 Haisheng ZhaoXijing MaoCuilin Zhu
Haisheng ZhaoXijing MaoCuilin Zhu Xiaohan ZouFanzhen Peng
Xiaohan ZouFanzhen Peng Wei YangBingjin LiGuangquan LiTongtong Ge*
Wei YangBingjin LiGuangquan LiTongtong Ge* Ranji Cui*
Ranji Cui*- Jilin Provincial Key Laboratory on Molecular and Chemical Genetic, Second Hospital of Jilin University, Changchun, China
Autism spectrum disorder (ASD) refers to a series of neurodevelopmental diseases characterized by two hallmark symptoms, social communication deficits and repetitive behaviors. Gamma-aminobutyric acid (GABA) is one of the most important inhibitory neurotransmitters in the central nervous system (CNS). GABAergic inhibitory neurotransmission is critical for the regulation of brain rhythm and spontaneous neuronal activities during neurodevelopment. Genetic evidence has identified some variations of genes associated with the GABA system, indicating an abnormal excitatory/inhibitory (E/I) neurotransmission ratio implicated in the pathogenesis of ASD. However, the specific molecular mechanism by which GABA and GABAergic synaptic transmission affect ASD remains unclear. Transgenic technology enables translating genetic variations into rodent models to further investigate the structural and functional synaptic dysregulation related to ASD. In this review, we summarized evidence from human neuroimaging, postmortem, and genetic and pharmacological studies, and put emphasis on the GABAergic synaptic dysregulation and consequent E/I imbalance. We attempt to illuminate the pathophysiological role of structural and functional synaptic dysregulation in ASD and provide insights for future investigation.
Introduction
Autism spectrum disorder (ASD) refers to a series of neurodevelopmental diseases characterized by two hallmark symptoms, social communication deficits and repetitive behaviors (Happe, 1999). Clinical observations suggest that patients with ASD were often comorbid with intellectual disability (ID), mood disorders, epilepsy, and intestinal disorders (Happé and Frith, 1996; Rudolph and Möhler, 2014; Richard et al., 2017). The prevalence of autism has gradually increased over time and with significantly higher frequency in male infants than in female infants (Fatemi et al., 2013). A number of investigations have demonstrated that genetic, epigenetic, environmental perinatal factors and their complicated interaction were implicated in the neurophysiological mechanisms of ASD (Belmonte et al., 2004; Hallmayer et al., 2011; Cattane et al., 2020). However, high clinical heterogeneity challenges both the clinical diagnosis and treatment and further investigations of the cellular and molecular pathogenesis of ASD.
Synaptic neurotransmission, including excitatory and inhibitory neurotransmission, is the neurophysiological basis of various functions of the brain. Aberrant neurotransmission, especially the imbalance of excitation and inhibition in the central nervous system (CNS), might be implicated in the ASD pathogenesis. Based on Genome-Wide Association Study (GWAS) and human genetic evidence, some synaptic genes encoding the synthesis, release, and binding receptor proteins of neurotransmitters have been reported to contribute to the pathogenesis of ASD (Kumar and Christian, 2009). Gamma-aminobutyric acid (GABA) is a pivotal inhibitory neurotransmitter in the CNS. GABAergic inhibitory neurotransmission is critical for the regulation of brain rhythm and spontaneous neuronal activities during brain development. The imbalance between excitation and GABAergic inhibition might be consequently contributed to the dysfunction of the CNS GABAergic system. Increasing evidence has shown that dysfunction of inhibitory neurotransmission is implicated in neurophysiological mechanisms of neuropsychiatry disorders, including schizophrenia, depression, and anxiety (Amin et al., 2006; Möhler, 2012; Duman et al., 2019). Especially, it was widely studied that abnormal neuronal excitatory firing in specific brain regions caused by the absence of GABAergic inhibition underlies the neurophysiological mechanism of epilepsy (Puts and Edden, 2012; Kaila et al., 2014). GABAergic dysfunction might provide a potential reasonable explanation for high comorbidity with epilepsy or increased susceptibility to epilepsy observed in patients with autism (Richard et al., 2017).
In this review, we summarized evidence from genetic, human neuroimaging, postmortem, and genetic and pharmacological studies focusing on GABAergic dysfunction and investigating the consequent excitatory/inhibitory (E/I) imbalance in ASD. We put emphasis on the convergent and neurophysiological alterations of how GABA is involved in ASD from different lines of evidence. We aimed to illuminate the potentiality of GABA and GABA receptor (GABA R) modulators as potential therapeutic targets for ASD.
Evidence From In Vivo Neuroimaging Studies
Human evidence from proton magnetic resonance spectroscopy (1H-MRS), neuroimaging, enzyme-linked immunosorbent assay (ELISA), and postmortem brain studies reported altered levels of GABA metabolites and the ratio of GABA to metabolites, such as glutamate and creatine (Rolf et al., 1993; Dhossche et al., 2002; Thatcher et al., 2009; Al-Otaish et al., 2018). A study showed that the electroencephalogram (EGG) phase reset of 54 patients with ASD showed reduced number and/or strength of thalamo-cortical connections comparing with healthy subjects (Thatcher et al., 2009). Earlier evidence showed increased serotonin levels and decreased levels of amino acids, namely, aspartic acid, glutamine, glutamic acid, and GABA, in patients with autism in comparison with controls (Rolf et al., 1993). However, contradictory results exist that young autism patients exhibited higher plasma GABA levels which decreased with age (Dhossche et al., 2002). Several evidence also reported higher plasma GABA levels and lower glutamate/GABA levels in autistic subjects than in controls (El-Ansary and Al-Ayadhi, 2014; Al-Otaish et al., 2018). Al Otaish et al. (2018) found a higher glutamate/glutamine ratio and lower levels of plasma glutamine in ASD patients. Another study focusing on alterations of anti-neuronal antibody levels including anti-glutamic acid decarboxylase antibodies (GAD), anti-glutamate receptor antibodies, and seven types of anti-ganglioside antibodies levels by ELISA found no significant difference between autistic and control subjects (Bayram et al., 2016).
Recently, amounts of evidence using magnetic resonance spectroscopy (MRS) found that the lower ratio of GABA/glutamate (Harada et al., 2011; Kubas et al., 2012; Drenthen et al., 2016; Ajram et al., 2017), N-acetylaspartate/creatine, GABA/creatine, and glutamate/creatine (Kubas et al., 2012) in the frontal lobes, lower GABA/creatine levels in the anterior cingulate cortex (ACC) of patients with ASD than normal controls (Kubas et al., 2012; Cochran et al., 2015). The binocular rivalry dynamics exhibited in normal controls, which depend on the balance of inhibitory and excitatory cortical dynamics, is absent in participants with autism; these findings indicate reduced GABAergic action in the autistic brain (Robertson et al., 2016). The researchers have drawn a conclusion that children with autism have lower sensorimotor GABA levels via a tactile task detecting amplitude discrimination and frequency discrimination under the presence of an adapting stimulus (Puts et al., 2017). ASD patients exhibited lower GABA concentrations in the sensorimotor cortex, and the GABA concentrations were positively correlated with self-reported tactile hypersensitivity in adults with ASD (Sapey-Triomphe et al., 2019). It has been shown that children with autism showed higher GABA concentration in the bilateral visual cortex (VIS) which was found to be related to more efficient search and social impairments (Edmondson et al., 2020). Intriguingly, these results suggest that GABAergic system dysfunction in ASD patients, especially in children, was associated with higher cognitive function, including somatosensory function, search and social behaviors.
A recent investigation found that there was a negative correlation between thalamic GABA and autism-spectrum quotient (AQ) in male participants with autism, while the thalamic GABA levels were positively correlated with AQ in female participants (Fung et al., 2021). These results indicate that GABA levels might correlate with autism symptom severity in a gender-specific way. Considering the high clinical heterogeneity of autism, the altered GABA system might be more pronounced for a specific subtype of clinical ASD. Drenthen et al. found higher Glu/creatine and lower GABA/glutamate concentrations in the high-functioning autism (HFA) participants, which are consistent with previous research results (Drenthen et al., 2016). Evidence showed that children and adolescents with HFA have reduced total N-acetylaspartate (tNAA) and total creatine, increased glutamate +glutamine (Glx)/tNAA, but unchanged Glx and comparable GABA levels between healthy and autism groups (Carvalho Pereira et al., 2018). Intriguingly, they also observed that smaller absolute and relative GABA levels were associated with poor communication skills of patients with autism (Carvalho Pereira et al., 2018). These results indicate a critical relationship of the altered GABA system with clinical autism, but the levels of GABA metabolites and the ratio of GABA to metabolites are not sufficient to be a clinical target for diagnosis due to the poor precision and accuracy.
Evidence From In Vitro Postmortem Studies
Results from postmortem studies have shown that altered GABAergic signaling is involved in the neurophysiological mechanisms of autism. The autistic group had increased GAD 67 mRNA expression in cerebellar interneurons and reduced glutamic acid decarboxylase 65(GAD 65) mRNA in the larger labeled cells in the cerebellar dentate nuclei (Yip et al., 2008, 2009). It suggests that a disturbance of the intrinsic cerebellar circuitry in the autistic brain might affect autism-related cognitive and behaviors by affecting the Purkinje cell (PC) output to other key brain regions. Individuals with autism showed a selective increase in the density of calbindin (CB)+, calretinin (CR)+, and parvalbumin (PV)+ interneurons in the hippocampus than those of normal controls (Lawrence et al., 2010). A recent study using immunohistochemical staining found significantly decreased protein levels of α2 subunit of GABA-A receptor in the axon initial segment of pyramidal cells in supragranular layers of prefrontal cortical areas of autistic participants (Hong et al., 2020). Fatemi et al. reported downregulation of different subunits of GABA-A and GABA-B receptors both in mRNA and protein levels (Fatemi et al., 2009a; Fatemi et al., 2009b; Fatemi et al., 2010; Blatt and Fatemi, 2011; Fatemi and Folsom, 2011). They reported GABBR1, GABBR2, GABRA1, and GABRB3 were significantly decreased in the cerebellum (Fatemi et al., 2009a; Fatemi et al., 2009b), GABRA1, GABRA2, GABRA3, and GABRB3 were significantly reduced in the parietal cortex, and GABRA1 was significantly altered in the superior frontal cortex (Fatemi et al., 2009a).
Evidence from quantitative receptor autoradiographic studies was carried out to determine the density and distribution of excitatory and inhibitory receptors (Blatt et al., 2001; Guptill et al., 2007; Oblak et al., 2009; Lawrence et al., 2010). The binding sites of benzodiazepine, a positive allosteric modulator on GABA-A R, were significantly reduced in the hippocampus and ACC of the autistic brain, while other receptors were not significantly changed (Oblak et al., 2009). Further evidence found that reduced [(3)H]flunitrazepam-labeled benzodiazepines(BDZ) sites in the autistic hippocampus were due to a decrease in the binding site number or number of receptors but not due to the binding affinity (Blatt et al., 2001; Guptill et al., 2007; Lawrence et al., 2010). Besides the hippocampus, the cerebellar region can receive inputs widely from the frontal cortex via the granular layer; a significant decrease of BDZ binding sites in the supragranular layers and infragranular layers of the cerebellar region was found (Oblak et al., 2009). These results indicate that a significant dysregulation of the GABAergic system, from the synthesis to receptors, in autistic brains, might provide potential targets for therapeutics and diagnosis of autism and related neuropsychiatric diseases.
Evidence From Genetic Studies
ASD and other neurodevelopmental disorders which are characterized by repetitive behaviors and social deficits are highly hereditable. Genetic evidence showed that single-gene mutations are rare in families with autism. Each common genetic variant causes a mild genetic effect on ASD risk, while the cumulative effect of common genetic variants contribute up to 15–50% to ASD (Vorstman et al., 2017). Rare genetic variants, including de novo and intrinsic variants, contribute 10–30% of the genetic contribution to the occurrence of ASD (Vorstman et al., 2017). Several evidence have observed cytogenetic abnormalities in chromosome 15q11–q13 region in individuals with autism (Buxbaum et al., 2002; Hogart et al., 2007; Guo et al., 2017). 15q11–q13 site contains coding regions of specific subunits of GABA-AR, including GABRB3, GABRA5, and GABRG3, which are strongly implicated in the pathogenesis of autism (Delahanty et al., 2011; Bonnet-Brilhault et al., 2016; Guo et al., 2017). Duplications of gene E3 ubiquitin ligase UBE3A and three non-imprinted GABA-AR genes which are located in 15q11.2–q13.1 (Dup15q syndrome) can induce autistic phenotype behavioral deficits (Bonnet-Brilhault et al., 2016). It was found that single-nucleotide polymorphisms (SNP)s of GABRB3 rs2081648, GABRA5 rs35586628, and GABRG3 rs208129 polymorphisms are related to symptom-based behavioral deficits in sociability (Noroozi et al., 2018). GABRA4 was also found to increase autism risk through interaction with GABRB1, indicating a complex gene–gene interaction in GABA receptor subunit genes involved in the ethology of autism (Ma et al., 2005). Thus, overall results suggest that GABA-related gene polymorphisms seem to be sufficient to induce the autistic behavioral phenotype; whether rescue of GABA-related gene deficits by genetic approach can improve behaviors is worth to be further discussed.
GABAergic Dysfunction in Animal Models of ASD
Although accumulating in vivo and in vitro evidence from human studies suggest dysfunction of the GABA system is associated with ASD and genetic evidence further confirmed it, the molecular and cellular mechanisms are still obscure. Recently, transgenic technology enables translating genetic variations into rodent models to further investigate the structural and functional synaptic dysfunction in ASD. Investigating convergent cellular signalings or GABAergic alterations of current genetic and pharmacological animal models contribute to further understanding the mechanisms of ASD. Current candidate genes induce dysfunction of GABAergic transmission by affecting transcription of GABA-A receptor(GABA-AR), presynaptic GABA release, formation of GABAergic synapse, and synaptic structure–mediated transmission (Figure 1).
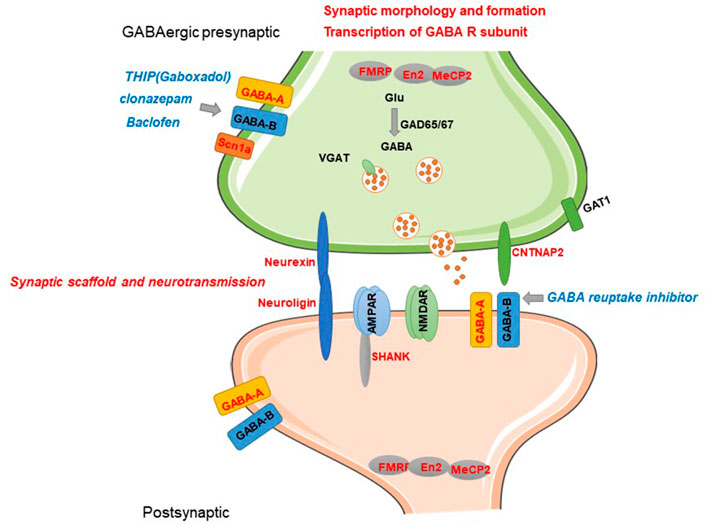
FIGURE 1. Current known candidate genes of ASD involved in GABAergic synaptic transmission and related mechanisms. Current candidate genes induce dysfunction of GABAergic transmission by affecting transcription of GABA-A subunits, presynaptic GABA release, formation of GABAergic synapse, and synaptic structure–mediated transmission. GABA-related candidate genes and regulatory role are represented by red letters. Pharmacological modulators of GABA-R reported to have alleviation efficacy are represented by blue letters.
GABA Receptor Mutant Animal Models
Direct evidence of GABA dysfunction in autism is from that mouse carrying mutant subunit of GABA receptors exhibited sociability impairments and repetitive behaviors which belong to core symptoms of ASD. Delahanty et al. (2011) first demonstrated that in vitro mutant β3 subunit-containing α1β3γ2 or α3β3γ2 GABA-AR showed reduced whole-cell current and functional receptor. In vivo evidence showed that a mouse model carrying β3 subunit GABA-AR in which serine residues 408 and 409 (S408/9) mutated to alanines (S408/9A) and S408/9A homozygotes exhibited core ASD symptoms, including increased repetitive behaviors and decreased social interaction, and altered dendrite spine structure and consequently decreased tonic inhibition and increased phasic inhibition (Vien et al., 2015). These results suggest specific GABA-AR defect exert a risk effect linking 15q11–q13 autism duplication region with autism. Impaired GABAergic signaling during brain development is a key event related to the neurodevelopmental disease. In addition, 15q duplication mice (a model for ASD with human chromosome 15q11–q13 paternal duplication) exhibited facilitated activity-induced long-term potentiation (LTP) of glutamate synapses onto layer 5 pyramidal neurons and reduced number of inhibitory synapses, indicating shifted E/I balance is implicated with autism (Saitow et al., 2020).
Single-Gene Models of ASD
GABAergic dysfunction could also be a downstream consequence of mutations in genes and not directly be involved in GABA transmission because the proper development and function of GABA synapses rely on numerous signaling and scaffolding proteins. Currently known candidate genes of ASD contain transcription factors, scaffolding proteins, receptors, and signaling pathway proteins (Vorstman et al., 2017) (Table 1). Accumulating evidence has shown some of the current candidate genes induce dysfunction of GABAergic transmission by affecting transcription of GABA-AR subunits, development of GABAergic synapse, synaptic structure–mediated transmission. GABA functions as an excitatory neurotransmitter during embryonic development (Ben-Ari, 2002). It was found that prenatal VPA exposure–induced autism model and the fragile X syndrome (Fragile X) ASD model have an abnormal excitatory GABA inhibitory shift during delivery, with elevated intracellular chloride levels and maintenance of higher levels of excitatory GABA, which promotes glutamic acid activity and gamma oscillatory activity (Tyzio et al., 2014). The CA3 hippocampal region of the BTBR T+tf/J mouse, an animal model of idiopathic autism, showed impaired coherent network oscillations at the early postnatal period, which might be attributed to a reduction of the intrinsic excitability of CA3 principal cells and a reduction of the shunting activity in GABAergic interneurons projecting to principal cells (Cellot et al., 2016). These results indicated that the transition of inhibitory to excitatory neurotransmission during early brain development might be pathological mechanisms of imbalance of the E/I ratio and GABA disturbance involved in autism. In contrast, it was found that neurofibromatosis type 1 (NF1) models display increased GABAergic transmission, while decreased GABA levels were observed in patients with ASD (Gonçalves et al., 2017). The inconsistent findings might be due to GABAergic transmission and disproportionately distribution of GABA receptors in a brain region–specific manner. Thus, specific brain regions and developmental time window play a critical role in understanding the mechanisms of GABA involved in autism.
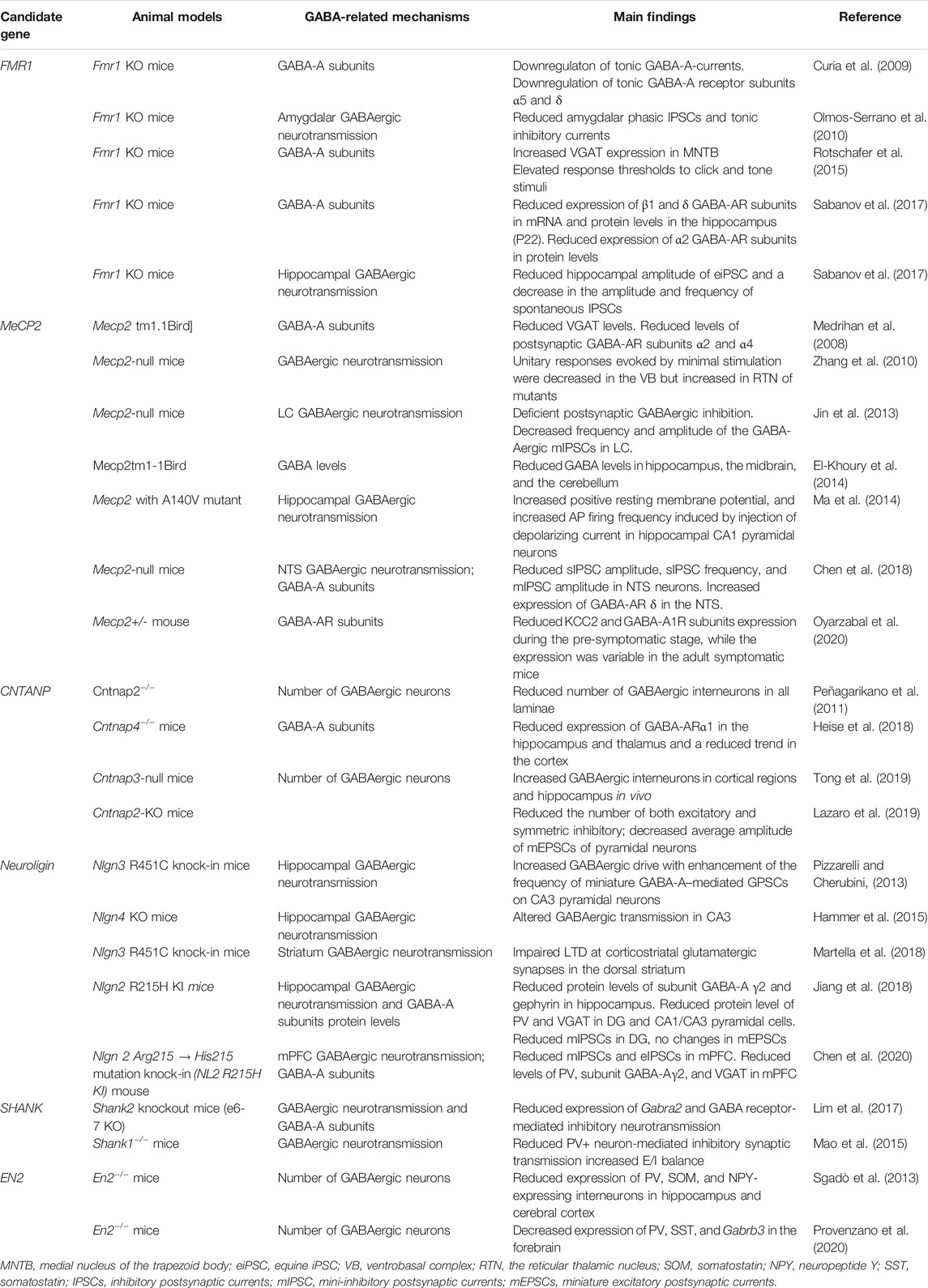
TABLE 1. Alterations of GABA system in single-gene models of ASD.
FMR1
Fragile mental retardation protein (FMRP), encoded by FMR1, is an extensively studied RNA-binding protein that is implicated in the pathogenesis of Fragile X, ASD, and other IDs (De Rubeis and Bagni, 2011). Research confirmed FMR1 and FMRP-targeting gene set associated with ASD and other neuropsychiatry diseases (Jansen et al., 2017). FMRP can affect synaptic phenotype as an mRNA-binding transitional suppressor via modulating the synthesis of synaptic proteins which implicated in synaptic transmission (Bagni and Zukin, 2019). Early studies have widely reported altered GABA-AR subunit expression levels in Fmr1 knockout (KO) mice. Fragile X mice exhibited reduced expression of α1, α3, and α4, β1 and β2, and γ1 and γ2, subunits of GABA-AR both in mRNA and protein levels comparing with wild-type mice. However, GAD65 and GAD67 expression levels were inversely increased in Fragile X mice (D’Hulst et al., 2006; Adusei et al., 2010). It was found that GABA-A β1 and β3, GABAB-R1, two Na+–K+–2Cl− cotransporter proteins (NKCC1 and NKCC2), gephyrin and ubiquilin, of Fmr1 KO mice were not significantly different from those of wild-type mice at any of the postnatal time points (Adusei et al., 2010). In addition, Fmr1 KO mice exhibited abnormal transmission in cognition and sociability-related brain regions. Centonze et al. found that Fmr1 KO mice have fewer GABAergic synapses whereas higher levels of spontaneous GABA inhibitory transmission in the striatum (Centonze et al., 2008). Phasic GABAergic inhibition was not significantly altered, while the tonic GABA-A currents were downregulated in Fmr1 KO mice, and this might be related to the lack in expression of GABA-AR subunits α5 and δ in the subiculum (Curia et al., 2009). Furthermore, both significant reduction of the phasic inhibitory postsynaptic currents (IPSCs) and tonic inhibitory currents were observed in the Fmr1 KO mice. Fmr1 KO mice showed neuronal hyperexcitability in the amygdala which can be rescued by the administration of GABA agonist gaboxadol (THIP) can rescue the amygdalar neurons via promoting the tonic inhibitory tone (Olmos-Serrano et al., 2010). Lack of RGS4, a regulator of G-protein signaling protein, can inhibit the susceptibility of FMR1 mice to audiogenic seizures. Treatment with GABA-B receptor antagonist is prone to induce seizures in Fmr1/RGS4 double-knockout mice than wild-type mice, whereas agonist of GABA-B baclofen can inhibit seizures in Fmr1/RGS4 double-knockout mice (Pacey et al., 2009). Similarly, baclofen was found to rescue the increased baseline and auditory-evoked high-frequency gamma (30–80 Hz) power in Fmr1 KO mice and improved the impaired working memory (Sinclair et al., 2017). These findings indicate impaired GABAergic transmission, especially in cognition and sociability-related brain regions, appears to be implicated in the contribution of gene FMR1 in neurodevelopmental and neuropsychiatry disorders.
MeCP2
MeCP2 is a structural chromosomal protein encoded by gene MeCP2 which is implicated in Rett syndrome (RTT) pathology (Amir et al., 1999) and subsequently was found to cause non-specific ASD and neurodevelopmental diseases (Tillotson and Bird, 2019). The loss of MeCP2 from a subset of forebrain GABAergic neurons also recapitulates many features of RTT (Chao et al., 2010). Earlier evidence showed that MeCP2-deficient GABAergic neurons of forebrain showed reduced inhibitory quantal size, which is consistent with a presynaptic reduction in GAD1 and GAD2 expression levels, and GABA immunoreactivity (Chao et al., 2010). It was found that MeCP2 disruption causes the defect of GABA release from presynapse and impaired GABAergic postsynaptic inhibition in locus coeruleus (LC) and NTS (Jin et al., 2013). MeCP2-null mice exhibited increased amplitude of spontaneous miniature and evoked excitatory postsynaptic current (EPSC)s without affecting the intrinsic excitability in the nucleus tractus solitarius (NTS), which is associated with decreased brain-derived neurotrophic factor (BDNF) availability in the primary afferent pathway, and these changes can be normalized by exogenous BDNF application (Kline et al., 2010).
Some evidence has demonstrated that the loss of MeCP2 induces reduced expression levels of vesicular GABA transporter (VGAT), GAD1, and GAD2, and altered expression of GABA-AR subunits (Medrihan et al., 2008; Chao et al., 2010; Chen et al., 2018; Oyarzabal et al., 2020). In particular, it was found that GABA-A subunit expression was developmentally decreased especially in the no-symptom stage in MeCP2+/− mouse brain (Oyarzabal et al., 2020). MeCP2 loss decreased the level of spontaneous glutamate receptor-mediated synaptic currents in hippocampal CA3 neurons and induced hyperexcitability in hippocampal neurons (Zhang et al., 2008; Calfa et al., 2011; Ma et al., 2014). Direct evidence was found from studies demonstrating that neurons lacking MeCP2 exhibited the defect of GABAergic transmission whereas not in the neighboring neurons with MeCP2 and forebrain-restricted loss of Mecp2 reduce the number of GABAergic synapses in the cortex (Zhang et al., 2014). These results indicate that MeCP2 loss induces defect in presynaptic GABA release, reduced number of GABAergic synapses, and impaired GABAergic inhibition.
Pharmacological stimulation of the GABAergic system exhibited restoration of abnormal neurotransmission and behavioral phenotype. GABA-A-R agonist THIP (gaboxadol) was reported to alleviate symptoms of RTT, including the motor dysfunction and social abnormalities (Zhong et al., 2017). GABA reuptake inhibitor can improve the life span of Mecp2-deficient mice (El-Khoury et al., 2014). Application of benzodiazepine-like modulator midazolam can transiently abolish the breathing defects of Mecp2-deficient mice (Voituron and Hilaire, 2011). Notably, mirtazapine, an antidepressant known to promote GABA release, was found to inhibit the hopping behaviors of Mecp2-null mice (Bittolo et al., 2016). These results confirmed that GABAergic neurotransmission defect appears to be involved in neurophysiological mechanisms of MeCP2 deficiency–induced abnormal behaviors.
Notably, sex difference of RTT has higher prevalence in girls than in boys and is significantly distinguished with ASD (Beattie, 2004). It was shown that RTT is the second most prevalent cause of intellectual disability in girls (Matagne et al., 2017). The maternal experience transiently enhances GAD67 in the auditory cortex and enhances PV-expressing neurons only in MeCP2 het mice; knockout of MeCP2 specifically in PV neurons is sufficient to impair learning behavior of pups, indicating experience-dependent plasticity might be impaired during development in adult MeCP2 het mice (Krishnan et al., 2017). Only restoration of the MeCP2 expression in GABAergic neurons of male MeCP2 null mice can increase inhibitory neurotransmission and subsequently improve ataxia and social impairments but not anxiety; these physiological and behavioral changes were not observed in female mice (Ure et al., 2016). These results suggest that MeCP2 is critical for the regulation of the GABAergic system, especially during neurodevelopment.
Nlgn2, 3, and 4
Neurexins (NRXNs) and neuroligins (NLGNs) are critical structural molecules involved in synapse formation and differentiation as cell adhesion molecules. NRXNs localized in presynapse can bind to postsynapitc NLGNs, bridge the synaptic cleft, and contribute to synapse stabilization (Blunk et al., 2014). In vivo and in vitro evidence have demonstrated that neuroligin-2 plays an important role in the development and maintenance of GABAergic synapses (Chen et al., 2011; Varley et al., 2011; Fekete et al., 2015; Uchigashima et al., 2016; Panzanelli et al., 2017). NLGN2 can precede KCC2 expression and regulate GABA functional switch in early postnatal development (Panzanelli et al., 2017). Notably, knockdown of Nlgn2 can reverse the inhibitory GABA to excitatory action in mature neurons, indicating NLGN2 involvement in maintenance of GABA function as an inhibitory neurotransmitter in mature neurons (Sun et al., 2013). NLGN2 can induce fast decay of mini inhibitory postsynaptic currents (mIPSC) by regulation of differential expression of postsynaptic GABA-AR subtypes (Fu and Vicini, 2009). Overexpression of NLGN1B and NLGN2A in newborn neurons of the hippocampus can increase the spine density and impaired spatial memory (Krzisch et al., 2017). These data suggest that neuroligins might affect the formation of neural connectivity via affecting the early postnatal transition of excitatory GABA.
Several evidence have shown that abnormal NLGN2 expression is associated with neuropsychiatric behaviors. Reduced NLGN2 expression in the prefrontal cortex (PFC) is involved in attention deficits induced by peripubertal stress which can be reversed by virus-mediated NLGN2 rescue (Tzanoulinou et al., 2016). Animal models using the genetic engineering method showed that Nlgn 2 R215H knock-in (KI) mouse exhibited schizophrenia-like behaviors and higher anxiety behaviors with significant reduction of mIPSCs in the dentate gyrus (DG), reduced subunit GABA-A γ2 expression levels, and reduced presynaptic PV and VGAT levels, while there were no changes of miniature excitatory postsynaptic currents (mEPSCs), vesicular glutamate transporter (VGLUT), and postsynaptic density protein 95 (PSD95) expression levels (Jiang et al., 2018; Van Zandt et al., 2019). NLGN2 of ACC is involved in KCC2-mediated abnormal GABAergic transmission induced aggressive behaviors (Jager et al., 2020).
Social deficits, enhanced spatial learning, increased locomotor activity, and increased aggressive behaviors were observed in Nlgn3 R451C mutant mice (Jaramillo et al., 2014; Burrows et al., 2015). It was found that elevated aggressive behaviors in Nlgn3 R451C mutant mice could be normalized by risperidone, a novel antipsychotic agent that antagonizes serotonin (5-hydroxytryptamine) 5-HT2 and dopamine D2 receptor (Burrows et al., 2015). Earlier findings demonstrated Nlgn3 R451C mutant mice exhibited enhanced GABAergic transmission with increased frequency of IPSCs while having no significant effect on excitatory transmission (Tabuchi et al., 2007). Both Nlgn3 R451C knock-in and Nlgn3 KO mutant impaired tonic endocannabinoid signaling (C et al., 20132020). Nlgn3 R451C knock-in mice appeared to show an increased GABAergic drive with enhancement of the frequency of miniature GABA-A–mediated giant depolarizing potentials (GPSCs) on CA3 pyramidal neurons (Pizzarelli and Cherubini, 2013). In addition, mice carrying R451C-Nlgn3 mutation showed impaired long-term depression (LTD) at corticostriatal glutamatergic synapses which could be rescued by exogenous activation of cannabinoid CB1 receptors or enhancement of the endocannabinoid tone (Martella et al., 2018). Mice and rats carrying Nlgn3 mutations showed reduced striatal glutamate indicating glutamate/GABA abnormalities in the corticostriatal circuitry (Horder et al., 2018).
Hippocampal Nlgn4 likely localized at excitatory synapses originating from PV-positive interneurons (Marro et al., 2019). ASD-related Nlgn4 mutations can alter the compositions of hippocampal perisomatic inhibitory synapses and impaired the synchronized oscillatory activity which might be implicated in the cognitive dysfunction of ASD (Hammer et al., 2015). Glycine is another widely acknowledged inhibitory neurotransmitter in the CNS. It was found that Nlgn4 KO mice exhibited impaired glycinergic transmission in the retina with fewer glycine receptors mediating fast glycinergic transmission and slower glycinergic mIPSCs (Hoon et al., 2011). A recent study confirmed that NLGN4 significantly impaired glycinergic transmission and reduced the numbers of glycinergic synapses while having no impact on glutamatergic synapses. Intriguingly, they found overexpression of NLGN4 causes impairments of excitatory transmission but not that of inhibitory transmission (Zhang et al., 2018). These findings confirmed the contribution of members of NLGN family to neurodevelopmental diseases via various molecular and cellular mechanisms.
CNTNAP2, 3, and 4
CNTNAP2 encodes contactin-associated protein-like 2 (Caspr2), a protein of the neurexin superfamily, and interacts with NLGN proteins, scaffolding proteins PSD95 and gephyrin (Bonnet-Brilhault et al., 2016). Human and animal studies have reported that mutations in CNTNAP2 cause a syndrome form of ASD with significant behavioral defects in social interaction and stereotypes (Peñagarikano et al., 2011). It was found that Cntnap2−/− mice showed ASD core symptoms, accompanied with significant reduced GABAergic neurons and reduced cortical neuronal synchrony; these changes can be rescued by risperidone treatment (Peñagarikano et al., 2011). Cntnap2 KO mice displayed remarkable reduced excitatory and inhibitory synaptic inputs onto pyramidal neurons of mPFC with normal dendritic arborization, and decreased average amplitude of mEPSCs in pyramidal neurons, while no significant changes in mIPSCs (Lazaro et al., 2019). However, contradictory results from a genome analysis found that rare heterozygous mutations in any of the CNTN or CNTNAP genes, including CNTNAP2, were not significantly involved in the plausible contribution to risk of autism (Murdoch et al., 2015).
Other members of CNTNAP family are reported to be related to autism (Tong et al., 2019). It was found that knockdown of CNTNAP3 expression led to the decreased dendritic length and branch. Intriguingly, the formation of excitatory synapse in CNTNAP3-knockdown mice was decreased, while that of the inhibitory synapse was increased (Tong et al., 2019). ASD-related mutations in CNTNAP3 (R1219X, P614A, and R786C) may be a loss-of-function mutation (Tong et al., 2019). Earlier evidence showed that CNTNAP4 might be relevant in autism (Cottrell et al., 2011). CNTNAP family contained five members from CNTNAP1 to CNTNAP5, while how CNTNAP5 exert function was obscure. These results indicate that CNTNAP has extensive effect on synapse formation and function; the specific molecular mechanisms of CNTNAP involved in synapse function and ASD need to be further investigated.
SCN1A
Haploinsufficiency of the SCN1A gene encoding brain voltage-gated sodium channel NaV1.1. is the genetic etiology of Dravet syndrome, a developmental disorder characterized by recurrent intractable seizures, cognitive deficit, and autistic-like behaviors (Han et al., 2012). Han S et al. reported that haploinsufficiency of the SCN1A gene impaired GABAergic neurotransmission by conditional deleting the NaV1.1 channels in forebrain interneurons which can be fully rescued by low-dose treatment with clonazepam, a positive allosteric modulator of GABA-AR (Han et al., 2012). The expression of the SCN1A gene was found significantly decreased in PFC of offspring of maternal immune activation (MIA) models, which also indicates impaired GABAergic transmission involved in pharmacological models of autism (Yang J. Q. et al., 2021).
SHANK1, 2, and 3
The Shank family of scaffold proteins, mainly containing members SHANK1 to SHANK3, can bind a variety of membrane and cytoplasmic proteins to form a postsynaptic scaffolding protein complex which is related to the formation and function of excitatory synapses (Sheng and Kim, 2000). Though de novo deletions, insertion, splicing mutations, and point mutations of SHNAK have been identified in ASD patients, in vivo mechanisms in which SHANK relate to ASD remain obscure. Animal evidence showed absence of shank altered to the expression of receptor subunits of GABA and GABAergic inhibitory synaptic transmission. SHANK1 protein is reported to be highly expressed in PV-positive fast-spiking inhibitory interneurons in the hippocampus (Mao et al., 2015). Shank1 mutant mice have impaired inhibitory synaptic outputs to hippocampal CA1 pyramidal neurons and consequently impaired excitatory and inhibitory balance (Mao et al., 2015). Shank1−/− and Shank3B−/− mice showed reduced PV expression in mPFC and somatosensory cortex and striatum (Filice et al., 2016). Shank2 knockout mice (e6-7 KO) showed a reduction in the expression levels of Gabra2 and GABA R-mediated inhibitory signaling, which can be reversed by treatment with an allosteric modulator for the GABA-AR (Lim et al., 2017). Shank2−/− and Shank3αβ−/− mice exhibited reduced levels of several cell surface glutamate receptors, such as GluN1 and GluA2, in the striatum and cortex (Heise et al., 2018). Considering that SHANK family proteins are mainly localized at the postsynaptic densities of excitatory synapses, there is still lack of evidence showing a direct relationship between SHANK and GABAergic transmission.
EN2
Several evidence have shown homeobox transcription factor Engrailed2 (En2) appears to be a susceptibility locus for ASD (Benayed et al., 2005; Brune et al., 2008; Wang et al., 2008; Benayed et al., 2009). Mouse carrying mutants in En2 exhibited similar morphological abnormalities in the cerebellar with autistic individuals (Gharani et al., 2004). Protein EN2 is mainly present in postnatal GABAergic neurons of the mouse hippocampus and cerebral cortex, and it was found that persistant high expression of EN2 during the prenatal and early postnatal period contributes to autism by affecting the maturation of Purkinje cell (PCs) in the cerebellum (James et al., 2013, 2014). EN2 expression levels alter the number of GABAergic interneurons (Sgadò et al., 2013; Provenzano et al., 2020). Evidence from an in vitro study found that En2 increases the complexity of the dendritic tree of GABAergic neurons and reduced the number of mature synapses as well as the area of postsynaptic densities (Soltani et al., 2017).
Pharmacological Models
Besides the single-gene models, pharmacological models were widely used to discuss the pathogenesis of ASD and identification of novel drug targets (Table 2). Valproic acid (VPA) as a broad-spectrum antiepileptic drug has a significant facilitative effect on the GABAergic neurotransmission system (Miyazaki et al., 1988; Lattanzi et al., 2019). Intraperitoneal administration of VPA can significantly increase GABA levels and upregulate the seizure thresholds (Nau and Löscher, 1982; Löscher and Vetter, 1984). Amounts of evidence have shown that prenatal exposure with 400–600 mg/kg VPA was found to induce significant autistic behaviors in offspring which was widely used as pharmacological rodent models for the ASD study (Nicolini and Fahnestock, 2018). Earlier evidence has shown that VPA prenatal exposure-induced ASD models have a significant reduction of PV+ inhibitory interneurons numbers in the parietal and occipital cortex (Gogolla et al., 2009), reduced GABA-A and GABA-B receptor expression, and reduced GABA levels (Kazlauskas et al., 2016), reduced GAD65 and GAD67 levels in the cerebellar, amygdala (Olexová et al., 2016; Chau et al., 2017), and cortex (Cusmano and Mong, 2014; Chau et al., 2017; Hou et al., 2018), and reduced GAT1 expression in the amygdala (Olexová et al., 2016). In contrast, contradictory results found that cortical GAD67 expression levels showed an increasing trend from adolescence to adulthood in VPA models (Hou et al., 2018). High-performance liquid chromatography (HPLC) results showed that the levels of glutamate, Gln, and GABA were significantly increased in the hippocampus of male VPA and thalidomide (THAL)-induced autism rats. Nuclear magnetic resonance (NMR) spectroscopy results showed increased levels of Gln and GABA in the VPA models (Zieminska et al., 2018). A recent study reported widely decreased expression of genes encoding different subunits of GABA-AR, including GABRA1, GABRA2, GABRA3, GABRB3, GABBR1, and GABBR2 (Yang Y. et al., 2021), which is consistent with an earlier study reporting reduced GABRA1, GABRA5, and GABRB2 levels in the cortex of VPA-induced autism models (Chau et al., 2017). Yang et al. found that the frequency and amplitude of spontaneous excitatory postsynaptic currents (sEPSCs) were comparable between control and VPA mice models, while the frequency of spontaneous inhibitory postsynaptic currents (sIPSCs) was decreased in VPA models (Yang J. Q. et al., 2021). It was reported that the normal expression of GABAρ3 in the cerebellum was increased linearly throughout the normal development process, while the VPA models exhibited reduced GABAρ3 levels in PCs and ependymal glial cells (EGCs) from lobule X of the cerebellum (Varman et al., 2018). Administration of both GABA-A and GABA-B receptor agonists can improve the social deficits (Kazlauskas et al., 2016; Yang Y. et al., 2021). Individual GABA-AR agonist, chlorpromazine or GABA-BR baclofen administration can alleviate autistic-like behaviors in the VPA model. Combined chlorpromazine with baclofen can alleviate sociability deficits, anxiety and repetitive behaviors (Yang Y. et al., 2021). This evidence suggests impaired GABAergic inhibitory transmission in prenatal VPA exposure- induced autism models.
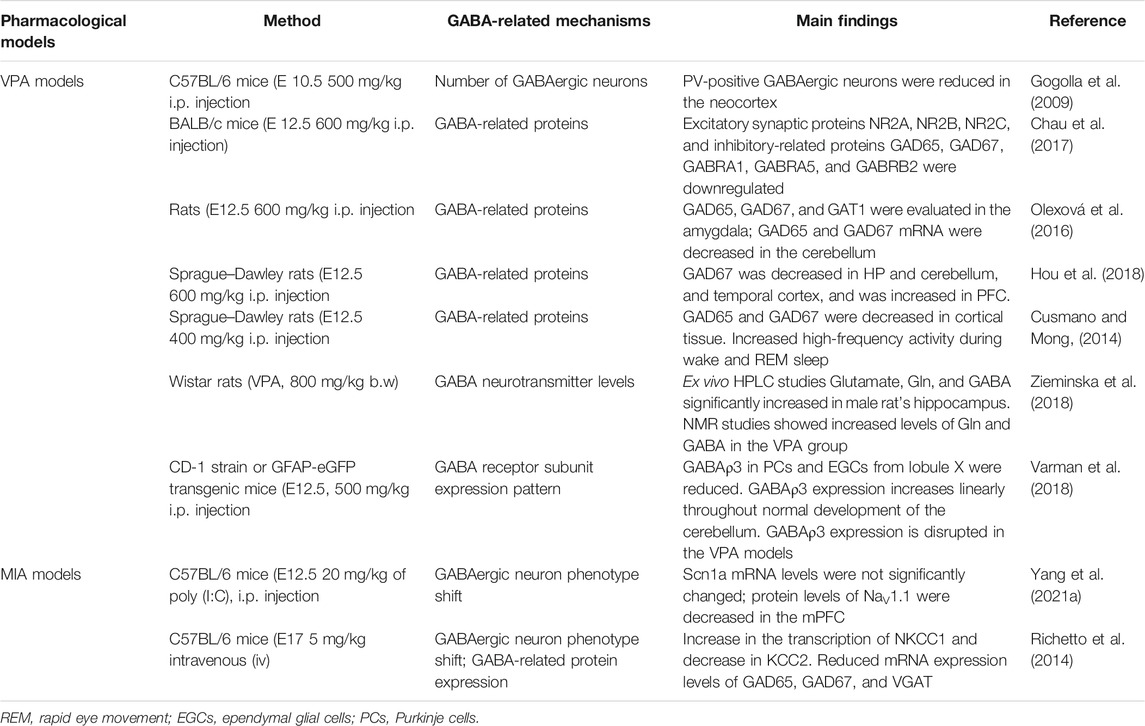
TABLE 2. Alterations of the GABA system in pharmacological models of ASD.
Another widely studied pharmacological model of ASD is the MIA model, which refers to immune activation by immunogen administration during pregnancy, such as lipopolysaccharide (LPS) or polyinosinic–polycytidylic acid (poly (I:C)) (Meyer, 2014; Haddad et al., 2020). The mRNA and protein levels of GAD65, GAD67, and GABA-A α2, 3, 4, 5 were reduced in the cortex of MIA models, and there was no significant difference in expression levels of GABA and glutamate-related subunits in the cortex or cerebellum (Zhang et al., 2021). Treatment with low-dose benzodiazepines clonazepam can completely reverse behavioral abnormalities of offspring of MIA models by enhancing GABAergic neurotransmission (Yang J. Q. et al., 2021). In addition, the mRNA expression levels of NKCC1 and KCC2 were increased in the peripubertal offspring of MIA models (Richetto et al., 2014). The presence of imbalanced NKCC1 and KCC2 expressions in the adult brain is typically taken as an index of an immature GABAergic phenotype (Ben-Ari et al., 2012). Thus, these results suggest prenatal maternal immune activation might have effect on long- lasting GABAergic changes relevant to autism and other neuropsychiatric disorders with prenatal infectious etiologies.
Potential Therapeutic Efficacy of GABA R Modulators in ASD
GABA-A receptors are a series of ion channel–type receptors that are sites of action for benzodiazepines, barbiturates, and anesthetics. GABA-B receptors are metabotropic receptors (Jembrek and Vlainic, 2015). Presynaptic GABA-B receptors act by inhibiting voltage-gated calcium channels (Krystal et al., 2002). As mentioned above, accumulating evidence has shown some of the current candidate genes mutant induce dysfunction of GABAergic transmission by affecting transcription of GABA-A subunits, development of GABAergic synapse, and synaptic structure–mediated transmission. Treatment with GABA agonists can alleviate autism-like behaviors by disinhibition of presynaptic GABAergic neurons. In addition, diuretic NKCC1 chloride importer antagonist bumetanide can reinforce GABAergic inhibition by decreasing Cl-; animal evidence showed that treatment with bumetanide reduces the severity of autism symptoms (Hadjikhani et al., 2018; James et al., 2019). The most widely studied GABA modulator is baclofen, a GABAB receptor agonist, which was reported to reverse social approach deficits and repetitive behaviors of BTBR T+ Itpr3tf/J (BTBR) mice, and also reduced the stereotyped jumping in C58/J (C58), another ASD model (Silverman et al., 2015). In addition, the hyperactivity, anxiety, aggression, and repetitive behaviors of the FMR1 KO mouse model were found to be completely rescued by a GABA-AR agonist, gaboxadol, which is also called OV101 or THIP and prefer to activate δ subunit-containing extrasynaptic GABA-AR (Cogram et al., 2019).
Up to now, there are several positive results from clinical trials on the role of GABA modulators on patients with autism. Brondino et al. summarized human clinical trials of pharmacological efficacy of GABA modulator on autism (Brondino et al., 2016). A randomized, controlled, phase 2 trial results showed arbaclofen failed to improve the abnormal behavioral phenotype of children and adults with fragile X syndrome (Berry-Kravis et al., 2012). A randomized, placebo-controlled, phase 2 study of 150 participants with ASD showed there was no difference in the primary outcome measure between the arbaclofen group and control group, while there was improvement on the clinician-rated clinical global impression of severity in a specified secondary analysis (Veenstra-VanderWeele et al., 2017). Baclofen as an adjuvant therapy can enhance the therapeutical effect of antipsychotic drugs, risperidone, in children with ASD, including improvement of irritability, lethargy, stereotypic behavior, hyperactivity, and inappropriate speech (Mahdavinasab et al., 2019). A study with a small sample found that treatment with bumetanide decreased the autistic behaviors in autistic infants (Lemonnier and Ben-Ari, 2010). In addition, an open-label trial pilot study showed 10 months of bumetanide treatment can improve emotion recognition and stimulate the activation of social and emotional perception-related brain regions during the perception of emotional faces in adolescents and young adults with autism (Hadjikhani et al., 2015). A recent investigation showed bumetanide significantly reduces the severity of ASD core symptoms which relate to a decreased GABA/glutamate ratio in both the insular cortex (IC) and visual cortex (VC) (Zhang et al., 2020). In conclusion, current evidence in clinical trials is limited by small sample sizes and short study follow-ups, complicated clinical heterogeneity, and inconsistent inclusion criteria. Thus, to date, there is insufficient evidence to suggest the use of GABA modulators in autistic patients. Medication safety is also an issue to consider in addition to efficacy.
Conclusion and Future Perspectives
Numerous ASD candidate genes regulate extensive functions of the CNS; thus, it is hard to converge specific molecular mechanisms of ASD according to current genetic and pathological evidence. However, a lot of ASD-related genetic variations from human genetic studies identified the involvement of synaptic genes which consolidate the hypothesis that synaptic structure or/and function dysfunction are involved in ASD. In addition, technology advances propel modeling rodent models of ASD and other neuropsychiatric disorders using manipulation of candidate genes or environmental factors. We summarized primary GABAergic inhibitory dysfunction of animal models of non-syndromic gene mutations (CNPANP, neuroligin, SHANK, and EN2) and syndromic gene mutations (FMR1, Mecp2, and SCN1A). Current evidence indicates GABA is related to ASD from two aspects. First, GABA receptor mutants in specific neurons sufficiently recapitulate core ASD symptoms. Duplications of gene E3 ubiquitin ligase UBE3A and three non-imprinted GABA-AR genes located in 15q11.2–q13.1 (Dup15q syndrome) induce autistic phenotype behavioral deficits (Bonnet-Brilhault et al., 2016). Besides, well-acknowledged ASD candidate genes were specifically expressed in GABAergic neurons and implicated in GABA function. Scn1a encodes sodium channel subunits that are upregulated in Pv+ fast-spiking interneurons. Pharmacological approaches promoting GABA transmission suppress behavioral symptoms of Dravet’s syndrome (Han et al., 2012). The structural chromosomal protein MeCP2, homeobox transcription factor En2, and RNA-binding protein FMRP target metabolism, synthesis and modification of synaptic proteins related with GABA synaptic function. Neuroligins might affect the formation of neural connectivity via affecting the early postnatal transition of excitatory GABA.
Pharmacological intervention with agonist targeting specific GABA receptors was found to partially alleviate ASD-related behavioral abnormalities in animal models and clinical trials. However, the therapeutic efficacy of clinical modulators of GABA receptors needs to be supported by clinical trials with larger sample sizes. These results collectively suggest that GABAergic synaptic transmission dysfunction, especially in specific neurons or circuitry during the developmental stage, might underlie the pathophysiology of ASD. However, increased GABAergic synaptic transmission is also reported in some animal studies, indicating impaired GABAergic transmission might be a critical etiology of imbalance of the E/I ratio involved in the development of ASD. Substantial questions as to the specific etiology of ASD remain unclear and require future investigation. First, researchers need to take the reliability of current behavioral assays of mice into consideration when mimicking the clinical ASD phenotype. Additionally, a complicated reciprocal relation exists between the excitation and inhibition, especially in early brain development. In consideration that GABA can shift from “excitatory” to “inhibitory” and play a critical role in subtle regulation of neuronal network during critical developmental stages, it cannot be easily ruled out that altered excitation might be secondary to reduced inhibition and GABA dysfunction. Thus, further investigations are needed to shed light on the pathogenesis of ASD and provide a comprehensive understanding of synaptopathology in ASD.
Author Contributions
HZ, XZ, FP and CZ wrote the manuscript. XM and GL made the graphics. WY, BL, TG, and RC provided the final revision.
Funding
This work was supported by National Key R&D Program of China (#2018YFC1311600). Jilin Science and Technology Agency funds in China (YDZJ202102CXJD077). Jilin Province Medical and Health Talents (2019SCZT007 and 2019SCZT013). Program of Jilin finance department (2019SRCJ017).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adusei, D. C., Pacey, L. K. K., Chen, D., and Hampson, D. R. (2010). Early Developmental Alterations in GABAergic Protein Expression in Fragile X Knockout Mice. Neuropharmacology 59, 167–171. doi:10.1016/j.neuropharm.2010.05.002
Ajram, L. A., Horder, J., Mendez, M. A., Galanopoulos, A., Brennan, L. P., and Wichers, R. H. (2017). Shifting Brain Inhibitory Balance and Connectivity of the Prefrontal Cortex of Adults with Autism Spectrum Disorder. Transl. Psychiatry 7, e1137. doi:10.1038/tp.2017.104
Al-Otaish, H., Al-Ayadhi, L., Bjørklund, G., Chirumbolo, S., Urbina, M. A., and El-Ansary, A. (2018). Relationship between Absolute and Relative Ratios of Glutamate, Glutamine and GABA and Severity of Autism Spectrum Disorder. Metab. Brain Dis. 33, 843–854. doi:10.1007/s11011-018-0186-6
Amin, Z., Mason, G. F., Cavus, I., Krystal, J. H., Rothman, D. L., and Epperson, C. N. (2006). The Interaction of Neuroactive Steroids and GABA in the Development of Neuropsychiatric Disorders in Women. Pharmacol. Biochem. Behav. 84, 635–643. doi:10.1016/j.pbb.2006.06.007
Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett Syndrome Is Caused by Mutations in X-Linked MECP2, Encoding Methyl-CpG-Binding Protein 2. Nat. Genet. 23, 185–188. doi:10.1038/13810
Bagni, C., and Zukin, R. S. (2019). A Synaptic Perspective of Fragile X Syndrome and Autism Spectrum Disorders(Review). Neuron, 1070–1088. doi:10.1016/j.neuron.2019.02.041
Bayram, A. K., Kardas, F., Demirci, E. O., Gokahmetoglu, S., Ozmen, S., Canpolat, M., et al. (2016). Lack of Serum Antineuronal Antibodies in Children with Autism. Bratisl Lek Listy 117, 77–79. doi:10.4149/bll_2016_015
Beattie, M. S. (2004). Inflammation and Apoptosis: Linked Therapeutic Targets in Spinal Cord Injury. Trends Mol. Med. 10, 580–583. doi:10.1016/j.molmed.2004.10.006
Belmonte, M. K., Allen, G., Beckel-Mitchener, A., Boulanger, L. M., Carper, R. A., and Webb, S. J. (2004). Autism and Abnormal Development of Brain Connectivity. J. Neurosci. 24, 9228–9231. doi:10.1523/jneurosci.3340-04.2004
Ben-Ari, Y. (2002). Excitatory Actions of Gaba during Development: the Nature of the Nurture. Nat. Rev. Neurosci. 3, 728–739. doi:10.1038/nrn920
Ben-Ari, Y., Khalilov, I., Kahle, K. T., and Cherubini, E. (2012). The GABA Excitatory/inhibitory Shift in Brain Maturation and Neurological Disorders. Neuroscientist 18, 467–486. doi:10.1177/1073858412438697
Benayed, R., Choi, J., Matteson, P. G., Gharani, N., Kamdar, S., Brzustowicz, L. M., et al. (2009). Autism-associated Haplotype Affects the Regulation of the Homeobox Gene, ENGRAILED 2. Biol. Psychiatry 66, 911–917. doi:10.1016/j.biopsych.2009.05.027
Benayed, R., Gharani, N., Rossman, I., Mancuso, V., Lazar, G., Kamdar, S., et al. (2005). Support for the Homeobox Transcription Factor Gene ENGRAILED 2 as an Autism Spectrum Disorder Susceptibility Locus. Am. J. Hum. Genet. 77, 851–868. doi:10.1086/497705
Berry-Kravis, E. M., Hessl, D., Rathmell, B., Zarevics, P., Cherubini, M., Walton-Bowen, K., et al. (2012). Effects of STX209 (Arbaclofen) on Neurobehavioral Function in Children and Adults with Fragile X Syndrome: a Randomized, Controlled, Phase 2 Trial. Sci. Transl. Med. 4, 152ra127. doi:10.1126/scitranslmed.3004214
Bittolo, T., Raminelli, C. A., Deiana, C., Baj, G., Vaghi, V., Ferrazzo, S., et al. (2016). Pharmacological Treatment with Mirtazapine Rescues Cortical Atrophy and Respiratory Deficits in MeCP2 Null Mice. Sci. Rep. 6, 19796. doi:10.1038/srep19796
Blatt, G. J., and Fatemi, S. H. (2011). Alterations in GABAergic Biomarkers in the Autism Brain: Research Findings and Clinical Implications. Anat. Rec. (Hoboken) 294, 1646–1652. doi:10.1002/ar.21252
Blatt, G. J., Fitzgerald, C. M., Guptill, J. T., Booker, A. B., Kemper, T. L., and Bauman, M. L. (2001). Density and Distribution of Hippocampal Neurotransmitter Receptors in Autism: an Autoradiographic Study. J. Autism Dev. Disord. 31, 537–543. doi:10.1023/a:1013238809666
Blunk, A. D., Akbergenova, Y., Cho, R. W., Lee, J., Walldorf, U., Xu, K., et al. (2014). Postsynaptic Actin Regulates Active Zone Spacing and Glutamate Receptor Apposition at the Drosophila Neuromuscular junction. Mol. Cel. Neurosci. 61, 241–254. doi:10.1016/j.mcn.2014.07.005
Bonnet-Brilhault, F., Alirol, S., Blanc, R., Bazaud, S., Marouillat, S., Thépault, R. A., et al. (2016). GABA/Glutamate Synaptic Pathways Targeted by Integrative Genomic and Electrophysiological Explorations Distinguish Autism from Intellectual Disability. Mol. Psychiatry 21, 411–418. doi:10.1038/mp.2015.75
Brondino, N., Fusar-Poli, L., Panisi, C., Damiani, S., Barale, F., and Politi, P. (2016). Pharmacological Modulation of GABA Function in Autism Spectrum Disorders: A Systematic Review of Human Studies. J. Autism Dev. Disord. 46, 825–839. doi:10.1007/s10803-015-2619-y
Brune, C. W., Korvatska, E., Allen-Brady, K., CookJr, E. H., Dawson, G., Devlin, B., et al. (2008). Heterogeneous Association between Engrailed-2 and Autism in the CPEA Network. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147B, 187–193. doi:10.1002/ajmg.b.30585
Burrows, E. L., Laskaris, L., Koyama, L., Churilov, L., Bornstein, J. C., Hill-Yardin, E. L., et al. (2015). A Neuroligin-3 Mutation Implicated in Autism Causes Abnormal Aggression and Increases Repetitive Behavior in Mice. Mol. Autism 6, 62. doi:10.1186/s13229-015-0055-7
Buxbaum, J. D., Silverman, J. M., Smith, C. J., Greenberg, D. A., Kilifarski, M., Reichert, J., et al. (2002). Association between a GABRB3 Polymorphism and Autism. Mol. Psychiatry 7, 311–316. doi:10.1038/sj.mp.4001011
Calfa, G., Hablitz, J. J., and Pozzo-Miller, L. (2011). Network Hyperexcitability in Hippocampal Slices from Mecp2 Mutant Mice Revealed by Voltage-Sensitive Dye Imaging. J. Neurophysiol. 105, 1768–1784. doi:10.1152/jn.00800.2010
Carvalho Pereira, A., Violante, I. R., Mouga, S., Oliveira, G., and Castelo-Branco, M. (2018). Medial Frontal Lobe Neurochemistry in Autism Spectrum Disorder Is Marked by Reduced N-Acetylaspartate and Unchanged Gamma-Aminobutyric Acid and Glutamate+GlutamineLevels. Jautism Dev. Disord. 48, 1467–1482. doi:10.1007/s10803-017-3406-8
Cattane, N., Richetto, J., and Cattaneo, A. (2020). Prenatal Exposure to Environmental Insults and Enhanced Risk of Developing Schizophrenia and Autism Spectrum Disorder: Focus on Biological Pathways and Epigenetic Mechanisms. Neurosci. Biobehav. Rev. 117, 253–278. doi:10.1016/j.neubiorev.2018.07.001
Cellot, G., Maggi, L., Di Castro, M. A., Catalano, M., Migliore, R., Migliore, M., et al. (2016). Premature Changes in Neuronal Excitability Account for Hippocampal Network Impairment and Autistic-like Behavior in Neonatal BTBR T+tf/J Mice. Sci. Rep. 6, 31696. doi:10.1038/srep31696
Centonze, D., Rossi, S., Mercaldo, V., Napoli, I., Ciotti, M. T., De Chiara, V., et al. (2008). Abnormal Striatal GABA Transmission in the Mouse Model for the Fragile X Syndrome. Biol. Psychiatry 63, 963–973. doi:10.1016/j.biopsych.2007.09.008
Chao, H. T., Chen, H., Samaco, R. C., Xue, M., Chahrour, M., Yoo, J., et al. (2010). Dysfunction in GABA Signalling Mediates Autism-like Stereotypies and Rett Syndrome Phenotypes. Nature 468, 263–269. doi:10.1038/nature09582
Chau, D. K., Choi, A. Y., Yang, W., Leung, W. N., and Chan, C. W. (2017). Downregulation of Glutamatergic and GABAergic Proteins in Valproric Acid Associated Social Impairment during Adolescence in Mice. Behav. Brain Res. 316, 255–260. doi:10.1016/j.bbr.2016.09.003
Chen, A. I., Nguyen, C. N., Copenhagen, D. R., Badurek, S., Minichiello, L., Ranscht, B., et al. (2011). TrkB (Tropomyosin-related Kinase B) Controls the Assembly and Maintenance of GABAergic Synapses in the Cerebellar Cortex. J. Neurosci. 31, 2769–2780. doi:10.1523/JNEUROSCI.4991-10.2011
Chen, C. Y., Di Lucente, J., Lin, Y. C., Lien, C. C., Rogawski, M. A., Maezawa, I., et al. (2018). Defective GABAergic Neurotransmission in the Nucleus Tractus Solitarius in Mecp2-Null Mice, a Model of Rett Syndrome. Neurobiol. Dis. 109, 25–32. doi:10.1016/j.nbd.2017.09.006
Chen, J., Dong, B., Feng, X., Jiang, D., Chen, G., Long, C., et al. (2020). Aberrant mPFC GABAergic Synaptic Transmission and Fear Behavior in Neuroligin-2 R215H Knock-In Mice. Brain Res. 1730, 146671. doi:10.1016/j.brainres.2020.146671
Cochran, D. M., Sikoglu, E. M., Hodge, S. M., Edden, R. A., Foley, A., Kennedy, D. N., et al. (2015). Relationship Among Glutamine, γ-Aminobutyric Acid, and Social Cognition in Autism Spectrum Disorders. J. Child. Adolesc. Psychopharmacol. 25, 314–322. doi:10.1089/cap.2014.0112
Cogram, P., Deacon, R., Warner-Schmidt, J. L., von Schimmelmann, M. J., Abrahams, B. S., and During, M. J. (2019). Gaboxadol Normalizes Behavioral Abnormalities in a Mouse Model of Fragile X Syndrome. Front. Behav. Neurosci. 13, 141. doi:10.3389/fnbeh.2019.00141
Cottrell, C. E., Bir, N., Varga, E., Alvarez, C. E., Bouyain, S., Zernzach, R., et al. (2011). Contactin 4 as an Autism Susceptibility Locus. Autism Res. 4, 189–199. doi:10.1002/aur.184
Curia, G., Papouin, T., Séguéla, P., and Avoli, M. (2009). Downregulation of Tonic GABAergic Inhibition in a Mouse Model of Fragile X Syndrome. Cereb. Cortex 19, 1515–1520. doi:10.1093/cercor/bhn159
Cusmano, D. M., and Mong, J. A. (2014). In Utero exposure to Valproic Acid Changes Sleep in Juvenile Rats: a Model for Sleep Disturbances in Autism. Sleep 37, 1489–1499. doi:10.5665/sleep.3998
D'Hulst, C., De Geest, N., Reeve, S. P., Van Dam, D., De Deyn, P. P., Hassan, B. A., et al. (2006). Decreased Expression of the GABAA Receptor in Fragile X Syndrome. Brain Res. 1121, 238–245. doi:10.1016/j.brainres.2006.08.115
De Rubeis, S., and Bagni, C. (2011). Regulation of Molecular Pathways in the Fragile X Syndrome: Insights into Autism Spectrum Disorders. J. Neurodev. Disord. 3, 257–269. doi:10.1007/s11689-011-9087-2
Delahanty, R. J., Kang, J. Q., Brune, C. W., Kistner, E. O., Courchesne, E., Cox, N. J., et al. (2011). Maternal Transmission of a Rare GABRB3 Signal Peptide Variant Is Associated with Autism. Mol. Psychiatry 16, 86–96. doi:10.1038/mp.2009.118
Dhossche, D., Applegate, H., Abraham, A., Maertens, P., Bland, L., Bencsath, A., et al. (2002). Elevated Plasma Gamma-Aminobutyric Acid (GABA) Levels in Autistic Youngsters: Stimulus for a GABA Hypothesis of Autism. Med. Sci. Monit. 8, PR1–6.
Drenthen, G. S., Barendse, E. M., Aldenkamp, A. P., van Veenendaal, T. M., Puts, N. A., Edden, R. A., et al. (2016). Altered Neurotransmitter Metabolism in Adolescents with High-Functioning Autism. Psychiatry Res. Neuroimaging 256, 44–49. doi:10.1016/j.pscychresns.2016.09.007
Duman, R. S., Sanacora, G., and Krystal, J. H. (2019). Altered Connectivity in Depression: GABA and Glutamate Neurotransmitter Deficits and Reversal by Novel Treatments. Neuron 102, 75–90. doi:10.1016/j.neuron.2019.03.013
Edmondson, D. A., Xia, P., McNally Keehn, R., Dydak, U., and Keehn, B. (2020). A Magnetic Resonance Spectroscopy Study of Superior Visual Search Abilities in Children with Autism Spectrum Disorder. Autism Res. 13, 550–562. doi:10.1002/aur.2258
El-Ansary, A., and Al-Ayadhi, L. (2014). GABAergic/Glutamatergic Imbalance Relative to Excessive Neuroinflammation in Autism Spectrum Disorders. J. Neuroinflammation 11, 189. doi:10.1186/s12974-014-0189-0
El-Khoury, R., Panayotis, N., Matagne, V., Ghata, A., Villard, L., and Roux, J. C. (2014). GABA and Glutamate Pathways Are Spatially and Developmentally Affected in the Brain of Mecp2-Deficient Mice. PLoS ONE 9, e92169. doi:10.1371/journal.pone.0092169
Fatemi, S. H., and Folsom, T. D. (2011). Dysregulation of Fragile × Mental Retardation Protein and Metabotropic Glutamate Receptor 5 in superior Frontal Cortex of Individuals with Autism: a Postmortem Brain Study. Mol. Autism 2, 6. doi:10.1186/2040-2392-2-6
Fatemi, S. H., Folsom, T. D., Kneeland, R. E., Yousefi, M. K., Liesch, S. B., and Thuras, P. D. (2013). Impairment of Fragile X Mental Retardation Protein-Metabotropic Glutamate Receptor 5 Signaling and its Downstream Cognates Ras-Related C3 Botulinum Toxin Substrate 1, Amyloid Beta A4 Precursor Protein, Striatal-Enriched Protein Tyrosine Phosphatase, and homer 1, in Autism: a Postmortem Study in Cerebellar Vermis and superior Frontal Cortex. Mol. Autism 4, 21. doi:10.1186/2040-2392-4-21
Fatemi, S. H., Folsom, T. D., Reutiman, T. J., and Thuras, P. D. (2009a). Expression of GABA(B) Receptors Is Altered in Brains of Subjects with Autism. Cerebellum 8, 64–69. doi:10.1007/s12311-008-0075-3
Fatemi, S. H., Reutiman, T. J., Folsom, T. D., Rooney, R. J., Patel, D. H., and Thuras, P. D. (2010). mRNA and Protein Levels for GABAAalpha4, Alpha5, Beta1 and GABABR1 Receptors Are Altered in Brains from Subjects with Autism. J. Autism Dev. Disord. 40, 743–750. doi:10.1007/s10803-009-0924-z
Fatemi, S. H., Reutiman, T. J., Folsom, T. D., and Thuras, P. D. (2009b). GABA(A) Receptor Downregulation in Brains of Subjects with Autism. J. Autism Dev. Disord. 39, 223–230. doi:10.1007/s10803-008-0646-7
Fekete, C. D., Chiou, T. T., Miralles, C. P., Harris, R. S., Fiondella, C. G., Loturco, J. J., et al. (2015). In Vivo clonal Overexpression of Neuroligin 3 and Neuroligin 2 in Neurons of the Rat Cerebral Cortex: Differential Effects on GABAergic Synapses and Neuronal Migration. J. Comp. Neurol. 523, 1359–1378. doi:10.1002/cne.23740
Filice, F., Vörckel, K. J., Sungur, A. Ö., Wöhr, M., and Schwaller, B. (2016). Reduction in Parvalbumin Expression Not Loss of the Parvalbumin-Expressing GABA Interneuron Subpopulation in Genetic Parvalbumin and Shank Mouse Models of Autism. Mol. Brain 9, 10. doi:10.1186/s13041-016-0192-8
Földy, C., Malenka, R. C., and Südhof, T. C. (2013). Autism-associated Neuroligin-3 Mutations Commonly Disrupt Tonic Endocannabinoid Signaling. Neuron 78, 498–509. doi:10.1016/j.neuron.2013.02.036
Fu, Z., and Vicini, S. (2009). Neuroligin-2 Accelerates GABAergic Synapse Maturation in Cerebellar Granule Cells. Mol. Cel. Neurosci. 42, 45–55. doi:10.1016/j.mcn.2009.05.004
Fung, L. K., Flores, R. E., Gu, M., Sun, K. L., James, D., Schuck, R. K., et al. (2021). Thalamic and Prefrontal GABA Concentrations but Not GABAA Receptor Densities Are Altered in High-Functioning Adults with Autism Spectrum Disorder. Mol. Psychiatry 26, 1634–1646. doi:10.1038/s41380-020-0756-y
Gharani, N., Benayed, R., Mancuso, V., Brzustowicz, L. M., and Millonig, J. H. (2004). Association of the Homeobox Transcription Factor, ENGRAILED 2, 3, with Autism Spectrum Disorder. Mol. Psychiatry 9, 474–484. doi:10.1038/sj.mp.4001498
Gogolla, N., Leblanc, J. J., Quast, K. B., Südhof, T. C., Fagiolini, M., and Hensch, T. K. (2009). Common Circuit Defect of Excitatory-Inhibitory Balance in Mouse Models of Autism. J. Neurodev. Disord. 1, 172–181. doi:10.1007/s11689-009-9023-x
Gonçalves, J., Violante, I. R., Sereno, J., Leitão, R. A., Cai, Y., Abrunhosa, A., et al. (2017). Testing the Excitation/inhibition Imbalance Hypothesis in a Mouse Model of the Autism Spectrum Disorder: In Vivo Neurospectroscopy and Molecular Evidence for Regional Phenotypes. Mol. Autism 8, 47. doi:10.1186/s13229-017-0166-4
Guo, H., Peng, Y., Hu, Z., Li, Y., Xun, G., Ou, J., et al. (2017). Genome-wide Copy Number Variation Analysis in a Chinese Autism Spectrum Disorder Cohort. Sci. Rep. 7, 44155. doi:10.1038/srep44155
Guptill, J. T., Booker, A. B., Gibbs, T. T., Kemper, T. L., Bauman, M. L., and Blatt, G. J. (2007). [3H]-flunitrazepam-labeled Benzodiazepine Binding Sites in the Hippocampal Formation in Autism: a Multiple Concentration Autoradiographic Study. J. Autism Dev. Disord. 37, 911–920. doi:10.1007/s10803-006-0226-7
Haddad, F. L., Patel, S. V., and Schmid, S. (2020). Maternal Immune Activation by Poly I:C as a Preclinical Model for Neurodevelopmental Disorders: A Focus on Autism and Schizophrenia. Neurosci. Biobehav. Rev. 113, 546–567. doi:10.1016/j.neubiorev.2020.04.012
Hadjikhani, N., Åsberg Johnels, J., Lassalle, A., Zürcher, N. R., Hippolyte, L., Gillberg, C., et al. (2018). Bumetanide for Autism: More Eye Contact, Less Amygdala Activation. Sci. Rep. 8, 3602. doi:10.1038/s41598-018-21958-x
Hadjikhani, N., Zürcher, N. R., Rogier, O., Ruest, T., Hippolyte, L., Ben-Ari, Y., et al. (2015). Improving Emotional Face Perception in Autism with Diuretic Bumetanide: a Proof-Of-Concept Behavioral and Functional Brain Imaging Pilot Study. Autism 19, 149–157. doi:10.1177/1362361313514141
Hallmayer, J., Cleveland, S., Torres, A., Phillips, J., Cohen, B., Torigoe, T., et al. (2011). Genetic Heritability and Shared Environmental Factors Among Twin Pairs with Autism. Arch. Gen. Psychiatry 68, 1095–1102. doi:10.1001/archgenpsychiatry.2011.76
Hammer, M., Krueger-Burg, D., Tuffy, L. P., Cooper, B. H., Taschenberger, H., Goswami, S. P., et al. (2015). Perturbed Hippocampal Synaptic Inhibition and γ-Oscillations in a Neuroligin-4 Knockout Mouse Model of Autism. Cell Rep. 13, 516–523. doi:10.1016/j.celrep.2015.09.011
Han, S., Tai, C., Westenbroek, R. E., Yu, F. H., Cheah, C. S., Potter, G. B., et al. (2012). Autistic-like Behaviour in Scn1a+/- Mice and rescue by Enhanced GABA-Mediated Neurotransmission. Nature 489 (7416), 385–390. doi:10.1038/nature11356
Happe, F. (1999). Autism: Cognitive Deficit or Cognitive Style. Trends Cogn. Sci. (Regul. Ed., 16–222. doi:10.1016/s1364-6613(99)01318-2
Harada, M., Taki, M. M., Nose, A., Kubo, H., Mori, K., Nishitani, H., et al. (2011). Non-invasive Evaluation of the GABAergic/glutamatergic System in Autistic Patients Observed by MEGA-Editing Proton MR Spectroscopy Using a Clinical 3 Tesla Instrument. J. Autism Dev. Disord. 41, 447–454. doi:10.1007/s10803-010-1065-0
Heise, C., Preuss, J. M., Schroeder, J. C., Battaglia, C. R., Kolibius, J., Schmid, R., et al. (2018). Heterogeneity of Cell Surface Glutamate and GABA Receptor Expression in Shank and CNTN4 Autism Mouse Models. Front. Mol. Neurosci. 11, 212. doi:10.3389/fnmol.2018.00212
Hogart, A., Nagarajan, R. P., Patzel, K. A., Yasui, D. H., and Lasalle, J. M. (2007). 15q11-13 GABAA Receptor Genes Are Normally Biallelically Expressed in Brain yet Are Subject to Epigenetic Dysregulation in Autism-Spectrum Disorders. Hum. Mol. Genet. 16, 691–703. doi:10.1093/hmg/ddm014
Hong, T., Falcone, C., Dufour, B., Amina, S., Castro, R. P., Regalado, J., et al. (2020). GABAARα2 Is Decreased in the Axon Initial Segment of Pyramidal Cells in Specific Areas of the Prefrontal Cortex in Autism. Neuroscience 437, 76–86. doi:10.1016/j.neuroscience.2020.04.025
Hoon, M., Soykan, T., Falkenburger, B., Hammer, M., Patrizi, A., Schmidt, K. F., et al. (2011). Neuroligin-4 Is Localized to Glycinergic Postsynapses and Regulates Inhibition in the Retina. Proc. Natl. Acad. Sci. U.S.A. 108, 3053–3058. doi:10.1073/pnas.1006946108
Horder, J., Petrinovic, M. M., Mendez, M. A., Bruns, A., Takumi, T., Spooren, W., et al. (2018). Glutamate and GABA in Autism Spectrum Disorder-A Translational Magnetic Resonance Spectroscopy Study in Man and Rodent Models. Transl. Psychiatry 8, 106. doi:10.1038/s41398-018-0155-1
Hou, Q., Wang, Y., Li, Y., Chen, D., Yang, F., and Wang, S. (2018). A Developmental Study of Abnormal Behaviors and Altered GABAergic Signaling in the VPA-Treated Rat Model of Autism. Front. Behav. Neurosci. 12, 182. doi:10.3389/fnbeh.2018.00182
Jager, A., Amiri, H., Bielczyk, N., van Heukelum, S., Heerschap, A., Aschrafi, A., et al. (2020). Cortical Control of Aggression: GABA Signalling in the Anterior Cingulate Cortex. Eur. Neuropsychopharmacol. 30, 5–16. doi:10.1016/j.euroneuro.2017.12.007
James, B. J., Gales, M. A., and Gales, B. J. (2019). Bumetanide for Autism Spectrum Disorder in Children: A Review of Randomized Controlled Trials. Ann. Pharmacother. 53, 537–544. doi:10.1177/1060028018817304
James, S. J., Shpyleva, S., Melnyk, S., Pavliv, O., and Pogribny, I. P. (2013). Complex Epigenetic Regulation of Engrailed-2 (EN-2) Homeobox Gene in the Autism Cerebellum. Transl. Psychiatry 3, e232. doi:10.1038/tp.2013.8
James, S. J., Shpyleva, S., Melnyk, S., Pavliv, O., and Pogribny, I. P. (2014). Elevated 5-hydroxymethylcytosine in the Engrailed-2 (EN-2) Promoter Is Associated with Increased Gene Expression and Decreased MeCP2 Binding in Autism Cerebellum. Transl. Psychiatry 4, e460. doi:10.1038/tp.2014.87
Jansen, A., Dieleman, G. C., Smit, A. B., Verhage, M., Verhulst, F. C., Polderman, T., et al. (2017). Gene-set Analysis Shows Association between FMRP Targets and Autism Spectrum Disorder. Eur. J. Hum. Genet. 25, 863–868. doi:10.1038/ejhg.2017.55
Jaramillo, T. C., Liu, S., Pettersen, A., Birnbaum, S. G., and Powell, C. M. (2014). Autism-related Neuroligin-3 Mutation Alters Social Behavior and Spatial Learning. Autism Res. 7, 264–272. doi:10.1002/aur.1362
Jembrek, M. J., and Vlainic, J. (2015). GABA Receptors: Pharmacological Potential and Pitfalls. Curr. Pharm. Des. 21, 4943–4959. doi:10.2174/1381612821666150914121624
Jiang, D. Y., Wu, Z., Forsyth, C. T., Hu, Y., Yee, S. P., and Chen, G. (2018). GABAergic Deficits and Schizophrenia-like Behaviors in a Mouse Model Carrying Patient-Derived Neuroligin-2 R215H Mutation. Mol. Brain 11, 31. doi:10.1186/s13041-018-0375-6
Jin, X., Cui, N., Zhong, W., Jin, X. T., and Jiang, C. (2013). GABAergic Synaptic Inputs of Locus Coeruleus Neurons in Wild-type and Mecp2-Null Mice. Am. J. Physiol. Cel. Physiol. 304, C844–C857. doi:10.1152/ajpcell.00399.2012
Kaila, K., Ruusuvuori, E., Seja, P., Voipio, J., and Puskarjov, M. (2014). GABA Actions and Ionic Plasticity in Epilepsy. Curr. Opin. Neurobiol. 26, 34–41. doi:10.1016/j.conb.2013.11.004
Kazlauskas, N., Campolongo, M., Lucchina, L., Zappala, C., and Depino, A. M. (2016). Postnatal Behavioral and Inflammatory Alterations in Female Pups Prenatally Exposed to Valproic Acid. Psychoneuroendocrinology 72, 11–21. doi:10.1016/j.psyneuen.2016.06.001
Kline, D. D., Ogier, M., Kunze, D. L., and Katz, D. M. (2010). Exogenous Brain-Derived Neurotrophic Factor Rescues Synaptic Dysfunction in Mecp2-Null Mice. J. Neurosci. 30, 5303–5310. doi:10.1523/JNEUROSCI.5503-09.2010
Krishnan, K., Lau, B. Y., Ewall, G., Huang, Z. J., and Shea, S. D. (2017). MECP2 Regulates Cortical Plasticity Underlying a Learned Behaviour in Adult Female Mice. Nat. Commun. 8, 14077. doi:10.1038/ncomms14077
Krystal, J. H., Sanacora, G., Blumberg, H., Anand, A., Charney, D. S., Marek, G., et al. (2002). Glutamate and GABA Systems as Targets for Novel Antidepressant and Mood-Stabilizing Treatments. Mol. Psychiatry 7 (Suppl. 1), S71–S80. doi:10.1038/sj.mp.4001021
Krzisch, M., Fülling, C., Jabinet, L., Armida, J., Gebara, E., Cassé, F., et al. (2017). Synaptic Adhesion Molecules Regulate the Integration of New Granule Neurons in the Postnatal Mouse Hippocampus and Their Impact on Spatial Memory. Cereb. Cortex 27, 4048–4059. doi:10.1093/cercor/bhw217
Kubas, B., Kułak, W., Sobaniec, W., Tarasow, E., Lebkowska, U., and Walecki, J. (2012). Metabolite Alterations in Autistic Children: a 1H MR Spectroscopy Study. Adv. Med. Sci. 57, 152–156. doi:10.2478/v10039-012-0014-x
Kumar, R. A., and Christian, S. L. (2009). Genetics of Autism Spectrum Disorders. Curr. Neurol. Neurosci. Rep. 9, 188–197. doi:10.1007/s11910-009-0029-2
Lattanzi, S., Trinka, E., Del Giovane, C., Nardone, R., Silvestrini, M., and Brigo, F. (2019). Antiepileptic Drug Monotherapy for Epilepsy in the Elderly: A Systematic Review and Network Meta-Analysis. Epilepsia 60, 2245–2254. doi:10.1111/epi.16366
Lawrence, Y. A., Kemper, T. L., Bauman, M. L., and Blatt, G. J. (2010). Parvalbumin-, Calbindin-, and Calretinin-Immunoreactive Hippocampal Interneuron Density in Autism. Acta Neurol. Scand. 121, 99–108. doi:10.1111/j.1600-0404.2009.01234.x
Lazaro, M. T., Taxidis, J., Shuman, T., Bachmutsky, I., Ikrar, T., Santos, R., et al. (2019). Reduced Prefrontal Synaptic Connectivity and Disturbed Oscillatory Population Dynamics in the CNTNAP2 Model of Autism. Cel Rep 27, 2567–2578. doi:10.1016/j.celrep.2019.05.006
Lemonnier, E., and Ben-Ari, Y. (2010). The Diuretic Bumetanide Decreases Autistic Behaviour in Five Infants Treated during 3 Months with No Side Effects. Acta Paediatr. 99, 1885–1888. doi:10.1111/j.1651-2227.2010.01933.x
Lim, C. S., Kim, H., Yu, N. K., Kang, S. J., Kim, T., Ko, H. G., et al. (2017). Enhancing Inhibitory Synaptic Function Reverses Spatial Memory Deficits in Shank2 Mutant Mice. Neuropharmacology 112, 104–112. doi:10.1016/j.neuropharm.2016.08.016
Löscher, W., and Vetter, M. (1984). Relationship between Drug-Induced Increases of GABA Levels in Discrete Brain Areas and Different Pharmacological Effects in Rats. Biochem. Pharmacol. 33, 1907–1914. doi:10.1016/0006-2952(84)90546-x
Ma, D. Q., Whitehead, P. L., Menold, M. M., Martin, E. R., Ashley-Koch, A. E., Mei, H., et al. (2005). Identification of Significant Association and Gene-Gene Interaction of GABA Receptor Subunit Genes in Autism. Am. J. Hum. Genet. 77, 377–388. doi:10.1086/433195
Ma, L. Y., Wu, C., Jin, Y., Gao, M., Li, G. H., Turner, D., et al. (2014). Electrophysiological Phenotypes of MeCP2 A140V Mutant Mouse Model. CNS Neurosci. Ther. 20, 420–428. doi:10.1111/cns.12229
Mahdavinasab, S. M., Saghazadeh, A., Motamed-Gorji, N., Vaseghi, S., Mohammadi, M. R., Alichani, R., et al. (2019). Baclofen as an Adjuvant Therapy for Autism: a Randomized, Double-Blind, Placebo-Controlled Trial. Eur. Child. Adolesc. Psychiatry 28, 1619–1628. doi:10.1007/s00787-019-01333-5
Mao, W., Watanabe, T., Cho, S., Frost, J. L., Truong, T., Zhao, X., et al. (2015). Shank1 Regulates Excitatory Synaptic Transmission in Mouse Hippocampal Parvalbumin-Expressing Inhibitory Interneurons. Eur. J. Neurosci. 41 (8), 1025–1035. doi:10.1111/ejn.12877
Marro, S. G., Chanda, S., Yang, N., Janas, J. A., Valperga, G., Trotter, J., et al. (2019). Neuroligin-4 Regulates Excitatory Synaptic Transmission in Human Neurons. Neuron 103, 617–626. e6. doi:10.1016/j.neuron.2019.05.043
Martella, G., Meringolo, M., Trobiani, L., De Jaco, A., Pisani, A., and Bonsi, P. (2018). The Neurobiological Bases of Autism Spectrum Disorders: the R451C-Neuroligin 3 Mutation Hampers the Expression of Long-Term Synaptic Depression in the Dorsal Striatum. Eur. J. Neurosci. 47, 701–708. doi:10.1111/ejn.13705
Matagne, V., Ehinger, Y., Saidi, L., Borges-Correia, A., Barkats, M., Bartoli, M., et al. (2017). A Codon-Optimized Mecp2 Transgene Corrects Breathing Deficits and Improves Survival in a Mouse Model of Rett Syndrome. Neurobiol. Dis. 99, 1–11. doi:10.1016/j.nbd.2016.12.009
Medrihan, L., Tantalaki, E., Aramuni, G., Sargsyan, V., Dudanova, I., Missler, M., et al. (2008). Early Defects of GABAergic Synapses in the Brain Stem of a MeCP2 Mouse Model of Rett Syndrome. J. Neurophysiol. 99, 112–121. doi:10.1152/jn.00826.2007
Meyer, U. (2014). Prenatal poly(i:C) Exposure and Other Developmental Immune Activation Models in Rodent Systems. Biol. Psychiatry 75, 307–315. doi:10.1016/j.biopsych.2013.07.011
Miyazaki, C., Matsuyama, K., Ichikawa, M., and Goto, S. (1988). Effect of Sodium Valproate (VPA) on Cerebral Amino Acids: Mechanism of Gamma-Aminobutyric Acid (GABA) Elevation and Possible Causal Relation of VPA-Induced Encephalopathy and Glutamine Level. Chem. Pharm. Bull. 36, 3589–3594. doi:10.1248/cpb.36.3589
Möhler, H. (2012). The GABA System in Anxiety and Depression and its Therapeutic Potential. Neuropharmacology 62, 42–53. doi:10.1016/j.neuropharm.2011.08.040
Murdoch, J. D., Gupta, A. R., Sanders, S. J., Walker, M. F., Keaney, J., Fernandez, T. V., et al. (2015). No Evidence for Association of Autism with Rare Heterozygous point Mutations in Contactin-Associated Protein-like 2 (CNTNAP2), or in Other Contactin-Associated Proteins or Contactins. Plos Genet. 11, e1004852. doi:10.1371/journal.pgen.1004852
Nau, H., and Löscher, W. (1982). Valproic Acid: Brain and Plasma Levels of the Drug and its Metabolites, Anticonvulsant Effects and Gamma-Aminobutyric Acid (GABA) Metabolism in the Mouse. J. Pharmacol. Exp. Ther. 220, 654–659.
Nicolini, C., and Fahnestock, M. (2018). The Valproic Acid-Induced Rodent Model of Autism. Exp. Neurol. 299, 217–227. doi:10.1016/j.expneurol.2017.04.017
Noroozi, R., Taheri, M., Ghafouri-Fard, S., Bidel, Z., Omrani, M. D., Moghaddam, A. S., et al. (2018). Meta-analysis of GABRB3 Gene Polymorphisms and Susceptibility to Autism Spectrum Disorder. J. Mol. Neurosci. 65, 432–437. doi:10.1007/s12031-018-1114-2
Oblak, A., Gibbs, T. T., and Blatt, G. J. (2009). Decreased GABAA Receptors and Benzodiazepine Binding Sites in the Anterior Cingulate Cortex in Autism. Autism Res. 2, 205–219. doi:10.1002/aur.88
Olexová, L., Štefánik, P., and Kršková, L. (2016). Increased Anxiety-like Behaviour and Altered GABAergic System in the Amygdala and Cerebellum of VPA Rats - an Animal Model of Autism. Neurosci. Lett. 629, 9–14.
Olmos-Serrano, J. L., Paluszkiewicz, S. M., Martin, B. S., Kaufmann, W. E., Corbin, J. G., and Huntsman, M. M. (2010). Defective GABAergic Neurotransmission and Pharmacological rescue of Neuronal Hyperexcitability in the Amygdala in a Mouse Model of Fragile X Syndrome. J. Neurosci. 30, 9929–9938. doi:10.1523/JNEUROSCI.1714-10.2010
Oyarzabal, A., Xiol, C., Castells, A. A., Grau, C., O'Callaghan, M., Fernández, G., et al. (2020). Comprehensive Analysis of GABAA-A1r Developmental Alterations in Rett Syndrome: Setting the Focus for Therapeutic Targets in the Time Frame of the Disease. Int. J. Mol. Sci. 21. doi:10.3390/ijms21020518
Pacey, L. K., Heximer, S. P., and Hampson, D. R. (2009). Increased GABA(B) Receptor-Mediated Signaling Reduces the Susceptibility of Fragile X Knockout Mice to Audiogenic Seizures. Mol. Pharmacol. 76, 18–24. doi:10.1124/mol.109.056127
Panzanelli, P., Früh, S., and Fritschy, J. M. (2017). Differential Role of GABAA Receptors and Neuroligin 2 for Perisomatic GABAergic Synapse Formation in the hippocampus. Brain Struct. Funct. 222, 4149–4161. doi:10.1007/s00429-017-1462-7
Peñagarikano, O., Abrahams, B. S., Herman, E. I., Winden, K. D., Gdalyahu, A., Dong, H., et al. (2011). Absence of CNTNAP2 Leads to Epilepsy, Neuronal Migration Abnormalities, and Core Autism-Related Deficits. Cell 147, 235–246. doi:10.1016/j.cell.2011.08.040
Pizzarelli, R., and Cherubini, E. (2013). Developmental Regulation of GABAergic Signalling in the hippocampus of Neuroligin 3 R451C Knock-In Mice: an Animal Model of Autism. Front. Cel. Neurosci. 7, 85. doi:10.3389/fncel.2013.00085
Provenzano, G., Gilardoni, A., Maggia, M., Pernigo, M., Sgadò, P., Casarosa, S., et al. (2020). Altered Expression of GABAergic Markers in the Forebrain of Young and Adult Engrailed-2 Knockout Mice. Genes (Basel) 11. doi:10.3390/genes11040384
Puts, N. A., and Edden, R. A. (2012). In Vivo magnetic Resonance Spectroscopy of GABA: a Methodological Review. Prog. Nucl. Magn. Reson. Spectrosc. 60, 29–41. doi:10.1016/j.pnmrs.2011.06.001
Puts, N., Wodka, E. L., Harris, A. D., Crocetti, D., Tommerdahl, M., Mostofsky, S. H., et al. (2017). Reduced GABA and Altered Somatosensory Function in Children with Autism Spectrum Disorder. Autism Res. 10, 608–619. doi:10.1002/aur.1691
Richard, A. E., Scheffer, I. E., and Wilson, S. J. (2017). Features of the Broader Autism Phenotype in People with Epilepsy Support Shared Mechanisms between Epilepsy and Autism Spectrum Disorder. Neurosci. Biobehav. Rev. 75, 203–233. doi:10.1016/j.neubiorev.2016.12.036
Richetto, J., Calabrese, F., Riva, M. A., and Meyer, U. (2014). Prenatal Immune Activation Induces Maturation-dependent Alterations in the Prefrontal GABAergic Transcriptome. Schizophr Bull. 40, 351–361. doi:10.1093/schbul/sbs195
Robertson, C. E., Ratai, E. M., and Kanwisher, N. (2016). Reduced GABAergic Action in the Autistic Brain. Curr. Biol. 26, 80–85. doi:10.1016/j.cub.2015.11.019
Rolf, L. H., Haarmann, F. Y., Grotemeyer, K. H., and Kehrer, H. (1993). Serotonin and Amino Acid Content in Platelets of Autistic Children. Acta Psychiatr. Scand. 87, 312–316. doi:10.1111/j.1600-0447.1993.tb03378.x
Rotschafer, S. E., Marshak, S., and Cramer, K. S. (2015). Deletion of Fmr1 Alters Function and Synaptic Inputs in the Auditory Brainstem. PLoS ONE 10, e0117266. doi:10.1371/journal.pone.0117266
Rudolph, U., and Möhler, H. (2014). GABAA Receptor Subtypes: Therapeutic Potential in Down Syndrome, Affective Disorders, Schizophrenia, and Autism. Annu. Rev. Pharmacol. Toxicol. 54, 483–507. doi:10.1146/annurev-pharmtox-011613-135947
Sabanov, V., Braat, S., D'Andrea, L., Willemsen, R., Zeidler, S., Rooms, L., et al. (2017). Impaired GABAergic Inhibition in the hippocampus of Fmr1 Knockout Mice. Neuropharmacology 116, 71–81. doi:10.1016/j.neuropharm.2016.12.010
Saitow, F., Takumi, T., and Suzuki, H. (2020). Change in Serotonergic Modulation Contributes to the Synaptic Imbalance of Neuronal Circuit at the Prefrontal Cortex in the 15q11-13 Duplication Mouse Model of Autism. Neuropharmacology 165, 107931. doi:10.1016/j.neuropharm.2019.107931
Sapey-Triomphe, L. A., Lamberton, F., Sonié, S., Mattout, J., and Schmitz, C. (2019). Tactile Hypersensitivity and GABA Concentration in the Sensorimotor Cortex of Adults with Autism. Autism Res. 12, 562–575. doi:10.1002/aur.2073
Sgadò, P., Genovesi, S., Kalinovsky, A., Zunino, G., Macchi, F., Allegra, M., et al. (2013). Loss of GABAergic Neurons in the hippocampus and Cerebral Cortex of Engrailed-2 Null Mutant Mice: Implications for Autism Spectrum Disorders. Exp. Neurol. 247, 496–505. doi:10.1016/j.expneurol.2013.01.021
Sheng, M., and Kim, E. (2000). The Shank Family of Scaffold Proteins. J. Cel. Sci. 113 (Pt 11), 1851–1856. doi:10.1242/jcs.113.11.1851
Silverman, J. L., Pride, M. C., Hayes, J. E., Puhger, K. R., Butler-Struben, H. M., Baker, S., et al. (2015). GABAB Receptor Agonist R-Baclofen Reverses Social Deficits and Reduces Repetitive Behavior in Two Mouse Models of Autism. Neuropsychopharmacology 40, 2228–2239. doi:10.1038/npp.2015.66
Sinclair, D., Featherstone, R., Naschek, M., Nam, J., Du, A., Wright, S., et al. (2017). GABA-B Agonist Baclofen Normalizes Auditory-Evoked Neural Oscillations and Behavioral Deficits in the Fmr1 Knockout Mouse Model of Fragile X Syndrome. eNeuro 4. doi:10.1523/ENEURO.0380-16.2017
Soltani, A., Lebrun, S., Carpentier, G., Zunino, G., Chantepie, S., Maïza, A., et al. (2017). Increased Signaling by the Autism-Related Engrailed-2 Protein Enhances Dendritic Branching and Spine Density, Alters Synaptic Structural Matching, and Exaggerates Protein Synthesis. PLoS ONE 12, e0181350. doi:10.1371/journal.pone.0181350
Sun, C., Zhang, L., and Chen, G. (2013). An Unexpected Role of Neuroligin-2 in Regulating KCC2 and GABA Functional Switch. Mol. Brain 6, 23. doi:10.1186/1756-6606-6-23
Tabuchi, K., Blundell, J., Etherton, M. R., Hammer, R. E., Liu, X., Powell, C. M., et al. (2007). A Neuroligin-3 Mutation Implicated in Autism Increases Inhibitory Synaptic Transmission in Mice. Science 318, 71–76. doi:10.1126/science.1146221
Thatcher, R. W., North, D. M., Neubrander, J., Biver, C. J., Cutler, S., and Defina, P. (2009). Autism and EEG Phase Reset: Deficient GABA Mediated Inhibition in Thalamo-Cortical Circuits. Dev. Neuropsychol. 34, 780–800. doi:10.1080/87565640903265178
Tillotson, R., and Bird, A. (2019). The Molecular Basis of MeCP2 Function in the Brain. J. Mol. Biol. doi:10.1016/j.jmb.2019.10.004
Tong, D. L., Chen, R. G., Lu, Y. L., Li, W. K., Zhang, Y. F., Lin, J. K., et al. (2019). The Critical Role of ASD-Related Gene CNTNAP3 in Regulating Synaptic Development and Social Behavior in Mice. Neurobiol. Dis. 130, 104486. doi:10.1016/j.nbd.2019.104486
Tyzio, R., Nardou, R., Ferrari, D. C., Tsintsadze, T., Shahrokhi, A., Eftekhari, S., et al. (2014). Oxytocin-mediated GABA Inhibition during Delivery Attenuates Autism Pathogenesis in Rodent Offspring. Science 343, 675–679. doi:10.1126/science.1247190
Tzanoulinou, S., García-Mompó, C., Riccio, O., Grosse, J., Zanoletti, O., Dedousis, P., et al. (2016). Neuroligin-2 Expression in the Prefrontal Cortex Is Involved in Attention Deficits Induced by Peripubertal Stress. Neuropsychopharmacology 41, 751–761. doi:10.1038/npp.2015.200
Uchigashima, M., Ohtsuka, T., Kobayashi, K., and Watanabe, M. (2016). Dopamine Synapse Is a Neuroligin-2-Mediated Contact between Dopaminergic Presynaptic and GABAergic Postsynaptic Structures. Proc. Natl. Acad. Sci. U.S.A. 113, 4206–4211. doi:10.1073/pnas.1514074113
Ure, K., Lu, H., Wang, W., Ito-Ishida, A., Wu, Z., He, L. J., et al. (2016). Restoration of Mecp2 Expression in GABAergic Neurons Is Sufficient to rescue Multiple Disease Features in a Mouse Model of Rett Syndrome. Elife 5, 35. doi:10.7554/eLife.14198
Van Zandt, M., Weiss, E., Almyasheva, A., Lipior, S., Maisel, S., and Naegele, J. R. (2019). Adeno-associated Viral Overexpression of Neuroligin 2 in the Mouse hippocampus Enhances GABAergic Synapses and Impairs Hippocampal-dependent Behaviors. Behav. Brain Res. 362, 7–20. doi:10.1016/j.bbr.2018.12.052
Varley, Z. K., Pizzarelli, R., Antonelli, R., Stancheva, S. H., Kneussel, M., Cherubini, E., et al. (2011). Gephyrin Regulates GABAergic and Glutamatergic Synaptic Transmission in Hippocampal Cell Cultures. J. Biol. Chem. 286, 20942–20951. doi:10.1074/jbc.M111.234641
Varman, D. R., Soria-Ortíz, M. B., Martínez-Torres, A., and Reyes-Haro, D. (2018). GABAρ3 Expression in Lobule X of the Cerebellum Is Reduced in the Valproate Model of Autism. Neurosci. Lett. 687, 158–163. doi:10.1016/j.neulet.2018.09.042
Veenstra-VanderWeele, J., Cook, E. H., King, B. H., Zarevics, P., Cherubini, M., Walton-Bowen, K., et al. (2017). Arbaclofen in Children and Adolescents with Autism Spectrum Disorder: A Randomized, Controlled, Phase 2 Trial. Neuropsychopharmacology 42, 1390–1398. doi:10.1038/npp.2016.237
Vien, T. N., Modgil, A., Abramian, A. M., Jurd, R., Walker, J., Brandon, N. J., et al. (2015). Compromising the Phosphodependent Regulation of the GABAAR β3 Subunit Reproduces the Core Phenotypes of Autism Spectrum Disorders. Proc. Natl. Acad. Sci. U.S.A. 112, 14805–14810. doi:10.1073/pnas.1514657112
Voituron, N., and Hilaire, G. (2011). The Benzodiazepine Midazolam Mitigates the Breathing Defects of Mecp2-Deficient Mice. Respir. Physiol. Neurobiol. 177, 56–60. doi:10.1016/j.resp.2011.02.002
Vorstman, J., Parr, J. R., Moreno-De-Luca, D., Anney, R., NurnbergerJr, J. I., and Hallmayer, J. F. (2017). Autism Genetics: Opportunities and Challenges for Clinical Translation. Nat. Rev. Genet. 18, 362–376. doi:10.1038/nrg.2017.4
Wang, L., Jia, M., Yue, W., Tang, F., Qu, M., Ruan, Y., et al. (2008). Association of the ENGRAILED 2 (EN2) Gene with Autism in Chinese Han Population. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147B, 434–438. doi:10.1002/ajmg.b.30623
Yang, J. Q., Yang, C. H., and Yin, B. Q. (2021a). Combined the GABA-A and GABA-B Receptor Agonists Attenuates Autistic Behaviors in a Prenatal Valproic Acid-Induced Mouse Model of Autism. Behav. Brain Res. 403, 113094. doi:10.1016/j.bbr.2020.113094
Yang, Y., Wang, B., Zhong, Z., Chen, H., Ding, W., and Hoi, M. (2021b). Clonazepam Attenuates Neurobehavioral Abnormalities in Offspring Exposed to Maternal Immune Activation by Enhancing GABAergic Neurotransmission. Biochem. Pharmacol. 192, 114711. doi:10.1016/j.bcp.2021.114711
Yip, J., Soghomonian, J. J., and Blatt, G. J. (2009). Decreased GAD65 mRNA Levels in Select Subpopulations of Neurons in the Cerebellar Dentate Nuclei in Autism: an In Situ Hybridization Study. Autism Res. 2, 50–59. doi:10.1002/aur.62
Yip, J., Soghomonian, J. J., and Blatt, G. J. (2008). Increased GAD67 mRNA Expression in Cerebellar Interneurons in Autism: Implications for Purkinje Cell Dysfunction. J. Neurosci. Res. 86, 525–530. doi:10.1002/jnr.21520
Zhang, B., Gokce, O., Hale, W. D., Brose, N., and Südhof, T. C. (2018). Autism-associated Neuroligin-4 Mutation Selectively Impairs Glycinergic Synaptic Transmission in Mouse Brainstem Synapses. J. Exp. Med. 215, 1543–1553. doi:10.1084/jem.20172162
Zhang, L., He, J., Jugloff, D. G., and Eubanks, J. H. (2008). The MeCP2-Null Mouse hippocampus Displays Altered Basal Inhibitory Rhythms and Is Prone to Hyperexcitability. Hippocampus 18, 294–309. doi:10.1002/hipo.20389
Zhang, L., Huang, C. C., Dai, Y., Luo, Q., Ji, Y., Wang, K., et al. (2020). Symptom Improvement in Children with Autism Spectrum Disorder Following Bumetanide Administration Is Associated with Decreased GABA/glutamate Ratios. Transl. Psychiatry 10, 9. doi:10.1038/s41398-020-0692-2
Zhang, W., Peterson, M., Beyer, B., Frankel, W. N., and Zhang, Z. W. (2014). Loss of MeCP2 from Forebrain Excitatory Neurons Leads to Cortical Hyperexcitation and Seizures. J. Neurosci. 34, 2754–2763. doi:10.1523/JNEUROSCI.4900-12.2014
Zhang, X., Ibi, M., Haga, R., Iwata, K., Matsumoto, M., Asaoka, N., et al. (2021). NOX1/NADPH Oxidase Affects the Development of Autism-like Behaviors in a Maternal Immune Activation Model. Biochem. Biophys. Res. Commun. 534, 59–66. doi:10.1016/j.bbrc.2020.11.070
Zhang, Z. W., Zak, J. D., and Liu, H. (2010). MeCP2 Is Required for normal Development of GABAergic Circuits in the Thalamus. J. Neurophysiol. 103, 2470–2481. doi:10.1152/jn.00601.2009
Zhong, W., Johnson, C. M., Cui, N., Oginsky, M. F., Wu, Y., and Jiang, C. (2017). Effects of Early-Life Exposure to THIP on Brainstem Neuronal Excitability in the Mecp2-Null Mouse Model of Rett Syndrome before and after Drug withdrawal(Article). Physiol. Rep.
Keywords: autism spectrum disorders, GABA, neurodevelopmental disorders, inhibitory neurotransmission, excitatory/inhibitory balance
Citation: Zhao H, Mao X, Zhu C, Zou X, Peng F, Yang W, Li B, Li G, Ge T and Cui R (2022) GABAergic System Dysfunction in Autism Spectrum Disorders. Front. Cell Dev. Biol. 9:781327. doi: 10.3389/fcell.2021.781327
Received: 22 September 2021; Accepted: 16 December 2021;
Published: 07 February 2022.
Edited by:
Chuang Wang, Ningbo University, ChinaReviewed by:
Doyoun Kim, Korea Research Institute of Chemical Technology (KRICT), South KoreaYihong Ma, Kumamoto University, Japan
Copyright © 2022 Zhao, Mao, Zhu, Zou, Peng, Yang, Li, Li, Ge and Cui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tongtong Ge, Z2V0dEBqbHUuZWR1LmNu; Ranji Cui, Y3VpcmFuamlAamx1LmVkdS5jbg==