Abstract
Dandruff is a prevalent chronic inflammatory skin condition of the scalp that has been associated with Malassezia yeasts. However, the microbial role has not been elucidated yet, and the etiology of the disorder remains poorly understood. Using high-throughput 16S rDNA and ITS1 sequencing, we characterized cutaneous bacterial and fungal microbiotas from healthy and dandruff subjects, comparing scalp and forehead (lesional and non-lesional skin sites). Bacterial and fungal communities from dandruff analyzed at genus level differed in comparison with healthy ones, presenting higher diversity and greater intragroup variation. The microbial shift was observed also in non-lesional sites from dandruff subjects, suggesting that dandruff is related to a systemic process that is not restricted to the site exhibiting clinical symptoms. In contrast, Malassezia microbiota analyzed at species level did not differ according to health status. A 2-step OTU assignment using combined databases substantially increased fungal assigned sequences, and revealed the presence of highly prevalent uncharacterized Malassezia organisms (>37% of the reads). Although clinical symptoms of dandruff manifest locally, microbial dysbiosis beyond clinically affected skin sites suggests that subjects undergo systemic alterations, which could be considered for redefining therapeutic approaches.
Introduction
Human skin is a complex ecosystem inhabited by a variety of microorganisms, including bacteria, fungi, archaea, and viruses (Kong, 2011). Our knowledge of the skin microbiome has increased substantially in recent years, driven by advances in sequencing technologies and in bioinformatics (Tomic-Canic et al., 2014). These approaches have shown interpersonal, topographical, and temporal variations of microbial communities (Grice et al., 2009; Caporaso et al., 2011; Findley et al., 2013; Oh et al., 2014, 2016).
Skin microbiome has positive impact on several aspects of human health, such as immune response modulation and protection against pathogens (Wanke et al., 2011; Naik et al., 2015). Microorganisms interact with host keratinocytes and innate immune system, stimulating the secretion of antimicrobial peptides, free fatty acids, cytokines and chemokines, which might lead to adaptive immune responses (Gallo and Nakatsuji, 2011; Fyhrquist et al., 2016).
Microbial communities from skin have also been associated with the development of skin disorders (Grice, 2014). Dandruff is one of the most common skin conditions, affecting approximately half of adult population worldwide (Piérard-Franchimont et al., 2006a). This inflammatory chronic disorder is related to skin barrier disruption, epidermal cellular proliferation and differentiation, as well as shifts in gene expression patterns, and in cytokine and lipid production (Kerr et al., 2011; Mills et al., 2012; Bonnist et al., 2014). It is characterized by erythema, itching and scaling on scalp (Schwartz et al., 2013), and is also related to alopecia (Piérard-Franchimont et al., 2006b). Dandruff also has social and psychological impact, affecting self-esteem, and confidence (Manuel and Ranganathan, 2011). It has been frequently associated with yeasts from Malassezia genus, which are also members of the healthy cutaneous microbiome (Saunders et al., 2012). However, the role Malassezia organisms play in the development of the symptoms has not been elucidated and the etiology of dandruff remains poorly understood.
We used next-generation sequencing (NGS) to analyze bacterial and fungal microbiota associated with skin from healthy and dandruff subjects. The comparison between lesional and non-lesional skin sites from dandruff subjects provided new perspectives for the understanding of this skin disorder, establishing steps toward a broader view of dandruff etiology and the role of the microbiome in the symptom development.
Materials and methods
Subjects and sample collection
The research protocol was approved by the UFABC Institutional Review Board (Protocol 732.172) and was conducted according to the principles expressed in the World Medical Association Declaration of Helsinki. All subjects provided written informed consent prior to any study-related procedures.
Thirteen patients with dandruff and 11 healthy subjects were enrolled, with the collaboration of “Instituto Superior de Medicina e Dermatologia—ISMD” (São Paulo, SP, Brazil). Volunteers were individuals from both genders, ages between 18 and 61 years old (Supplementary Table 1). All participants provided information regarding health status, medical history, and daily habits. Volunteers were non-smokers, did not have cutaneous diseases except dandruff, did not receive antibiotics or systemic antifungals 1 month prior to sampling, and did not use anti-dandruff shampoos and chemical products (dyeing, bleaching, permanent waving, straightening etc.) on scalp and hair at least 2 weeks prior to sampling.
Volunteers were asked to use a neutral shampoo 3 times a week during 2 weeks prior to sampling to standardize the scalp condition. They were advised not to wash their scalp 2 days before the sampling procedure, and not to use other hair products on the scalp (Clavaud et al., 2013).
Dandruff severity was measured as previously described (Clavaud et al., 2013). Scalp was divided into eight sections and dandruff scores ranging from 0 to 5 were assigned to each area by comparison with reference pictures. The values were averaged to obtain the final score. Samples from scalp (vertex of the head) and forehead (central area) were obtained using sterile cotton swabs soaked in a solution containing 0.15M NaCl and 0.1% Tween 20, as previously described (Paulino et al., 2006). Forehead from dandruff subjects did not show any sign of desquamation or inflammation. Swabs were placed in microcentrifuge tubes, transported in dry ice, and stored at −80°C. Cotton swabs with no skin contact submitted to the same procedures were used as negative controls.
DNA extraction, PCR amplifications, and high-throughput sequencing
Total genomic DNA was extracted from the swabs by using Power Soil DNA Isolation Kit (MOBIO Laboratories Inc., Carlsbad, CA, USA) according to the manufacturer's instructions. The head of each swab was cut from the handle and placed into a tube provided by the kit, which contained beads to efficiently disrupt cell walls.
Primers 520F (Claesson et al., 2009) (5′-AYTGGGYDTAAAGNG-3′) and 907R (Lane et al., 1985) (5′-CCGTCAATTCMTTTRA-3′) were used to amplify by PCR a fragment containing the V4 hypervariable region of the 16S rRNA gene from bacteria. For fungi 18S-F/5.8S-1R ITS1 primers were used (Findley et al., 2013). Sequencing was performed using MiSeq Illumina Platform with Illumina paired-end MiSeq Reagent Kit v2 (2 × 250) following Illumina's standard protocol. Only reads obtained with forward primers were considered in the analyses as they showed higher sequencing quality. Single reads were submitted to size filtering using Seqyclean software (https://bitbucket.org/izhbannikov/seqyclean). Sequences shorter than 200 bp were excluded from the analyses. Quality filtering was done using Phred quality score (≥20; Ewing et al., 1998). Size and quality filtering were performed using Qiime pipeline (Caporaso et al., 2010).
The dataset has been deposited in the EBI Metagenomics database (project number PRJEB16723).
Taxonomic assignment
Bacterial taxonomic assignment was performed considering 97% identity and 95% coverage by comparing the sequences with Greengenes database v 13_8 (http://greengenes.lbl.gov/) using Uclust software (Edgar, 2010), implemented by Qiime. Fungal community analyses were done at genus level through BLAST (Altschul et al., 1990) using QIIME against a database of ITS1 reference sequences manually curated from UNITE database v 2015-03-02 (https://unite.ut.ee/), with 70% similarity threshold. Singletons were removed from the analysis. A database of Malassezia sequences were constructed using all the sequences from Malassezia organisms in the UNITE database that were manually curated (reference sequences—Kõljalg et al., 2013), combined with Genbank sequences presenting ≥97% similarity and ≥95% coverage in relation to those selected from the Unite database. Analyses at species level for Malassezia organisms were performed considering 97% similarity threshold and ≥95% coverage. Fungal unassigned sequences were clustered using CD-HIT (Li and Godzik, 2006; 97% similarity threshold), and clusters with relative abundance ≥1% in at least one sample were compared through BLAST against the complete Genbank database. This step of analysis also considered three Malassezia species described recently: M. brasiliensis, M. pscittasci (Cabañes et al., 2016) and M. arunalokei (Honnavar et al., 2016), which were not included in the Malassezia sequence database built with UNITE and Genbank.
Statistical analysis
Non-metrical Multidimensional Scaling (nmMDS) using Bray-Curtis similarity distances (Bray and Curtis, 1957) based on Log (X+1) transformed data were performed to assess the relationships between bacterial communities from different samples. Analysis of Similarities (ANOSIM; Clarke, 1993) was applied to verify differences based on body site and health condition. ANOSIM global R value ranges from 1 to −1 (R ~ 0 indicates the same level of variation within and between groups). ANOSIM test was performed using α = 0.05 for statistical significance. Average similarity within groups was calculated using SIMPER (Clarke, 1993). Diversity was measured using Shannon-Weaver diversity index. These analyses were performed using Primer 6 (Clarke and Gorley, 2006). To estimate depth of sequence sampling, rarefaction curves were obtained for each sample using PAST 3.03 (Hammer et al., 2001) and EstimateS 9.1.0 (Colwell, 2005). For mean comparisons two-way ANOVA test was performed using two-tailed p-values and α = 0.05. Linear discriminant analysis (LDA) effect size tool-LEfSe (Segata et al., 2011) was used to identify Operational Taxonomical Units (OTUs) with differential relative abundance comparing healthy and dandruff subject samples from each body site. For Kruskal-Wallis test, α = 0.05 was used as a cut-off for statistical significance. LDA score was calculated for OTUs with p ≤ 0.05 (LDA score threshold ≥2.00). Differentially abundant OTUs were used to calculate the microbial dysbiosis index (MD-index), defined as the log of (total abundance of OTUs increased in dandruff) over (total abundance of OTUs decreased in dandruff) (Gevers et al., 2014).
Results
Data collection and sequence analysis
We analyzed bacterial and fungal communities in 48 skin samples from 11 healthy and 13 dandruff individuals. Scalp and forehead samples were studied to assess variation across two sebaceous body areas and to enable comparison between lesional and non-lesional skin sites in dandruff subjects.
In silico analyses were performed to select primer sets for bacteria and fungi considering specificity and coverage. Amplicons containing the V4 hypervariable region of 16S rRNA gene from bacteria and the ITS1 region from fungi were sequenced using Illumina MiSeq platform.
We obtained ~2.3 million reads for bacteria and 1.4 million for fungi after quality and size filtering (Supplementary Table 2). Approximately 88% of the bacterial sequences were assigned using Greengenes database, and 612 Operational Taxonomic Units (OTUs) were found. Chloroplast and mitochondria sequences (nearly 8% of the reads) were removed from the analysis. For fungi, only approximately half of the sequences (51.4%) were assigned using UNITE database. Therefore, we performed a second step of analysis consisting of sequence clustering and assignment using GenBank to allow a larger and more reliable panorama of the fungal communities. This strategy increased the percentage of assigned sequences (>83%). A total of 274 fungal OTUs were found. Moreover, the method allowed the detection of contaminant sequences assigned to non-fungal taxonomic groups (7.8% of the reads). They correspond to plants and were removed from the analyses. Rarefaction curves were generated to estimate depth of sequence sampling (Supplementary Figures 1, 2).
Taxonomic composition of microbial communities
Actinobacteria, Firmicutes, and Proteobacteria were the most abundant bacterial phyla. At genus level, Propionibacterium, Staphylococcus, and Corynebacterium were found to be the three most abundant genera in both healthy and dandruff subjects (Figure 1A). Malassezia sp. comprised the vast majority of fungi in almost all of the samples (~96% of the reads; Figure 1B); therefore, we also performed taxonomic assignment at Malassezia species level. Nine Malassezia formally described species were detected (M. restricta, M. globosa, M. sympodialis, M. dermatis, M. japonica, M. obtusa, M. pachydermatis, M. sloofiae, and M. furfur), as well as an uncharacterized phylotype previously described (LCP-2008a—Genbank accession number EU192362). M. restricta was the most abundant species. After the 2-step OTU assignment for fungi, other uncharacterized Malassezia organisms were found, corresponding to more than 37% of the reads. Their sequences are dissimilar from all Malassezia type strains, thus were not assigned to any formally described species. The uncharacterized Malassezia sequences formed three subgroups (≥95% sequence similarity), two of them being highly abundant: Malassezia sp. subgroup 1 was found in all 48 samples, comprising 26.72% of the sequences; and subgroup 2 was found in 46 samples (9.6% of the sequences; Figure 1C).
Figure 1
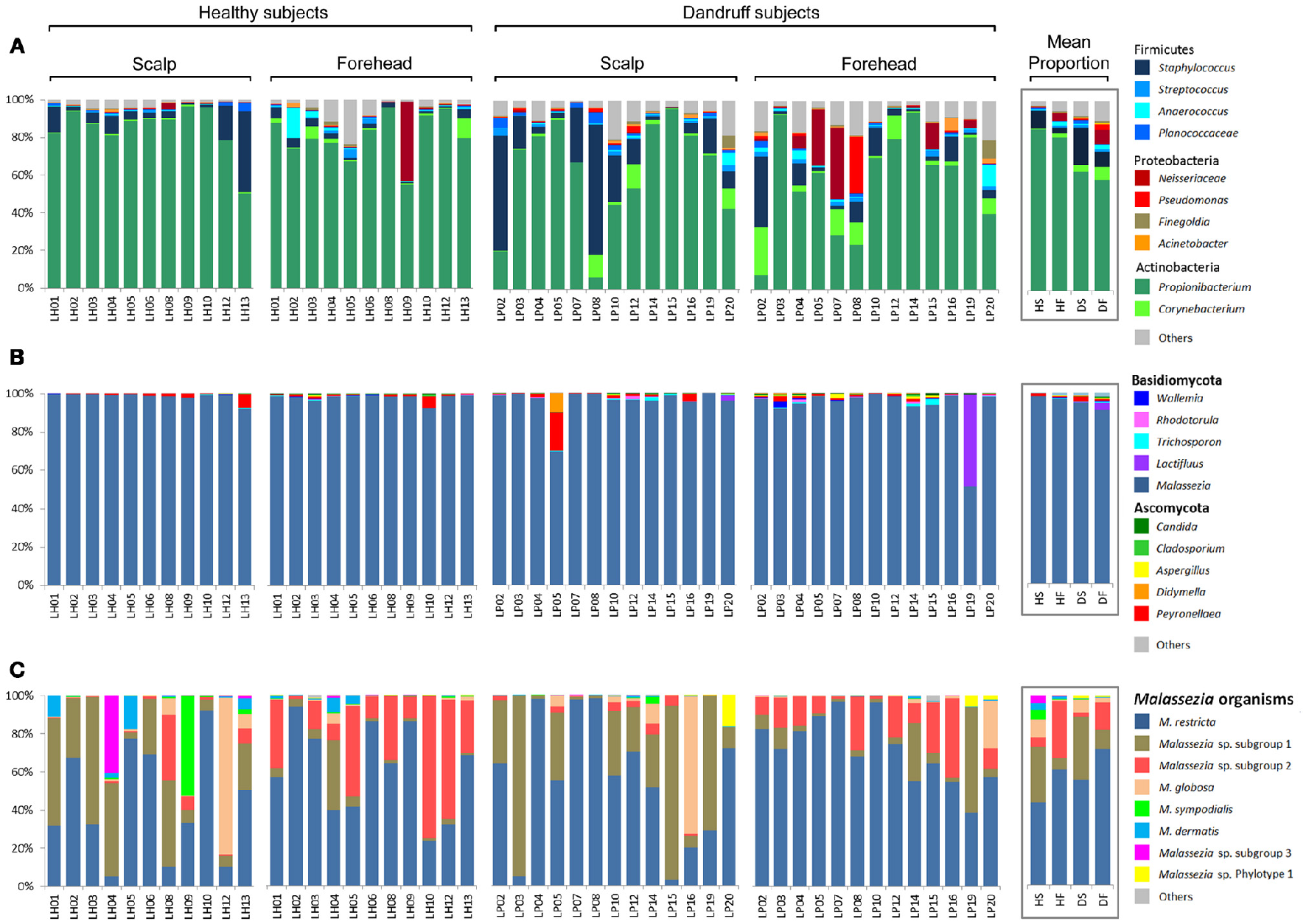
Skin microbial composition in healthy and dandruff subjects. Relative abundance of (A) Bacteria at genus level; (B) Fungi at genus level; and (C)Malassezia at species level. Mean proportions according to health status and body site are shown in the box. HS, scalp samples from healthy subjects; HF, forehead samples from healthy subjects; DS, scalp samples from dandruff subjects; DF, forehead samples from dandruff subjects.
Microbial communities in health and dandruff
Statistical analyses using non-metrical Multidimensional Scaling and ANOSIM revealed that healthy bacterial and fungal communities at genus level clustered according to body site, indicating that scalp and forehead from healthy subjects harbor distinct microbiotas (bacteria: p = 0.023, R = 0.122; fungi: p = 0.005, R = 0.188; Figures 2A,B). Within each body site group, scalp samples had higher intra-group similarity than forehead samples, as revealed by similarity percentage (SIMPER analysis) (Figures 2D,E).
Figure 2
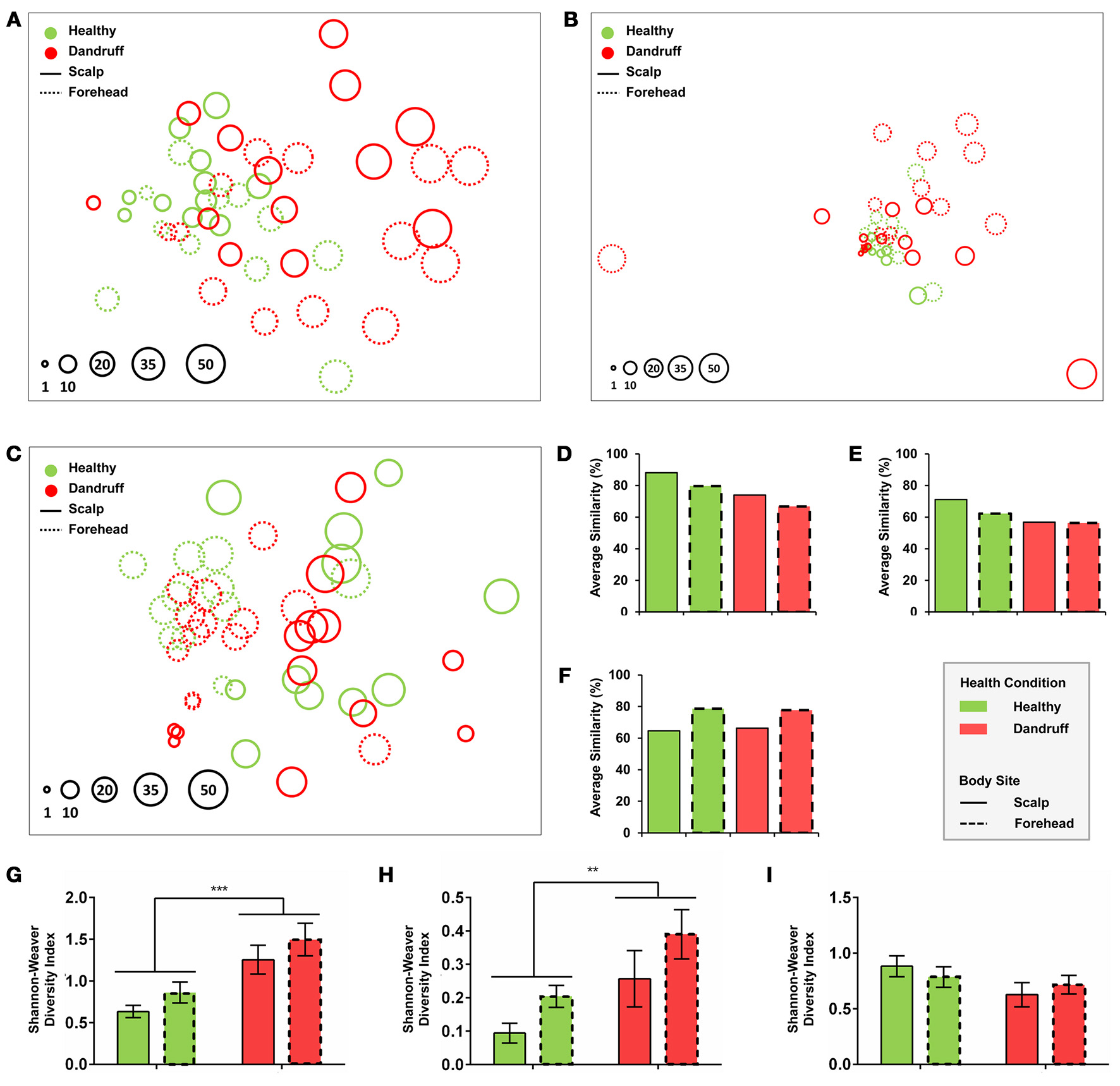
Community and diversity analyses. Non-Metrical Multidimensional Scaling (nmMDS) of (A) Bacteria at genus level, (B) Fungi at genus level, and (C)Malassezia at species level showing 48 samples from healthy and dandruff subjects. Each circle represents one sample, and diameters are proportional to the Shannon-Weaver diversity (lowest value indexed as 1). Average similarity (SIMPER) for (D) Bacteria at genus level, (E) Fungi at genus level and (F)Malassezia at species level. Mean diversity based on Shannon-Weaver Index for (G) Bacteria at genus level, (H) Fungi at genus level, and (I)Malassezia at species level. Significance was determined by Two-way ANOVA Test. Bars represent Mean ± SEM. **p < 0.005; ***p < 0.001.
Bacterial and fungal communities at genus level were more dissimilar among samples from dandruff subjects as compared with healthy subjects (Figures 2A,B,D,E). Despite the clinical differences between samples from scalp (lesional site) and forehead (non-lesional site) from dandruff subjects, the variations between the sites resembles the differences observed between scalp and forehead from healthy subjects, indicating that dandruff condition is related to dysbiosis in both clinically involved and uninvolved skin sites. Analysis using Shannon-Weaver index revealed that diversity was significantly higher in dandruff samples than in healthy ones for both bacteria (p = 0.0002; Figure 2G) and fungi (p = 0.0095; Figure 2H) at genus level. Diversity showed no correlation with number of reads from each sample from both bacteria (p = 0.4284) and fungi (p = 0.7219).
Malassezia microbiota at species level also differed between body sites in healthy subjects (p = 0.001, R = 0.393; Figure 2C). However, contrasting with genus level analyses, in dandruff subjects the average similarity among samples did not differ in relation to healthy subjects for both scalp and forehead samples (Figure 2F). Shannon-Weaver diversity indexes obtained for healthy and dandruff subjects were not statistically different either (Figure 2I), suggesting that the diversity of Malassezia community at species level was not associated with dandruff condition.
Differential abundance of bacterial and fungal taxa
We also searched for specific OTUs that could be differentially abundant between groups using Linear discriminant analysis effect size tool (LEfSe). Considering bacteria and fungi at genus level, remarkably more OTUs were found to be overrepresented in scalp and forehead samples from dandruff subjects in comparison with healthy ones (Figures 3A–D). These findings corroborate the occurrence of dysbiosis in lesional as well as non-lesional body sites. Bacterial genera Pseudomonas, Leptotrichia, Micrococcus, Selenomonas, Erwinia, Enhydrobacter, and Bartonellaceae were significantly more abundant in dandruff subject samples in both skin sites, whereas Propionibacterium was underrepresented (Figures 3A,B). In the case of fungi, dandruff samples from both sites were enriched in genera Candida, Aspergillus, and Filobasidium (Figures 3C,D). Analysis of Malassezia microbiota at species level did not reveal OTUs overrepresented in dandruff subjects, either in scalp or forehead samples. Two OTUs of low prevalence were found to be underrepresented in dandruff, one was an uncharacterized Malassezia subgroup (Figures 3E,F).
Figure 3
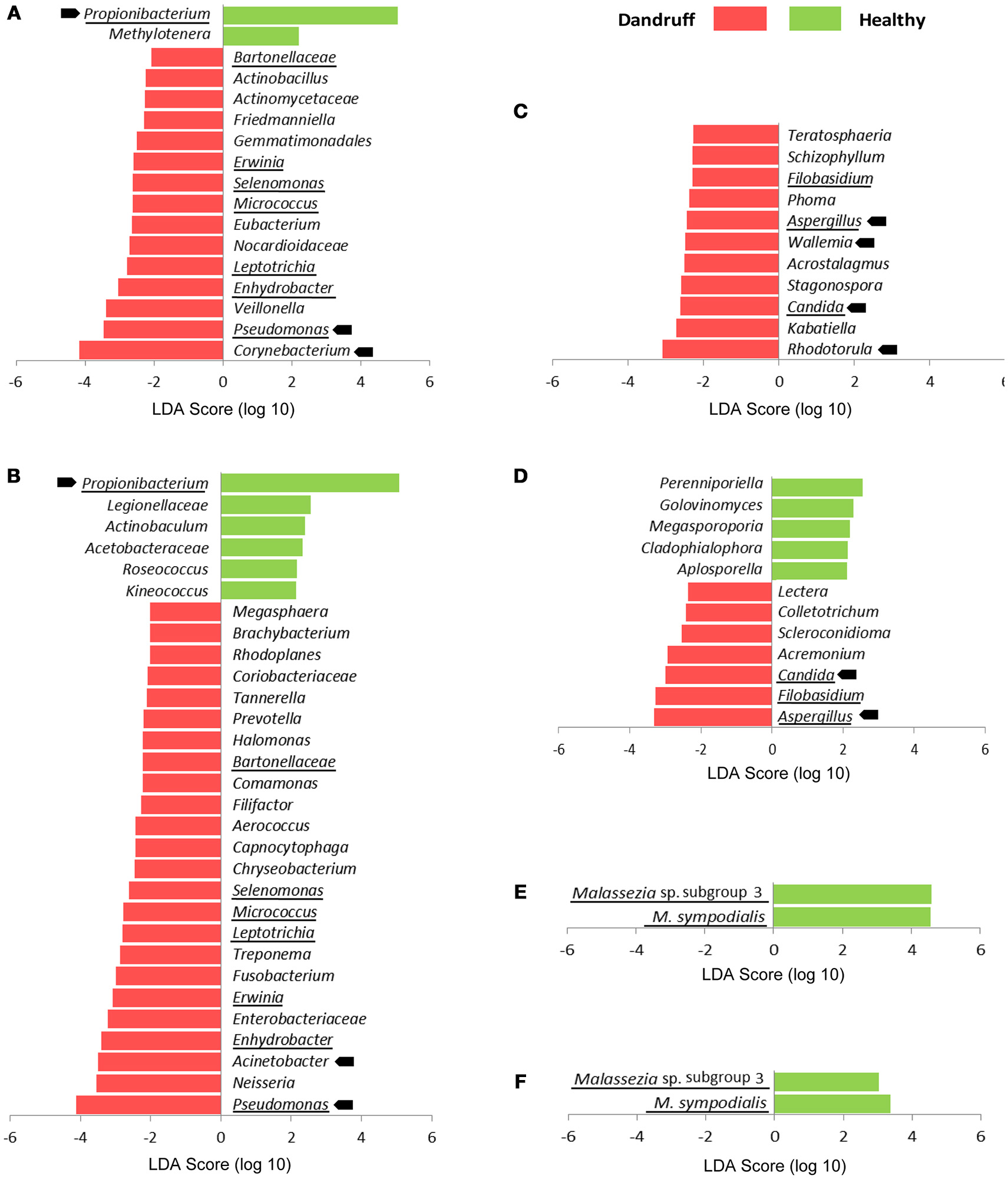
Differentially abundant OTUs stratified by body site and health status. (A) Bacteria at genus level from scalp samples; (B) Bacteria at genus level from forehead samples; (C) Fungi at genus level from scalp samples; (D) Fungi at genus level from forehead samples. (E)Malassezia at species level from scalp samples; (F)Malassezia at species level from forehead samples. Abundant OTUs are indicated by arrows; OTUs differentially abundant in both skin sites are underlined. LDA score threshold was set to ≥2.0. Kruskal-Wallis p < 0.05 was considered statistically significant.
Microbial dysbiosis index (MD-index), based on differentially abundant bacterial OTUs, differed according to health status. Samples from healthy subjects showed overall lower MD-indexes as well as lower diversity for both scalp and forehead, when compared with samples from dandruff subjects (Figures 4A,B). Average MD-index was significantly higher in dandruff samples than in healthy ones (p < 0.0001; Figure 4C). MD-index could not be calculated for fungi because there were no underrepresented OTUs in scalp samples; and in the case of forehead, some samples lacked some of the underrepresented OTUs.
Figure 4
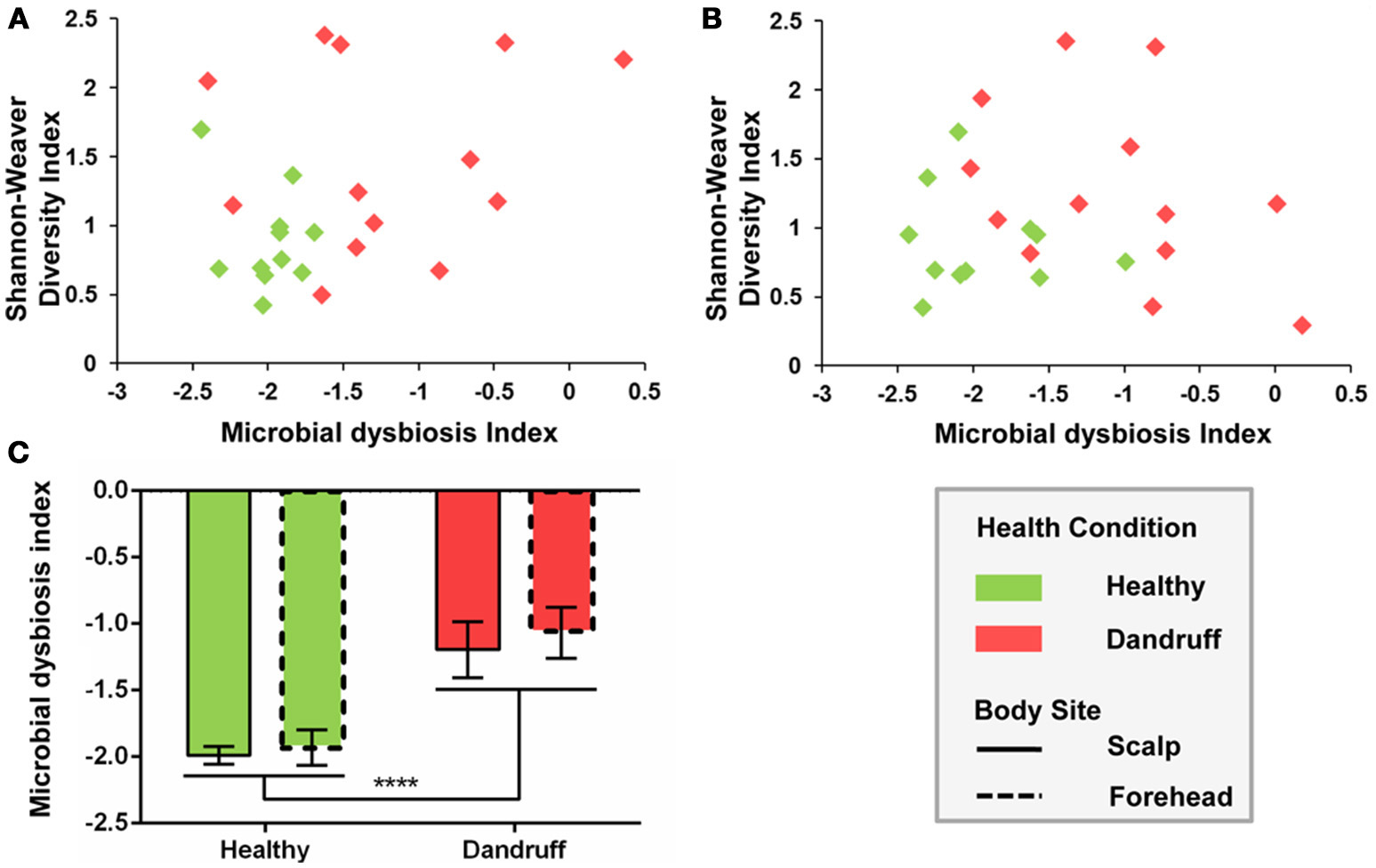
Microbial dysbiosis index (MD-index) for bacterial communities according to health status. (A) Scatter plot of MD-index vs. Shannon-Weaver diversity for scalp samples; (B) Scatter plot of MD-index vs. Shannon-Weaver diversity for forehead samples; (C) Average MD-index. Significance was determined by Two-way ANOVA Test. Bars represent Mean ± SEM. ****p < 0.0001.
Discussion
Microbial communities inhabiting the human body and their relation with diseases have been the subject of many studies and intense team efforts (The Human Microbiome Project Consortium, 2012a,b; The Integrative HMP (iHMP) Research Network Consortium, 2014). In the case of dermatopathologies, the association with microbiome imbalances is often not well-established. Many skin diseases and disorders have been primarily associated with one specific group of microorganisms, e.g., acne vulgaris (Das and Reynolds, 2014), rosacea (Casas et al., 2012), seborrheic dermatitis, and dandruff (Hay, 2011); although the possible microbial role in the development of symptoms is not completely understood.
Despite its high prevalence, the shortage of effective treatments, and the impact of symptoms (Piérard-Franchimont et al., 2006a), the etiology of dandruff remains poorly understood. Knowledge regarding seborrheic dermatitis, a related condition that has been often considered a more severe form of dandruff affecting body areas other than the scalp (Piérard-Franchimont et al., 2006a; Schwartz et al., 2013), is also limited. The association of these conditions with Malassezia yeasts has been reported (Gemmer et al., 2002; DeAngelis et al., 2005; Tajima et al., 2008; Clavaud et al., 2013); however, it is still unclear whether they are actually the causal agent of dandruff, and if there are other microorganisms involved in the pathogenic process.
Here, we report the characterization of bacterial and fungal microbiome from healthy and dandruff subjects. Lesional and non-lesional skin sites were examined, revealing that bacterial and fungal communities at genus level are imbalanced in dandruff subjects, even in non-lesional sites (Figures 5A,B). These findings suggest that the process associated with dandruff may be systemic, and not restricted to the skin site exhibiting the symptoms. An instability of the skin microenvironment at systemic level, possibly due to immune response alterations, might lead to dysbiosis, disruption of skin barrier (Gallo and Nakatsuji, 2011; Hooper et al., 2012) and to clinical manifestation of dandruff in specific skin sites. This could have implications for therapeutic approaches: since dandruff manifests locally, treatment is normally not systemic, which may explain difficulties in controlling symptoms. In the future, studies of microbial communities from a greater number of body sites should be performed to further support the hypothesis and to evaluate the extent of the alterations in the body. Dysbiosis has been associated with various immune-related conditions; however, most cases involve loss of microbial diversity, as consequence of the expansion of potentially harmful microorganisms or loss of beneficial species (Petersen and Round, 2014). Specifically regarding skin disorders, a loss of bacterial diversity has been reported in patients with atopic dermatitis (Kong et al., 2012) and psoriasis (Alekseyenko et al., 2013), in comparison with healthy subjects. Our data suggest that a different phenomenon occurs in dandruff, as microbial diversity was higher than in healthy subjects. The skin environment in dandruff might become less selective for microbial growth, which could also account for the decrease of intragroup similarly of dandruff bacterial and fungal communities.
Figure 5
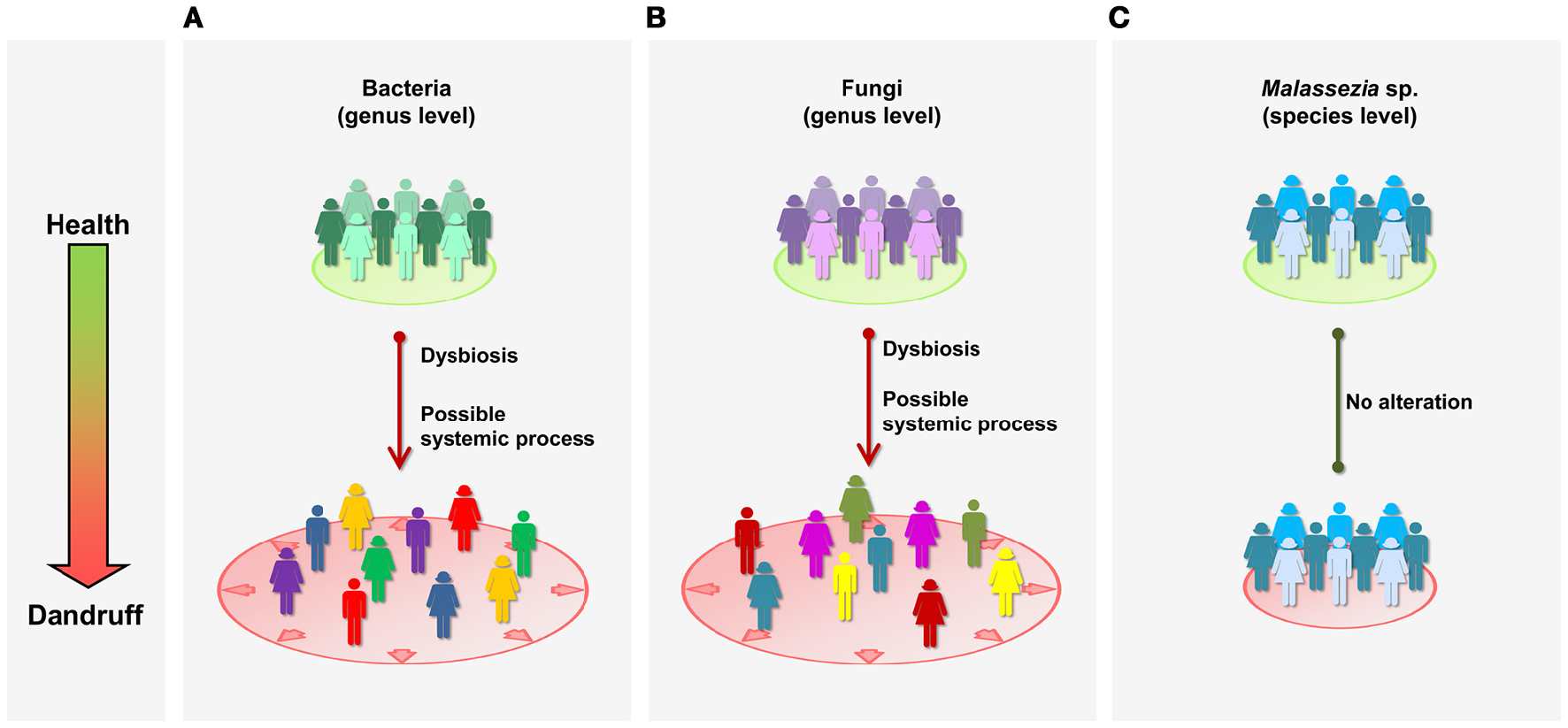
Schematic representation of microbial communities in health and dandruff. (A) Bacteria at genus level; (B) Fungi at genus level; and (C)Malassezia at species level. Different colors represent distinct communities; greater distance between figures represent less intra-group similarity. “Systemic” refers to general processes in the body, as opposed to specific local sites.
Although fungal community is dominated by Malassezia genus, consistent with previous studies of healthy skin (Findley et al., 2013; Oh et al., 2014), we found no evidence of association between Malassezia and dandruff. Moreover, in contrast with microbial shifts observed at genus level, Malassezia microbiota at species level was not altered in dandruff, suggesting it is not affected by possible systemic alterations taking place in dandruff subjects (Figure 5C). It is surprising that Malassezia communities from scalp samples were similar in diversity and composition to one another regardless of whether they are from healthy or dandruff subjects.
Bacterial and fungal microbiome associated with dandruff has been previously studied in scalp samples (lesional site) through cloning and Sanger sequencing, showing that the ratios between abundant bacterial and fungal organisms shifted in dandruff compared with healthy subjects, especially Malassezia restricta, Propionibacterium sp., and Staphylococcus sp. (Clavaud et al., 2013; Wang et al., 2015). More recently, Xu et al. (2016) showed that host factors such as skin sebum and water content are related to dandruff. Analyzing scalp microbiome by NGS, the authors found that dandruff is more closely related to bacteria than to fungi, and reported no significant association with Malassezia at species level, although some OTUs showed positive or negative correlation with dandruff (Xu et al., 2016). Our results corroborate the relationship between dandruff and bacteria but not with Malassezia at species level, and showed for the first time increased diversity and dissimilarity among dandruff samples, affecting also asymptomatic skin sites.
A bioinformatics strategy based on sequence clustering and OTU assignment using combined databases increased the percentage of fungal assigned sequences, and revealed the presence of uncharacterized Malassezia organisms. Previously, we have reported Malassezia sequences that were dissimilar from any described species in healthy, psoriasis and seborrheic dermatitis subjects (Paulino et al., 2006, 2008; Soares et al., 2015). A more comprehensive analysis shown here emphasized the importance of such uncharacterized organisms, found in large proportions in most samples regardless of the health status. Such findings highlight the significance of developing strategies to assess unassigned DNA sequences, in order to ensure a reliable view of fungal communities. Further, studies are needed, including the isolation and complete characterization of these organisms, as well as phylogenetic analysis using DNA regions other than ITS1, which contains large insertions and deletions that restrict the reliability of the sequence alignment.
Data presented here indicate that dandruff is associated with bacterial and fungal dysbiosis and suggests a systemic process affecting even asymptomatic skin sites. Findings showed that microbiome analyses, particularly of fungal communities, require strategies to assess unclassified sequences, otherwise useful information might be lost. Further, studies of larger cohorts and more extensive topographical analysis, immune response, functional characterization of microbial communities, and bacterial-fungal interactions might generate novel hypotheses and provide new perspectives on the pathogenesis of dandruff and other skin diseases, as well as to expand our understanding of healthy skin microbiota.
Statements
Author contributions
LP, CC, and LB conceived the study; RS, VD, PC, and AB performed the experiments; RS, PC, AB, and LP analyzed the data; RS, PC, RD, CC, LB, and LP interpreted and discussed the results; RS and LP wrote the paper.
Acknowledgments
We thank the employees and medical team from Instituto Superior de Medicina e Dermatologia-ISMD, São Paulo, Brazil. We are also grateful to Suzi Dal Belo and Daphine Antonia Clemente from L'Oréal Research and Innovation Brazil for their assistance in the clinical characterization of dandruff, Daniel H. Littwin for language revision, and to all volunteers who collaborated with this work. This work was supported by a grant from L'Oréal. RD, CC, and LB are employees of L'Oréal Research and Innovation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fcimb.2016.00157/full#supplementary-material
References
1
Alekseyenko A. V. Perez-Perez G. I. De Souza A. Strober B. Gao Z. Bihan M. et al . (2013). Community differentiation of the cutaneous microbiota in psoriasis. Microbiome. 1–31. 10.1186/2049-2618-1-31
2
Altschul S. F. Gish W. Miller W. Myers E. W. Lipman D. J. (1990). Basic local alignment search tool. J. Mol. Biol.215, 403–410. 10.1016/S0022-2836(05)80360-2
3
Bonnist E. Y. Pudney P. D. Weddell L. A. Campbell J. Baines F. L. Paterson S. E. et al . (2014). Understanding the dandruff scalp before and after treatment: an in vivo Raman spectroscopic study. Int. J. Cosmet. Sci.36, 347–354. 10.1111/ics.12132
4
Bray J. R. Curtis J. T. (1957). An ordination of the upland forest communities of southern wisconsin. Ecol. Monogr.27, 325–349. 10.2307/1942268
5
Cabañes F. J. Coutinho S. D. Puig L. Bragulat M. R. Castellá G. (2016). New lipid-dependent Malassezia species from parrots. Rev. Iberoam. Micol.33, 92–99. 10.1016/j.riam.2016.03.003
6
Caporaso J. G. Kuczynski J. Stombaugh J. Bittinger K. Bushman F. D. Costello E. K. et al . (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods7, 335–336. 10.1038/nmeth.f.303
7
Caporaso J. G. Lauber C. L. Costello E. K. Berg-Lyons D. Gonzalez A. Stombaugh J. et al . (2011). Moving pictures of the human microbiome. Genome Biol.12:R50. 10.1186/gb-2011-12-5-r50
8
Casas C. Paul C. Lahfa M. Livideanu B. Lejeune O. Alvarez-Georges S. et al . (2012). Quantification of demodex folliculorum by pcr in rosacea and its relationship to skin innate immune activation. Exp. Dermatol.21, 906–910. 10.1111/exd.12030
9
Claesson M. J. O'sullivan O. Wang Q. Nikkila J. Marchesi J. R. Smidt H. et al . (2009). Comparative analysis of pyrosequencing and a phylogenetic microarray for exploring microbial community structures in the human distal intestine. PLoS ONE4:e6669. 10.1371/journal.pone.0006669
10
Clarke K. R. (1993). Non-parametric multivariate analyses of changes in community structure. Aus. J. Ecol.18, 117–143. 10.1111/j.1442-9993.1993.tb00438.x
11
Clarke K. R. Gorley R. N. (2006). PRIMER v6: User Manual/Tutorial. Plymouth: Plymouth Marine Laboratory.
12
Clavaud C. Jourdain R. Bar-Hen A. Tichit M. Bouchier C. Pouradier F. et al . (2013). Dandruff is associated with disequilibrium in the proportion of the major bacterial and fungal populations colonizing the scalp. PLoS ONE8:e58203. 10.1371/annotation/bcff4a59-10b7-442a-8181-12fa69209e57
13
Colwell R. K. (2005). EstimateS: Statistical Estimation of Species Richness and Shared Species from Samples. User's Guide and application. Available online at: http://viceroy.eeb.uconn.edu/EstimateS/EstimateSPages/EstSUsersGuide/EstimateSUsersGuide.htm (Accessed 09, 2014).
14
Das S. Reynolds R. V. (2014). Recent advances in acne pathogenesis: implications for therapy. Am. J. Clin. Dermatol.15, 479–488. 10.1007/s40257-014-0099-z
15
DeAngelis Y. M. Gemmer C. M. Kaczvinsky J. R. Kenneally D. C. Schwartz J. R. Dawson T. L. Jr. (2005). Three etiologic facets of dandruff and seborrheic dermatitis: Malassezia fungi, sebaceous lipids, and individual sensitivity. J. Investig. Dermatol. Symp. Proc.10, 295–297. 10.1111/j.1087-0024.2005.10119.x
16
Edgar R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics26, 2460–2461. 10.1093/bioinformatics/btq461
17
Ewing B. Hillier L. Wendl M. C. Green P. (1998). Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res.8, 175–185. 10.1101/gr.8.3.175
18
Findley K. Oh J. Yang J. Conlan S. Deming C. Meyer J. A. et al . (2013). Topographic diversity of fungal and bacterial communities in human skin. Nature498, 367–370. 10.1038/nature12171
19
Fyhrquist N. Salava A. Auvinen P. Lauerma A. (2016). Skin Biomes. Curr. Allergy Asthma Rep.16, 40. 10.1007/s11882-016-0618-5
20
Gallo R. L. Nakatsuji T. (2011). Microbial symbiosis with the innate immune defense system of the skin. J. Invest. Dermatol.131, 1974–1980. 10.1038/jid.2011.182
21
Gemmer C. M. DeAngelis Y. M. Theelen B. Boekhout T. Dawson T. L. Jr. (2002). Fast, noninvasive method for molecular detection and differentiation of Malassezia yeast species on human skin and application of the method to dandruff microbiology. J. Clin. Microbiol.40, 3350–3357. 10.1128/JCM.40.9.3350-3357.2002
22
Gevers D. Kugathasan S. Denson L. A. Vázquez-Baeza Y. Van Treuren W. Ren B. et al . (2014). The treatment-naive microbiome in new-onset Crohn's disease. Cell Host Microbe15, 382–392. 10.1016/j.chom.2014.02.005
23
Grice E. A. (2014). The skin microbiome: potential for novel diagnostic and therapeutic approaches to cutaneous disease. Semin. Cutan. Med. Surg.33, 98–103. 10.12788/j.sder.0087
24
Grice E. A. Kong H. H. Conlan S. Deming C. B. Davis J. Young A. C. et al . (2009). Topographical and temporal diversity of the human skin microbiome. Science324, 1190–1192. 10.1126/science.1171700
25
Hammer O. Harper D. A. T. Ryan P. D. (2001). Paleontological statistics software package for education and data analysis. Palaeontol. Electron.4, 9.
26
Hay R. J. (2011). Malassezia, dandruff and seborrhoeic dermatitis: an overview.Br. J. Dermatol.165(Suppl. 2), 2–8. 10.1111/j.1365-2133.2011.10570.x
27
Honnavar P. Prasad G. S. Ghosh A. Dogra S. Handa S. Rudramurthy S. M. (2016). Malassezia arunalokei sp. nov., a novel yeast species isolated from seborrheic dermatitis patients and healthy individuals from India. J. Clin. Microbiol.54, 1826–1834. 10.1128/JCM.00683-16
28
Hooper L. V. Littman D. R. Macpherson A. J. (2012). Interactions between the microbiota and the immune system. Science336, 1268–1273. 10.1126/science.1223490
29
Kerr K. Darcy T. Henry J. Mizoguchi H. Schwartz J. R. Morrall S. et al . (2011). Epidermal changes associated with symptomatic resolution of dandruff: biomarkers of scalp health. Int. J. Dermatol.50, 102–113. 10.1111/j.1365-4632.2010.04629.x
30
Kõljalg U. Nilsson R. H. Abarenkov K. Tedersoo L. Taylor A. F. Bahram M. et al . (2013). Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol.22, 5271–5277. 10.1111/mec.12481
31
Kong H. H. (2011). Skin microbiome: genomics-based insights into the diversity and role of skin microbes. Trends Mol. Med.17, 320–328. 10.1016/j.molmed.2011.01.013
32
Kong H. H. Oh J. Deming C. Conlan S. Grice E. A. Beatson M. A. et al . (2012). Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res.22, 850–859. 10.1101/gr.131029.111
33
Lane D. J. Pace B. Olsen G. J. Stahl D. A. Sogin M. L. Pace N. R. (1985). Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc. Natl. Acad. Sci. U.S.A.82, 6955–6959. 10.1073/pnas.82.20.6955
34
Li W. Godzik A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics22, 1658–1659. 10.1093/bioinformatics/btl158
35
Manuel F. Ranganathan S. (2011). A new postulate on two stages of dandruff: a clinical perspective. Int. J. Trichol.3, 3–6. 10.4103/0974-7753.82117
36
Mills K. J. Hu P. Henry J. Tamura M. Tiesman J. P. Xu J. (2012). Dandruff/seborrhoeic dermatitis is characterized by an inflammatory genomic signature and possible immune dysfunction: transcriptional analysis of the condition and treatment effects of zinc pyrithione. Br. J. Dermatol166(Suppl. 2), 33–40. 10.1111/j.1365-2133.2012.10863.x
37
Naik S. Bouladoux N. Linehan J. L. Han S. J. Harrison O. J. Wilhelm C. et al . (2015). Commensal-dendritic-cell interaction specifies a unique protective skin immune signature. Nature520, 104–108. 10.1038/nature14052
38
Oh J. Byrd A. L. Deming C. Conlan S. Kong H. H. Segre J. A. (2014). Biogeography and individuality shape function in the human skin metagenome. Nature514, 59–64. 10.1038/nature13786
39
Oh J. Byrd A. L. Park M. Kong H. H. Segre J. A. (2016). Temporal stability of the human skin microbiome. Cell165, 854–866. 10.1016/j.cell.2016.04.008
40
Paulino L. C. Tseng C. H. Blaser M. J. (2008). Analysis of Malassezia microbiota in healthy superficial human skin and in psoriatic lesions by multiplex real-time PCR. FEMS Yeast Res.8, 460–471. 10.1111/j.1567-1364.2008.00359.x
41
Paulino L. C. Tseng C. H. Strober B. E. Blaser M. J. (2006). Molecular analysis of fungal microbiota in samples from healthy human skin and psoriatic lesions. J. Clin. Microbiol.44, 2933–2941. 10.1128/JCM.00785-06
42
Petersen C. Round J. L. (2014). Defining dysbiosis and its influence on host immunity and disease. Cell. Microbiol.16, 1024–1033. 10.1111/cmi.12308
43
Piérard-Franchimont C. Xhauflaire-Uhoda E. Loussouarn G. Saint Léger D. Piérard G. E. (2006b). Dandruff-associated smouldering alopecia: a chronobiological assessment over 5 years. Clin. Exp. Dermatol.31, 23–26. 10.1111/j.1365-2230.2005.02026.x
44
Piérard-Franchimont C. Xhauflaire-Uhoda E. Piérard G. E. (2006a). Revisiting dandruff. Int. J. Cosmet. Sci.28, 311–318. 10.1111/j.1467-2494.2006.00326.x
45
Saunders C. W. Scheynius A. Heitman J. (2012). Malassezia fungi are specialized to live on skin and associated with dandruff, eczema, and other skin diseases. PLoS Pathog.8:e1002701. 10.1371/journal.ppat.1002701
46
Schwartz J. R. Messenger A. G. Tosti A. Todd G. Hordinsky M. Hay R. J. et al . (2013). A comprehensive pathophysiology of dandruff and seborrheic dermatitis - towards a more precise definition of scalp health. Acta Derm. Venereol.93, 131–137. 10.2340/00015555-1382
47
Segata N. Izard J. Waldron L. Gevers D. Miropolsky L. Garrett W. S. et al . (2011). Metagenomic biomarker discovery and explanation. Genome Biol.12:R60. 10.1186/gb-2011-12-6-r60
48
Soares R. C. Zani M. B. Arruda A. C. Arruda L. H. Paulino L. C. (2015). Malassezia intra-specific diversity and potentially new species in the skin microbiota from Brazilian healthy subjects and seborrheic dermatitis patients. PLoS ONE10:e0117921. 10.1371/journal.pone.0117921
49
Tajima M. Sugita T. Nishikawa A. Tsuboi R. (2008). Molecular analysis of Malassezia microflora in seborrheic dermatitis patients: comparison with other diseases and healthy subjects. J. Invest. Dermatol.128, 345–351. 10.1038/sj.jid.5701017
50
The Human Microbiome Project Consortium (2012a). A framework for human microbiome research. Nature486, 215–221. 10.1038/nature11209
51
The Human Microbiome Project Consortium (2012b). Structure, function and diversity of the healthy human microbiome. Nature486, 207–214. 10.1038/nature11234
52
The Integrative HMP (iHMP) Research Network Consortium (2014). The Integrative Human Microbiome Project: dynamic analysis of microbiome-host omics profiles during periods of human health and disease. Cell Host Microbe16, 276–289. 10.1016/j.chom.2014.08.014
53
Tomic-Canic M. Perez-Perez G. I. Blumenberg M. (2014). Cutaneous microbiome studies in the times of affordable sequencing. J. Dermatol. Sci.75, 82–87. 10.1016/j.jdermsci.2014.05.001
54
Wang L. Clavaud C. Bar-Hen A. Cui M. Gao J. Liu Y. et al . (2015). Characterization of the major bacterial-fungal populations colonizing dandruff scalps in Shanghai, China, shows microbial disequilibrium. Exp. Dermatol.24, 398–400. 10.1111/exd.12684
55
Wanke I. Steffen H. Christ C. Krismer B. Götz F. Peschel A. et al . (2011). Skin commensals amplify the innate immune response to pathogens by activation of distinct signaling pathways. J. Invest. Dermatol.131, 382–390. 10.1038/jid.2010.328
56
Xu Z. Wang Z. Yuan C. Liu X. Yang F. Wang T. et al . (2016). Dandruff is associated with the conjoined interactions between host and microorganisms. Sci. Rep.6:24877. 10.1038/srep24877
Summary
Keywords
dandruff, skin, microbiota, Malassezia , dysbiosis, next generation sequencing
Citation
Soares RC, Camargo-Penna PH, de Moraes VCS, De Vecchi R, Clavaud C, Breton L, Braz ASK and Paulino LC (2016) Dysbiotic Bacterial and Fungal Communities Not Restricted to Clinically Affected Skin Sites in Dandruff. Front. Cell. Infect. Microbiol. 6:157. doi: 10.3389/fcimb.2016.00157
Received
30 August 2016
Accepted
02 November 2016
Published
17 November 2016
Volume
6 - 2016
Edited by
Lorenza Putignani, Bambino Gesù Children's Hospital (IRCCS), Italy
Reviewed by
Valerio Iebba, Sapienza University of Rome, Italy; Aaron Conrad Ericsson, University of Missouri, USA
Updates
Copyright
© 2016 Soares, Camargo-Penna, de Moraes, De Vecchi, Clavaud, Breton, Braz and Paulino.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luciana C. Paulino luciana.paulino@ufabc.edu.br
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.