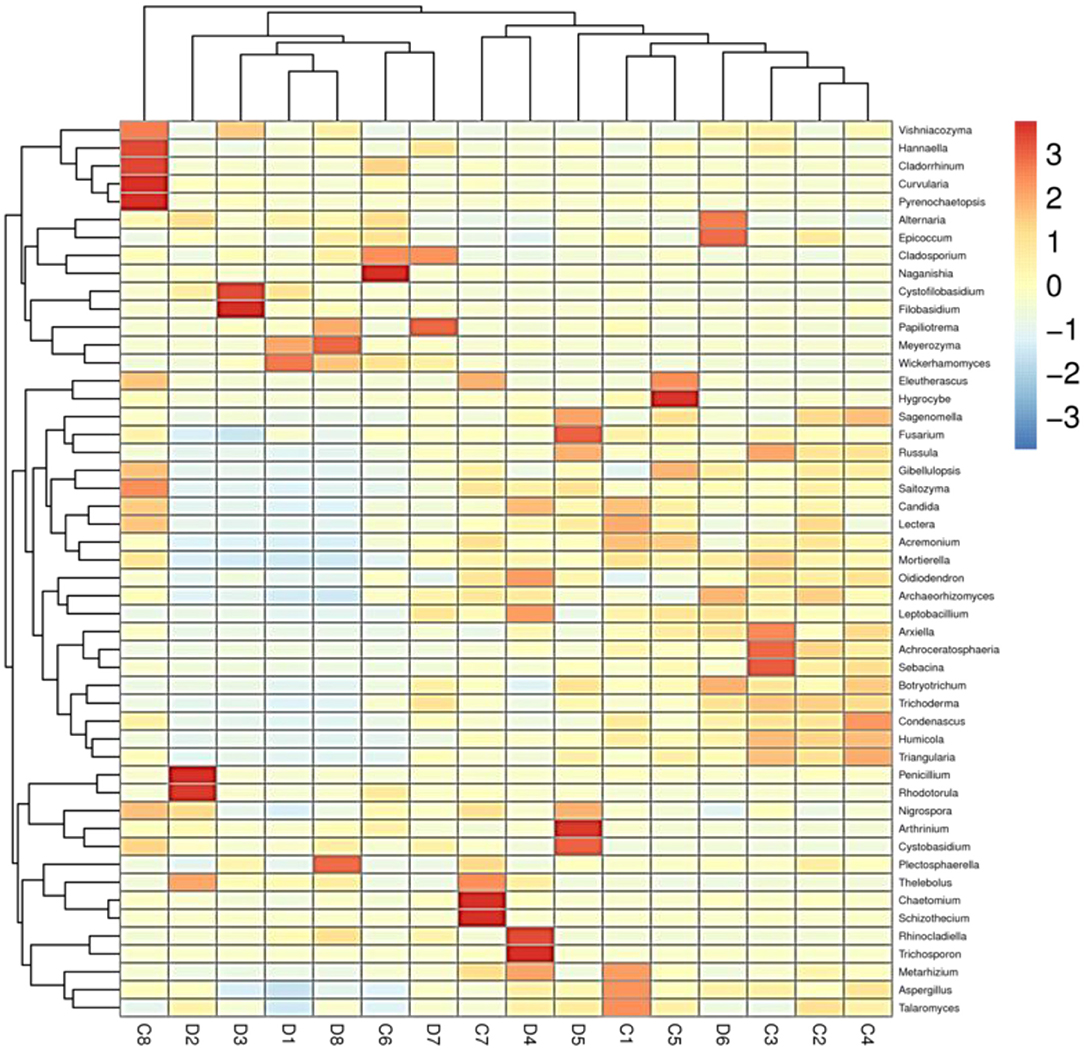
Figure 5. Heatmap at genus level of each sample (top 50).
The microbial community significant differences between the two groups were analyzed by Metastats software. At the phylum level, the richness of Rozellomycota, Zoopagomycota, Mortierellomycota, and Kickxellomycota in the diarrheal group significantly decreased than the healthy group (p < 0.05; Figure 6). Furthermore, of the 95 fungal genera with significant differences, the relative abundance of 56 fungal genera increased significantly in the diarrheal group. In comparison, the relative abundance of the remaining 39 fungal genera decreased significantly (Supplementary Table 1). Notably, 28 fungal genera were detected only in the diarrhea group, while 37 fungal genera in the healthy group disappeared in the diarrhea group, showing a significant diarrhea correlation (Figure 7). LEfSe was analyzed for significant differences between healthy and diarrheal groups. One order (Saccharomycetales), one family (Phaffomycetaceae), and two genera (Wickerhamomyces and Meyerozyma) were significantly more abundant in the diarrheal group. The relative abundances of three orders (Agaricales, Pezizales, and Sordariales) and three families (Nectriaceae, Bolbitiaceae, and Chaetomiaceae) were significantly higher in the healthy group than in the diarrheal group (Figures 8A,B).
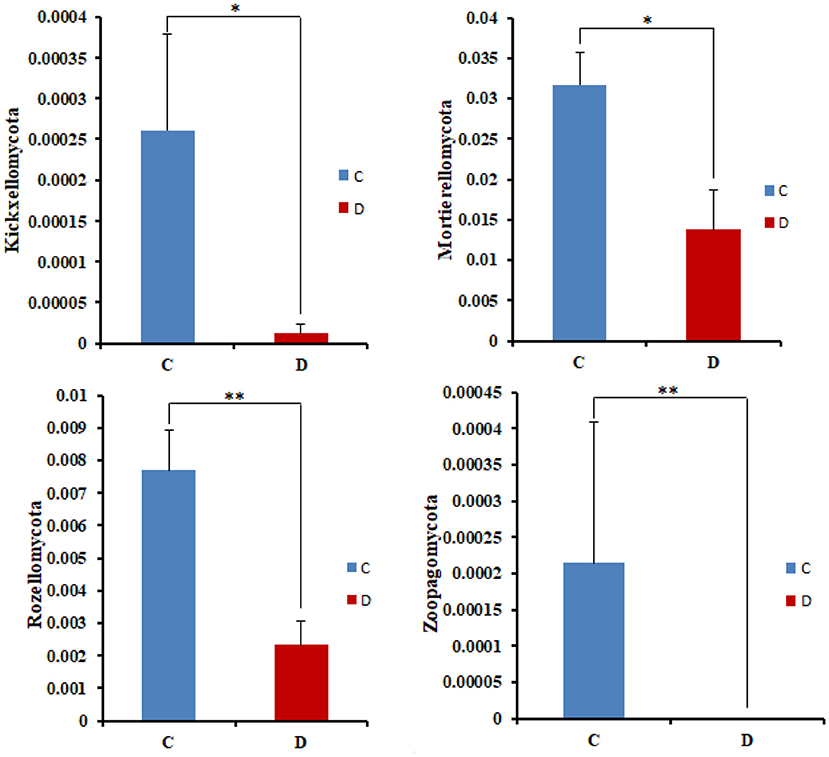
Figure 6. Comparison with the fungal community structure at phylum level in the two groups. *p < 0.05, **p < 0.01.
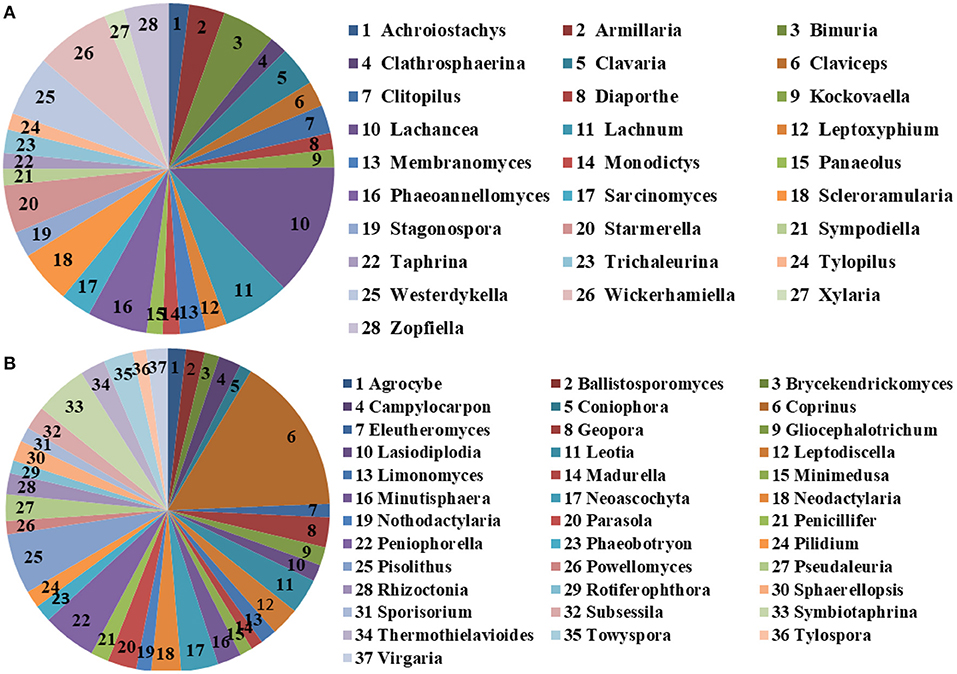
Figure 7. The unique fungal genera of the two groups. (A) Genera detected only in the diarrheal group. (B) Genera detected only in the healthy group. The sector area represents the abundance of these genera.
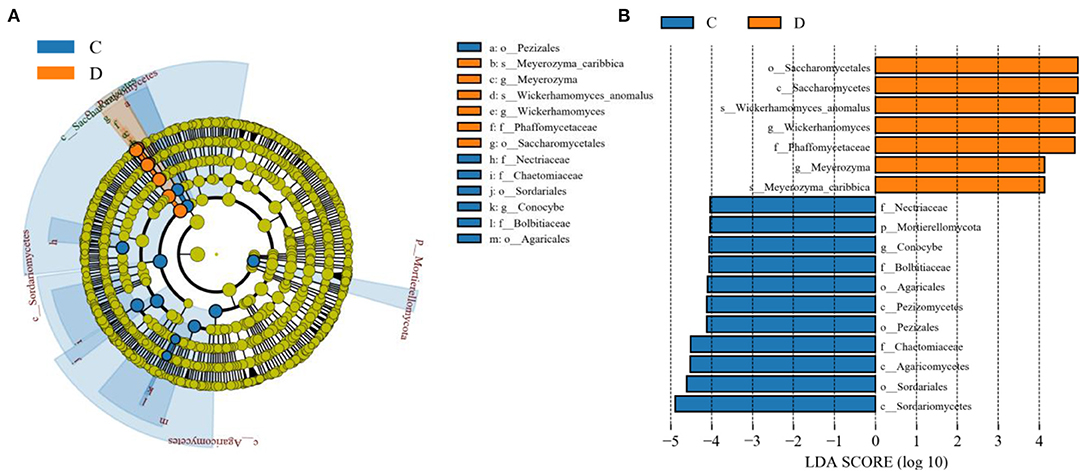
Figure 8. Effect size (LEfSe) analysis of intergroup samples. (A) LEfSe cladogram of fungal communities between the two groups. (B) Distribution histogram based on line discriminant analysis (LDA) value (LDA scores > 4).
Correlation Network Analysis
A species correlation network with 937 edges and 78 nodes was drawn based on the python programming language. The top 48 genera with the highest correlation are shown in Figure 9. Wickerhamomyces was negatively associated with Triangularia (0.8794), Sebacina (0.8794), Humicola (0.8294), Condenascus (0.8147), and Lepiota (0.7994), positively associated with Meyerozyma (0.6941) and Rhinocladiella (0.5451). Alternaria was positively related to Cystofilobasidium (0.5059), Arthrinium (0.6284), and Curvularia (0.5636). Penicillium was positively associated with Talaromyces (0.6471), Aspergillus (0.6265), Triangularia (0.5941), Sebacina (0.6324), Russula (0.6029), Condenascus (0.6324), Acremonium (0.5971), Achroceratosphaeria (0.5206), Humicola (0.6441), Trichoderma (0.5588), and Botryotrichum (0.5235). Cystofilobasidium was positively associated with Arthrinium (0.6431). Filobasidium was positively associated with Meyerozyma (0.5647) and Rhinocladiella (0.6377) (Supplementary Table 2).
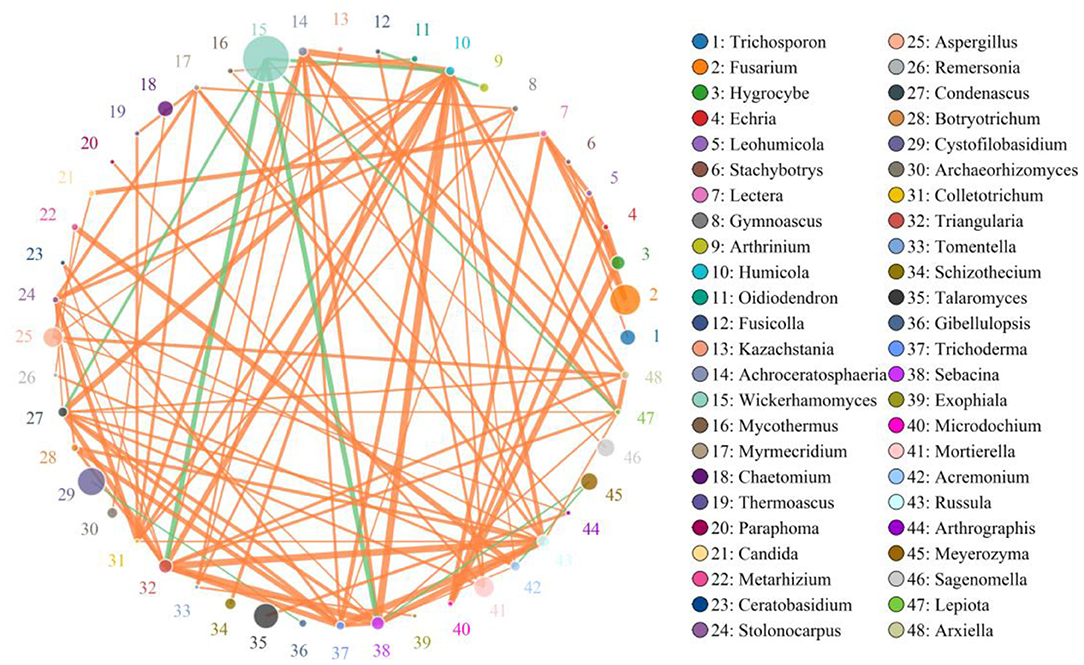
Figure 9. Network diagram of fungi at the genus level in the gut of Baer's pochard. The circles with different colors represent different genera, and the circle size represents the average abundance of each genera. The thickness of the line between two circles represents the strength of the correlation. The orange line represents the positive correlation, while the green line represents the negative correlation.
Discussion
Baer's pochard is one of the most endangered birds in the world (18). The International Union for Conservation of Nature created the International Single Species Action Plan (ISSAP) for Baer's pochard in 2014. China has an important distribution of Baer's pochard, with a population of about 600–800, which plays a key role in protecting Baer's pochard world population. The National wetland park (Shangqiu, China) is an important wintering ground and resting place for migratory birds. Baer's pochard was first observed in the wetland park in 2017. The largest population was over 150 observed in winter 2018, representing 15% of the global population. The wetland park was designated as “an important breeding site for Baer's pochard” in September 2019, indicating that it is essential for keeping birds in the park, and critical for the survival of the population. The health and breeding of Baer's pochard in these breeding sites are of great significance for population recovery and expansion.
A variety of pathogens such as bacteria, viruses, and fungi can cause animal diarrhea-related diseases, leading to animal nutrition absorption disorders, vomiting, diarrhea, dehydration, and other clinical symptoms (34). Wildlife is more vulnerable to the threat of diarrhea due to the lack of timely, effective veterinary intervention. Studies have confirmed that the gut microbiome plays an important role in host growth, development, and immune response (35, 36). Changes in the gut microbial community not only lead to gastrointestinal diseases, such as metabolic syndrome, obesity, and diarrhea (37, 38), but also cause diseases outside the digestive system, including central nervous system disorders (39), cardiovascular disease (40), etc. The gut microbial imbalance may also result in functional diarrhea (41, 42). Beneficial microbes can form a biological barrier by competing for intestinal mucosa cells, preventing pathogenic and conditional pathogenic microbes from invading. When the balance of gut microbiota is broken, the pathogenic microbes and their toxins increase relatively, which will damage the intestinal mucosa barrier, increase its permeability. Intestinal pathogenic microbes and their antigens can activate mast cells, and the sensitized mast cells can release histamine, serotonin, prostaglandin, trypsin, and other active substances to enhance smooth muscle contraction, increase intestinal peristalsis, and eventually result in diarrhea (43, 44). Moreover, long-term diarrhea can further aggravate gut microbial dysbiosis and diarrhea. Therefore, the investigation of the composition and structure of gut microbiota is conducive to mastering the pathogenesis of diarrhea and formulating effective prevention and treatment measures. The analysis of gut microbiota, which is closely related to the occurrence and development of diseases, mainly focused on bacteria. The research on fungi, especially the gut fungal alterations of wild protected animals under different health states, has rarely been reported.
The main fungal genera known to cause diarrhea are Candida, Eurotium, Cryptococcus, Mucor, Histoplasma, and Fusarium (13, 14, 45–48). Moreover, infection with other pathogenic microorganisms, abuse of antibiotics, or reduced immune function can disrupt the inter-constraints between normal gut resident microorganisms, resulting in fungus-associated diarrhea (49, 50). Given the importance of the gut fungal community in dysbacteriosis associated diarrhea, it is crucial to learn more about its composition and structure. In the current study, we systematically investigated the gut fungal alterations in Baer's pochard associated with diarrhea and explored the effect of diarrhea on the gut fungal diversity of the Baer's pochards. Results showed that diarrhea significantly decreased the gut fungal diversity and altered the fungal community composition and structure of Baer's pochard. LEfSe and Metastats analyses between healthy and diarrheal groups found significant differences in fungi from phyla to species that may be closely related to the health status of Baer's pochard. Our study could provide molecular data for further protection of this endangered water bird, and provide a scientific theoretical basis for population recovery and the formulation of effective protection policies and measures.
This study demonstrated that Ascomycota, Basidiomycota, and Mortierellomycota were dominant phyla in the gut of healthy and diarrheal Baer's pochard. The above phyla were also found to be preponderant in the gut mycobiota of ghost moth, silkworm, human, dog, and cattle (51–56). Among lower abundance representatives of fungi, Mortierellomycota was found with a higher abundance in healthy soil (57). Rozellomycota and Chytridiomycota dominate the aquatic fungal community, and many of these could be parasitic within the cells of fungi, plants, or animals, especially waterfowl (58, 59). Glomeromycota and Mucoromycota have proved to be important arbuscular mycorrhizal fungi (60). Genus Basidiobolus (Phylum Zoopagomycota) has a rich range of secondary metabolic genes involved in producing terpene cyclase, surfactin-like, and siderophore (61). The abundance of the fungal phyla mentioned above decreased significantly in the diarrheal group, revealing that diarrhea had a significant effect on the gut mycobiota.
At the genus levels, the composition and relative abundance of the gut fungal flora in the healthy and diarrheal groups also showed significant differences. The relative abundance of Wickerhamomyces, Alternaria, Penicillium, Cystofilobasidium, and Filobasidium in the diarrheal group was significantly higher than in the healthy group. Wickerhamomyces was reported to be significantly increased in ulcerative colitis patients and animals with colitis induced by dextran sulfate sodium (62, 63). Two genera were positively correlated to Wickerhamomyces, i.e., Meyerozyma and Rhinocladiella. Meyerozyma was dramatically increased in type 2 short bowel syndrome rats (64). Remarkably, Meyerozyma caribbica, which was recognized as a gut fungi associated with diarrhea, has been confirmed to cause onychomycosis (65). Many fungi in the genus Rhinocladiella (e.g., Rhinocladiella mackenziei, Rhinocladiella aquaspersa, Rhinocladiella similis) are pathogens of various diseases in humans and animals (66–68). A variety of secondary metabolites produced by small-spored Alternaria species have chronic or acute toxic effects of mutagenic, carcinogenicity, and teratogenicity in humans and animals (69). Fungi in the genus Penicillium, such as Penicillium expansum, Penicillinum citrinum, and Penicillium islandicum, could produce some harmful mycotoxins and carcinogenic compounds, such as verrucosidin, ochratoxin A, citrinine, patulin, citreoviridin, penicillic acid, and other toxic secondary metabolites, and then cause humans and animals various diseases (70–72). Cystofilobasidiaceae family was overrepresented in Crohn's disease (73). Filobasidium magnum and Filobasidium uniguttulatum were pathogens of otomycosis and bovine mastitis (74, 75). The appearance and changes in the above fungi in the gut of Baer's pochard were reported for the first time. LEfSe and Metastats analyses between healthy and diarrheal groups further identified several fungi closely associated with diarrhea. Abundances of Wickerhamomyces, Meyerozyma, and Rhinocladiella with positive correlations were significantly increased in the diarrheal group. LEfSe result showed that the relative abundance of Wickerhamomyces anomalus and Meyerozyma caribbica were significantly higher in the diarrheal group. It has been reported that Wickerhamomyces anomalus is a zoonotic pathogen that can cause severe iatrogenic infections and multiple clinical symptoms in animals (76, 77). Meyerozyma caribbica is a gut fungus associated with diarrhea (65). The significant increase of Rhinocladiella similis in the diarrheal group can cause chromoblastomycosis in humans (66, 78). Based on the analysis of the above results, it was speculated that these three fungi are the main pathogens causing diarrhea in the study subjects.
The microbial interaction (e.g., bacteria–fungi interaction) is important for keeping microbial homeostasis (79). Bacteria or bacterial metabolites can regulate the pathogenic properties and virulence of gut fungi. In contrast, fungi can protect themselves by secreting certain substances, forming biofilms, or building connections with other bacteria (80). Moreover, gut fungi are involved in the construction of the host gut microflora and the development of the immune system and then affect gut function and the physiological functions of other parenteral organs such as the liver, lungs, and brain (81). Fungal dysbiosis may increase allergies and inflammation, especially in the gut (82). Xi et al. (83) reported that the dominant genera in the diarrheal Baer's pochard were Planococcus, Psychrobacter, and Exiguobacterium. The abundances of beneficial bacteria such as Alloprevotella, Veillonella, Parabacteroides, Prevotellaceae, uncultured_bacterium_f_Lachnospiraceae, Carnobacterium, Ruminococcaceae_UCG-002, and Ruminococcaceae_UCG-014 were significantly reduced in the gut of diarrheal Baer's pochard (83). Meyerozyma guilliermondii in the human gut can induce liver synthesis of more prostaglandin E2 to develop chronic alcoholic hepatic steatosis. Oral administration of water-insoluble polysaccharides could significantly increase the abundance of Lachnospiraceae and decrease the abundance of Meyerozyma guilliermondii (84). Psychrobacter and Penicillium were identified to coexist in a clinical microbiology laboratory of a university teaching hospital (85). Both Penicillium spp. (Penicillium olsonii, Penicillium brevicompactum, and Penicillium cavernicola), and Psychrobacter sp. (Psychrobacter glacincola) are capable of producing highly active protease and lipase (86), indicating that the two microorganisms may have synergistic action in the related function. To clarify how complex interactions between bacteria and fungi affect host health states, studies of the community composition of bacteria and fungi in the host gut, the association between bacterial and fungal taxa in the microbiome, and the relationship between bacterial–fungal disorders and host disease are required. Moreover, the effects of some limiting factors in this study, such as gender, age, and sample size, on the results also need to be considered in subsequent studies.
Conclusion
In conclusion, this study analyzed the relationship between diarrhea and the diversity and composition of gut mycobiota in healthy and diarrheal Baer's pochard based on the high-throughput sequencing technique. Results showed significantly decreased gut fungal diversity of diarrheal Baer's pochard, and its composition also changed significantly. Moreover, the present study identified several fungi that may be associated with the occurrence and development of diarrhea in Baer's pochard, laying the theoretical basis for monitoring and formulating prevention and control measures for the infectious diseases of Baer's pochard.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/sra/PRJNA753894.
Ethics Statement
The animal study was reviewed and approved by Ethics Committee of the Shangqiu Normal University.
Author Contributions
LX conceived and designed the experiments and wrote the manuscript. YS, ZL, and JZ contributed to the sample collection and reagent preparation. XQ analyzed the data. JH revised the manuscript. All authors reviewed the manuscript.
Funding
This study was supported by the Screening and Application of Anti-Foodborne Campylobacter Phage (Key Scientific Research Projects of Higher Education Institutions in Henan Province: 21B230008) and the study of PYY3-36 Active Immunization on Nutritional Physiology of Rats (Key Scientific Research Projects of Higher Education Institutions in Henan Province: 15A230028).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2021.756486/full#supplementary-material
References
1. Paul SS, Chatterjee RN, Raju MV, Prakash B, Rao SV, Yadav SP, et al. Gut microbial composition differs extensively among Indian native chicken breeds originated in different geographical locations and a commercial broiler line, but breed-specific, as well as across-breed core microbiomes, are found. Microorganisms. (2021) 9:391. doi: 10.3390/microorganisms9020391
2. Tolnai E, Fauszt P, Fidler G, Pesti-Asboth G, Szilagyi E, Stagel A, et al. Nutraceuticals induced changes in the broiler gastrointestinal tract microbiota. mSystems. (2021) 6:e01124. doi: 10.1128/mSystems.01124-20
3. Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. (2012) 489:220–30. doi: 10.1038/nature11550
4. Chen ZB, Malhi NK. Endothelium-gut communication: IGF-1Rs crosstalk with microbiota. EMBO Rep. (2021) 22:e52896. doi: 10.15252/embr.202152896
5. Julio-Pieper M, López-Aguilera A, Eyzaguirre-Velásquez J, Olavarría-Ramírez L, Ibacache-Quiroga C, Bravo J, et al. Gut susceptibility to viral invasion: contributing roles of diet, microbiota and enteric nervous system to mucosal barrier preservation. Int J Mol Sci. (2021) 22:4734. doi: 10.3390/ijms22094734
6. Gutierrez MW, Arrieta MC. The intestinal mycobiome as a determinant of host immune and metabolic health. Curr Opin Microbiol. (2021) 62:8–13. doi: 10.1016/j.mib.2021.04.004
7. Mahtab N, Zhou L, Zhang F, Wang W. Seasonal variations in the gut fungal communities of Hooded Crane (Grus monacha) at wintering and stopover sites in China. Animals (Basel). (2021) 11:941. doi: 10.3390/ani11040941
8. Mims TS, Abdallah QA, Stewart JD, Watts SP, White CT, Rousselle TV, et al. The gut mycobiome of healthy mice is shaped by the environment and correlates with metabolic outcomes in response to diet. Commun Biol. (2021) 4:281. doi: 10.1038/s42003-021-01820-z
9. Estifan E, Laxina I, Adib S, Suh JS, Baddoura W. A case of Cryptococcal hepatitis in an HIV patient with a negative serum cryptococcal antigen. Cureus. (2019) 11:e6496. doi: 10.7759/cureus.6496
10. Zhao X, Wang L, Wei N, Zhang J, Ma W, Zhao H, et al. Epidemiological and clinical characteristics of healthcare-associated infection in elderly patients in a large Chinese tertiary hospital: a 3-year surveillance study. BMC Infect Dis. (2020) 20:121. doi: 10.1186/s12879-020-4840-3
11. Li K, Mehmood K, Zhang H, Jiang X, Shahzad M, Dong X, et al. Characterization of fungus microbial diversity in healthy and diarrheal yaks in Gannan region of tibet autonomous. Prefecture Acta Trop. (2018) 182:14–26. doi: 10.1016/j.actatropica.2018.02.017
12. Sciavilla P, Strati F, Di-Paola M, Modesto M, Vitali F, Cavalieri D, et al. Gut microbiota profiles and characterization of cultivable fungal isolates in IBS patients. Appl Microbiol Biotechnol. (2021) 105:3277–88. doi: 10.1007/s00253-021-11264-4
13. Hong G, Li Y, Yang M, Li G, Qian W, Xiong H, et al. Gut fungal dysbiosis and altered bacterial-fungal interaction in patients with diarrhea-predominant irritable bowel syndrome: an explorative study. Neurogastroenterol Motil. (2020) 32:e13891. doi: 10.1111/nmo.13891
14. Donnelly KA, Wellehan JFX, Quesenberry K. Gastrointestinal disease associated with non-albicans Candida species in six birds. J Avian Med Surg. (2019) 33:413–8. doi: 10.1647/2018-419
15. Keller B, Kuder H, Visscher C, Siesenop U, Kamphues J. Yeasts in liquid swine diets: identification methods, growth temperatures and gas-formation potential. J Fungi (Basel). (2020) 6:337. doi: 10.3390/jof6040337
16. Cavalheiro M, Pereira D, Formosa-Dague C, Leitão C, Pais P, Ndlovu E, et al. From the first touch to biofilm establishment by the human pathogen Candida glabrata: a genome-wide to nanoscale view. Commun Biol. (2021) 4:886. doi: 10.1038/s42003-021-02412-7
17. Khosravi AR, Shokri H, Ziglari T, Naeini AR, Mousavi Z, Hashemi H. Outbreak of severe disseminated aspergillosis in a flock of ostrich (Struthio camelus). Mycoses. (2008) 51:557–9. doi: 10.1111/j.1439-0507.2008.01504.x
18. Tong X. Baer's pochard duck at risk of extinction. Science. (2020) 369:928. doi: 10.1126/science.abd2087
19. Mishra P, Tulsani NJ, Jakhesara SJ, Dafale NA, Patil NV, Purohit HJ, et al. Exploring the eukaryotic diversity in rumen of Indian camel (Camelus dromedarius) using 18S rRNA amplicon sequencing. Arch Microbiol. (2020) 202:1861–72. doi: 10.1007/s00203-020-01897-w
20. Cao Y, Wang L, Ke S, Villafuerte-Gálvez JA, Pollock NR, Barrett C, et al. Fecal microbiota combined with host immune factors distinguish clostridioides difficile infection from asymptomatic carriage. Gastroenterology. (2021) 160:2328–39. doi: 10.1053/j.gastro.2021.02.069
21. Sun B, Xia Y, Garber PA, Amato KR, Gomez A, Xu X, et al. Captivity is associated with gut mycobiome composition in Tibetan Macaques (Macaca thibetana). Front Microbiol. (2021) 12:665853. doi: 10.3389/fmicb.2021.665853
22. Li Y, Hu X, Yang S, Zhou J, Qi L, Sun X, et al. Comparison between the fecal bacterial microbiota of healthy and diarrheic captive musk deer. Front Microbiol. (2018) 9:300. doi: 10.3389/fmicb.2018.00300
23. Wang B, Deng B, Yong F, Zhou H, Qu C, Zhou Z. Comparison of the fecal microbiomes of healthy and diarrheic captive wild boar. Microb Pathog. (2020) 147:104377. doi: 10.1016/j.micpath.2020.104377
24. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. (2014) 15:2114–20. doi: 10.1093/bioinformatics/btu170
25. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal. (2011) 17:10–2. doi: 10.14806/ej.17.1.200
26. Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. (2011) 27:2957–63. doi: 10.1093/bioinformatics/btr507
27. Edgar RC. Accuracy of taxonomy prediction for 16S rRNA and fungal ITS sequences. PeerJ. (2018) 6:e4652. doi: 10.7717/peerj.4652
28. Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: a versatile open source tool for metagenomics. PeerJ. (2016) 4:e2584. doi: 10.7717/peerj.2584
29. Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. (2019) 37:852–7. doi: 10.1038/s41587-019-0209-9
30. Ko DR, Chung SP, You JS, Cho S, Park Y, Chun B, et al. Effects of paraquat ban on herbicide poisoning-related mortality. Yonsei Med J. (2017) 58:859–66. doi: 10.3349/ymj.2017.58.4.859
31. Bogado Pascottini O, Spricigo JFW, Van Schyndel SJ, Mion B, Rousseau J, Weese JS, et al. Effects of parity, blood progesterone, and non-steroidal anti-inflammatory treatment on the dynamics of the uterine microbiota of healthy postpartum dairy cows. PLoS ONE. (2021) 16:e0233943. doi: 10.1371/journal.pone.0233943
32. Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. (2011) 12:R60. doi: 10.1186/gb-2011-12-6-r60
33. White JR, Nagarajan N, Pop M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput Biol. (2009) 5:e1000352. doi: 10.1371/journal.pcbi.1000352
34. Yang Q, Huang X, Zhao S, Sun W, Yan Z, Wang P, et al. Structure and function of the fecal microbiota in diarrheic neonatal piglets. Front Microbiol. (2017) 8:502. doi: 10.3389/fmicb.2017.00502
35. Cosola C, Rocchetti MT, Gesualdo L. Gut microbiota, the immune system, and cytotoxic T lymphocytes. Methods Mol Biol. (2021) 2325:229–41. doi: 10.1007/978-1-0716-1507-2_16
36. Qi R, Sun J, Qiu X, Zhang Y, Wang J, Wang Q, et al. The intestinal microbiota contributes to the growth and physiological state of muscle tissue in piglets. Sci Rep. (2021) 11:11237. doi: 10.1038/s41598-021-90881-5
37. Sangster W, Hegarty JP, Schieffer KM, Wright JR, Hackman J, Toole DR, et al. Bacterial and fungal mcrobiota changes distinguish C. difficile infection from other forms of diarrhea: results of a prospective inpatient study. Front Microbiol. (2016) 7:789. doi: 10.3389/fmicb.2016.00789
38. Schoeler M, Caesar R. Dietary lipids, gut microbiota and lipid metabolism. Rev Endocr Metab Disord. (2019) 20:461–72. doi: 10.1007/s11154-019-09512-0
39. Jiang C, Li G, Huang P, Liu Z, Zhao B. The gut microbiota and Alzheimer's disease. J Alzheimers Dis. (2017) 58:1–15. doi: 10.3233/JAD-161141
40. Wang ZN, Zhao YZ. Gut microbiota derived metabolites in cardiovascular health and disease. Protein Cell. (2018) 5:416–31. doi: 10.1007/s13238-018-0549-0
41. Altomare A, Di Rosa C, Imperia E, Emerenziani S, Cicala M, Guarino MPL. Diarrhea predominant-irritable bowel syndrome (IBS-D): effects of different nutritional patterns on intestinal dysbiosis and symptoms. Nutrients. (2021) 13:1506. doi: 10.3390/nu13051506
42. Li Y, Xia S, Jiang X, Feng C, Gong S, Ma J, et al. Gut microbiota and diarrhea: an updated review. Front Cell Infect Microbiol. (2021) 11:625210. doi: 10.3389/fcimb.2021.625210
43. Guo J, Ren C, Han X, Huang W, You Y, Zhan J. Role of IgA in the early-life establishment of the gut microbiota and immunity: implications for constructing a healthy start. Gut Microbes. (2021) 13:1–21. doi: 10.1080/19490976.2021.1908101
44. Kim ET, Lee SJ, Kim TY, Lee HG, Atikur RM, Gu BH, et al. Dynamic changes in fecal microbial communities of neonatal dairy calves by aging and diarrhea. Animals (Basel). (2021) 11:1113. doi: 10.3390/ani11041113
45. Lee SC, Billmyre RB, Li A, Carson S, Sykes SM, Huh EY, et al. Analysis of a food-borne fungal pathogen outbreak: virulence and genome of a Mucor circinelloides isolate from yogurt. MBio. (2014) 5:e01390–14. doi: 10.1128/mBio.01390-14
46. de Abreu DPB, Machado CH, Makita MT, Botelho CFM, Oliveira FG, da Veiga CCP, et al. Intestinal lesion in a dog due to Cryptococcus gattii type VGII and review of published cases of Canine gastrointestinal cryptococcosis. Mycopathologia. (2017) 182:597–602. doi: 10.1007/s11046-016-0100-x
47. Yao Y, Long M. The biological detoxification of deoxynivalenol: a review. Food Chem Toxicol. (2020) 145:111649. doi: 10.1016/j.fct.2020.111649
48. Dahiya D, Kichloo A, Singh J, Albosta M, Wani F. Histoplasmosis and inflammatory bowel disease: a case report. World J Gastrointest Endosc. (2021) 13:24–32. doi: 10.4253/wjge.v13.i1.24
49. Stewart DB, Wright JR, Fowler M, McLimans CJ, Tokarev V, Amaniera I, et al. Integrated meta-omics reveals a fungus-associated bacteriome and distinct functional pathways in Clostridioides difficile infection. mSphere. (2019) 4:e00454–19. doi: 10.1128/mSphere.00454-19
50. Wu Y, Wang L, Luo R, Chen H, Nie C, Niu J, et al. Effect of a multispecies probiotic mixture on the growth and incidence of diarrhea, immune function, and fecal microbiota of pre-weaning dairy calves. Front Microbiol. (2021) 12:681014. doi: 10.3389/fmicb.2021.681014
51. Foster ML, Dowd SE, Stephenson C, Steiner JM, Suchodolski JS. Characterization of the fungal microbiome (mycobiome) in fecal samples from dogs. Vet Med Int. (2013) 2013:658373. doi: 10.1155/2013/658373
52. Hamad I, Ranque S, Azhar EI, Yasir M, Jiman-Fatani AA, Tissot-Dupont H, et al. Culturomics and amplicon-based metagenomic approaches for the study of fungal population in human gut microbiota. Sci Rep. (2017) 7:16788. doi: 10.1038/s41598-017-17132-4
53. Chen B, Du K, Sun C, Vimalanathan A, Liang X, Li Y, et al. Gut bacterial and fungal communities of the domesticated silkworm (Bombyx mori) and wild mulberry-feeding relatives. ISME J. (2018) 12:2252–62. doi: 10.1038/s41396-018-0174-1
54. Liu G, Zheng X, Long H, Rao Z, Cao L, Han R. Gut bacterial and fungal communities of the wild and laboratory-reared Thitarodes larvae, host of the Chinese medicinal fungus Ophiocordyceps sinensis on Tibetan plateau. Insects. (2021) 12:327. doi: 10.3390/insects12040327
55. Palumbo F, Squartini A, Barcaccia G, Macolino S, Pornaro C, Pindo M, et al. A multi-kingdom metabarcoding study on cattle grazing Alpine pastures discloses intra-seasonal shifts in plant selection and faecal microbiota. Sci Rep. (2021) 11:889. doi: 10.1038/s41598-020-79474-w
56. Salamon D, Sroka-Oleksiak A, Gurgul A, Arent Z, Szopa M, Bulanda M, et al. Analysis of the gut mycobiome in adult patients with type 1 and type 2 diabetes using next-generation sequencing (NGS) with increased sensitivity-pilot study. Nutrients. (2021) 13:1066. doi: 10.3390/nu13041066
57. Yuan J, Wen T, Zhang H, Zhao ML, Penton CR, Thomashow LS, et al. Predicting disease occurrence with high accuracy based on soil macroecological patterns of Fusarium wilt. ISME J. (2020) 14:2936–50. doi: 10.1038/s41396-020-0720-5
58. Corsaro D, Michel R, Walochnik J, Venditti D, Müller KD, Hauröderet B, et al. Molecular identification of Nucleophaga terricolae sp. nov (Rozellomycota), and new insights on the origin of the Microsporidia. Parasitol Res. (2016) 115:3003–11. doi: 10.1007/s00436-016-5055-9
59. Zhang Z, Deng Q, Cao X, Zhou Y, Song C. Patterns of sediment fungal community dependent on farming practices in aquaculture ponds. Front Microbiol. (2021) 12:542064. doi: 10.3389/fmicb.2021.542064
60. Yang SZ, Gao X, Meng JH, Zhang AY, Zhou YM, Long M, et al. Metagenomic analysis of bacteria, fungi, bacteriophages, and helminths in the gut of giant pandas. Front Microbiol. (2018) 9:1717. doi: 10.3389/fmicb.2018.01717
61. Tabima JF, Trautman IA, Chang Y, Wang Y, Mondo S, Kuo A, et al. Phylogenomic analyses of non-dikarya fungi supports horizontal gene transfer driving diversification of secondary metabolism in the amphibian gastrointestinal symbiont, Basidiobolus. G3 (Bethesda). (2020) 10:3417–33. doi: 10.1534/g3.120.401516
62. Qiu X, Zhang F, Yang X, Wu N, Jiang W, Li X, et al. Changes in the composition of intestinal fungi and their role in mice with dextran sulfate sodium-induced colitis. Sci Rep. (2015) 5:10416. doi: 10.1038/srep10416
63. Xu J, Chen N, Song Y, Wu Z, Wu N, Zhang Y, et al. Alteration of fungal microbiota after 5-ASA treatment in UC Patients. Inflamm Bowel Dis. (2020) 26:380–90. doi: 10.1093/ibd/izz207
64. Hu X, Cheng W, Fan S, Huang Y, Chen X, Jiang Z, et al. Therapeutic potential of an intestinotrophic hormone, glucagon-like peptide 2, for treatment of type 2 short bowel syndrome rats with intestinal bacterial and fungal dysbiosis. BMC Infect Dis. (2021) 21:583. doi: 10.1186/s12879-021-06270-w
65. Montoya AM, Luna-Rodríguez CE, Bonifaz A, Treviño-Rangel RJ, Rojas OC, González GM. Physiological characterization and molecular identification of some rare yeast species causing onychomycosis. J Mycol Med. (2021) 31:101121. doi: 10.1016/j.mycmed.2021.101121
66. Coelho RA, Joffe LS, Alves GM, Figueiredo-Carvalho MHG, Brito-Santos F, Amaral ACF, et al. A screening of the MMV Pathogen Box® reveals new potential antifungal drugs against the etiologic agents of chromoblastomycosis. PLoS ONE. (2020) 15:e0229630. doi: 10.1371/journal.pone.0229630
67. Zain Mushtaq M, Zafar Mahmood SB, Nasir N, Saad Rashid M, Irshad M, Habib K, et al. Cerebral phaeohyphomycosis due to Rhinocladiella mackenziei in an immunocompetent patient: a case report and review of literature. Curr Med Mycol. (2020) 6:65–8. doi: 10.18502/cmm.6.3.4497
68. Heidrich D, Pagani DM, Koehler A, de Oliveira Alves K, Scroferneker ML. Effect of melanin biosynthesis inhibition in the antifungal susceptibility of chromoblastomycosis agents. Antimicrob Agents Chemother. (2021) 10:AAC.00546–00521. doi: 10.1128/AAC.00546-21
69. Crudo F, Aichinger G, Mihajlovic J, Dellafiora L, Varga E, Puntscher H, et al. Gut microbiota and undigested food constituents modify toxin composition and suppress the genotoxicity of a naturally occurring mixture of Alternaria toxins in vitro. Arch Toxicol. (2020) 94:3541–52. doi: 10.1007/s00204-020-02831-1
70. Shiratori N, Kobayashi N, Tulayakul P, Sugiura Y, Takino M, Endo M, et al. Occurrence of Penicillium brocae and Penicillium citreonigrum, which produce a mutagenic metabolite and a mycotoxin citreoviridin, respectively, in selected commercially available rice grains in Thailand. Toxins (Basel). (2017) 9:194. doi: 10.3390/toxins9060194
71. El-Samawaty AEMA, El-Wakil DA, Alamery S, Mahmoud MMH. Potency of plant extracts against Penicillium species isolated from different seeds and fruits in Saudi Arabia. Saudi J Biol Sci. (2021) 28:3294–302. doi: 10.1016/j.sjbs.2021.02.074
72. Valente S, Piombo E, Schroeckh V, Meloni GR, Heinekamp T, Brakhage A, et al. CRISPR-Cas9-based discovery of the Verrucosidin biosynthesis gene cluster in Penicillium polonicum. Front Microbiol. (2021) 12:660871. doi: 10.3389/fmicb.2021.660871
73. Liguori G, Lamas B, Richard ML, Brandi G, da Costa G, Hoffmann TW, et al. Fungal dysbiosis in mucosa-associated microbiota of Crohn's disease Patients. J Crohns Colitis. (2016) 10:296–305. doi: 10.1093/ecco-jcc/jjv209
74. Fadda ME, Pisano MB, Scaccabarozzi L, Mossa V, Deplano M, Moroni P, et al. Use of PCR-restriction fragment length polymorphism analysis for identification of yeast species isolated from bovine intramammary infection. J Dairy Sci. (2013) 96:7692–7. doi: 10.3168/jds.2013-6996
75. Aboutalebian S, Mahmoudi S, Okhovat A, Khodavaisy S, Mirhendi H. Otomycosis due to the rare fungi Talaromyces purpurogenus, Naganishia albida and Filobasidium magnum. Mycopathologia. (2020) 185:569–75. doi: 10.1007/s11046-020-00439-8
76. Yilmaz-Semerci S, Demirel G, Taştekin A. Wickerhamomyces anomalus blood stream infection in a term newborn with pneumonia. Turk J Pediatr. (2017) 59:349–51. doi: 10.24953/turkjped.2017.03.021
77. Meade E, Savage M, Slattery M, Garvey M. Investigation of alternative therapeutic and biocidal options to combat antifungal-resistant zoonotic fungal pathogens isolated from companion animals. Infect Dis Rep. (2021) 13:348–66. doi: 10.3390/idr13020034
78. de Andrade TS, de Almeida AMZ, Basano SA, Takagi EH, Szeszs M, Melhem MSC, et al. Chromoblastomycosis in the Amazon region, Brazil, caused by Fonsecaea pedrosoi, Fonsecaea nubica, and Rhinocladiella similis: clinicopathology, susceptibility, and molecular identification. Med Mycol. (2020) 58:172–80. doi: 10.1093/mmy/myz034
79. Ley RE, Lozupone CA, Hamady M, Knight R, Gordon JI. Worlds within worlds: evolution of the vertebrate gut microbiota. Nat Rev Microbiol. (2008) 6:776–88. doi: 10.1038/nrmicro1978
80. Seelbinder B, Chen J, Brunke S, Vazquez-Uribe R, Santhaman R, Meyer AC, et al. Antibiotics create a shift from mutualism to competition in human gut communities with a longer-lasting impact on fungi than bacteria. Microbiome. (2020) 8:133. doi: 10.1186/s40168-020-00899-6
81. Bernardes EVT, Pettersen VK, Gutierrez MW, Lapointe IL, Jendzjowsky NG, Cavin JB, et al. Intestinal fungi are causally implicated in microbiome assembly and immune development in mice. Nat Commun. (2020) 11:2577. doi: 10.1038/s41467-020-16431-1
82. Iliev ID, Funari VA, Taylor KD, Nguyen Q, Reyes CN, Strom SP, et al. Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science. (2012) 336:1314–7. doi: 10.1126/science.1221789
83. Xi L, Song Y, Han J, Qin X. Microbiome analysis reveals the significant changes in gut microbiota of diarrheic Baer's Pochards (Aythya baeri). Microb Pathog. (2021) 157:105015. doi: 10.1016/j.micpath.2021.105015
84. Sun SS, Wang K, Sun L, Cheng BS, Qiao SS, Xu X, et al. Therapeutic manipulation of gut microbiota by polysaccharides of Wolfiporia cocos reveals the contribution of the gut fungi-induced PGE2 to alcoholic hepatic steatosis. Gut Microbes. (2020) 12:1830693. doi: 10.1080/19490976.2020.1830693
85. Nagano Y, Walker J, Loughrey A, Millar C, Goldsmith C, Rooney P, et al. Identification of airborne bacterial and fungal species in the clinical microbiology laboratory of a university teaching hospital employing ribosomal DNA (rDNA) PCR and gene sequencing techniques. Int J Environ Health Res. (2009) 19:187–99. doi: 10.1080/09603120802474229
Keywords: Baer's pochard, gut microbiota, ITS, composition, diarrhea
Citation: Xi L, Qin X, Song Y, Han J, Li Z and Zhang J (2021) Gut Microbial Alterations in Diarrheal Baer's Pochards (Aythya baeri). Front. Vet. Sci. 8:756486. doi: 10.3389/fvets.2021.756486
Received: 10 August 2021; Accepted: 03 September 2021;
Published: 14 October 2021.
Edited by:
Mujeeb Ur Rehman, Livestock and Dairy Development Department, PakistanReviewed by:
Huan Li, Lanzhou University, ChinaMengyue Guo, Chinese Academy of Medical Sciences and Peking Union Medical College, China
Hui Zhang, South China Agricultural University, China
Copyright © 2021 Xi, Qin, Song, Han, Li and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Xi, eGlsaV8wODA4QDEyNi5jb20=; Jincheng Han, aGppbmNoZW5nQDEyNi5jb20=