Abstract
Helicobacter pylori lives within the mucus layer of the human stomach, in close proximity to gastric epithelial cells. While a great deal is known about the effects of H. pylori on human cells and the specific bacterial products that mediate these effects, relatively little work has been done to investigate alterations in H. pylori that may be triggered by bacterial contact with human cells. In this review, we discuss the spectrum of changes in bacterial physiology and morphology that occur when H. pylori is in contact with gastric epithelial cells. Several studies have reported that cell contact causes alterations in H. pylori gene transcription. In addition, H. pylori contact with gastric epithelial cells promotes the formation of pilus-like structures at the bacteria–host cell interface. The formation of these structures requires multiple genes in the cag pathogenicity island, and these structures are proposed to have an important role in the type IV secretion system-dependent process through which CagA enters host cells. Finally, H. pylori contact with epithelial cells can promote bacterial replication and the formation of microcolonies, phenomena that are facilitated by the acquisition of iron and other nutrients from infected cells. In summary, the gastric epithelial cell surface represents an important niche for H. pylori, and upon entry into this niche, the bacteria alter their behavior in a manner that optimizes bacterial proliferation and persistent colonization of the host.
Introduction
Helicobacter pylori is highly adapted for colonization of the human stomach, and is found in about half of all humans worldwide (Amieva and El-Omar, 2008; Atherton and Blaser, 2009; Cover and Blaser, 2009). H. pylori associates specifically with gastric mucosal tissue in the stomach or the duodenum, but not with intestinal or squamous-type epithelium (Wyatt et al., 1987, 1990; Carrick et al., 1989). The reasons for a specific association between H. pylori and gastric epithelium are not well understood. One possibility is that H. pylori utilizes specific components of gastric mucus or other factors released by gastric epithelial cells as nutritional sources. In addition, H. pylori may have a competitive advantage compared to other bacteria in the environment overlying gastric epithelial cells, but may lack this advantage in other sites.
Within the stomach, H. pylori can occupy a range of different microenvironments. The bacteria are typically most abundant within the gastric antrum, but can also be found within the corpus. H. pylori is found predominantly within the gastric mucus layer (Hazell et al., 1986), but occasionally can be internalized by gastric epithelial cells (Dubois and Boren, 2007); invasion beyond the epithelial layer is considered to be a rare event. Within the gastric mucus layer, the bacteria can be found relatively close to the gastric lumen or deep within gastric glands, and can be either free-swimming (Hazell et al., 1986; Schreiber et al., 2004; Celli et al., 2009) or attached to gastric epithelial cells (Hessey et al., 1990). At any given time, the proportion of adherent H. pylori is lower than the proportion of non-adherent organisms. Adherent bacteria localize preferentially to intercellular junctions (Hazell et al., 1986), but also can adhere to non-junctional sites. Relatively little is known about the dynamics of bacterial attachment to gastric epithelial cells. For example, it is not known whether adherent organisms remain attached for short durations and then detach, or whether adherent bacteria remain permanently attached and are eventually shed along with the gastric epithelial cells.
Because H. pylori is found in close proximity to gastric epithelial cells, there are numerous opportunities for the bacteria to cause alterations in gastric epithelial cell architecture and function. Many of the changes in gastric epithelial cells caused by H. pylori are attributable to the actions of two secreted bacterial proteins, VacA and CagA. VacA is a pore-forming toxin that is secreted by the bacteria through an autotransporter pathway. The cellular alterations caused by VacA include increased permeability of the plasma membrane, changes in endosomal structure and function, changes in mitochondrial membrane permeability, and cell death (Montecucco and de Bernard, 2003; Cover and Blanke, 2005; Rieder et al., 2005; Jones et al., 2010). CagA is an effector protein that is translocated directly from bacteria into host cells through the action of a type IV secretion system (Hatakeyama, 2004; Bourzac and Guillemin, 2005; Rieder et al., 2005; Backert et al., 2010; Fischer, 2011; Tegtmeyer et al., 2011; Terradot and Waksman, 2011). Both cagA and genes encoding components of this type IV secretion system are contained within a 40 kb chromosomal region known as the cag pathogenicity island (PAI). There is heterogeneity among H. pylori strains, such that strains may contain an intact cag PAI, may contain fragments of this PAI, or may completely lack this region (Olbermann et al., 2010). Upon entry into gastric epithelial cells, CagA interacts with multiple intracellular target proteins and causes a wide array of alterations in cellular signaling, leading to changes in cell shape, increased cellular motility and cellular invasiveness, alterations in monolayer polarity and permeability, and increased cellular proliferation (Hatakeyama, 2004; Bourzac and Guillemin, 2005; Rieder et al., 2005; Backert et al., 2010; Tegtmeyer et al., 2011). Because CagA activates signaling pathways associated with carcinogenesis, it has been termed a “bacterial oncoprotein” (Hatakeyama, 2004). The actions of VacA and CagA on epithelial cells have been described in detail in other reviews (Montecucco and de Bernard, 2003; Hatakeyama, 2004; Bourzac and Guillemin, 2005; Cover and Blanke, 2005; Rieder et al., 2005; Backert et al., 2010; Jones et al., 2010; Tegtmeyer et al., 2011) and will not be discussed in detail here.
The effects of H. pylori on gastric epithelial cells and the specific bacterial factors that mediate these effects have previously been described in great detail. In contrast, relatively little work has been done to investigate alterations in H. pylori that may be triggered by bacterial contact with human cells. In this article, we review the multiple ways in which contact with gastric epithelial cells causes alterations in H. pylori.
Transcriptional Regulation of H. pylori Genes in Response to Bacterial Contact with Gastric Epithelial Cells
Several studies have reported that H. pylori adherence to gastric epithelial cells triggers alterations in H. pylori gene transcription (Joyce et al., 2001; van Amsterdam et al., 2003; Kim et al., 2004; Gieseler et al., 2005). These alterations have been detected using a variety of approaches, including transcriptional reporter assays, quantitative real-time PCR (RT-PCR), and array-based hybridization methods.
To identify bacterial genes that are differentially expressed upon bacterial contact with gastric epithelial cells, one study analyzed a plasmid library derived from H. pylori strain 1061, containing random chromosomal fusions to a promoterless cat gene, which allowed chloramphenicol resistance to be used as a marker of gene expression (van Amsterdam et al., 2003). Twenty-one unique clones exhibited increased resistance to chloramphenicol in the presence of HM02 gastric epithelial cells, compared to the level of chloramphenicol resistance in the absence of these cells. Most of the clones were not characterized in detail, but one that exhibited a marked increase in chloramphenicol resistance contained a fusion to vacA (encoding the secreted toxin VacA; van Amsterdam et al., 2003). RT-PCR confirmed that bacterial contact with gastric epithelial cells resulted in increased levels of vacA transcription. These results provide evidence that vacA expression is upregulated upon bacterial contact with gastric epithelial cells.
Effects of cell contact on H. pylori gene transcription have also been assessed by using transcriptional reporter assays to monitor the expression of selected genes (Joyce et al., 2001). The transcriptional reporters were constructed by fusing nine putative promoter regions from the cag PAI with ureB (encoding the B subunit of urease). These reporters were introduced into the hpn locus of an H. pylori strain (C57) in which the endogenous ureB locus had been disrupted. When co-cultured with HEp-2 cells, two of the reporter strains [containing promoters upstream from cagP (cag15) and cagG (cag21)] exhibited increased UreB expression in comparison to when the bacteria were cultured in medium alone (Joyce et al., 2001). The other reporter strains (containing putative promoter regions of seven other cag genes) did not exhibit any significant increase in UreB expression following attachment to HEp-2 cells. A recent report, which describes the use of a high-throughput approach to analyze the transcriptome of H. pylori, verified that a transcriptional start site is present upstream from cagP, but a transcriptional start site was not identified immediately upstream from cagG (Sharma et al., 2010). Instead, it was reported that cagG is transcribed within an operon that originates further upstream (with cagC as the first gene), or as part of a suboperon (with cagF as the first gene) (Sharma et al., 2010). Multiple genes within these operons might be upregulated upon attachment of H. pylori to epithelial cells.
Macroarray hybridization methods also have been used to detect changes in H. pylori gene transcription following bacterial attachment to AGS gastric epithelial cells (Kim et al., 2004). When H. pylori strain 69a was co-cultured with these cells, the transcription of 22 H. pylori genes was upregulated and transcription of 21 genes was downregulated. Regulation of a subset of these genes was confirmed by PCR-based methods. The list of genes confirmed to be regulated included one in the cag PAI (cag3), two encoding outer membrane proteins (omp6 and omp11), and genes encoding proteins involved in chemotaxis and motility (flaA and flgB), transport and binding functions (tonB), metabolic functions (sodB), and transcription- and translation-related functions (Kim et al., 2004).
Another study used quantitative RT-PCR to compare transcription levels of five H. pylori genes following infection of AGS cells with eight different H. pylori strains (Gieseler et al., 2005). Changes in gene expression in individual strains were reproducible, but many results were strain-specific. For example, upon infection of AGS cells, cagA mRNA was upregulated in one strain and downregulated in two other strains (Gieseler et al., 2005). Similar variation was also observed in the transcriptional regulation of katA (a catalase important for oxidative defense), napA (neutrophil activating protein), ureA (a urease subunit), and vacA. Therefore, there may be variation among H. pylori strains in the genes that are differentially expressed following contact with gastric epithelial cells.
Collectively, these reports provide evidence that H. pylori contact with gastric epithelial cells leads to alterations in bacterial gene transcription. Notably, there is very little overlap among the genes that have been identified. Some of the variability may be attributed to variation in study design. Differences in the choice of bacterial strains or cell lines also contribute to the variability. Despite the considerable variability in results, some cohesive elements can be found among these reports. For example, genes encoding virulence factors (including vacA and genes in the cag PAI) have been identified in several studies (Joyce et al., 2001; van Amsterdam et al., 2003; Boonjakuakul et al., 2004, 2005; Kim et al., 2004; Gieseler et al., 2005; Scott et al., 2007; Castillo et al., 2008). Alterations in host cells induced by these virulence factors are predicted to alter the gastric environment in a manner that is favorable for H. pylori.
At present, very little is known about the mechanisms through which contact with epithelial cells leads to alterations in H. pylori gene transcription. One possibility is that specific factors (including ions, small molecules, or peptides) are released or secreted by the epithelial cells and sensed by adherent H. pylori. Conversely, if specific factors are bound, internalized, or metabolized by the epithelial cells, there may be a reduced concentration of these factors, and the reduced concentration might be sensed by adherent H. pylori. A second possibility is that binding of H. pylori to components on the surface of host cells might trigger alterations in the bacteria. A third possibility is that epithelial cells might release factors that can be used as nutrients by H. pylori, thereby stimulating bacterial metabolism and growth. Although multiple variables, including pH and concentrations of various metals or other ions are known to have an effect on H. pylori gene transcription (Merrell et al., 2003a,b; Loh et al., 2007), thus far the factors that stimulate transcriptional alterations in adherent H. pylori have not been identified. Similarly, the signaling pathways that may be involved in mediating cell contact-induced alterations in H. pylori gene transcription have yet to be identified.
There has been considerable interest in identifying genes that are upregulated during H. pylori colonization of mammalian hosts, compared to bacterial growth in vitro (Graham et al., 2002; Boonjakuakul et al., 2004, 2005; Scott et al., 2007; Castillo et al., 2008). One approach involved the use of recombination-based in vivo expression technology (RIVET) analysis to detect H. pylori gene expression in mice (Castillo et al., 2008). Among the six identified bacterial promoters that were induced in the host compared to in vitro conditions, three were predicted to regulate genes with potential roles in host colonization. Specifically, the promoter region upstream of cagZ (which likely controls expression of three genes in the cag PAI), the promoter predicted to regulate mobA, mobB, and mobD genes (which have roles in horizontal gene transfer of plasmid DNA), and the region upstream of a vacA paralog (HP0289, which encodes an autotransporter protein of unknown function) were all found to be upregulated in the host. Analysis of deletion mutant strains revealed that cagZ, mobA, mobB, and mobD are important for H. pylori colonization of mice (Castillo et al., 2008). Another study analyzed H. pylori gene expression during infection of rhesus macaques, and reported that the expression of seven genes, including several from an operon within the cag PAI, was increased early in infection of rhesus macaques in comparison to H. pylori growth in vitro at stationary phase (Boonjakuakul et al., 2005). In contrast, the transcription of many other genes, including five genes from the cag PAI, was decreased in vivo compared to stationary phase in vitro (Boonjakuakul et al., 2005). Although the experiments with animal models provide important insight into the differential expression of H. pylori genes in vivo and in vitro, it is not possible to discern whether the observed changes in gene expression in vivo result from bacterial contact with gastric epithelial cells or from bacterial exposure to other environmental conditions.
Assembly of Pili Following H. pylori Contact with Gastric Epithelial Cells
In addition to the observed alterations in H. pylori gene transcription that occur upon bacterial contact with gastric epithelial cells, alterations in bacterial cell morphology also occur. Specifically, extracellular structures termed “pili” are formed, and extend from the surface of the bacteria to the surface of the gastric epithelial cells (Figure 1). Multiple studies have reported that these structures are produced when H. pylori is co-cultured with gastric epithelial cells, whereas the structures are produced infrequently when H. pylori is cultured in the absence of epithelial cells (Rohde et al., 2003; Kwok et al., 2007; Jimenez-Soto et al., 2009; Shaffer et al., 2011). Initial studies noted that pili were synthesized by a wild-type H. pylori strain when in contact with gastric epithelial cells, but not by a mutant strain lacking the cag PAI (Rohde et al., 2003). Several individual genes within the H. pyloricag PAI have been reported to be required for pilus formation (Rohde et al., 2003; Tanaka et al., 2003; Kwok et al., 2007; Shaffer et al., 2011). Since the cag PAI encodes components of a T4SS that translocates CagA into host cells (Fischer et al., 2001), these pili are considered to be bacterially encoded structures associated with the T4SS, rather than protrusions from the gastric epithelial cell.
Figure 1
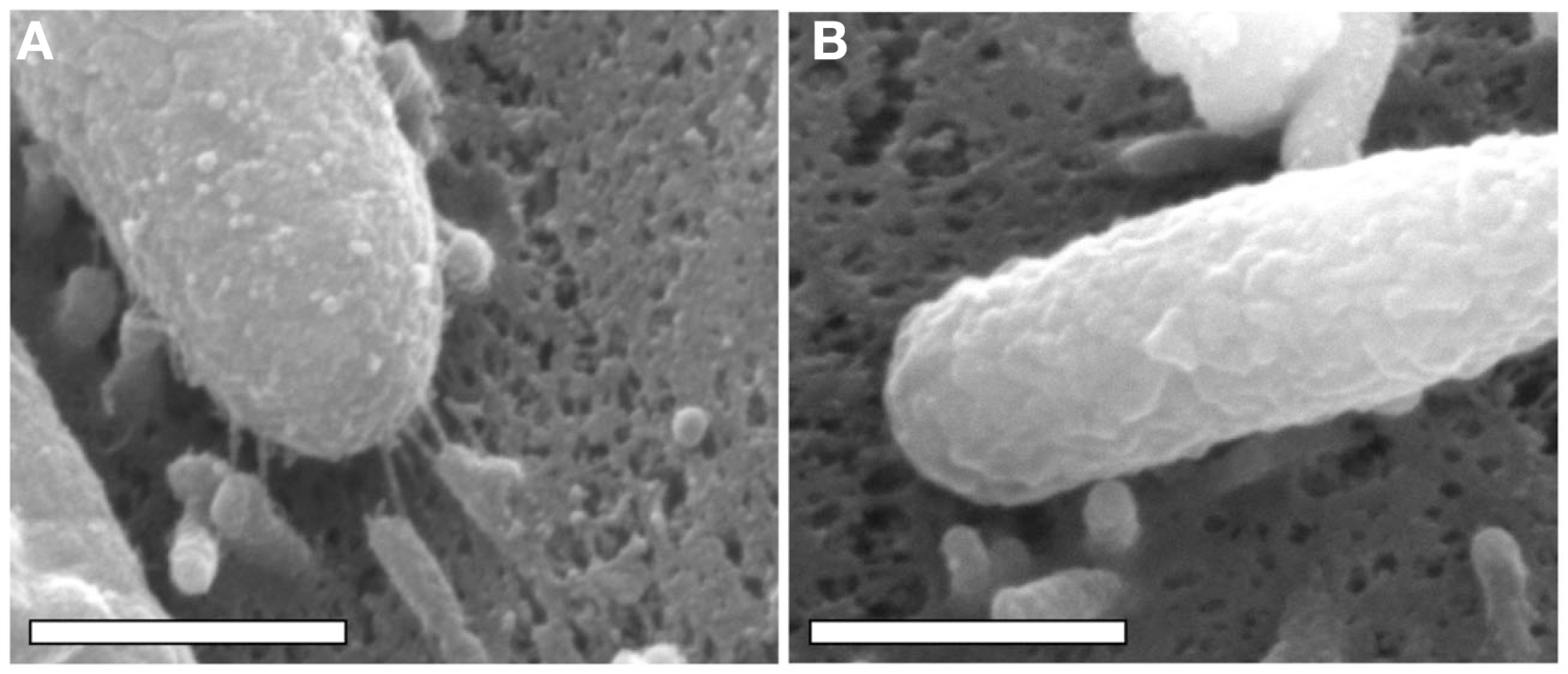
Scanning electron microscopy analysis of bacterial cells in contact with AGS human gastric epithelial cells. H. pylori strain 26695 and a mutant strain of 26695 lacking the entire cag PAI (26695 Δcag PAI) were co-cultured with AGS cells, and the cells were visualized by scanning electron microscopy, as described previously (Shaffer et al., 2011). Scanning electron micrographs of (A)H. pylori 26695 adhering to an AGS cell, (B)H. pylori 26695 Δcag PAI adhering to an AGS cell. In (A), pili are visible at the interface between the bacteria and the host cell. Magnification bars indicate 500 nm.
Similar pili are features of the T4SSs of several bacterial species, including the Agrobacterium tumefaciens VirB/VirD4 system, T4SSs of Legionella and Brucella, and T4SSs associated with plasmid-encoded conjugation systems (Frost et al., 1986, 1994; Lai and Kado, 1998, 2000; Eisenbrandt et al., 1999; Lai et al., 2000; Aly and Baron, 2007; Alvarez-Martinez and Christie, 2009; Fronzes et al., 2009). The Agrobacterium VirB/VirD4 system serves as a model for understanding T4SS assembly and architecture (Alvarez-Martinez and Christie, 2009; Fronzes et al., 2009). The pili encoded by this system, known as T-pili, are about 10 nm in diameter (Lai and Kado, 2000; Lai et al., 2000; Aly and Baron, 2007; Alvarez-Martinez and Christie, 2009; Fronzes et al., 2009), which allows them to be distinguished from other types of pili (3 nm diameter) and flagella (15 nm diameter). Plasmid-encoded conjugative pili are also 8–12 nm in diameter (Frost et al., 1986, 1994; Eisenbrandt et al., 1999; Alvarez-Martinez and Christie, 2009; Fronzes et al., 2009). Agrobacterium T-pili are comprised of a major component (VirB2) and a minor component (VirB5) that is localized to the tips of the pili (Aly and Baron, 2007; Alvarez-Martinez and Christie, 2009; Fronzes et al., 2009). VirB2 association with VirB5 is required for pilus production (Krall et al., 2002; Yuan et al., 2005), and VirB5 also controls the length of the pilus (Aly and Baron, 2007). In addition to having a role in DNA and protein translocation, these proteins may have a role in facilitating bacterial contact with host cells (Yeo et al., 2003; Hwang and Gelvin, 2004; Alvarez-Martinez and Christie, 2009). T-pilus biogenesis requires multiple virB genes, but not virD4 (Fullner et al., 1996; Lai et al., 2000; Fronzes et al., 2009).
Several Cag proteins have been detected as constituents of H. pyloricag T4SS-associated pili, using immunogold labeling and electron microscopic methods. These include CagY, CagT, CagX, and CagL, as well as the effector protein CagA (Rohde et al., 2003; Tanaka et al., 2003; Kwok et al., 2007; Jimenez-Soto et al., 2009). Immunoelectron microscopy analyses revealed that CagY is present on the sides of the pili in patches or continuous extensions, and it was proposed that CagY represents a pilus sheath (Rohde et al., 2003). CagY is characterized by numerous repeat units; variation in these repeat units may provide a means for evading host immune defenses (Liu et al., 1999; Aras et al., 2003; Delahay et al., 2008). CagT was detected at the base of the pili in one study and along the length of the pili in another study (Rohde et al., 2003; Tanaka et al., 2003). Since CagT, CagX, and CagY are homologs of T4SS components (VirB7, VirB9, and VirB10, respectively) that comprise a core complex spanning the inner and outer membranes in T4SSs of other bacterial species, it is somewhat surprising that these proteins have been localized to the pilus in H. pylori. Each of these proteins has been localized to a H. pylori membrane fraction, and there is evidence that CagT, CagX, and CagY are constituents of a protein complex (Kutter et al., 2008); these observations suggest that these proteins might comprise a T4SS core complex in H. pylori.
Helicobacter pylori CagC exhibits weak homology to VirB2, the major structural component of Agrobacterium pili, and therefore it has been proposed that CagC may serve the same function in the H. pyloricag T4SS (Andrzejewska et al., 2006). Although CagC was detected on the surface of H. pylori (Andrzejewska et al., 2006), this protein has not been definitively localized to H. pylori pili. CagL is proposed to have a role corresponding to the VirB5 minor pilus subunit of Agrobacterium T-pili (Backert et al., 2008). CagL exhibits weak sequence homology to the VirB5 ortholog of Brucella spp., but it lacks any substantial sequence relatedness to Agrobacterium VirB5 (Backert et al., 2008). CagL contains a conserved LQxR motif at the C terminus, which is similar to the motif found in VirB5 of Brucella suis and plasmid-encoded conjugation systems (Backert et al., 2008).
The reported dimensions of H. pyloricag T4SS-associated pili vary considerably among different studies. One study reported that the structures measure about 14 nm in width (Shaffer et al., 2011), which is similar to the diameters of pili in T4SSs of other bacterial species (Alvarez-Martinez and Christie, 2009; Fronzes et al., 2009). Another study reported that the structures were either 45 or 70 nm in width, depending on whether or not a sheath was present (Rohde et al., 2003). The difference in reported dimensions among studies is not attributable to a difference in H. pylori strains, since H. pylori strain 26695 was used in all of these studies. Potentially the discrepancy in dimensions is attributable to differences in electron microscopy methods. For example, in some studies, images of the pili were generated using a method in which the structures were coated with carbon, whereas in other studies, images were generated using a method in which the structures were coated with a relatively thinner layer of gold (Rohde et al., 2003; Kwok et al., 2007; Jimenez-Soto et al., 2009; Shaffer et al., 2011). Another factor that may help to account for different results among the studies is that the bacteria were not consistently cultured under the same conditions. Some studies visualized exclusively structures that were present when H. pylori was co-cultured with gastric epithelial cells, whereas other studies also visualized structures that were present when the bacteria were cultured in the absence of epithelial cells.
While multiple lines of evidence indicate that the assembly of H. pylori pili requires genes in the cag PAI (Rohde et al., 2003; Kwok et al., 2007; Shaffer et al., 2011), the composition of these pili remains incompletely characterized and the complete set of genes required for pilus biogenesis has not been defined. Biochemical analysis of these pili has been difficult because the assembly of these structures requires co-culture of H. pylori with gastric epithelial cells; pili are produced infrequently when H. pylori is grown in the absence of gastric epithelial cells (Rohde et al., 2003; Tanaka et al., 2003; Kwok et al., 2007; Shaffer et al., 2011). Several individual genes within the H. pylori cag PAI, including cagI, cagL, cagT, cagX, and cagY are reported to be required for biogenesis of pili (Rohde et al., 2003; Tanaka et al., 2003; Kwok et al., 2007; Shaffer et al., 2011), and these genes are also required for CagA translocation into host cells (Fischer et al., 2001; Shaffer et al., 2011). In many cases, the evidence supporting a role of these genes in pilus production was reported as “data not shown” and complemented mutant strains were not analyzed. One study reported that cagα/virB11 ATPase was required for pilus formation, and another study reported that it was not required (Tanaka et al., 2003; Kwok et al., 2007). Since at least fourteen genes encoded within the cag PAI are essential for CagA translocation (Fischer et al., 2001), it seems probable that several additional Cag proteins might be required for pilus formation. It is notable that pilus biogenesis requires at least two genes (cagI and cagL) that lack obvious homologs in T4SSs of other bacterial species (Shaffer et al., 2011), and CagL has been detected as a structural component of H. pylori pili (Kwok et al., 2007). This suggests that there are unique features of the H. pylori T4SS compared to T4SSs in other bacteria.
The dimensions of appendages on the surface of bacteria can be regulated by a variety of processes. For example, molecular rulers such as YscP of Yersinia pestis and FliK flagellar protein of Salmonella spp. control the dimensions of T3SS needles and flagella, respectively (Makishima et al., 2001; Journet et al., 2003; Mota et al., 2005). As a clue into the mechanisms by which assembly of H. pyloricag pili might be regulated, it is of interest that a ΔcagH mutant strain formed pili that were thicker and longer than the pili formed by a wild-type strain (Shaffer et al., 2011). This suggests that CagH has a role in regulating pilus dimensions. The mechanism by which CagH regulates pilus dimensions is not known, but CagH contains a flagellar hook (FlgK) domain closely related to the corresponding domain in FlgK from Burkholderia spp. (Shaffer et al., 2011). Possibly CagH has a role in terminating pilus assembly, analogous to the role of FlgK in terminating FlgK assembly. Alternatively, CagH may serve as a molecular ruler, analogous to FliK and YscP, to control the dimensions of cag T4SS pili in H. pylori.
An important finding was the discovery that CagL, a component of H. pylori pili, binds to integrins on the host cell surface (Kwok et al., 2007; Saha et al., 2010; Tegtmeyer et al., 2010; Conradi et al., 2012). Initially it was reported that CagL binding to α5β1 integrin was mediated by an RGD motif within CagL (Kwok et al., 2007), but a subsequent report suggested that this binding may occur independently of the RGD motif (Jimenez-Soto et al., 2009). In addition to CagL, several other Cag proteins, including CagI, CagY, and CagA are capable of binding β1 integrin (Jimenez-Soto et al., 2009). Integrins are primarily localized to the basolateral surfaces of polarized epithelial cells, and therefore, it is not clear how H. pylori on the apical surface of an intact gastric epithelial cell monolayer would contact integrins. One possibility is that H. pylori may alter the localization of this receptor, resembling the process by which enteropathogenic Escherichia coli perturbs the basolateral localization of β1 integrin (Muza-Moons et al., 2003). Alternatively, H. pylori may gain access to integrins by disrupting the integrity of the gastric epithelial monolayer (Amieva et al., 2003).
Since pili are formed when H. pylori is in contact with gastric epithelial cells but rarely in the absence of eukaryotic cells (Rohde et al., 2003; Kwok et al., 2007; Shaffer et al., 2011), the formation of these structures must be tightly regulated and there must be a stimulus that triggers pilus formation. Similarly, assembly of the Agrobacterium T4SS is regulated and is stimulated by factors such as acetosyringone (Stachel et al., 1986). One hypothesis is that the interaction of H. pylori with host cell integrins might provide a stimulus for pilus formation. Alternatively, other components of the plasma membrane or environmental conditions found at the plasma membrane surface may be relevant. α5β1 integrin associates with cholesterol-rich microdomains or lipid rafts (Hutton et al., 2010), and depletion of host cholesterol results in an abrogation of H. pyloricag T4SS function (Lai et al., 2008; Hutton et al., 2010). Therefore, it is possible that multiple components of lipid rafts may promote assembly of cag T4SS-associated pili.
The binding of H. pylori adhesins to receptors on host cells may also influence assembly or action of the T4SS. For example, the activity of the T4SS is enhanced by the presence of the BabA adhesin (Ishijima et al., 2011). This suggests that adherence of H. pylori to gastric epithelial cells through BabA or other adhesins might be required for pilus biogenesis.
The functional role of pili associated with the cag T4SS is not yet fully understood. One hypothesis is that the pili are directly responsible for CagA translocation. In support of this hypothesis, CagA has been detected by immunogold labeling at the tip of the pili (Kwok et al., 2007; Jimenez-Soto et al., 2009), and several H. pylori mutant strains that fail to form pili lack the ability to translocate CagA into host cells (Fischer et al., 2001; Tanaka et al., 2003; Kwok et al., 2007; Shaffer et al., 2011). If pili are directly responsible for CagA translocation into cells, it is unclear whether CagA passes through a channel within the pilus or along the outside of the pilus. Another hypothesis is that CagA translocation into cells occurs through a pilus-independent process, and the pili merely serve to anchor H. pylori to host cells. For example, when H. pylori adheres to epithelial cells, host phospholipids such as phosphatidylserine are externalized to the outer leaflet of the membrane, and this mislocalization of phosphatidylserine may facilitate the delivery of CagA into host cells through a pilus-independent process (Murata-Kamiya et al., 2010).
Helicobacter pylori stimulates production of IL-8 by epithelial cells through a cag PAI-dependent pathway (Fischer et al., 2001), but the role of pili in this process also remains poorly defined. H. pylori-induced IL-8 secretion can occur through both a CagA-dependent process and a CagA-independent process, and the latter phenomenon is attributed to intracellular entry of peptidoglycan (Viala et al., 2004; Brandt et al., 2005). Since intracellular entry of peptidoglycan is dependent on an intact cag T4SS (Viala et al., 2004), it is possible that this process may require the presence of pili. If so, it is unclear whether peptidoglycan is translocated through the pilus, or whether the pilus serves alternate functions, such as anchoring H. pylori to host cells or permeabilizing the epithelial cells to allow intracellular entry of peptidoglycan.
Formation of H. pylori Microcolonies on the Epithelial Cell Surface
Investigations of H. pylori and polarized epithelial cell systems have shown that bacterial contact with epithelial cells can stimulate growth of the bacteria. In these experiments, H. pylori preferentially attached to intercellular junctions on the apical surface of cells and then formed microcolonies at these sites (Tan et al., 2009). Time-lapse microscopy experiments indicated that the microcolonies arose as a result of binary fission (Tan et al., 2009). H. pylori cultured under the same conditions in the absence of epithelial cells were not capable of replicating.
Isogenic cagA mutants were unable to replicate on the apical surface of polarized epithelial cells, which indicates that actions of CagA are required for bacterial replication in this site (Tan et al., 2009). Interestingly, cagA mutant strains, while defective in ability to replicate on the apical cell surface, were able to replicate on the basolateral surface. Since CagA can cause alterations in cell polarity (Amieva et al., 2003), it was proposed that CagA-induced disruption of cell polarity contributes to H. pylori growth on the apical surface (Tan et al., 2009). In support of this hypothesis, disruption of cell polarity with inhibitors of atypical protein kinase C or Parb1 enabled a cagA mutant strain to grow on the surface of polarized cells (Tan et al., 2009). Apical co-infection of polarized epithelial cells with cagA mutant bacteria and wild-type bacteria did not rescue the growth defect of the cagA mutant bacteria, which suggests that cellular alterations induced by CagA may occur in a localized manner.
As a potential mechanism for the observed growth of H. pylori on the apical surface of polarized epithelial cells, it has been proposed that the bacteria can derive nutrients from the epithelial cells, and that acquisition of such nutrients permits growth of adherent bacteria. Thus far, acquisition of iron has been studied in greatest detail. One series of experiments showed that although cagA mutant bacteria were unable to grow on the apical surface of polarized epithelial cells, the addition of exogenous ferric chloride to the apical medium partially rescued this defect (Tan et al., 2011). Likewise, in comparison to wild-type bacteria, an isogenic vacA mutant strain demonstrated decreased microcolony formation on the apical surface of polarized epithelial cells, and addition of iron to the apical chamber rescued this defect (Tan et al., 2011). This leads to the hypothesis that CagA and VacA facilitate acquisition of iron from host cells. As a possible mechanism, it was shown that both CagA and VacA contribute to increased basolateral uptake and transcytosis of transferrin as a source of iron (Tan et al., 2011; Figure 2). Mislocalization of the transferrin receptor from the basolateral to the apical surface of polarized cells was also observed. Silencing of transferrin receptor expression resulted in reduced H. pylori growth on the apical surface (Tan et al., 2011). The ability of CagA to increase transferrin uptake was dependent on intact CagA EPIYA motifs, which are sites where CagA undergoes tyrosine phosphorylation within host cells. Although VacA is involved in apical mislocalization of the transferrin receptor to sites of bacterial attachment, the addition of exogenous VacA to cells did not stimulate transferrin receptor mislocalization. This suggests that the observed alterations in transferrin receptor localization are dependent on delivery of VacA by intact bacteria.
Figure 2
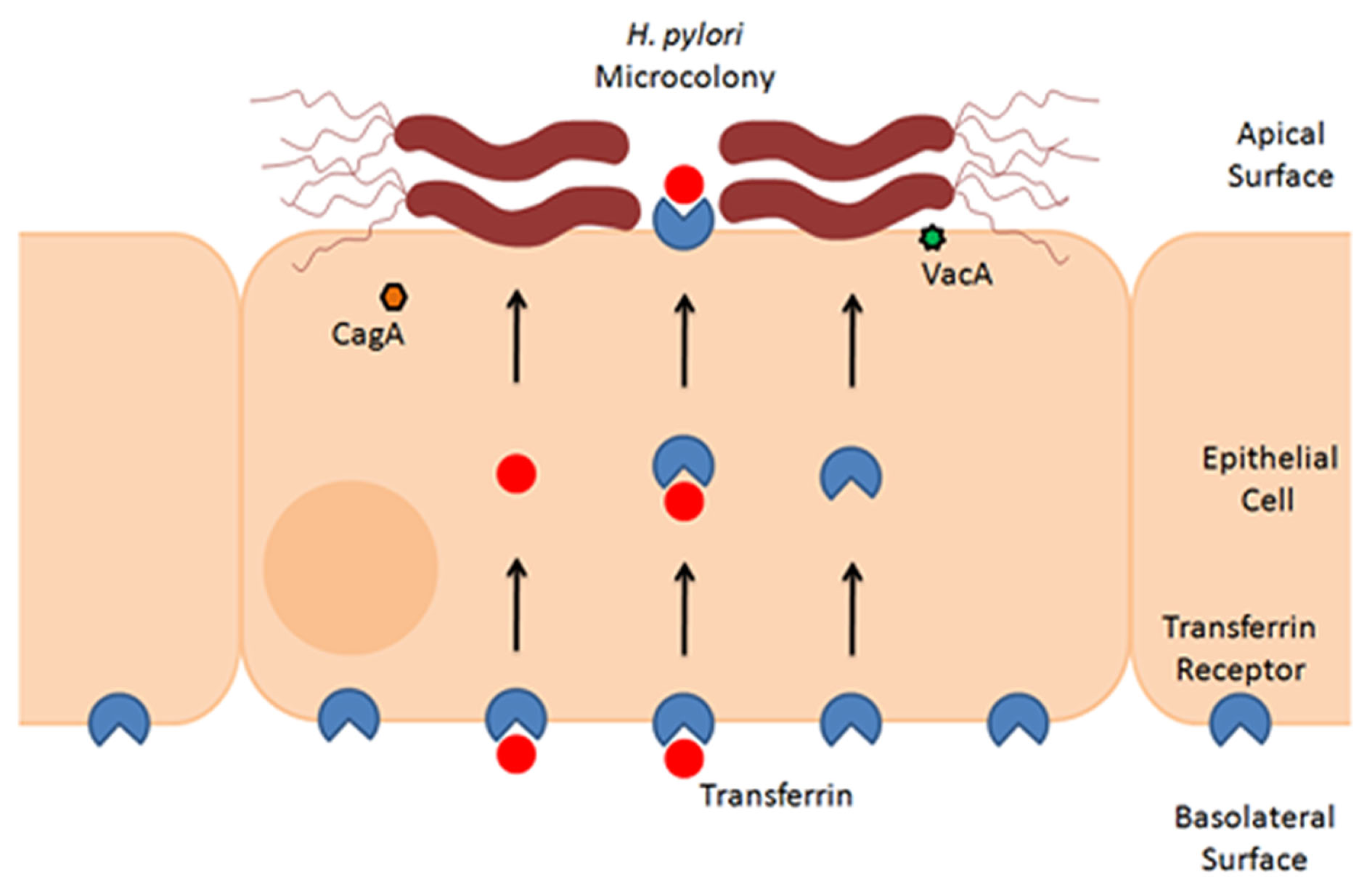
Formation of H. pylori microcolonies on the surface of epithelial cells. This schematic figure illustrates that H. pylori can form microcolonies on the surface of polarized epithelial monolayers. Microcolony formation is dependent in part on acquisition of iron from host cells, through a process that involves actions of CagA and VacA (Tan et al., 2011). Experimental studies indicate that H. pylori stimulates basolateral uptake and transcytosis of transferrin (red symbols), as well as mislocalization of the transferrin receptor (blue symbols) from the basolateral to the apical surface of polarized cells (Tan et al., 2011). Therefore, a current model proposes that H. pylori acquires iron from cells by utilization of holotransferrin (i.e., transferrin saturated with iron) (Tan et al., 2011).
These studies using polarized epithelial monolayers provide important insights into the process whereby H. pylori can obtain iron from host cells, and reveal that CagA and VacA have important roles in this process. It is notable that the addition of iron to the apical medium only partially rescued the ability of a ΔcagA mutant to grow on the apical surface of polarized cells (Tan et al., 2011). This leads to the hypothesis that the bacteria may acquire a variety of other nutrients besides iron from host cells.
Conclusion
The surface of gastric epithelial cells represents an important niche for H. pylori. The literature discussed in this review, focusing on alterations in H. pylori that occur upon bacterial contact with these cells, provides a glimpse of the complex interactions that occur at the bacteria–host cell interface. Alterations in H. pylori gene transcription upon bacterial contact with gastric epithelial cells have not yet been investigated in detail, but the finding that transcription of vacA and several cag PAI genes may be altered provides an initial framework for understanding changes that the bacteria undergo in this environment (Joyce et al., 2001; van Amsterdam et al., 2003; Boonjakuakul et al., 2004, 2005; Kim et al., 2004; Gieseler et al., 2005; Scott et al., 2007; Castillo et al., 2008). Upon contact with gastric epithelial cells, the bacteria do not only change their transcriptional profile, but also form distinct pilus-like structures. Changes in bacterial transcription and the formation of these pili probably facilitate the delivery of bacterial effector molecules, such as VacA and CagA, into host cells. These effector molecules cause cellular changes that result in an increased availability of iron and other nutrients to the bacteria. Upon uptake of these nutrients, H. pylori alters its behavior by replicating and forming microcolonies on the apical cell surface. We speculate that specific components on the surface of gastric epithelial cells (or specific environmental conditions present at the host–pathogen interface) are sensed by H. pylori, and that the bacteria accordingly modulate their morphology and ultrastructure to adapt to this environment. Such alterations may optimize the ability of H. pylori to proliferate and may promote persistent colonization of the host.
Statements
Acknowledgments
This work was supported by the National Institutes of Health (R01 AI068009, AI039657, and CA116087) and the Department of Veterans Affairs.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Alvarez-Martinez C. E. Christie P. J. (2009). Biological diversity of prokaryotic type IV secretion systems. Microbiol. Mol. Biol. Rev.73, 775–808.10.1128/MMBR.00023-09
2
Aly K. A. Baron C. (2007). The VirB5 protein localizes to the T-pilus tips in Agrobacterium tumefaciens. Microbiology153, 3766–3775.10.1099/mic.0.2007/010462-0
3
Amieva M. R. El-Omar E. M. (2008). Host-bacterial interactions in Helicobacter pylori infection. Gastroenterology134, 306–323.10.1053/j.gastro.2007.11.009
4
Amieva M. R. Vogelmann R. Covacci A. Tompkins L. S. Nelson W. J. Falkow S. (2003). Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science300, 1430–1434.10.1126/science.1081919
5
Andrzejewska J. Lee S. K. Olbermann P. Lotzing N. Katzowitsch E. Linz B. Achtman M. Kado C. I. Suerbaum S. Josenhans C. (2006). Characterization of the pilin ortholog of the Helicobacter pylori type IV cag pathogenicity apparatus, a surface-associated protein expressed during infection. J. Bacteriol.188, 5865–5877.10.1128/JB.00060-06
6
Aras R. A. Fischer W. Perez-Perez G. I. Crosatti M. Ando T. Haas R. Blaser M. J. (2003). Plasticity of repetitive DNA sequences within a bacterial (Type IV) secretion system component. J. Exp. Med.198, 1349–1360.10.1084/jem.20030381
7
Atherton J. C. Blaser M. J. (2009). Coadaptation of Helicobacter pylori and humans: ancient history, modern implications. J. Clin. Invest.119, 2475–2487.10.1172/JCI38605
8
Backert S. Fronzes R. Waksman G. (2008). VirB2 and VirB5 proteins: specialized adhesins in bacterial type-IV secretion systems?Trends Microbiol.16, 409–413.10.1016/j.tim.2008.07.001
9
Backert S. Tegtmeyer N. Selbach M. (2010). The versatility of Helicobacter pylori CagA effector protein functions: the master key hypothesis. Helicobacter15, 163–176.10.1111/j.1523-5378.2010.00797.x
10
Boonjakuakul J. K. Canfield D. R. Solnick J. V. (2005). Comparison of Helicobacter pylori virulence gene expression in vitro and in the Rhesus macaque. Infect. Immun.73, 4895–4904.10.1128/IAI.73.8.4895-4904.2005
11
Boonjakuakul J. K. Syvanen M. Suryaprasad A. Bowlus C. L. Solnick J. V. (2004). Transcription profile of Helicobacter pylori in the human stomach reflects its physiology in vivo. J. Infect. Dis.190, 946–956.10.1086/423142
12
Bourzac K. M. Guillemin K. (2005). Helicobacter pylori-host cell interactions mediated by type IV secretion. Cell. Microbiol.7, 911–919.
13
Brandt S. Kwok T. Hartig R. Konig W. Backert S. (2005). NF-kappaB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc. Natl. Acad. Sci. U.S.A.102, 9300–9305.10.1073/pnas.0409873102
14
Carrick J. Lee A. Hazell S. Ralston M. Daskalopoulos G. (1989). Campylobacter pylori, duodenal ulcer, and gastric metaplasia: possible role of functional heterotopic tissue in ulcerogenesis. Gut30, 790–797.10.1136/gut.30.6.790
15
Castillo A. R. Woodruff A. J. Connolly L. E. Sause W. E. Ottemann K. M. (2008). Recombination-based in vivo expression technology identifies Helicobacter pylori genes important for host colonization. Infect. Immun.76, 5632–5644.10.1128/IAI.00627-08
16
Celli J. P. Turner B. S. Afdhal N. H. Keates S. Ghiran I. Kelly C. P. Ewoldt R. H. McKinley G. H. So P. Erramilli S. Bansil R. (2009). Helicobacter pylori moves through mucus by reducing mucin viscoelasticity. Proc. Natl. Acad. Sci. U.S.A.106, 14321–14326.10.1073/pnas.0903438106
17
Conradi J. Huber S. Gaus K. Mertink F. Royo Gracia S. Strijowski U. Backert S. Sewald N. (2012). Cyclic RGD peptides interfere with binding of the Helicobacter pylori protein CagL to integrins α(V)β (3) and α (5)β (1). Amino Acids. [Epub ahead of print].
18
Cover T. L. Blanke S. R. (2005). Helicobacter pylori VacA, a paradigm for toxin multifunctionality. Nat. Rev. Microbiol.3, 320–332.10.1038/nrmicro1095
19
Cover T. L. Blaser M. J. (2009). Helicobacter pylori in health and disease. Gastroenterology136, 1863–1873.10.1053/j.gastro.2009.01.073
20
Delahay R. M. Balkwill G. D. Bunting K. A. Edwards W. Atherton J. C. Searle M. S. (2008). The highly repetitive region of the Helicobacter pylori CagY protein comprises tandem arrays of an alpha-helical repeat module. J. Mol. Biol.377, 956–971.10.1016/j.jmb.2008.01.053
21
Dubois A. Boren T. (2007). Helicobacter pylori is invasive and it may be a facultative intracellular organism. Cell. Microbiol9, 1108–1116.10.1111/j.1462-5822.2007.00921.x
22
Eisenbrandt R. Kalkum M. Lai E. M. Lurz R. Kado C. I. Lanka E. (1999). Conjugative pili of IncP plasmids, and the Ti plasmid T pilus are composed of cyclic subunits. J. Biol. Chem.274, 22548–22555.10.1074/jbc.274.32.22548
23
Fischer W. (2011). Assembly and molecular mode of action of the Helicobacter pylori cag type IV secretion apparatus. FEBS J.278, 1203–1212.10.1111/j.1742-4658.2011.08036.x
24
Fischer W. Puls J. Buhrdorf R. Gebert B. Odenbreit S. Haas R. (2001). Systematic mutagenesis of the Helicobacter pylori cag pathogenicity island: essential genes for CagA translocation in host cells and induction of interleukin-8. Mol. Microbiol.42, 1337–1348.10.1046/j.1365-2958.2001.02714.x
25
Fronzes R. Christie P. J. Waksman G. (2009). The structural biology of type IV secretion systems. Nat. Rev. Microbiol.7, 703–714.10.1038/nrmicro2218
26
Frost L. S. Ippen-Ihler K. Skurray R. A. (1994). Analysis of the sequence and gene products of the transfer region of the F sex factor. Microbiol. Rev.58, 162–210.
27
Frost L. S. Lee J. S. Scraba D. G. Paranchych W. (1986). Two monoclonal antibodies specific for different epitopes within the amino-terminal region of F pilin. J. Bacteriol.168, 192–198.
28
Fullner K. J. Lara J. C. Nester E. W. (1996). Pilus assembly by Agrobacterium T-DNA transfer genes. Science273, 1107–1109.10.1126/science.273.5278.1107
29
Gieseler S. Konig B. Konig W. Backert S. (2005). Strain-specific expression profiles of virulence genes in Helicobacter pylori during infection of gastric epithelial cells and granulocytes. Microbes Infect. 7, 437–447.10.1016/j.micinf.2004.11.018
30
Graham J. E. Peek R. M. Jr. Krishna U. Cover T. L. (2002). Global analysis of Helicobacter pylori gene expression in human gastric mucosa. Gastroenterology123, 1637–1648.10.1053/gast.2002.36589
31
Hatakeyama M. (2004). Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat. Rev. Cancer4, 688–694.10.1038/nrc1433
32
Hazell S. L. Lee A. Brady L. Hennessy W. (1986). Campylobacter pyloridis and gastritis: association with intercellular spaces and adaptation to an environment of mucus as important factors in colonization of the gastric epithelium. J. Infect. Dis.153, 658–663.10.1093/infdis/153.4.658
33
Hessey S. J. Spencer J. Wyatt J. I. Sobala G. Rathbone B. J. Axon A. T. Dixon M. F. (1990). Bacterial adhesion and disease activity in Helicobacter associated chronic gastritis. Gut31, 134–138.10.1136/gut.31.2.134
34
Hutton M. L. Kaparakis-Liaskos M. Turner L. Cardona A. Kwok T. Ferrero R. L. (2010). Helicobacter pylori exploits cholesterol-rich microdomains for induction of NF-kappaB-dependent responses and peptidoglycan delivery in epithelial cells. Infect. Immun. 78, 4523–4531.10.1128/IAI.00439-10
35
Hwang H. H. Gelvin S. B. (2004). Plant proteins that interact with VirB2, the Agrobacterium tumefaciens pilin protein, mediate plant transformation. Plant Cell16, 3148–3167.10.1105/tpc.104.026476
36
Ishijima N. Suzuki M. Ashida H. Ichikawa Y. Kanegae Y. Saito I. Boren T. Haas R. Sasakawa C. Mimuro H. (2011). BabA-mediated adherence is a potentiator of the Helicobacter pylori type IV secretion system activity. J. Biol. Chem.286, 25256–25264.10.1074/jbc.M111.233601
37
Jimenez-Soto L. F. Kutter S. Sewald X. Ertl C. Weiss E. Kapp U. Rohde M. Pirch T. Jung K. Retta S. F. Terradot L. Fischer W. Haas R. (2009). Helicobacter pylori type IV secretion apparatus exploits beta1 integrin in a novel RGD-independent manner. PLoS Pathog. 5, e1000684.10.1371/journal.ppat.1000684
38
Jones K. R. Whitmire J. M. Merrell D. S. (2010). A tale of two toxins: Helicobacter pylori CagA and VacA modulate host pathways that impact disease. Front. Microbiol.1:115.10.3389/fmicb.2010.00115
39
Journet L. Agrain C. Broz P. Cornelis G. R. (2003). The needle length of bacterial injectisomes is determined by a molecular ruler. Science302, 1757–1760.10.1126/science.1091422
40
Joyce E. A. Gilbert J. V. Eaton K. A. Plaut A. Wright A. (2001). Differential gene expression from two transcriptional units in the cag pathogenicity island of Helicobacter pylori. Infect. Immun. 69, 4202–4209.10.1128/IAI.69.7.4202-4209.2001
41
Kim N. Marcus E. A. Wen Y. Weeks D. L. Scott D. R. Jung H. C. Song I. S. Sachs G. (2004). Genes of Helicobacter pylori regulated by attachment to AGS cells. Infect. Immun.72, 2358–2368.10.1128/IAI.72.12.7374-7378.2004
42
Krall L. Wiedemann U. Unsin G. Weiss S. Domke N. Baron C. (2002). Detergent extraction identifies different VirB protein subassemblies of the type IV secretion machinery in the membranes of Agrobacterium tumefaciens. Proc. Natl. Acad. Sci. U.S.A.99, 11405–11410.10.1073/pnas.172390699
43
Kutter S. Buhrdorf R. Haas J. Schneider-Brachert W. Haas R. Fischer W. (2008). Protein subassemblies of the Helicobacter pylori cag type IV secretion system revealed by localization and interaction studies. J. Bacteriol.190, 2161–2171.10.1128/JB.01341-07
44
Kwok T. Zabler D. Urman S. Rohde M. Hartig R. Wessler S. Misselwitz R. Berger J. Sewald N. Konig W. Backert S. (2007). Helicobacter exploits integrin for type IV secretion and kinase activation. Nature449, 862–866.10.1038/nature06187
45
Lai C. H. Chang Y. C. Du S. Y. Wang H. J. Kuo C. H. Fang S. H. Fu H. W. Lin H. H. Chiang A. S. Wang W. C. (2008). Cholesterol depletion reduces Helicobacter pylori CagA translocation and CagA-induced responses in AGS cells. Infect. Immun.76, 3293–3303.10.1128/IAI.00365-08
46
Lai E. M. Chesnokova O. Banta L. M. Kado C. I. (2000). Genetic and environmental factors affecting T-pilin export and T-pilus biogenesis in relation to flagellation of Agrobacterium tumefaciens. J. Bacteriol.182, 3705–3716.10.1128/JB.182.2.327-336.2000
47
Lai E. M. Kado C. I. (1998). Processed VirB2 is the major subunit of the promiscuous pilus of Agrobacterium tumefaciens. J. Bacteriol.180, 2711–2717.
48
Lai E. M. Kado C. I. (2000). The T-pilus of Agrobacterium tumefaciens. Trends Microbiol.8, 361–369.10.1016/S0966-842X(00)01802-3
49
Liu G. McDaniel T. K. Falkow S. Karlin S. (1999). Sequence anomalies in the Cag7 gene of the Helicobacter pylori pathogenicity island. Proc. Natl. Acad. Sci. U.S.A.96, 7011–7016.10.1073/pnas.96.26.14694
50
Loh J. T. Torres V. J. Cover T. L. (2007). Regulation of Helicobacter pylori cagA expression in response to salt. Cancer Res.67, 4709–4715.10.1158/0008-5472.CAN-06-4746
51
Makishima S. Komoriya K. Yamaguchi S. Aizawa S. I. (2001). Length of the flagellar hook and the capacity of the type III export apparatus. Science291, 2411–2413.10.1126/science.1058366
52
Merrell D. S. Goodrich M. L. Otto G. Tompkins L. S. Falkow S. (2003a). pH-regulated gene expression of the gastric pathogen Helicobacter pylori. Infect. Immun. 71, 3529–3539.10.1128/IAI.71.6.3529-3539.2003
53
Merrell D. S. Thompson L. J. Kim C. C. Mitchell H. Tompkins L. S. Lee A. Falkow S. (2003b). Growth phase-dependent response of Helicobacter pylori to iron starvation. Infect. Immun.71, 6510–6525.s10.1128/IAI.71.6.3529-3539.2003
54
Montecucco C. de Bernard M. (2003). Molecular and cellular mechanisms of action of the vacuolating cytotoxin (VacA) and neutrophil-activating protein (HP-NAP) virulence factors of Helicobacter pylori. Microbes Infect. 5, 715–721.10.1016/S1286-4579(03)00124-2
55
Mota L. J. Journet L. Sorg I. Agrain C. Cornelis G. R. (2005). Bacterial injectisomes: needle length does matter. Science307, 1278.10.1126/science.1107679
56
Murata-Kamiya N. Kikuchi K. Hayashi T. Higashi H. Hatakeyama M. (2010). Helicobacter pylori exploits host membrane phosphatidylserine for delivery, localization, and pathophysiological action of the CagA oncoprotein. Cell Host Microbe7, 399–411.10.1016/j.chom.2010.04.005
57
Muza-Moons M. M. Koutsouris A. Hecht G. (2003). Disruption of cell polarity by enteropathogenic Escherichia coli enables basolateral membrane proteins to migrate apically and to potentiate physiological consequences. Infect. Immun.71, 7069–7078.10.1128/IAI.71.12.7069-7078.2003
58
Olbermann P. Josenhans C. Moodley Y. Uhr M. Stamer C. Vauterin M. Suerbaum S. Achtman M. Linz B. (2010). A global overview of the genetic and functional diversity in the Helicobacter pylori cag pathogenicity island. PLoS Genet. 6, e1001069.10.1371/journal.pgen.1001069
59
Rieder G. Fischer W. Haas R. (2005). Interaction of Helicobacter pylori with host cells: function of secreted and translocated molecules. Curr. Opin. Microbiol.8, 67–73.10.1016/j.mib.2004.12.004
60
Rohde M. Puls J. Buhrdorf R. Fischer W. Haas R. (2003). A novel sheathed surface organelle of the Helicobacter pylori cag type IV secretion system. Mol. Microbiol.49, 219–234.10.1046/j.1365-2958.2003.03549.x
61
Saha A. Backert S. Hammond C. E. Gooz M. Smolka A. J. (2010). Helicobacter pylori CagL activates ADAM17 to induce repression of the gastric H, K-ATPase alpha subunit. Gastroenterology139, 239–248.10.1053/j.gastro.2010.03.036
62
Schreiber S. Konradt M. Groll C. Scheid P. Hanauer G. Werling H. O. Josenhans C. Suerbaum S. (2004). The spatial orientation of Helicobacter pylori in the gastric mucus. Proc. Natl. Acad. Sci. U.S.A.101, 5024–5029.10.1073/pnas.0308686101
63
Scott D. R. Marcus E. A. Wen Y. Oh J. Sachs G. (2007). Gene expression in vivo shows that Helicobacter pylori colonizes an acidic niche on the gastric surface. Proc. Natl. Acad. Sci. U.S.A.104, 7235–7240.10.1073/pnas.0610468104
64
Shaffer C. L. Gaddy J. A. Loh J. T. Johnson E. M. Hill S. Hennig E. E. McClain M. S. McDonald W. H. Cover T. L. (2011). Helicobacter pylori exploits a unique repertoire of type IV secretion system components for pilus assembly at the bacteria-host cell interface. PLoS Pathog. 7, e1002237.10.1371/journal.ppat.1002237
65
Sharma C. M. Hoffmann S. Darfeuille F. Reignier J. Findeiss S. Sittka A. Chabas S. Reiche K. Hackermuller J. Reinhardt R. Stadler P. F. Vogel J. (2010). The primary transcriptome of the major human pathogen Helicobacter pylori. Nature464, 250–255.10.1038/nature08756
66
Stachel S. E. Nester E. W. Zambryski P. C. (1986). A plant cell factor induces Agrobacterium tumefaciens vir gene expression. Proc. Natl. Acad. Sci. U.S.A.83, 379–383.10.1073/pnas.83.2.379
67
Tan S. Noto J. M. Romero-Gallo J. Peek R. M. Jr. Amieva M. R. (2011). Helicobacter pylori perturbs iron trafficking in the epithelium to grow on the cell surface. PLoS Pathog. 7, e1002050.10.1371/journal.ppat.1002050
68
Tan S. Tompkins L. S. Amieva M. R. (2009). Helicobacter pylori usurps cell polarity to turn the cell surface into a replicative niche. PLoS Pathog. 5, e1000407.10.1371/journal.ppat.1000407
69
Tanaka J. Suzuki T. Mimuro H. Sasakawa C. (2003). Structural definition on the surface of Helicobacter pylori type IV secretion apparatus. Cell. Microbiol.5, 395–404.10.1046/j.1462-5822.2003.00286.x
70
Tegtmeyer N. Hartig R. Delahay R. M. Rohde M. Brandt S. Conradi J. Takahashi S. Smolka A. J. Sewald N. Backert S. (2010). A small fibronectin-mimicking protein from bacteria induces cell spreading and focal adhesion formation. J. Biol. Chem.285, 23515–23526.10.1074/jbc.M109.096214
71
Tegtmeyer N. Wessler S. Backert S. (2011). Role of the cag-pathogenicity island encoded type IV secretion system in Helicobacter pylori pathogenesis. FEBS J.278, 1190–1202.10.1111/j.1742-4658.2011.08035.x
72
Terradot L. Waksman G. (2011). Architecture of the Helicobacter pylori Cag-type IV secretion system. FEBS J.278, 1213–1222.10.1111/j.1742-4658.2011.08037.x
73
van Amsterdam K. van Vliet A. H. Kusters J. G. Feller M. Dankert J. van der Ende A. (2003). Induced Helicobacter pylori vacuolating cytotoxin VacA expression after initial colonisation of human gastric epithelial cells. FEMS Immunol. Med. Microbiol.39, 251–256.10.1016/S0928-8244(03)00226-8
74
Viala J. Chaput C. Boneca I. G. Cardona A. Girardin S. E. Moran A. P. Athman R. Memet S. Huerre M. R. Coyle A. J. DiStefano P. S. Sansonetti P. J. Labigne A. Bertin J. Philpott D. J. Ferrero R. L. (2004). Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat. Immunol.5, 1166–1174.10.1038/ni1131
75
Wyatt J. I. Rathbone B. J. Dixon M. F. Heatley R. V. (1987). Campylobacter pyloridis and acid induced gastric metaplasia in the pathogenesis of duodenitis. J. Clin. Pathol.40, 841–848.10.1136/jcp.40.8.841
76
Wyatt J. I. Rathbone B. J. Sobala G. M. Shallcross T. Heatley R. V. Axon A. T. Dixon M. F. (1990). Gastric epithelium in the duodenum: its association with Helicobacter pylori and inflammation. J. Clin. Pathol.43, 981–986.10.1136/jcp.43.12.981
77
Yeo H. J. Yuan Q. Beck M. R. Baron C. Waksman G. (2003). Structural and functional characterization of the VirB5 protein from the type IV secretion system encoded by the conjugative plasmid pKM101. Proc. Natl. Acad. Sci. U.S.A.100, 15947–15952.10.1073/pnas.2535211100
78
Yuan Q. Carle A. Gao C. Sivanesan D. Aly K. A. Hoppner C. Krall L. Domke N. Baron C. (2005). Identification of the VirB4-VirB8-VirB5-VirB2 pilus assembly sequence of type IV secretion systems. J. Biol. Chem.280, 26349–26359.10.1074/jbc.M502347200
Summary
Keywords
Helicobacter pylori , VacA, CagA, type IV secretion, cag pathogenicity island, iron, gastric cancer
Citation
Johnson EM, Gaddy JA and Cover TL (2012) Alterations in Helicobacter pylori Triggered by Contact with Gastric Epithelial Cells. Front. Cell. Inf. Microbio. 2:17. doi: 10.3389/fcimb.2012.00017
Received
10 December 2011
Accepted
06 February 2012
Published
28 February 2012
Volume
2 - 2012
Edited by
D. Scott Merrell, Uniformed Services University, USA
Reviewed by
Robert Maier, University of Georgia, USA; Jay V. Solnick, University of California Davis, USA
Copyright
© 2012 Johnson, Gaddy and Cover.
This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Timothy L. Cover, Division of Infectious Diseases, Vanderbilt University School of Medicine, A2200 Medical Center North, Nashville, TN 37232, USA. e-mail: timothy.l.cover@vanderbilt.edu
†Elizabeth M. Johnson and Jennifer A. Gaddy have contributed equally to this work.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.