 Jui-Chieh Yin1†
Jui-Chieh Yin1† Chun-Hsien Fei1†
Chun-Hsien Fei1† Yen-Chen Lo2,3†Yu-Yuan Hsiao4Jyun-Cyuan Chang1
Yen-Chen Lo2,3†Yu-Yuan Hsiao4Jyun-Cyuan Chang1 Jay C. Nix5
Jay C. Nix5 Yuan-Yu Chang2
Yuan-Yu Chang2 Lee-Wei Yang2,6*
Lee-Wei Yang2,6* I-Hsiu Huang1,7*
I-Hsiu Huang1,7* Shuying Wang1,7*
Shuying Wang1,7*- 1Department of Microbiology and Immunology, College of Medicine, National Cheng Kung University, Tainan, Taiwan
- 2Institute of Bioinformatics and Structural Biology, National Tsing Hua University, Hsinchu, Taiwan
- 3Bioinformatics Program, Taiwan International Graduate Program, Academia Sinica, Taipei, Taiwan
- 4Department of Biological Science and Technology, National Chiao Tung University, Hsinchu, Taiwan
- 5Molecular Biology Consortium, Advanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, CA, USA
- 6Physics Division, National Center for Theoretical Sciences, Hsinchu, Taiwan
- 7Center of Infectious Disease and Signaling Research, National Cheng Kung University, Tainan, Taiwan
Sortases function as cysteine transpeptidases that catalyze the covalent attachment of virulence-associated surface proteins into the cell wall peptidoglycan in Gram-positive bacteria. The substrate proteins targeted by sortase enzymes have a cell wall sorting signal (CWSS) located at the C-terminus. Up to date, it is still not well understood how sortases with structural resemblance among different classes and diverse species of bacteria achieve substrate specificity. In this study, we focus on elucidating the molecular basis for specific recognition of peptide substrate PPKTG by Clostridium difficile sortase B (Cd-SrtB). Combining structural studies, biochemical assays and molecular dynamics simulations, we have constructed a computational model of Cd-SrtBΔN26–PPKTG complex and have validated the model by site-directed mutagensis studies and fluorescence resonance energy transfer (FRET)-based assay. Furthermore, we have revealed that the fourth amino acid in the N-terminal direction from cleavage site of PPKTG forms specific interaction with Cd-SrtB and plays an essential role in configuring the peptide to allow more efficient substrate-specific cleavage by Cd-SrtB.
Introduction
Bacterial surface proteins are crucial virulence factors that mediate adhesion to the host as the first step establishing an infection. The sortase family of cysteine transpeptidases catalyzes the anchoring of a wide variety of virulence-associated surface proteins to the cell wall peptidoglycan (Spirig et al., 2011; Cascioferro et al., 2014; Bradshaw et al., 2015). Sortases, unique to Gram-positive bacteria, recognize and cleave the C-terminal cell wall sorting signal motif (CWSS) of substrate proteins (Schneewind et al., 1992, 1993; Paterson and Mitchell, 2004). Based on the primary sequences and their roles in biological functions, sortases are classified into six classes: A, B, C, D, E, and F. Class A sortases (SrtAs) are present in almost all Gram-positive bacteria. The first identified and best-known class A enzyme is the Staphylococcus aureus SrtA (Sa-SrtA), which anchors at least 19 LPXTG-containing surface proteins (Mazmanian et al., 1999; Perry et al., 2002; Spirig et al., 2011; Bradshaw et al., 2015). Sa-SrtA mutants exhibited a severe reduced adherence to epithelial cells and virulence in animal models (Flock et al., 1987; Mazmanian et al., 2000; Clancy et al., 2010). SrtAs are commonly called housekeeping sortases, whereas the remaining five classes are the accessory sortases. Class B sortases (SrtBs) recognize the NXXTN motif rather than the classical LPXTG motif and have distinct functions (Comfort and Clubb, 2004; Dramsi et al., 2005); some members of this group are involved in iron acquisition, whereas sortase B of Streptococcus pyogenes is involved in pili assembly (Kang et al., 2011). Class C sortases (SrtCs) are essential for pili polymerization in many species (Huang et al., 2010), such as Enterococcus faecalis (Kline et al., 2009), Corynebacterium diphtheria (Ton-That and Schneewind, 2003; Gaspar and Ton-That, 2006), Streptococcus agalactiae (Dramsi et al., 2006; Cozzi et al., 2012), and Streptococcus pneumonia (Fälker et al., 2008; LeMieux et al., 2008; Manzano et al., 2008). In addition, SrtC is required for aerial hyphae formation in Streptomyces coelicolor (Di Berardo et al., 2008). Class D sortases (SrtDs) are similar to SrtAs and perform a housekeeping role; they most frequently present in Bacillus species and are involved in spore formation (Marraffini and Schneewind, 2006). Recent studies have reported that Clostridium perfringens SrtD is structurally and catalytically distinct from Bacillus anthracis SrtD, suggesting that C. perfringens SrtD may display a different aspect of the SrtD family (Marraffini and Schneewind, 2006; Suryadinata et al., 2015). Class E and F sortases are mainly identified in Actinobacteria; they share a limited primary sequence homology with other sortases and their functions remain undetermined (Comfort and Clubb, 2004; Dramsi et al., 2005; Spirig et al., 2011).
In the genome of toxigenic C. difficile strain 630, only one functional sortase, the SrtB gene, is present (Donahue et al., 2014). C. difficile is a Gram-positive, anaerobic, and spore-forming bacterium that can colonize the gut if the normal intestinal microbiota is disrupted (Kelly and LaMont, 1998). C. difficile infection (CDI) is highly associated with antibiotic therapy and has been recognized as the leading cause of antibiotic-associated diarrhea, making it a major public health threat worldwide (Henrich et al., 2009; Bagdasarian et al., 2015). In the United States alone, CDI causes approximately 15,000–20,000 deaths annually, and CDI-associated hospitalizations among the general population doubled from 31 to 61 per 100,000 from 2008 to 2010 (Viseur et al., 2011). Furthermore, the CDI risk is high in patients receiving antibiotic treatments because their gastrointestinal flora is unfavorably altered. CDI manifestations can include asymptomatic colonization, mild to severe chronic diarrhea, pseudomembranous colitis, and death because of multiple organ failure (Kelly and LaMont, 2008). At present, metronidazole and vancomycin are mainly administered for treating CDI. However, up to 25% of patients treated for CDI experience recurrences after discontinuing antibiotic therapy (Bartlett et al., 1980; Tedesco et al., 1985; Leffler and Lamont, 2009; Surawicz et al., 2013). The increase in treatment failure or multiple relapses have raised a concern. An alternative therapy, fecal microbiota transplantation, has been used to restore healthy gut flora in patients with recurrent CDI (Rohlke and Stollman, 2012; Dodin and Katz, 2014; Duke and Fardy, 2014). Fecal transplantation is highly effective; however, it is still not widely accepted. In the last decade, sortase has been identified as a promising anti-infective therapeutic target (Zong et al., 2004b; Maresso et al., 2007; Suree et al., 2009; Oh et al., 2010; Jacobitz et al., 2014; Zhang et al., 2014), thus offering an encouraging avenue toward the development of drugs against CDI.
There have been many structural and functional studies on sortases from various Gram-positive pathogens (Spirig et al., 2011; Cascioferro et al., 2014; Bradshaw et al., 2015), and studies on C. difficile sortases were reported recently (Donahue et al., 2014; van Leeuwen et al., 2014; Chambers et al., 2015). Sa-SrtA has been extensively studied, and the catalytic mechanism underlying how Sa-SrtA anchors the surface protein to cell wall has been reported (Mazmanian et al., 1999; Perry et al., 2002). The membrane-bound Sa-SrtA scans and recognizes the LPXTG sequence of the CWSS, and a nucleophilic attack from the active thiol group of sortase cysteine residue to the peptide bond between the threonine and glycine of the LPXTG motif results in the formation of a thioester intermediate (Mazmanian et al., 1999; Ton-That et al., 1999; Perry et al., 2002). The sortase–acyl intermediate is then resolved by the nucleophilic attack of a free amino group within lipid II, resulting in the release of the surface protein from the sortase onto the cross bridge of the newly formed peptidoglycan (Frankel et al., 2005). This substrate release restores the enzyme active site, allowing the sortase to process more substrates (Frankel et al., 2005).
Recent studies have demonstrated that C. difficile SrtB (Cd-SrtB) can recognize and cleave (S/P)PXTG between threonine and glycine; however, Cd-SrtB cannot recognize the sequence LPXTG and NPQTN, corresponding to the recognition motifs for Sa-SrtA and Sa-SrtB, respectively (van Leeuwen et al., 2014; Chambers et al., 2015). It remains unclear how structurally similar sortases achieve substrate specificity. Thus far, the available structures of sortase–substrate complexes are limited to the nuclear magnetic resonance structure of Sa-SrtA bound to an LPATG substrate analog (Suree et al., 2009), a crystal structure of a Sa-SrtA mutant complexed with LPETG (Zong et al., 2004a), and a crystal structure of Sa-SrtB covalently bound to an NPQTN analog (Jacobitz et al., 2014). Therefore, studying and comparing new structures of sortases and sortase–substrate complexes from a wide range of organisms will enhance our understanding of how sortases recognize their respective substrates. In this study, we determined the crystal structure of the catalytically active SrtB from C. difficile and constructed a probable model of the Cd-SrtB–PPKTG complex by computer modeling and molecular dynamics simulations to gain structural insights into the substrate specificity for Cd-SrtB.
Materials and Methods
Protein Overexpression and Purification
The SrtBΔN26 from C. difficile 630 was cloned into a pMCSG7 vector by using a ligation-independent cloning method (Aslanidis and de Jong, 1990) and transformed into E. coli BL21 (DE3). A recombinant 6xHis-tagged SrtBΔN26 protein was induced by adding 0.5 mM isopropyl-β-D-thiogalactopyranoside when the cells reached an O.D.600 of 0.5, and further incubated at 37°C for 4 h. Cells were centrifuged at 8000 rpm for 30 min at 4°C, resuspended in buffer A (20 mM HEPES pH 7.4, 200 mM NaCl and 20 mM imidazole), and disrupted by sonication on ice. Moreover, the supernatant was loaded into an Ni-NTA column (GE Healthcare Life Sciences) and contaminant proteins were eliminated through a washing procedure by using 60 mM imidazole in buffer A. SrtBΔN26 proteins were eluted with 300 mM imidazole in buffer A. Fractions containing SrtBΔN26 proteins were pooled and further purified through HiLoad 26/600 Superdex™ 75 size-exclusion chromatography (GE Healthcare Life Sciences). Subsequently, the proteins were dialyzed in buffer B (10 mM HEPES pH 7.4 and 150 mM NaCl) and stored at 4°C for further use.
FRET-Based Assay
The peptide substrate of Cd-SrtBΔN26, PPKTG was conjugated using a fluorophore, 5-[(2-aminoethyl) amino] naphthalene-1-sulfonic acid, and a quencher, 4-([4-(dimethylamino) phenyl] azo) benzoic acid. To determine the suitable concentrations of Cd-SrtBΔN26 and fluorescently labeled PPKTG peptide in the assay, a matrix of various enzyme and substrate concentrations in the total volume of 100 μL in FRET buffer (10 mM HEPES pH 7.4 and 150 mM NaCl) was reacted in a 96-well black polystyrene plate and was incubated at 37°C for 48 h. The fluorescence signal was monitored at an excitation/emission wavelength of 340/490 nm and recorded every hour during the first 8 h and then at 24, 36, and 48 h by using a Spectra-Max M3 plate reader (Molecular Devices). The optimal concentrations of Cd-SrtBΔN26 and fluorogenic peptide used in our reactions are 240 and 20 μM, respectively. Stock solutions of MTSET and AAEK1 were dissolved in the FRET buffer, and curcumin was dissolved in DMSO. Serial dilutions of inhibitors at the millimolar range were added into the FRET buffer. All experiments were conducted in triplicate. The data are presented as means and standard errors. The statistical significance of the inhibitory effect on enzymatic activity was calculated using GraphPad Prism software (GraphPad Software). Two-tailed unpaired Student t-tests revealed significant differences between Cd-SrtBΔN26 + PPKTG and Cd-SrtBΔN26 + PPKTG + inhibitors at different concentrations (*p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001).
Crystallization
Purified Cd-SrtBΔN26 proteins were concentrated to 8–10 mg/mL for crystallization trials. For sparse matrix screening, numerous commercial kits (Hampton Research and Emeralds BioSystems) were used for performing the crystallization setup of the vapor diffusion method by using a high-throughput platform (Digilab Genomic Solutions). Cd-SrtBΔN26 crystals were observed in sitting drops containing 0.5 μL of protein and 0.5 μL of various crystallization solutions at 25°C within 1 week. Diffraction quality crystals were obtained using the hanging drop method by mixing 1 μL of protein (10 mg/ml in 10 mM HEPES pH 7.4 and 150 mM NaCl) and 1 μL of solution (0.1 M citric acid pH 3.5, 24% PEG 3350 and 0.1 M glycine). Prior to data collection, the crystals were directly mounted on loops from mother liquor and flash-frozen in liquid nitrogen without an additional cryoprotectant treatment.
X-Ray Data Collection and Processing
Diffraction data were collected on beamline BL13B1 of the National Synchrotron Radiation Research Center (NSRRC; Hsinchu, Taiwan) and beamline 4.2.2 of the Advanced Light Source (Berkeley, CA, USA). Most of our sortase crystals did not diffract beyond 3 Å resolution. The best crystal diffracted to 2.67 Å resolution and native data were collected at BL13B1 of the NSRRC. Ninety frames were collected, each with 1° oscillation and were exposed for 30 s at the wavelength of 1.0 Å with the crystal-to-detector distance of 400 mm at a temperature of 100 K. The data were indexed, integrated, and scaled using HKL2000 (Otwinowski and Minor, 1997). The initial data were scaled to 2.67 Å resolution, but the I/σI decreased to 1.67 at the highest resolution shell (2.77−2.67 Å) suggesting that the data were effective at a resolution of approximately 2.8 Å. The crystallographic parameters and data collection statistics were summarized in Table 1.
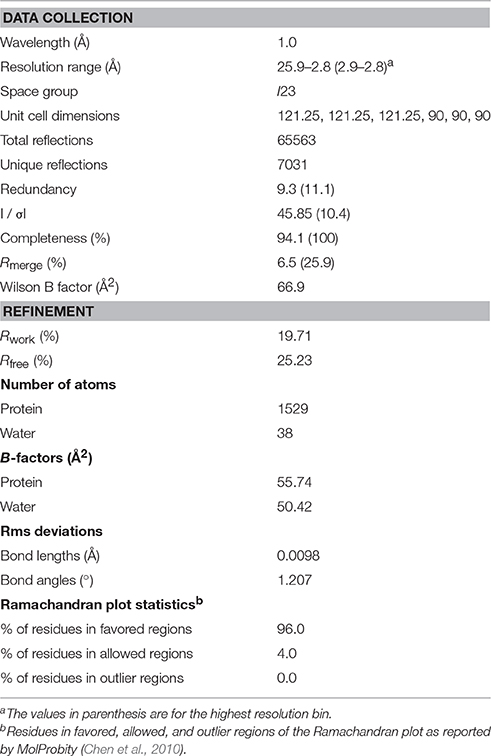
Table 1. Crystallographic data and refinement statistics.
Structure Determination and Refinement
The crystal structure of Cd-SrtBΔN26 was solved by molecular replacement method by program Phaser-MR (Mccoy et al., 2007) with the structure of Sa-SrtB (PDB 1QWZ) (Zong et al., 2004a) as a search model. Initially, the structure was determined at 3.5 Å resolution, and a polyalanine model was constructed. With the availability of better native datasets at higher resolutions, the model was manually rebuilt using COOT (Emsley et al., 2010) guided by 2Fo-Fc and Fo-Fc density maps. Computational refinement was conducted using REFMAC (Murshudov et al., 2011) and PHENIX (Adams et al., 2010), with 5% of the data flagged for cross-validation. We first carried out the refinement at 2.67 Å resolution, but the statistics were poor. The structural quality was improved when we systematically excluded the weak inflections by truncating data at different resolutions. Iterative model rebuilding and refinement were conducted. The final refinement statistics for the structural model at 2.8 Å resolution were summarized in Table 1. Coordinates and structure factors with the identifier 5GYJ have been deposited in the Protein Data Bank.
MD Simulations
The peptide of sequence NPQC co-crystalized with S. aureus SrtB structure (PDB 4LFD) was positioned in the catalytic pocket of C. difficile SrtB by superimposing of S. aureus SrtB onto C. difficile SrtB (Cd-SrtB). It was then mutated into a set of peptides of our interest, including PPKT and NPQT. PPKTG and NPQTN were modeled by adding one more G and N, respectively, in the C-terminus using the package VMD 1.9.2 (Humphrey et al., 1996). We further replaced the P4 residue in PPKTG to give SPKTG and NPKTG. Missing loops of 27–28 (ML), 162–167 (ESDYDY), 210–216 (TYEFDDA), and 225 (I) in the Cd-SrtB were modeled by UCSF CHIMERA (Yang et al., 2012). Cd-SrtB−peptide complexes were solvated in TIP3P water molecules of 8Å thickness in all directions of a rectangular box (Jorgensen et al., 1983). One hundred and fifty millimolar sodium chloride were added as counter-ions to neutralize the system. Energy minimization and explicit-solvent MD simulations were performed by NAMD 2.10 package (Phillips et al., 2005; Huang and MacKerell, 2013) with CHARMM36 force field (Huang and MacKerell, 2013). Non-bonded interactions were carried out using a cut-off distance of 12 Å, with a switching distance of 10 Å. With periodic boundary conditions, the Particle Mesh Ewald method was employed for calculations of electrostatic energy (Darden et al., 1993). The Cd-SrtB, crystal waters and peptides (PPKTG, SPKTG, NPKTG, and NPQTN) were first restrained to the positions reported in the X-ray crystallography and then gradually released, first on the side chains and then the entire peptide and protein. No hydrogen atom is restrained at all time. After a 1.25 ns canonical ensemble (NVT) heating process and a short isothermal–isobaric (NPT) equilibration, the whole system was allowed for a productive run for 9 ns in a NPT ensemble at 310K and 1 atm, respectively controlled by solvent friction and Nosé-Hoover Langevin piston (Feller et al., 1995). MD simulations trajectories are further analyzed by VMD, MDAnalysis toolkit (Michaud-Agrawal et al., 2011) and in-house programs coded in python.
Contact Frequency Analysis for the P4 Residue of Different Substrate Peptides
To understand the role of the P4 residue in the substrate peptides, we analyzed the contact frequency of the P4 residue of a peptide with Cd-SrtBΔN26. At every frame, a residue in Cd-SrtBΔN26 situating within 4.0 Å from the P4 residue of the substrate peptides is marked as a contact. For each contacting residue, the contacting percentage is defined as the number of frames that the residue is in contact divided by total number of frames in MD simulations.
Root-Mean Square Fluctuations (RMSF) Analysis for Peptide Residues
To calculate the root-mean square fluctuations (RMSF) for a residue, we first iteratively superimposed the peptides in all MD snapshots to their mean positions by Kabsch's approach (Kabsch, 1976) until the process converges. RMSF of a residue is calculated as , where N is the number of heavy atoms in this residue; M is the total number of frames; Xi, k is the i-th heavy atom in the k-th frame and is the mean position for atom i over all the frames.
Results
Catalytic Activity of the Recombinant Purified Cd-SrtBΔN26
The recombinant 6xHis-tagged C. difficile sortase enzyme with a deletion of 26 residues at the N-terminal transmembrane domain, designated as Cd-SrtBΔN26, was overexpressed in Escherichia coli BL21 (DE3) and purified using an Ni-NTA affinity column (Supplementary Figure 1A). Size exclusion chromatography revealed that Cd-SrtBΔN26 was eluted at a volume corresponding to an apparent molecular weight of approximately 24 kDa (Supplementary Figure 1B), suggesting that Cd-SrtBΔN26 exists as a monomer in solution.
To confirm whether the recombinant purified Cd-SrtBΔN26 retains protease activity, we constructed a fluorescently labeled peptide to observe the fluorescence signal after Cd-SrtBΔN26 cleaves the substrate peptide in vitro (Figure 1A). In this construct, the known peptide substrate PPKTG (van Leeuwen et al., 2014) is sandwiched between a fluorophore and quencher. When the peptide remains intact, the intrinsic fluorescence is considerably reduced because of the proximity between the fluorescence donor and quenching acceptor. When the peptide is cleaved, the un-quenched fluorophore gives an enhanced fluorescent signal. To assess whether the previously described sortase inhibitors can inhibit the catalytic activity of Cd-SrtBΔN26, MTSET, AAEK1, and curcumin (Maresso et al., 2007; Hu et al., 2013; Donahue et al., 2014) (Supplementary Figure 2) were examined using the fluorescence resonance energy transfer (FRET)-based assay. The concentration-dependent inhibitory effects of MTSET (Figure 1B), AAEK1 (Figure 1C), and curcumin (Figure 1D) on the cleavage activity of the recombinant Cd-SrtBΔN26 were observed as the fluorescence signals were reduced when the inhibitors were added to the reactions comprised of Cd-SrtBΔN26 and fluorogenic peptides. The results further verified the recombinant Cd-SrtBΔN26 is catalytically active.
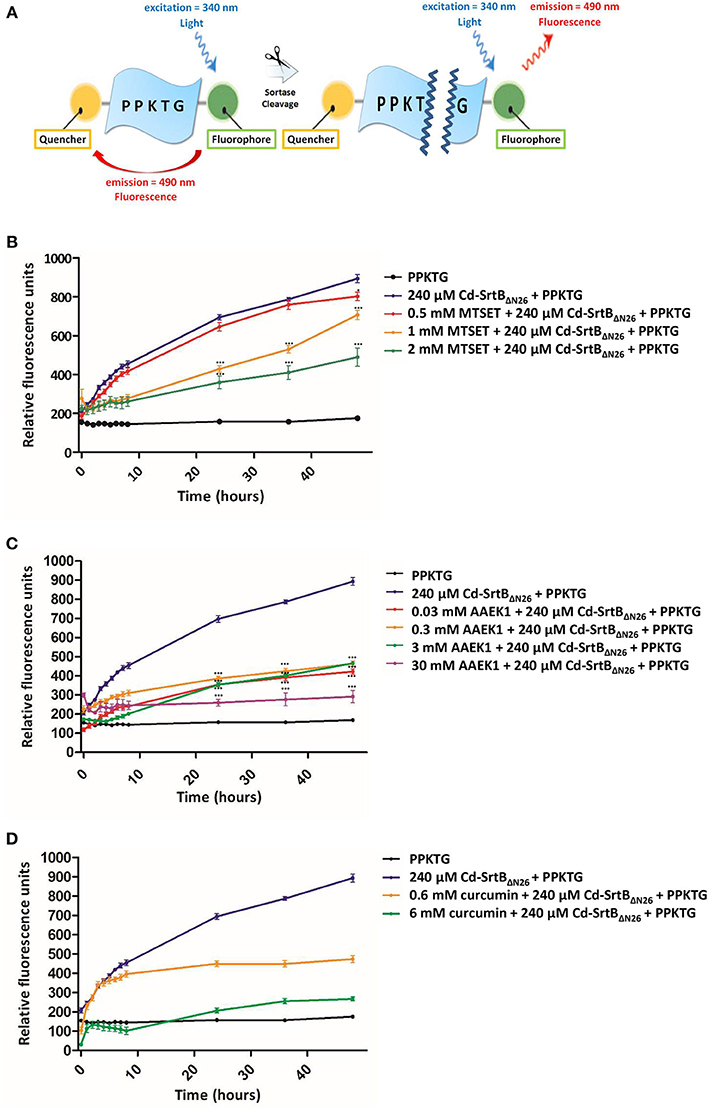
Figure 1. Enzymatic activity and inhibition of Cd-SrtBΔN26 by using a FRET-based assay. (A) Schematic representation of the peptide substrate PPKTG sandwiched between Edans and Dabcyl as the fluorophore quencher, respectively. (B–D) Catalytic activity of Cd-SrtBΔN26 and the effect of inhibitors monitored using the FRET-based assay. PPKTG was incubated with recombinant purified SrtBΔN26. The increase in the relative fluorescence signal was observed when PPKTG was cleaved by Cd-SrtBΔN26. The enzymatic cleavage of Cd-SrtBΔN26 was inhibited by adding (B) 0.5, 1, and 2 mM of MTSET; (C) 0.03, 0.3, 3, and 30 mM of AAEK1; and (D) 0.6 and 6 mM of curcumin.
Crystal Structure of Cd-SrtBΔN26
Cd-SrtBΔN26 comprises 198 residues with a 6xHis tag at the C-terminus. Cd-SrtBΔN26 crystallized in space group I23, with the unit cell parameters a = b = c = 121.25 Å and α = β = γ = 90°. The crystal structure of Cd-SrtBΔN26 was solved at 2.8 Å resolution by using the molecular replacement method, revealing one molecule in the crystallographic asymmetric unit. Most of the electron density was visible and interpretable for reliable model building. However, the density map for residues 27 and 28, 162–167, 210–216, and 225 as well as the C-terminal 6xHis tag was disordered. The crystallographic data and refinement statistics are summarized in Table 1. Validation of the Cd-SrtBΔN26 structure by using the MolProbity program (Chen et al., 2010) revealed no phi–psi angles in the disallowed region of the Ramachandran map.
The Cd-SrtBΔN26 structure possesses the sortase-unique protein fold, comprising eight β-strands (β1–β8), three α-helices (H1, H4, and H5), two 310-helices (H2 and H3), and several loops (Figure 2A). Resembling other sortase structures (Frankel et al., 2007; Kang et al., 2011; Jacobitz et al., 2014), the central β-barrel of Cd-SrtBΔN26 is formed by strands β1, β2, β5, and β6 on one side and by strands β3, β4, β7, and β8 on the other side. The characteristic N-terminal helix bundle, absent in SrtA structures and unique to SrtB, is composed of a 13-residue α-helix (H1), a 310-helix (H2), and a loop. The other 310-helix (H3) and a short α-helix (H4) are positioned between β4 and β5; H5 is inserted between the longest β-strand β6 and β7. The loop connecting β7 and β8 that has been postulated for accommodating peptidoglycan substrate binding was not visible in our structure, implying the flexibility of the large loop. Consistent with previous studies on sortase structures, β4, β7, and β8 of the β-barrel forming the Cys–His–Arg triad of Cd-SrtBΔN26 appears in a concave surface (Figure 2B). The catalytic residues Cys209 and His116 are located slightly beyond the C-terminal ends of β7 and β4, whereas Arg217 is anchored at the beginning of β8 (Figure 2C). A crystal structure of a catalytically inactive C. difficile mutant SrtBΔN32, C226A (PDB 4UX7) was published (Chambers et al., 2015). Superimposition of the catalytic residues of SrtB structures from C. difficile (PDB 5GYJ and 4UX7) (Chambers et al., 2015), S. aureus (PDB 1NG5) (Zhang et al., 2004), B. anthracis (PDB 1RZ2) (Zhang et al., 2004), and S. pyogenes (PDB 3PSQ) (Kang et al., 2011) shows the conservation of the active site (Supplementary Figure 3).
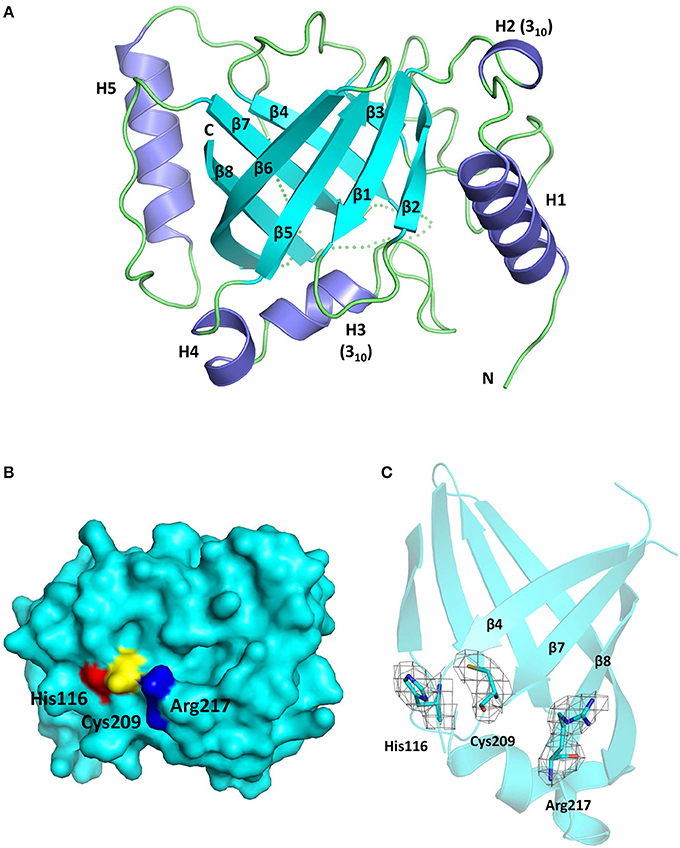
Figure 2. Crystal structure of Cd-SrtBΔN26. (A) Ribbon diagram of the SrtBΔN26 structure comprising three α-helices (H1, H4, and H5), two 310-helices (H2 and H3), and eight β-strands (β1–β8). Helices and β-strands are colored in purple and cyan, respectively. The flexible loops are presented in dashes. (B) Surface representation of Cd-SrtBΔN26 structure. The catalytic residues Cys209, His116, and Arg217 are colored in yellow, red, and blue, respectively. (C) Part of the overall omit map contoured at 1.0 sigma revealing the side-chain density of the catalytic Cys–His–Arg triad.
Cd-SrtBΔN26 is structurally equivalent to Cd-SrtBΔN32, C226A (Chambers et al., 2015). However, visualizing the sulfhydryl group of the catalytic cysteine residue in Cd-SrtBΔN26 is essential for facilitating our understanding of the substrate-specific catalysis of Cd-SrtB.
In silico Model of the Cd-SrtBΔN26–PPKTG Complex
To gain structural insights into how Cd-SrtBΔN26 recognizes PPKTG, we performed computational modeling based on the crystal structure of the Sa-SrtB–NPQT* complex (PDB 4LFD) (Jacobitz et al., 2014) for predicting the Cd-SrtBΔN26–PPKTG structure. In the Sa-SrtB–NPQT* structure, the substrate-binding pocket is delineated by a groove near the active site residues within the strands β4 and β7 and within loops β2/β3, β6/β7, and β7/β8. The NPQT* peptide was bound to Sa-SrtB in an “L-shaped” structure via hydrophobic interactions with Leu96, Tyr128, Tyr181, and Ile182 and via hydrogen bonds with Asn92, Thr177, Glu224, and Arg233. Moreover, the almost superimposable hydrophobic residues from S. aureus with the corresponding residues from C. difficile underlies the importance of their function (Supplementary Figure 4) and imply that the Cd-SrtB substrate may be positioned in a similar pattern within the hydrophobic groove. Therefore, we superimposed the structure of Sa-SrtB–NPQT* onto Cd-SrtBΔN26, mutated NPQT* to PPKT and added a glycine in the C-terminus in silico using the software VMD 1.9.2 (Humphrey et al., 1996) as an initial model of the Cd-SrtBΔN26–PPKTG complex. In addition, the missing residues and loops in the Cd-SrtBΔN26 structure, including the N- and C-terminal residues (27, 28, and 225), and residues located on the β6/β7 (162–167) and β7/β8 (210–216) loops were modeled using UCSF CHIMERA (Yang et al., 2012). To refine the docking pose of the PPKTG in the catalytic pocket of Cd-SrtBΔN26, energy minimization and MD simulations were conducted by gradually releasing the restraints on PPKTG, first on the side chains and then on the entire peptide, whereas the Cd-SrtBΔN26 residues and crystal waters were restrained to their atomic positions in the Cd-SrtBΔN26 structure at all times during the simulations.
The results from the computational modeling and unrestrained MD simulations suggest that PPKTG stays in the active site, forming a L-shape with a bend toward the N-terminus, resembling the structure of Sa-SrtB–NPQT* complex (Jacobitz et al., 2014) (Figure 3A). The sulfhydryl group of Cys209 is 5.0 Å from the carbonyl carbon of the threonine residue at the P1 position (Schechter and Berger, 1967), which is slightly further apart as compared with that of NPQT* in Sa-SrtB (Figure 3B). The side chain of Arg217 is hydrogen bonded to the hydroxyl oxygen of P1 Thr. The P2 Lys forms a hydrogen bond with Ser163 and salt-bridge interactions with Asp164. Moreover, the prolyl ring of P4 Pro noncovalently interacts with the aromatic ring of Tyr167. To assess whether Ser163 and Tyr167 are involved in substrate interactions as predicted in the structural model, two mutants that replace Ser163 and Tyr167 with alanine were generated in Cd-SrtBΔN26 by site-directed mutagenesis. By performing the FRET-based assay, we observed that the cleavage activity of mutants Cd-SrtBΔN26, S163A and Cd-SrtBΔN26, Y167A was substantially reduced compared to wild-type Cd-SrtBΔN26 (Figure 3C). The results indicate that the alanine substitution of Ser163 and Tyr167 did affect the interactions between Cd-SrtBΔN26 and PPTKG, resulting in the reduced florescence signals.
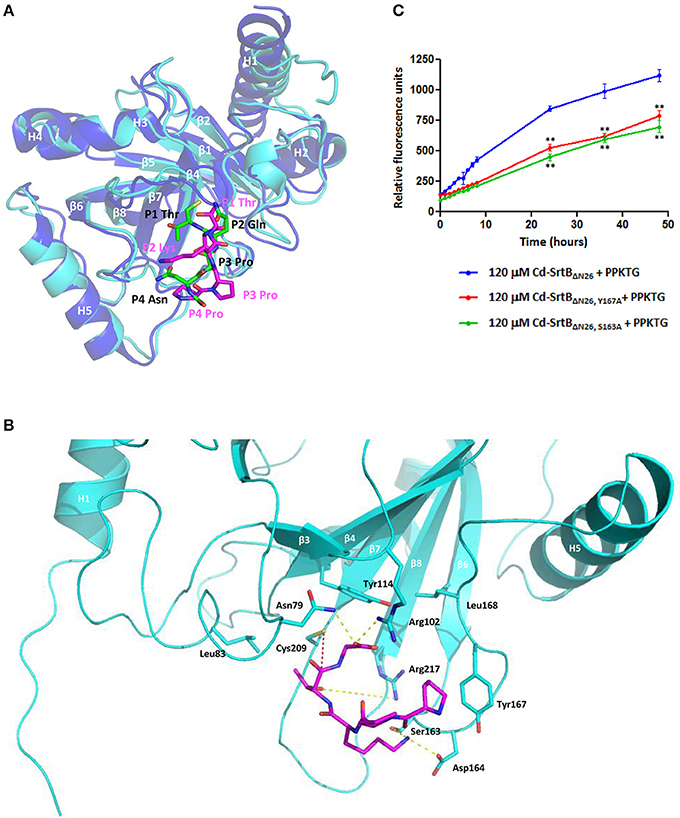
Figure 3. A computational model of the Cd-SrtBΔN26–PPKTG complex. (A) Superimposed structures of the Sa-SrtB–NPQT* and Cd-SrtBΔN26–PPKTG models. Blue, Sa-SrtB. Cyan, Cd-SrtBΔN26. NPQT* and PPKTG are presented in green and magenta, respectively. (B) The atomic interactions between PPKTG and Cd-SrtBΔN26 in the structural model. The hydrogen bond is colored in yellow. The distance from the sulfhydryl group of Cys209 to the carbonyl carbon of P1 Thr is represented in red. (C) Catalytic activity of Cd-SrtBΔN26 with mutation monitored using the FRET-based assay. PPKTG was incubated with recombinant purified SrtBΔN26, SrtBΔN26, Y167A, and SrtBΔN26, S163A. The increase in the relative fluorescence signal was observed when PPKTG was cleaved by Cd-SrtBΔN26. The enzymatic cleavage of Cd-SrtBΔN26 was inhibited with Y167A and S163A mutations.
Specificity Determinants of Substrate Peptides
To have a better understanding of specific recognition of the substrate peptide PPKTG by Cd-SrtB, we also constructed models of Cd-SrtBΔN26–SPKTG, Cd-SrtBΔN26–NPKTG, and Cd-SrtBΔN26–NVQTG complexes in the same way as constructing model of Cd-SrtBΔN26–PPKTG complex. Subsequently, we performed molecular dynamics (MD) simulations to analyze the residues in Cd-SrtBΔN26 and the contact frequency of those residues with different substrate peptides (Supplementary Figure 5). It is observed that the P4 residues in PPKTG and SPKTG are stabilized by residues of Cd-SrtBΔN26 located in binding pocket (Tyr101–Arg102, Ser163–Tyr167 for PPKTG, and Asp164–167, Phe213–Asp214 for SPKTG) (Figure 4). Our results show that the P4 residue of PPKTG in Cd-SrtBΔN26–PPKTG complex interacts with Asp166 for about 80% of the simulations time (10 ns) and forms hydrogen bonds with Asp166; while the P4 residues in NPKTG and NVQTG do not specifically interact with any residue in Cd-SrtBΔN26 (Figure 4 and Supplementary Figure 5). Moreover, we assumed that a peptide would be subjected to a conformation that allows Cd-SrtB to achieve a better catalytic efficiency if the distance (DISCys-Thr) between the sulfhydryl group of cysteine residue in Cd-SrtB and the carboxyl carbon of threonine residue in peptide is relatively short (Donahue et al., 2014; Chambers et al., 2015). We therefore examined distance distributions for four peptides of interest throughout 10 ns simulations. The distances are found to be 5.91 ± 0.53 Å, 5.81 ± 0.75 Å, 6.94 ± 0.74 Å, and 7.11 ± 0.77 Å for Cd-SrtBΔN26–PPKTG, Cd-SrtBΔN26–SPKTG, Cd-SrtBΔN26–NPKTG, and Cd-SrtBΔN26–NVQTG, respectively (Figure 5). To further explore the role of the P4 residues of peptides in substrate specificity, we calculated their root-mean square fluctuations (RMSFs) of the peptides in Cd-SrtBΔN26–PPKTG, Cd-SrtBΔN26–SPKTG, Cd-SrtBΔN26–NPKTG, and Cd-SrtBΔN26–NVQTG. The RMSFs are 0.19, 0.38, 0.48, and 0.58 Å for PPKTG SPKTG, NPKTG and NVQTG, respectively. The higher stability of P4 residues in (P/S)PKTG seen in our dynamic simulations correlates to the shorter DISCys-Thr and previously shown higher reaction activity (Donahue et al., 2014; Chambers et al., 2015).
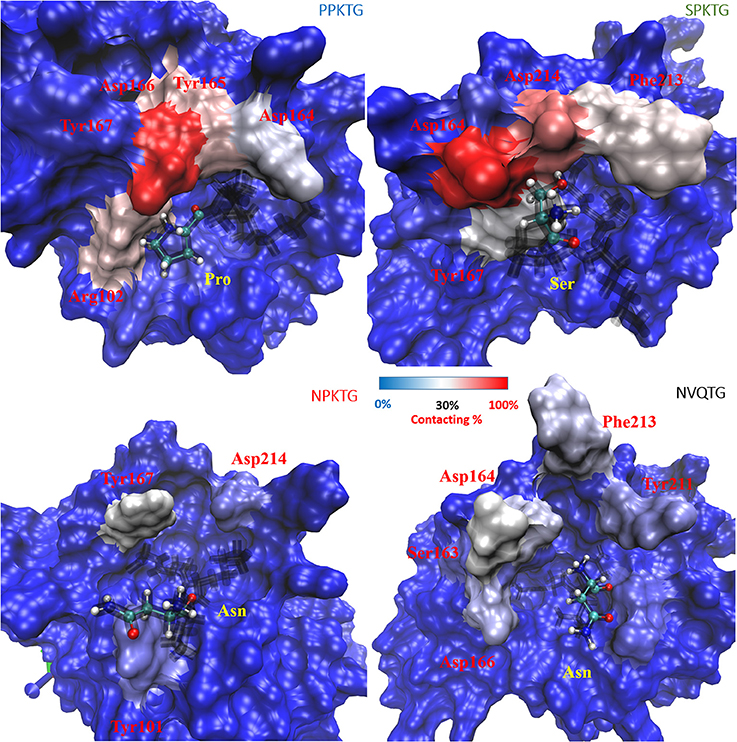
Figure 4. Contact frequency (to the P4 residue of substrate peptides) for residues in Cd-SrtBΔN26. Cd-SrtBΔN26 are shown in surface representation and opaque for the cases of Cd-SrtBΔN26–PPKTG (upper-left), Cd-SrtBΔN26–SPKTG (upper-right), Cd-SrtBΔN26–NPKTG (lower-left) and Cd-SrtBΔN26–NVQTG (lower-right). Different peptides are shown as black line, with the P4 residue labeled by ball-and-stick in CPK. The increased contact frequency for the P4 residues in different peptides is colored from blue to red for the purpose of easier visualization.
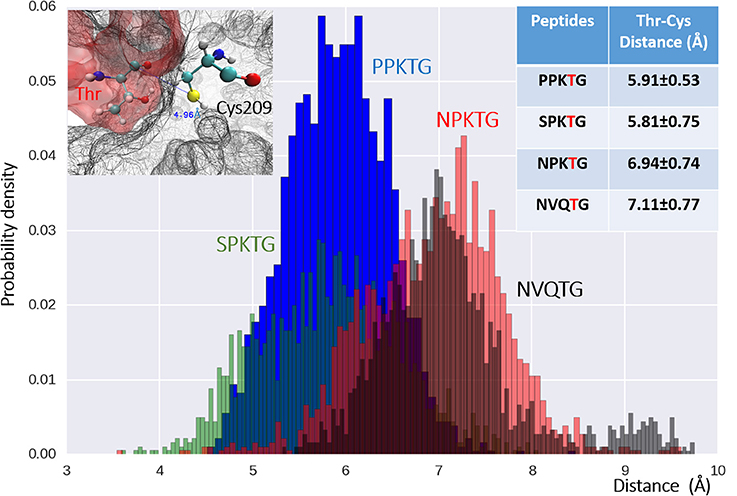
Figure 5. The distribution of distances between Cys209 (on SrtBΔN26) and Thr4 (on the peptides) for four examined peptides. Distances between the catalytic Cys209 in Cd-SrtBΔN26 (upper-left) and Thr4, the P4 residue in the peptides of Cd-SrtBΔN26–PPKTG (blue), Cd- SrtBΔN26–SPKTG (green), Cd-SrtBΔN26–NPKTG (red) and Cd-SrtBΔN26–NVQTG (black) of all the snapshots in simulations, are plotted in histogram. Mean distance and deviation for each peptide are provided in the table (upper-right). X-axis is the distance from the sulfur atom of Cys209 to the carboxyl carbon of the Thr in the peptides.
Discussion
In this work, we have presented the crystal structure of the catalytically active SrtB from C. difficile and provided a plausible interaction scheme to understand how SrtB recognizes the unique (S/P)PXTG motif.
The P4 residue of the sortase substrate is likely to be the specificity determinant. Based on our computational model of Cd-SrtBΔN26–PPKTG complex, we are able to identify that the hydrophobic residue Tyr167 in Cd-SrtBΔN26 forms specific interaction with P4 Pro in PPKTG, confirmed by site-directed mutagenesis and FRET-based assay (Figure 3). This hydrophobic interaction between sortase and substrate has not been seen in the current available crystal structures and may be unique to the Cd-SrtBΔN26–PPKTG complex. In the structure of Sa-SrtB–NPQT* complex, P4 Asn in NPQT* is hydrogen bonded to the carbonyl backbone of Thr177 within the β6/β7 loop in Sa-SrtB (Jacobitz et al., 2014). The residue Thr177 in Sa-SrtB is structurally equivalent to that of Ser163 in Cd-SrtBΔN26. However, structural superposition shows Ser163 in Cd-SrtBΔN26 is too far away to interact with P4 Asn in Sa-SrtB–NPQT*. Moreover, we also identified that Ser163 interacts with P2 Lys in PPKTG. As Tyr167A and Ser163A mutants exhibited reduced hydrolytic activity (Figure 3), we concluded that these two residues play important roles in specific substrate-binding and that the abolishment of the specific interactions affects the cleavage activity by Cd-SrtBΔN26. Taken together, the structural analyses have provided partial explanation why Cd-SrtBΔN26 does not recognize the NPQTN sorting signal. However, the actual crystal structure of Cd-SrtB–(S/P)PKT* is required to disclose the atomic interactions of the complex.
The simulation studies on the structural models of Cd-SrtBΔN26–PPKTG, Cd-SrtBΔN26–SPKTG, Cd-SrtBΔN26–NPKTG, and Cd-SrtBΔN26–NVQTG complexes suggest that the stability of P4 residue may have an effect on the position P1 residue and DISCys-Thr (Figures 4, 5). It seems reasonable to imply that the P4 Pro in PPKTG plays a role in configuring the substrate peptide to a preferred conformation, permitting Cd-SrtB to perform a more efficient cleavage. Furthermore, PPKTG and SPKTG that have a better Cd-SrtBΔN26 hydrolytic activity than NPKTG and NVQTG peptides (Donahue et al., 2014; Chambers et al., 2015) are found to have comparatively high contacting frequency with Cd-SrtBΔN26 via their P4 residue and a shorter DISCys-Thr throughout the simulations. It suggests the stabilization of P4 residue by surrounding loops near the active site can refrain the mobility of substrate peptides and therefore result in a shorter DISCys-Thr prompted for catalysis. The specificity determinant that associates with P4-led peptide conformation provides a molecular basis for specific recognition of PPKTG by Cd-SrtB.
Author Contributions
LY, IH, and SW conceived and designed the experiments. JY, CF, YL, JC, and YC performed he experiments. JY, YL, YH, JN, LY, IH, and SW analyzed the data. JY, CF, YL, LY, IH, and SW prepared the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the technical services provided by the Synchrotron Radiation Protein Crystallography Facility of the National Core Facility Program for Biotechnology, Ministry of Science and Technology, and the National Synchrotron Radiation Research Center, a national user facility supported by the Ministry of Science and Technology, Taiwan (R.O.C.). We are grateful to the National Center for High-Performance Computing at Hsinchu, Taiwan, for computer time and the use of their facilities. This work was supported by grants MOST 104-2113-M-007-019 to LY, MOST 102-2320-B-006-023-MY3 to IH, and MOST 103-2311-B-006-006 to SW from Ministry of Science and Technology, Taiwan.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fcimb.2016.00160/full#supplementary-material
References
Adams, P. D., Afonine, P. V., Bunkoczi, G., Chen, V. B., Davis, I. W., Echols, N., et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221. doi: 10.1107/S0907444909052925
Aslanidis, C., and de Jong, P. J. (1990). Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res. 18, 6069–6074. doi: 10.1093/nar/18.20.6069
Bagdasarian, N., Rao, K., and Malani, P. N. (2015). Diagnosis and treatment of Clostridium difficile in adults: a systematic review. JAMA 313, 398–408. doi: 10.1001/jama.2014.17103
Bartlett, J. G., Tedesco, F. J., Shull, S., Lowe, B., and Chang, T. (1980). Symptomatic relapse after oral vancomycin therapy of antibiotic-associated pseudomembranous colitis. Gastroenterology 78, 431–434.
Bradshaw, W. J., Davies, A. H., Chambers, C. J., Roberts, A. K., Shone, C. C., and Acharya, K. R. (2015). Molecular features of the sortase enzyme family. FEBS J. 282, 2097–2114. doi: 10.1111/febs.13288
Cascioferro, S., Totsika, M., and Schillaci, D. (2014). Sortase A: an ideal target for anti-virulence drug development. Microb. Pathog. 77, 105–112. doi: 10.1016/j.micpath.2014.10.007
Chambers, C. J., Roberts, A. K., Shone, C. C., and Acharya, K. R. (2015). Structure and function of a Clostridium difficile sortase enzyme. Sci. Rep. 5:9449. doi: 10.1038/srep09449
Chen, V. B., Arendall, W. B., Headd, J. J. III, Keedy, D. A., Immormino, R. M., Kapral, G. J., et al. (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21. doi: 10.1107/S0907444909042073
Clancy, K. W., Melvin, J. A., and McCafferty, D. G. (2010). Sortase transpeptidases: insights into mechanism, substrate specificity, and inhibition. Biopolymers 94, 385–396. doi: 10.1002/bip.21472
Comfort, D., and Clubb, R. T. (2004). A comparative genome analysis identifies distinct sorting pathways in gram-positive bacteria. Infect. Immun. 72, 2710–2722. doi: 10.1128/IAI.72.5.2710-2722.2004
Cozzi, R., Prigozhin, D., Rosini, R., Abate, F., Bottomley, M. J., Grandi, G., et al. (2012). Structural basis for group B streptococcus pilus 1 sortases C regulation and specificity. PLoS ONE 7:e49048. doi: 10.1371/journal.pone.0049048
Darden, T., York, D., and Pedersen, L. (1993). Particle mesh Ewald - An N.Log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092. doi: 10.1063/1.464397
Di Berardo, C., Capstick, D. S., Bibb, M. J., Findlay, K. C., Buttner, M. J., and Elliot, M. A. (2008). Function and redundancy of the chaplin cell surface proteins in aerial hypha formation, rodlet assembly, and viability in Streptomyces coelicolor. J. Bacteriol. 190, 5879–5889. doi: 10.1128/JB.00685-08
Dodin, M., and Katz, D. E. (2014). Faecal microbiota transplantation for Clostridium difficile infection. Int. J. Clin. Pract. 68, 363–368. doi: 10.1111/ijcp.12320
Donahue, E. H., Dawson, L. F., Valiente, E., Firth-Clark, S., Major, M. R., Littler, E., et al. (2014). Clostridium difficile has a single sortase, SrtB, that can be inhibited by small-molecule inhibitors. BMC Microbiol. 14:219. doi: 10.1186/s12866-014-0219-1
Dramsi, S., Caliot, E., Bonne, I., Guadagnini, S., Prévost, M. C., Kojadinovic, M., et al. (2006). Assembly and role of pili in group B streptococci. Mol. Microbiol. 60, 1401–1413. doi: 10.1111/j.1365-2958.2006.05190.x
Dramsi, S., Trieu-Cuot, P., and Bierne, H. (2005). Sorting sortases: a nomenclature proposal for the various sortases of Gram-positive bacteria. Res. Microbiol. 156, 289–297. doi: 10.1016/j.resmic.2004.10.011
Duke, P. S., and Fardy, J. (2014). Recurrent Clostridium difficile infection treated with home fecal transplantation: a case report. J. Med. Case Rep. 8, 393. doi: 10.1186/1752-1947-8-393
Emsley, P., Lohkamp, B., Scott, W. G., and Cowtan, K. (2010). Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501. doi: 10.1107/S0907444910007493
Fälker, S., Nelson, A. L., Morfeldt, E., Jonas, K., Hultenby, K., Ries, J., et al. (2008). Sortase-mediated assembly and surface topology of adhesive pneumococcal pili. Mol. Microbiol. 70, 595–607. doi: 10.1111/j.1365-2958.2008.06396.x
Feller, S. E., Zhang, Y. H., Pastor, R. W., and Brooks, B. R. (1995). Constant-pressure molecular-dynamics simulation - The Langevin piston method. J. Chem. Phys. 103, 4613–4621. doi: 10.1063/1.470648
Flock, J. I., Fröman, G., Jönsson, K., Guss, B., Signäs, C., Nilsson, B., et al. (1987). Cloning and expression of the gene for a fibronectin-binding protein from Staphylococcus aureus. EMBO J. 6, 2351–2357.
Frankel, B. A., Kruger, R. G., Robinson, D. E., Kelleher, N. L., and McCafferty, D. G. (2005). Staphylococcus aureus sortase transpeptidase SrtA: insight into the kinetic mechanism and evidence for a reverse protonation catalytic mechanism. Biochemistry 44, 11188–11200. doi: 10.1021/bi050141j
Frankel, B. A., Tong, Y., Bentley, M. L., Fitzgerald, M. C., and McCafferty, D. G. (2007). Mutational analysis of active site residues in the Staphylococcus aureus transpeptidase SrtA. Biochemistry 46, 7269–7278. doi: 10.1021/bi700448e
Gaspar, A. H., and Ton-That, H. (2006). Assembly of distinct pilus structures on the surface of Corynebacterium diphtheriae. J. Bacteriol. 188, 1526–1533. doi: 10.1128/JB.188.4.1526-1533.2006
Henrich, T. J., Krakower, D., Bitton, A., and Yokoe, D. S. (2009). Clinical risk factors for severe Clostridium difficile-associated disease. Emerging Infect. Dis. 15, 415–422. doi: 10.3201/eid1503.080312
Hu, P., Huang, P., and Chen, W. M. (2013). Curcumin inhibits the sortase a activity of the Streptococcus mutans UA159. Appl. Biochem. Biotechnol. 171, 396–402. doi: 10.1007/s12010-013-0378-9
Huang, I. H., Dwivedi, P., and Ton-That, H. (2010). Bacterial Pili and Fimbriae. Chichester: John Wiley & Sons Ltd.
Huang, J., and MacKerell, A. D. Jr. (2013). CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J. Comput. Chem. 34, 2135–2145. doi: 10.1002/jcc.23354
Humphrey, W., Dalke, A., and Schulten, K. (1996). VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38. doi: 10.1016/0263-7855(96)00018-5
Jacobitz, A. W., Wereszczynski, J., Yi, S. W., Amer, B. R., Huang, G. L., Nguyen, A. V., et al. (2014). Structural and computational studies of the Staphylococcus aureus sortase B-substrate complex reveal a substrate-stabilized oxyanion hole. J. Biol. Chem. 289, 8891–8902. doi: 10.1074/jbc.M113.509273
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., and Klein, M. L. (1983). Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935. doi: 10.1063/1.445869
Kabsch, W. (1976). A solution for the best rotation to relate two sets of vectors. Acta Cryst. 32, 922–923. doi: 10.1107/S0567739476001873
Kang, H. J., Coulibaly, F., Proft, T., and Baker, E. N. (2011). Crystal structure of Spy0129, a Streptococcus pyogenes class B sortase involved in pilus assembly. PLoS ONE 6:e15969. doi: 10.1371/journal.pone.0015969
Kelly, C. P., and LaMont, J. T. (1998). Clostridium difficile infection. Annu. Rev. Med. 49, 375–390. doi: 10.1146/annurev.med.49.1.375
Kelly, C. P., and LaMont, J. T. (2008). Clostridium difficile - More difficult than ever. N. Engl. J. Med. 359, 1932–1940. doi: 10.1056/NEJMra0707500
Kline, K. A., Kau, A. L., Chen, S. L., Lim, A., Pinkner, J. S., Rosch, J., et al. (2009). Mechanism for sortase localization and the role of sortase localization in efficient pilus assembly in Enterococcus faecalis. J. Bacteriol. 191, 3237–3247. doi: 10.1128/JB.01837-08
Leffler, D. A., and Lamont, J. T. (2009). Treatment of Clostridium difficile-associated disease. Gastroenterology 136, 1899–1912. doi: 10.1053/j.gastro.2008.12.070
LeMieux, J., Woody, S., and Camilli, A. (2008). Roles of the sortases of Streptococcus pneumoniae in assembly of the RlrA pilus. J. Bacteriol. 190, 6002–6013. doi: 10.1128/JB.00379-08
Manzano, C., Contreras-Martel, C., El Mortaji, L., Izoré, T., Fenel, D., Vernet, T., et al. (2008). Sortase-mediated pilus fiber biogenesis in Streptococcus pneumoniae. Structure 16, 1838–1848. doi: 10.1016/j.str.2008.10.007
Maresso, A. W., Wu, R., Kern, J. W., Zhang, R., Janik, D., Missiakas, D. M., et al. (2007). Activation of inhibitors by sortase triggers irreversible modification of the active site. J. Biol. Chem. 282, 23129–23139. doi: 10.1074/jbc.M701857200
Marraffini, L. A., and Schneewind, O. (2006). Targeting proteins to the cell wall of sporulating Bacillus anthracis. Mol. Microbiol. 62, 1402–1417. doi: 10.1111/j.1365-2958.2006.05469.x
Mazmanian, S. K., Liu, G., Ton-That, H., and Schneewind, O. (1999). Staphylococcus aureus sortase, an enzyme that anchors surface proteins to the cell wall. Science 285, 760–763. doi: 10.1126/science.285.5428.760
Mazmanian, S. K., Liu, G., Jensen, E. R., Lenoy, E., and Schneewind, O. (2000). Staphylococcus aureus sortase mutants defective in the display of surface proteins and in the pathogenesis of animal infections. Proc. Natl. Acad. Sci. U.S.A. 97, 5510–5515. doi: 10.1073/pnas.080520697
Mccoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C., and Read, R. J. (2007). Phaser crystallographic software. J. Appl. Cryst. 40, 658–674. doi: 10.1107/S0021889807021206
Michaud-Agrawal, N., Denning, E. J., Woolf, T. B., and Beckstein, O. (2011). MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 32, 2319–2327. doi: 10.1002/jcc.21787
Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., et al. (2011). REFMAC5 for the refinement of macromolecular crystal structures. Acta Cryst. 67, 355–367. doi: 10.1107/s0907444911001314
Oh, K. B., Nam, K. W., Ahn, H., Shin, J., Kim, S., and Mar, W. (2010). Therapeutic effect of (Z)-3-(2,5-dimethoxyphenyl)-2-(4-methoxyphenyl) acrylonitrile (DMMA) against Staphylococcus aureus infection in a murine model. Biochem. Biophys. Res. Commun. 396, 440–444. doi: 10.1016/j.bbrc.2010.04.113
Otwinowski, Z., and Minor, W. (1997). Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. doi: 10.1016/S0076-6879(97)76066-X
Paterson, G. K., and Mitchell, T. J. (2004). The biology of Gram-positive sortase enzymes. Trends Microbiol. 12, 89–95. doi: 10.1016/j.tim.2003.12.007
Perry, A. M., Ton-That, H., Mazmanian, S. K., and Schneewind, O. (2002). Anchoring of surface proteins to the cell wall of Staphylococcus aureus. III. Lipid II is an in vivo peptidoglycan substrate for sortase-catalyzed surface protein anchoring. J. Biol. Chem. 277, 16241–16248. doi: 10.1074/jbc.M109194200
Phillips, J. C., Braun, R., Wang, W., Gumbart, J., Tajkhorshid, E., Villa, E., et al. (2005). Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802. doi: 10.1002/jcc.20289
Rohlke, F., and Stollman, N. (2012). Fecal microbiota transplantation in relapsing Clostridium difficile infection. Therap. Adv. Gastroenterol. 5, 403–420. doi: 10.1177/1756283X12453637
Schechter, I., and Berger, A. (1967). On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 27, 157–162. doi: 10.1016/S0006-291X(67)80055-X
Schneewind, O., Mihaylova-Petkov, D., and Model, P. (1993). Cell wall sorting signals in surface proteins of gram-positive bacteria. EMBO J. 12, 4803–4811.
Schneewind, O., Model, P., and Fischetti, V. A. (1992). Sorting of protein A to the staphylococcal cell wall. Cell 70, 267–281. doi: 10.1016/0092-8674(92)90101-H
Spirig, T., Weiner, E. M., and Clubb, R. T. (2011). Sortase enzymes in Gram-positive bacteria. Mol. Microbiol. 82, 1044–1059. doi: 10.1111/j.1365-2958.2011.07887.x
Surawicz, C. M., Brandt, L. J., Binion, D. G., Ananthakrishnan, A. N., Curry, S. R., Gilligan, P. H., et al. (2013). Guidelines for diagnosis, treatment, and prevention of Clostridium difficile infections. Am. J. Gastroenterol. 108, 478–498. doi: 10.1038/ajg.2013.4
Suree, N., Liew, C. K., Villareal, V. A., Thieu, W., Fadeev, E. A., Clemens, J. J., et al. (2009). The structure of the Staphylococcus aureus sortase-substrate complex reveals how the universally conserved LPXTG sorting signal is recognized. J. Biol. Chem. 284, 24465–24477. doi: 10.1074/jbc.M109.022624
Suryadinata, R., Seabrook, S. A., Adams, T. E., Nuttall, S. D., and Peat, T. S. (2015). Structural and biochemical analyses of a Clostridium perfringens sortase D transpeptidase. Acta Crystallogr. D Biol. Crystallogr. 71, 1505–1513. doi: 10.1107/S1399004715009219
Tedesco, F. J., Gordon, D., and Fortson, W. C. (1985). Approach to patients with multiple relapses of antibiotic-associated pseudomembranous colitis. Am. J. Gastroenterol. 80, 867–868.
Ton-That, H., Liu, G., Mazmanian, S. K., Faull, K. F., and Schneewind, O. (1999). Purification and characterization of sortase, the transpeptidase that cleaves surface proteins of Staphylococcus aureus at the LPXTG motif. Proc. Natl. Acad. Sci. U.S.A. 96, 12424–12429. doi: 10.1073/pnas.96.22.12424
Ton-That, H., and Schneewind, O. (2003). Assembly of pili on the surface of Corynebacterium diphtheriae. Mol. Microbiol. 50, 1429–1438. doi: 10.1046/j.1365-2958.2003.03782.x
van Leeuwen, H. C., Klychnikov, O. I., Menks, M. A., Kuijper, E. J., Drijfhout, J. W., and Hensbergen, P. J. (2014). Clostridium difficile sortase recognizes a (S/P)PXTG sequence motif and can accommodate diaminopimelic acid as a substrate for transpeptidation. FEBS Lett. 588, 4325–4333. doi: 10.1016/j.febslet.2014.09.041
Viseur, N., Lambert, M., Delmee, M., Van Broeck, J., and Catry, B. (2011). Nosocomial and non-nosocomial Clostridium difficile infections in hospitalised patients in Belgium: compulsory surveillance data from 2008 to 2010. Euro Surveill. 16, pii: 20000.
Yang, Z., Lasker, K., Schneidman-Duhovny, D., Webb, B., Huang, C. C., Pettersen, E. F., et al. (2012). UCSF Chimera, MODELLER, and IMP: an integrated modeling system. J. Struct. Biol. 179, 269–278. doi: 10.1016/j.jsb.2011.09.006
Zhang, J., Liu, H., Zhu, K., Gong, S., Dramsi, S., Wang, Y. T., et al. (2014). Antiinfective therapy with a small molecule inhibitor of Staphylococcus aureus sortase. Proc. Natl. Acad. Sci. U.S.A. 111, 13517–13522. doi: 10.1073/pnas.1408601111
Zhang, R., Wu, R., Joachimiak, G., Mazmanian, S. K., Missiakas, D. M., Gornicki, P., et al. (2004). Structures of sortase B from staphylococcus aureus and Bacillus anthracis reveal catalytic amino acid triad in the active site. Structure 12, 1147–1156. doi: 10.1016/j.str.2004.06.001
Zong, Y. N., Bice, T. W., Ton-That, H., Schneewind, O., and Narayana, S. V. (2004a). Crystal structures of Staphylococcus aureus sortase A and its substrate complex. J. Biol. Chem. 279, 31383–31389. doi: 10.1074/jbc.M401374200
Keywords: Clostridium difficile, sortase, substrate specificity, crystal structure, fluorescence resonance energy transfer
Citation: Yin J-C, Fei C-H, Lo Y-C, Hsiao Y-Y, Chang J-C, Nix JC, Chang Y-Y, Yang L-W, Huang I-H and Wang S (2016) Structural Insights into Substrate Recognition by Clostridium difficile Sortase. Front. Cell. Infect. Microbiol. 6:160. doi: 10.3389/fcimb.2016.00160
Received: 04 October 2016; Accepted: 07 November 2016;
Published: 22 November 2016.
Edited by:
Justin Merritt, Oregon Health and Science University, USAReviewed by:
William D. Picking, University of Kansas, USAXingmin Sun, University of South Florida, USA
Copyright © 2016 Yin, Fei, Lo, Hsiao, Chang, Nix, Chang, Yang, Huang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lee-Wei Yang, bHd5YW5nQGxpZmUubnRodS5lZHUudHc=
I-Hsiu Huang, aWhzaXVodWFuZ0BtYWlsLm5ja3UuZWR1LnR3
Shuying Wang, c3N3YW5nMjNAbWFpbC5uY2t1LmVkdS50dw==
†These authors have contributed equally to this work.