 Priyanka Upadhyai
Priyanka Upadhyai Debalina Bose
Debalina Bose Neha Quadri
Neha Quadri- 1Department of Medical Genetics, Kasturba Medical College, Manipal, Manipal Academy of Higher Education, Manipal, Karnataka, India
- 2Advanced Technology Development Centre, Indian Institute of Technology Kharagpur, Kharagpur, West Bengal, India
The primary cilium is a microtubule-based sensory cell organelle templated by a modified parent centriole that mediates mechanotransduction and response to biochemical cues such as morphogens to regulate organismal development and homeostasis. Given that the cilium is a specialized microdomain devoid of its translation machinery, it relies on the endomembrane pathway for the delivery of proteins and other biomolecules to it. This review provides a comprehensive insight into how membrane trafficking modulators such as Rab and Rab-like proteins, and the exocyst complex control transport to the primary cilia, in turn regulating various aspects of their assembly and function. We integrate findings from in vitro and animal models and draw on human diseases associated with the dysfunction of Rabs or exocyst that exhibit phenotypes overlapping with those of ciliopathies, which further support their relevance to cilia biogenesis and maintenance.
1 Introduction
Cilia are conserved microtubular cell organelles indispensable for organismal development and tissue homeostasis. They were present in the last eukaryotic common ancestor (LECA) and have evolved a variety of structural specializations congruent with their functional adaptations throughout metazoa (Carvalho-Santos et al., 2011; Cavalier-Smith, 2022; Leung et al., 2025). Despite their architectural diversity, they retain universal features, such as an axonemal backbone with nine-fold microtubular symmetry that is templated by the mature parent centriole or basal body and is encased within the ciliary membrane that is contiguous with the plasma membrane. Cilia can be categorized into two broad groups: motile and primary cilia.
Motile cilia contain an axoneme with a central pair of microtubules in addition to the nine outer microtubule doublets (9 + 2) and are involved in cell functions such as fluid propulsion or cell motility. In more complex metazoans, they are present in the brain, middle ear, lungs, male and female reproductive tracts, and sperm, modulating functions such as the flow of cerebrospinal fluid, hearing, left-right patterning, mucus clearance, and mobility of the ova and sperm. Primary cilia are present in many postmitotic eukaryotic cells. They are non-motile due to the absence of the central pair of microtubules in their axonemes (9 + 0), except those at the left-right organizer in vertebrate embryos (Amack, 2022). Defects in ciliary structure or function cause disorders termed ciliopathies, which often manifest as congenital multisystemic abnormalities with phenotypes such as kidney cysts, retinal degeneration, and skeletal defects (Reiter and Leroux, 2017).
Cilia are largely devoid of translation machinery, except for mouse ependymal cells (Hao et al., 2021). During their assembly, membrane expansion is coordinated with axonemal extension; consequently, they rely on importing building blocks, such as tubulin (Craft et al., 2015), with the help of the intraflagellar transport (IFT) machinery (Lacey and Pigino, 2024). Proteins destined for the cilium contain ciliary targeting sequences (CTSs) and are transported to the cilium or periciliary membrane at its base by polarized vesicle trafficking (Nachury et al., 2010; Hsiao et al., 2012). This involves the biogenesis, trafficking, tethering, and fusion of vesicles that are orchestrated by a battery of factors, such as coat proteins, Ras-related protein in brain (Rab) guanosine triphosphatases (GTPases), multisubunit tethering complexes (MTCs), soluble N-ethyl-maleimide-sensitive factor (NSF) attachment protein receptors (SNAREs), motor proteins, and adaptors. The delivery of proteins into the ciliary compartment involves CTS recognition by specific mechanisms. Apart from some CTSs, for example, the Ax(S/A)xQ sequence recognized by the BBSome during the ciliary trafficking of somatostatin receptor 3 (Sstr3) (Jin et al., 2010), CTSs and corresponding trafficking module for most cilia proteins remain to be elucidated.
While membrane and soluble proteins (<40 kDa) can be passively translocated across the ciliary diffusion barrier, their delivery into the ciliary compartment is selectively modulated by the IFT-BBSome machinery, for example IFT-B regulates the transport of radial spokes in Chlamydomonas flagella (Lechtreck et al., 2022). Alternatively, importins can function as ciliary trafficking receptors for example, importin-β1 serves as the receptor for the transport of a Crumbs3 isoform to the primary cilium (Fan et al., 2007). Mechanisms of ciliary protein targeting have been extensively reviewed elsewhere (Jensen and Leroux, 2017; Lu and Madugula, 2018).
Here we review the Rabs, Rab-like proteins, and the exocyst complex, which is an MTC involved in vesicle tethering and exocytosis primarily at the plasma membrane, on how they regulate trafficking to the cilium, thereby controlling its biogenesis, maintenance, and function. Mutations in genes encoding these proteins or their regulatory factors may lead to disorders with abnormal cilia, underscoring their significance in cilia assembly and related pathologies (Table 1).
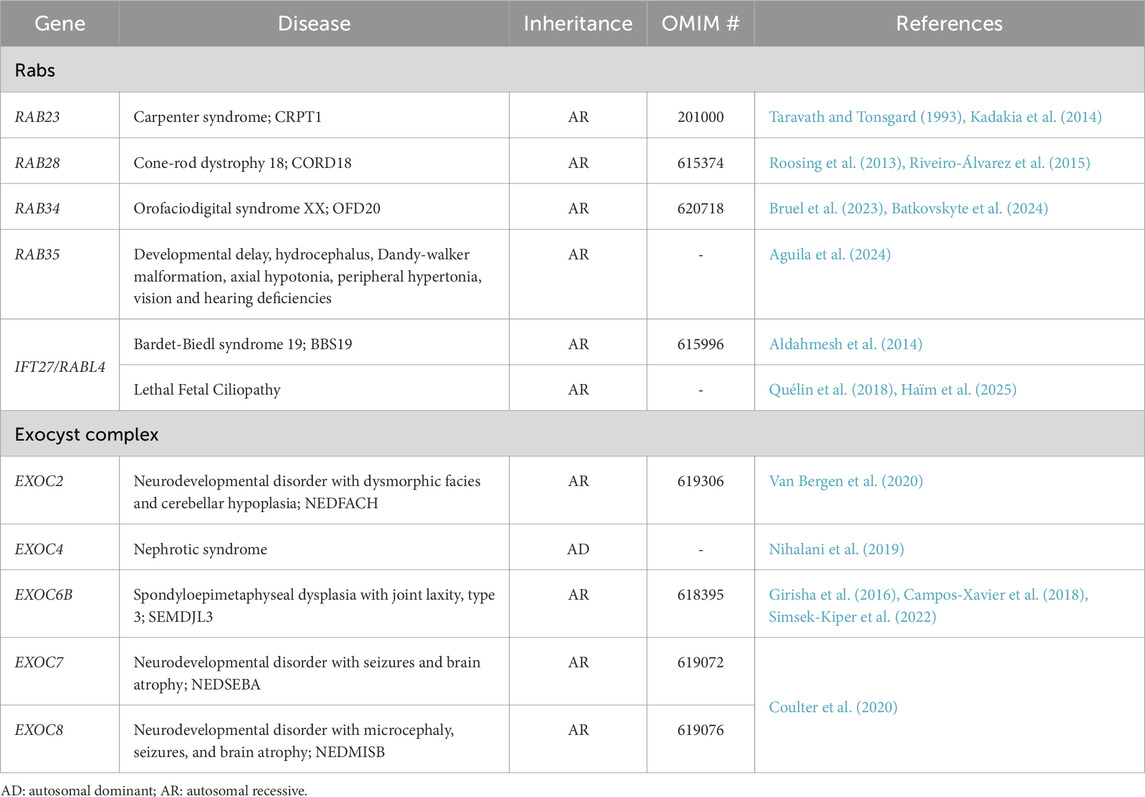
Table 1. Genetic disorders linked to cilia-associated Rabs and the exocyst complex.
2 Cilia structure and compositional uniqueness
The ciliary compartment is a specialized domain, enriched with biomolecules required for its assembly and functions. It is subdivided into distinct functional subdomains: ciliary pocket, a periciliary membrane invagination separating the membranes of some cilia from the adjoining plasma membrane (Molla-Herman et al., 2010); basal body anchoring it to the cell body through distal appendages (DAs) (Tanos et al., 2013; Ye et al., 2014) and subdistal appendages (sDAs) (Mazo et al., 2016; Chong et al., 2020); transition zone (TZ), the axonemal region most proximal to the basal body containing Y-shaped linkers that connect it to the ciliary membrane at sites known as ciliary necklace (Park and Leroux, 2022). The TZ consists of at least 15 proteins and is organized into the Meckle Syndrome (MKS), Nephronophthisis (NPHP), and Core-scaffolding modules (Park and Leroux, 2022). In metazoans, the subdomain proximal to it is termed the Inversin (INV) compartment; it has a distinct composition; it is devoid of Y-links and interacts with the TZ physically (Otto et al., 2003) and functionally (Warburton-Pitt et al., 2012). The distal appendage proteins (DAPs) form a cone-shaped gate in the transition fibers (TFs) that are modified DAs at the cilium base (Reiter et al., 2012), wherein CEP83, CEP89, CEP164, and SCLT1 form pinwheel spokes and FBF1 is embedded in the matrix (Yang et al., 2018). The ciliary tip is the distalmost end of the cilium where components of signaling pathways, such as, Glioma (Gli) and Sufu from the Hedgehog (Hh) pathway, aggregate in a signal-dependent manner (Haycraft et al., 2005; Chen et al., 2011) and retrograde cargo is loaded (Ye et al., 2018b). When G-protein-coupled-receptors (GPCRs) fail to be retrieved back to the cell via the BBSome (Ye et al., 2018a), they accumulate at the cilia tip before being removed by extracellular vesicles (Nager et al., 2017; Phua et al., 2017).
The cilium houses receptors and components of many signaling pathways, including Hh, GPCR, receptor tyrosine kinase, calcium, and transforming growth factor pathways, that enable it to integrate and transduce a plethora of extracellular signals, including developmental morphogens and mechanical cues (Mill et al., 2023; Hilgendorf et al., 2024). The spatiotemporal modulation of ciliary composition is integral to its dynamic signaling output and is regulated by the gating properties of the TZ and TFs (Chih et al., 2011; Jensen et al., 2015; Yang et al., 2018). In addition, protein distribution in the ciliary membrane (Hu et al., 2010) and along the axoneme (Ghossoub et al., 2013) is modulated by macromolecular scaffolds formed by the Septin proteins.
Investigation of primary cilia from various types of human and rodent cells (Ostrowski et al., 2002; Liu et al., 2007; Mayer et al., 2009; Ishikawa et al., 2012; Narita et al., 2012), motile cilia from multiciliated vertebrates (Sim et al., 2020) and unicellular species (Smith et al., 2005; Subota et al., 2014; McCafferty et al., 2024) shed light on their unique proteomes. These studies demonstrate that different ciliary subdomains also vary in their membrane lipid composition, which in turn is distinct from that of the plasma membrane. While the ciliary membrane has high levels of phosphatidylinositol-4 phosphate (PI4P) (Chávez et al., 2015; Garcia-Gonzalo et al., 2015), the TZ (Conduit et al., 2024) and plasma membrane (Kanemaru et al., 2022) are enriched with phosphatidylinositol 3, 4, 5 triphosphate (PI(3,4,5)P3) and phosphatidylinositol 4,5 bisphosphate (PI(4,5)P2), respectively. This unique lipid profile of the ciliary membrane subdomains is achieved by the opposing actions of phosphatases, for example, inositol polyphosphate 5 phosphatase E (INPP5E) (Bielas et al., 2009; Jacoby et al., 2009) and kinases, for example, type Iγ phosphatidylinositol 4-phosphate 5-kinase (PIPKIγ) (Xu et al., 2016). INPP5E localizes at the axoneme and its activity leads to the hydrolysis of 5-phosphate in PI(4,5)P2, leading to PI4P enrichment in the ciliary membrane; PI(4,5)P2 accumulates at the TZ owing to PIPKIγ localization at the basal body (Conduit and Vanhaesebroeck, 2020). The selective compartmentalization of phosphoinositides plays a pivotal role in modulating ciliary signaling. Tulp3, a PI(4,5)P2-interacting protein binds IFT-A complex to promote trafficking of a subset of ciliary GPCRs, such as Sstr3, Mchr1 (Mukhopadhyay et al., 2010) and Gpr161 (Chávez et al., 2015; Garcia-Gonzalo et al., 2015). Subsequently, Tulp3 has been shown to regulate a generalized multistep process for the ciliary uptake for integral membrane proteins (Badgandi et al., 2017). The spatial organization of phosphoinositides in the cilium also helps in regulating its morphology, for example, ribosome profiling of cilia regeneration in Chlamydomonas revealed serine palmitoyltransferase as an essential modulator of cilia morphology and biogenesis, as ceramides produced by it bind with IFT particles and motor proteins to mediate axoneme and ciliary membrane interaction (Wu et al., 2022).
3 Rabs in cilia trafficking
Rab proteins belong to the Ras superfamily of small GTPases, with 11 and 70 members identified in budding yeast and humans, respectively. They regulate various steps of intracellular trafficking, such as vesicle budding, transport, tethering, and membrane fusion. Functionally, they cycle between active GTP-bound and inactive GDP-bound forms (Müller and Goody, 2018). In their active state, they are engaged at the membrane via C-terminal prenylation and interact with specific effector proteins. The guanine nucleotide exchange factors (GEFs), promote the exchange of GDP for GTP, conversely, GTPase-activating proteins (GAPs) catalyze GTP hydrolysis, returning Rabs to their inactive cytosolic state. Structurally Rabs contain a canonical GTP-binding domain that consists of five conserved guanine moiety binding motifs (G1-G5), Rab family specific motif (RabF), Rab subfamily special motif, geranylgeranylation motif, a hypervariable C-terminal domain, a C-terminal interacting motif, switch regions, and complementary determining regions (Pereira-Leal and Seabra, 2000; Pereira-Leal and Seabra, 2001; Pereira-Leal et al., 2003; Stein et al., 2012; Li et al., 2014). Rabs control the directed traffic of post-Golgi or endocytic vesicles modulating ciliary vesicle formation, centriolar uncapping, basal body maturation, cilia membrane extension, IFT and cargo trafficking to the cilium.
3.1 Rab8-Rab11
3.1.1 Cilia assembly
Rab8 modulates the long-distance transport of trans-Golgi cargo (Wandinger-Ness and Zerial, 2014) and together with Rabin8, its GEF, and XM_037557 or TBC1D30, its GAP it modulates cilia biogenesis (Nachury et al., 2007; Yoshimura et al., 2007). Initiation of cilia assembly involves the transport particle protein complex II (TRAPPII) MTC and active Rab11GTP, a regulator of recycling endocytic vesicles to deliver Rabin8 to the preciliary vesicles (PCVs), which in turn recruits Rab8 (Knödler et al., 2010; Westlake et al., 2011). Interaction of TRAPPC14, a subunit of TRAPPII with Rabin8, Fbf1, and Cep83 promotes the engagement of the PCVs at the parent centriole (Cuenca et al., 2019). This is further augmented by Rab8 interaction with the distal centriolar proteins Cep164 (Schmidt et al., 2012), Ahi1 (Hsiao et al., 2009), and Talpid3 (Kobayashi et al., 2014). Fip3, a Rab11 effector binds Rabin8 to promote PCV trafficking, as well as stabilizes the Rab11-Rabin8 complex (Vetter et al., 2015; Walia et al., 2019). Recent imaging studies highlight the dynamic membrane conversion events in the Rab11-Rabin8-Rab8 cascade, wherein Rab11 and Rabin8 are depleted upon Rab8 loading (Saha et al., 2024). Moreover, Rab11 localization in the mature cilium was found to occur in a Rab8-dependent manner (Saha et al., 2024). Localization of Rab8 to the basal body is also facilitated by Chibby (Cby) which is recruited through its interaction with Cep164 (Burke et al., 2014) and Efa6a, a GEF for Arf6 GTPase (Partisani et al., 2021).
Ehd1, an Eps15 homology domain (Ehd) protein is a membrane-shaping factor that is recruited to PCVs and modulates their fusion into the ciliary vesicle (CV), which then encapsulates the distal surface of the parent centriole (Lu et al., 2015). This promotes localization of TZ proteins, Rpgrip1l, Tmem67 and Cep290 to the CV (Lu et al., 2015). Cep290 recruits Daz interacting zinc finger protein 1 (Dzip1), which in turn binds Rab8 and Cby to promote TZ assembly followed by CV extension (Zhang et al., 2015; Wu et al., 2020). Some studies suggest that Rab8 might be dispensable for the initial docking of ciliary vesicles, functioning only in subsequent stages of cilia membrane extension (Lu et al., 2015). Supporting this Rab8 was found to be non-essential for cilia formation in zebrafish and mammals (Sato et al., 2014; Aljiboury et al., 2023).
3.1.2 Ciliary trafficking
Rab8 is implicated in the trafficking of polycystin-1 (PC1) (Ward et al., 2011), polycystin-2 (PC2) (Hoffmeister et al., 2011), the C-terminal fragment of fibrocystin (Follit et al., 2010), Smoothened (Smo), a transmembrane Hh receptor, Kim1, an apical membrane protein to the ciliary membrane (Figure 1). It assists in the transport of Dishevelled, a core planar cell polarity (PCP) component to the basal body (Zilber et al., 2013) and that of EB1, a cytosolic microtubule-binding protein into the cilium (Boehlke et al., 2010). The enrichment of Ift20 at the basal body also depends on Rab8 (Maharjan et al., 2020). The Golgi to cilia transport of rhodopsin, a GPCR occurs through directed rhodopsin transport carriers to the rod outer segment of vertebrate photoreceptors and involves the action of the Rab11–Fip3–Rabin8 dual effector complex and the interaction of Arf4 with Asap1, its GAP (Deretic et al., 1995; Mazelova et al., 2009; Bachmann-Gagescu et al., 2011; Wang et al., 2012; Vetter et al., 2015; Wang and Deretic, 2015). The transport of Kif17, a soluble kinesin-2 motor protein, Crumbs3 and retinitis pigmentosa 2 to the cilia membrane utilizes Importin-β2 and transportin 1 (TNPO1) that are conserved receptors in the nucleocytoplasmic trafficking machinery (Fan et al., 2007; Dishinger et al., 2010; Hurd et al., 2011; Kee et al., 2012) Based on studies with the known CTSs of fibrocystin, photoreceptor retinol dehydrogenase, rhodopsin and retinitis pigmentosa 2, a dynamic ternary complex of TNPO1-Rab8-CTS was deduced that can modulate the selective entry and retention of ciliary membrane proteins (Madugula and Lu, 2016). Once inside the cilium, GTP hydrolysis converts Rab8 to its inactive GDP-bound state leading to the release of its cargo. The Rab8GDP is ubiquitinated and pre-emptively degraded by the protein quality control machinery (Takahashi et al., 2019) to prevent its accumulation which could exert harmful effects (Nachury et al., 2007; Yoshimura et al., 2007).
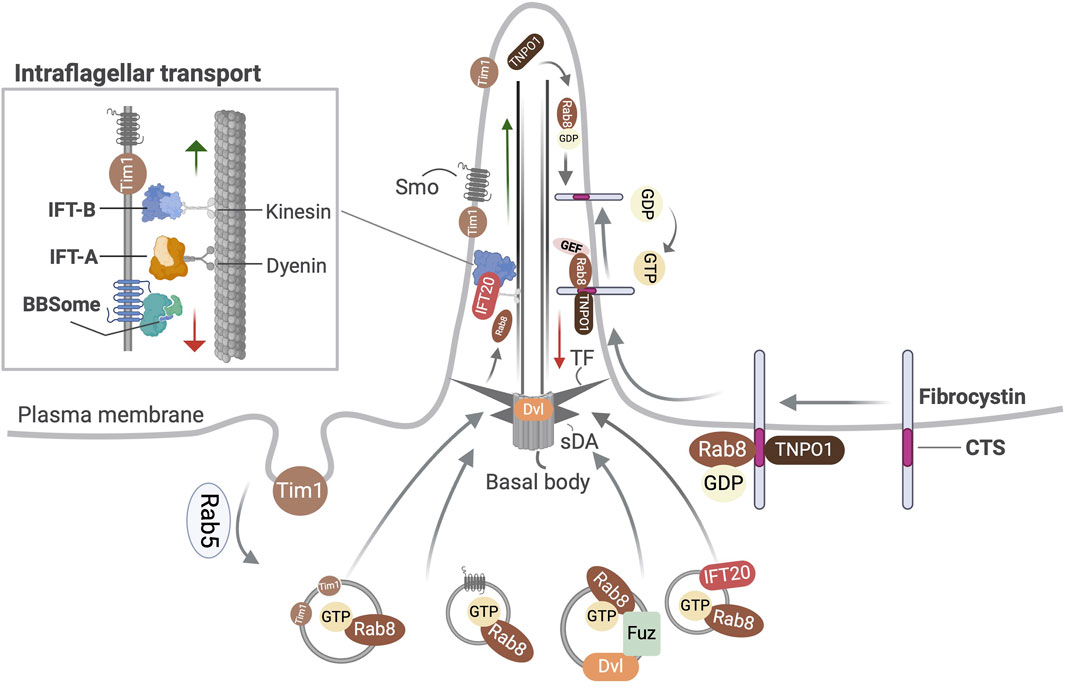
Figure 1. Overview of the transport functions of Rab8 related to primary cilia. Shown is Rab8 dependent transport of Smo, Dishevelled (Dvl), Tim1, and IFT20 into the primary cilium. In ciliated cells Fuzzy (Fuz) mediates the recruitment of Rab8 and Dvl to the primary cilium via the polarized trafficking route. Ciliary localization of fibrocystin involves the formation of a ternary complex with Rab8-TNPO1 at its ciliary targeting signal (CTS). TF, transition fibers; sDA, subdistal appendages.
Abnormal rab-8 expression in C. elegans compromises cilia structure and membrane transport (Kaplan et al., 2010). Dominant negative (T22N) form of rab8 in Xenopus laevis rod photoreceptors results in retinal degeneration and accumulation of tubulovesicular structures at the cilium base in surviving outer rod segments (Moritz et al., 2001). Interestingly, rab8 was dispensable while rab11 and rab35 were required for centrosome positioning and cilia formation during the development of the Kupffer’s vesicle (left-right organizer) in zebrafish (Aljiboury et al., 2023). In mammals, Rab8 occurs as Rab8a and Rab8b, which are isoforms encoded by different genes (Armstrong et al., 1996). Rab8b null mice are normal; Rab8a and Rab8b double knockout (KO) mice are also viable and largely normal except for the mis-localization of apical markers in selected tissues and do not show any ciliary abnormalities (Sato et al., 2014). This suggests that their ciliary functions may be redundant with other Rabs and likely explains the absence of human diseases associated with them. However, pathogenic variants in genes encoding Rab8 interacting proteins have been implicated in human disorders. Abnormal hexanucleotide repeat expansions in C9orf72 that together with SMCR8 function as Rab8-GAP are implicated in approximately half the genetic cases of amyotrophic lateral sclerosis and frontotemporal dementia via the suppression of ciliogenesis and Hh signaling (Tang et al., 2024). Mutations in OCRL1 that encodes for inositol-5-phosphatase, another Rab8 effector cause the X-linked oculocerebrorenal syndrome of Lowe (OCRL) or Lowe syndrome which manifests with abnormal primary cilia (Hou et al., 2011; Coon et al., 2012).
Mutations in Leucine rich repeat kinase 2 (LRRK2) that is a major regulator of both idiopathic (Nalls et al., 2014) and genetic forms of Parkinson’s Disease (PD) (Zimprich et al., 2004) cause hyperactivation of its kinase activity and are implicated in phosphorylation of Rab8a, Rab10 and Rab12 in cultured human and mouse cells, and mice (Ito et al., 2016; Steger et al., 2016; Thirstrup et al., 2017). Activating mutations in LRRK2 cause aberrant accumulation of phosphorylated Rab8a at the centrosome leading to abnormal centrosomal cohesion and positioning, and defective cell polarity and migration (Madero-Pérez et al., 2018).
3.2 Rab10
Rab10 belongs to the same subfamily as Rab8 and is implicated in intracellular transport activities ranging from polarized exocytosis to endo-phagocytic processes (Chua and Tang, 2018). It localizes at the trans-Golgi, endoplasmic reticulum (ER), and at the base of primary cilia in renal epithelia. It colocalizes with Exoc3 and Exoc4, which are exocyst complex subunits, and directly interacts with Exoc4 at the basal body (Babbey et al., 2010) (Figure 2). Earlier studies suggest that Rab10 may be required for cilia biogenesis, as its depletion impaired ciliation more strongly compared to the combined inhibition of Rab8a and Rab8b, and the cumulative silencing of all three had the strongest effect (Sato et al., 2014). In contrast, RAB8A/B and RAB10 KO in hTERT-RPE cells had no effect on ciliogenesis (Oguchi et al., 2020). Another study suggested that Rab10 antagonizes primary ciliogenesis (Dhekne et al., 2018). Dennd2b, a Rab10 GEF that localizes at the basal body was found to suppress ciliogenesis by modulating Rab10-dependent recruitment of CP110, whereas it controlled cilia length by RhoA activation (Kumar et al., 2022).
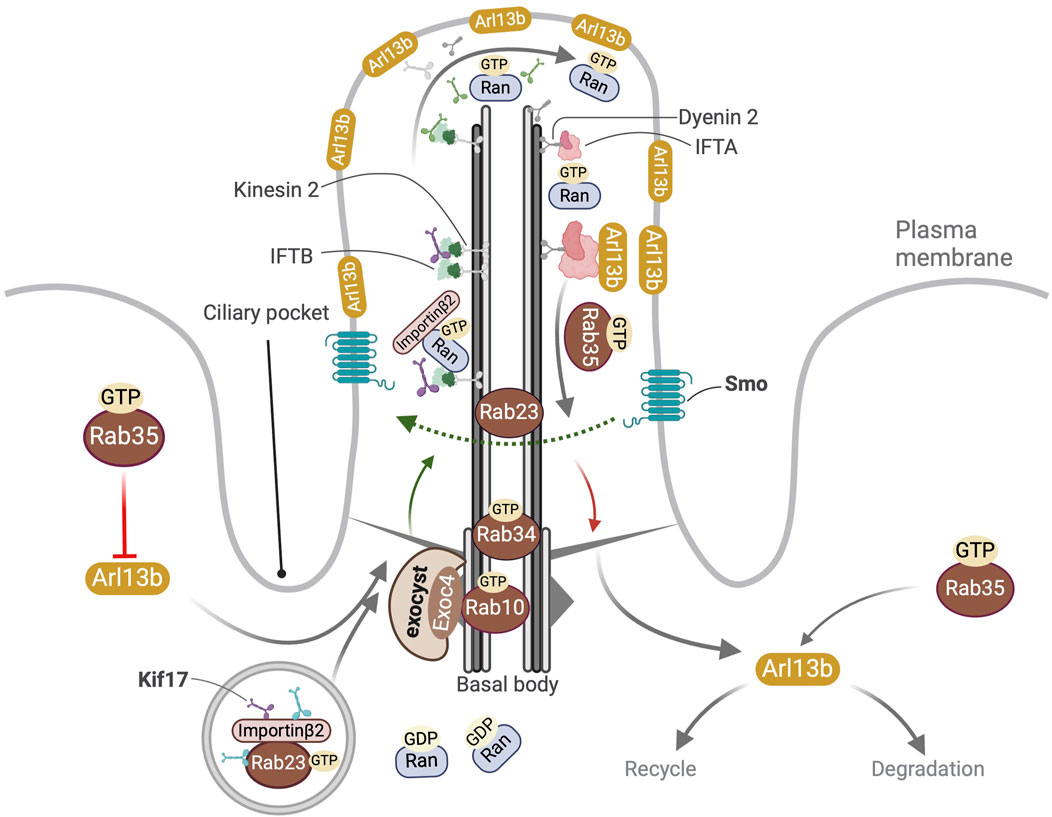
Figure 2. Overview of ciliary localization and functions of Rab10, Rab23, Rab34 and Rab35. Localization of GTP bound Rab10 and Rab34 at the basal body is shown. Active Rab10GTP directly interacts with the exocyst component Exoc4. In the cytosol Rab23GTP interacts with Kif17 and promotes its binding to importin-β2 to form a tripartite complex that then enters the cilia owing to the RanGTP-RanGDP gradient between the primary cilium and the cytosol. In the cilium RanGTP binds importin-β2 leading to the release of Kif17 from Rab23 and the anterograde transport of Kif17 to the primary cilia tip. Rab23 also promotes recycling of ciliary Smo. Rab35GTP controls the ciliary membrane composition by (a) inhibiting the import of Arl13b into the cilium (b) promoting the export of Arl13b out of the cilium (c) modulation of endocytic processes targeting Arl13b for degradation or recycling to non-ciliary destinations near the ciliary pocket.
Genetic disorders with Rab10 involvement include PD, wherein hyperactivated LRRK2 increases Rab10 phosphorylation, which in turn promotes its association with Rab interacting lysosomal protein-like 1 (RILPL1), a known suppressor of ciliogenesis (Dhekne et al., 2018) and accentuates the removal of ciliary membrane proteins (Schaub and Stearns, 2013). Disruption of DENND2B in a patient with subtelomeric de novo balanced translocation was associated with severe intellectual disability, muscular hypotonia, seizures, bilateral sensorineural hearing loss, submucous cleft palate, unilateral cystic kidney dysplasia, and other anomalies (Göhring et al., 2010).
3.3 Rab12
Rab12 is a key substrate of LRRK2 in the mouse brain that regulates the balance of ciliogenesis by controlling the centrosome/centriole homeostasis and inhibiting primary ciliogenesis. This depends on its interaction with LRRK2 and the phosphorylation of Rab10 (Dhekne et al., 2023). While Rab12 KO mice are viable with no obvious developmental anomalies, examination of their brain slices revealed an increased frequency of ciliated cells and longer cilia in the striatum, even though the mean cilia volume remained unchanged; similar results were obtained from Rab12 KO-derived primary astrocyte cultures in vitro (Li et al., 2024). This study showed that the overexpression of wild-type Rab12 led to centrosome amplification and aberrant cilia that resemble short “spikes” consisting of an enlarged lumen surrounding a defective axoneme. The overexpressed Rab12 promoted the colocalization of phosphorylated Rab10 and RILPL1 with endogenous LRRK2 at the amplified centrosomes (Dhekne et al., 2023). Similar results were obtained following Rab12 overexpression in RPE cells. Accordingly, when LRRK2 was inhibited in astrocytes where a hyperactivating form of Rab12 was expressed, it abolished Rab10 phosphorylation and prevented the Rab12 overactivation-dependent deregulation of primary ciliation (Li et al., 2024). This study also showed that in primary cultures of astrocytes derived from mice carrying LRRK2-G2019S, a common PD mutation, the disruption of Rab12 restored primary ciliation and centrosome homeostasis. Therefore, targeting the Rab12-LRRK2 complex could offer an attractive strategy for the amelioration of PD.
3.4 Other Rabs and primary cilia in the pathogenesis of PD
Other Rabs that are substrates of LRRK2 include Rab3, Rab35, and Rab43 (Steger et al., 2017). The pathology of PD involves the loss of dopaminergic neurons in the substantia nigra pars compacta region of the brain that projects into the dorsal striatum (Surmeier et al., 2014). Studies have shown that hyperactivating LRRK2 mutations induce loss of cilia and Hh signaling in the cholinergic and parvalbumin interneurons and astrocytes in the mouse dorsal striatum, leading to the decreased expression of glia-derived neurotrophic factors, namely GDNF (Khan et al., 2021; Khan et al., 2024) and Neurturin (Lin et al., 2025) that are neuroprotective and essential for the survival of dopamine-synthesizing neurons.
3.5 Rab28
Rab28 carries a C-terminal CAAX motif instead of the common geranylgeranylation motif present in other Rab family members (Brauers et al., 1996) that gets prenylated and is required for its membrane retention at specific subcellular locations. It was identified as a ciliary protein in rat retina where it localized at the basal body and ciliary rootlet of photoreceptors (Roosing et al., 2013). In C. elegans its active GTP-bound form localizes at the periciliary membrane and requires the prenyl-binding protein Pde6d, a molecular chaperone, the BBSome and Arl-3 for import into the cilium and bidirectional IFT (Jensen et al., 2016; Akella et al., 2020). Ciliary Rab28 and the BBSome negatively regulate extracellular vesicles in a cilia-dependent manner in the sensory organs of C. elegans (Akella et al., 2020).
Biallelic RAB28 null and hypomorphic alleles cause the rare cone-rod dystrophy (CRD; OMIM #615374) (Roosing et al., 2013; Riveiro-Álvarez et al., 2015). CRD is characterized primarily by the loss of cone photoreceptors or sometimes the loss of both cone and rod photoreceptors. It manifests with decreased visual acuity, color vision abnormalities, photo aversion, decreased sensitivity in the central visual field, progressive loss of peripheral vision, and night blindness (Hamel, 2007). Rab28 KO mice exhibit progressive retina degeneration phenocopying the clinical features of CRD (Ying et al., 2018). These mice showed that Rab28 is required for shedding and phagocytosis of cone outer segment discs (Ying et al., 2018). In zebrafish rab28 is required for both dawn and dusk outer segment phagocytosis peaks but not its basal levels (Carter et al., 2020; Moran et al., 2022).
A study with a homozygous missense single nucleotide variants (SNVs) in RAB28 in a pair of siblings from a consanguineous family reported significant retinal degeneration and postaxial polydactyly (PAP) wherein the transcript levels of Rab28 were unperturbed but its ciliary localization was abrogated (Jespersgaard et al., 2020). How Rab28 dysfunction causes PAP remains uncertain; however, this finding suggests that it may have a broader role in ciliopathies beyond CRD.
3.6 Rab34
Rab34 was first discovered in the Golgi apparatus (Wang and Hong, 2002). A CRISPR-based in vitro screen identified it as a positive regulator of primary ciliogenesis and Hh signaling (Pusapati et al., 2018). It was later identified as a ciliary protein using Ift27 proximity biotinylation in MEFs (Stuck et al., 2021). It modulates early cilia biogenesis by facilitating the fusion of PCVs into CVs (Xu et al., 2018). An extensive screen suggested that it was essential for serum starvation-dependent ciliogenesis in various cell lines e.g., hTERT-RPE1, NIH/3T3 cells, and MCF10A but unlike other Rabs its N-terminal and not the switch II region was required for ciliogenesis (Oguchi et al., 2020; Oguchi et al., 2022). Rab34 localizes at the basal body, and its GTP-bound form enters and resides in the cilium (Xu et al., 2018) (Figure 2). It localizes on the ciliary sheath in a specialized subdomain of assembling intracellular cilia and is pivotal for orchestrating initial steps in cilia membrane formation specifically in the intracellular but not the extracellular cilia biogenesis pathway (Ganga et al., 2021; Stuck et al., 2021).
Rab34−/− mice die perinatally, exhibiting polydactyly and craniofacial malformations (Dickinson et al., 2016; Xu et al., 2018). Rab34 plays both cilia dependent and independent roles in osteogenesis; its silencing in MC3T3-E1 murine preosteoblast cells strongly attenuated ciliation and Hh signaling that impaired cell proliferation, and osteoblast differentiation, in addition to the cilia-independent attenuation of type I collagen trafficking from the ER to Golgi apparatus (Yamaguchi et al., 2024). Recently, biallelic pathogenic nonsynonymous SNVs in RAB34 have been linked with orofacial digital syndrome XX (OFDXX; OMIM #620718), wherein the patients manifest with features such as polydactyly/syndactyly, shortening of long bones, cleft lip and palate, micrognathia, cerebral, cardiac and anorectal anomalies (Bruel et al., 2023; Batkovskyte et al., 2024). Despite the RAB34 pathogenic mutations being discerned as strong loss of function alleles that suppressed its expression and impaired primary ciliogenesis, the localization of the mutant RAB34 proteins at the basal body remained unperturbed (Bruel et al., 2023). Thus, a complete understanding of the molecular pathogenesis underlying OFDXX is still awaited.
3.7 Rab23
Rab23 was first identified in vertebrate embryogenesis for its function in dorsalization as an antagonist of Sonic hedgehog (Shh) that facilitates ventral cell fate specification (Chiang et al., 1996; Spörle et al., 1996). It localizes at the plasma membrane and endocytic vesicles, functioning downstream of Patched (Ptch) and Smo, which are Hh receptors but upstream of Gli transcription factors (Evans et al., 2003; Eggenschwiler et al., 2006). Rab23 activation requires Inturned and Fuzzy, which are GEFs and PCP pathway components functioning downstream of CV formation, and potentially parallel to Rab8 during primary cilia assembly (Gerondopoulos et al., 2019). Rab23 is required for cilia biogenesis (Yoshimura et al., 2007), where it controls the recycling of ciliary Smo (Boehlke et al., 2010) (Figure 2). Beyond the Shh pathway, it is also required for the ciliary trafficking of Kif17, a kinesin-2 motor protein by forming a complex with it and its nuclear transport adaptor Importin-β2 (Lim and Tang, 2015). It also promotes the IFT-B anterograde trafficking and Kif17-dependent targeting of D1-type dopaminergic receptors to the primary cilium (Leaf and Von Zastrow, 2015). It is also implicated in the ciliary targeting of other cargo, for example, subunits of olfactory cyclic nucleotide-gated channels (Jenkins et al., 2006). It indirectly controls the velocity of anterograde IFT as the latter depends on the number of active Kif17 motors and its dynamic equilibrium with Kif3ab motors when loaded on microtubules (Milic et al., 2017). Outside the cilium active Rab23 facilitates Kif17 migration to spindle poles; Rab23-Kif17-cargo complex modulates spindle organization via tubulin acetylation and drives actin cytoskeleton-mediated spindle migration during oocyte meiosis (Wang H. H. et al., 2019).
Pathogenic SNVs in RAB23 are associated with Carpenter Syndrome (CS) a rare genetic disorder characterized by craniosynostosis of the face and skull, polydactyly, brachydactyly, heart and eye defects, structural and functional central nervous system (CNS) abnormalities (OMIM #201000) (Taravath and Tonsgard, 1993; Kadakia et al., 2014). Despite its well-established role in ciliogenesis, whether RAB23 pathogenic variants cause abnormal ciliation in CS patients remains to be elucidated. Interestingly Rab23 null mice (open brain) succumb mid-gestation due to Hh hyperactivation in the neural tube (Eggenschwiler et al., 2001).
3.8 Rab35
Rab35 is a conserved plasma membrane and endosomal protein with many essential cellular functions, for example, in the final steps of cytokinesis, endocytic cargo recycling, cytoskeleton modulation, and cell polarity (Kouranti et al., 2006; Klinkert et al., 2016). Its localization at the ciliary membrane in its active GTP-bound state is controlled by Dennd1b, its GEF, and Tbc1d10a, its GAP (Kuhns et al., 2019). This study also showed that Rab35 directly interacts with Arl13b, modulating ciliary membrane composition by augmenting the ciliary transport of Smo, PI(4,5)P2 and antagonizing the targeting of Arl13b and in turn that of INPP5E (Figure 2).
Rab35 was indispensable for ciliation in the left-right organizer in zebrafish, acting upstream of Rab11 for the apical membrane targeting of CFTR receptor in this context (Aljiboury et al., 2023). Recent studies identified MiniBAR, a dual Rab35 and Rac1 effector, which co-localizes with them at the vesicles destined for the primary cilia, controlling the ciliary targeting of proteins, such as Ift88 and Arl13b and regulating ciliogenesis through modulation of the actin cytoskeleton (Serres et al., 2023). This study also showed that MiniBAR depletion causes left-right asymmetry defects and ciliopathy-like features in zebrafish.
A recent study reported a single patient with a neurodevelopmental disorder harboring a missense mutation in RAB35 which likely locks it in an inactive conformation, delaying cytokinesis, activating Arf6, and antagonizing primary ciliogenesis (Aguila et al., 2024). Consistent with its cellular roles, Rab35 KO mice are embryonic lethal with cardiac edema; its conditional depletion in the kidney and ureter causes hydronephrosis and manifests with reduced primary cilia length, actin cytoskeleton disruption, abnormal cell polarization with loss of tight junctions, reduced adherens junctions, defective Arf6 epithelial polarity, cell death, and compromised differentiation (Clearman et al., 2024).
3.9 Rab19
In polarized MDCK cells, Rab19 localizes at vesicles accumulating at the apical cell surface and subsequently peripheral to the site of cortical actin clearing during ciliation (Jewett et al., 2021). This study showed that its depletion abrogated actin clearing above the centrosome blocking primary cilia assembly. Rab19-mediated orchestration of ciliary membrane growth occurred both in extracellular and intracellular pathways of ciliogenesis (Jewett et al., 2021). In its absence, cilia assembly stopped at the CV stage, even though the localization of Ift88, Ift140, and Rpgrip1l were unperturbed (Jewett et al., 2021). This study also showed that Rab19 interacts with Tbc1d4, a Rab-GAP as well as with every subunit of the homotypic fusion and vacuole protein sorting (HOPS) complex, a MTC that mediates vesicle and membrane fusion in late endosomes and lysosomes (van der Beek et al., 2019; Jewett et al., 2021). Subsequent work revealed that ablation of HOPS blocked basal body linked cortical actin clearing indirectly by retaining Rab19 on enlarged endosomes-lysosomes away from the basal body (Hoffman and Prekeris, 2023). Nevertheless, the direct involvement of HOPS and Rab19 interaction in primary cilia biogenesis remains inconclusive.
3.10 Rab5
Rab5 is a principal regulator of early endocytosis that is involved in early endosome biogenesis and fusion, multivesicular body formation, and endosomal trafficking (Gorvel et al., 1991; Bucci et al., 1992; Zeigerer et al., 2012). It was linked to primary cilia by studies in C. elegans that discerned it at the endocytic vesicles targeted to the periciliary membrane compartment at the ciliary base and a minor population of it was also noted in some axonemes (Kaplan et al., 2010; van der Vaart et al., 2015). It colocalized with Ocrl1 on internalized cilia-derived endosomes (Coon et al., 2012). Nevertheless, whether it is directly involved in primary ciliogenesis is uncertain as dominant negative Rab5 attenuated the expression of Kim1 at the apical cell surface with no effect on the transport of ciliary proteins, such as Smo and EB1, and Kim1 silencing also did not affect cilia biogenesis (Boehlke et al., 2010).
4 Rab-like GTPases
4.1 Rabl2
Rabl2 is an atypical Rab-like GTPase devoid of the C-terminal isoprenylation motif present in canonical Rabs (Homma et al., 2021). It is recruited to the basal body through interactions with Cep19 (Nishijima et al., 2017; Zhou et al., 2022), Cep164 and Cep83 (Dateyama et al., 2019) (Figure 3). It binds GTP by GEF-independent high-turnover nucleotide exchange cycle, and in this state interacts with the IFT-B1 core subcomplex (Taschner et al., 2016) via the Ift74/Ift81 heterodimer to recruit the IFT-B holo-complex from a pool of pre-docked IFT-B complexes, into the cilium (Kanie et al., 2017). Thus, the Cep19-Rabl2-IFT-B complex mediates the ciliary uptake of IFT moieties. The stable KO of Cep19 or Rabl2 decreased the frequency of IFT trains in motion but had no effect on IFT velocities (Kanie et al., 2017). The Ift74/Ift81 heterodimer serves as a Rabl2-specific GAP (Boegholm et al., 2023). Rabl2 has been suggested to promote the ciliary targeting of GPCRs such as Gpr161 and Htr6 (Dateyama et al., 2019). One model proposed that it indirectly regulates the BBSome-mediated export across the TZ by binding IFT-B particles (Duan et al., 2021). The authors in this study suggested that this is required for fine-tuning of signals; Rabl2−/− MEFs are unable to activate the Hh signaling in response to stimulation (Duan et al., 2021). Subsequent studies showed that Ift25/Ift27 and Rabl2 bind the Ift74/Ift81 heterodimer in a mutually exclusive manner (Zhou et al., 2022). In this work the authors suggested that Rabl2GTP-Cep19 at the basal body promotes the recruitment of IFT-B particles lacking Ift25/Ift27; following the hydrolysis of the bound GTP a majority of the Rabl2 is replaced by Ift25/Ift27, and any remaining Rabl2GTP enters the cilium undergoing ciliary transportation while remaining associated with IFT-B machinery (Figure 3). Parallelly GPCRs are imported by Tulp3-IFT-A machinery (Zhou et al., 2022). Further they suggested that GPCRs are exported across the TZ by the BBSome which is connected to IFT-B particles via Lztfl1 binding it as well as the Ift25/Ift27 heterodimer (Figure 3) (Zhou et al., 2022).
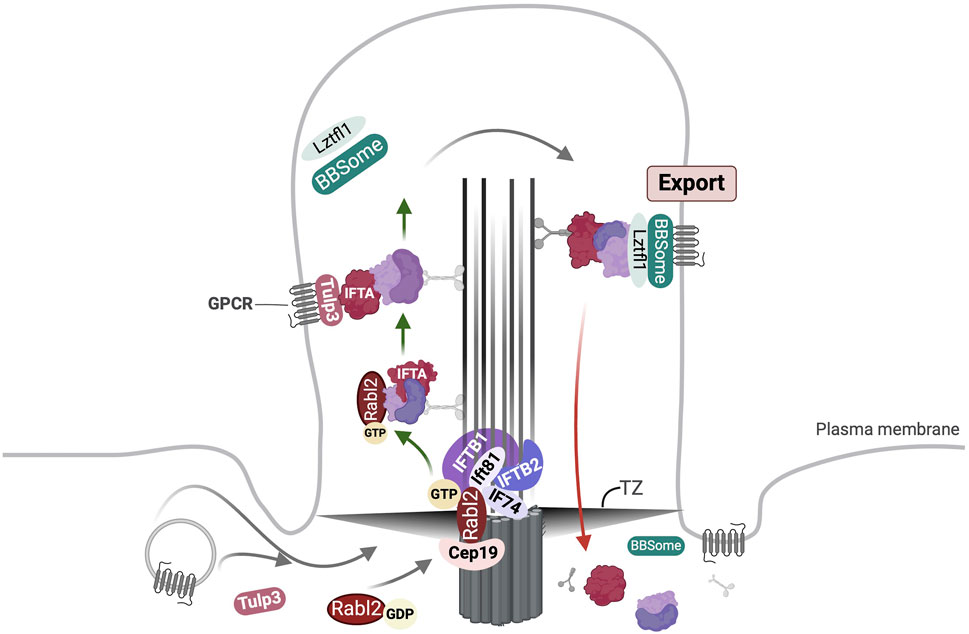
Figure 3. Overview of Rabl2 mediated ciliary transport. Rabl2GTP is engaged at the basal body via interaction with Cep19 and Ift74/Ift81 heterodimer. The Cep19-Rabl2-Ift74/Ift81 complex facilitates the ciliary uptake of IFT particles lacking Ift25/Ift27. Following this majority of Rabl2 undergoes hydrolysis of bound GTP resulting in its replacement by Ift25/Ift27. However, some Rabl2GTP may remain associated with the IFT-B trains and enter the cilium. Simultaneously, import of GPCRs is mediated by Tulp3 bound to IFT-A complex. Following anterograde transport the IFT trains are remodelled and motor proteins are exchanged at the ciliary tip (not shown). The export of GPCRs across the transition zone (TZ) is driven by BBSome bound to the IFT-B complex via Lztfl1 binding to Ift25/Ift27.
Rabl2 mutant Chlamydomonas lack flagella (Nishijima et al., 2017). Initially identified as an essential regulator of male fertility through a mutagenesis screen in mice, Rabl2 interacts with several IFT subunits, for example, Ift27, Ift81, and Ift172 for cargo delivery into the sperm tail (Lo et al., 2012). Consistent with this Rabl2 mutant mice (mot) carrying the missense mutation (D73G) showed compromised sperm motility and were infertile (Yi Lo et al., 2016). The Rabl2mot mice exhibited ciliopathy features such as adult-onset obesity and fatty livers owing to defective glucose and lipid metabolism. Similar features, such as infertility, obesity, retinal degeneration, and polydactyly are also exhibited by Rabl2 null mice despite overtly normal ciliation (Kanie et al., 2017; Duan et al., 2021).
Human patients with mutations in RABL2 are not known likely due to the functional redundancy between RABL2A and RABL2B (Kanie et al., 2017). However, a deletion in RABL2A has been identified as a risk factor for infertility among Australian men (Jamsai et al., 2014). Analysis of rare and potentially deleterious SNVs in RABL2 using mice has underscored its important role in cilia-mediated regulation of growth, left-right patterning, neural tube formation, limb development, and sperm motility (Ding et al., 2020). However, this study did not comment on the primary cilia architecture or frequency of ciliated cells in mutant Rabl2 tissues though the sperm in the latter resembled that of Rabl2mot mice.
4.2 Rabl4/Ift27
Initially, Ift27 was discovered as part of the IFT-B core in Chlamydomonas by biochemical studies (Lucker et al., 2005). Later it was identified as an ortholog of Rab-like 4 (Rabl4). It retains four characteristic RabF motifs, which together with phylogenetic analysis suggested that it belonged to the ancestral Rab family in the LECA (Diekmann et al., 2011). It forms a nucleotide independent heterodimer with Ift25 (Bhogaraju et al., 2011) that localizes at the basal body and undergoes IFT in various organisms (Qin et al., 2007; Wang et al., 2009; Bhogaraju et al., 2011; Huet et al., 2014). Ift27 binding to Ift25 is required for the latter’s entry to the cilium but not its stability (Eguether et al., 2014). The Ift25/Ift27 heterodimer interacts with the Ift74/Ift81 heterodimer which tethers it to the IFT-B1 core subcomplex (Bhogaraju et al., 2011; Taschner et al., 2014). GTP binding to Ift27 is not required for the assembly of the Ift25/Ift27 heterodimer but promotes the engagement of the latter to IFT-B1 and its ciliary cycling (Eguether et al., 2014; Huet et al., 2014; Liu et al., 2023).
The requirement of Ift27 for cilia biogenesis is context dependent. In Trypanosomes Ift27 silencing impedes cell growth and flagellum formation (Huet et al., 2014). In contrast, Ift27 is dispensable for cilia assembly in mice as its ablation results in neonatal lethality without structural ciliary anomalies except in the sperm flagella (Zhang et al., 2017). Nevertheless, Ift27−/− mice display pleiotropic structural defects in the skeleton, heart, lung, and brain that are reminiscent of defective Hh signaling (Eguether et al., 2014). Studies in mice cells suggested that Ift27 independent of the IFT-B complex associates and stabilizes a nucleotide-free form of Arl6/Bbs3, followed by GTP loading and Arl6 activation that triggers the assembly of the BBSome-Arl6 coat on membrane surfaces (Jin et al., 2010), mediating the export of the BBSome (Liew et al., 2014). Ift25/Ift27 heterodimer associates with the BBSome via Lztfl1, a BBSome modulator and is required for the export of the BBSome, Lztfl1 and its cargoes, such as Ptch1, Smo and Gpr161 out of the cilium (Eguether et al., 2014). In Chlamydomonas as in mammals IFT27 is not required for IFT (Sun et al., 2021). Here LZTFL1 stabilizes IFT25/IFT27 that is essential for BBSome recycling at the cilia tips (Sun et al., 2021), independent of IFT27 nucleotide state and facilitates BBSome-dependent phospholipase D ciliary export (Liu et al., 2023).
While hypomorphic variants in IFT27 cause Bardet-Biedl syndrome 19 (BBS19; OMIM #615996), a rare autosomal recessive condition characterized by ciliopathy characteristics, such as severe intellectual disability, polydactyly, renal failure, obesity, retinitis pigmentosa, and hypogonadism (Aldahmesh et al., 2014; Schaefer et al., 2019; Zhou et al., 2021); loss-of-function variants in IFT27 have been associated with a lethal fetal ciliopathy with short ribs, hypodysplastic kidneys, imperforate anus and situs invertus with severely impaired ciliogenesis in patient-derived kidney sections (Quélin et al., 2018; Haïm et al., 2025).
4.3 Rabl5/Ift22
A ciliary role was first anticipated for Rab-like 5 (Rabl5) when ifta-2, its ortholog in C. elegans was found to localize in ciliated sensory neurons (Schafer et al., 2006). While ifta-2 null worms did not exhibit typical ciliary defects they showed abnormal insulin-IGF-1-like signaling. Rabl5 is conserved across many ciliated organisms (Schafer et al., 2006; Silva et al., 2012). Studies showed that IFT22, a Rabl5 ortholog in C. reinhardtii associated with IFT-B complex, specifically with the IFT81/IFT74 heterodimer (Silva et al., 2012; Taschner et al., 2014). While in trypanosomes its recruitment to IFT-B1-1 subcomplex is essential for flallega formation; IFT and flagellation did not depend on its nucleotide state (Wachter et al., 2019). Work in C. reinhardtii suggests that active IFT22 interacts with GTP bound Arl6 to recruit the BBSome to the basal body (Xue et al., 2020). This study also revealed that ciliary uptake and cycling of the BBSome does not depend on IFT22. In contrast Chlamydomonas and Trypanosoma brucei (Adhiambo et al., 2009; Silva et al., 2012; Wachter et al., 2019) IFT22 disruption did not affect ciliogenesis or ciliary signaling in hTERT-RPE cells (Takei et al., 2018).
5 Exocyst in membrane trafficking
The exocyst is a conserved ∼ 750 kDa heterooctameric complex that belongs to the Complexes Associated with Tethering Containing Helical Rods (CATCHR) class of MTCs that modulate tethering and fusion of post-Golgi and recycling endocytic vesicles at the plasma membrane (Ungermann and Kümmel, 2019). Its holo-complex comprises of eight subunits: Sec3, Sec5, Sec6, Sec8, Sec10, Sec15, Exo70 and Exo84 in budding yeast that are termed EXOC1-8 in mammals (Hsu et al., 1996; TerBush et al., 1996). Each exocyst subunit contains the characteristic CATCHR domain and CorEx (core of exocyst) motif, an N-terminal coiled-coil followed by a rod composed of short antiparallel helical bundles (Mei et al., 2018). The mammalian exocyst is an octamer that is dynamically assembled from two subcomplexes (SC), SC1 (EXOC1-4) and SC2 (EXOC5-8) that are pre-assembled at the vesicles and likely arrive together at the plasma membrane (Ahmed et al., 2018; Maib and Murray, 2022).
Exocyst recruitment to secretory vesicles involves Sec15/Exoc6 binding Sec4p/Rab33b (Guo et al., 1999) and Sec2 its GEF (Medkova et al., 2006). Myo2, the yeast myosin V binds both Exoc6 and Rab33b (Jin et al., 2011). For vesicle delivery to polarized membranes, the conversion of PI4P to PI(4,5)P2 by Arf6 triggers exocyst recruitment and both SC1 and SC2 independently bind to the membrane PI(4,5)P2 (Maib and Murray, 2022). Studies in yeast showed that Sec3/Exoc1 (Zhang et al., 2008) and Exo70/Exoc7 (He et al., 2007) bind PI(4,5)P2 in the target membranes to engage the exocyst at the site of exocytosis. Recent studies also showed that the exocyst not only mediates SNARE assembly (Sivaram et al., 2005; Dubuke et al., 2015; Yue et al., 2017) but functions downstream driving complete membrane fusion and mixing of vesicle contents (Lee et al., 2024).
5.1 Exocyst and primary ciliogenesis
The exocyst is required to support numerous cellular functions, such as cell polarity (Polgar et al., 2015), migration (Zuo et al., 2006), autophagy (Bodemann et al., 2011), and cytokinesis (Gromley et al., 2005). It was first shown to localize to the primary cilium in MDCK cells (Rogers et al., 2004) and the ciliary stalk of photoreceptors in the frog retina (Mazelova et al., 2009). Using live imaging Rivera-Molina et. al., showed that in the presence of serum, the exocyst was enriched at the ciliary pocket of internal cilia; upon serum withdrawal, as the cilia are recycled to the cell surface, the exocyst was redistributed at its base (Rivera-Molina et al., 2021). In hTERT-RPE1 cells, SEPTIN9 a filamentous GTPase was shown to activate RhoA GTPase that in turn recruited the exocyst to the ciliary base and the depletion of either SEPTIN9 or the exocyst abolished the TZ and cilia assembly (Safavian et al., 2023).
While some exocyst subunits Exoc2, Exoc4 (Fogelgren et al., 2011; Seixas et al., 2016) and Exoc6 (Feng et al., 2012) localize to the axoneme, others such as Exoc3 (Yeaman et al., 2001), Exoc5 (Seixas et al., 2016), Exoc6b (Guleria et al., 2024), Exoc7 and Exoc8 (Seixas et al., 2016) occur at the basal body and periciliary regions (Figure 4). Some exocyst components also interact with ciliary proteins: Exoc5 binds Arl13b, Ift20, Ift88, and PC2 (Fogelgren et al., 2011); Exoc2 and Exoc3 directly interact with Arl13b and genetic interaction was reported between Arl13b and Exoc5 (Seixas et al., 2016). Ift20 interacts with Exoc4 and Exoc7 (Monis et al., 2017). The exocyst was also shown to cooperate with primary cilia in extracellular vesicle production in MDCK cells (Zuo et al., 2019a).
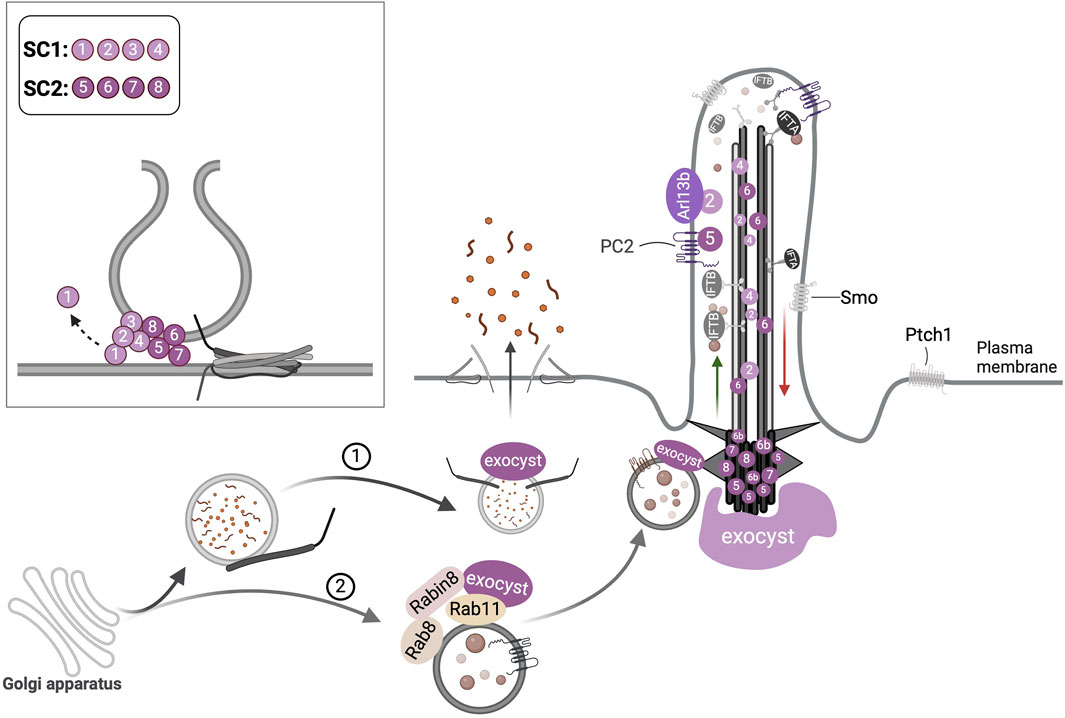
Figure 4. Ciliary transport functions of the exocyst complex. (1) Post-golgi vesicles in the secretory pathway are tethered at the plasma membrane by the exocyst that mediates exocytosis. (2) Vesicles with ciliary cargo are targeted to the cilium by the interaction of Exoc6, an exocyst subunit (not shown here) with Rab11 and Rabin8. The exocyst localizes at the primary cilium and periciliary regions; its subunits Exoc2, Exoc4, and Exoc6 have been detected at the axoneme, and others such as Exoc5, Exoc6b, Exoc7, and Exoc8 have been noted at the basal body. The direct binding of Arl13b to Exoc2 and PC2 to Exoc5 is illustrated. The inset depicts the formation of the heterooctameric exocyst complex at the target membranes by the dynamic assembly of its subcomplexes (SC), SC-1 and SC-2 which mediates tethering of the vesicles, SNARE assembly followed by membrane fusion.
Studies in vitro and animal models underscore the essentiality of the exocyst for ciliogenesis (Table 2). Many studies targeted Exoc5 as it is indispensable for exocyst assembly. In zebrafish, exoc5 morphants phenocopied pc2 depletion, and the manipulation of Exoc5 in MDCK cells led to aberrant ciliary assembly and mechanosensation defects reminiscent of Autosomal dominant polycystic kidney disease (ADPKD) (Fogelgren et al., 2011). An exoc5 loss of function mutant in zebrafish showed gross abnormalities of the brain, retina, and heart consistent with ciliopathies (Lobo et al., 2017). The complete ablation of exoc5 in zebrafish resulted in severe cardiac dysfunction and succumbed 4 days post fertilization; Exoc5 endocardial cell-specific silencing in mice showed dysmorphic aortic valves abrogating ciliogenesis and allied ciliary signaling (Fulmer et al., 2019). In MDCK cells the knockdown of Exoc5 or mutating its CTS suppressed ciliogenesis and its overexpression led to elevated ciliation (Zuo et al., 2019b). Other studies also recapitulated abnormal ciliation following Exoc5 knockdown in MDCK cells, though lumen formation was unaffected in 3-dimensional cultures; mice with kidney-specific knockdown of Exoc5 exhibited PCP defects with suppressed primary ciliation (Polgar et al., 2015). Seixas et al. showed that Exoc5 conditional inhibition in mouse kidneys caused high mortality and the surviving animals had a range of abnormalities including enlarged kidneys with cysts, significantly fewer and shorter cilia and overall reduced Arl13b protein levels (Seixas et al., 2016). The Exoc5 CTS was required for proper exocyst function in ciliation and its mutation led to defective cystogenesis and tubulogenesis in MDCK cells (Zuo et al., 2019b).
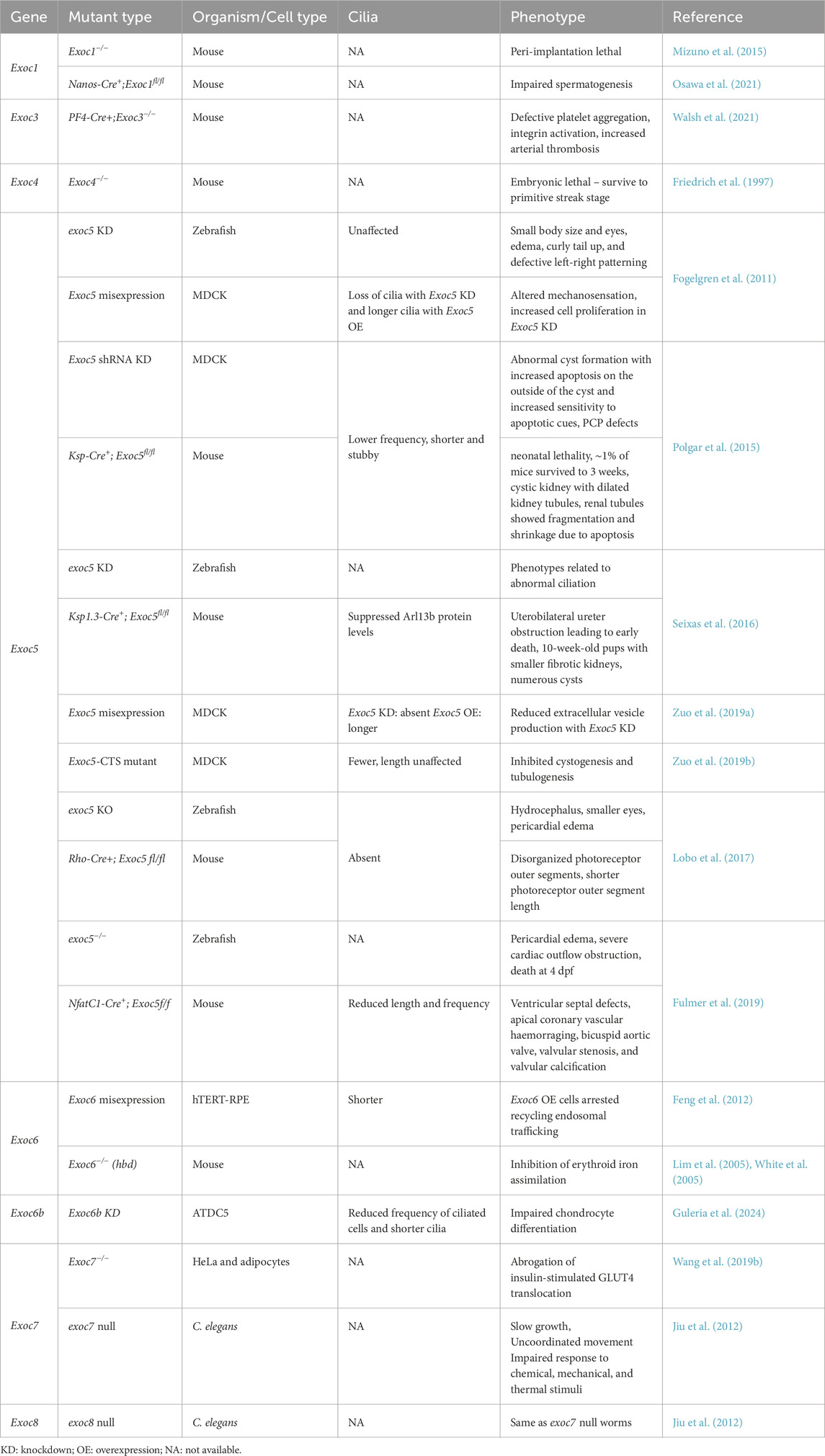
Table 2. Exocyst and cilia in organismal development.
Studies in MDCK cells, zebrafish, and mice also revealed that Cdc42, a small GTPase that interacts with the exocyst colocalizes with Exoc5 at the primary cilium and acts in concert with it for polarized secretion and primary ciliogenesis (Zhang et al., 2001; Zuo et al., 2011). Accordingly, exoc5 morphant zebrafish have small eyes (Fogelgren et al., 2011) and show loss of photoreceptor cilia, similar to that observed following cdc42 silencing (Choi et al., 2013). Cdc42 depletion in the mouse kidney led to polycystic kidney disease, aberrant cystogenesis with fewer and abnormal cilia surrounding the cysts (Choi et al., 2013). The knockdown of Tuba, a Cdc42-specific GEF suppressed primary ciliation in MDCK cells and tuba morphant zebrafish exhibited severe renal abnormalities and defective cilia (Baek et al., 2016). Exoc6 silencing in hTERT-RPE1 cells produced shorter cilia (Feng et al., 2012). Inhibition of Exoc6b, a paralog of Exoc6 in ATDC5 pre-chondrocytes strongly suppressed both the frequency of ciliated cells, cilia length, and deregulated chondrocyte differentiation (Guleria et al., 2024). In C. elegans exoc7 and exoc8 loss of function mutants showed behavioral defects akin to that observed with dysfunctional ciliogenesis albeit without any structural ciliary defects (Jiu et al., 2012).
5.2 Exocyst in ciliary trafficking
The delivery of fibrocystin and PC2 to the ciliary membrane depends on the exocyst (Monis et al., 2017). Similarly, rhodopsin targeting to outer photoreceptor segments requires exocyst function. Accordingly, retina-specific Exoc5 KO in mice led to the loss of photoreceptor outer segments and their associated cilia, as well as impaired vision (Lobo et al., 2017). Exoc6 directly binds Rabin8 in hTERT-RPE1 cells indicating the involvement of the exocyst in vesicle targeting during cilia assembly (Feng et al., 2012). In mammalian cells, the trafficking of soluble ciliary proteins, such as Gli2/3 and Lkb1 to the primary cilium was dependent on the exocyst and involved Rab14, Rab18, Rab23, and Arf4 (Niedziółka et al., 2024).
5.3 Exocyst and ciliopathies
Rare pathogenic mutations in several exocyst subunits are linked to monogenic diseases with abnormal cilia. EXOC2 biallelic mutations cause a neurodevelopmental disorder with dysmorphic facies and cerebellar hypoplasia (NEDFACH; OMIM #619306), wherein patient-derived fibroblasts EXOC2 expression was strongly suppressed leading to impaired vesicle fusion and exocytosis, and defective ARL13B localization in the cilium (Van Bergen et al., 2020). Homozygous null mutations in EXOC6B are linked to spondyloepimetaphyseal dysplasia with joint laxity type 3 (SEMD-JL3; OMIM #618395) that manifest largely with skeletal anomalies, such as hyperlaxity of joints and multiple joint dislocation, although some SEMD-JL3 patients do exhibit structural brain abnormalities and cognitive deficits (Girisha et al., 2016; Campos-Xavier et al., 2018; Simsek-Kiper et al., 2022). Our studies showed that EXOC6B mutant patient-derived fibroblast cells were defective in primary ciliogenesis (Simsek-Kiper et al., 2022). Similarly silencing Exoc6b in ATDC5 cells inhibited cilia assembly and led to abnormal chondrocyte differentiation (Guleria et al., 2024). Partial loss of function mutations in EXOC7 cause neurodevelopmental disorder with seizures and brain atrophy (NEDSEBA; OMIM #619072). Truncating mutations in EXOC8 are linked to neurodevelopmental disorder with microcephaly, seizures, and brain atrophy (NEDMISB; OMIM #619076). Exoc7 null mice are embryonic lethal (Coulter et al., 2020). The exoc7 loss of function zebrafish manifest ciliopathy features, such as edema and small eyes, as well as showed microcephaly owing to elevated apoptosis that led to a reduction in the number of neural progenitors in the telencephalon of mutant zebrafish (Coulter et al., 2020).
6 Future perspectives
Here we reviewed the involvement of Rabs, Rab-like factors, and the exocyst in ciliary trafficking and biogenesis. Based on the existing findings we note the below:
The Rab8-Rabin8-Rab11 cascade is implicated in early ciliogenesis. While Rab8 has been extensively studied we know comparatively little about the essentiality of Rab11 in this process. Engineering mutant Rab11 locked in GTP and GDP bound conformations by gene editing in vitro and animal models may help to shed light on this.
Activating mutations in LRRK2 linked to PD have been shown to involve several Rab substrates that function at least in part at the cilium, including Rab8, Rab10, and Rab12. More studies are required to uncover not only all the Rabs involved but also to elucidate their precise roles. Furthermore, it is unclear why LRRK2-dependent cilia loss is cell-type specific, affecting astrocyte and cholinergic interneuron cilia but not those in medium spiny neurons of the dorsal striatum (Khan et al., 2024; Lin et al., 2025). It would be interesting to examine this problem from the lens of the types of ciliary proteins differentially trafficked in cells sensitive to versus those that are not to LRRK2 over activating mutations.
For many years RAB23 mutations have been linked to CS which manifests with ciliopathy-like features. However, ciliation and trafficking abnormalities in them remain to be elucidated. The tissue-specific analysis of cilia in major organ systems affected in CS, for example, the CNS, skeleton, and heart may help to shed light on ciliary defects and pathologically relevant RAB23 cargo. In terms of pathomechanisms, future work could focus on the genotype-phenotype correlation of RAB28 mutations linked with CRD and PAP.
Secretomics in HeLa cells has shown that the exocyst is required for constitutive secretion (Pereira et al., 2023). It would be interesting for future studies to examine if exocyst-dependent ciliary trafficking is required in this process.
Conditional depletion of Exoc5 in podocytes, a unique cell type in the kidney glomerulus damages its structure leading to proteinuria and renal failure, without primary cilia involvement (Nihalani et al., 2019). In this study, the authors argue that podocyte-specific Ift88 silencing also does not lead to renal abnormalities. This is contrary to another study that reported a homozygous missense mutation in IFT139 that encodes for TTC21B, an IFT-A retrograde complex subunit in a patient with Focal segmental glomerulosclerosis wherein IFT139 silencing was suggested to result in abnormal primary cilia with aberrant aggregation of IFT-B components at the tips (Huynh Cong et al., 2014). Further studies will be needed to resolve the physiological relevance of the exocyst and cilia axis in podocytes.
The contribution of cilia is unclear in several exocyst-linked genetic diseases, for example, EXOC7 and EXOC8 linked NEDSEBA and NEDMISB, respectively. More detailed analysis is warranted to examine the molecular pathomechanisms in these disorders, including the disruption of primary ciliogenesis in them.
It is likely that not only the exocyst but other MTCs such as HOPs also interplay with distinct Rabs to modulate primary ciliogenesis. Future studies should examine these interactions to unveil their unique requirements in ciliary trafficking.
Finally, improving our understanding of the crosstalk and combinatorial action between different constituents of the membrane trafficking machinery will deepen our understanding of the sophisticated biological mechanisms underlying cilia biogenesis and function, as well as their clinical implications.
Author contributions
PU: Conceptualization, Formal Analysis, Funding acquisition, Methodology, Project administration, Resources, Software, Supervision, Visualization, Writing – original draft, Writing – review and editing. DB: Data curation, Writing – original draft, Investigation, Writing – review and editing. NQ: Data curation, Visualization, Writing – review and editing, Formal Analysis, Investigation.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Priyanka Upadhyai is principal investigator for the research grants entitled ‘Investigating EXOC6B function and the pathomechanisms underlying its associated skeletal dysplasia’ (no. IIRPSG-2024-01-00376) and ‘Investigating the crosstalk between primary cilia and autophagy in chondrogenesis and its modulation by Fibroblast growth factor (FGF) signaling in FGFR3 related skeletal dysplasias in vitro’ (2020-0107/CMB/ADHOC-BMS) funded by the Indian Council for Medical Research, Government of India.
Acknowledgments
All figures have been created with BioRender.com. We also acknowledge the work of many researchers which could not be cited here due to space constraints.
Conflict of interest
The authors declare that the present review article was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adhiambo, C., Blisnick, T., Toutirais, G., Delannoy, E., and Bastin, P. (2009). A novel function for the atypical small G protein Rab-like 5 in the assembly of the trypanosome flagellum. J. cell Sci. 122, 834–841. doi:10.1242/jcs.040444
Aguila, A., Salah, S., Kulasekaran, G., Shweiki, M., Shaul-Lotan, N., Mor-Shaked, H., et al. (2024). A neurodevelopmental disorder associated with a loss-of-function missense mutation in RAB35. J. Biol. Chem. 300, 107124. doi:10.1016/j.jbc.2024.107124
Ahmed, S. M., Nishida-Fukuda, H., Li, Y., Mcdonald, W. H., Gradinaru, C. C., and Macara, I. G. (2018). Exocyst dynamics during vesicle tethering and fusion. Nat. Commun. 9, 5140. doi:10.1038/s41467-018-07467-5
Akella, J. S., Carter, S. P., Nguyen, K., Tsiropoulou, S., Moran, A. L., Silva, M., et al. (2020). Ciliary Rab28 and the BBSome negatively regulate extracellular vesicle shedding. eLife 9, e50580. doi:10.7554/eLife.50580
Aldahmesh, M. A., Li, Y., Alhashem, A., Anazi, S., Alkuraya, H., Hashem, M., et al. (2014). IFT27, encoding a small GTPase component of IFT particles, is mutated in a consanguineous family with Bardet-Biedl syndrome. Hum. Mol. Genet. 23, 3307–3315. doi:10.1093/hmg/ddu044
Aljiboury, A. A., Ingram, E., Krishnan, N., Ononiwu, F., Pal, D., Manikas, J., et al. (2023). Rab8, Rab11, and Rab35 coordinate lumen and cilia formation during zebrafish left-right organizer development. PLoS Genet. 19, e1010765. doi:10.1371/journal.pgen.1010765
Amack, J. D. (2022). Structures and functions of cilia during vertebrate embryo development. Mol. Reprod. Dev. 89, 579–596. doi:10.1002/mrd.23650
Armstrong, J., Thompson, N., Squire, J. H., Smith, J., Hayes, B., and Solari, R. (1996). Identification of a novel member of the Rab8 family from the rat basophilic leukaemia cell line, RBL.2H3. J. Cell Sci. 109 (Pt 6), 1265–1274. doi:10.1242/jcs.109.6.1265
Babbey, C. M., Bacallao, R. L., and Dunn, K. W. (2010). Rab10 associates with primary cilia and the exocyst complex in renal epithelial cells. Am. J. Physiol. Ren. Physiol. 299, F495–F506. doi:10.1152/ajprenal.00198.2010
Bachmann-Gagescu, R., Phelps, I. G., Stearns, G., Link, B. A., Brockerhoff, S. E., Moens, C. B., et al. (2011). The ciliopathy gene cc2d2a controls zebrafish photoreceptor outer segment development through a role in Rab8-dependent vesicle trafficking. Hum. Mol. Genet. 20, 4041–4055. doi:10.1093/hmg/ddr332
Badgandi, H. B., Hwang, S. H., Shimada, I. S., Loriot, E., and Mukhopadhyay, S. (2017). Tubby family proteins are adapters for ciliary trafficking of integral membrane proteins. J. Cell Biol. 216, 743–760. doi:10.1083/jcb.201607095
Baek, J.-I., Kwon, S.-H., Zuo, X., Choi, S. Y., Kim, S.-H., and Lipschutz, J. H. (2016). Dynamin binding protein (tuba) deficiency inhibits ciliogenesis and nephrogenesis in vitro and in vivo. J. Biol. Chem. 291, 8632–8643. doi:10.1074/jbc.M115.688663
Batkovskyte, D., Komatsu, M., Hammarsjö, A., Pooh, R., Shimokawa, O., Ikegawa, S., et al. (2024). Compound heterozygous variants in RAB34 in a rare skeletal ciliopathy syndrome. Clin. Genet. 105, 87–91. doi:10.1111/cge.14419
Bhogaraju, S., Taschner, M., Morawetz, M., Basquin, C., and Lorentzen, E. (2011). Crystal structure of the intraflagellar transport complex 25/27. EMBO J. 30, 1907–1918. doi:10.1038/emboj.2011.110
Bielas, S. L., Silhavy, J. L., Brancati, F., Kisseleva, M. V., Al-Gazali, L., Sztriha, L., et al. (2009). Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat. Genet. 41, 1032–1036. doi:10.1038/ng.423
Bodemann, B. O., Orvedahl, A., Cheng, T., Ram, R. R., Ou, Y.-H., Formstecher, E., et al. (2011). RalB and the exocyst mediate the cellular starvation response by direct activation of autophagosome assembly. Cell 144, 253–267. doi:10.1016/j.cell.2010.12.018
Boegholm, N., Petriman, N. A., Loureiro-López, M., Wang, J., Vela, M. I. S., Liu, B., et al. (2023). The IFT81-IFT74 complex acts as an unconventional RabL2 GTPase-activating protein during intraflagellar transport. EMBO J. 42, e111807. doi:10.15252/embj.2022111807
Boehlke, C., Bashkurov, M., Buescher, A., Krick, T., John, A.-K., Nitschke, R., et al. (2010). Differential role of Rab proteins in ciliary trafficking: rab23 regulates smoothened levels. J. Cell Sci. 123, 1460–1467. doi:10.1242/jcs.058883
Brauers, A., Schürmann, A., Massmann, S., Mühl-Zürbes, P., Becker, W., Kainulainen, H., et al. (1996). Alternative mRNA splicing of the novel GTPase Rab28 generates isoforms with different C-termini. Eur. J. Biochem. 237, 833–840. doi:10.1111/j.1432-1033.1996.0833p.x
Bruel, A.-L., Ganga, A. K., Nosková, L., Valenzuela, I., Martinovic, J., Duffourd, Y., et al. (2023). Pathogenic RAB34 variants impair primary cilium assembly and cause a novel oral-facial-digital syndrome. Hum. Mol. Genet. 32, 2822–2831. doi:10.1093/hmg/ddad109
Bucci, C., Parton, R. G., Mather, I. H., Stunnenberg, H., Simons, K., Hoflack, B., et al. (1992). The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway. Cell 70, 715–728. doi:10.1016/0092-8674(92)90306-w
Burke, M. C., Li, F. Q., Cyge, B., Arashiro, T., Brechbuhl, H. M., Chen, X., et al. (2014). Chibby promotes ciliary vesicle formation and basal body docking during airway cell differentiation. J. Cell Biol. 207, 123–137. doi:10.1083/jcb.201406140
Campos-Xavier, B., Rogers, R. C., Niel-Bütschi, F., Ferreira, C., Unger, S., Spranger, J., et al. (2018). Confirmation of spondylo-epi-metaphyseal dysplasia with joint laxity, EXOC6B type. Am. J. Med. Genet. A 176, 2934–2935. doi:10.1002/ajmg.a.40631
Carter, S. P., Moran, A. L., Matallanas, D., Mcmanus, G. J., Blacque, O. E., and Kennedy, B. N. (2020). Genetic deletion of zebrafish Rab28 causes defective outer segment shedding, but not retinal degeneration. Front. Cell Dev. Biol. 8, 136. doi:10.3389/fcell.2020.00136
Carvalho-Santos, Z., Azimzadeh, J., Pereira-Leal, J. B., and Bettencourt-Dias, M. (2011). Evolution: tracing the origins of centrioles, cilia, and flagella. J. Cell Biol. 194, 165–175. doi:10.1083/jcb.201011152
Cavalier-Smith, T. (2022). Ciliary transition zone evolution and the root of the eukaryote tree: implications for opisthokont origin and classification of kingdoms Protozoa, Plantae, and Fungi. Protoplasma 259, 487–593. doi:10.1007/s00709-021-01665-7
Chávez, M., Ena, S., Van Sande, J., De Kerchove D'exaerde, A., Schurmans, S., and Schiffmann, S. N. (2015). Modulation of ciliary phosphoinositide content regulates trafficking and sonic hedgehog signaling output. Dev. Cell 34, 338–350. doi:10.1016/j.devcel.2015.06.016
Chiang, C., Litingtung, Y., Lee, E., Young, K. E., Corden, J. L., Westphal, H., et al. (1996). Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 383, 407–413. doi:10.1038/383407a0
Chih, B., Liu, P., Chinn, Y., Chalouni, C., Komuves, L. G., Hass, P. E., et al. (2011). A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat. Cell Biol. 14, 61–72. doi:10.1038/ncb2410
Choi, S. Y., Chacon-Heszele, M. F., Huang, L., Mckenna, S., Wilson, F. P., Zuo, X., et al. (2013). Cdc42 deficiency causes ciliary abnormalities and cystic kidneys. J. Am. Soc. Nephrol. 24, 1435–1450. doi:10.1681/ASN.2012121236
Chong, W. M., Wang, W.-J., Lo, C.-H., Chiu, T.-Y., Chang, T.-J., Liu, Y.-P., et al. (2020). Super-resolution microscopy reveals coupling between mammalian centriole subdistal appendages and distal appendages. Elife 9, e53580. doi:10.7554/eLife.53580
Chen, Y., Yue, S., Xie, L., Pu, X.-H., Jin, T., Cheng, S. Y., et al. (2011). Dual phosphorylation of suppressor of fused (Sufu) by PKA and GSK3β regulates its stability and localization in the primary cilium. J. Biolog. Chem. 286 (15), 13502–13511.
Chua, C. E. L., and Tang, B. L. (2018). Rab 10—a traffic controller in multiple cellular pathways and locations. J. Cell. Physiology 233, 6483–6494. doi:10.1002/jcp.26503
Clearman, K. R., Timpratoom, N., Patel, D., Rains, A. B., Haycraft, C. J., Croyle, M. J., et al. (2024). Rab35 is required for embryonic development and kidney and ureter homeostasis through regulation of epithelial cell junctions. J. Am. Soc. Nephrol. 35, 719–732. doi:10.1681/ASN.0000000000000335
Conduit, S. E., Pearce, W., Bhamra, A., Bilanges, B., Bozal-Basterra, L., Foukas, L. C., et al. (2024). A class I PI3K signalling network regulates primary cilia disassembly in normal physiology and disease. Nat. Commun. 15, 7181. doi:10.1038/s41467-024-51354-1
Conduit, S. E., and Vanhaesebroeck, B. (2020). Phosphoinositide lipids in primary cilia biology. Biochem. J. 477, 3541–3565. doi:10.1042/BCJ20200277
Coon, B. G., Hernandez, V., Madhivanan, K., Mukherjee, D., Hanna, C. B., Barinaga-Rementeria Ramirez, I., et al. (2012). The Lowe syndrome protein OCRL1 is involved in primary cilia assembly. Hum. Mol. Genet. 21, 1835–1847. doi:10.1093/hmg/ddr615
Coulter, M. E., Musaev, D., Degennaro, E. M., Zhang, X., Henke, K., James, K. N., et al. (2020). Regulation of human cerebral cortical development by EXOC7 and EXOC8, components of the exocyst complex, and roles in neural progenitor cell proliferation and survival. Genet. Med. 22, 1040–1050. doi:10.1038/s41436-020-0758-9
Craft, J. M., Harris, J. A., Hyman, S., Kner, P., and Lechtreck, K. F. (2015). Tubulin transport by IFT is upregulated during ciliary growth by a cilium-autonomous mechanism. J. Cell Biol. 208, 223–237. doi:10.1083/jcb.201409036
Cuenca, A., Insinna, C., Zhao, H., John, P., Weiss, M. A., Lu, Q., et al. (2019). The C7orf43/TRAPPC14 component links the TRAPPII complex to Rabin8 for preciliary vesicle tethering at the mother centriole during ciliogenesis. J. Biol. Chem. 294, 15418–15434. doi:10.1074/jbc.RA119.008615
Dateyama, I., Sugihara, Y., Chiba, S., Ota, R., Nakagawa, R., Kobayashi, T., et al. (2019). RABL2 positively controls localization of GPCRs in mammalian primary cilia. J. Cell Sci. 132, jcs224428. doi:10.1242/jcs.224428
Deretic, D., Huber, L. A., Ransom, N., Mancini, M., Simons, K., and Papermaster, D. S. (1995). Rab8 in retinal photoreceptors may participate in rhodopsin transport and in rod outer segment disk morphogenesis. J. cell Sci. 108, 215–224. doi:10.1242/jcs.108.1.215
Dhekne, H. S., Tonelli, F., Yeshaw, W. M., Chiang, C. Y., Limouse, C., Jaimon, E., et al. (2023). Genome-wide screen reveals Rab12 GTPase as a critical activator of Parkinson’s disease-linked LRRK2 kinase. eLife 12, e87098. doi:10.7554/eLife.87098
Dhekne, H. S., Yanatori, I., Gomez, R. C., Tonelli, F., Diez, F., Schüle, B., et al. (2018). A pathway for Parkinson’s Disease LRRK2 kinase to block primary cilia and Sonic hedgehog signaling in the brain. eLife 7, e40202. doi:10.7554/eLife.40202
Dickinson, M. E., Flenniken, A. M., Ji, X., Teboul, L., Wong, M. D., White, J. K., et al. (2016). High-throughput discovery of novel developmental phenotypes. Nature 537, 508–514. doi:10.1038/nature19356
Diekmann, Y., Seixas, E., Gouw, M., Tavares-Cadete, F., Seabra, M. C., and Pereira-Leal, J. B. (2011). Thousands of Rab GTPases for the cell biologist. PLoS Comput. Biol. 7, e1002217. doi:10.1371/journal.pcbi.1002217
Ding, X., Fragoza, R., Singh, P., Zhang, S., Yu, H., and Schimenti, J. C. (2020). Variants in RABL2A causing male infertility and ciliopathy. Hum. Mol. Genet. 29, 3402–3411. doi:10.1093/hmg/ddaa230
Dishinger, J. F., Kee, H. L., Jenkins, P. M., Fan, S., Hurd, T. W., Hammond, J. W., et al. (2010). Ciliary entry of the kinesin-2 motor KIF17 is regulated by importin-beta2 and RanGTP. Nat. Cell Biol. 12, 703–710. doi:10.1038/ncb2073
Duan, S., Li, H., Zhang, Y., Yang, S., Chen, Y., Qiu, B., et al. (2021). Rabl2 GTP hydrolysis licenses BBSome-mediated export to fine-tune ciliary signaling. EMBO J. 40, e105499. doi:10.15252/embj.2020105499
Dubuke, M. L., Maniatis, S., Shaffer, S. A., and Munson, M. (2015). The exocyst subunit Sec6 interacts with assembled exocytic SNARE complexes. J. Biol. Chem. 290, 28245–28256. doi:10.1074/jbc.M115.673806
Eggenschwiler, J. T., Bulgakov, O. V., Qin, J., Li, T., and Anderson, K. V. (2006). Mouse Rab23 regulates hedgehog signaling from smoothened to Gli proteins. Dev. Biol. 290, 1–12. doi:10.1016/j.ydbio.2005.09.022
Eggenschwiler, J. T., Espinoza, E., and Anderson, K. V. (2001). Rab23 is an essential negative regulator of the mouse Sonic hedgehog signalling pathway. Nature 412, 194–198. doi:10.1038/35084089
Eguether, T., San Agustin, J. T., Keady, B. T., Jonassen, J. A., Liang, Y., Francis, R., et al. (2014). IFT27 links the BBSome to IFT for maintenance of the ciliary signaling compartment. Dev. Cell 31, 279–290. doi:10.1016/j.devcel.2014.09.011
Evans, T. M., Ferguson, C., Wainwright, B. J., Parton, R. G., and Wicking, C. (2003). Rab23, a negative regulator of hedgehog signaling, localizes to the plasma membrane and the endocytic pathway. Traffic 4, 869–884. doi:10.1046/j.1600-0854.2003.00141.x
Fan, S., Fogg, V., Wang, Q., Chen, X. W., Liu, C. J., and Margolis, B. (2007). A novel Crumbs3 isoform regulates cell division and ciliogenesis via importin beta interactions. J. Cell Biol. 178, 387–398. doi:10.1083/jcb.200609096
Feng, S., Knödler, A., Ren, J., Zhang, J., Zhang, X., Hong, Y., et al. (2012). A Rab8 guanine nucleotide exchange factor-effector interaction network regulates primary ciliogenesis. J. Biol. Chem. 287, 15602–15609. doi:10.1074/jbc.M111.333245
Fogelgren, B., Lin, S. Y., Zuo, X., Jaffe, K. M., Park, K. M., Reichert, R. J., et al. (2011). The exocyst protein Sec10 interacts with Polycystin-2 and knockdown causes PKD-phenotypes. PLoS Genet. 7, e1001361. doi:10.1371/journal.pgen.1001361
Follit, J. A., Li, L., Vucica, Y., and Pazour, G. J. (2010). The cytoplasmic tail of fibrocystin contains a ciliary targeting sequence. J. Cell Biol. 188, 21–28. doi:10.1083/jcb.200910096
Friedrich, G. A., Hildebrand, J. D., and Soriano, P. (1997). The secretory protein Sec8 is required for paraxial mesoderm formation in the mouse. Dev. Biol. 192, 364–374. doi:10.1006/dbio.1997.8727
Fulmer, D., Toomer, K., Guo, L., Moore, K., Glover, J., Moore, R., et al. (2019). Defects in the exocyst-cilia machinery cause bicuspid aortic valve disease and aortic stenosis. Circulation 140, 1331–1341. doi:10.1161/CIRCULATIONAHA.119.038376
Ganga, A. K., Kennedy, M. C., Oguchi, M. E., Gray, S., Oliver, K. E., Knight, T. A., et al. (2021). Rab34 GTPase mediates ciliary membrane formation in the intracellular ciliogenesis pathway. Curr. Biol. 31, 2895–2905.e7. doi:10.1016/j.cub.2021.04.075
Garcia-Gonzalo, F. R., Phua, S. C., Roberson, E. C., Garcia, G., Abedin, M., Schurmans, S., et al. (2015). Phosphoinositides regulate ciliary protein trafficking to modulate hedgehog signaling. Dev. Cell 34, 400–409. doi:10.1016/j.devcel.2015.08.001
Gerondopoulos, A., Strutt, H., Stevenson, N. L., Sobajima, T., Levine, T. P., Stephens, D. J., et al. (2019). Planar cell polarity effector proteins inturned and Fuzzy form a Rab23 GEF complex. Curr. Biol. 29, 3323–3330.e8. doi:10.1016/j.cub.2019.07.090
Ghossoub, R., Hu, Q., Failler, M., Rouyez, M. C., Spitzbarth, B., Mostowy, S., et al. (2013). Septins 2, 7 and 9 and MAP4 colocalize along the axoneme in the primary cilium and control ciliary length. J. Cell Sci. 126, 2583–2594. doi:10.1242/jcs.111377
Girisha, K. M., Kortüm, F., Shah, H., Alawi, M., Dalal, A., Bhavani, G. S., et al. (2016). A novel multiple joint dislocation syndrome associated with a homozygous nonsense variant in the EXOC6B gene. Eur. J. Hum. Genet. 24, 1206–1210. doi:10.1038/ejhg.2015.261
Göhring, I., Tagariello, A., Endele, S., Stolt, C. C., Ghassibé, M., Fisher, M., et al. (2010). Disruption of ST5 is associated with mental retardation and multiple congenital anomalies. J. Med. Genet. 47, 91–98. doi:10.1136/jmg.2009.069799
Gorvel, J.-P., Chavrier, P., Zerial, M., and Gruenberg, J. (1991). rab5 controls early endosome fusion in vitro. Cell 64, 915–925. doi:10.1016/0092-8674(91)90316-q
Gromley, A., Yeaman, C., Rosa, J., Redick, S., Chen, C.-T., Mirabelle, S., et al. (2005). Centriolin anchoring of exocyst and SNARE complexes at the midbody is required for secretory-vesicle-mediated abscission. Cell 123, 75–87. doi:10.1016/j.cell.2005.07.027
Guleria, V. S., Quadri, N., Prasad, K., Das, R., and Upadhyai, P. (2024). Early insights into the role of Exoc6B associated with spondyloepimetaphyseal dysplasia with joint laxity type 3 in primary ciliogenesis and chondrogenic differentiation in vitro. Mol. Biol. Rep. 51, 274. doi:10.1007/s11033-023-09114-9
Guo, W., Roth, D., Walch-Solimena, C., and Novick, P. (1999). The exocyst is an effector for Sec4p, targeting secretory vesicles to sites of exocytosis. Embo J. 18, 1071–1080. doi:10.1093/emboj/18.4.1071
Haïm, D., Roux, N., Boutaud, L., Verlin, L., Quélin, C., Moncler, C., et al. (2025). Complete loss of IFT27 function leads to a phenotypic spectrum of fetal lethal ciliopathy associated with altered ciliogenesis. Eur. J. Hum. Genet. 33, 387–392. doi:10.1038/s41431-025-01810-3
Haycraft, C. J., Banizs, B., Aydin-Son, Y., Zhang, Q., Michaud, E. J., Yoder, B. K., et al. (2005). Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS. genetics. 1 (4), e53.
Huynh Cong, E., Bizet, A. A., Bizet, A. A., Woerner, S., Gribouval, O., Filhol, E., et al. (2014). A homozygous missense mutation in the ciliary gene TTC21B causes familial FSGS. J. Am. Soc. Nephrol. 25 (11), 2435–2443.
Hao, K., Chen, Y., Yan, X., and Zhu, X. (2021). Cilia locally synthesize proteins to sustain their ultrastructure and functions. Nat. Commun. 12, 6971. doi:10.1038/s41467-021-27298-1
He, B., Xi, F., Zhang, X., Zhang, J., and Guo, W. (2007). Exo70 interacts with phospholipids and mediates the targeting of the exocyst to the plasma membrane. Embo J. 26, 4053–4065. doi:10.1038/sj.emboj.7601834
Hilgendorf, K. I., Myers, B. R., and Reiter, J. F. (2024). Emerging mechanistic understanding of cilia function in cellular signalling. Nat. Rev. Mol. Cell Biol. 25, 555–573. doi:10.1038/s41580-023-00698-5
Hoffman, H. K., and Prekeris, R. (2023). HOPS-dependent lysosomal fusion controls Rab19 availability for ciliogenesis in polarized epithelial cells. J. Cell Sci. 137, jcs261047. doi:10.1242/jcs.261047
Hoffmeister, H., Babinger, K., Gürster, S., Cedzich, A., Meese, C., Schadendorf, K., et al. (2011). Polycystin-2 takes different routes to the somatic and ciliary plasma membrane. J. Cell Biol. 192, 631–645. doi:10.1083/jcb.201007050
Homma, Y., Hiragi, S., and Fukuda, M. (2021). Rab family of small GTPases: an updated view on their regulation and functions. Febs J. 288, 36–55. doi:10.1111/febs.15453
Hou, X., Hagemann, N., Schoebel, S., Blankenfeldt, W., Goody, R. S., Erdmann, K. S., et al. (2011). A structural basis for Lowe syndrome caused by mutations in the Rab-binding domain of OCRL1. Embo J. 30, 1659–1670. doi:10.1038/emboj.2011.60
Hsiao, Y. C., Tong, Z. J., Westfall, J. E., Ault, J. G., Page-Mccaw, P. S., and Ferland, R. J. (2009). Ahi1, whose human ortholog is mutated in Joubert syndrome, is required for Rab8a localization, ciliogenesis and vesicle trafficking. Hum. Mol. Genet. 18, 3926–3941. doi:10.1093/hmg/ddp335
Hsiao, Y. C., Tuz, K., and Ferland, R. J. (2012). Trafficking in and to the primary cilium. Cilia 1, 4. doi:10.1186/2046-2530-1-4
Hsu, S.-C., Ting, A. E., Hazuka, C. D., Davanger, S., Kenny, J. W., Kee, Y., et al. (1996). The mammalian brain rsec6/8 complex. Neuron 17, 1209–1219. doi:10.1016/s0896-6273(00)80251-2
Hu, Q., Milenkovic, L., Jin, H., Scott, M. P., Nachury, M. V., Spiliotis, E. T., et al. (2010). A Septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science 329, 436–439. doi:10.1126/science.1191054
Huet, D., Blisnick, T., Perrot, S., and Bastin, P. (2014). The GTPase IFT27 is involved in both anterograde and retrograde intraflagellar transport. Elife 3, e02419. doi:10.7554/eLife.02419
Hurd, T. W., Fan, S., and Margolis, B. L. (2011). Localization of retinitis pigmentosa 2 to cilia is regulated by Importin beta2. J. Cell Sci. 124, 718–726. doi:10.1242/jcs.070839
Ishikawa, H., Thompson, J., Yates, J. R., and Marshall, W. F. (2012). Proteomic analysis of mammalian primary cilia. Curr. Biol. 22, 414–419. doi:10.1016/j.cub.2012.01.031
Ito, G., Katsemonova, K., Tonelli, F., Lis, P., Baptista, M. A., Shpiro, N., et al. (2016). Phos-tag analysis of Rab10 phosphorylation by LRRK2: a powerful assay for assessing kinase function and inhibitors. Biochem. J. 473, 2671–2685. doi:10.1042/BCJ20160557
Jacoby, M., Cox, J. J., Gayral, S., Hampshire, D. J., Ayub, M., Blockmans, M., et al. (2009). INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat. Genet. 41, 1027–1031. doi:10.1038/ng.427
Jamsai, D., Lo, J. C. Y., Mclachlan, R. I., and O'bryan, M. K. (2014). Genetic variants in the RABL2A gene in fertile and oligoasthenospermic infertile men. Fertil. Steril. 102, 223–229. doi:10.1016/j.fertnstert.2014.04.007
Jenkins, P. M., Hurd, T. W., Zhang, L., Mcewen, D. P., Brown, R. L., Margolis, B., et al. (2006). Ciliary targeting of olfactory CNG channels requires the CNGB1b subunit and the kinesin-2 motor protein, KIF17. Curr. Biol. 16, 1211–1216. doi:10.1016/j.cub.2006.04.034
Jensen, V. L., Carter, S., Sanders, A. a.W. M., Li, C., Kennedy, J., Timbers, T. A., et al. (2016). Whole-organism developmental expression profiling identifies RAB-28 as a novel ciliary GTPase associated with the BBSome and intraflagellar transport. PLoS Genet. 12, e1006469. doi:10.1371/journal.pgen.1006469
Jensen, V. L., and Leroux, M. R. (2017). Gates for soluble and membrane proteins, and two trafficking systems (IFT and LIFT), establish a dynamic ciliary signaling compartment. Curr. Opin. Cell Biol. 47, 83–91. doi:10.1016/j.ceb.2017.03.012
Jensen, V. L., Li, C., Bowie, R. V., Clarke, L., Mohan, S., Blacque, O. E., et al. (2015). Formation of the transition zone by Mks5/Rpgrip1L establishes a ciliary zone of exclusion (CIZE) that compartmentalises ciliary signalling proteins and controls PIP 2 ciliary abundance. EMBO J. 34, 2537–2556. doi:10.15252/embj.201488044
Jespersgaard, C., Hey, A. B., Ilginis, T., Hjortshøj, T. D., Fang, M., Bertelsen, M., et al. (2020). A missense mutation in RAB28 in a family with cone-rod dystrophy and postaxial polydactyly prevents localization of RAB28 to the primary cilium. Investigative Ophthalmol. and Vis. Sci. 61, 29. doi:10.1167/iovs.61.2.29
Jewett, C. E., Soh, A. W. J., Lin, C. H., Lu, Q., Lencer, E., Westlake, C. J., et al. (2021). RAB19 directs cortical remodeling and membrane growth for primary ciliogenesis. Dev. Cell 56, 325–340.e8. doi:10.1016/j.devcel.2020.12.003
Jin, H., White, S. R., Shida, T., Schulz, S., Aguiar, M., Gygi, S. P., et al. (2010). The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell 141, 1208–1219. doi:10.1016/j.cell.2010.05.015
Jin, Y., Sultana, A., Gandhi, P., Franklin, E., Hamamoto, S., Khan, A. R., et al. (2011). Myosin V transports secretory vesicles via a Rab GTPase cascade and interaction with the exocyst complex. Dev. Cell 21, 1156–1170. doi:10.1016/j.devcel.2011.10.009
Jiu, Y., Jin, C., Liu, Y., Holmberg, C. I., and Jäntti, J. (2012). Exocyst subunits Exo70 and Exo84 cooperate with small GTPases to regulate behavior and endocytic trafficking in C. elegans. PLOS ONE 7, e32077. doi:10.1371/journal.pone.0032077
Kadakia, S., Helman, S. N., Healy, N. J., Saman, M., and Wood-Smith, D. (2014). Carpenter syndrome: a review for the craniofacial surgeon. J. Craniofacial Surg. 25, 1653–1657. doi:10.1097/SCS.0000000000001121
Kanemaru, K., Shimozawa, M., Kitamata, M., Furuishi, R., Kayano, H., Sukawa, Y., et al. (2022). Plasma membrane phosphatidylinositol (4,5)-bisphosphate is critical for determination of epithelial characteristics. Nat. Commun. 13, 2347. doi:10.1038/s41467-022-30061-9
Kanie, T., Abbott, K. L., Mooney, N. A., Plowey, E. D., Demeter, J., and Jackson, P. K. (2017). The CEP19-RABL2 GTPase complex binds IFT-B to initiate intraflagellar transport at the ciliary base. Dev. Cell 42, 22–36.e12. doi:10.1016/j.devcel.2017.05.016
Kaplan, O. I., Molla-Herman, A., Cevik, S., Ghossoub, R., Kida, K., Kimura, Y., et al. (2010). The AP-1 clathrin adaptor facilitates cilium formation and functions with RAB-8 in C. elegans ciliary membrane transport. J. Cell Sci. 123, 3966–3977. doi:10.1242/jcs.073908
Kee, H. L., Dishinger, J. F., Blasius, T. L., Liu, C. J., Margolis, B., and Verhey, K. J. (2012). A size-exclusion permeability barrier and nucleoporins characterize a ciliary pore complex that regulates transport into cilia. Nat. Cell Biol. 14, 431–437. doi:10.1038/ncb2450
Khan, S. S., Jaimon, E., Lin, Y.-E., Nikoloff, J., Tonelli, F., Alessi, D. R., et al. (2024). Loss of primary cilia and dopaminergic neuroprotection in pathogenic LRRK2-driven and idiopathic Parkinson’s disease. Proc. Natl. Acad. Sci. 121, e2402206121. doi:10.1073/pnas.2402206121
Khan, S. S., Sobu, Y., Dhekne, H. S., Tonelli, F., Berndsen, K., Alessi, D. R., et al. (2021). Pathogenic LRRK2 control of primary cilia and Hedgehog signaling in neurons and astrocytes of mouse brain. Elife 10, e67900. doi:10.7554/eLife.67900
Klinkert, K., Rocancourt, M., Houdusse, A., and Echard, A. (2016). Rab35 GTPase couples cell division with initiation of epithelial apico-basal polarity and lumen opening. Nat. Commun. 7, 11166. doi:10.1038/ncomms11166
Knödler, A., Feng, S., Zhang, J., Zhang, X., Das, A., Peränen, J., et al. (2010). Coordination of Rab8 and Rab11 in primary ciliogenesis. Proc. Natl. Acad. Sci. 107, 6346–6351.
Kobayashi, T., Kim, S., Lin, Y. C., Inoue, T., and Dynlacht, B. D. (2014). The CP110-interacting proteins Talpid3 and Cep290 play overlapping and distinct roles in cilia assembly. J. Cell Biol. 204, 215–229. doi:10.1083/jcb.201304153
Kouranti, I., Sachse, M., Arouche, N., Goud, B., and Echard, A. (2006). Rab35 regulates an endocytic recycling pathway essential for the terminal steps of cytokinesis. Curr. Biol. 16, 1719–1725. doi:10.1016/j.cub.2006.07.020
Kuhns, S., Seixas, C., Pestana, S., Tavares, B., Nogueira, R., Jacinto, R., et al. (2019). Rab35 controls cilium length, function and membrane composition. EMBO Rep. 20, e47625. doi:10.15252/embr.201847625
Kumar, R., Francis, V., Kulasekaran, G., Khan, M., Armstrong, G. a.B., and Mcpherson, P. S. (2022). A cell-based GEF assay reveals new substrates for DENN domains and a role for DENND2B in primary ciliogenesis. Sci. Adv. 8, eabk3088. doi:10.1126/sciadv.abk3088
Lacey, S. E., and Pigino, G. (2024). The intraflagellar transport cycle. Nat. Rev. Mol. Cell Biol. 26, 175–192. doi:10.1038/s41580-024-00797-x
Leaf, A., and Von Zastrow, M. (2015). Dopamine receptors reveal an essential role of IFT-B, KIF17, and Rab23 in delivering specific receptors to primary cilia. Elife 4, e06996. doi:10.7554/eLife.06996
Lechtreck, K. F., Liu, Y., Dai, J., Alkhofash, R. A., Butler, J., Alford, L., et al. (2022). Chlamydomonas ARMC2/PF27 is an obligate cargo adapter for intraflagellar transport of radial spokes. Elife 11, e74993. doi:10.7554/eLife.74993
Lee, C., Lepore, D., Lee, S.-H., Kim, T. G., Buwa, N., Lee, J., et al. (2024). Exocyst stimulates multiple steps of exocytic SNARE complex assembly and vesicle fusion. Nat. Struct. Mol. Biol. 32, 150–160. doi:10.1038/s41594-024-01388-2
Leung, M. R., Sun, C., Zeng, J., Anderson, J. R., Niu, Q., Huang, W., et al. (2025). Structural diversity of axonemes across mammalian motile cilia. Nature 637, 1170–1177. doi:10.1038/s41586-024-08337-5
Li, F., Yi, L., Zhao, L., Itzen, A., Goody, R. S., and Wu, Y. W. (2014). The role of the hypervariable C-terminal domain in Rab GTPases membrane targeting. Proc. Natl. Acad. Sci. U. S. A. 111, 2572–2577. doi:10.1073/pnas.1313655111
Li, X., Zhu, H., Huang, B. T., Li, X., Kim, H., Tan, H., et al. (2024). RAB12-LRRK2 complex suppresses primary ciliogenesis and regulates centrosome homeostasis in astrocytes. Nat. Commun. 15, 8434. doi:10.1038/s41467-024-52723-6
Liew, G. m., Ye, F., Nager, A. r., Murphy, J. P., Lee, J. s., Aguiar, M., et al. (2014). The intraflagellar transport protein IFT27 promotes BBSome exit from cilia through the GTPase ARL6/BBS3. Dev. Cell 31, 265–278. doi:10.1016/j.devcel.2014.09.004
Lim, J. E., Jin, O., Bennett, C., Morgan, K., Wang, F., Trenor Iii, C. C., et al. (2005). A mutation in Sec15l1 causes anemia in hemoglobin deficit (hbd) mice. Nat. Genet. 37, 1270–1273. doi:10.1038/ng1659
Lim, Y. S., and Tang, B. L. (2015). A role for Rab23 in the trafficking of Kif17 to the primary cilium. J. Cell Sci. 128, 2996–3008. doi:10.1242/jcs.163964
Lin, Y.-E., Jaimon, E., Tonelli, F., and Pfeffer, S. R. (2025). Pathogenic LRRK2 mutations cause loss of primary cilia and neurturin in striatal parvalbumin interneurons. Life Sci. Alliance 8, e202402922. doi:10.26508/lsa.202402922
Liu, Q., Tan, G., Levenkova, N., Li, T., Pugh, E. N., Rux, J. J., et al. (2007). The proteome of the mouse photoreceptor sensory cilium complex. Mol. Cell Proteomics 6, 1299–1317. doi:10.1074/mcp.M700054-MCP200
Liu, Y.-X., Zhang, R.-K., and Fan, Z.-C. (2023). RABL4/IFT27 in a nucleotide-independent manner promotes phospholipase D ciliary retrieval via facilitating BBSome reassembly at the ciliary tip. J. Cell. Physiology 238, 549–565. doi:10.1002/jcp.30945
Lo, J. C. Y., Jamsai, D., O’connor, A. E., Borg, C., Clark, B. J., Whisstock, J. C., et al. (2012). RAB-like 2 has an essential role in male fertility, sperm intra-flagellar transport, and tail assembly. PLoS Genet. 8, e1002969. doi:10.1371/journal.pgen.1002969
Lobo, G. P., Fulmer, D., Guo, L., Zuo, X., Dang, Y., Kim, S.-H., et al. (2017). The exocyst is required for photoreceptor ciliogenesis and retinal development. J. Biol. Chem. 292, 14814–14826. doi:10.1074/jbc.M117.795674
Lu, L., and Madugula, V. (2018). Mechanisms of ciliary targeting: entering importins and Rabs. Cell Mol. Life Sci. 75, 597–606. doi:10.1007/s00018-017-2629-3
Lu, Q., Insinna, C., Ott, C., Stauffer, J., Pintado, P. A., Rahajeng, J., et al. (2015). Early steps in primary cilium assembly require EHD1/EHD3-dependent ciliary vesicle formation. Nat. Cell Biol. 17, 228–240. doi:10.1038/ncb3109
Lucker, B. F., Behal, R. H., Qin, H., Siron, L. C., Taggart, W. D., Rosenbaum, J. L., et al. (2005). Characterization of the intraflagellar transport complex B core: direct interaction of the IFT81 and IFT74/72 subunits. J. Biol. Chem. 280, 27688–27696. doi:10.1074/jbc.M505062200
Madero-Pérez, J., Fdez, E., Fernández, B., Lara Ordóñez, A. J., Blanca Ramírez, M., Gómez-Suaga, P., et al. (2018). Parkinson disease-associated mutations in LRRK2 cause centrosomal defects via Rab8a phosphorylation. Mol. Neurodegener. 13 (3), 3. doi:10.1186/s13024-018-0235-y
Madugula, V., and Lu, L. (2016). A ternary complex comprising transportin1, Rab8 and the ciliary targeting signal directs proteins to ciliary membranes. J. Cell Sci. 129, 3922–3934. doi:10.1242/jcs.194019
Maharjan, Y., Lee, J. N., Kwak, S. A., Dutta, R. K., Park, C., Choe, S. K., et al. (2020). TMEM135 regulates primary ciliogenesis through modulation of intracellular cholesterol distribution. EMBO Rep. 21, e48901. doi:10.15252/embr.201948901
Maib, H., and Murray, D. H. (2022). A mechanism for exocyst-mediated tethering via Arf6 and PIP5K1C-driven phosphoinositide conversion. Curr. Biol. 32, 2821–2833.e6. doi:10.1016/j.cub.2022.04.089
Mayer, U., Küller, A., Daiber, P. C., Neudorf, I., Warnken, U., Schnölzer, M., et al. (2009). The proteome of rat olfactory sensory cilia. Proteomics 9, 322–334. doi:10.1002/pmic.200800149
Mazelova, J., Astuto-Gribble, L., Inoue, H., Tam, B. M., Schonteich, E., Prekeris, R., et al. (2009). Ciliary targeting motif VxPx directs assembly of a trafficking module through Arf4. EMBO J. 28, 183–192. doi:10.1038/emboj.2008.267
Mazo, G., Soplop, N., Wang, W. J., Uryu, K., and Tsou, M. F. (2016). Spatial control of primary ciliogenesis by subdistal appendages alters sensation-associated properties of cilia. Dev. Cell 39, 424–437. doi:10.1016/j.devcel.2016.10.006
Mccafferty, C. L., Papoulas, O., Lee, C., Bui, K. H., Taylor, D. W., Marcotte, E. M., et al. (2024). An amino acid-resolution interactome for motile cilia identifies the structure and function of ciliopathy protein complexes. Dev. Cell 60, 965–978.e3. doi:10.1016/j.devcel.2024.11.019
Medkova, M., France, Y. E., Coleman, J., and Novick, P. (2006). The rab exchange factor Sec2p reversibly associates with the exocyst. Mol. Biol. Cell 17, 2757–2769. doi:10.1091/mbc.e05-10-0917
Mei, K., Li, Y., Wang, S., Shao, G., Wang, J., Ding, Y., et al. (2018). Cryo-EM structure of the exocyst complex. Nat. Struct. Mol. Biol. 25, 139–146. doi:10.1038/s41594-017-0016-2
Milic, B., Andreasson, J. O. L., Hogan, D. W., and Block, S. M. (2017). Intraflagellar transport velocity is governed by the number of active KIF17 and KIF3AB motors and their motility properties under load, Proc. Natl. Acad. Sci. U. S. A., 114, E6830–E6838. doi:10.1073/pnas.1708157114
Mill, P., Christensen, S. T., and Pedersen, L. B. (2023). Primary cilia as dynamic and diverse signalling hubs in development and disease. Nat. Rev. Genet. 24, 421–441. doi:10.1038/s41576-023-00587-9
Mizuno, S., Takami, K., Daitoku, Y., Tanimoto, Y., Dinh, T. T., Mizuno-Iijima, S., et al. (2015). Peri-implantation lethality in mice carrying megabase-scale deletion on 5qc3.3 is caused by Exoc1 null mutation. Sci. Rep. 5, 13632. doi:10.1038/srep13632
Molla-Herman, A., Ghossoub, R., Blisnick, T., Meunier, A., Serres, C., Silbermann, F., et al. (2010). The ciliary pocket: an endocytic membrane domain at the base of primary and motile cilia. J. Cell Sci. 123, 1785–1795. doi:10.1242/jcs.059519
Monis, W. J., Faundez, V., and Pazour, G. J. (2017). BLOC-1 is required for selective membrane protein trafficking from endosomes to primary cilia. J. Cell Biol. 216, 2131–2150. doi:10.1083/jcb.201611138
Moran, A. L., Carter, S. P., Kaylor, J. J., Jiang, Z., Broekman, S., Dillon, E. T., et al. (2022). Dawn and dusk peaks of outer segment phagocytosis, and visual cycle function require Rab28. FASEB J. 36, e22309. doi:10.1096/fj.202101897R
Moritz, O. L., Tam, B. M., Hurd, L. L., Peränen, J., Deretic, D., and Papermaster, D. S. (2001). Mutant rab8 impairs docking and fusion of rhodopsin-bearing post-golgi membranes and causes cell death of TransgenicXenopus rods. Mol. Biol. Cell 12, 2341–2351. doi:10.1091/mbc.12.8.2341
Mukhopadhyay, S., Wen, X., Chih, B., Nelson, C. D., Lane, W. S., Scales, S. J., et al. (2010). TULP3 bridges the IFT-A complex and membrane phosphoinositides to promote trafficking of G protein-coupled receptors into primary cilia. Genes Dev. 24, 2180–2193. doi:10.1101/gad.1966210
Müller, M. P., and Goody, R. S. (2018). Molecular control of Rab activity by GEFs, GAPs and GDI. Small GTPases 9, 5–21. doi:10.1080/21541248.2016.1276999
Nachury, M. V., Loktev, A. V., Zhang, Q., Westlake, C. J., Peränen, J., Merdes, A., et al. (2007). A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 129, 1201–1213. doi:10.1016/j.cell.2007.03.053
Nachury, M. V., Seeley, E. S., and Jin, H. (2010). Trafficking to the ciliary membrane: how to get across the periciliary diffusion barrier? Annu. Rev. Cell Dev. Biol. 26, 59–87. doi:10.1146/annurev.cellbio.042308.113337
Nager, A. R., Goldstein, J. S., Herranz-Pérez, V., Portran, D., Ye, F., Garcia-Verdugo, J. M., et al. (2017). An actin network dispatches ciliary GPCRs into extracellular vesicles to modulate signaling. Cell 168, 252–263. e214. doi:10.1016/j.cell.2016.11.036
Nalls, M. A., Pankratz, N., Lill, C. M., Do, C. B., Hernandez, D. G., Saad, M., et al. (2014). Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 46, 989–993. doi:10.1038/ng.3043
Narita, K., Kozuka-Hata, H., Nonami, Y., Ao-Kondo, H., Suzuki, T., Nakamura, H., et al. (2012). Proteomic analysis of multiple primary cilia reveals a novel mode of ciliary development in mammals. Biol. Open 1, 815–825. doi:10.1242/bio.20121081
Niedziółka, S. M., Datta, S., Uśpieński, T., Baran, B., Skarżyńska, W., Humke, E. W., et al. (2024). The exocyst complex and intracellular vesicles mediate soluble protein trafficking to the primary cilium. Commun. Biol. 7, 213. doi:10.1038/s42003-024-05817-2
Nihalani, D., Solanki, A. K., Arif, E., Srivastava, P., Rahman, B., Zuo, X., et al. (2019). Disruption of the exocyst induces podocyte loss and dysfunction. J. Biol. Chem. 294, 10104–10119. doi:10.1074/jbc.RA119.008362
Nishijima, Y., Hagiya, Y., Kubo, T., Takei, R., Katoh, Y., and Nakayama, K. (2017). RABL2 interacts with the intraflagellar transport-B complex and CEP19 and participates in ciliary assembly. Mol. Biol. Cell 28, 1652–1666. doi:10.1091/mbc.E17-01-0017
Oguchi, M. E., Homma, Y., and Fukuda, M. (2022). The N-terminal Leu-Pro-Gln sequence of Rab34 is required for ciliogenesis in hTERT-RPE1 cells. Small GTPases 13, 77–83. doi:10.1080/21541248.2021.1894910
Oguchi, M. E., Okuyama, K., Homma, Y., and Fukuda, M. (2020). A comprehensive analysis of Rab GTPases reveals a role for Rab34 in serum starvation-induced primary ciliogenesis. J. Biol. Chem. 295, 12674–12685. doi:10.1074/jbc.RA119.012233
Osawa, Y., Murata, K., Usui, M., Kuba, Y., Le, H. T., Mikami, N., et al. (2021). EXOC1 plays an integral role in spermatogonia pseudopod elongation and spermatocyte stable syncytium formation in mice. Elife 10, e59759. doi:10.7554/eLife.59759
Ostrowski, L. E., Blackburn, K., Radde, K. M., Moyer, M. B., Schlatzer, D. M., Moseley, A., et al. (2002). A proteomic analysis of human cilia: identification of novel components. Mol. Cell Proteomics 1, 451–465. doi:10.1074/mcp.m200037-mcp200
Otto, E. A., Schermer, B., Obara, T., O'toole, J. F., Hiller, K. S., Mueller, A. M., et al. (2003). Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat. Genet. 34, 413–420. doi:10.1038/ng1217
Park, K., and Leroux, M. R. (2022). Composition, organization and mechanisms of the transition zone, a gate for the cilium. EMBO Rep. 23, e55420. doi:10.15252/embr.202255420
Partisani, M., Baron, C. L., Ghossoub, R., Fayad, R., Pagnotta, S., Abélanet, S., et al. (2021). EFA6A, an exchange factor for Arf6, regulates early steps in ciliogenesis. J. Cell Sci. 134, jcs249565. doi:10.1242/jcs.249565
Pereira, C., Stalder, D., Anderson, G. S. F., Shun-Shion, A. S., Houghton, J., Antrobus, R., et al. (2023). The exocyst complex is an essential component of the mammalian constitutive secretory pathway. J. Cell Biol. 222, e202205137. doi:10.1083/jcb.202205137
Pereira-Leal, J. B., and Seabra, M. C. (2000). The mammalian Rab family of small GTPases: definition of family and subfamily sequence motifs suggests a mechanism for functional specificity in the Ras superfamily. J. Mol. Biol. 301, 1077–1087. doi:10.1006/jmbi.2000.4010
Pereira-Leal, J. B., and Seabra, M. C. (2001). Evolution of the Rab family of small GTP-binding proteins. J. Mol. Biol. 313, 889–901. doi:10.1006/jmbi.2001.5072
Pereira-Leal, J. B., Strom, M., Godfrey, R. F., and Seabra, M. C. (2003). Structural determinants of Rab and Rab Escort Protein interaction: rab family motifs define a conserved binding surface. Biochem. Biophys. Res. Commun. 301, 92–97. doi:10.1016/s0006-291x(02)02963-7
Phua, S. C., Chiba, S., Suzuki, M., Su, E., Roberson, E. C., Pusapati, G. V., et al. (2017). Dynamic remodeling of membrane composition drives cell cycle through primary cilia excision. Cell 168, 264–279.e15. doi:10.1016/j.cell.2016.12.032
Polgar, N., Lee, A. J., Lui, V. H., Napoli, J. A., and Fogelgren, B. (2015). The exocyst gene Sec10 regulates renal epithelial monolayer homeostasis and apoptotic sensitivity. Am. J. Physiol. Cell Physiol. 309, C190–C201. doi:10.1152/ajpcell.00011.2015
Pusapati, G. V., Kong, J. H., Patel, B. B., Krishnan, A., Sagner, A., Kinnebrew, M., et al. (2018). CRISPR screens uncover genes that regulate target cell sensitivity to the morphogen sonic hedgehog. Dev. Cell 44, 113–129.e8. doi:10.1016/j.devcel.2017.12.003
Qin, H., Wang, Z., Diener, D., and Rosenbaum, J. (2007). Intraflagellar transport protein 27 is a small G protein involved in cell-cycle control. Curr. Biol. 17, 193–202. doi:10.1016/j.cub.2006.12.040
Quélin, C., Loget, P., Boutaud, L., Elkhartoufi, N., Milon, J., Odent, S., et al. (2018). Loss of function IFT27 variants associated with an unclassified lethal fetal ciliopathy with renal agenesis. Am. J. Med. Genet. Part A 176, 1610–1613. doi:10.1002/ajmg.a.38685
Reiter, J. F., Blacque, O. E., and Leroux, M. R. (2012). The base of the cilium: roles for transition fibres and the transition zone in ciliary formation, maintenance and compartmentalization. EMBO Rep. 13, 608–618. doi:10.1038/embor.2012.73
Reiter, J. F., and Leroux, M. R. (2017). Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 18, 533–547. doi:10.1038/nrm.2017.60
Riveiro-Álvarez, R., Xie, Y. A., López-Martínez, M., Gambin, T., Pérez-Carro, R., Ávila-Fernández, A., et al. (2015). New mutations in the RAB28 gene in 2 Spanish families with cone-rod dystrophy. JAMA Ophthalmol. 133, 133–139. doi:10.1001/jamaophthalmol.2014.4266
Rivera-Molina, F. E., Xi, Z., Reales, E., Wang, B., and Toomre, D. (2021). Exocyst complex mediates recycling of internal cilia. Curr. Biol. 31, 5580–5589.e5. doi:10.1016/j.cub.2021.09.067
Rogers, K. K., Wilson, P. D., Snyder, R. W., Zhang, X., Guo, W., Burrow, C. R., et al. (2004). The exocyst localizes to the primary cilium in MDCK cells. Biochem. Biophysical Res. Commun. 319, 138–143. doi:10.1016/j.bbrc.2004.04.165
Roosing, S., Rohrschneider, K., Beryozkin, A., Sharon, D., Weisschuh, N., Staller, J., et al. (2013). Mutations in RAB28, encoding a farnesylated small GTPase, are associated with autosomal-recessive cone-rod dystrophy. Am. J. Hum. Genet. 93, 110–117. doi:10.1016/j.ajhg.2013.05.005
Safavian, D., Kim, M. S., Xie, H., El-Zeiry, M., Palander, O., Dai, L., et al. (2023). Septin-mediated RhoA activation engages the exocyst complex to recruit the cilium transition zone. J. Cell Biol. 222, e201911062. doi:10.1083/jcb.201911062
Saha, I., Insinna, C., and Westlake, C. J. (2024). Rab11-Rab8 cascade dynamics in primary cilia and membrane tubules. Cell Rep. 43, 114955. doi:10.1016/j.celrep.2024.114955
Sato, T., Iwano, T., Kunii, M., Matsuda, S., Mizuguchi, R., Jung, Y., et al. (2014). Rab8a and Rab8b are essential for several apical transport pathways but insufficient for ciliogenesis. J. Cell Sci. 127, 422–431. doi:10.1242/jcs.136903
Schaefer, E., Delvallée, C., Mary, L., Stoetzel, C., Geoffroy, V., Marks-Delesalle, C., et al. (2019). Identification and characterization of known biallelic mutations in the IFT27 (BBS19) gene in a novel family with bardet-biedl syndrome. Front. Genet. 10, 21. doi:10.3389/fgene.2019.00021
Schafer, J. C., Winkelbauer, M. E., Williams, C. L., Haycraft, C. J., Desmond, R. A., and Yoder, B. K. (2006). IFTA-2 is a conserved cilia protein involved in pathways regulating longevity and dauer formation in Caenorhabditis elegans. J. cell Sci. 119, 4088–4100. doi:10.1242/jcs.03187
Schaub, J. R., and Stearns, T. (2013). The Rilp-like proteins Rilpl1 and Rilpl2 regulate ciliary membrane content. Mol. Biol. cell 24, 453–464. doi:10.1091/mbc.E12-08-0598
Schmidt, K. N., Kuhns, S., Neuner, A., Hub, B., Zentgraf, H., and Pereira, G. (2012). Cep164 mediates vesicular docking to the mother centriole during early steps of ciliogenesis. J. Cell Biol. 199, 1083–1101. doi:10.1083/jcb.201202126
Seixas, C., Choi, S. Y., Polgar, N., Umberger, N. L., East, M. P., Zuo, X., et al. (2016). Arl13b and the exocyst interact synergistically in ciliogenesis. Mol. Biol. Cell 27, 308–320. doi:10.1091/mbc.E15-02-0061
Serres, M. P., Shaughnessy, R., Escot, S., Hammich, H., Cuvelier, F., Salles, A., et al. (2023). MiniBAR/GARRE1 is a dual Rac and Rab effector required for ciliogenesis. Dev. Cell 58, 2477–2494.e8. doi:10.1016/j.devcel.2023.09.010
Silva, D. A., Huang, X., Behal, R. H., Cole, D. G., and Qin, H. (2012). The RABL5 homolog IFT22 regulates the cellular pool size and the amount of IFT particles partitioned to the flagellar compartment in Chlamydomonas reinhardtii. Cytoskelet. Hob. 69, 33–48. doi:10.1002/cm.20546
Sim, H. J., Yun, S., Kim, H. E., Kwon, K. Y., Kim, G. H., Yun, S., et al. (2020). Simple method to characterize the ciliary proteome of multiciliated cells. J. Proteome Res. 19, 391–400. doi:10.1021/acs.jproteome.9b00589
Simsek-Kiper, P. O., Jacob, P., Upadhyai, P., Taşkıran, Z. E., Guleria, V. S., Karaosmanoglu, B., et al. (2022). Biallelic loss-of-function variants in EXOC6B are associated with impaired primary ciliogenesis and cause spondylo-epi-metaphyseal dysplasia with joint laxity type 3. Hum. Mutat. 43, 2116–2129. doi:10.1002/humu.24478
Sivaram, M. V., Saporita, J. A., Furgason, M. L., Boettcher, A. J., and Munson, M. (2005). Dimerization of the exocyst protein Sec6p and its interaction with the t-SNARE Sec9p. Biochemistry 44, 6302–6311. doi:10.1021/bi048008z
Smith, J. C., Northey, J. G., Garg, J., Pearlman, R. E., and Siu, K. W. (2005). Robust method for proteome analysis by MS/MS using an entire translated genome: demonstration on the ciliome of Tetrahymena thermophila. J. Proteome Res. 4, 909–919. doi:10.1021/pr050013h
Spörle, R., Günther, T., Struwe, M., and Schughart, K. (1996). Severe defects in the formation of epaxial musculature in open brain (opb) mutant mouse embryos. Development 122, 79–86. doi:10.1242/dev.122.1.79
Steger, M., Diez, F., Dhekne, H. S., Lis, P., Nirujogi, R. S., Karayel, O., et al. (2017). Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. eLife 6, e31012. doi:10.7554/eLife.31012
Steger, M., Tonelli, F., Ito, G., Davies, P., Trost, M., Vetter, M., et al. (2016). Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. elife 5, e12813. doi:10.7554/eLife.12813
Stein, M., Pilli, M., Bernauer, S., Habermann, B. H., Zerial, M., and Wade, R. C. (2012). The interaction properties of the human Rab GTPase family–comparative analysis reveals determinants of molecular binding selectivity. PLoS One 7, e34870. doi:10.1371/journal.pone.0034870
Stuck, M. W., Chong, W. M., Liao, J.-C., and Pazour, G. J. (2021). Rab34 is necessary for early stages of intracellular ciliogenesis. Curr. Biol. 31, 2887–2894.e4. doi:10.1016/j.cub.2021.04.018
Subota, I., Julkowska, D., Vincensini, L., Reeg, N., Buisson, J., Blisnick, T., et al. (2014). Proteomic analysis of intact flagella of procyclic Trypanosoma brucei cells identifies novel flagellar proteins with unique sub-localization and dynamics. Mol. Cell Proteomics 13, 1769–1786. doi:10.1074/mcp.M113.033357
Sun, W.-Y., Xue, B., Liu, Y.-X., Zhang, R.-K., Li, R.-C., Xin, W., et al. (2021). Chlamydomonas LZTFL1 mediates phototaxis via controlling BBSome recruitment to the basal body and its reassembly at the ciliary tip. Proc. Natl. Acad. Sci. 118, e2101590118. doi:10.1073/pnas.2101590118
Surmeier, D. J., Graves, S. M., and Shen, W. (2014). Dopaminergic modulation of striatal networks in health and Parkinson’s disease. Curr. Opin. Neurobiol. 29, 109–117. doi:10.1016/j.conb.2014.07.008
Takahashi, T., Minami, S., Tsuchiya, Y., Tajima, K., Sakai, N., Suga, K., et al. (2019). Cytoplasmic control of Rab family small GTPases through BAG6. EMBO Rep. 20, e46794. doi:10.15252/embr.201846794
Takei, R., Katoh, Y., and Nakayama, K. (2018). Robust interaction of IFT70 with IFT52–IFT88 in the IFT-B complex is required for ciliogenesis. Biol. Open 7, bio033241. doi:10.1242/bio.033241
Tang, D., Bao, H., and Qi, S. (2024). The C9orf72-SMCR8 complex suppresses primary ciliogenesis as a RAB8A GAP. Autophagy 20, 1205–1207. doi:10.1080/15548627.2024.2311541
Tanos, B. E., Yang, H. J., Soni, R., Wang, W. J., Macaluso, F. P., Asara, J. M., et al. (2013). Centriole distal appendages promote membrane docking, leading to cilia initiation. Genes Dev. 27, 163–168. doi:10.1101/gad.207043.112
Taravath, S., and Tonsgard, J. H. (1993). Cerebral malformations in Carpenter syndrome. Pediatr. Neurol. 9, 230–234. doi:10.1016/0887-8994(93)90092-q
Taschner, M., Kotsis, F., Braeuer, P., Kuehn, E. W., and Lorentzen, E. (2014). Crystal structures of IFT70/52 and IFT52/46 provide insight into intraflagellar transport B core complex assembly. J. Cell Biol. 207, 269–282. doi:10.1083/jcb.201408002
Taschner, M., Weber, K., Mourão, A., Vetter, M., Awasthi, M., Stiegler, M., et al. (2016). Intraflagellar transport proteins 172, 80, 57, 54, 38, and 20 form a stable tubulin-binding IFT-B2 complex. EMBO J. 35, 773–790. doi:10.15252/embj.201593164
Terbush, D. R., Maurice, T., Roth, D., and Novick, P. (1996). The Exocyst is a multiprotein complex required for exocytosis in Saccharomyces cerevisiae. EMBO J. 15, 6483–6494. doi:10.1002/j.1460-2075.1996.tb01039.x
Thirstrup, K., Dächsel, J. C., Oppermann, F. S., Williamson, D. S., Smith, G. P., Fog, K., et al. (2017). Selective LRRK2 kinase inhibition reduces phosphorylation of endogenous Rab10 and Rab12 in human peripheral mononuclear blood cells. Sci. Rep. 7, 10300. doi:10.1038/s41598-017-10501-z
Ungermann, C., and Kümmel, D. (2019). Structure of membrane tethers and their role in fusion. Traffic 20, 479–490. doi:10.1111/tra.12655
Van Bergen, N. J., Ahmed, S. M., Collins, F., Cowley, M., Vetro, A., Dale, R. C., et al. (2020). Mutations in the exocyst component EXOC2 cause severe defects in human brain development. J. Exp. Med. 217, e20192040. doi:10.1084/jem.20192040
Van Der Beek, J., Jonker, C., Van Der Welle, R., Liv, N., and Klumperman, J. (2019). CORVET, CHEVI and HOPS – multisubunit tethers of the endo-lysosomal system in health and disease. J. Cell Sci. 132, jcs189134. doi:10.1242/jcs.189134
Van Der Vaart, A., Rademakers, S., and Jansen, G. (2015). DLK-1/p38 MAP kinase signaling controls cilium length by regulating RAB-5 mediated endocytosis in Caenorhabditis elegans. PLoS Genet. 11, e1005733. doi:10.1371/journal.pgen.1005733
Vetter, M., Stehle, R., Basquin, C., and Lorentzen, E. (2015). Structure of Rab11–FIP3–rabin8 reveals simultaneous binding of FIP3 and Rabin8 effectors to Rab11. Nat. Struct. & Mol. Biol. 22, 695–702. doi:10.1038/nsmb.3065
Wachter, S., Jung, J., Shafiq, S., Basquin, J., Fort, C., Bastin, P., et al. (2019). Binding of IFT22 to the intraflagellar transport complex is essential for flagellum assembly. EMBO J. 38, e101251. doi:10.15252/embj.2018101251
Walia, V., Cuenca, A., Vetter, M., Insinna, C., Perera, S., Lu, Q., et al. (2019). Akt regulates a Rab11-effector switch required for ciliogenesis. Dev. Cell 50, 229–246.e7. doi:10.1016/j.devcel.2019.05.022
Walsh, T. G., Li, Y., Williams, C. M., Aitken, E. W., Andrews, R. K., and Poole, A. W. (2021). Loss of the exocyst complex component EXOC3 promotes hemostasis and accelerates arterial thrombosis. Blood Adv. 5, 674–686. doi:10.1182/bloodadvances.2020002515
Wandinger-Ness, A., and Zerial, M. (2014). Rab proteins and the compartmentalization of the endosomal system. Cold Spring Harb. Perspect. Biol. 6, a022616. doi:10.1101/cshperspect.a022616
Wang, H. H., Zhang, Y., Tang, F., Pan, M. H., Wan, X., Li, X. H., et al. (2019a). Rab23/Kif17 regulate meiotic progression in oocytes by modulating tubulin acetylation and actin dynamics. Development 146, dev171280. doi:10.1242/dev.171280
Wang, J., and Deretic, D. (2015). The Arf and Rab11 effector FIP3 acts synergistically with ASAP1 to direct Rabin8 in ciliary receptor targeting. J. Cell Sci. 128, 1375–1385. doi:10.1242/jcs.162925
Wang, J., Morita, Y., Mazelova, J., and Deretic, D. (2012). The Arf GAP ASAP1 provides a platform to regulate Arf4-and Rab11-Rab8-mediated ciliary receptor targeting. Embo J. 31, 4057–4071. doi:10.1038/emboj.2012.253
Wang, S., Crisman, L., Miller, J., Datta, I., Gulbranson, D. R., Tian, Y., et al. (2019b). Inducible Exoc7/Exo70 knockout reveals a critical role of the exocyst in insulin-regulated GLUT4 exocytosis. J. Biol. Chem. 294, 19988–19996. doi:10.1074/jbc.RA119.010821
Wang, T., and Hong, W. (2002). Interorganellar regulation of lysosome positioning by the Golgi apparatus through Rab34 interaction with Rab-interacting lysosomal protein. Mol. Biol. Cell 13, 4317–4332. doi:10.1091/mbc.e02-05-0280
Wang, Z., Fan, Z.-C., Williamson, S. M., and Qin, H. (2009). Intraflagellar transport (IFT) protein IFT25 is a phosphoprotein component of IFT complex B and physically interacts with IFT27 in Chlamydomonas. PLoS One 4, e5384. doi:10.1371/journal.pone.0005384
Warburton-Pitt, S. R., Jauregui, A. R., Li, C., Wang, J., Leroux, M. R., and Barr, M. M. (2012). Ciliogenesis in Caenorhabditis elegans requires genetic interactions between ciliary middle segment localized NPHP-2 (inversin) and transition zone-associated proteins. J. Cell Sci. 125, 2592–2603. doi:10.1242/jcs.095539
Ward, H. H., Brown-Glaberman, U., Wang, J., Morita, Y., Alper, S. L., Bedrick, E. J., et al. (2011). A conserved signal and GTPase complex are required for the ciliary transport of polycystin-1. Mol. Biol. Cell 22, 3289–3305. doi:10.1091/mbc.E11-01-0082
Westlake, C. J., Baye, L. M., Nachury, M. V., Wright, K. J., Ervin, K. E., Phu, L., et al. (2011). Primary cilia membrane assembly is initiated by Rab11 and transport protein particle II (TRAPPII) complex-dependent trafficking of Rabin8 to the centrosome. Proc. Natl. Acad. Sci. 108, 2759–2764. doi:10.1073/pnas.1018823108
White, R. A., Boydston, L. A., Brookshier, T. R., Mcnulty, S. G., Nsumu, N. N., Brewer, B. P., et al. (2005). Iron metabolism mutant hbd mice have a deletion in Sec15l1, which has homology to a yeast gene for vesicle docking. Genomics 86, 668–673. doi:10.1016/j.ygeno.2005.09.015
Wu, D., Huang, J., Zhu, H., Chen, Z., Chai, Y., Ke, J., et al. (2022). Ciliogenesis requires sphingolipid-dependent membrane and axoneme interaction, Proc. Natl. Acad. Sci. U. S. A., 119, e2201096119, doi:10.1073/pnas.2201096119
Wu, Z., Pang, N., Zhang, Y., Chen, H., Peng, Y., Fu, J., et al. (2020). CEP290 is essential for the initiation of ciliary transition zone assembly. PLoS Biol. 18, e3001034. doi:10.1371/journal.pbio.3001034
Xu, Q., Zhang, Y., Wei, Q., Huang, Y., Hu, J., and Ling, K. (2016). Phosphatidylinositol phosphate kinase PIPKIγ and phosphatase INPP5E coordinate initiation of ciliogenesis. Nat. Commun. 7, 10777. doi:10.1038/ncomms10777
Xu, S., Liu, Y., Meng, Q., and Wang, B. (2018). Rab34 small GTPase is required for Hedgehog signaling and an early step of ciliary vesicle formation in mouse. J. Cell Sci. 131, jcs213710. doi:10.1242/jcs.213710
Xue, B., Liu, Y. X., Dong, B., Wingfield, J. L., Wu, M., Sun, J., et al. (2020). Intraflagellar transport protein RABL5/IFT22 recruits the BBSome to the basal body through the GTPase ARL6/BBS3. Proc. Natl. Acad. Sci. U. S. A. 117, 2496–2505. doi:10.1073/pnas.1901665117
Yamaguchi, H., Barrell, W. B., Faisal, M., Liu, K. J., and Komatsu, Y. (2024). Ciliary and non-ciliary functions of Rab34 during craniofacial bone development. Biochem. Biophysical Res. Commun. 724, 150174. doi:10.1016/j.bbrc.2024.150174
Yang, T. T., Chong, W. M., Wang, W.-J., Mazo, G., Tanos, B., Chen, Z., et al. (2018). Super-resolution architecture of mammalian centriole distal appendages reveals distinct blade and matrix functional components. Nat. Commun. 9, 2023. doi:10.1038/s41467-018-04469-1
Ye, F., Nager, A. R., and Nachury, M. V. (2018a). BBSome trains remove activated GPCRs from cilia by enabling passage through the transition zone. J. Cell Biol. 217, 1847–1868. doi:10.1083/jcb.201709041
Ye, F., Nager, A. R., and Nachury, M. V. (2018b). BBSome trains remove activated GPCRs from cilia by enabling passage through the transition zone. J. Cell Biol. 217, 1847–1868. doi:10.1083/jcb.201709041
Ye, X., Zeng, H., Ning, G., Reiter, J. F., and Liu, A. (2014). C2cd3 is critical for centriolar distal appendage assembly and ciliary vesicle docking in mammals. Proc. Natl. Acad. Sci. U. S. A. 111, 2164–2169. doi:10.1073/pnas.1318737111
Yeaman, C., Grindstaff, K. K., Wright, J. R., and Nelson, W. J. (2001). Sec6/8 complexes on trans-Golgi network and plasma membrane regulate late stages of exocytosis in mammalian cells. J. Cell Biol. 155, 593–604. doi:10.1083/jcb.200107088
Yi Lo, J. C., O’connor, A. E., Andrews, Z. B., Lo, C., Tiganis, T., Watt, M. J., et al. (2016). RABL2 is required for hepatic fatty acid homeostasis and its dysfunction leads to steatosis and a diabetes-like state. Endocrinology 157, 4732–4743. doi:10.1210/en.2016-1487
Ying, G., Boldt, K., Ueffing, M., Gerstner, C. D., Frederick, J. M., and Baehr, W. (2018). The small GTPase RAB28 is required for phagocytosis of cone outer segments by the murine retinal pigmented epithelium. J. Biol. Chem. 293, 17546–17558. doi:10.1074/jbc.RA118.005484
Yoshimura, S., Egerer, J., Fuchs, E., Haas, A. K., and Barr, F. A. (2007). Functional dissection of Rab GTPases involved in primary cilium formation. J. Cell Biol. 178, 363–369. doi:10.1083/jcb.200703047
Yue, P., Zhang, Y., Mei, K., Wang, S., Lesigang, J., Zhu, Y., et al. (2017). Sec3 promotes the initial binary t-SNARE complex assembly and membrane fusion. Nat. Commun. 8, 14236. doi:10.1038/ncomms14236
Zeigerer, A., Gilleron, J., Bogorad, R. L., Marsico, G., Nonaka, H., Seifert, S., et al. (2012). Rab5 is necessary for the biogenesis of the endolysosomal system in vivo. Nature 485, 465–470. doi:10.1038/nature11133
Zhang, B., Zhang, T., Wang, G., Wang, G., Chi, W., Jiang, Q., et al. (2015). GSK3β-Dzip1-Rab8 cascade regulates ciliogenesis after mitosis. PLoS Biol. 13, e1002129. doi:10.1371/journal.pbio.1002129
Zhang, X., Bi, E., Novick, P., Du, L., Kozminski, K. G., Lipschutz, J. H., et al. (2001). Cdc42 interacts with the exocyst and regulates polarized secretion. J. Biol. Chem. 276, 46745–46750. doi:10.1074/jbc.M107464200
Zhang, X., Orlando, K., He, B., Xi, F., Zhang, J., Zajac, A., et al. (2008). Membrane association and functional regulation of Sec3 by phospholipids and Cdc42. J. Cell Biol. 180, 145–158. doi:10.1083/jcb.200704128
Zhang, Y., Liu, H., Li, W., Zhang, Z., Shang, X., Zhang, D., et al. (2017). Intraflagellar transporter protein (IFT27), an IFT25 binding partner, is essential for male fertility and spermiogenesis in mice. Dev. Biol. 432, 125–139. doi:10.1016/j.ydbio.2017.09.023
Zhou, Z., Katoh, Y., and Nakayama, K. (2022). CEP19-RABL2-IFT-B axis controls BBSome-mediated ciliary GPCR export. Mol. Biol. Cell 33, ar126. doi:10.1091/mbc.E22-05-0161
Zhou, Z., Qiu, H., Castro-Araya, R.-F., Takei, R., Nakayama, K., and Katoh, Y. (2021). Impaired cooperation between IFT74/BBS22–IFT81 and IFT25–IFT27/BBS19 causes Bardet-Biedl syndrome. Hum. Mol. Genet. 31, 1681–1693. doi:10.1093/hmg/ddab354
Zilber, Y., Babayeva, S., Seo, J. H., Liu, J. J., Mootin, S., and Torban, E. (2013). The PCP effector Fuzzy controls cilial assembly and signaling by recruiting Rab8 and Dishevelled to the primary cilium. Mol. Biol. Cell 24, 555–565. doi:10.1091/mbc.E12-06-0437
Zimprich, A., Biskup, S., Leitner, P., Lichtner, P., Farrer, M., Lincoln, S., et al. (2004). Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607. doi:10.1016/j.neuron.2004.11.005
Zuo, X., Fogelgren, B., and Lipschutz, J. H. (2011). The small GTPase Cdc42 is necessary for primary ciliogenesis in renal tubular epithelial cells. J. Biol. Chem. 286, 22469–22477. doi:10.1074/jbc.M111.238469
Zuo, X., Kwon, S. H., Janech, M. G., Dang, Y., Lauzon, S. D., Fogelgren, B., et al. (2019a). Primary cilia and the exocyst are linked to urinary extracellular vesicle production and content. J. Biol. Chem. 294, 19099–19110. doi:10.1074/jbc.RA119.009297
Zuo, X., Lobo, G., Fulmer, D., Guo, L., Dang, Y., Su, Y., et al. (2019b). The exocyst acting through the primary cilium is necessary for renal ciliogenesis, cystogenesis, and tubulogenesis. J. Biol. Chem. 294, 6710–6718. doi:10.1074/jbc.RA118.006527
Keywords: primary cilia, Rabs, Rab-like proteins, exocyst, ciliary trafficking, ciliopathies
Citation: Upadhyai P, Bose D and Quadri N (2025) A tale of Rabs and the exocyst complex in ciliary trafficking and biogenesis. Front. Cell Dev. Biol. 13:1574638. doi: 10.3389/fcell.2025.1574638
Received: 11 February 2025; Accepted: 21 April 2025;
Published: 06 May 2025.
Edited by:
Venkata Vivek Reddy Palicharla, University of Texas Southwestern Medical Center, United StatesReviewed by:
Tomoharu Kanie, University of Oklahoma Health Sciences Center, United StatesIssei Shimada, Nagoya City University, Japan
Irene Ojeda Naharros, University of California, San Francisco, United States
Copyright © 2025 Upadhyai, Bose and Quadri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Priyanka Upadhyai, cHJpeWFua2EudUBtYW5pcGFsLmVkdQ==