 Mingna Duan1†
Mingna Duan1† Yafang Liu1†Jun Wu2†Shuang Liu1
Yafang Liu1†Jun Wu2†Shuang Liu1 Shaobo Tang1Dandan Jiang3Quanfu Zhang4*Wei Gu5,6*
Shaobo Tang1Dandan Jiang3Quanfu Zhang4*Wei Gu5,6* Xing Yang1*
Xing Yang1*- 1Integrated Laboratory of Pathogenic Biology, College of Preclinical Medicine, Dali University, Dali, China
- 2Department of Ophthalmology, Dali Bai Autonomous Prefecture People’s Hospital, Dali, China
- 3School of Public Health, Dali University, Dali, China
- 4Department of Gastroenterology, Clinical Medical College, The First Affiliated Hospital of Chengdu Medical College, Chengdu, China
- 5Department of Infection, The First Affiliated Hospital of Dali University, Dali, China
- 6The Key Laboratory of Infectious Diseases of Yunnan Provincial Education Department, Dali, China
Fleas serve as hosts to a diverse array of pathogens, which present significant medical and veterinary concerns for human and livestock health. The mitochondrial genome (mtDNA) has long been regarded as a classical model in biogenetics and species evolution research. However, the availability of mitochondrial genome data for fleas remains scarce. In this study, we sequenced Macrostylophora euteles specimens collected from the Yunnan plague focus and Citellophilus tesquorum sungaris specimens from Jilin plague focus. The obtained sequences were compared to the sequences of 24 flea species retrieved from the NCBI database, focusing on base composition, evolution rates, nucleotide polymorphism and phylogenetic analysis. All fleas analyzed contained a total of 37 genes. Gene sequences exhibited remarkable stability, with no evidence of gene rearrangement. Additionally, the base composition demonstrated a pronounced AT bias. Results from both methodologies and across the two datasets consistently indicated strong monophyly for the superfamilies Ceratophylloidea and Pulicoidea, as well as for the family Pulicidae. In contrast, the superfamily Hystrichopsylloidea, along with the families Ceratophyllidae, Leptopsyllidae and Ctenophthalmidae, were identified as paraphyletic. This research provides valuable molecular data to support taxonomic and phylogenetic studies of fleas.
1 Introduction
Fleas are small, blood-feeding insects characterized by their smooth and tough body surface, typically ranging in color from brown to black-brown. These insects possess remarkable jumping abilities and hold medical significance. They belong to the phylum Arthropoda, class Insecta, and order Siphonaptera (23). To date, approximately 3,000 species and subspecies across 19 families have been identified globally (21). Fleas serve as primary vectors for the transmission of plague, making them critical indicators for identifying the early stages of plague epidemic. The majority of flea species primarily infest rodents, with only about 5% parasitizing birds (30). A wide range of zoonotic pathogens has been identified in fleas, including Yersinia pestis, Rickettsia typhi, Rickettsia felis, Bartonella henselae, Bartonella quintana, and Francisella tularensis (11, 14). Additionally, fleas function as intermediate hosts for Dipylidium caninum (41). Annually, these parasites and associated pathogens result in over $15 billion in global economic losses and have raised significant international public health concerns (36).
In recent years, there has been a notable rise in both emerging and re-emerging flea-borne diseases due to environmental changes and shifts in human behavior, posing significant challenges to public health (14). Consequently, the precise identification of flea species and systematic classification research are crucial for the prevention and control of these diseases. However, reaching a consensus on classification remains challenging, particularly concerning the number of families and included species (17). Traditional morphological identification methods require extensive professional experience and are often constrained by subtle morphological variations within species (27). With the advancements in genetic technology, molecular biological techniques have become increasingly prevalent for accurate identification and classification of fleas, thereby addressing some of the limitations associated with traditional morphology.
Macrostylophora euteles (Jordan and Rothschild, 1911 in wu (53)) is the first identified species within the genus Macrostylophora of the Ceratophyllidae family. This species primarily inhabits the provinces of Yunnan and Sichuan in China, with documented occurrences also reported in Myanmar and Thailand (10). Citellophilus tesquorum sungaris also referred to as Citellophilus sungaris sungaris (Jordan, 1929 in Wu (53)), a member of the Citellophilus genus within the same family, predominantly thrives in Inner Mongolia, Jilin and Harbin regions of China, as well as in Russia and Mongolia (34). Notably, it serves as a key vector in marmot- or ground squirrel-associated plague foci in northeastern China (13). As primary vectors of plague, fleas provide an early warning signal for assessing plague outbreaks and play a crucial role in maintaining the natural focus of plague and the continuity of the biological community (39).
As a classical model for structural, functional and evolutionary studies, the mitochondrial genome holds significant value in the fields of biogenetics and species evolution (18, 25). To address the scarcity of mitochondrial genome resources for fleas and to provide molecular insights into flea prevention and control, as well as to enhance our understanding of phylogenetic relationships, we conducted a study involving the sequencing of the complete mitogenomes of M. euteles and C. tesquorum sungaris. We also analyzed their genetic characteristics and phylogenetic relationships.
2 Methods
2.1 Sample collection and DNA extraction
The adult fleas of the M. euteles were collected from the body surface of Callosciurus erythraeus in Xiangyun County, Yunnan Province (25° 33′N, 100° 77′E), China. Meanwhile, specimens of C. tesquorum sungaris were obtained from Cricetulus barabensis in Qian’an County, Jilin Province (44° 99′N, 124° 12′E), China. All flea specimens were preserved in EP tubes containing 95% ethanol, transported to the laboratory for identification by professionals based on morphological characteristics (53), and subsequently stored at −20°C for future use. Prior to DNA extraction, the specimens underwent cleaning with sterile double distilled water followed by drying on sterile filter paper. Genomic DNA was then separated using a DNA Extraction Kit (TIANGEN BIOTECH, Beijing, China) according to the manufacturer’s instructions.
2.2 Mitochondrial sequencing and assembly
Sequencing was conducted using second-generation sequencing (NGS) on the Illumina NovaSeq platform to construct gene libraries, perform paired-end sequencing of these libraries to obtain raw gene fragment data, and conduct preliminary assembly of quality-controlled clean data to generate all possible scaffolds. Mitochondrial genomes from closely related species with available sequences were used as reference datasets, and the resulting assembled sequences were compared using BLAST for de novo assembly. After generating raw data in FASTQ format, we calculated the number of reads, total base count, and GC content. Data filtering criteria primarily involved utilizing AdapterRemoval version 2 (43) software for removing 3’end adapters, thereby eliminating contamination and ensuring high-quality data. High-quality data underwent de novo assembly using SPAdes v3.15.5 (38), followed by correction using Pilon v1.19 (50) to obtain the final mitochondrial sequence.
2.3 Gene annotation and data analysis
The mitogenome assembly was performed using A5-miseq v20150522 (8). The mitochondrial genomes of M. euteles and C. tesquorum sungaris were annotated via the MITOS WebServer1 (3). Circular maps of the mitogenomes were generated using the CGView Server2 (46). Relative synonymous codon usage (RSCU) was evaluated with CodonW v1.4.2, while nucleotide composition analysis was conducted using DNA star v11.1. The skewness of the relative base composition was calculated using the formulas GC-skew = (G − C)/(G + C) and AT-skew = (A − T)/(A + T) (37). The DnaSP v6.12.3 (42) was utilized to conduct analyses of evolutionary rates and nucleotide diversity. The former was evaluated through the ratios of non-synonymous (Ka) to synonymous (Ks) substitutions (Ka/Ks), while the latter was assessed using a sliding window approach with a window size of 100 and a step size of 25.
2.4 Phylogenetic relationships analysis
To elucidate the phylogenetic relationships within the order Siphonaptera, mitochondrial genome sequences from 26 flea species were employed to construct phylogenetic trees. This dataset included two newly sequenced species from this study and an additional 24 species obtained from the NCBI database (Table 1). Boreus elegans (HQ696579) was designated as outgroup, and 13 protein-coding genes (PCGs) were utilized for the phylogenetic analysis of these 27 species. The amino acid and nucleotide sequences were aligned using MAFFT v7.520 (20), followed by trimming of ambiguous regions with Gblocks v0.91b (47). Subsequently, the sequences were concatenated in PhyloSuite v1.2.3 (54) to produce two distinct datasets: one comprising the amino acid sequence dataset (PCGaa) and the other consisting of the nucleotide sequence dataset (PCGnt).
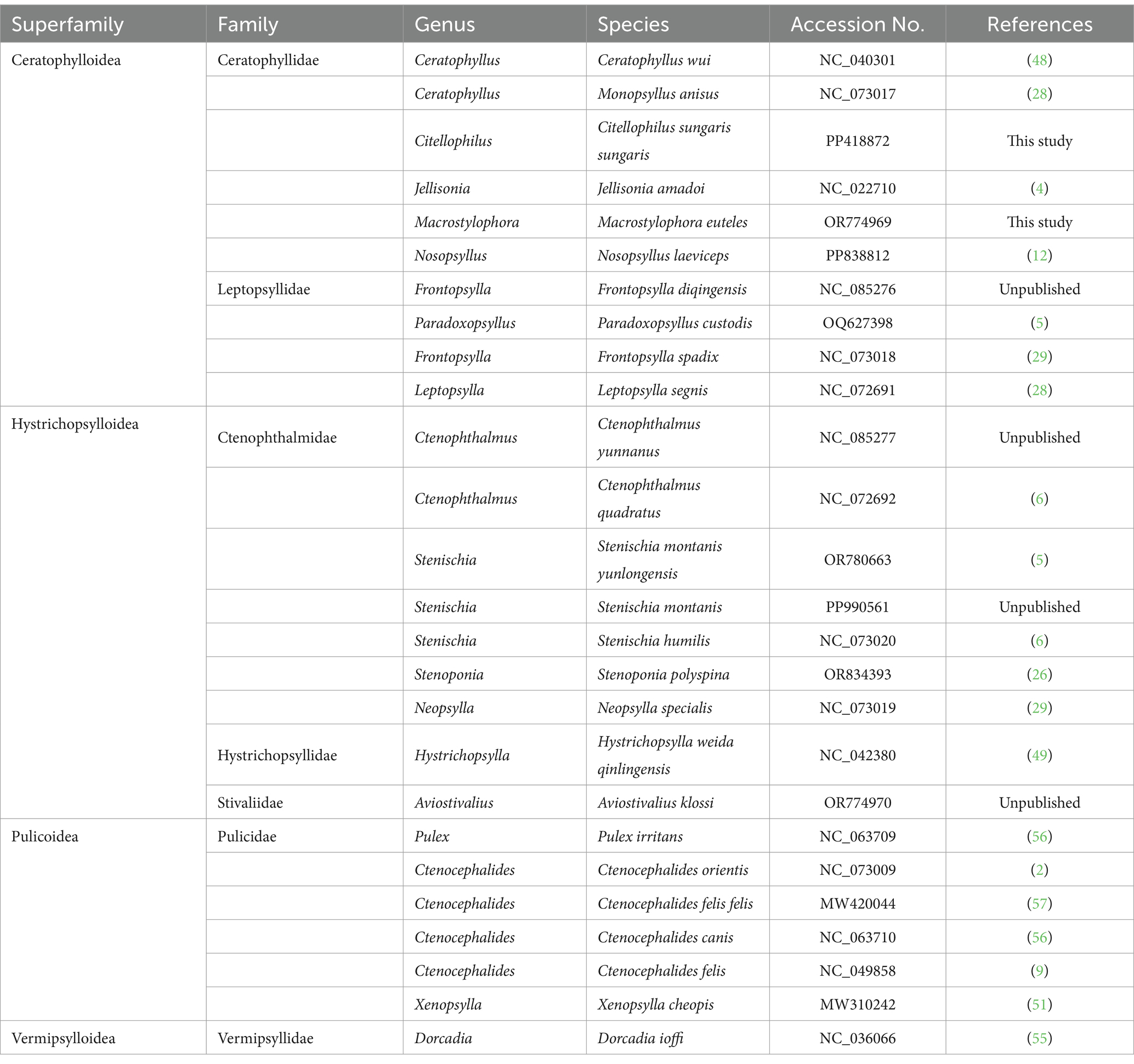
Table 1. Taxonomic information and GenBank accession numbers of 26 mitogenomes of Siphonaptera were selected for characterization and phylogenetic analysis.
Phylogenetic relationships were evaluated using maximum likelihood (ML) analysis with IQ-TREE v2.3.6 (35) and Bayesian inference (BI) with MrBayes v3.2.7 (40). For the ML analysis, the optimal model for the nucleotide dataset of PCGs was determined to be GTR + F + I + G4, while the most appropriate model for the amino acid dataset of PCGs was identified as mtART + F + I + G4 using ModelFinder (19), with a bootstrap value of 1,000 replicates. The BI analysis involved four simultaneous Markov Chain Monte Carlo (MCMC) runs over two million generations, sampling every 1,000 generations, with the first 25% of data discarded as burn-in. The final phylogenetic tree was visualized and refined utilizing FigTree v1.4.43 and the Interactive Tree of Life (iTOL) v6.9.1 (22).
3 Results
3.1 Mitogenome structure and nucleotide composition
In this study, we conducted a thorough analysis of the mitochondrial genomes of two newly sequenced flea species alongside 24 fleas previously published in the NCBI database. In the newly sequenced species M. euteles and C. tesquorum sungaris, a total of 37 genes were identified, comprising 13 PCGs, 22 transfer RNA (tRNA) genes, and 2 ribosomal RNA (rRNA) genes (Supplementary Figure S1). Of these 37 genes, 23 are situated on the heavy strand (H strand), while the remaining 14 are located on the light strand (L strand) (Supplementary Table S1). The mitochondrial genomes of M. euteles and C. tesquorum sungaris measure 16,027 and 15,345 bp in length, respectively. The average mitochondrial genome length for Siphonaptera is 16,963 bp, with Ctenocephalides orientis exhibiting the longest genome at 22,189 bp and Stenoponia polyspina having the shortest at merely 14,933 bp (Table 2). Using cox1 as a reference, we observed that the mitochondrial gene arrangements in 26 flea species are consistent with the putative ancestral mitochondrial genome arrangement of Drosophila yakuba and previously sequenced Mecoptera species, providing no evidence of gene rearrangement.

Table 2. The base features of Siphonaptera mitochondrial genomes.
The mitogenomes of fleas demonstrate a pronounced AT bias, with AT content varying from 76.71% in Ceratophyllus wui to 83.21% in C. orientis, resulting in an average AT content of 79.26%. The AT-skew ranged from −0.0511 in C. orientis to 0.0236 in Leptopsylla segnis, while GC-skew spanned from −0.2682 in M. euteles to 0.2477 in L. segnis. Notably, L. segnis was the only species exhibiting a positive GC-skew, whereas all other species showed negative values (Table 2).
3.2 PCGs, evolution rate and nucleotide diversity
The total length of 13 PCGs in M. euteles and C. tesquorum sungaris was 11,114 bp and 11,030 bp, respectively. Notably, C. tesquorum sungaris exhibited the shortest PCGs among the 26 flea species analyzed. The AT content within the PCGs of all examined flea species was significantly high, with Xenopsylla cheopis displaying the highest level at 80.31%. The AT-skew of PCGs ranged from −0.1557 to −0.1174, indicating a consistent negative trend. Conversely, GC-skew varied between −0.0060 and 0.0440, remaining positive except for Neopsylla specialis and L. segnis (Table 2). Analysis of RSCU in M. euteles and C. tesquorum sungaris revealed 27 and 26 high-frequency codons (RSCU>1), respectively. In both species, UUA was the most frequently used codon, while the least frequently used codons were CGC in M. euteles and UCG in C. tesquorum sungaris (Figure 1). Within the order Siphonaptera, the majority of PCGs utilized the canonical ATN as their start codon, with ATG being the most prevalent. Specifically, all 26 flea species examined used ATG as the initiating codon for cox3, nad4, and nad4l. In contrast, nonstandard start codons such as AAA, GCA, GTG, and GTA were identified in cox1, while TTG was observed in atp6 and atp8 (Figure 2A). Regarding stop codons, TAA was predominantly utilized by the 13 PCGs (Figure 2B).
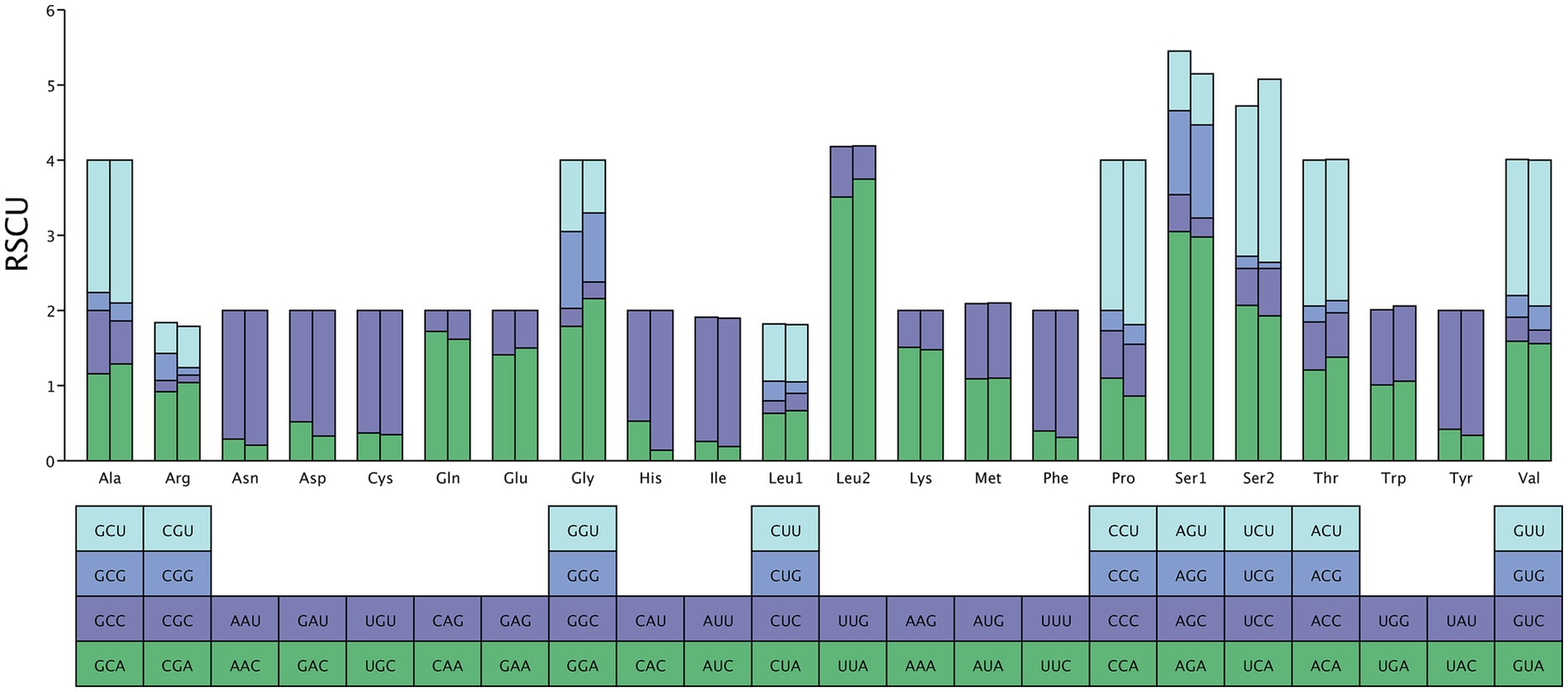
Figure 1. The relative codon usage of two newly sequenced mitochondrial genome, with M. euteles on the left and C. tesquorum sungaris on the right.
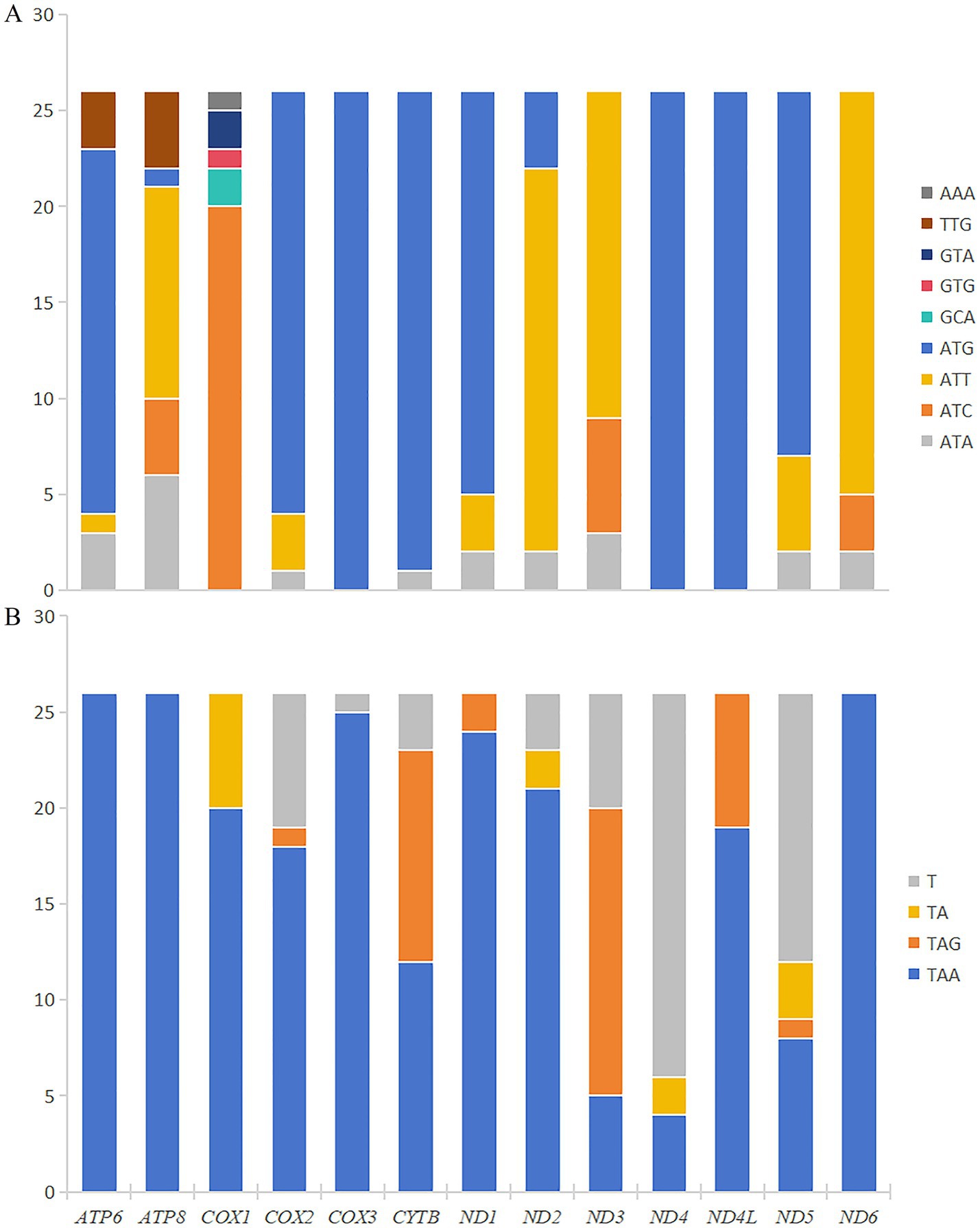
Figure 2. Comparison of start codons (A) and stop codons (B) of PCGs of 26 species.
The evolutionary rates and nucleotide diversity of 13 PCGs in the mitochondria of 26 flea species were evaluated through the analysis of Ka/Ks ratios and Pi values. The findings indicated that the average Ka/Ks ratios for each PCG ranged from 0.079 to 0.802, with all values remaining below 1, thereby suggesting that these genes have experienced purifying selection throughout their evolutionary history. Notably, atp8 exhibited the highest rate of evolution, reflecting a relatively low intensity of purifying selection pressure. In contrast, cox1 displayed the lowest mean Ka/Ks ratio and underwent significant purifying selection (Figure 3). Significantly higher nucleotide sequence variability (Pi values > 0.30) was observed in atp8 and nad2 genes, while markedly lower sequence variability (Pi values < 0.18) was noted in cox1 and cox2 genes (Figure 4).
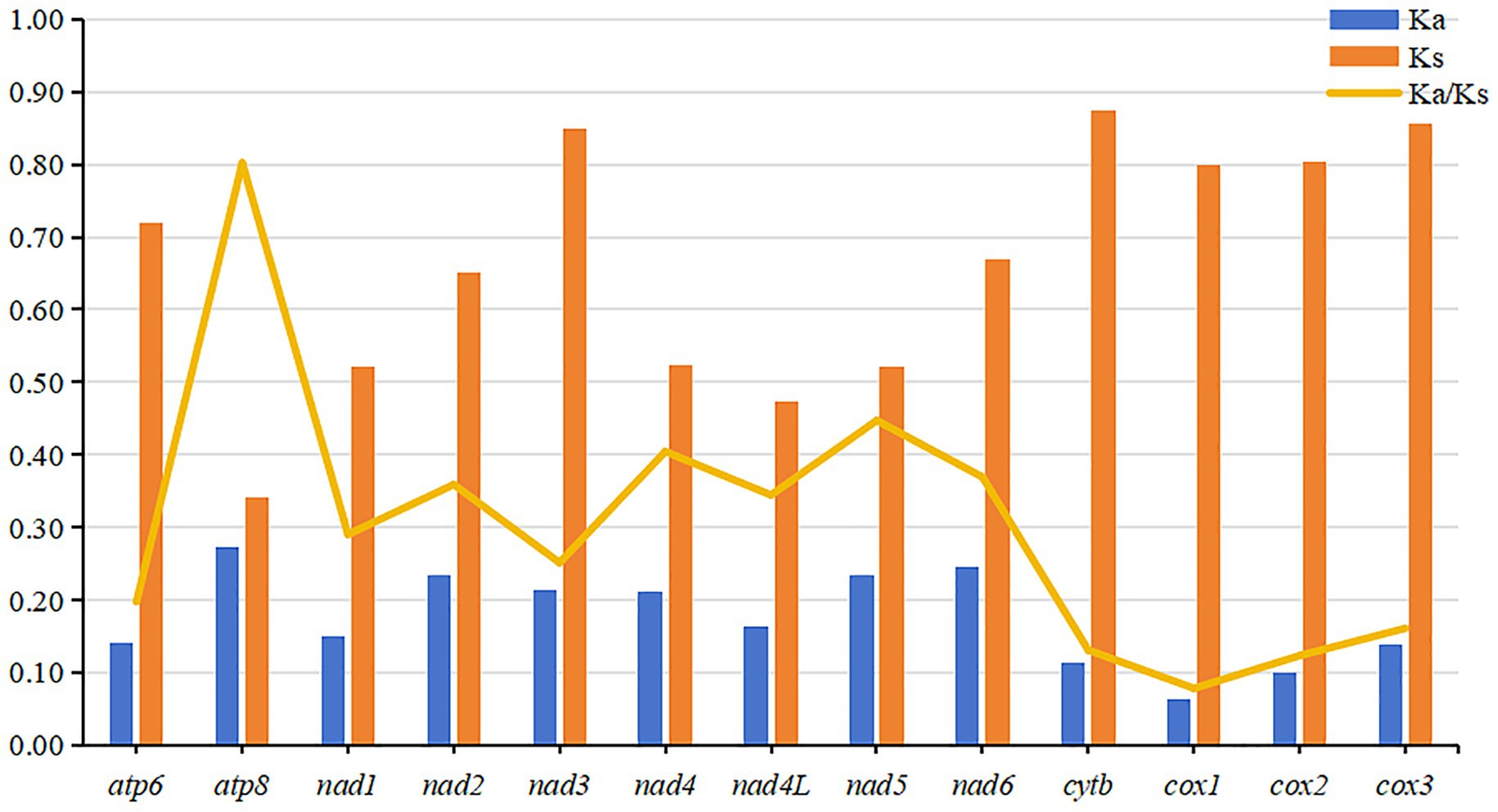
Figure 3. The evolutionary rates, represented by the ratio of nonsynonymous to synonymous substitutions (Ka/Ks), of 13 mitochondrial protein-coding genes (PCGs) across 26 flea species.
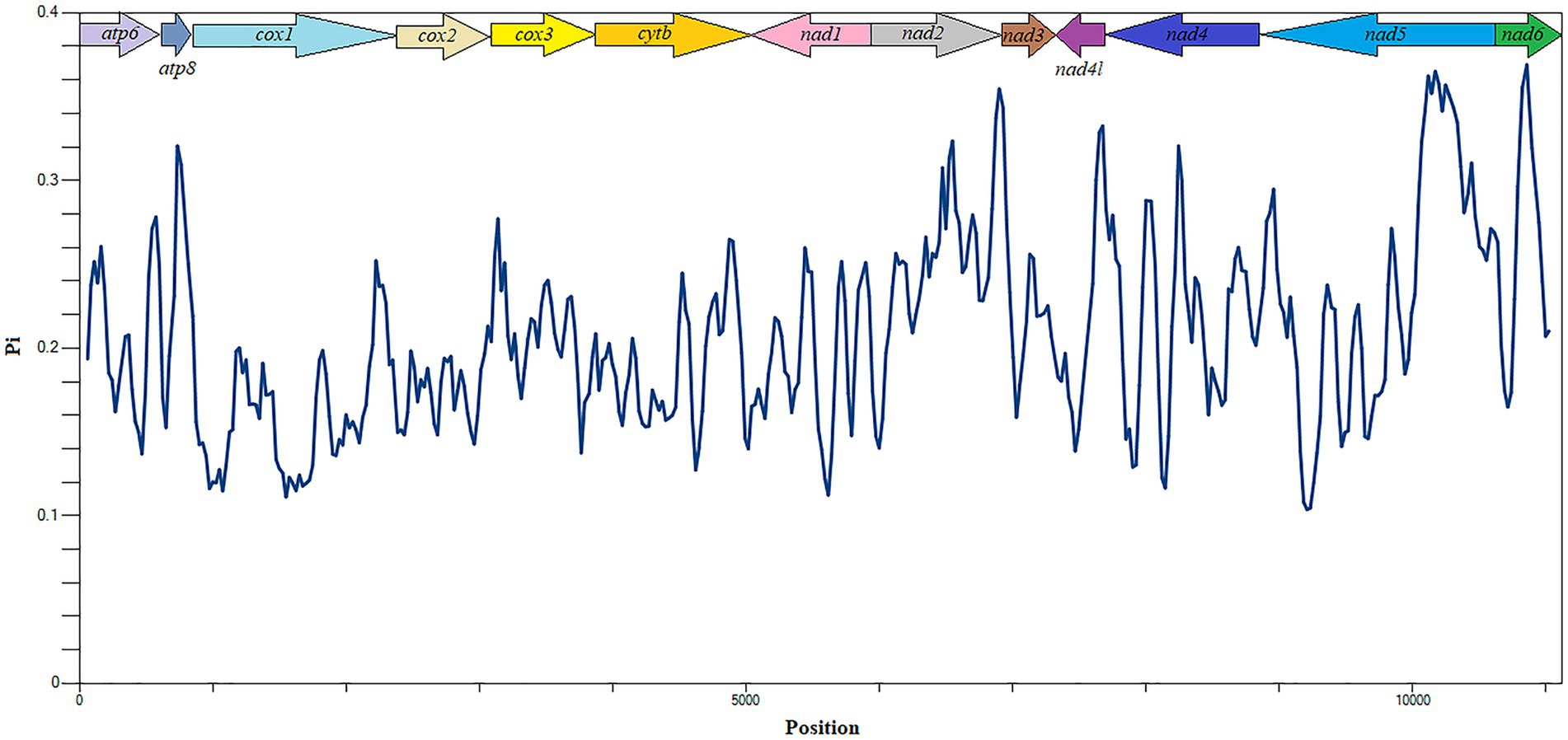
Figure 4. Nucleotide diversity analysis of 13 PCGs in Siphonaptera mitochondrial genomes.
3.3 Transfer RNA and ribosomal RNA genes
We performed a comprehensive analysis of 22 tRNA genes from the mitogenomes of 26 species. Our observations revealed that the sizes of these tRNAs ranged from 54 to 74 bp, with total lengths of tRNA genes varying between 1,405 and 1,439 bp, and AT content fluctuating between 78.84 and 81.12% (Table 2). Among the analyzed tRNA genes, 14 were encoded by the H strand, while the remaining 8 were encoded by the L strand.
The ribosomal RNA (rRNA) genes include the large ribosomal subunit RNA (rrnL) and the small ribosomal subunit RNA (rrnS). In all 26 flea species, the rrnL genes was located between the leucine transfer RNA (tRNA-Leu, trnL1 (UAG)) and valine transfer RNA (tRNA-Val, trnV), with a size ranging from 1,196 to 1,317 bp. The AT content varied from 80.43 to 83.76%. Conversely, the rrnS gene was situated between the trnV gene and control region, measuring between 774 and 799 bp, with AT content ranging from 78.97 to 82.94% (Table 2).
3.4 Phylogenetic inference
Based on the nucleotide and amino acid sequences of 13 PCGs from 26 flea species across seven families within the order Siphonaptera, phylogenetic analysis using BI and ML methods generated four distinct tree topologies (Figures 5, 6). In the amino acid tree, all families were divided into two primary clades. The first clade comprised six families (Bpp = 0.991, Bv = 36): Ceratophyllidae, Leptopsyllidae, Ctenophthalmidae, Hystrichopsyllidae, Stivaliidae and Vermipsyllidae. The second clade consisted solely of Pulicidae (Bpp = 1, Bv = 100). Notably, in the nucleotide tree, Stivaliidae clustered with Pulicidae, but this arrangement was only supported in the ML tree (Bv = 36). Across all phylogenetic trees, the superfamilies Ceratophylloidea and Pulicoidea, as well as the family Pulicidae, consistently exhibited strong monophyly with support values of 100 in the ML analysis and 1 in the BI analysis. Conversely, the superfamily Hystrichopsylloidea, along with the families Ceratophyllidae, Leptopsyllidae and Ctenophthalmidae, were identified as paraphyletic. However, due to the limited representation of species within the families Pygiopsyllidae, Hystrichopsyllidae, and Vermipsyllidae (each containing only one species), their monophyly could not be conclusively determined. The four topologies indicated that M. euteles was most closely related to Paradoxopsyllus custodis (PCGnt: Bpp = 1, Bv = 100; PCGaa: Bpp = 1; Bv = 100), while C. tesquorum sungaris was closely associated with (Monopsyllus anisus + Ceratophyllus wui) (PCGnt: Bpp = 1, Bv = 98; PCGaa: Bpp = 1, Bv = 85). Additionally, the insertion P. custodis contributed to the paraphyly observed in the family Ceratophyllidae.
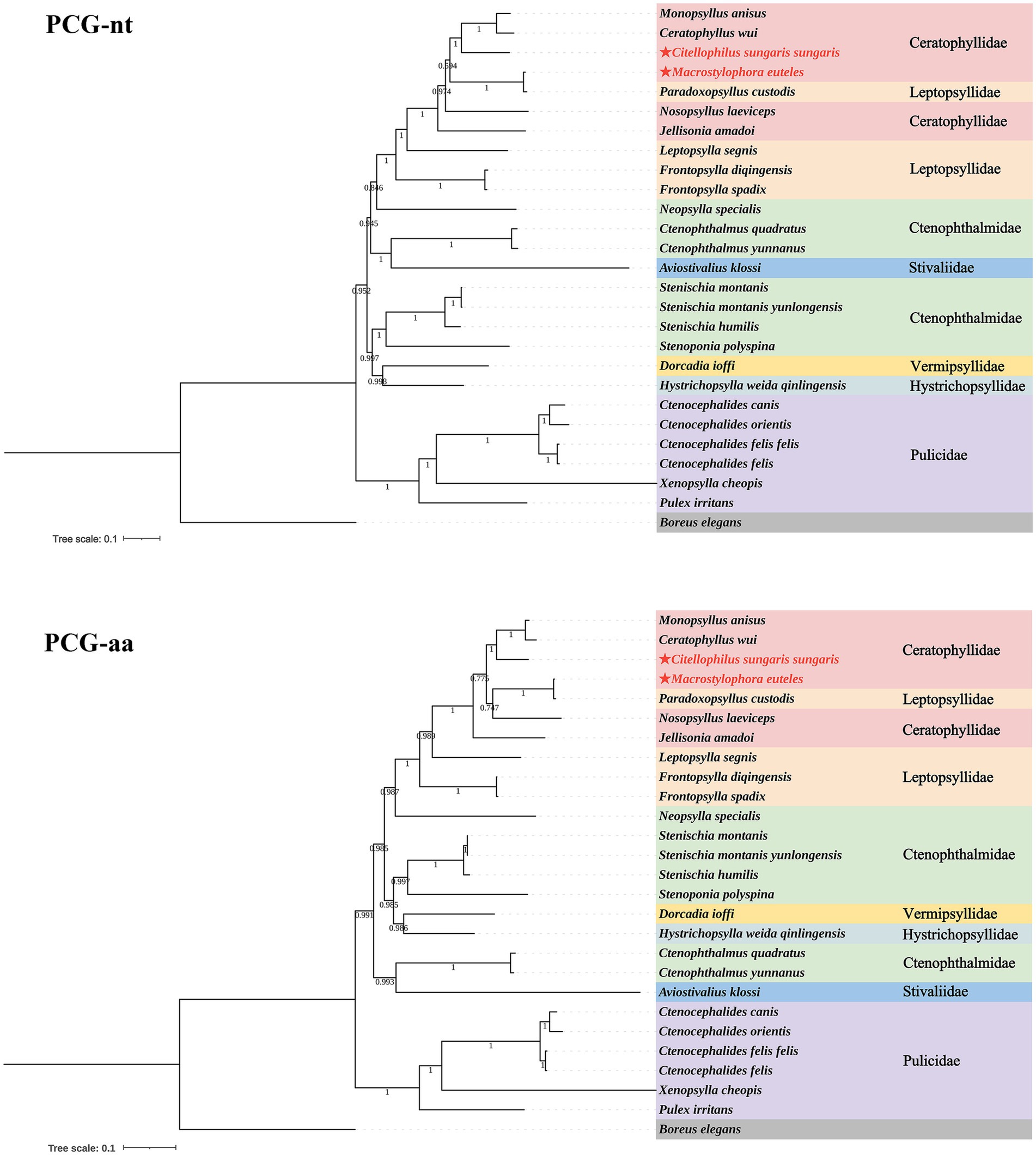
Figure 5. Phylogenetic trees for 26 species within the order Siphonaptera were constructed utilizing the Bayesian inference (BI) method.
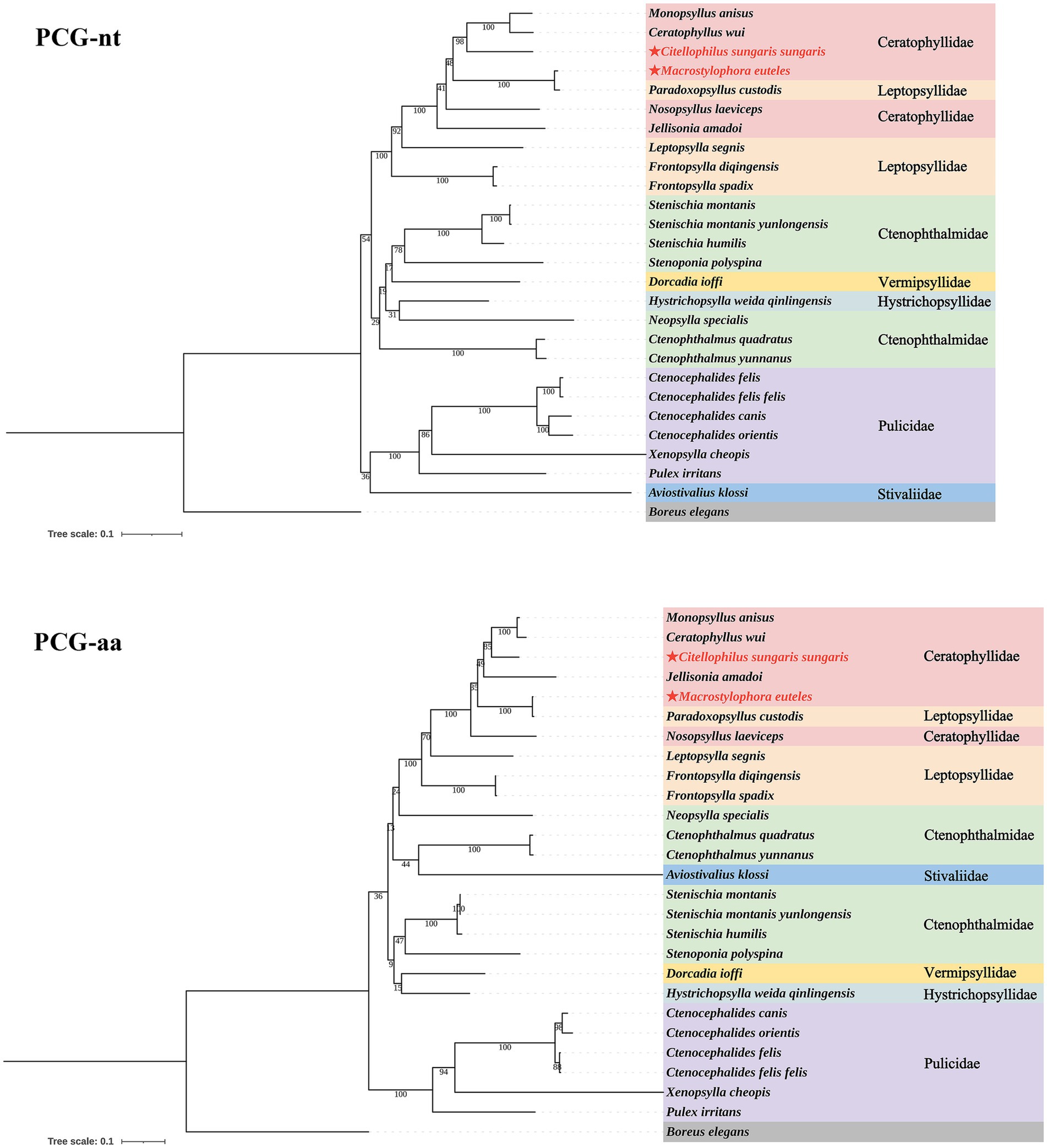
Figure 6. Phylogenetic trees for 26 species within the order Siphonaptera were constructed utilizing the maximum likelihood (ML) method.
4 Discussion
The precise identification of fleas is a critical component in the study of these parasites and the diagnosis of flea-borne diseases. In this study, we sequenced the mitogenomes of M. euteles and C. tesquorum sungaris. By integrating data from the NCBI database, we conducted a comparative analysis of the mitogenomes of 26 flea species to elucidate the characteristics of Siphonaptera mitogenomes. The gene order in the flea mitogenome is conserved and aligns with that of the presumed ancestral mitogenome of insects (7). Among 26 flea species, the proportion of AT bases in their mitogenomes exceeds that of GC bases, indicating a pronounced AT bias. In Mecopterida, the sequences of Siphonaptera exhibited a higher AT content compared to those of Diptera and Mecoptera (45). The AT skew values were negative across all species, with the exception of L. segnis (AT-skew = 0.0236) and Neopsylla specialis (AT-skew = 0.0001). Similarly, the GC skew values were negative for all species except L. segnis (GC-skew = 0.2477). This observed strand bias could be attributed to asymmetric mutation pressures during DNA replication and/or transcription, possibly involving the deamination of adenine and cytosine nucleotides (15).
ATN is regarded as the predominant start codon in the class Insecta. In this study, ATN was identified as the most frequent start codon, whereas TTG was observed in atp6 and atp8, and other rare start codons were found in cox1. Common stop codons in Siphonaptera are TAA and TAG; however, some species utilize incomplete stop codons such as T or TA, which can be converted to the complete stop codon TAA through polyadenylation post-transcription (1). The ratio of non-synonymous substitution rate (Ka) to synonymous substitution rate (Ks) exceeded 1, suggesting that the associated protein-coding genes were subject to positive selection. This positive selection pressure enabled the species to continually enhance its environmental adaptability. A Ka/Ks ratio of 1 indicates that protein-coding genes evolved neutrally, unaffected by natural selection. Conversely, a Ka/Ks ratio less than 1 signifies that protein-coding genes are under negative selection, which serves to constrain mitochondrial gene mutations, ensuring the stable function of proteins involved in mitochondrial oxidative phosphorylation (16). The Ka/Ks ratios for all 13 protein-coding genes were below 1, indicating that these genes maintained functional stability across the 26 flea species during evolution.
Evident inconsistencies were observed in the treatment of superfamily relationships across several previously published taxonomic classifications (24, 31–33, 44). This study confirmed the monophyly of the superfamilies Ceratophylloidea and Pulicoidea, whereas the superfamily Hystrichopsylloidea was found to be paraphyletic. These findings are consistent with a recent phylogenetic analysis based on five molecular markers (58) as well as mitochondrial genome data (5, 26). Whiting et al. (52) conducted the first comprehensive analysis of fleas using a four-locus molecular matrix, supporting the monophyly of the families Ceratophyllidae and Pulicidae. In contrast, Leptopsyllidae, Hystrichopsyllidae, and Ctenophthalmidae were identified as forming paraphyletic assemblages. Recent phylogenetic analyses employing maximum likelihood methods and utilizing five molecular markers (18S rDNA, 28S rDNA, EF-1α, cox1, and cox2) indicated that the families Leptopsyllidae, Ceratophyllidae, Pulicidae, and Pygiopsyllidae form monophyletic groups, whereas the family Ctenophthalmidae was found to be paraphyletic (58). However, most recent phylogenetic studies based on mitochondrial data of Siphonaptera suggest that the family Ceratophyllidae may be paraphyletic (12, 26), consistent with the findings reported herein. It should be noted that the current study encompasses only approximately 1% of known flea species. To comprehensively evaluate the phylogenetic relationships within the order Siphonaptera and achieve more robust conclusions, it is imperative to decode a greater number of mitochondrial genomes from this order.
5 Conclusion
In this study, we conducted the first comprehensive analysis of the mitochondrial genomes of Macrostylophora euteles and Citellophilus tesquorum sungaris. The phylogenetic tree constructed based on the mitochondrial genome dataset exhibited distinct topology compared to previous studies, indicating the necessity for additional genomic data to elucidate the phylogeny of fleas more accurately. This research will provide valuable molecular data to support taxonomic and phylogenetic studies of fleas.
Data availability statement
All data and materials of the study are included in the manuscript and the Supplementary material. The complete mitochondrial genome data of M. euteles and C. tesquorum sungaris have been stored on the NCBI website under the accession number OR774969 and PP418872.
Ethics statement
The animal study was approved by the Laboratory Animal Management Committee of Dali University (Approval No. 2022-P2-280). The study was conducted in strict compliance with both local legislation and institutional guidelines.
Author contributions
MD: Conceptualization, Data curation, Writing – original draft, Writing – review & editing. YL: Conceptualization, Data curation, Writing – original draft, Writing – review & editing. JW: Conceptualization, Funding acquisition, Writing – original draft, Writing – review & editing. SL: Data curation, Writing – review & editing. ST: Data curation, Writing – review & editing. DJ: Funding acquisition, Methodology, Writing – review & editing. QZ: Project administration, Supervision, Writing – review & editing. WG: Project administration, Supervision, Writing – review & editing. XY: Funding acquisition, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by the Special Basic Cooperative Research Programs of Yunnan Provincial Undergraduate Universities’ Association (Grant No. 202401BA070001-005), the Yunnan Fundamental Research Projects (Grant No. 202401AT070084), the Special Basic Cooperative Research Programs of Yunnan Provincial Undergraduate Universities’ Association (Grant No. 202301BA070001-045), and Scientific Research Fund of Yunnan Education Department (Grant No. 2024J0857).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fvets.2025.1558328/full#supplementary-material
Footnotes
References
1. Beckenbach, AT, and Stewart, JB. Insect mitochondrial genomics 3: the complete mitochondrial genome sequences of representatives from two Neuropteroid orders: a dobsonfly (order Megaloptera) and a Giant lacewing and an Owlfly (order Neuroptera). Genome. (2009) 52:31–8. doi: 10.1139/G08-098
2. Beliavskaia, A, Tan, KK, Sinha, A, Husin, NA, Lim, FS, Loong, SK, et al. Metagenomics of culture isolates and insect tissue illuminate the evolution of Wolbachia, Rickettsia and Bartonella symbionts in Ctenocephalides Spp. Fleas. Microb Genom. (2023) 9:1045. doi: 10.1099/mgen.0.001045
3. Bernt, M, Donath, A, Jühling, F, Externbrink, F, Florentz, C, Fritzsch, G, et al. Mitos: improved De Novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. (2013) 69:313–9. doi: 10.1016/j.ympev.2012.08.023
4. Cameron, SL. The complete mitochondrial genome of a flea, Jellisonia Amadoi (Siphonaptera: Ceratophyllidae). Mitochondrial DNA. (2015) 26:289–90. doi: 10.3109/19401736.2013.825779
5. Chen, B, Duan, M, Liu, S, Liu, Y, Tang, S, Jiang, D, et al. The complete mitochondrial genome and phylogenetic implications of Paradoxopsyllus Custodis and Stenischia Montanis Yunlongensis. Sci Rep. (2024) 14:31555. doi: 10.1038/s41598-024-84175-9
6. Chen, B, Liu, YF, Lu, XY, Jiang, DD, Wang, X, Zhang, QF, et al. Complete mitochondrial genome of Ctenophthalmus Quadratus and Stenischia Humilis in China provides insights into fleas phylogeny. Front Vet Sci. (2023) 10:1255017. doi: 10.3389/fvets.2023.1255017
7. Clary, DO, and Wolstenholme, DR. The mitochondrial DNA molecular of Drosophila Yakuba: nucleotide sequence, Gene Organization, and genetic code. J Mol Evol. (1985) 22:252–71. doi: 10.1007/BF02099755
8. Coil, D, Jospin, G, and Darling, AE. A5-Miseq: an updated pipeline to assemble microbial genomes from Illumina Miseq data. Bioinformatics. (2015) 31:587–9. doi: 10.1093/bioinformatics/btu661
9. Driscoll, TP, Verhoeve, VI, Gillespie, JJ, Johnston, JS, Guillotte, ML, Rennoll-Bankert, KE, et al. A chromosome-level assembly of the cat flea genome uncovers rampant gene duplication and genome size plasticity. BMC Biol. (2020) 18:70. doi: 10.1186/s12915-020-00802-7
10. Durden, LA, and Beaucournu, JC. Three new species of fleas belonging to the genus Macrostylophora from the three-striped ground squirrel, Lariscus Insignis, in Java. Med Vet Entomol. (2014) 28:398–406. doi: 10.1111/mve.12059
11. Eisen, RJ, and Gage, KL. Transmission of flea-borne zoonotic agents. Annu Rev Entomol. (2012) 57:61–82. doi: 10.1146/annurev-ento-120710-100717
12. Fu, YT, Xun, Y, Peng, YY, Zhang, Y, and Wu, X. The complete mitochondrial genome of the rodent flea Nosopsyllus Laeviceps: genome description, comparative analysis, and phylogenetic implications. Parasit Vectors. (2024) 17:253. doi: 10.1186/s13071-024-06329-y
13. Gong, ZD, Yu, X, Liu, QY, Ye, RY, Lu, L, Xu, L, et al. Ecological-geographic landscapes of natural plague foci in China vi. Biological characteristics of natural vectors of Yesinia Pestis. Chin J Epidemiol. (2012) 33:818–22. doi: 10.3760/cma.j.issn.0254-6450.2012.08.014
14. Hamzaoui, BE, Zurita, A, Cutillas, C, and Parola, P. Fleas and flea-borne diseases of North Africa. Acta Trop. (2020) 211:105627. doi: 10.1016/j.actatropica.2020.105627
15. Hassanin, A, Léger, N, and Deutsch, J. Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of metazoa, and consequences for phylogenetic inferences. Syst Biol. (2005) 54:277–98. doi: 10.1080/10635150590947843
16. Hurst, LD. The Ka/Ks ratio: diagnosing the form of sequence evolution. Trends Genet. (2002) 18:486–7. doi: 10.1016/S0168-9525(02)02722-1
17. Iannino, F, Sulli, N, Maitino, A, Pascucci, I, Pampiglione, G, and Salucci, S. Fleas of dog and cat: species, biology and flea-borne diseases. Vet Ital. (2017) 53:277–88. doi: 10.12834/VetIt.109.303.3
18. Jiang, M, Ni, Y, Zhang, J, Li, J, and Liu, C. Complete mitochondrial genome of Mentha Spicata L. reveals multiple chromosomal configurations and Rna editing events. Int J Biol Macromol. (2023) 251:126257. doi: 10.1016/j.ijbiomac.2023.126257
19. Kalyaanamoorthy, S, Minh, BQ, Wong, TKF, von Haeseler, A, and Jermiin, LS. Modelfinder: fast model selection for accurate phylogenetic estimates. Nat Methods. (2017) 14:587–9. doi: 10.1038/nmeth.4285
20. Katoh, K, and Standley, DM. Mafft multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. (2013) 30:772–80. doi: 10.1093/molbev/mst010
21. Lareschi, M. Siphonaptera (order): fleas In: SL Gardner and SA Gardner, editors. Concepts in animal parasitology. Lincoln, Nebraska: United States (2024). 756–70.
22. Letunic, I, and Bork, P. Interactive tree of life (Itol) V6: recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. (2024) 52:W78–82. doi: 10.1093/nar/gkae268
23. Lewis, RE. Fleas (Siphonaptera) In: RP Lane and RW Crosskey, editors. Medical insects and arachnids. Dordrecht: Springer Netherlands (1993). 529–75.
24. Lewis, RE. Notes on the geographical distribution and host preferences in the order Siphonaptera. Part 8. New taxa described between 1984 and 1990, with a current classification of the order. J Med Entomol. (1993) 30:239–56. doi: 10.1093/jmedent/30.1.239
25. Li, J, Chen, Y, Liu, Y, Wang, C, Li, L, and Chao, Y. Complete mitochondrial genome of Agrostis Stolonifera: insights into structure, codon usage, repeats, and Rna editing. BMC Genomics. (2023) 24:466. doi: 10.1186/s12864-023-09573-1
26. Lin, X, Pu, J, and Dong, W. The first Mitogenome of the subfamily Stenoponiinae (Siphonaptera: Ctenophthalmidae) and implications for its phylogenetic position. Sci Rep. (2024) 14:18179. doi: 10.1038/s41598-024-69203-y
27. Linardi, PM, and Santos, JL. Ctenocephalides Felis Felis vs. Ctenocephalides Canis (Siphonaptera: Pulicidae): some issues in correctly identify these species. Rev Bras Parasitol Vet. (2012) 21:345–54. doi: 10.1590/S1984-29612012000400002
28. Liu, YF, Chen, B, Lu, XY, Jiang, DD, Wang, T, Geng, L, et al. Complete Mitogenomes characterization and phylogenetic analyses of Ceratophyllus Anisus and Leptopsylla Segnis. Front Vet Sci. (2023) 10:1218488. doi: 10.3389/fvets.2023.1218488
29. Liu, YF, Chen, B, Lu, XY, Liu, S, Jiang, DD, Wang, X, et al. Analysis of complete Mitogenomes and phylogenetic relationships of Frontopsylla spadix and Neopsylla Specialis. Front Vet Sci. (2023) 10:1250381. doi: 10.3389/fvets.2023.1250381
30. Maleki-Ravasan, N, Solhjouy-Fard, S, Beaucournu, JC, Laudisoit, A, and Mostafavi, E. The fleas (Siphonaptera) in Iran: diversity, host range, and medical importance. PLoS Negl Trop Dis. (2017) 11:e0005260. doi: 10.1371/journal.pntd.0005260
31. Mardon, DK. On the relationships, classification, Aedeagal morphology and zoogeography of the genera of Pygiopsyllidae (Insecta: Siphonaptera). Aust J Zool Suppl Ser. (1978) 26:1–69. doi: 10.1071/AJZS064
32. Medvedev, S. Morphological basis of the classification of fleas (Siphonaptera). Entomol Rev. (1994) 73:30–51.
33. Medvedev, S. Classification of fleas (order Siphonaptera) and its theoretical foundations. Entomol Rev. (1998) 78:1080–93.
34. Medvedev, SG, Kotti, BK, and Verzhutsky, DB. Diversity of fleas (Siphonaptera), vectors of plague pathogens: the flea Citellophilus Tesquorum (Wagner, 1898), a parasite of ground squirrels of the genus Spermophilus. Entmol Rev. (2019) 99:565–79. doi: 10.1134/S0013873819050014
35. Minh, BQ, Schmidt, HA, Chernomor, O, Schrempf, D, Woodhams, MD, von Haeseler, A, et al. Iq-tree 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. (2020) 37:1530–4. doi: 10.1093/molbev/msaa015
36. Nisbet, AJ, and Huntley, JF. Progress and opportunities in the development of vaccines against mites, fleas and Myiasis-causing flies of veterinary importance. Parasite Immunol. (2006) 28:165–72. doi: 10.1111/j.1365-3024.2006.00803.x
37. Perna, NT, and Kocher, TD. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J Mol Evol. (1995) 41:353–8. doi: 10.1007/BF01215182
38. Prjibelski, A, Antipov, D, Meleshko, D, Lapidus, A, and Korobeynikov, A. Using Spades De Novo assembler. Curr Protoc Bioinformatics. (2020) 70:e102. doi: 10.1002/cpbi.102
39. Reis, DASFDL, Filgueira Bezerra, M, Sobreira Bezerra, DASM, and Leal, NC. Rodent hosts and flea vectors in Brazilian plague foci: a review. Integr Zool. (2021) 16:810–9. doi: 10.1111/1749-4877.12480
40. Ronquist, F, Teslenko, M, van der Mark, P, Ayres, DL, Darling, A, Höhna, S, et al. Mrbayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. (2012) 61:539–42. doi: 10.1093/sysbio/sys029
41. Rousseau, J, Castro, A, Novo, T, and Maia, C. Dipylidium Caninum in the twenty-first century: epidemiological studies and reported cases in companion animals and humans. Parasit Vectors. (2022) 15:131. doi: 10.1186/s13071-022-05243-5
42. Rozas, J, Ferrer-Mata, A, Sánchez-DelBarrio, JC, Guirao-Rico, S, Librado, P, Ramos-Onsins, SE, et al. Dnasp 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol. (2017) 34:3299–302. doi: 10.1093/molbev/msx248
43. Schubert, M, Lindgreen, S, and Orlando, L. Adapterremoval V2: rapid adapter trimming, identification, and read merging. BMC Res Notes. (2016) 9:88. doi: 10.1186/s13104-016-1900-2
44. Smit, F. The fleas of New Zealand (Siphonapter). J R Soc N Z. (1979) 9:143–232. doi: 10.1080/03036758.1979.10419413
45. Song, F, Li, H, Jiang, P, Zhou, X, Liu, J, Sun, C, et al. Capturing the phylogeny of Holometabola with mitochondrial genome data and Bayesian site-heterogeneous mixture models. Genome Biol Evol. (2016) 8:1411–26. doi: 10.1093/gbe/evw086
46. Stothard, P, and Wishart, DS. Circular genome visualization and exploration using Cgview. Bioinformatics. (2005) 21:537–9. doi: 10.1093/bioinformatics/bti054
47. Talavera, G, and Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. (2007) 56:564–77. doi: 10.1080/10635150701472164
48. Tan, L, Guan, X, Zhang, L, Zhu, F, and Lei, C. The complete mitochondrial genome of the flea Ceratophyllus Wui (Siphonaptera: Ceratophyllidae). Mitochondrial DNA B Resour. (2018) 3:401–2. doi: 10.1080/23802359.2018.1456370
49. Tan, L, Yao, X, Liu, J, Lei, C, Huang, Q, and Hu, B. The complete mitochondrial genome of the flea Hystrichopsylla Weida Qinlingensis (Siphonaptera: Hystrichopsylla). Mitochondrial DNA B Resour. (2023) 8:501–3. doi: 10.1080/23802359.2022.2053367
50. Walker, BJ, Abeel, T, Shea, T, Priest, M, Abouelliel, A, Sakthikumar, S, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. (2014) 9:e112963. doi: 10.1371/journal.pone.0112963
51. Wei, F, Jia, X, Wang, Y, Yang, Y, Wang, J, Gao, C, et al. The complete mitochondrial genome of Xenopsylla Cheopis (Siphonaptera: Pulicidae). Mitochondrial DNA B Resour. (2022) 7:170–1. doi: 10.1080/23802359.2021.2017368
52. Whiting, MF, Whiting, AS, Hastriter, MW, and Dittmar, K. A molecular phylogeny of fleas (Insecta: Siphonaptera): origins and host associations. Cladistics. (2008) 24:677–707. doi: 10.1111/j.1096-0031.2008.00211.x
54. Xiang, CY, Gao, F, Jakovlić, I, Lei, HP, Hu, Y, Zhang, H, et al. Using Phylosuite for molecular phylogeny and tree-based analyses. iMeta. (2023) 2:e87. doi: 10.1002/imt2.87
55. Xiang, HT, Wen, FQ, and Wang, GL. The complete nucleotide sequence of the mitochondrial genome of Dorcadia Ioffi (Siphonaptera: Vermipsyllidae). Mitochondrial DNA B Resour. (2017) 2:389–90. doi: 10.1080/23802359.2017.1347901
56. Zhang, Y, Fu, Y-T, Yao, C, Deng, Y-P, Nie, Y, and Liu, G-H. Mitochondrial Phylogenomics provides insights into the taxonomy and phylogeny of fleas. Parasit Vectors. (2022) 15:223. doi: 10.1186/s13071-022-05334-3
57. Zhang, Y, Nie, Y, Deng, YP, Liu, GH, and Fu, YT. The complete mitochondrial genome sequences of the cat flea Ctenocephalides Felis Felis (Siphonaptera: Pulicidae) support the hypothesis that C. felis isolates from China and USA were the same C. F. Felis subspecies. Acta Trop. (2021) 217:105880. doi: 10.1016/j.actatropica.2021.105880
Keywords: fleas, Macrostylophora euteles , Citellophilus tesquorum sungaris, mitochondrial genome, gene structure, phylogenetic
Citation: Duan M, Liu Y, Wu J, Liu S, Tang S, Jiang D, Zhang Q, Gu W and Yang X (2025) The complete mitochondrial genomes of Macrostylophora euteles and Citellophilus tesquorum sungaris and the phylogenetics of known Siphonaptera mitogenomes. Front. Vet. Sci. 12:1558328. doi: 10.3389/fvets.2025.1558328
Edited by:
Nicola Pugliese, University of Bari Aldo Moro, ItalyReviewed by:
Michael Hastriter, Brigham Young University, United StatesMireille Harimalala, Institut Pasteur de Madagascar, Madagascar
Copyright © 2025 Duan, Liu, Wu, Liu, Tang, Jiang, Zhang, Gu and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Quanfu Zhang, MjYyMjA0NTgwN0BxcS5jb20=; Wei Gu, Z3c3NzdAMTYzLmNvbQ==; Xing Yang, eWFuZzA4MjIwMDEzQDE2My5jb20=
†These authors have contributed equally to this work and share first authorship