 Diana I. Ayala*
Diana I. Ayala* Daniel S. GrumNicholas P. EvansKay N. RussoEmily A. KimminauBenjamin R. TribleManohar M. LahotiCurtis L. NovakTheodore P. Karnezos
Daniel S. GrumNicholas P. EvansKay N. RussoEmily A. KimminauBenjamin R. TribleManohar M. LahotiCurtis L. NovakTheodore P. Karnezos- Purina Animal Nutrition Center, Land O' Lakes, Gray Summit, MO, United States
Focal Ulcerative Dermatitis (FUDS) is an emerging dermatological disease that affects cage-free laying flocks, it is characterized by the development of a lesion on the dorsum of the birds; FUDS is sporadic in nature and can result in a drop in egg production and up to 50% of cumulative mortality. A total of two cage-free flocks (flock 1: no history of FUDS; flock 2: birds affected with FUDS) from a commercial laying hen operation in the mid-west U.S. were sampled in this study. The microbial composition of skin, cloacal, cecal, and ileal samples from each bird was characterized through next generation sequencing (NGS). Results identified Staphylococcus aureus and Staphylococcus agnetis as the potential causative agents of FUDS, being the most predominant in FUDS positive birds. These results were confirmed by plating, with both staphylococci as the only pathogens isolated from lesions of FUDS positive birds. A total of 68 confirmed Staphylococcus isolates from skin and environmental samples were further analyzed by whole genome sequencing (WGS) for the presence of antimicrobial resistance (AMR) genes and virulence factors that could have contributed to the development of FUDS. Forty-four-point one-two percent of the isolates had between one and four acquired AMR genes encoding for macrolides, lincosamides, spectrogramines, and beta-lactams resistance. Six classes of virulence factors associated with adherence, enzyme, immune evasion, secretion system, toxin, and iron uptake were identified. The antimicrobial effect of 4 proprietary Bacillus Direct Fed Microbial (DFM) combinations was evaluated against the Staphylococcus aureus and Staphylococcus agnetis isolates, by agar well-diffusion (AWD) assay and competitive exclusion (CE) on broth culture. Through this antimicrobial screening, a particular two-strain combination of Bacillus pumilus was identified as the most effective inhibitor of both staphylococci. A customized Bacillus pumilus product is being used at different farms with history of FUDS resulting in the successful inhibition of both Staphylococcus aureus and Staphylococcus agnetis, decreasing FUDS mortalities, and improving harvestable eggs.
Introduction
In the U.S. consumer and retailer demand on how laying hens are managed has shifted over the past decade, with increased interest in the use of alternative production systems over conventional systems (1). In 2019, 81.6% of egg-laying birds in the U.S. were housed in conventional cages while 13.3% were cage-free (2). On December 1, 2020, the total U.S. cage-free flock had 80.1 million hens, a 9.3 million-head increase from the same date in 2019 (3). These numbers are estimated to increase with over half of the U.S hen housing expected to become cage-free by 2025 (4). Due to the nature of their production design, cage-free housing systems have reportedly higher incidence of bacterial infections (5, 6). Focal Ulcerative Dermatitis Syndrome (FUDS) is one of the emerging diseases cage-free laying flocks are experiencing. FUDS was first described in 2009 in a laying hen operation in Midwest US, the syndrome is characterized by the development of lesions on the dorsum of the birds, just cranial to the uropygial gland. The onset of the condition typically occurs between 24 and 50 weeks of age, lacks seasonality, and is mostly observed in houses with slat floors (7). Outbreaks are relatively sporadic in nature; however, when present can result in up to 50% cumulative mortality.
Typical FUDS lesions are reminiscent of swine exudative epidermitis, a disease caused by Staphylococcus hyicus. Staphylococci are ubiquitous in poultry farm environments and are part of the normal skin and mucous membrane microbiota of birds; however, when the integrity of the skin or other mucosal membranes is compromised, some Staphylococcus species can become opportunistic pathogens causing localized or systemic infections (8, 9). S. hyicus strains have been implicated as secondary causative agents of pre-existing dermatoses (10), they produce exfoliative toxins that induce dermatitis resulting in thickening of the skin (11). Although little research has been done on the etiology of FUDS, a correlation between disease and S. hyicus abundance has been observed (7). The production losses due to Staphylococcus infections in poultry (egg laying or meat birds) have been associated with lameness, drop in egg production (9, 12), increased mortality, and condemnation of carcasses at slaughter facilities (8).
Antibiotics have been widely used in the poultry industry to promote growth (13) and control pathogens including Staphylococcus spp., thus maximizing production. However, the increased use of antibiotics in livestock production has led to a rapid spread of antimicrobial resistance (AMR) among bacterial isolates and growing public concern about AMR effects in human health (14), resulting in a slow decline in antibiotic use in animal production (13). These trends, together with increased demand by retailers and consumers for antibiotic-free poultry production, has highlighted the importance of developing alternative technologies to reduce/inhibit bacterial pathogens without negative impacts on animal performance.
Direct fed microbials (DFM) have emerged as a viable alternative to the use of antibiotics in poultry farming (15, 16). They are live microorganisms that when fed in adequate amounts confer benefits to the host (17); among the benefits are improved immunity, enhanced growth, and an overall increase in laying performance (14). Multiple bacterial genera have been used in poultry production, with Lactobacillus, Bacillus, and Bifidobacterium being the most common. Each DFM strain confers a specific action impacting the host and microbial ecology of the gastro-intestinal track. Their mode of action includes the production of antimicrobial compounds (i.e., bacteriocins, organic acids), competitive exclusion mechanisms, and the production of beneficial fermentation products such as volatile fatty acids (18). This study aimed to characterize the cecal, ileal, cloacal, and skin microbiota of FUDS positive and FUDS negative laying hens through next generation sequencing (NGS). The primary goals were to (1) identify the pathogens causing FUDS, (2) isolate the pathogens, (3) identify association among isolates, and 4) develop a DFM combination inhibitory against the FUDS causative agents.
Methodology
Flock descriptions and lesion scoring
A commercial laying hen operation in the mid-west United States with a history of FUDS was sampled for this study. Within the operation, two cage-free flocks were selected: (1) flock 1; no history of the disease (Control) and (2) flock 2; birds affected by FUDS. Both flocks originated from the same hatchery, housed in a similar cage-free production system, and were sampled at 73 weeks of age. A total of 20 birds from the control flock and 39 birds from the FUDS affected flock [18 birds showing no symptoms of FUDS (FUDS−) and 21 birds showing visible symptoms of FUDS (FUDS +)] were sampled for this study.
Prior to sample collection, birds from the FUDS affected flock were scored for lesions according to the size of the lesion located cranial to the uropygial gland as illustrated in Figure 1. Lesions were assessed for size and involvement of the underlying tissue with scores ranging from 0 to 3: 0, no lesion; 1, lesion size < 2 cm; 2, lesion size 2–6 cm; 3, lesions >6 cm.
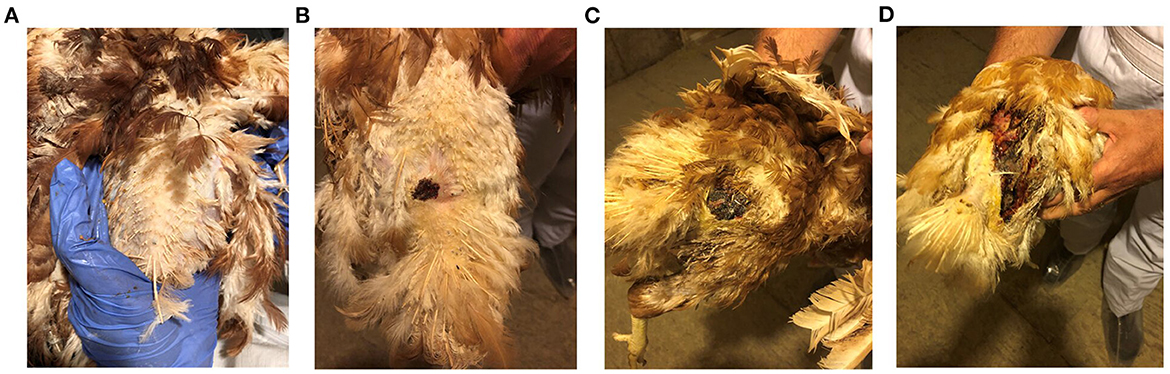
Figure 1. FUDS lesion scoring. Scores were assessed based on size and involvement in the underlying tissue. Scores ranged from 0 to 3: (A) 0, no lesion; (B) 1, lesion size < 2 cm; (C) 2, lesion size 2–6 cm; (D) 3, lesions > 6 cm.
Sample collection for microbiome analysis
A total of four sample types were collected from each bird in this study: skin, cloaca, cecum, and ileum. Samples were collected aseptically to avoid cross contamination. Skin samples were taken using a flocked swab (Puritan, Guilford, ME, USA) that was rotated and swabbed on a 2 cm2 area of affected skin cranial to the uropygial gland for FUDS + birds and the same skin area for Control and FUDS–birds. Cloacal samples were collected by inserting a flocked swab ~1 cm into the cloaca, the swab was rotated to allow the collection of enough cloacal material. For cecum and ileum sample collection, the thoraco-abdominal cavity was opened to reveal the gastrointestinal tract. A 1 cm opening was made in the middle of the ileum and a flocked swab was then rotated inside the lumen. The process was repeated with a new swab for the collection of cecal contents. Following sample collection, all swabs were placed into sterile microcentrifuge tubes containing 1 mL of DNA/RNA shield (Zymo Research, Irvine CA), and immediately placed on ice after collection, and shipped overnight to the Emerging Technology Center at Purina Animal Nutrition Center (ETC: PANC) for further processing.
16S microbiome DNA extraction and library preparation
Total microbial community DNA was extracted from all sample types and analyzed by using the Quick-DNA Fecal/Soil Microbe miniprep (Zymo Research Corp, Irvine, CA, USA) following manufacturer's recommendations. Pure DNA was quantified using a Qubit 2.0 fluorometer. DNA samples at a 5 ng/μl concentration were used to prepare libraries using the Illumina 16S-metagenomics library preparation protocol. The V3–V4 hyper variable region of bacterial 16S rRNA gene was amplified by using the primers 341F (5′-CCTACGGGNGGCWGCAG-3′) and 805R (5′-GACTACHVGGGTATCTAATCC-3′) containing Illumina adaptors as illustrated in Klindworth et al. (19). The PCR amplicons were checked in agarose gels and purified using AMPure XP beads (Beckman Coulter Inc) as per manufacturer's recommendations. Purified amplicons were then indexed by using the Nextera UD index set (Illumina, San Diego, CA, USA). Libraries were quantified in triplicate using a Qubit 2.0 fluorometer with a dsDNA HS Assay kit (Invitrogen, Carlsbad, CA, USA), pooled at equal concentrations (4 nM) to generate equivalent number of raw reads, and diluted to a final concentration of 6 pM. Amplicon libraries were spiked with 5% PhiX control (Illumina, San Diego, CA, USA) according to manufacturer's recommendations. Samples were sequenced using a MiSeq Reagent kit v3 (600 cycle) on an Illumina MiSeq platform (Illumina, San Diego, CA, USA).
16S data analysis and bioinformatics
Raw sequence data were analyzed using quantitative insights into microbial ecology (QIIME2) pipeline (20). Using the MiSeq reporter software (Illumina, San Diego, CA, USA), amplification primers and Illumina adapters were trimmed, samples were demultiplexed, and fastq.gz files generated. Raw sequence data were filtered and processed using the DADA2 pipeline on QIIME2. The 16S rRNA gene sequences were clustered into amplicon sequence variants (ASVs) and taxonomy was assigned based on the comparison against the SILVA database (21). Species richness and diversity indices were calculated using QIIME. Beta diversity was determined using the weighted UniFrac distance with a permutational multivariate analysis of variance (PERMANOVA) to determine differences between microbial communities based on phylogenic relatedness of whole communities.
Statistical analysis
The linear discriminant analysis (LDA) effect size (LEfSe) (22) was used in this study to identify statistically significant taxa to characterize the differences among FUDS+, FUDS-, and Control groups. The non-parametric factorial Kruskal-Wallis sum rank test was used by the LEfSe method to identify operational taxonomic unit (OTU) abundance differences between two groups. Pairwise tests among groups were then performed by using the Wilcoxon rank-sum test, followed by the LDA on abundances to estimate the effect size of each differentially significant abundant OTU. OTUs were considered significantly different when differences had p < 0.05 and an LDA score (log10) > 3.5. A second LEfSe analysis was performed on Staphylococcus and Lactobacillus to identify specific species associated with the health status of the birds, OTUs were considered significantly different when p < 0.05 and an LDA score (log10) was > 3.5.
Sample collection for bacterial isolation
A total of 10 FUDS+, 10 FUDS-, and 10 Control birds were selected based on the presence/absence of skin lesions for sample collection. Feathers were removed around the preen gland and a 5 cm2 surface of the skin was swabbed using pre-moistened sponge swabs (Whirl-Pak, Madison, WI). In addition, a total of 3 environmental sample types, per flock, including from on top of the nest boxes, perches, and scratch areas were collected. A 5 cm2 surface was swabbed at an even interval and distributed throughout the houses. Samples were shipped overnight to the ETC: PANC for culture and bacterial isolation.
Bacterial isolation and growth conditions
Upon arrival at ETC: PANC, samples were homogenized in a stomacher (Stomacher® 400, Seward) for 3 min at 230 rpm. A 1 mL aliquot was collected and added to 9 mL of Baird Staphylococcus enrichment broth base (Sigma-Aldrich, St. Louis, MO) supplemented with 0.1 mL Potassium Tellurite solution (Sigma-Aldrich, St. Louis, MO), tubes were incubated aerobically at 37°C for 24 h. After incubation, samples were serially diluted in 9 mL of buffered peptone water (BPW, Sigma-Aldrich, St. Louis, MO), spread plated onto CHROMagar Staphylococcus agar plates (CHROMagar, Paris, France), and incubated aerobically at 37°C for 24 h. Based on colony morphology, up to 4 representative colonies were selected from each plate, streaked onto tryptic soy agar plates (TSA, Sigma-Aldrich, St. Louis, MO), and incubated at 37°C for 24 h. A single well-isolated colony was selected from each TSA plate, grown in 9 mL of tryptic soy broth (TSB, Sigma-Aldrich, St. Louis, MO) and incubated at 37°C for 24 h (overnight culture). A 1.5 mL aliquot of the overnight culture was used for gDNA extraction and a 500 μl aliquot was added to 50% glycerol and stored at −80°C for further testing.
16S PCR amplification for bacterial speciation
A 1.5 mL aliquot of the overnight culture was used for gDNA extraction using the Invitrogen PureLink DNA extraction kit (Thermo Fisher Scientific, Waltham, MA, USA). DNA samples were quantified using a Qubit 2.0 fluorometer (Life Technologies, CA, United States) and diluted to a 20 ng/μl for PCR amplification.
A 1,483 base pair (bp) region of the 16S ribosomal subunit was PCR amplified using forward primer 5'-AGAGTTTGATCCTGGCTCAG and reverse primer 5'-GGTTACCTTGTTACGACTT. The PCR amplification was carried out using ZymoTaq PreMix according to the manufacturer's directions (Zymo Research, Irvine CA). Following the reaction, PCR products were purified using a DNA Clean and Concentrator kit (Zymo Research, Irvine CA), according to the manufacturer's instructions. Purified products were then sent to the Core Sequencing Facility of the University of Missouri for sanger sequencing. The taxonomic ID for each raw sequence data was assigned based on the results of a National Center for Biotechnology Information (NCBI) basic local alignment search tool (BLAST) inquiry (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
Whole-genome sequencing
A total of 68 PCR confirmed Staphylococcus isolates from the environmental and skin samples of FUDS+ and FUDS- birds were selected for further genotypic analysis. Staphylococcus isolates were grown overnight in 9 mL of TSB and incubated at 37°C for 24 h. Total genomic DNA was isolated using the Invitrogen Purelink DNA Extraction kit. Pure gDNA was quantified using a Qubit 2.0 fluorometer and used for library preparation with the Nextera XT v2.0 kit (San Diego, CA, United States) as per manufacturer's recommendations. DNA libraries were paired-end sequenced using the 2 × 300 bp v3 sequencing kit on an Illumina MiSeq platform. Raw reads were preprocessed and filtered using Trimmomatic version 0.36 (23), followed by de novo assembly using SPAdes version 11 (24).
WGS bioinformatic analysis
Assembled genomes were used for the identification of virulence factors and potential antimicrobial resistance (AMR) genes by comparing them to the Virulence Finder and ResFinder databases, respectively (25, 26). Alignments were identified as positive when percentage of identity was 95% or higher. A phylogenetic tree was generated using the concatenated alignment in RAxML (Randomized Axelerated Maximum Likelihood) (27).
DFMs selection and growth conditions
A total of 4 proprietary Bacillus DFM combinations from a set of >500 novel DFMs were selected for this study based on their antagonistic effect against a set of bacterial pathogens including Salmonella, Staphylococcus aureus, E. coli, among others. Samples of 1 g of each lyophilized DFM combination were grown individually in 9 mL of Luria-Bertani (LB, ThermoFisher Scientific, Waltham, MA) broth at 37°C for 24 h. Overnight cultures were used for agar-well diffusion assay, as described below, and were serially diluted in 9 mL of BPW and plated aerobically onto LB agar plates (ThermoFisher Scientific, Waltham, MA) at 37°C for 24 h for enumeration.
Pathogen inhibition screening by agar-well diffusion assay
The agar well-diffusion method, described by Vinderola and others (28) with slight modifications (29), was used to determine the antimicrobial activity of a set of Bacillus DFM combinations from a stock culture collection, maintained at ETC: PANC, against Staphylococcus isolates. S. agnetis and S. aureus isolates recovered from the skin of FUDS+ birds were incubated overnight in TSB for 18–24 h at 37°C and then diluted to 106 CFU/mL. A 100 μl of the last dilution was swabbed onto Nutrient agar (Sigma-Aldrich, St. Louis, MO) plates to create a lawn to a final concentration of 105 CFU/mL. Plates were dried for 5 min in a biosafety cabinet, then 6-mm wide wells were made in the agar and each well-duplicate was filled with 100 μl aliquots of one of 4 proprietary Bacillus DFM combinations (108 CFU/mL) and a single well with 100 μl of uninoculated TSB broth (control). Plates were incubated at 4°C for 2 h to allow suspensions to diffuse in the agar followed by a 24 h incubation at 37°C. Antimicrobial activity was assessed by measuring the clear zone of inhibition around each well. DFM combinations were ranked for their overall antimicrobial activity [described by Ayala and others (29)] by calculating the sum of the DFM individual inhibition (mm) scores across all Staphylococcus isolates tested.
Competitive exclusion broth culture assay
Staphylococcus and Bacillus DFM cultures were grown overnight as described above, co-inoculated at 105 and 106 CFU/mL respectively in Nutrient broth (ThermoFisher Scientific, Waltham, MA) and incubated with agitation (130 rpm) at 37°C for 24 h (29). After incubation, co-cultures were serially diluted and plated onto CHROMagar Staphylococcus agar plates and incubated at 37°C for 24 h for Staphylococcus enumeration. Antimicrobial activity was assessed by the reduction of Staphylococcus (log10 CFU/mL) with respect to control samples (Staphylococcus cultures without DFM) after 24 h of co-inoculation. DFM cultures were ranked for their antagonistic effect by summing the overall reductions (log10 CFU/mL) across all Staphylococcus isolates tested.
Results
Lesion score
Birds from control and FUDS–groups had a lesion score of 0 while birds from the FUDS+ group had lesion scores ranging from 1 to 3 (1 = 14 birds, 2 = 5 birds, and 3 = 2 birds), as shown in Figures 1A–D.
16S PCR amplification for bacterial speciation
The taxonomic identification of Staphylococcus isolates from environmental samples collected from the Control; and FUDS affected flocks is shown in Table 1. A total of 11 Staphylococcus species were recovered from both environments, Staphylococcus simulans was the most commonly isolated, found in all environmental samples from both flocks (Table 1).
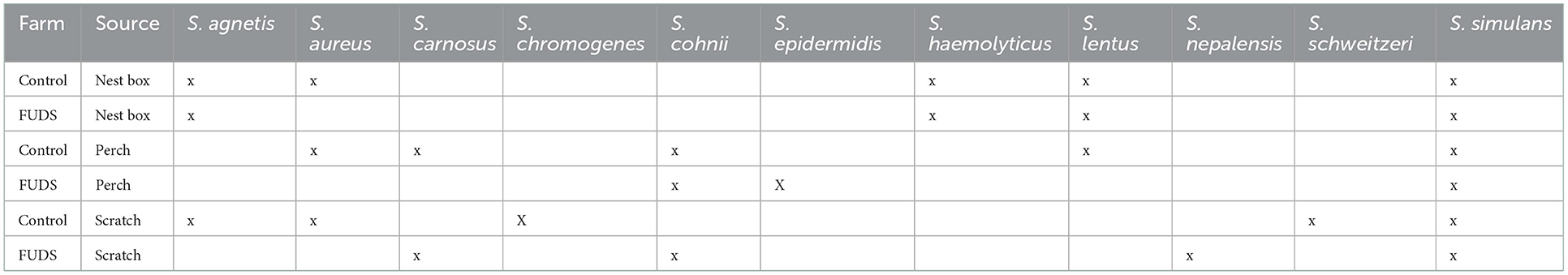
Table 1. Taxonomic identification of Staphylococcus isolates from environmental samples collected from the Control and FUDS affected flocks.
The taxonomic identification of Staphylococcus isolates from skin swabs collected from the Control and FUDS affected flocks is shown in Table 2. A total of 10 Staphylococcus species were identified, with skin swabs from non-affected birds showing more Staphylococcus species variation. Staphylococcus aureus and Staphylococcus agnetis were the only two Staphylococcus species identified in the FUDS + skin swabs (Table 2).

Table 2. Taxonomic identification of Staphylococcus isolates from skin swabs collected from the FUDS–and FUDS + birds from control and FUDS affected flocks.
16S metagenomics analysis
The complete 16S rRNA data set analyzed in this study is available in the NCBI Sequence Read Archive repository with accession BioProject ID PRJNA884648.
Microbial composition by sample type
The composition of cecum, ileum, cloaca, and skin bacterial communities were explored through principal coordinate analysis (PCoA) plots using Bray-Curtis index. As shown in the PCoA plot (Figure 2), samples tended to cluster by sample type and sub-clustered by flock of origin with a uniformly divergent microbial composition observed in control and FUDS affected. Within FUDS affected flock, samples clustered by disease state (Figure 2). For the FUDS + birds, the skin samples showed a large deviation from the control and of FUDS–birds, however the ileum samples were not strongly clustered by flock and/or disease state. The weighted UniFrac distance metrics were used to determine the variation among microbial communities from cecum, ileum, cloaca, and skin. In accordance with the PCoA plots, the microbiota from each sample type was found compositionally distinct from the microbiota in other sample types (p < 0.001).
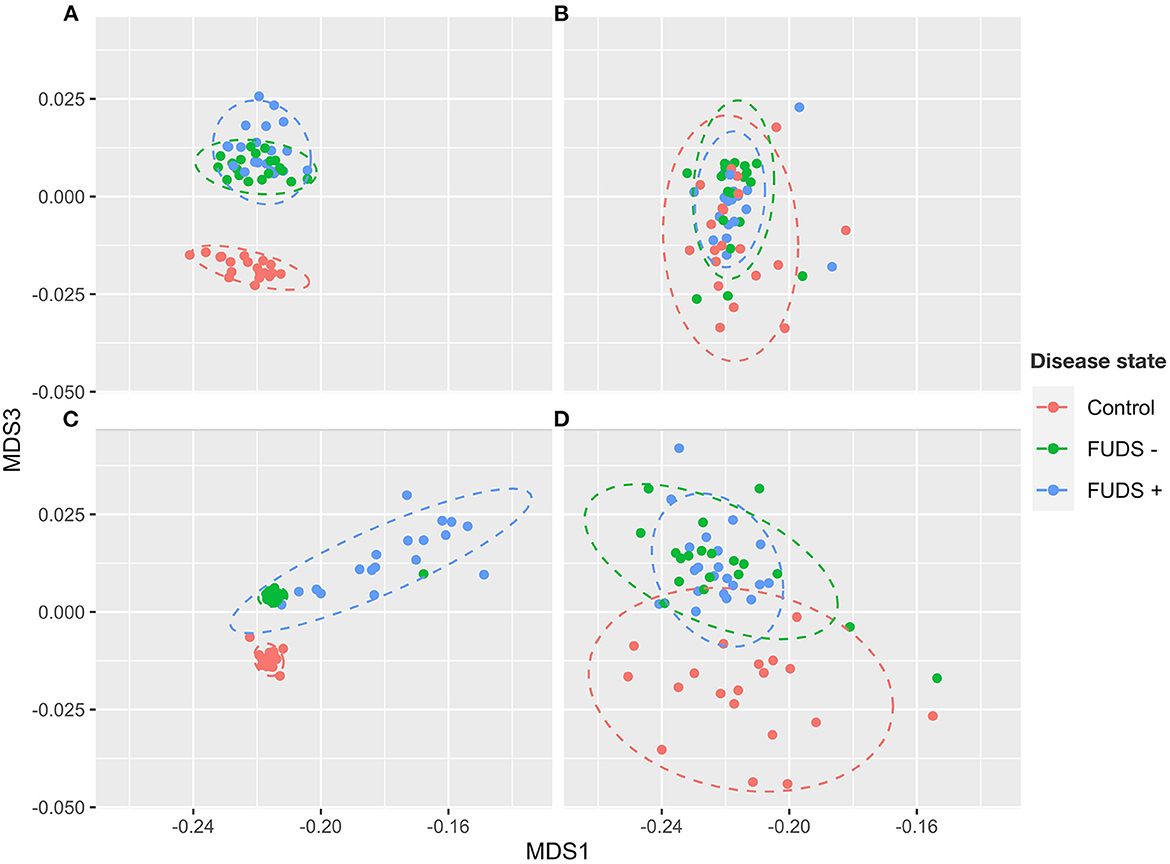
Figure 2. Principal component analysis (PCoA) for Control, FUDS –, and FUDS + flocks by sample type. (A) Cecum; (B) ileum; (C) skin; and (D) cloaca. The plot was generated using the Bray-Curtis metrics.
Relative abundance by phyla
At the phylum level for all flocks, the six most predominant phyla by sample type are shown in Figure 3. Ileum samples were dominated by Firmicutes, Proteobacteria, Bacteroidetes, Actinobacteria, Cyanobacteria, and Fusobacteria, with these phyla representing 99% of the total abundance, and Firmicutes alone comprising 97% of total bacteria present. Cecum samples were primarily composed by Firmicutes, Bacteroidetes, Proteobacteria, Fusobacteria, Cyanobacteria, and Verrucomicrobia, with these phyla representing 96% of all bacteria present. Cloacal samples were dominated by Firmicutes, Fusobacteria, Proteobacteria, Bacteroidetes, Actinobacteria, and Synergistetes, with these six phyla representing 98% of the total relative abundance. Skin samples were primarily composed by Firmicutes, Bacteroidetes, Proteobacteria, Actinobacteria, Fusobacteria, and Cyanobacteria; these phyla comprised 98% of the total bacterial community present.
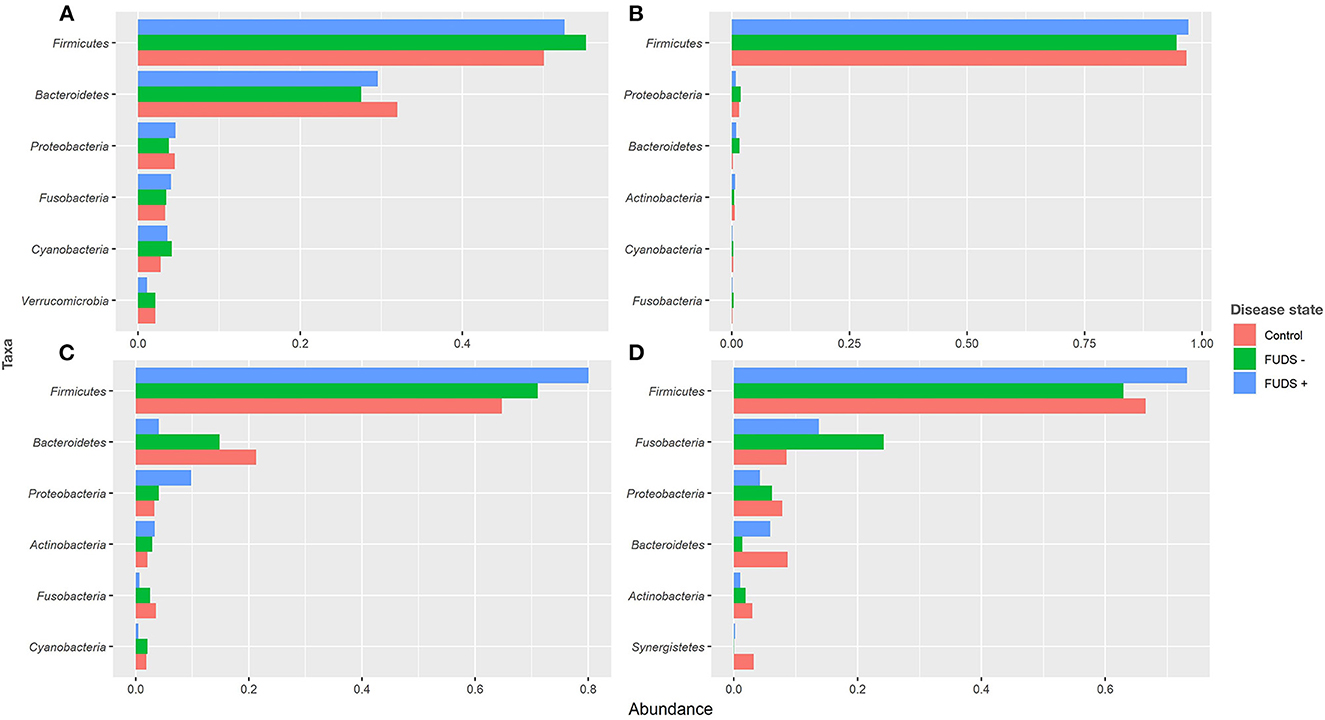
Figure 3. Relative abundance plot of top six taxa at phylum level for Control, FUDS –, and FUDS + flocks by sample type. (A) Cecum; (B) ileum; (C) skin; and (D) cloaca.
Phyla relative abundance comparisons among flocks
Firmicutes and Bacteroidetes were the two most abundant phyla in cecum across flocks. Firmicutes was found at 52.62, 55.27, 50.12% in FUDS+, FUDS-, and control flocks, respectively while Bacteroidetes was found at 29.59, 27.52, and 32.97 for FUDS+, FUDS-, and control flocks, respectively.
Ileum samples were dominated by Firmicutes with similar relative abundances across flocks and disease state. It was found at 97.10, 94.56, and 96.67% for FUDS+, FUDS- and control flocks, respectively.
Cloacal samples had notable abundance differences across samples. Main differences were observed for the top four phyla: Firmicutes, Fusobacteria, Proteobacteria, and Bacteroidetes. FUDS+ birds had the highest relative abundance of Firmicutes representing 73.18, 62.92, and 66.52% for FUDS +, FUDS-, and control birds, respectively. Fusobacteria was found at 13.73, 24.23, and 8.52% across FUDS+, FUDS- and control birds, respectively. Proteobacteria displayed a mild trend with FUDS+ birds having the lowest relative abundance when compared to asymptomatic and healthy birds. Proteobacteria were present at 4.21, 6.12, and 7.86% in FUDS+, FUDS- and control birds, respectively.
Skin samples microbiota was dominated by Firmicutes, Bacteroidetes, and Proteobacteria. Across disease state, the skin microbiome from sick birds had higher relative abundances of Firmicutes and Proteobacteria, and lower relative abundances of Bacteroidetes when compared with healthy birds. Firmicutes relative abundances were at 80.10, 71.11, and 64.79% for FUDS+, FUDS- and control birds, respectively. Bacteroidetes was found at 4.11, 14.84, and 21.31% for FUDS+, FUDS- and control birds, respectively. The relative abundance of Proteobacteria was found at 9.87, 4.08, and 3.33% for FUDS+, FUDS- and control birds, respectively.
Relative abundance by genera
At the genus level for all flocks, the six most predominant genera by sample type are shown in Figure 4. Ileum samples were primarily composed by Lactobacillus, Rombustia, Turnibacter, Clostridium, Tyzzerella, and Streptococcus, with these genera representing 91% of total bacterial population. Cecum samples were dominated by Bacteroides, Lactobacillus, Fusobacterium, Ruminococcaceae UCG.005, Alistipes, and Lachnospiraceae, with these genera representing 36% of total bacteria. The top six genera from cloacal samples were Fusobacterium, Turnibacter, Lactobacillus, Rombustia, Clostridium, and Bacteroides, with these genera representing 63% of the total bacteria present. Skin samples were mainly composed by Staphylococcus, Lactobacillus, Bacteroides, Turnibacter, Rombustia, and Ruminococcus, with these genera representing 48% of the total skin microbiota.
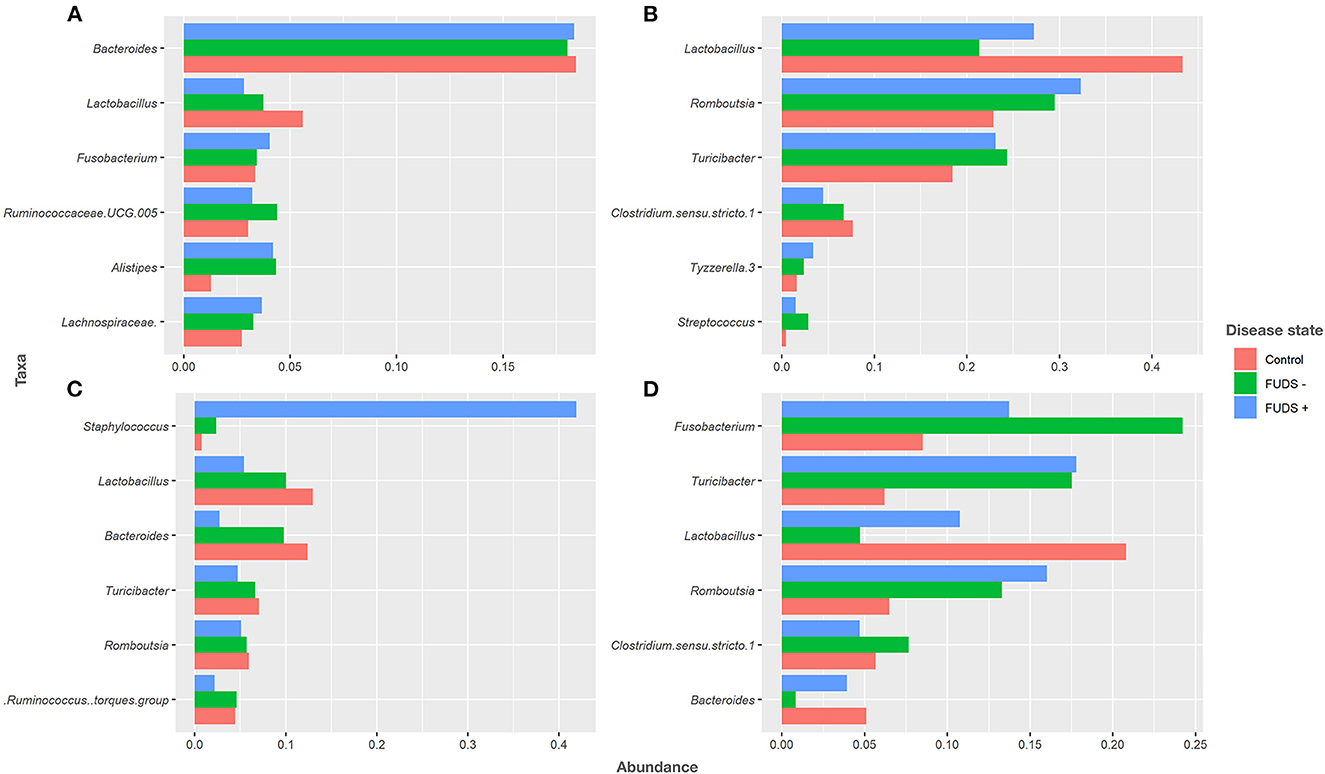
Figure 4. Relative abundance plot of top six taxa at genus level for Control, FUDS –, and FUDS + flocks by sample type. (A) Cecum; (B) ileum; (C) skin; and (D) cloaca.
Skin samples taxa relative abundance
Looking specifically at the lesion, Staphylococcus abundance was significantly greater (p < 0.001) in the skin of FUDS + birds compared to FUDS–birds, highlighting a potential association with the development of FUDS. The relative abundance by genus from skin samples for control and FUDS flocks is shown in Figure 5. Staphylococcus spp. abundance increased as FUDS lesion progressed. In samples from the control flock (lesion score = 0), Staphylococcus spp. represented 0.7% of total relative abundance whereas, in the samples from the FUDS+ flock, Staphylococcus spp. concentration was 2.1, 41.6, 45.6, and 60.8%, at lesion scores 0, 1, 2, and 3, respectively. Staphylococcus diverged into two similarly abundant OTU groups when accessed at the species level. However, Silva taxonomic identification was able to speciate Staphylococcus agnetis for one OTU group, whereas the second group was not identified below the genus level. Further analysis via NCBI blast of the sequences associated with the second Staphylococcus group identified them as Staphylococcus aureus. The predominant Staphylococcus aureus OTU was present at 0.71, 2.06, 21.65, and 37.79% for lesion scores 0, 1, 2, and 3, respectively. Staphylococcus agnetis was present at 0.04, 19.97, 18.56, and 23.13% for lesion scores 0, 1, 2, and 3 respectively.
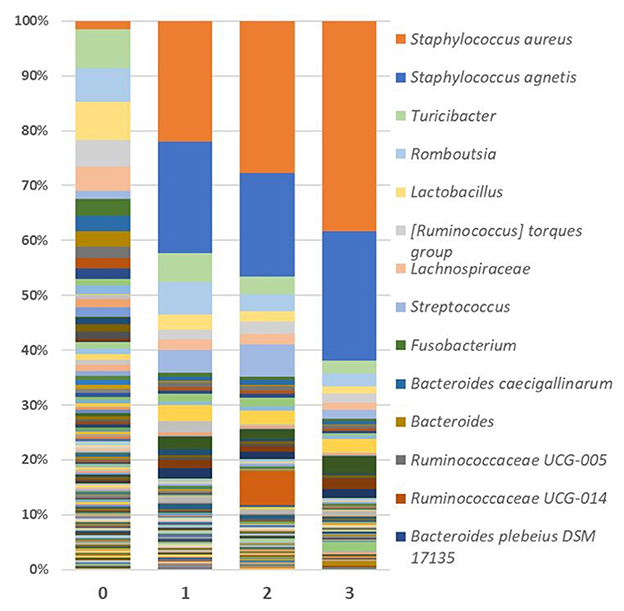
Figure 5. Relative abundance as average percentage of bacterial taxa populations for skin samples grouped by lesion score. The predominant Staphylococcus aureus OTU was present at 0.709, 2.063, 21.647, and 37.787% for lesion scores 0, 1, 2, and 3, respectively. Staphylococcus agnetis was present at 0.043, 19.969, 18.564, and 23.125% for lesion scores 0, 1, 2, and 3, respectively.
Genera relative abundance comparisons among flocks
Bacteroides and Lactobacillus were the two most abundant genus in cecum samples across flocks. Lactobacillus relative abundance was found at increased levels in control (5.62%), followed by FUDS- (3.77%), and FUDS + (2.82%) birds, respectively. Bacteroides relative abundances were similar among flocks, found at 18.35, 18.03, and 18.04% in FUDS+, FUDS- and control birds, respectively.
Lactobacillus was the most abundant taxon in the ileum samples with increased abundance in the control samples. Lactobacillus was found at 27.24, 21.35, and 43.38% in FUDS+, FUDS-, and control birds, respectively. Romboutsia trended downward following disease state, being the highest in FUDS+ birds at 32.36%, followed by 29.474% in FUDS -, and 18.437% in control birds.
Cloacal samples displayed the most differences across flocks and disease state with Fusobactereium, Turicibacter and Lactobacillus being the top three most abundant taxa. Fusobacterium was present at 13.72, 24.23, and 8.46% in FUDS+, FUDS- and control birds, respectively. Turicibacter was notably different among flocks, but not across disease state within a flock. Turicibacter made up 17.78%, and 17.52% of FUDS+ and FUDS- birds' microbial composition, while only comprising 6.27% of control bird cloacal microbiota. Lactobacillus was also differentially abundant across flocks and disease state, with its peak representation in the control flock. Lactobacillus was found at 10.75, 4.74, and 21.61% in FUDS+, FUDS- and control birds, respectively.
Skin samples were dominated by Staphylococcus, Lactobacillus and Bacteroides, with notable trends across disease state and flocks. Staphylococcus comprised 41.96% of FUDS+, 2.35% of FUDS-, and 0.77% of control skin samples. Lactobacillus exhibited its lowest representation in FUDS+ samples. Lactobacillus was present at 5.39, 10.02, and 13.05% in FUDS+, FUDS-, and control samples, respectively. Bacteroides displayed a similar trend as Lactobacillus among disease states. Bacteroides was found at 2.72, 9.79, 12.01% in FUDS+, FUDS-, and control samples, respectively.
Staphylococcus relative abundance by sample type
Staphylococcus abundance in the skin was found at 41.96, 2.63, and 0.77%, and for FUDS+, FUDS -, and control flocks, respectively. In the ileum it was at 0.032, 0.052, and 0.011% for FUDS+, FUDS-, and control flocks, respectively. Staphylococcus in the cloaca was found at 0.058, 0.075, and 0.654%, for FUDS+, FUDS-, and control flocks, respectively. The lowest Staphylococcus abundance was found in the cecum, found at 0.001, 0.003, and 0% for FUDS+, FUDS-, and control flocks, respectively.
Linear discriminate analysis effect size analysis
The LEfSe analysis was performed to identify differential microbial features between the Control and FUDS affected flocks (Figure 6). For the skin, the genus Staphylococcus was significantly enriched (p < 0.05) in FUDS affected flock and Bacteroides and Lactobacillus significantly enriched (p < 0.05) in the Control (healthy) flock. A second LEfSe analysis was performed only on Staphylococcus to identify specific species associated with the health status of the birds (Figure 7). S. aureus and S. agnetis were identified as the only two taxa associated with FUDS + birds (p < 0.05), while S. cohnii was significantly enriched in the Control flock. Lactobacillus was found as a significant taxon associated with the healthy group in all sample types analyzed in this study. Further LEfSe analysis on the differential Lactobacillus spp. showed a high diversity of Lactobacillus associated with healthy birds, with L. alvi, L. gallinarum, L. johnsonii, and L. acidophilus differentially abundant in the control flock as compared to the FUDS affected flock.
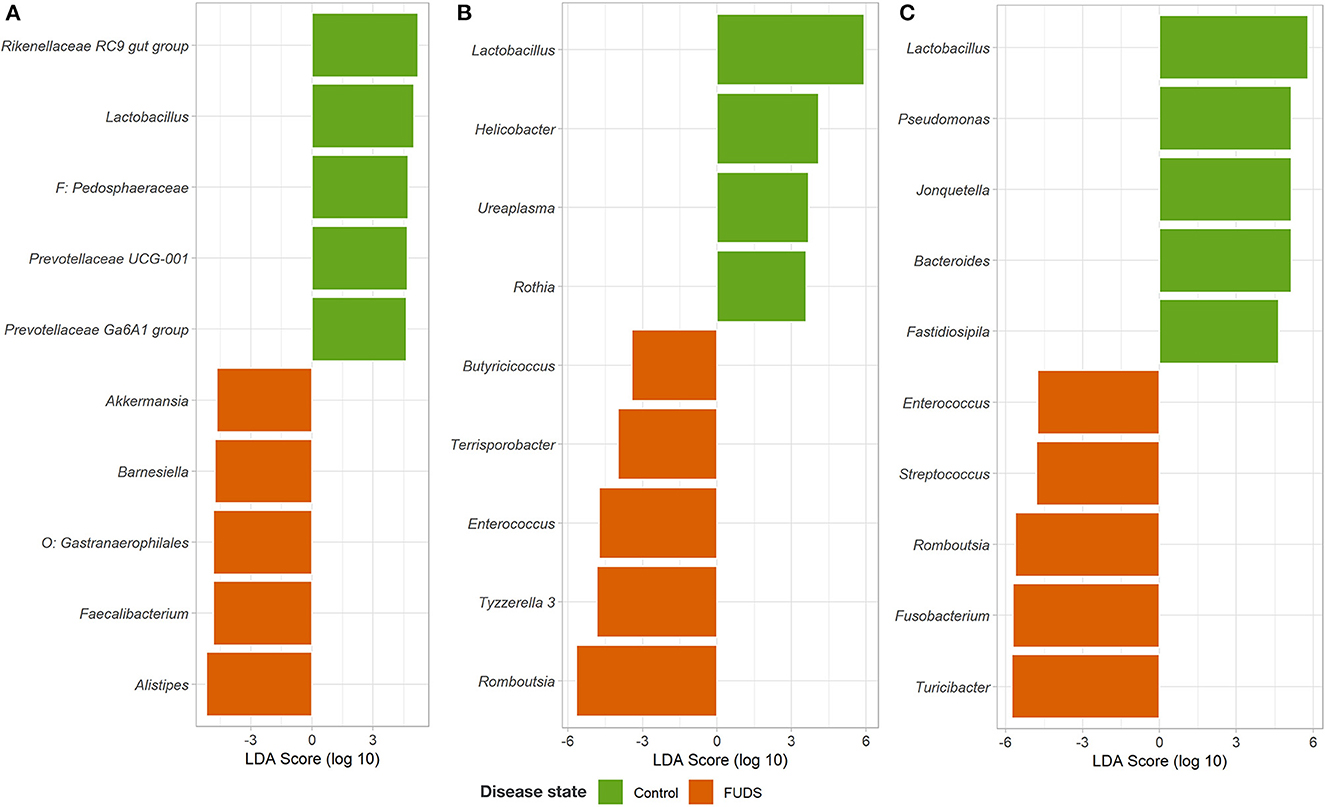
Figure 6. Linear discriminant analysis (LDA) effect size (LEfSe) for microbial differential abundance between Control and FUDS affected flocks. (A) Cecum, (B) ileum, and (C) cloaca.
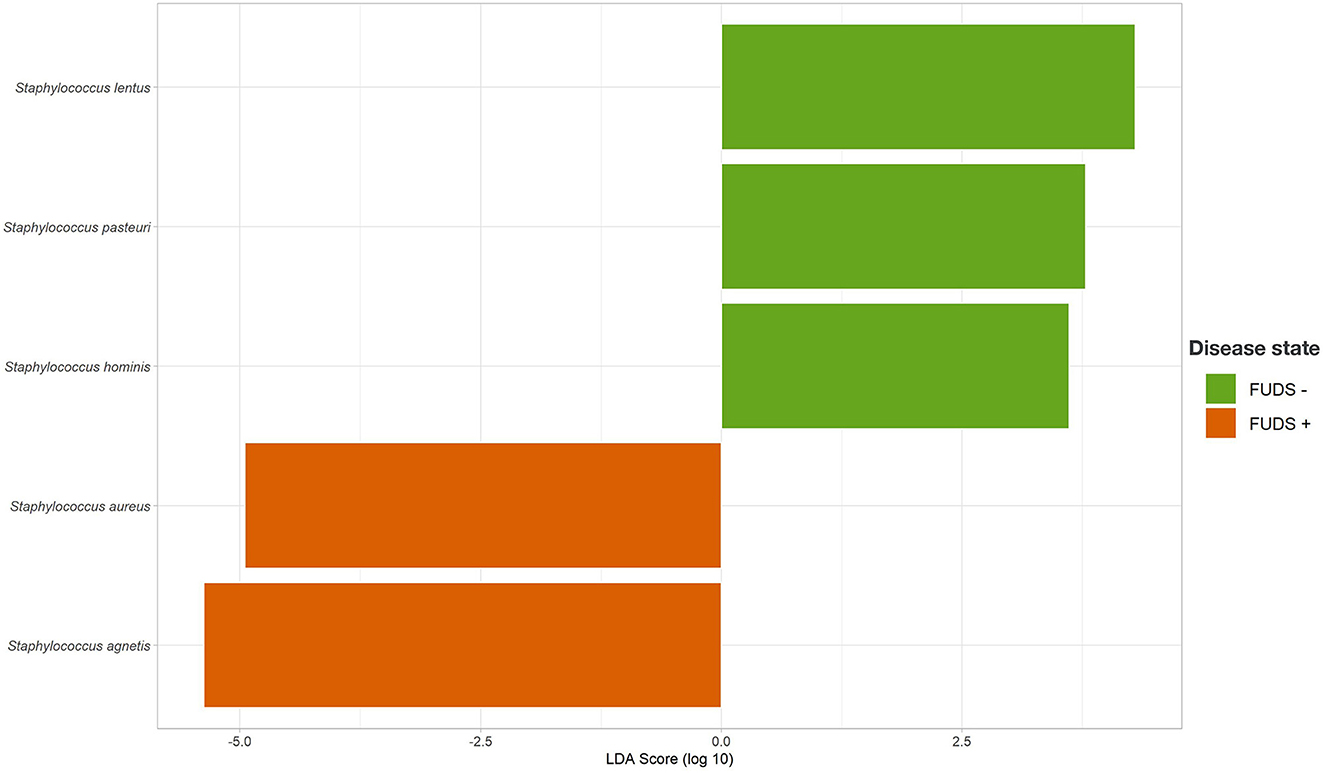
Figure 7. Linear discriminant analysis (LDA) effect size (LEfSe) for Staphylococcus differential abundance between FUDS + and FUDS–flocks.
Whole genome sequence analysis
The WGS data set analyzed in this study is available in the NCBI Sequence Read Archive repository with accession BioProject ID PRJNA884648. Staphylococcus species of all 68 isolates selected were confirmed by WGS, three isolates taxonomically identified by 16S PCR amplification as S. fleuretti, S. simulans, and S. pasteuri were identified by WGS as S. sciuri, S. aureus, and S. warneri, respectively (Table 3, Figure 8).
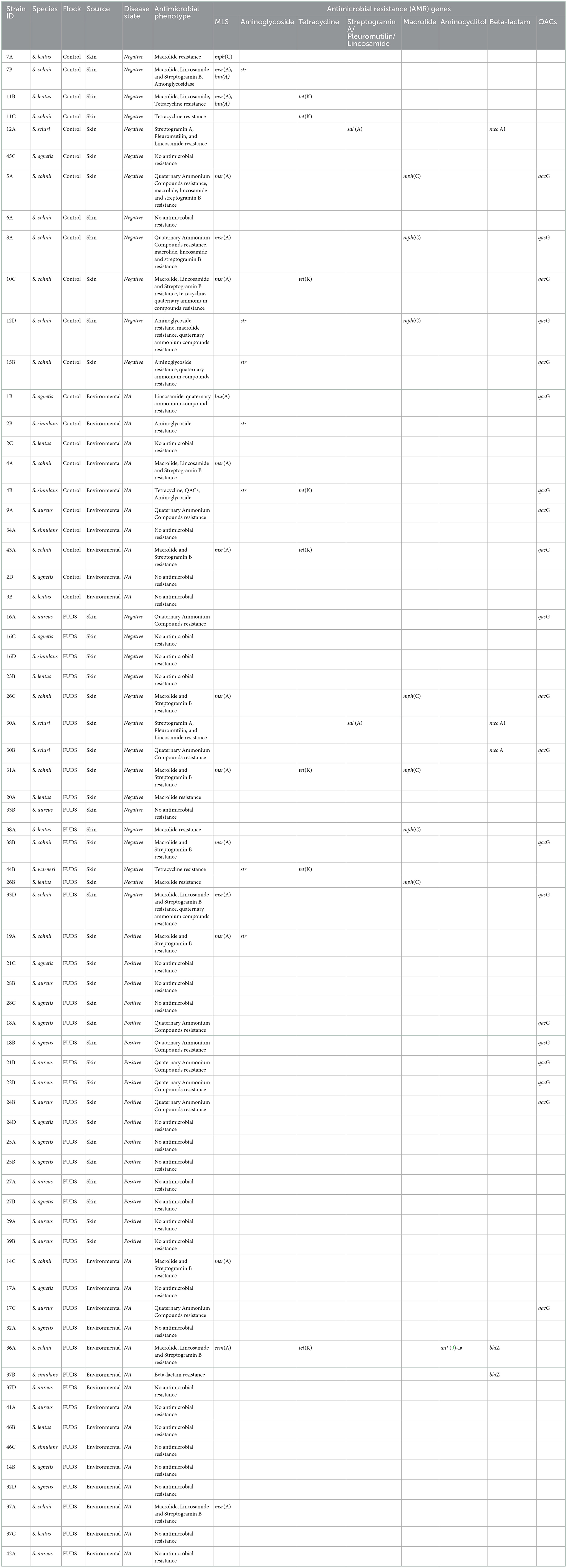
Table 3. Whole genome sequencing analysis of staphylococci isolates from control and FUDS affected flocks, bioinformatics analysis for identificantion of AMR genes.
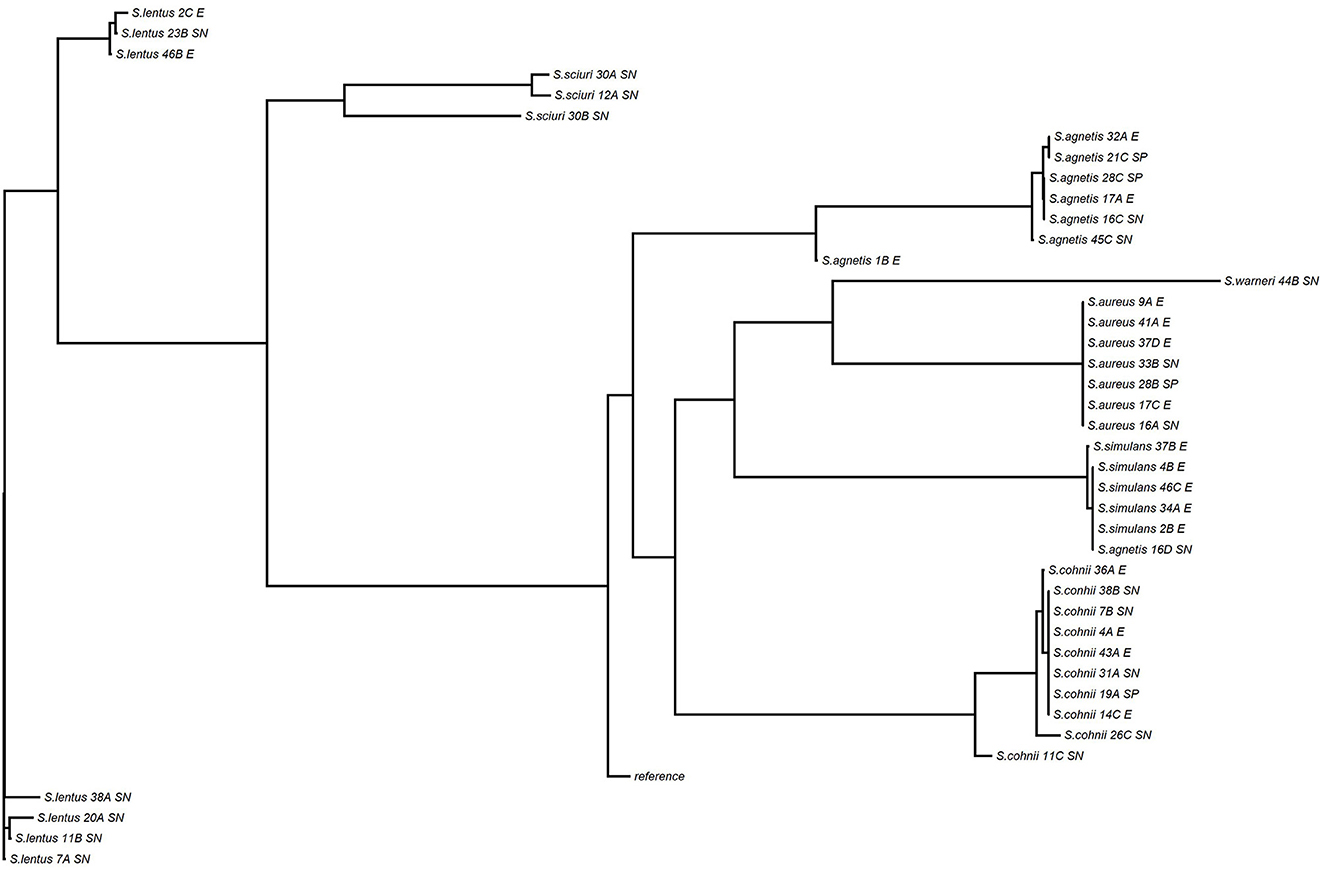
Figure 8. Phylogenetic tree of Staphylococcus isolates from skin and environmental samples of Control and FUDS flocks.
Characterization of antimicrobial resistance and disinfectant resistance
A total of 30 Staphylococcus isolates (44.12%) out of the 68 sequenced had between one and four acquired AMR genes encoding for macrolides, lincosamides, spectrogramines, aminoglycosides, tetracyclines, and beta-lactams resistance (Table 3). The msrA gene was the most common AMR acquired gene identified in the set, encoding for macrolide, lincosamide and streptogramin B resistance. It was carried by 14 isolates (13 S. cohnii isolates, 1 S. lentus) and was found alone (n = 6) or in combination with mphC (n = 3), tetK (n = 2), lnuA (n = 2), and spr (n = 1). The tetK and mphC genes encoding the tetracycline efflux protein and the macrolide phosphotransferase C protein, respectively, were the second and third most common acquired AMR genes identified carried by 9 (5 S. cohnii, 2 S. lentus, 1 S. warneri, and 1 S. simulans) and 8 (5 S. cohnii, 3 S. lentus) isolates, respectively. The tetK gene was found alone (n = 1), or in combination with msrA (n = 4), str (n = 2), and mphC (n = 2). The mphC gene was found alone (n = 2), or in combination with msrA (n = 4), tetK (n = 1), and str (n = 1).
Control flock
A total of 22 Staphylococcus isolates (32.35%) out of the 68 sequenced were isolated from skin (n = 12; 54.45%) and from environment (n = 10; 45.45%). Ten (83.33 %) of the skin isolates had AMR genes in their genomes, with the mphC, msrA, and lnuA genes associated with MLS (macrolide, lincosamide and streptogramin B) resistance (n = 6), as the most prevalent; 2 isolates (16.67%) did not have AMR genes in their genomes. Five (50%) of the environmental isolates had AMR genes associated with MLS, aminoglycoside, and tetracycline resistance, the 5 remaining environmental isolates had no AMR genes in their genomes (Table 3).
FUDS affected flock
A total of 46 Staphylococcus isolates (67.64%) out of the 68 sequenced were isolated from FUDS–skin (n = 15; 32.61%), FUDS + skin (n = 16; 34.78%), and from environment (n = 15; 32.61%). Differences in AMR genes content was observed among FUDS–and FUDS + skin isolates. A total of 11 (73.33%) FUDS–skin isolates had AMR resistance genes associated with MLS and tetracycline resistance, with the msrA as the most common; the 4 (26.77%) remaining isolates had no AMR resistance genes in their genomes. FUDS + skin isolates were dominated by S. aureus and S. agnetis, all S. aureus (n = 7) and S. agnetis (n = 8) selected for WGS had no AMR genes in their genomes. One isolate, S. cohnii, from FUDS+ skin samples had the msrA gene. The environmental isolates selected were composed of S. aureus (n = 4), S. agnetis (n = 4), S. cohnii (n = 3), S. simulans (n = 2), S. lentus (2). A total of 11 (73.33%) of the selected isolates had no AMR genes in their genomes, similarly to the skin + isolates, S. aureus and S. agnetis isolates did not have AMR genes in their genomes. The 4 (26.67%) remaining environmental isolates had AMR genes associated with MLS, tetracycline, and beta-lactam resistance (Table 3).
Disinfectant resistance
A total of 20 isolates (29.41%), out of the 68 sequenced, had the qacG gene encoding resistance to quaternary ammonium compounds (QACs). The QACs resistant isolates were composed of 5 (25%) from skin in the control group, 4 (20%) from environment of control group, 5 (25%) from FUDS + skin, 5 (25%) from FUDS–skin, and 1 (5%) from FUDS house environment. Eight (40%) isolates had the quaG gene alone, the 12 (60%) remaining had the quaG gene in combination with one (n = 5), or two (n = 7) acquired AMR genes. The most common phenotype identified in the isolates exhibiting QACs resistance was msrA- mphC- qacG (n = 3), msrA- tetK- qacG (n = 2), msrA- qacG (n = 2) (Table 3).
Identification of Staphylococcus virulence factors
Six classes of Staphylococcus VF were identified including adherence, enzyme, immune evasion, secretion system, toxin, and iron uptake. In addition, VF with >50% homology to non-Staphylococcus species were identified; the latter included the following 7 VF classes: adhesion proteins, regulation, surface protein anchoring, anti-phagocytosis, intracellular survival, serum resistance, and phagosome arresting (Supplementary Table 1).
The VF class with most genes identified were enzyme (n = 171) and immune evasion, followed by adherence (n = 84), and toxin (n = 84). Among the enzymes-encoding genes, geh and lip, encoding for lipase, were the most prevalent (n = 54), followed by sspA (n = 39), encoding for serine V8 protease, and hysA (n = 26), encoding for hyaluronate lyase (Supplementary Table 1). The geh and lip genes were identified in all Staphylococcus species analyzed in this study except for S. sciuri and S. lentus, sspA was found across all Staphylococcus species, and hysA only in S. aureus and S. agnetis isolates. In the immune evasion category, the most common VF included capB (n = 36), encoding for polyglutamic acid capsule, adsA (n = 35) encoding for adenosine synthase (A), and galE (n = 22) encoding for polysaccharide capsule. The capB was found in all Staphylococcus species analyzed in this study except for S. aureus, S. sciuri and S. lentus; adsA was only present in S. aureus, S. agnetis, and S. simulans, and galE present in all species except for S. aureus, S. sciuri and S. warneri.
In the category of adherence, the ebp gene encoding for elastin binding protein was the most common (n = 19), followed by icaA, icaB, and icaC, genes associated with intercellular adhesion (n = 17), other genes found were fnbB (n = 13) encoding for fibronectin binding protein, cna (n = 13) associated with collagen adhesion, and atl (n = 10) encoding for autolysin. The ebp was found only in S. aureus, and S. cohnii; the icaA, icaB, and icaC genes were only present in S. aureus, S. sciuri; the cna genes was present only in S. agnetis, and the atl only present in S. aureus. In the category of toxin, the set genes encoding for exotoxin were the most common (n = 25), followed by hlgA, hlgB, and hlgC genes associated with gamma hemolysin (n = 14), and hlb (n = 14) encoding for hemolysin B. The set genes were found only in S. aureus and S. agnetis. The hlgA, hlgB, and hlgC genes were found only in S. aureus; adsA was only present in S. aureus, S. agnetis, and S. simulans, and galE present in all species except for S. aureus, S. sciuri and S. warneri.
Antimicrobial effect by agar-well diffusion assay
The inhibitory results of the four proprietary Bacillus DFM combinations against S. aureus and S. agnetis isolated from skin lesions of birds affected with FUDS are shown in Table 4. All DFM tested in this study were found inhibitory against both Staphylococcus aureus and Staphylococcus agnetis. S. aureus inhibition ranged from 6 to 22 mm with DFM Combo 4 and DFM Combo 1, respectively. S. agnetis inhibition ranged from 14.5 to 25 mm with DFM Combo 4 and DFM Combo 1, respectively. Bacillus DFM Combo 1 produced the highest inhibitory effect with clear zones of inhibition ranging from 20 to 22 mm for S. aureus isolates and 22–25 mm for S. agnetis isolates (Table 4). DFM Combo 4 was the least inhibitory with zones of inhibitions ranging from 6 to 11.5 mm for S. aureus isolates and 14.5 to 16 mm for S. agnetis isolates recovered from the lesions.
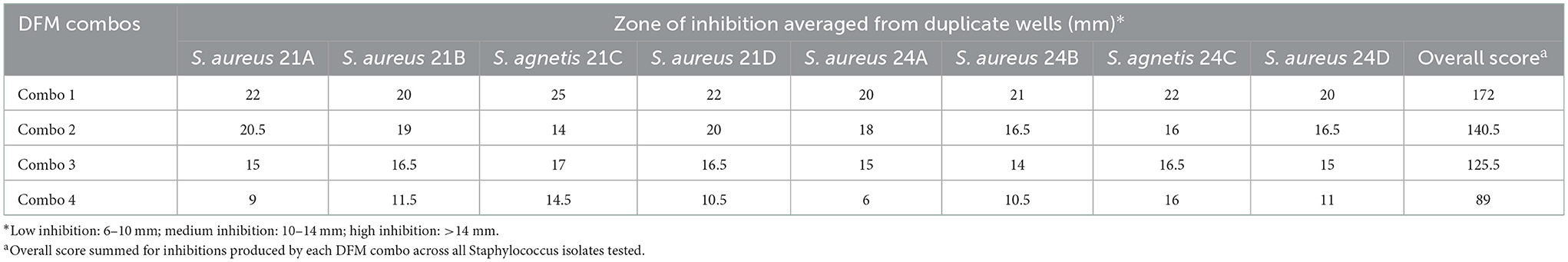
Table 4. Antimicrobial activity, by agar-well diffusion, of proprietary DFM combination against S. agnetis and S. aureus isolated from skin of FUDS + birds.
Antimicrobial effect by competitive exclusion on broth
The antimicrobial effect of the 4 Bacillus DFM combinations by competitive exclusion on broth is shown in Table 5. All DFM combos were found inhibitory, S. aureus reductions ranged from 0.24 to 1.55 log10 CFU/mL with DFM Combo 4 and Combo 2, respectively. S. agnetis and S. aureus reductions (log10 CFU/mL) were evaluated 24 h after co-inoculation with respect to control samples. Reductions of S. agnetis ranged from −0.05 to 1.17 log10 CFU/mL with DFM Combo 4 and Combo 2, respectively. Reductions were summed up across all Staphylococcus isolates and DFM combos were ranked from highest to lowest antimicrobial effect, DFM combo 2 showed the highest inhibitions across all Staphylococcus isolates tested.
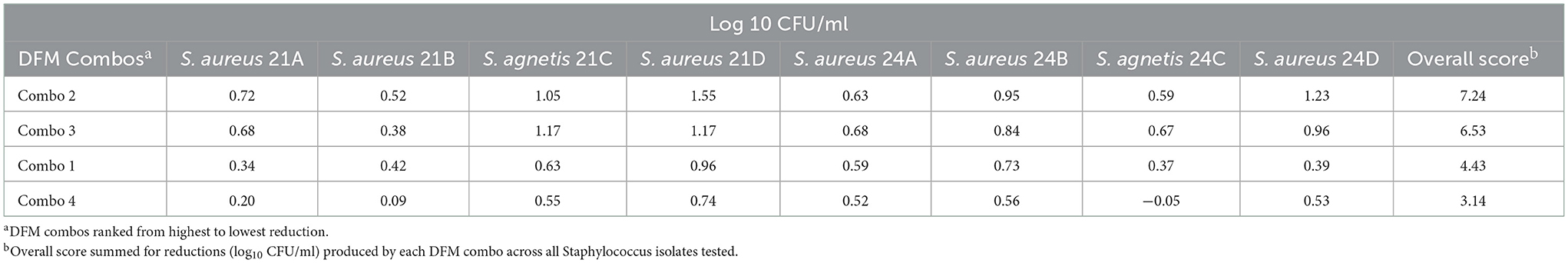
Table 5. S. agnetis and S. aureus reduction (log10 CFU/ml) by DFM combos co-cultivated 24 h on nutrient broth.
Agar-well diffusion (AWD) overall score was added to the competitive exclusion (CE) overall score to rank the DFM combinations with highest antimicrobial effect (Table 6). DFM Combo 1 had the greatest effect across all isolates tested.
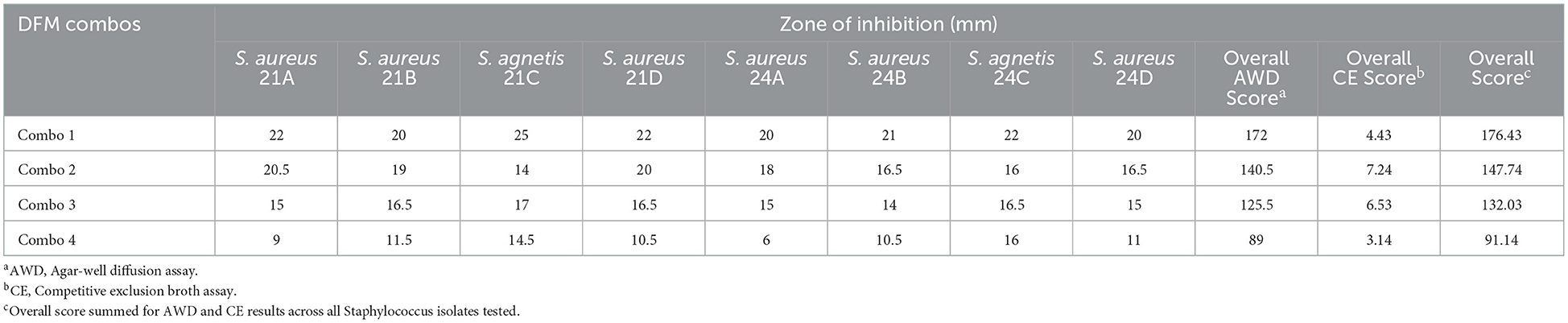
Table 6. Combined well-diffusion and competitive exclusion scores of DFM screened across all Staphylococcus isolates tested.
Discussion
The transition from conventional to cage-free production systems has presented some challenges for the egg producers, one of these challenges is the increase of bacterial infections which have been described as one of the most common causes of mortality in birds raised in cage free and litter-based systems (5). Skin infections in commercial poultry have severe impacts on growth, egg production, feed conversion, and overall performance. Members of the Staphylococcus genus have been described as common causative agents of these conditions (30, 31). Sources of infectious agents include soil, litter, cross-contamination among infected birds, and fomites such as contaminated equipment.
In this study, next generation sequencing was used to characterize the microbiome composition of sites in the gastrointestinal tract (GIT) and skin of laying hens with signs of focal ulcerative dermatitis (FUDS) and compare it to the microbiome composition of healthy birds with the overall objective of identifying the causative agents of FUDS and potential routes of infection. The comparison of the microbial community structure between healthy and sick populations is crucial to identify what constitutes a healthy gut/skin microbiota and identify potential dysbiosis, thereby providing opportunities to modulate it and improve overall animal health (32). The microbial composition can be affected by many factors including age, breed, diet, sample type (i.e., GIT site), disease state, housing system, and management. A significant variation in the microbial population was observed in this study attributed to health status of the flock and sample type. For example, a genus identified at significantly higher abundance in the skin and confirmed as present in the FUDS + birds was Staphylococcus. Staphylococcus abundance in the skin was found at 2.63 and 0.77% in FUDS–and control flocks, respectively, in comparison with FUDS + flock (>41.96%).
Staphylococci are ubiquitous in poultry farms and hatcheries and are natural inhabitants of the chicken skin and mucous membranes but have also been associated with secondary infections (12). The 16S metagenomics and isolation results in this study showed a high diversity of Staphylococcus species on skin and environmental samples from FUDS negative and control flocks as compared to the FUDS positive group. In agreement with the ubiquitous nature of Staphylococci in the microbial population of poultry houses, the environmental samples analyzed in this study showed high diversity of Staphylococcus species including S. cohnii, S. lentus, S. simulans, S. agnetis, and S. aureus. Recent studies have associated the abundance of coagulase-negative staphylococci (CoNS) such as S. conhii and S. simulans with healthy skin in humans and mice models (33–35). In 2020, Brown et al. (34) reported that the commensal S. simulans effectively blocked methicillin-resistant Staphylococcus aureus (MRSA) quorum sensing in an infection model in mice. The observed resistance to pathogenic colonization was a result of the production of an autoinducing peptide that acted as MRSA S. aureus accessory gene regulator (agr) inhibitor protecting the host from invasive infection. In this study, S. simulans was isolated from all environmental sample types and was also isolated from skin samples of FUDS negative and control groups. This result could suggest a potential protective effect of this species in the development of the disease.
Staphylococcus cohnii was another commensal Staphylococci identified on samples from the skin of FUDS negative and control birds and not present on FUDS positive skin samples. It was also identified by the LEfSe analysis as the most enriched taxon in the Control flock (healthy) not present in the FUDS flock. Similar to S. simulans, S. cohnii has been associated with healthy skin. In a skin microbiome analysis of mice with dermatitis found that commensal S. cohnii isolated from sick mice had preventive and therapeutic effects on S. aureus mediated skin inflammation (33). Further investigation is needed to determine the potential protective effect of commensal Staphylococci found on control and FUDS- samples and not isolated from FUDS + skin samples, on the development of FUDS.
The Lactobacillus genus was found ubiquitously in all sample types associated with the healthy birds. Our findings are similar to those observed by Ngunjiri et al. (36) who studied the effect of age and body site in the microbial composition of commercial layer chickens. The authors found Lactobacillus ubiquitous in the trachea, nasal, ileum, and cecum samples analyzed. Lactobacillus was the most significant taxon in the cloaca and ileum of the control flock, suggesting a potential beneficial/protective effect for the birds. Lactobacillus has been commonly ascribed a protective effect through the production of lactic acid that promotes the inhibition of pathogenic bacteria.
The contribution of the skin microbiota to the development of FUDS was analyzed through 16S metagenomic sequencing, and the results showed a higher Staphylococcus load (>60% of all bacteria present) as skin lesions progressed from lesion score 0 to 3 than on FUDS negative and control samples. The dysbiosis revealed was characterized by increased abundance of S. aureus and S. agnetis. These results were confirmed by plate count enumeration with S. aureus and S. agnetis being the only Staphylococci isolated from the skin of FUDS positive birds. Our results agree with those observed by Ito et al. (33) in their comparative skin microbiome analysis in mice to identify commensal strains with protective effect of inflammatory skin diseases. The authors observed that the severity of dermatitis in mice was associated with an increased abundance of S. aureus and Corynebacterium mastitidis and negatively associated with the abundance of S. cohnii (33).
Staphylococcus aureus has been associated with the development of multiple localized infections in poultry including bumblefoot, joint infections, osteomyelitis, and skin abscesses (9, 37, 38). It is also the most common non-clostridial bacterial pathogen associated with gangrenous dermatitis, a systemic infection in chickens (31). In our study, S. aureus isolates were recovered from all environmental samples and skin samples from FUDS + birds which demonstrates broad distribution of the pathogen in the poultry house tested. Meyer et al. (9) reported that S. aureus was the main bacterial pathogen isolated from different tissues (lungs, liver, wattle lesions, among others) from multiple commercial layer hen flocks, experiencing an increased mortality and a drop in egg production between April and November of 2018. Even though, S. aureus was the predominant bacteria isolated, a challenge study, to elucidate the mechanisms by which S. aureus could have been causing the systemic infection, was not able to replicate the disease, highlighting the potential role of other infectious agents in the development of the disease (9). In our study, S. aureus and S. agnetis were found in combination in skin samples from FUDS + birds, suggesting a potential synergistic interaction of both staphylococci in the pathogenesis of the disease.
Staphylococcus agnetis, a coagulase-variable Staphylococcus, was identified as a separate species in 2012, associated with clinical and subclinical cases of bovine mastitis (39). Since its discovery, it has been recognized as the causative agent of poultry diseases such as bacterial chondronecrosis with osteomyelitis (BCO) (40), valvular endocarditis and septicemia in broiler breeder flocks (41). In our study, S. agnetis isolates were recovered from environmental samples from control (nest box, scratch) and FUDS (nest box) houses; similar to S. aureus, it was positively correlated with the severity of the lesion with increased abundance in samples from FUDS + birds with a lesion score 3. S. agnetis isolates were found clustered together, suggesting a single clone circulating in the farm environment.
FUDS is an emerging disease of cage-free laying flocks, cases have been identified in both Bovans and Hy-Line browns. Previous bacterial analyses on swabs from FUDS lesions identified S. hyicus, a skin commensal bacterium associated with opportunistic infections, as the main pathogen present in lesions (42). In our study, 16S microbiome analysis and bacterial isolation of isolates from the skin, identified S. agnetis and S. aureus in FUDS positive birds. The WGS of S. agnetis isolates recovered confirmed them as S. agnetis. As identified by Taponen and others (39), S. agnetis isolates are closely related to S. hyicus and S. chromogenes, their differentiation using phenotypic methods and 16S PCR amplification has not been successful leading to misidentification of these species (8). In addition, it is hypothesized that S. agnetis diverged from S. hyicus and that some of the infections attributed to S. hyicus might have been caused by S. agnetis (8).
Further characterization to identify antimicrobial resistance genes and virulent factors that could have contributed to the development of FUDS was performed on a selected set of Staphylococcus isolates recovered in this study. The use of antibiotics in livestock for disease control and growth promotion has led to the emergence of AMR genes and a worldwide public health concern, due to their potential dissemination to commensal bacteria (43). Antimicrobial resistance genes were widely spread in the screened isolates, with 44.12% of them harboring between one and four acquired AMR genes, they were found equally distributed in control and FUDS flocks, in the skin and environmental samples. Macrolide, lincosamide and spectrogramine resistance was the most common, characterized by the presence of the msrA and mphC genes in the isolates' genotypes, the msrA gene was harbored by 46.66% of the isolates (13 S. cohnii isolates, 1 S. lentus). Our results are in agreement with those observed by Zmantar and others (44) in a study to determine the antimicrobial susceptibility and resistance to quaternary ammonium compounds (QACs) in clinical S. aureus and coagulase negative Staphylococcus (CoNS). Authors found the msrA gene as one of the most prevalent, identified in 41% of CoNS, and in 10.2% of S. aureus strains. Surprisingly, in our study, all S. aureus and 93.33% (n = 14/15) of S. agnetis isolates did not carry any AMR genes in their genotypes. The msrA gene encodes for an ATP-dependent efflux pump that transports erythromycin and streptogramin B from the cell inducing resistance to these antibiotics; and has been mostly associated to Staphylococcus and recently discovered in other genera, including Streptococcus, Enterococcus and Pseudomonas (45).
The tetK gene was the second most common AMR gene identified in our study, harbored by 30% of the isolates. It encodes a tetracycline efflux pump that confers resistance, this resistance is commonly observed in staphylococci isolates regardless of source due to the widespread of tetK/tetM genes (46). Ali Syed et al. (47) found similar results, with the tetK/tetM genes present in 58.8% of staphylococci from farmed chickens. Similar to our results, the authors found the genes harbored by CoNS. The wide distribution of AMR genes among staphylococci not associated with the development of FUDS constitutes a potential risk of transmission of AMR genes to pathogenic staphylococci or commensal bacteria, highlighting the importance of monitoring antibiotic resistance in poultry facilities.
The harboring of disinfectant-resistance genes was also analyzed in this study. Disinfectants are widely used in poultry farming and consist of two or more active components with quaternary ammonium compounds (QACs) the most frequently used component (48). The application of subinhibitory concentrations has reduced the bactericidal effect of sanitizers, leading to an increased resistance to QACs-based sanitizers and a potential increase of antibiotic resistance. Resistance to QACs is associated to the presence of the qac (qacA, qacB, qacC, qacD, and qacG) and smr genes. In this study, 29.41% of the isolates sequenced had the qacG gene present, 40% of the isolates (exclusively S. aureus and S. agnetis) had the qacG gene present and no other AMR gene. The remaining 60% of the Staphylococci isolates screened, had the qacG gene present in combination in one or two AMR genes with resistance to macrolides the most common. El Zayed Zaki et al. (49) evaluated the prevalence of qac and smr genes in clinical isolates of S. aureus from hospital-acquired infections and their susceptibility to QACs-based sanitizers and antibiotics. Authors found the qacG gene as one of the most prevalent genes, present in 17.8% of the isolates, the results were strongly correlated with reduced susceptibility to QACs-based sanitizers. In our study, S. aureus and S. agnetis from FUDS + birds did not exhibit resistance to QACs.
The analysis of virulence is an important step when evaluating interventions to inhibit the causative agents of a disease. It is well-established that staphylococci express a wide variety of virulence factors that allow them to evade the immune response and increase their infectivity (50). Staphylococci isolates screened in this study harbored a number of virulence genes encoding for enzyme, immune evasion, adherence, and toxins that could have an additive effect in the development of FUDS. The geh and lip genes encoding for lipase were the most prevalent enzyme-encoding genes identified, both were found widely spread in Staphylococcus isolates screened in this study. Lipases are important factors for colonization and persistence of staphylococci strains in the skin and have a potential role in bacterial growth by the release of host fatty acids during pathogenesis. Delekta et al. (51) demonstrated that the inactivation of the geh gene reduced the ability of S. aureus strains to utilize host LDLs as a source of exogenous fatty acids, with a potential impact on pathogenesis (51). The hysA gene encoding for hyaluronate lyase was another common virulence factor identified, only present in S. aureus and S. agnetis. Hyaluronidases are bacterial enzymes that cleave the β-1,4 glycosidic bond of hyaluronic acid, a natural substance abundant in the skin, joints, and tissues. These virulence factors are associated with a rapid pathogenic dissemination through tissues causing increased lesion size during infection. Tissues with high concentrations of hyaluronic acid have often been found infected with S. aureus (52). Ibberson et al. (52) demonstrated a reduction in tissue damage of the lung of mice when challenged using a S. aureus hysA mutant strain, emphasizing the role of hysA in S. aureus bacterial infection. In our study, the hysA gene was widely spread among S. aureus and S. agnetis isolates from FUDS + birds, suggesting a potential role in their dissemination, pathogenicity, and developing of FUDS.
Staphylococci harbors cell wall proteins involved in adhesion together with tissue and immune defense evasion (50). Cell wall proteins interact directly or indirectly with host receptors to initiate attachment to epithelial cells. This interaction between pathogen and host cells has been found to be species and strain dependent (53). The S. aureus protein A encoded by the spA gene has been commonly associated with immune response evasion. In our study, it was found in 3 S. aureus isolates (2 from FUDS environmental samples and 1 from FUDS + skin sample). Other proteins, such as the fnbB, encoding for fibronectin binding proteins and cna encoding for collagen adhesin were found only in S. agnetis isolates (n = 13). Poulsen et al. (41) analyzed the genomes of three S. agnetis isolates from birds with valvular endocarditis and identified the fnbB gene together with other six virulence factors associated with fibronectin binding proteins. The presence of these genes highlights a potential role in cell adhesion, diffusion, and invasion of host tissues. The production of toxins is another important factor that contributes to pathogenesis by the disruption of eukaryotic membranes, interfering with receptors and causing cell lysis (50). S. aureus and S. agnetis in this study, harbored genes encoding for exotoxin (set genes), alpha and beta-porin forming toxins, and exfoliative toxin type A. These toxins have been implicated in the development of Staphylococcus-induced dermatoses, such as acantholytic dermatitis in humans (10), and gangrenous dermatitis in poultry (31). Our results concur with those observed by Al-Rubaye et al. (40) who identified S. agnetis as a causative agent of lameness in chickens, and the authors also identified a repertoire of virulence determinants including exfoliative toxin A, fibronectin-binding proteins, and several others associated with adherence, toxin biosynthesis, host immune evasion, and secretion systems. Our findings suggest that the development of FUDS occur as a result of a combination of “inside-out” mechanisms by which both staphylococci translocate across the host tissues using the cooperative virulence factors they express and proliferate in the skin developing typical FUDS lesions. Exposed lesions are a source of cross-contamination to other animals and the environment.
Once pathogens were identified and fully characterized the next step was the development of a customized DFM product to inhibit/reduce their growth. The developed product was based on the use of proprietary novel direct fed microbials. DFM's can be a natural alternative to the subtherapeutic use of antibiotics in animal production. There is an increased interest in their use to reduce the spread of antimicrobial resistance while supporting an improved production efficiency. In this study, four proprietary Bacillus combinations were developed based on previous antagonistic screening against multiple pathogens including Staphylococcus spp. The antimicrobial effect of the combinations was determined based on the overall reduction of S. aureus and S. agnetis isolates by agar well diffusion and competitive exclusion antimicrobial screenings. DFM combo 1, a combination of two Bacillus pumilus, was found the most effective at inhibiting the growth of both staphylococci isolated from the skin of diseased birds as compared to the other combinations tested. As observed in other studies, there was a strong association between Bacillus species and antagonistic effect, with highest inhibitions achieved with the Bacillus pumilus combination in DFM combo 1, followed by combo 2 containing two B. pumilus, a B. amyloliquifaciens, and a B. subtilis strain.
Bacilli are endospore-forming bacteria widely used in DFM preparations due to their ability to survive high temperatures, acidic environments, and desiccation, thus increasing their viability during manufacturing and pelleting processes and enhancing their stability in the host' GIT (54, 55). They are known to promote gut health by the production of enzymes, antimicrobial peptides, and other metabolites that inhibit/reduce the growth of bacterial pathogens (56). In our study, the B. pumilus combination exerted the improved inhibitions with zones of inhibition ranging between 20–22 mm, and 22–25 mm, for S. aureus and S. agnetis strains, respectively. These inhibitions were higher than those observed by Ouoba and others (57) who reported that, in an agar spot diffusion assay, B. pumilus strains B6 and B10 inhibited indicator S. aureus isolates with zones of inhibition < 3 mm, 24 h after incubation (57).
Similar to other Bacilli, B. pumilus produce antimicrobial peptides such as bacteriocins and other compounds including amicoumacins with antimicrobial, antifungal, and anti-inflammatory properties (58). Aunpad and Bangchang (59) reported the enhanced antimicrobial effect of the bacteriocin pumilicin 4, against methicillin-resistant S. aureus (MRSA). In 2012, Hashimoto and others (60) reported a strong anti-MRSA effect by derivatives of amicoumacins A and B with minimum inhibitory concentrations (MICs) between 0.6 and 256 ug/ml allowing no visible growth of MRSA. In our study, the co-culture of B. pumilus combo 1 with Staphylococcus aureus and Staphylococcus agnetis allowed for reductions between 0.34–0.96 log10 CFU/ml, and 0.37–0.63 log10 CFU/ml, respectively, after 24 h of co-inoculation. It is important to highlight that the concentrations of the Bacillus pumilus tested were found at 108CFU/ml, 24 h after co-inoculation.
The control of pathogens is one of the major interests for the poultry industry from an economic and public health point of view. The Bacillus DFM supplementation of layer hen diets has shown improved laying production by enhancing gut health, increasing intestinal fermentation and mineral assimilation, and improving the overall daily feed intake (14, 61, 62). The exact mechanism by which Bacillus DFM supplementation inhibits pathogens is not well-understood; the direct inhibition by the production of antimicrobial metabolites or competitive exclusion have been suggested as the main modes of action, other mechanisms such as the enhancement of the intestinal mucosal layer to avoid microbial diffusion across the membrane has also been suggested (14). The targeted B. pumilus combination developed for this study, inhibited both pathogens in vitro and is being used at different farms with history of FUDS with successful results at: (1) inhibiting the synergistic effect of S. agnetis and S. aureus, (2) decreasing FUDS mortalities, and (3) improving harvestable eggs (data not shown). Further analysis will be performed to determine the effect of the Bacillus DFM supplementation on a potential shift of the microbial population of laying hens at farms previously afflicted with FUDS.
Conclusion
Staphylococcus aureus and Staphylococcus agnetis were identified as the causative agents of FUDS in laying hens. Both pathogens were found as statistically significant bacterial species only present in the skin of FUDS + birds, this result was confirmed by plating with both staphylococci as the only pathogens isolated from the skin. Both S. aureus and S. agnetis had a high number of virulence factors associated with adherence, toxin production, and immune evasion that could have contribute to their pathogenesis and the development of FUDS. The customized Bacillus pumilus product developed in this study was found successful at inhibiting both pathogens in vitro and is currently being fed at poultry facilities with chickens afflicted with FUDS with improved results at reducing mortality and enhanced egg production.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
DA, DG, and BT performed all experiments in the study. NE collected samples for analysis, scored birds based on lesion progression, and wrote the sampling collection methodology. DA, ML, and DG contributed to the bioinformatics analyses. TK and CN conceived the study and developed the study design. DA wrote the first draft of the manuscript. DG, BT, KR, and EK contributed to writing. All authors contributed to the editing of the final version of the manuscript.
Funding
The authors declare that this study received funding from Purina Animal Nutrition. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Conflict of interest
DA, DG, NE, KR, EK, BT, ML, CN, and TK were employed by Purina Animal Nutrition Center, Land O' Lakes.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2023.1110573/full#supplementary-material
References
1. Brannan KE, Anderson KE. Examination of the impact of range, cage-free, modified systems, and conventional cage environments on the labor inputs committed to bird care for three brown egg layer strains. J Appl Poult Res. (2021) 30:1–7. doi: 10.1016/j.japr.2020.100118
2. United States Department of Agriculture. National Agricultural Statistics Service (USDA-NASS); Washington DC: Chicken and Eggs (2019). p. 1948–9064.
3. United States Department of Agriculture. USDA Cage-Free Shell-Egg Report: Chickens and Eggs 2020 Summary (February 2021). USDA, National Agricultural Statistics Service (2020).
4. van Goor A, Redweik GAJ, Stromberg ZR, Treadwell CG, Xin H, Mellata M. Microbiome and biological blood marker changes in hens at different laying stages in conventional and cage free housings. Poult Sci. (2020) 99:2362–74. doi: 10.1016/j.psj.2020.01.011
5. Lay DC, Fulton RM, Hester PY, Karcher DM, Kjaer JB, Mench JA, et al. Hen welfare in different housing systems. Poult Sci. (2011) 90:278–94. doi: 10.3382/ps.2010-00962
6. Hartcher KM, Jones B. The welfare of layer hens in cage and cage-free housing systems. Worlds Poult Sci J. (2017) 73:767–81. doi: 10.1017/S0043933917000812
7. Gingerich E. The Current State of Pullet and Layer Health in the United States. In: Proceedings of Midwest Poultry Federation (2020). Available online at: https://midwestpoultry.com/wp-content/uploads/2020/08/Gingerich-Eric.pdf (accessed August 12–14, 2020).
8. Szafraniec GM, Szeleszczuk P, Dolka B. A review of current knowledge on staphylococcus agnetis in poultry. Animals. (2020) 10:1–19. doi: 10.3390/ani10081421
9. Meyer, M, Bobeck, E, Sato, Y, El-Gazzar, B,. Comparison of Naturally Occurring vs. Experimental Infection of Staphylococcus aureus Septicemia in Laying Hens in Two Different Age Groups. Available online at: http://meridian.allenpress.com/avian-diseases/article-pdf/65/2/310/2868161/i0005-2086-65-2-310.pdf (accessed October 7, 2021).
10. Chénier S, Lallier L. Acantholytic folliculitis and epidermitis associated with Staphylococcus hyicus in a line of white leghorn laying chickens. Vet Pathol. (2012) 49:284–7. doi: 10.1177/0300985811415705
11. Ahrens P, Andresen LO. Cloning and sequence analysis of genes encoding Staphylococcus hyicus exfoliative toxin types A, B, C, and D. J Bacteriol. (2004) 186:1833–7. doi: 10.1128/JB.186.6.1833-1837.2004
12. Fries-Craft K, Meyer MM, Sato Y, El-Gazzar M, Bobeck EA. Age and Staphylococcus aureus inoculation route differentially alter metabolic potential and immune cell populations in laying hens. Front Vet Sci. (2021) 8:1–13. doi: 10.3389/fvets.2021.653129
13. Johnson TA, Sylte MJ, Looft T. In-feed bacitracin methylene disalicylate modulates the turkey microbiota and metabolome in a dose-dependent manner. Sci Rep. (2019) 9:8212. doi: 10.1038/s41598-019-44338-5
14. Jha R, Das R, Oak S, Mishra P. Probiotics (Direct-fed microbials) in poultry nutrition and their effects on nutrient utilization, growth and laying performance, and gut health: a systematic review. Animals. (2020) 10:1–19. doi: 10.3390/ani10101863
15. Krysiak K, Konkol D, Korczyński M. Review overview of the use of probiotics in poultry production. Animals. (2021) 11:1620. doi: 10.3390/ani11061620
16. Shivaramaiah S, Pumford NR, Morgan MJ, Wolfenden RE, Wolfenden AD, Torres-Rodriguez A, et al. Evaluation of bacillus species as potential candidates for direct-fed microbials in commercial poultry. Poult Sci. (2011) 90:1574–80. doi: 10.3382/ps.2010-00745
17. Fuller R. History and development of probiotics. In: Probiotics. Netherlands: Springer (1992). p. 1–8.
18. Hernandez-Patlan D, Solis-Cruz B, Pontin KP, Hernandez-Velasco X, Merino-Guzman R, Adhikari B, et al. Impact of a bacillus direct-fed microbial on growth performance, intestinal barrier integrity, necrotic enteritis lesions, and ileal microbiota in broiler chickens using a laboratory challenge model. Front Vet Sci. (2019) 6:108. doi: 10.3389/fvets.2019.00108
19. Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. (2013) 41:e1. doi: 10.1093/nar/gks808
20. Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. (2019) 37:852–7. doi: 10.1038/s41587-019-0209-9
21. Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. (2013) 41:D590–6. doi: 10.1093/nar/gks1219
22. Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. (2011) 12:R60. doi: 10.1186/gb-2011-12-6-r60
23. Bolger AM, Lohse M, Usadel B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics. (2014) 30:2114–20. doi: 10.1093/bioinformatics/btu170
24. Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. (2012) 19:455–77. doi: 10.1089/cmb.2012.0021
25. Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, et al. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. (2012) 67:2640–4. doi: 10.1093/jac/dks261
26. Chen L, Yang J, Yu J, Yao Z, Sun L, Shen Y, et al. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. (2005) 33: D325–8. doi: 10.1093/nar/gki008
27. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. (2014) 30:1312–3. doi: 10.1093/bioinformatics/btu033
28. Vinderola G, Capellini B, Villarreal F, Suárez V, Quiberoni A, Reinheimer J. Usefulness of a set of simple in vitro tests for the screening and identification of probiotic candidate strains for dairy use. LWT. (2008) 41:1678–88. doi: 10.1016/j.lwt.2007.10.008
29. Ayala DI, Cook PW, Franco JG, Bugarel M, Kottapalli KR, Loneragan GH, et al. A systematic approach to identify and characterize the effectiveness and safety of novel probiotic strains to control foodborne pathogens. Front Microbiol. (2019) 10:1108. doi: 10.3389/fmicb.2019.01108
30. Szafraniec GM, Szeleszczuk P, Dolka B. Review on skeletal disorders caused by Staphylococcus spp. in poultry. Vet Quart. (2022) 42:21–40. doi: 10.1080/01652176.2022.2033880
31. Gornatti-Churria CD, Crispo M, Shivaprasad HL, Uzal FA. Gangrenous dermatitis in chickens and turkeys. J Vet Diagn Invest. (2018) 30:188–96. doi: 10.1177/1040638717742435
32. Carrasco JMD, Casanova NA, Miyakawa MEF. Microbiota, gut health and chicken productivity: What is the connection? Microorganisms. (2019) 7:374. doi: 10.3390/microorganisms7100374
33. Ito Y, Sasaki T, Li Y, Tanoue T, Sugiura Y, Skelly AN, et al. Staphylococcus cohnii is a potentially biotherapeutic skin commensal alleviating skin inflammation. Cell Rep. (2021) 35:109052. doi: 10.1016/j.celrep.2021.109052
34. Brown MM, Kwiecinski JM, Cruz LM, Shahbandi A, Todd DA, Cech NB, et al. Novel peptide from commensal Staphylococcus simulans blocks methicillin-resistant Staphylococcus aureus quorum sensing and protects host skin from damage. Antimicrob Agents Chemother. (2020) 64:e00172–20. doi: 10.1128/AAC.00172-20
35. Byrd AL, Belkaid Y, Segre JA. The human skin microbiome. Nat Rev Microbiol. (2018) 16:143–55. doi: 10.1038/nrmicro.2017.157
36. Ngunjiri JM, Taylor KJM, Abundo MC, Jang H, Elaish M, Mahesh KC, et al. Farm stage, bird age, and body site dominantly affect the quantity, taxonomic composition, and dynamics of respiratory and gut microbiota of commercial layer chickens. Appl Environ Microbiol. (2019) 85:e03137-18. doi: 10.1128/AEM.03137-18
37. Jiang T, Mandal RK, Wideman RF, Khatiwara A, Pevzner I, Kwon YM. Molecular survey of bacterial communities associated with bacterial chondronecrosis with osteomyelitis (BCO) in broilers. PLoS ONE. (2015) 10:e0124403. doi: 10.1371/journal.pone.0124403
38. Andreasen CB. Staphylococcosis. In:Swayne DE, , editor. 14th ed. Diseases of Poultry. Hoboken, NJ: John Wiley and Sons, Ltd. (2020). p. 995–1003.
39. Taponen S, Supré K, Piessens V, van Coillie E, de Vliegher S, Koort JMK. Staphylococcus agnetis sp. nov, a coagulasevariable species from bovine subclinical and mild clinical mastitis. Int J Syst Evol Microbiol. (2011) 62:61–5. doi: 10.1099/ijs.0.028365-0
40. Alrubaye AAK, Ekesi NS, Hasan A, Koltes DA, Wideman RF, Rhoads DD. Chondronecrosis with osteomyelitis in broilers: further defining a bacterial challenge model using standard litter flooring and protection with probiotics. Poult Sci. (2020) 99:6474–80. doi: 10.1016/j.psj.2020.08.067
41. Poulsen LL, Thøfner I, Bisgaard M, Olsen RH, Christensen JP, Christensen H. Staphylococcus agnetis, a potential pathogen in broiler breeders. Vet Microbiol. (2017) 212:1–6. doi: 10.1016/j.vetmic.2017.10.018
42. Lossie G, Beilke E, Gingerich E, Wakenell P. A unique case of ulcerative dermatitis in a flock of cage free, commercial, brown layers. In: Annual Proceedings of American Association of Avian Pathologists. (2016). p. 5–6. Available online at: https://aaap.memberclicks.net/assets/2016_Annual_Meeting/aaap%20proceedings%202016%20v2.pdf (accessed October 7, 2020).
43. Peralta-Sánchez JM, Martín-Platero AM, Ariza-Romero JJ, Rabelo-Ruiz M, Zurita-González MJ, Baños A, et al. Egg production in poultry farming is improved by probiotic bacteria. Front Microbiol. (2019) 10:1042. doi: 10.3389/fmicb.2019.01042
44. Zmantar T, Kouidhi B, Miladi H, Bakhrouf A. Detection of macrolide and disinfectant resistance genes in clinical Staphylococcus aureus and coagulase-negative staphylococci. BMC Res Notes. (2011) 4:453. doi: 10.1186/1756-0500-4-453
45. Ojo KK, Striplin MJ, Ulep CC, Close NS, Zittle J, Luis H, et al. Staphylococcus efflux msr(A) gene characterized in Streptococcus, Enterococcus, Corynebacterium, and Pseudomonas isolates. Antimicrob Agents Chemother. (2006) 50:1089–91. doi: 10.1128/AAC.50.3.1089-1091.2006
46. Leroy S, Christieans S, Talon R. Tetracycline gene transfer in staphylococcus xylosus in situ during sausage fermentation. Front Microbiol. (2019) 10:392. doi: 10.3389/fmicb.2019.00392
47. Syed MA, Ullah H, Tabassum S, Fatima B, Woodley TA, Ramadan H, et al. Staphylococci in poultry intestines: a comparison between farmed and household chickens. Poult Sci. (2020) 99:4549–57. doi: 10.1016/j.psj.2020.05.051
48. Maertens H, Demeyere K, de Reu K, Dewulf J, Vanhauteghem D, van Coillie E, et al. Effect of subinhibitory exposure to quaternary ammonium compounds on the ciprofloxacin susceptibility of Escherichia coli strains in animal husbandry. BMC Microbiol. (2020) 20:155. doi: 10.1186/s12866-020-01818-3
49. el Sayed Zaki M. Molecular study of resistance of Staphylococcus aureus to antiseptic quaternary ammonium compounds. J Glob Antimicrob Resist. (2019) 17:94–7. doi: 10.1016/j.jgar.2018.11.022
50. González-Martín M, Corbera JA, Suárez-Bonnet A, Tejedor-Junco MT. Virulence factors in coagulase-positive Staphylococci of veterinary interest other than Staphylococcus aureus. Vet Quart. (2020) 40:118–31. doi: 10.1080/01652176.2020.1748253
51. Delekta PC, Shook JC, Lydic TA, Mulks MH, Hammer ND. Staphylococcus aureus utilizes host-derived lipoprotein particles as sources of fatty acids. J Bacteriol. (2018) 200:e00728-17. doi: 10.1128/JB.00728-17
52. Ibberson CB, Jones CL, Singh S, Wise MC, Hart ME, Zurawski D, et al. Staphylococcus aureus hyaluronidase is a CodY-regulated virulence factor. Infect Immun. (2014) 82:4253–64. doi: 10.1128/IAI.01710-14
53. Garciarena CD, McHale TM, Watkin RL, Kerrigan SW. Coordinated molecular cross-talk between staphylococcus aureus, endothelial cells and platelets in bloodstream infection. Pathogens. (2015) 4:869–82. doi: 10.3390/pathogens4040869
54. Nishiyama T, Nakagawa K, Imabayashi T, Iwatani S, Yamamoto N, Tsushima N. Probiotic bacillus subtilis c-3102 improves eggshell quality after forced molting in aged laying hens. J. Poult. Sci. (2021) 58:230–7. doi: 10.2141/jpsa.0200081
55. Susanti D, Volland A, Tawari N, Baxter N, Gangaiah D, Plata G, et al. Multi-omics characterization of host-derived Bacillus spp. probiotics for improved growth performance in poultry. Front Microbiol. (2021) 12:747845. doi: 10.3389/fmicb.2021.747845
56. Grant A, Gay CG, Lillehoj HS. Bacillus spp. as direct-fed microbial antibiotic alternatives to enhance growth, immunity, and gut health in poultry. Avian Pathol. (2018) 47:339–51. doi: 10.1080/03079457.2018.1464117
57. Ouoba LII, Diawara B, Jespersen L, Jakobsen M. Antimicrobial activity of Bacillus subtilis and Bacillus pumilus during the fermentation of African locust bean (Parkia biglobosa) for Soumbala production. J Appl Microbiol. (2007) 102:963–70. doi: 10.1111/j.1365-2672.2006.03156.x
58. Tran C, Cock IE, Chen X, Feng Y. Antimicrobial bacillus: metabolites and their mode of action. Antibiotics. (2022) 11:88. doi: 10.3390/antibiotics11010088
59. Aunpad RN-BK. Pumilicin 4, a novel bacteriocin with anti-MRSA and anti-VRE activity produced by newly isolated bacteria bacillus pumilus strain WAPB4. Curr Microbiol. (2007) 55:308–13. doi: 10.1007/s00284-006-0632-2
60. Hashimoto M, Taguchi T, Nishida S, Ueno K, Koizumi K, Aburada M, et al. Isolation of 8-phosphate ester derivatives of amicoumacins: structure-activity relationship of hydroxy amino acid moiety. J Antibiot. (2007) 60:752–6. doi: 10.1038/ja.2007.99
61. Abd El-Hack ME, El-Saadony MT, Shafi ME, Qattan SYA, Batiha GE, Khafaga AF, et al. Probiotics in poultry feed: a comprehensive review. J Anim Physiol Anim Nutr. (2020) 104:1835–50. doi: 10.1111/jpn.13454
Keywords: commercial laying hens, gut microbiota, skin microbiota, FUDS, DFM development
Citation: Ayala DI, Grum DS, Evans NP, Russo KN, Kimminau EA, Trible BR, Lahoti MM, Novak CL and Karnezos TP (2023) Identification and characterization of the causative agents of Focal Ulcerative Dermatitis in commercial laying hens. Front. Vet. Sci. 10:1110573. doi: 10.3389/fvets.2023.1110573
Received: 29 November 2022; Accepted: 13 January 2023;
Published: 08 February 2023.
Edited by:
Bruno Solis-Cruz, Faculty of Higher Studies Cuautitlán, National Autonomous University of Mexico, MexicoReviewed by:
Roberto Senas Cuesta, University of Arkansas, United StatesMuhammad Zahoor, University of Agriculture, Dera Ismail Khan, Pakistan
Copyright © 2023 Ayala, Grum, Evans, Russo, Kimminau, Trible, Lahoti, Novak and Karnezos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Diana I. Ayala,  ZGF5YWxhQGxhbmRvbGFrZXMuY29t
ZGF5YWxhQGxhbmRvbGFrZXMuY29t