 Pengpeng Xiao1,2
Pengpeng Xiao1,2 Jicheng Han1,2Ying Zhang3Chenghui Li1,2Xiaofang Guo4Shubo Wen2Mingyao Tian2,5Yiquan Li1,2Maopeng Wang2,6Hao Liu2,7Jingqiang Ren2,8Hongning Zhou4Huijun Lu2,5*Ningyi Jin1,2*
Jicheng Han1,2Ying Zhang3Chenghui Li1,2Xiaofang Guo4Shubo Wen2Mingyao Tian2,5Yiquan Li1,2Maopeng Wang2,6Hao Liu2,7Jingqiang Ren2,8Hongning Zhou4Huijun Lu2,5*Ningyi Jin1,2*- 1Yanbian University Medical College, Yanji, China
- 2Institute of Military Veterinary, Academy of Military Medical Sciences, Changchun, China
- 3College of Veterinary Medicine, College of Animal Science, Jilin University, Changchun, China
- 4Yunnan Institute of Parasitic Diseases, Simao, China
- 5Jiangsu Co-innovation Center for Prevention and Control of Important Animal Infectious Diseases and Zoonoses, Yangzhou, China
- 6Institute of Virology, Wenzhou University, Wenzhou, China
- 7School of Life Sciences and Engineering, Foshan University, Foshan, China
- 8Division of Economic Animal Epidemic, Institute of Special Economic Animal and Plant Sciences, Changchun, China
More than 6,000 mosquitoes of six species from six sites were collected and tested for their virome using metagenomics sequencing and bioinformatic analysis. The identified viral sequences belonged to more than 50 viral families. The results were verified by PCR of selected viruses in all mosquitoes, followed by phylogenetic analysis. In the present study, we identified the partial dengue virus (DENV), Zika virus (ZIKV), and Japanese encephalitis virus (JEV) sequences in mosquitoes. Metagenomic analysis and the PCR amplification revealed three DENV sequences, one of which encodes a partial envelope protein. Two ZIKV sequences both encoding partial nonstructural protein 3 and one JEV sequence encoding the complete envelope protein were identified. There was variability in the viral titers of the newly isolated virus JEV-China/YN2016-1 of different passage viruses. The newly identified Zika virus gene from ZIKV-China/YN2016-1 was an Asian genotype and shared the highest nucleotide sequence identity (97.1%) with a ZIKV sequence from Thailand isolated in 2004. Phylogenetic analysis of ZIKV-China/YN2016-1 and ZIKV-China/YN2016-2 with known Flavivirus genes indicated that ZIKV has propagated in Yunnan province, China.
Introduction
Mosquitoes are important hosts and insect vectors for a range of infectious viruses including Japanese encephalitis virus (JEV), Zika virus (ZIKV), dengue virus (DENV), and Getah virus (GETV), which pose a significant threat to human health and represent an economic burden worldwide (Pham et al., 2005; Klungthong et al., 2007). Mosquitoes acquire viruses when consuming blood from hosts undergoing viremia. The viruses then replicate and propagate in the insect host before being introduced into further victims during biting and blood feeding (Ritchie et al., 2007; Motooka et al., 2015). Mosquitoes feed on a wide range of hosts including humans and other vertebrates, invertebrates, and plants (Shi et al., 2014). Yunnan province in China harbors a diverse range of mosquito-borne viruses (Feng et al., 2015); therefore, regional surveillance is imperative. The detection of viruses in mosquitoes is usually performed by using reverse transcription polymerase chain reaction (RT-PCR) and nested PCR approaches (Almeida et al., 2005; Houghton, 2009; Li et al., 2015; Sim et al., 2015). However, compared with Illumina sequencing (Alquezar-Planas et al., 2013; Cholleti et al., 2016; Ergünay et al., 2017), these traditional methods are time-consuming and labor-intensive to detect low-level viromes in mosquito vectors (He et al., 2013; Miesen et al., 2016). Therefore, metagenomic analysis of mosquitoes is likely to be of great value to avoid missing the detection of viruses with high infectivity and pathogenicity as well as the detection of previously unknown viruses.
The present study aimed to build a valid surveillance method to monitor the distribution of Flaviviridae from mosquitoes in Yunnan province, China and to provide useful insights into viral isolation, prevention, and control. Diverse and abundant viromes from mosquitoes isolated in Yunnan province were investigated using metagenomic sequencing and nested PCR. The presence of DENV, JEV, and ZIKV viruses was confirmed, and JEV was isolated. This preliminary exploration of the metagenomes of mosquito-borne viruses lays the foundation for further research on the territorial distribution, diversity, and surveillance of mosquito-borne viruses in China and other countries.
Materials and Methods
Mosquito Collection
More than 6,000 living or freshly dead female mosquitoes were collected in Yunnan province, China, during August and September 2016 (Figure 1). Sample I comprised Culex tritaeniorhynchus (C. tritaeniorhynchus). Sample II was a mixture of Armigeres obturbans (Ar. obturbans) and Aedes albopictus (Ae. albopictus). Sample III was a collection of Anopheles sinensis (An. sinensis), Uranotaenia hebes (U. hebes), and Armigeres durhami (Ar. durhami) (Table 1). The mosquito samples were grouped based on species. C. tritaeniorhynchus was the predominant transmitting vector of JEV, and Ar. obturbans and Ae. albopictus were the predominant transmitting vectors of DENV and ZIKV (Byrd et al., 2013). The identified species of mosquitoes were collected separately and stored at −80°C. This research was approved by the Ethics Committee and the Research Ethics Committee of Yanbian University Medical College. All experiments involving active viruses were performed in a biosafety level 3 laboratory.
Figure 1. Distribution of sample collection sites in Yunnan province, China, 2016. The sample collection sites are labeled with red stars.
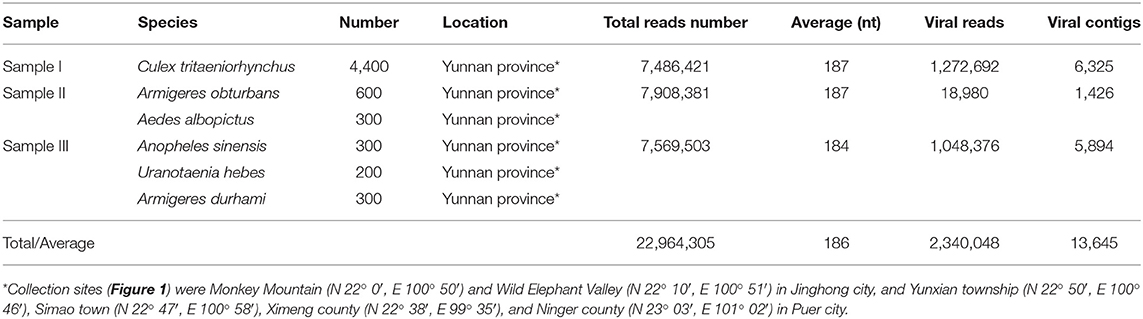
Table 1. Mosquito samples used in the metagenomic analysis and data from Illumina sequencing.
Preparation of Mosquito Samples
Briefly, each mosquito sample was mixed and ground using a tissue grinder with SM buffer (50 mM Tris, 10 mM MgSO4, 0.1 M NaCl, pH 7.5) (MeilunBio, Dalian, China). To remove mosquito debris and other materials, the mixed samples were centrifuged at 13,000 × g for 20 min, and the supernatants were used to extract the viral nucleic acid.
Extraction of Viral Nucleic Acid and Reverse Transcription
To remove the free nucleic acid and eliminate the contaminating host genomic DNA, 14 U Turbo DNase (Ambion, Austin, TX, USA), 25 U Benzonase Nuclease (Novagen, San Diego, CA, USA), 20 U RNase I (Fermentas, Ontario, Canada), and 10 × DNase buffer (Ambion) were added to 127 μL of the supernatants to a final volume of 150 μL, followed by digestion at 37°C for 1 h. The viral nucleic acid in the obtained products was extracted using a virus nucleic acid isolation kit (Bioer Technology, Hangzhou, China) according to the manufacturer's instructions. The total viral nucleic acids were reverse-transcribed using anchored random primers and Superscript III reverse transcriptase (Invitrogen, Carlsbad, CA, USA). The anchored random primers (Table 2) were added separately to the viral nucleic acid and incubated at 75°C for 5 min and then placed on ice for 5 min for denaturation. To obtain the reverse-transcribed product, 40 U of RNase OUT (Invitrogen, Carlsbad, USA), 200 U of SuperScript III reverse transcriptase (Invitrogen), 1 μL of 0.1 M dithiothreitol (DTT) (Invitrogen), 1 μL of 10 mM dNTPs (TaKaRa, Dalian China), 4 μL of 5 × first-strand buffer (Invitrogen), and RNase-free H2O (TaKaRa) were added to a final volume of 20 μL and incubated at 25°C for 10 min, followed by 50°C for 60 min and then 75°C for 10 min.

Table 2. Barcode DNA used in the metagenomic analysis (He et al., 2013).
Synthesis of Double-Strand cDNA (dscDNA)
Before the synthesis of dscDNA, 1 μL of RNase H (TaKaRa) was added to the obtained products to degrade the free RNA. To synthesize dscDNA, anchored random primers were added and incubated 75°C for 5 min and then on ice for 5 min for denaturation. Then, 1 μL of Klenow fragment (TaKaRa), 1 μL of 10 mM dNTPs (TaKaRa), 2 μL of 10 × Klenow buffer (TaKaRa), and 6 μL of dd H2O (TaKaRa) were added and the samples were incubated at 37°C for 60 min, followed by 75°C for 10 min. Then, 0.5 μL of exonuclease I (TaKaRa), 1 μL of shrimp alkaline phosphatase (SAP, TaKaRa, Dalian, China), 5 μL of 10 × phosphatase buffer (TaKaRa), and 24 μL of DEPC H2O (TaKaRa) were added and incubated at 37°C for 60 min followed by 75°C for 10 min to eliminate the phosphates and the free single-strand nucleic acid in the dscDNA reaction.
Sequence-Independent Single-Primer Amplification (SISPA) and Purification of PCR Products
To increase the quantity of the viral nucleic acids, SISPA was applied to amplify the dscDNA. The 50 μL reaction comprised 10 μL of dscDNA, 2 μL of barcode primer (Table 2), 1 μL of AccuPrime Taq DNA polymerase (Invitrogen), 5 μL of 10 × AccuPrime buffer I (Invitrogen), and 32 μL dd H2O (TaKaRa). The PCR condition comprised 95°C for 3 min; 40 circles of 95°C for 20 s, 54°C for 20 s, and 68°C for 70 s; and a final extension at 68°C for 7 min. The obtained PCR products were purified using a PCR purification kit (QIAGEN, Hilden, Germany) and eluted in 30 μL of TE buffer (100 mM Tris-HCl, 10 mM EDTA, pH 8.0) (Promega, Madison, USA).
Metaviral Sequencing
The purified PCR products from the three samples were sent to the Wuhan Genome Institute (BGI, Shenzhen, China) for Illumina sequencing. Briefly, to obtain ~180 bp DNA fragments, the purified PCR products were ultrasonicated, and dATP and Klenow fragment were added to produce 3′-dA overhangs. To establish genomic DNA libraries, DNA fragments were bound to Illumina adaptors and amplified using PCR with adaptor primers. Amplicons were ligated to flow cells to which fluorescently labeled dNTPs were added. DNA sequences were identified by using the sequencing-by-synthesis method (SBS, Illumina). Base calling was monitored by the program GAPipeline (BGI), with default settings. No-calling reads and adaptor sequences were eliminated. The remaining sequences were assembled into contigs using SOAPdenovo software (BGI, Shenzhen, China). Contigs and sequences longer than 100 bp were defined as significant data for further in silico analysis.
Computational Analysis
The contigs and sequences were aligned using the blastx and blastn with the nonredundant and viral reference sequences in the GenBank database. (https://www.ncbi.nlm.nih.gov/genbank/) BLAST hits with an E-value ≤ 10e−5 were considered significant. After eliminating the bacterial and eukaryotic sequences, we analyzed the virus-like sequences.
Identification of Detected Viruses
Based on the alignment outcomes of the viral contigs and the match position of the viral contigs with the corresponding viruses in GenBank, specific primers were designed and synthesized to identify the detected viruses. Viral nucleic acids were extracted by Bioer Technology (Hangzhou, China) and amplified using the designed primers and a PCR Master Mix (Tiangen, Beijing, China). To avoid false positives in Illumina sequencing, viral identification was performed three times in an independent manner.
Phylogenetic Analysis
The obtained PCR products were sequenced, and the sequences were aligned against sequences of the representative viruses using CLUSTAL W version 2.0. (Accession numbers are shown in the phylogenetic trees). To achieve better accuracy in phylogenetic analysis, neighbor-joining phylogenetic trees were produced using MEGA 7 with 1,000 bootstrap replicates (Wang et al., 2012; Li, 2015).
Cell Culture
The hamster cell line BHK-21 (conserved in our laboratory) was employed in this study. BHK-21 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (HyClone, Logan, UT, USA) with 10% fetal bovine serum (FBS) (HyClone) and 1% penicillin and streptomycin (Pen Strep) (HyClone), and incubated at 37°C in 5% CO2.
Isolation of Viruses
Viral isolation was conducted with the positive ground mosquito supernatants. Briefly, the ground mosquito supernatants were diluted seven-fold with DMEM containing 2% FBS and cultured with BHK-21 cells for 5–7 days. Cultures were examined daily for evidence of a virally induced cytopathic effect (CPE). Cultures without a CPE were blind-passaged three times.
Identification of the Antiviral Effect Using an Indirect Immunofluorescence Assay (IFA)
BHK-21 cells were seeded in 12-well plates, and after 24 h of culture, they were rinsed with phosphate-buffered saline (PBS) (Solarbio, Beijing, China) and fixed using methanol (Solarbio) for 10 min at room temperature. The cells were washed three times with PBS and then incubated in blocking buffer [3% bovine serum albumin (BSA) (BOSTER, Beijing, China) in Tris-buffered saline (Solarbio) and Tween20 (Solarbio)] for 1 h at room temperature. The cells were then incubated with an anti-E monoclonal antibody (Abcam, Cambridge, UK) (1:20) in PBS with 3% BSA overnight at 4°C. After the cells were washed three times in PBS, they were incubated with a fluorescently labeled secondary antibody—fluorescein isothiocyanate-conjugated goat antimouse antibody (ZSGB-Bio, Beijing, China)—in the dark at 37°C for 1 h. The nuclei were washed with PBS three times and stained with 4,6-diamidino-2-phenylindole (DAPI) (Solarbio). Slides were imaged under a fluorescence microscope (Olympus, Tokyo, Japan).
Observation by Negative-Stain Electron Microscopy
BHK-21 cells with suspected JEV-induced CPE were collected by repeated freezing and thawing three times, followed by centrifugation at 10,000 × g for 15 min. The obtained virus suspension was centrifuged at 60,000 × g for 4 h and the supernatant was removed gently and discarded. Subsequently, the viral precipitate was resuspended with an isometric mixture of 6.1% (v/v) pH 7.2 glutaraldehyde (HEDEBIO, Beijing, China) fixative and DMEM, 25 μL of which were added to the copper grid. After desiccation, one drop of 3% phosphotungstic acid (JINDU, Shanghai, China) was added for negative staining. Before observation under an electron microscope (FEI, Hillsboro, USA), the grid was placed in an incubator (SANYO, Osaka, Japan) at 37°C for desiccation.
Amplification and Phylogenetic Analysis of JEV Complete Genome
The JEV strain was isolated, and then the complete sequence was amplified using RT-PCR. The designed primers are displayed in Table 3. The complete JEV genome was assembled by PCR products using SeqMan version 7.1 (DNAStar, Madison, USA) with the default value. Subsequently, phylogenetic analysis was performed based on the complete sequence of JEV with other representative JEV strains reported in China and neighboring countries.

Table 3. Primer pairs used for the complete JEV genome.
Detection of the Viral Titer of Different Passages
The viral titers of different passages of JEV (passage 5, passage 10, passage 20, and passage 30) were evaluated in BHK-21 cells in triplicate, with DMEM as a control. Cells were incubated with serial ten-fold dilutions of JEV or DMEM for 120 h at 37°C. The viral titer TCID50 (50% tissue culture infective dose) was calculated by using the Reed–Muench method (Krah, 1991), according to the cytopathic effect (CPE) in cells caused by viruses. It was evaluated based on the changes in cell culture morphology under a light microscope (OLYMPUS, Tokyo, Japan) that were observed at 100 × magnification. The CPE of JEV or DMEM at each dilution was evaluated in eight replicates.
Variability of Envelope (E) Genes of Different Passage Viruses
Viral RNA was extracted from the supernatant using an RNA viral kit according to the manufacturer's protocol (Bioer Technology) after a CPE was induced in BHK-21 cells by different passage viruses (P5, P10, P20, and P30). The purified RNA was used as the template for cDNA synthesis using the SuperScript™ III first-strand synthesis system (Invitrogen) with the reverse primer JWR1 5′-AGATCCTGTGTTCTTCCTCACCACCA-3′, according to the manufacturer's instructions. The envelope (E) genes of different passage viruses were amplified using the paired primers shown in Table 4 and sequenced. The alignment of the four E genes was performed using MEGA 7.0.

Table 4. Primer pairs used for identification by means of nested PCR.
Statistical Analysis
The data were analyzed using SPSS 17.0 software. Group comparisons for viral titers were carried out by using the t-test for independent means. Experiments were performed in triplicate and a minimum of three independent experiments were evaluated. The value of P < 0.05 was considered statistically significant.
Genbank Accession Numbers
The data from Illumina sequencing have been deposited in the GenBank Sequence Reads Archive under accession numbers SRR7204303 to SRR7204305. The GenBank accession numbers of DENV-China/YN2016-1, DENV-China/YN2016-2, DENV-China/YN2016-3, ZIKV-China/YN2016-1, and ZIKV-China/YN2016-2 sequences are MG751801–MG751805. The GenBank accession numbers for the E gene and the complete genome of JEV-China/YN2016-1 are MG644382 and MH385014.
Results
Metaviral Sequencing and the Virome of Mosquitoes
To obtain the clean data of the virome of the mosquitoes, contaminating host sequences and barcode DNA were eliminated. A total of 22,964,305 reads were acquired by Illumina sequencing, with averaging read lengths of 186 nt (Table 1). The amount of viral sequences was 17.00, 0.24, and 13.85% in sample I, II, and III respectively and the viruses could be clearly classified (Figure 2). Among them, 87.4% were vertebrate viruses, 5.4% were plant viruses, and 1% were fungal viruses; insect viruses and phages accounted for 0.8 and 0.6%, respectively (Figure 2A). Most viral sequences in Sample I were from C. tritaeniorhynchus, the predominant JEV-transmitting vector in China, followed by A. obturbans. A. albopictus is the predominant ZIKV-transmitting vector. Viral sequences were classified at the family level (Figure 2B), and Flaviviridae was the predominant viral family, together with certain other important viral families such as Circoviridae, Anelloviridae, Parvoviridae, and Peribunyaviridea. Additionally, numerous unclassified viral sequences, presumably belonging to novel viruses not previously identified in mosquitoes, were identified and are worthy of further study.
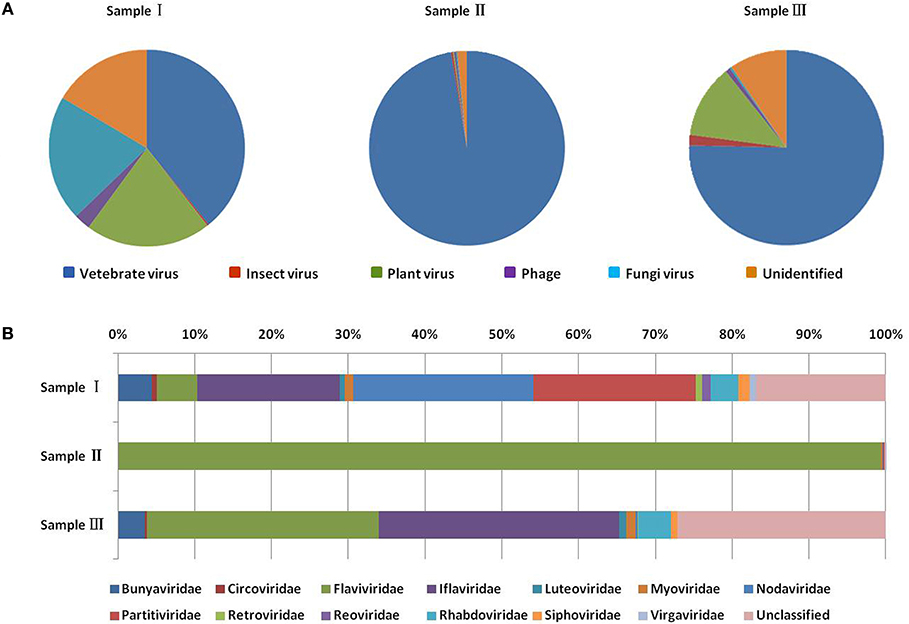
Figure 2. Categories of viral hosts and families of viral sequences in the three mosquito samples. Viral sequences were sorted according to the viral host category (A). The proportions of fungal viruses are too small to be seen in the figure. Viral sequences are classified at the family level (B). Families with <10 reads are not shown. Different host categories and families are indicated by different colors.
PCR Verification of the Metavirome Results
Viral sequences were assembled into contigs using SOAPdenovo. From the 13,645 assembled viral contigs, 32 DENV-like contigs with a read coverage of 184 × (236–1408 nt), 13 ZIKV-like contigs with a read coverage of 34 × (194–652 nt), and 26 JEV-like contigs with a read coverage of 52 × (306–1,564 nt) were obtained. The DENV-like contigs shared 92–98% nt identity with known DENV sequences. The ZIKV-like contigs shared 95–99% nt identity with known ZIKV sequences. The JEV-like contigs shared 96–99% nt identity with known JEV sequences. To confirm the outcome of metavirome sequencing, specific primers were designed and synthesized to amplify the identified viruses (Table 4). The following were the results of PCR identification in mosquito samples.
PCR Amplification of Dengue Virus
Dengue virus belongs to Flaviviridae, and 3,044 cases of dengue were reported in China in 2005–2012, among which 134 cases were reported in Yunnan province. Currently, there is no DENV-5 case reported in China. There have been DENV 1–4 outbreaks in China repeatedly and alternately. However, DENV-1 is predominant. (Xiong and Chen, 2014). Based on the alignment results of viral contigs, viral PCR amplicons from samples II shared a high identity with the dengue virus (DENV) genes, indicating that they belong to insect viral species. The results were verified by nested RT-PCR, which identified a 1,231 nt sequence (DENV-China/YN2016-1) encoding a partial nonstructural protein 4A, 4B, and 5 gene; a 491 nt sequence (DENV-China/YN2016-2) encoding a partial E protein, and a 939 nt sequence (DENV-China/YN2016-3) encoding a partial nonstructural protein 5 gene from DENV. Phylogenetic analysis showed that the newly identified DENV sequences were genotype IV. In addition, DENV-China/YN2016-2 was highly homologous to a DENV sequence from Thailand that was isolated in 1991, sharing 95.3% nt identity. This suggested that the viruses represented a DENV variant (Figure 3A). Interestingly, the partial E gene in DENV-China/YN2016-2 may represent a new genotype IV gene in the DENV variant strain.
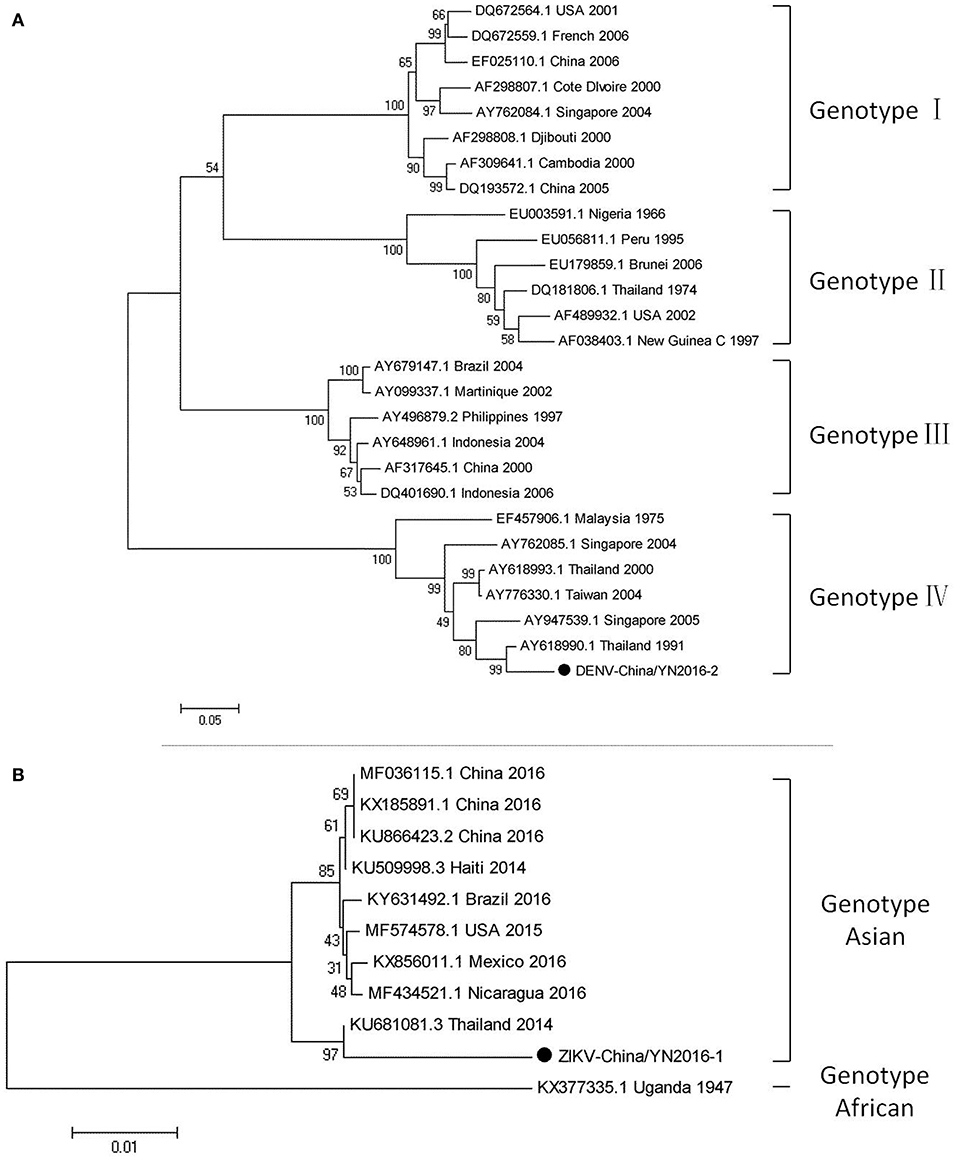
Figure 3. Phylogenetic trees of DENV and ZIKV. Phylogenetic trees based on the E gene of DENV (A) and the nonstructural protein NS3 of ZIKV (B). The trees were constructed using the p-distance-based neighbor-joining method in MEGA 7.0 software. Bootstrap values were calculated with 1,000 replicates. Black solid circles indicate the genes identified in this study.
PCR Amplification of Zika Virus
The first imported case of ZIKV, pertaining to Flaviviridae, was confirmed in China in February 2016 (Liu et al., 2016; Zhang et al., 2016). A total of 28 cases were reported in Zhejiang province, China in September 2016 (Deng et al., 2016; Li et al., 2016). In addition, 19 cases were reported in Guangdong province, China in 2016 (Sun et al., 2017). In line with the alignment results of viral contigs, viral PCR amplicons from sample II shared a high identity with ZIKV, also belonging to insect viral species. These results were also confirmed by means of nested RT-PCR, which identified 694 and 470 nt sequences, both encoding partial nonstructural protein 3 from ZIKV. These sequences were named ZIKV-China/YN2016-1 and ZIKV-China/YN2016-2, respectively. Phylogenetic analysis showed that the newly identified Zika virus sequence ZIKV-China/YN2016-1 was from an Asian genotype. The nucleotide identity analysis revealed that it shared the highest nucleotide sequence identity (97.1%) with a ZIKV sequence from Thailand that was isolated in 2004 (Figure 3B).
PCR Amplification of JEV
Since 1951, 33,900 cases of JEV, which belongs to Flaviviridae, were reported in China. The most obvious feature for the territory distribution of JE in China in the 1970s or 2010 is that a small number of cases were reported in west and north China, whereas a high number of cases occurred in east and southwest China. Yunnan province is still a highly epidemic area for JEV. First reported in 1949, JEV has existed in China for more than 60 years. The prevalence of JEV is genotype I and III in China, and genotype I is predominant in Yunnan province (Zheng et al., 2012). According to the alignment results of viral contigs, viral PCR amplicons from sample I shared a high identity with JEV genes, indicating that they belong to insect viral species. The results were verified by means of nested RT-PCR, which identified a 1,500 nt sequence (JEV-China/YN2016-1) encoding a complete E protein. Phylogenetic analysis showed that the newly identified JEV sequence, JEV-China/YN2016-1, was genotype I (Figure 4). Nucleotide identity analysis revealed that it shared the highest nucleotide sequence identity (98.8%) with a JEV sequence from Laos that was isolated in 2009.
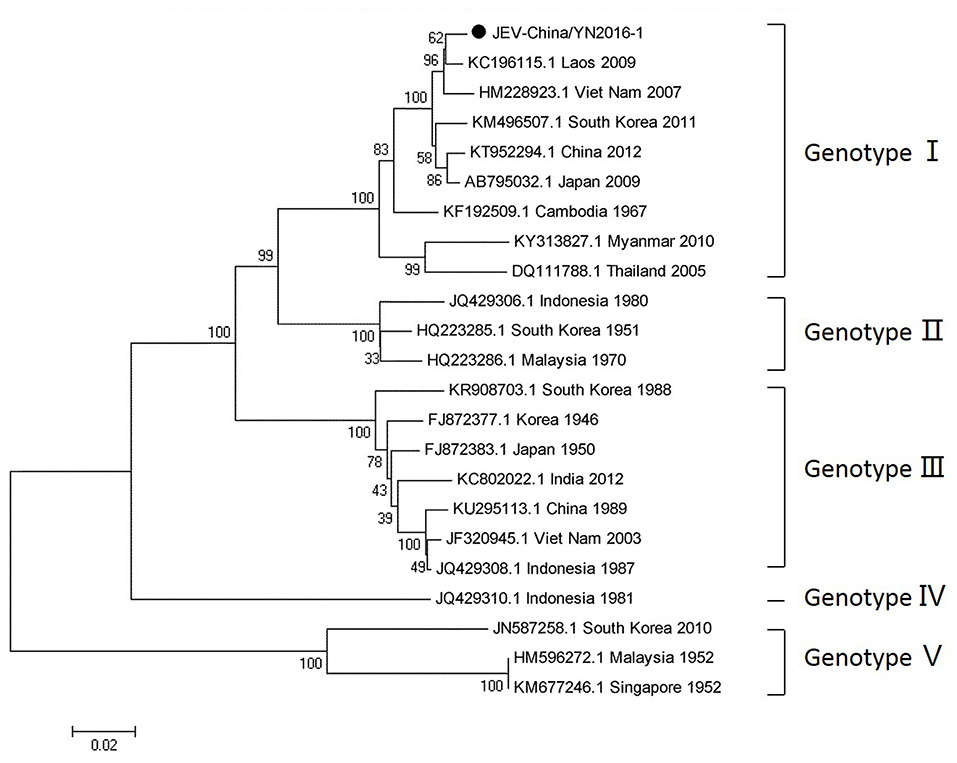
Figure 4. Phylogenetic trees of the JEV E gene. Phylogenetic trees based on the E gene of JEV. The trees were constructed using the p-distance-based neighbor-joining method in MEGA 7.0 software. Bootstrap values were calculated with 1,000 replicates. Black solid circles indicate the genes identified in this study.
Viral Identification of JEV and Phylogenetic Analysis of its Complete Genome
After inoculation into BHK-21 cells, the obtained JEV strain induced an obvious CPE. Monolayer cells appeared rounded, with bulging, aggregation, shedding, and incomplete destruction of a single loose layer, with gaps between cells. The severity of the CPE was time-dependent. JEV was propagated in BHK-21 cells for 24 h. The levels of intracellular virus were measured by IFA using an E-specific monoclonal antibody. As expected, mock-infected cells showed no E expression, while JEV-infected cells were positive for E expression. Viral particles were observed using negative-stain electron microscopy. The particles were rounded with a diameter of ~30–40 nm. Moreover, there were tiny protrusions on the surface, similar to JEV particles (Figure 5). The phylogenetic analysis of the complete JEV genome showed that JEV-China/YN2016-1 still shared the highest nucleotide sequence identity (97.6%) with a JEV sequence from Laos that was isolated in 2009 (Figure 6).
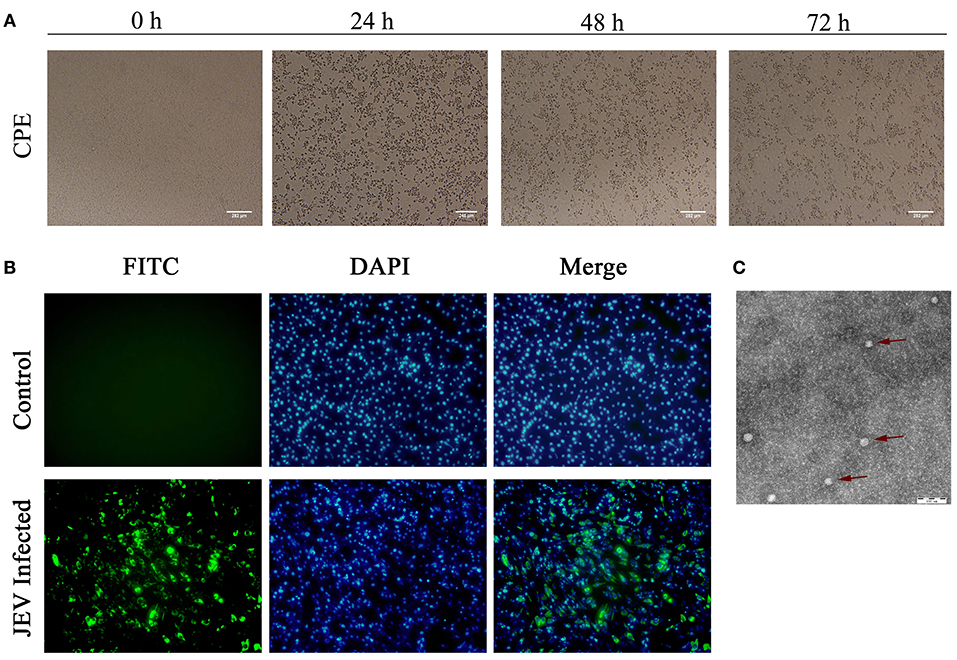
Figure 5. Identification of JEV-China/YN2016-1 isolated in Ximeng county of Yunnan province by CPE, IFA, and negative-stain electron microscopy. The CPE of BHK-21 cells infected with JEV-China/YN2016-1 at 24, 48, and 72 h (A). The indirect immunofluorescence assay (IFA) of the strain JEV-China/YN2016-1 using an anti-E monoclonal antibody (Abcam, Cambridge, UK) and FITC-conjugated goat antimouse antibody (ZSGB-Bio, Beijing, China) (B). Negative-stain electron microscopy of JEV-China/YN2016-1 particles (C).
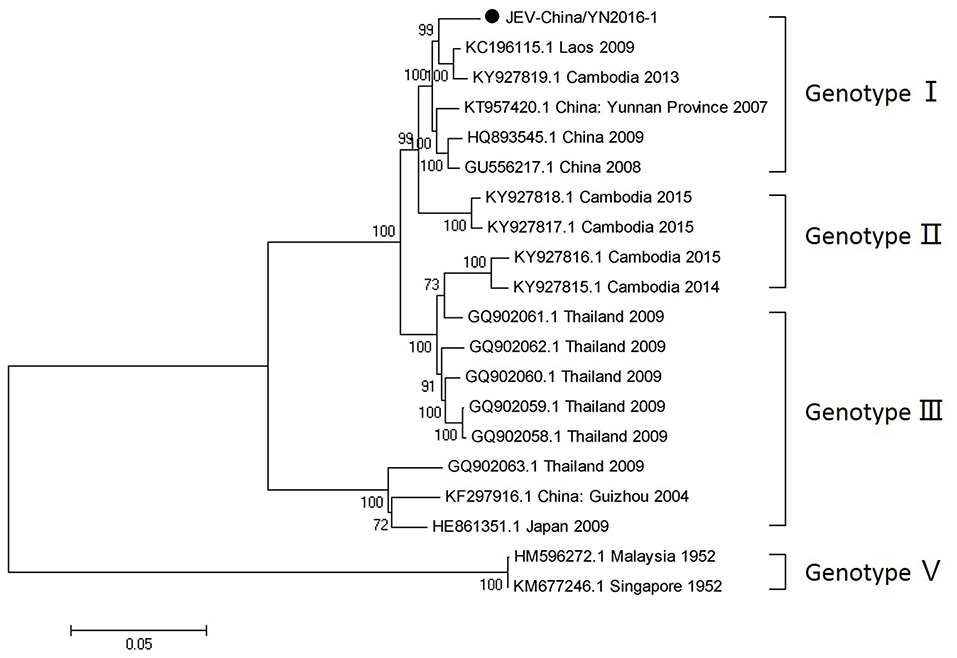
Figure 6. Phylogenetic trees of the complete JEV genome. Phylogenetic trees based on the complete genome of JEV. The trees were constructed using the p-distance-based neighbor-joining method in MEGA 7.0 software. Bootstrap values were calculated with 1,000 replicates. Black solid circles indicate the complete genome of JEV isolated in this study.
Viral Titer of JEV-China/YN2016-1 of Different Passage Viruses
In BHK-21 cells, the averages of viral titers of JEV-China/YN2016-1 of different passage viruses were 2.82 × 104 TCID50/0.1 mL (P5), 7.59 × 105 TCID50/0.1 mL (P10), 3.16 × 105 TCID50/0.1 mL (P20), and 2.14 × 104 TCID50/0.1 mL (P30). This showed that there was variability in viral titers. To test the evolution of viruses, a JEV strain with GenBank accession number JQ086762.1 was set as the control. Its averages of viral titers were 3.73 × 105 TCID50/0.1 mL (P5), 4.26 × 105 TCID50/0.1 mL (P10), 3.87 × 105 TCID50/0.1 mL (P20), and 4.14 × 105 TCID50/0.1 mL (P30). This indicated that the viral titers of the control were stable.
Variability of JEV-China/YN2016-1 E Gene of Different Passage Viruses
To avoid enzyme errors or sequencing errors, DNA polymerase Ex Taq (TaKaRa, Dalian, China), Fly DNA Polymerase (Transgen Biotech, Beijing, China), and Taq Master Mix (Tiangen, Beijing, China) were used three times to amplify the E gene of the JEV strain and the control at different viral passages. The PCR products of each viral passage were all sequenced, with consistent results in repeated trials. The JEV-China/YN2016-1 E genes of different passage viruses (P5, P10, P20, and P30) were aligned using MegAlign version 7.1 (DNAStar, Madison, USA) with the default value. Nucleotide identity analysis showed that P5 shared 99.1–99.2% nucleotide identity with the other passage viruses (P10, P20, and P30). P10 possessed 100% nucleotide identity with P20, and they both had 99.9% nucleotide identity with P30. By nucleotide alignment, 13 nucleotide sites in the E gene appeared with variation. Amino acid identity analysis showed that P5 shared 99.8% identity with P10 and P20, which shared 100% identity with each other. Interestingly, P5 and P30 had the same amino acid sequences, with no variation. By means of amino acid sequences alignment against those of P10 and P20, one amino acid site (position 138 aa) of E proteins was mutated, with a change of amino acid K to E.
Discussion
Mosquitoes are the intermediate hosts of numerous viruses that infect humans, animals, insects, plants, and other species, and play a significant role in the prevalence of many infectious diseases (Zhang et al., 2013; Fisher et al., 2015; Shi et al., 2015). Before 2007, virus identification by traditional virological methods required several decades and involved laboratories worldwide (Krah, 1991). Compared with the traditional method, metagenomic sequencing by Illumina sequencing combined with a high throughput analysis is more efficient, resulting in the identification of many viruses from different species in recent years by several laboratories (Wilson et al., 2015). Moreover, metagenomic sequencing has revealed the high abundance of viruses in mosquitoes (Bolling et al., 2015; Fauver et al., 2016; Shi et al., 2017). Metagenomic sequencing has also contributed to the discovery of novel viruses and their identification and characterization (Carissimo et al., 2016; Frey et al., 2016). In the present study, more than 50 viral families were detected by Illumina sequencing in mosquito samples. However, there was a considerable difference in the percentage of viral sequences obtained from the three mosquito groups. It could be related to the biology of the vectors (Atoni et al., 2018; Xia et al., 2018; Zakrzewski et al., 2018). Still, new findings of viral distribution and evolution in Yunnan province were obtained.
RT-PCR and nested-PCR were used to confirm the results of Illumina sequencing. The detection of DENV in the mosquito samples showed that the virus still exists in Yunnan province, and many strains of DENV have been uncovered by previous studies (Guo et al., 2013; Shi et al., 2014; Zhang et al., 2014; Wang et al., 2016). However, the detection of new DENV strains is important, as it suggests a new prevalence of DENV in this area. Moreover, the identification of the Zika virus genes is significant, suggesting that increased environmental surveillance is required because of its strong infectivity to humans, especially pregnant women and infants. Meanwhile, the presence of unidentified viruses in the Illumina sequencing results may ascribed to inadequate mosquito sampling and limited collection sites.
ZIKV and DENV were detected simultaneously, indicating that these viruses co-circulate in Yunnan province and suggesting that co-infection by the viruses is possible. Zika virus pathogenesis can be enhanced by preexisting dengue virus (Bardina et al., 2017). The simultaneous detection of the genes of two viruses may represent a new challenge to the control of ZIKV. We performed the isolation of DENV and ZIKV viruses in Vero and C6/36 cells besides BHK-21 cells as well as the suckling-mouse brain was also used in viral isolation. It was unfortunate that we could not isolate DENV and ZIKV.
The detection and isolation of JEV indicated that the virus still exists in Yunnan province. Analyzing the variant sites in the E gene and the consequences for the sequence of the encoded protein, we discovered that amino acid 138 in the E proteins of viruses at P5 and P30 is K (lysine). However, amino acid 138 in the E proteins of viruses at P10 and P20 is E (glutamic). Lysine at position 138 in the E protein is an attenuated viral site (Ni et al., 1994). The results showed that JEV-China/YN2016-1 has undergone mutations when passaged in BHK-21 cells, suggesting the possibility of increased threat to human health induced by JEV variation. The phylogenetic analysis based on the complete genome and the E gene of JEV-China/YN2016-1 showed that it shared the highest nucleotide sequence identity with a JEV sequence from Laos that was isolated in 2009, indicating the possible threat of JEV transboundary transmission.
This is the first report of the detection of Zika virus genes in Yunnan province, China. The newly identified DENV-China/YN2016-2 is a novel genotype IV gene in the DENV mutant strain. Metagenomic analysis of the viromes of mosquitoes in Yunnan province, combined with nested PCR, revealed that the distribution of viruses is dependent on both the mosquito species and the geographical location. The present work explored only a small portion of the virome of mosquitoes prevailing in this region of China. A much larger study on mosquitoes in other provinces in China and from other countries is needed to assess the diversity of mosquito-borne viruses. In conclusion, our findings provide a useful insight into the viral isolation and the characterization of Zika and dengue viruses and could hence be applied to larger studies in the future.
Author Contributions
PX, HJL, and NJ conceived and designed the experiments. PX, JH, CL, SW, YL, MW, and NJ performed the experiments. PX, HL, JR, and NJ analyzed the data. YZ, XG, HZ, HJL, and MT contributed reagents, materials, and analysis tools. PX and NJ wrote the paper. All authors read and approved the final version of the manuscript.
Funding
This work was supported by the National Program on Key Research Project of China [grant numbers 2016YFD0500401, 2017YFD0501803], the Special Fund for Agro-scientific Research in the Public Interest [grant number 201203082], the National Natural Science Foundation of China [grant number 31272573], the Yunnan Academician Workstation [grant number 2018IC151], and the National Natural Science Foundation [grant number 3180130343].
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Almeida, R. S., Spilki, F. R., Roehe, P. M., and Arns, C. W. (2005). Detection of Brazilian bovine respiratory syncytial virus strain by a reverse transcriptase-nested-polymerase chain reaction in experimentally infected calves. Vet. Microbiol. 105, 131–135. doi: 10.1016/j.vetmic.2004.11.004
Alquezar-Planas, D. E., Mourier, T., Bruhn, C. A., Hansen, A. J., Vitcetz, S. N., Mork, S., et al. (2013). Discovery of a divergent HPIV4 from respiratory secretions using second and third generation metagenomic sequencing. Sci. Rep. 3:2468. doi: 10.1038/srep02468
Atoni, E., Yujuan, W., Samuel, K., Cecilia, W., Ali, Z., Vincent, O., et al. (2018). Metagenomic virome analysis of Culex mosquitoes from Kenya and China. Viruses 10:30. doi: 10.3390/v10010030
Bardina, S. V., Bunduc, P., Tripathi, S., Duehr, J., Frere, J. J., Brown, J. A., et al. (2017). Enhancement of Zika virus pathogenesis by preexisting antiflavivirus immunity. Science 356, 175–180. doi: 10.1126/science.aal4365
Bolling, B. G., Scott, C. W., Robert, B. T., and Nikos, V. (2015). Insect-specific virus discovery: significance for the arbovirus community. Viruses 7, 4911–4928. doi: 10.3390/v7092851
Byrd, C. M., Grosenbach, D. W., Berhanu, A., Dai, D., Jones, K. F., Cardwell, K. B., et al. (2013). Novel benzoxazole inhibitor of dengue virus replication that targets the NS3 helicase. Antimicrob. Agents Chemother. 57, 1902–1912. doi: 10.1128/AAC.02251-12
Carissimo, G., Karin, E., Julie, R., Inge, H., Mawlouth, D., Diawo, D., et al. (2016). Identification and characterization of two novel RNA viruses from Anopheles gambiae species complex mosquitoes. PLoS ONE 11:e0153881. doi: 10.1371/journal.pone.0153881
Cholleti, H., Hayer, J., Abilio, A. P., Mulandane, F. C., Verner-Carlsson, J., Falk, K. I., et al. (2016). Discovery of novel viruses in mosquitoes from the zambezi valley of mozambique. PLoS ONE 11:e0162751. doi: 10.1371/journal.pone.0162751
Deng, Y. Q., Zhao, H., Li, X. F., Zhang, N. N., Liu, Z. Y., Jiang, T., et al. (2016). Isolation, identification and genomic characterization of the Asian lineage Zika virus imported to China. Sci. China Life Sci. 59, 428–430. doi: 10.1007/s11427-016-5043-4
Ergünay, K., Brinkmann, A., Litzba, N., Gunay, F., Kar, S., Oter, K., et al. (2017). A novel rhabdovirus, related to Merida virus, in field-collected mosquitoes from Anatolia and Thrace. Arch. Virol. 162, 1903–1911. doi: 10.1007/s00705-017-3314-4
Fauver, J. F., Nathan, D. G., Benjamin, J. K., James, W. L., Steven, M. L., Lawrence, S. F., et al. (2016). West African Anopheles gambiae mosquitoes harbor a taxonomically diverse virome including new insect-specific flaviviruses, mononegaviruses, and totiviruses. Virology 498, 288–299. doi: 10.1016/j.virol.2016.07.031
Feng, Y., He, B., Fu, S., Yang, W., Zhang, Y., Tu, C., et al. (2015). Isolation and identification of the Akabane virus from mosquitoes in Yunnan Province, China. Bing Du Xue. Bao 31, 51–57.
Fisher, R. G., Smith, D. M., Murrell, B., Slabbert, R., Kirby, B. M., Edson, C., et al. (2015). Next generation sequencing improves detection of drug resistance mutations in infants after PMTCT failure. J. Clin. Virol. 62, 48–53. doi: 10.1016/j.jcv.2014.11.014
Frey, K. G., Tara, B., Theron, H., Carlos, J. S., Guillermo, P., Vishwesh, P. M., et al. (2016). Bioinformatic characterization of mosquito viromes within the Eastern United States and Puerto Rico: discovery of novel viruses. Evol. Bioinform. Online 12 (Suppl 2):1–12. doi: 10.4137/EBO.S38518
Guo, X., Zhao, Q., Wu, C., Zuo, S., Zhang, X., Jia, N., et al. (2013). First isolation of dengue virus from Lao PDR in a Chinese traveler. Virol. J. 10:70. doi: 10.1186/1743-422X-10-70
He, B., Li, Z., Yang, F., Zheng, J., Feng, Y., Guo, H., et al. (2013). Virome profiling of bats from Myanmar by metagenomic analysis of tissue samples reveals more novel Mammalian viruses. PLoS ONE 8:e61950. doi: 10.1371/journal.pone.0061950
Houghton, M. (2009). The long and winding road leading to the identification of the hepatitis C virus. J. Hepatol. 51, 939–948. doi: 10.1016/j.jhep.2009.08.004
Klungthong, C., Gibbons, R. V., Thaisomboonsuk, B., Nisalak, A., Kalayanarooj, S., Thirawuth, V., et al. (2007). Dengue virus detection using whole blood for reverse transcriptase PCR and virus isolation. J. Clin. Microbiol. 45, 2480–2485. doi: 10.1128/JCM.00305-07
Krah, D. L. (1991). A simplified multiwell plate assay for the measurement of hepatitis A virus infectivity. Biologicals 19, 223–227. doi: 10.1016/1045-1056(91)90039-M
Li, H., Cao, Y. X., He, X. X., Fu, S. H., Lyu, Z., He, Y., et al. (2015). Real-time RT-PCR assay for the detection of tahyna virus. Biomed. Environ. Sci. 28, 374–377. doi: 10.3967/bes2015.052
Li, J. F. (2015). A fast neighbor joining method. Genet. Mol. Res. 14, 8733–8743. doi: 10.4238/2015.July.31.22
Li, X. F., Han, J. F., Shi, P. Y., and Qin, C. F. (2016). Zika virus: a new threat from mosquitoes. Sci. China Life Sci. 59, 440–442. doi: 10.1007/s11427-016-5020-y
Liu, L., Wu, W., Zhao, X., Xiong, Y., Zhang, S., Liu, X., et al. (2016). Complete genome sequence of Zika virus from the first imported case in mainland China. Genome Announc. 4:e00291–16. doi: 10.1128/genomeA.00618-16
Miesen, P., Ivens, A., Buck, A. H., and van Rij, R. P. (2016). Small RNA Profiling in Dengue virus 2-infected aedes mosquito cells reveals viral piRNAs and novel host miRNAs. PLoS Negl. Trop. Dis. 10:e0004452. doi: 10.1371/journal.pntd.0004452
Motooka, D., Nakamura, S., Hagiwara, K., and Nakaya, T. (2015). Viral detection by high-throughput sequencing. Methods Mol. Biol. 1236, 125–134. doi: 10.1007/978-1-4939-1743-3_11
Ni, H., Burns, N. J., Chang, G. J., Zhang, M. J., Wills, M. R., Trent, D. W., et al. (1994). Comparison of nucleotide and deduced amino acid sequence of the 5' non-coding region and structural protein genes of the wild-type Japanese encephalitis virus strain SA14 and its attenuated vaccine derivatives. J. Gen. Virol. 75, 1505–1510. doi: 10.1099/0022-1317-75-6-1505
Pham, T. N., Macparland, S. A., Coffin, C. S., Lee, S. S., Bursey, F. R., and Michalak, T. I. (2005). Mitogen-induced upregulation of hepatitis C virus expression in human lymphoid cells. J. Gen. Virol. 86, 657–666. doi: 10.1099/vir.0.80624-0
Ritchie, S. A., van den Hurk, A. F., Zborowski, P., Kerlin, T. J., Banks, D., Walker, J. A., et al. (2007). Operational trials of remote mosquito trap systems for Japanese encephalitis virus surveillance in the Torres Strait, Australia. Vector Borne Zoonotic Dis. 7, 497–506. doi: 10.1089/vbz.2006.0643
Shi, C., Liu, Y., Hu, X., Xiong, J., Zhang, B., and Yuan, Z. (2015). A metagenomic survey of viral abundance and diversity in mosquitoes from Hubei province. PLoS ONE 10:e0129845. doi: 10.1371/journal.pone.0129845
Shi, J., Duan, Z., Sun, J., Wu, M., Wang, B., Zhang, J., et al. (2014). Identification and validation of a novel microRNA-like molecule derived from a cytoplasmic RNA virus antigenome by bioinformatics and experimental approaches. Virol. J. 11:121. doi: 10.1186/1743-422X-11-121
Shi, M., Peter, N., Jay, N., John, E., Allison, I., and Edward, C. H. (2017). High-resolution metatranscriptomics reveals the ecological dynamics of mosquito-associated RNA viruses in western Australia. J. Virol. 91, e00680–17. doi: 10.1128/JVI.00680-17
Sim, S., Aw, P. P., Wilm, A., Teoh, G., Hue, K. D., Nguyen, N. M., et al. (2015). Tracking dengue virus intra-host genetic diversity during human-to-mosquito transmission. PLoS Negl. Trop. Dis. 9:e0004052. doi: 10.1371/journal.pntd.0004052
Sun, J., Wu Zhong, H., Guan, D., Zhang, H., Tan, Q., et al. (2017). Returning ex-patriot Chinese to Guangdong, China, increase the risk for local transmission of Zika virus. J. Infect. 75, 356–367. doi: 10.1016/j.jinf.2017.07.001
Wang, B., Yang, H., Feng, Y., Zhou, H., Dai, J., Hu, Y., et al. (2016). The distinct distribution and phylogenetic characteristics of dengue virus serotypes/genotypes during the 2013 outbreak in Yunnan, China: Phylogenetic characteristics of 2013 dengue outbreak in Yunnan, China. Infect. Genet. Evol. 37, 1–7. doi: 10.1016/j.meegid.2015.10.022
Wang, J., Guo, M. Z., and Xing, L. L. (2012). FastJoin, an improved neighbor-joining algorithm. Genet. Mol. Res. 11, 1909–1922. doi: 10.4238/2012.July.19.10
Wilson, M. R., Shanbhag, N. M., Reid, M. J., Singhal, N. S., Gelfand, J. M., Sample, H. A., et al. (2015). Diagnosing balamuthia mandrillaris encephalitis with metagenomic deep sequencing. Ann. Neurol. 78, 722–730. doi: 10.1002/ana.24499
Xia, H., Yujuan, W., Chenyan, S., Evans, A., Lu, Z., and Zhiming, Y. (2018). Comparative metagenomic profiling of viromes associated with four common mosquito species in China. Virol. Sin. 33, 59–66. doi: 10.1007/s12250-018-0015-4
Xiong, Y., and Chen, Q. (2014). Epidemiology of dengue fever in China since 1978. Nan Fang Yi Ke Da Xue Xue Bao 34, 1822–1825.
Zakrzewski, M., Gordana, R., Jonathan, D., Lutz, K., Yee, S. P., Igor, F., et al. (2018). Mapping the virome in wild-caught Aedes aegypti from Cairns and Bangkok. Sci. Rep. 8:4690. doi: 10.1038/s41598-018-22945-y
Zhang, F. C., Zhao, H., Li, L. H., Jiang, T., Hong, W. X., Wang, J., et al. (2014). Severe dengue outbreak in Yunnan, China, 2013. Int. J. Infect. Dis. 27, 4–6. doi: 10.1016/j.ijid.2014.03.1392
Zhang, X., Sheng, J., Plevka, P., Kuhn, R. J., Diamond, M. S., and Rossmann, M. G. (2013). Dengue structure differs at the temperatures of its human and mosquito hosts. Proc. Natl. Acad. Sci. U.S.A. 110, 6795–6799. doi: 10.1073/pnas.1304300110
Zhang, Y., Chen, W., Wong, G., Bi, Y., Yan, J., Sun, Y., et al. (2016). Highly diversified Zika viruses imported to China, 2016. Protein Cell 7, 461–464. doi: 10.1007/s13238-016-0274-5
Keywords: metagenomic analysis, mosquito, virome, virus detection, phylogenetic analysis
Citation: Xiao P, Han J, Zhang Y, Li C, Guo X, Wen S, Tian M, Li Y, Wang M, Liu H, Ren J, Zhou H, Lu H and Jin N (2018) Metagenomic Analysis of Flaviviridae in Mosquito Viromes Isolated From Yunnan Province in China Reveals Genes From Dengue and Zika Viruses. Front. Cell. Infect. Microbiol. 8:359. doi: 10.3389/fcimb.2018.00359
Received: 14 March 2018; Accepted: 24 September 2018;
Published: 24 October 2018.
Edited by:
Rachel L. Roper, The Brody School of Medicine at East Carolina University, United StatesReviewed by:
José Eduardo Levi, Universidade de São Paulo, BrazilQiangming Sun, Institute of Medical Biology (CAMS), China
Copyright © 2018 Xiao, Han, Zhang, Li, Guo, Wen, Tian, Li, Wang, Liu, Ren, Zhou, Lu and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huijun Lu, aHVpanVuX2x1QDEyNi5jb20=
Ningyi Jin, bmluZ3lpa0AxMjYuY29t