 Kristin R. Baker1,2*
Kristin R. Baker1,2* Bimal Jana1,2
Bimal Jana1,2 Anna Mette Hansen3
Anna Mette Hansen3 Hanne Mørck Nielsen4
Hanne Mørck Nielsen4 Henrik Franzyk3*Luca Guardabassi1,5
Henrik Franzyk3*Luca Guardabassi1,5- 1Department of Veterinary and Animal Sciences, Faculty of Health and Medical Sciences, University of Copenhagen, Frederiksberg, Denmark
- 2Department of Biomedical Sciences, Ross University School of Veterinary Medicine, Basseterre, Saint Kitts and Nevis
- 3Department of Drug Design and Pharmacology, Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark
- 4Department of Pharmacy, Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark
- 5Department of Pathobiology and Population Sciences, Royal Veterinary College, Hatfield, United Kingdom
Synthetic peptidomimetics may be designed to mimic functions of antimicrobial peptides, including potentiation of antibiotics, yet possessing improved pharmacological properties. Pairwise screening of 42 synthetic peptidomimetics combined with the antibiotics azithromycin and rifampicin in multidrug-resistant (MDR) Escherichia coli ST131 and Klebsiella pneumoniae ST258 led to identification of two subclasses of α-peptide/β-peptoid hybrids that display synergy with azithromycin and rifampicin (fractional inhibitory concentration indexes of 0.03–0.38). Further screening of the best three peptidomimetics in combination with a panel of 21 additional antibiotics led to identification of peptidomimetics that potentiated ticarcillin/clavulanate and erythromycin against E. coli, and clindamycin against K. pneumoniae. The study of six peptidomimetics was extended to Pseudomonas aeruginosa, confirming synergy with antibiotics for five of them. The most promising compound, H-(Lys-βNPhe)8-NH2, exerted only a minor effect on the viability of mammalian cells (EC50 ≥ 124–210 μM), and thus exhibited the highest selectivity toward bacteria. This compound also synergized with rifampicin and azithromycin at sub-micromolar concentrations (0.25–0.5 μM), thereby inducing susceptibility to these antibiotics at clinically relevant concentrations in clinical MDR isolates. This peptidomimetic lead and its analogs constitute promising candidates for efficient repurposing of rifampicin and azithromycin against Gram-negative pathogens.
Introduction
Traditional antimicrobial therapies have become ineffective in treating infections caused by multidrug-resistant (MDR) Gram-negative pathogens. Thus, new therapeutic strategies are required to manage these infections (Doi et al., 2017; World Health Organisation, 2017). Emergence of resistance to mono-drug therapies has drawn attention to combination therapies with new agents that limit resistance development or overcome resistance mechanisms (Kalan and Wright, 2011; Brown, 2015; Ribeiro et al., 2015; Brown and Wright, 2016; Melander and Melander, 2017; Singh et al., 2017; Moon and Huang, 2018). In this regard, co-application of certain peptides has been found to constitute a promising means for overcoming intrinsic resistance to certain classes of antimicrobials (e.g., macrolides, lincosamides and rifamycins) in Gram-negative pathogens due to their ability to increase the permeability of the outer membrane (Gill et al., 2015; Baker et al., 2018).
Despite the potential utility of peptides as potentiators of antibiotics, their clinical use is hampered by unfavorable pharmacological properties. In particular, many peptides also affect the viability of mammalian cells and possess inherently low stability toward proteolytic degradation, which (particularly for linear peptides) represents a major obstacle for their development into drugs (Ghosh and Haldar, 2015; Mojsoska and Jenssen, 2015; Molchanova et al., 2017a). While most naturally occurring peptides consist exclusively of α-amino acid residues, peptidomimetics incorporate unnatural amino acids or mimics thereof, resulting in alternative backbones that resist enzymatic cleavage (Molchanova et al., 2017a). In addition, end modifications (Jahnsen et al., 2015) may broaden the chemical space covered. These features of peptidomimetics enable design of compounds with fine-tuned physicochemical properties that confer enhanced antibacterial activity as well as an improved pharmacological profile (Méndez-Samperio, 2014). Moreover, certain peptidomimetics have proved capable of potentiating traditional antibiotics (Renau et al., 2002; Goldberg et al., 2013; Jammal et al., 2015; Lainson et al., 2017), suggesting a possible role in combination therapy. Despite displaying several features that are superior to those of peptides, their therapeutic potential relies on technological advances to enable reduced production cost, improved bioavailability, and minimized toxicity toward host cells (Molchanova et al., 2017a). Nonetheless, recent advances in synthesis of peptidomimetics has enabled investigation of diverse compound arrays, thus facilitating identification of peptidomimetic antibiotic adjuvants with suitable pharmacological properties.
Benefiting from already available peptidomimetic arrays (Hein-Kristensen et al., 2011; Jahnsen et al., 2012, 2014, 2015; Liu et al., 2013), we performed a search for peptidomimetics capable of circumventing the intrinsically low susceptibility to certain antibiotics in Gram-negative pathogens. To address potential toxicity issues, which are considered the most critical factor currently limiting the therapeutic use of peptidomimetics, the screening process was designed so that only compounds that potentiate antibiotics at sub-micromolar nontoxic concentrations were pursued. Thus, screening of pairwise peptidomimetic-antibiotic combinations led to identification of α-peptide/β-peptoid hybrids capable of inducing susceptibility to azithromycin and rifampicin (below their putative clinical breakpoints) in clinical MDR isolates of Escherichia coli, K. pneumoniae and P. aeruginosa.
Materials and Methods
Bacterial Strains, Antibiotics and Media
Bacterial strains used in this study comprise MDR clinical isolates of E. coli ST131 (Cerquetti et al., 2010) and K. pneumoniae ST258 (Jana et al., 2017), and American type culture collection (ATCC) strains: ATCC 25922 (E. coli), ATCC 13883 (K. pneumoniae), and ATCC 27853 (P. aeruginosa). Additional clinical isolates of E. coli, K. pneumoniae, and P. aeruginosa resistant to β-lactams (including carbapenems and cephalosporins) were provided by Laurent Poirel, Université de Fribourg, Switzerland. The following growth media was routinely used for bacterial culturing: Luria-Burtani (LB), cation-adjusted Mueller-Hinton agar (MHA) and broth (MHB II). All assays were performed in cation-adjusted MHA or MHB II. Antibiotics were purchased from Sigma-Aldrich and the peptidomimetic collection (Table S1) was curated and provided by Henrik Franzyk. This collection contained peptoids, α-peptide/β-peptoid hybrids, and a variety of end-group modifications, thereby representing a wide range of physicochemical and structural properties, including varied cationic character, length and hydrophobicity. Stock solutions were prepared by dissolving the compounds in deionized water, while test solutions were obtained by further dilution with MHB II medium.
Peptidomimetic Synthesis
α-Peptide/β-peptoid peptidomimetics were synthesized on a Rink amide resin (loading: 0.5–0.7 mmol/g; 0.05–0.1 mmol scale) in Teflon reactors (10 mL) by standard Fmoc-based solid-phase synthesis using the appropriate dimeric building blocks (Bonke et al., 2008; Jahnsen et al., 2014). Fmoc deprotection was performed with excess 20% piperidine in DMF (2 × 10 min, each time with 5 mL; shaking at room temperature). After Fmoc deprotection (and after coupling as well) the resin was washed with DMF, MeOH, and DCM (each 3 × 3 min with 5 mL). Coupling was performed with the appropriate building block (2.0 equiv. for loading, 2.5 equiv for the first two elongations, and 3.0 equiv. for subsequent elongations) and PyBOP/DIPEA (1:2 equiv. relative to the building block) in DMF (2–3 mL) for >2 h under shaking at room temperature. Capping was applied after the fourth coupling via treatment with Ac2O–DIPEA–NMP 1:2:3 (5 mL, 10 min at room temperature). Final Fmoc deprotection was followed by attachment of any N-terminal end group: the carboxylic acid (5 equiv.) corresponding to the desired moiety was added under conditions identical to those applied for the above coupling procedure used for the dimeric building blocks. Cleavage and simultaneous side chain deprotection: Excess TFA-CH2Cl2 90:10 (2 × 30 min under shaking at room temperature). The filtrate was collected and the resin was eluted with CH2Cl2 (2 mL). The combined filtrates were concentrated in vacuo, and co-evaporated three times with toluene. The crude products were purified by using preparative HPLC, and the resulting pure fractions were freeze-dried as previously described (Jahnsen et al., 2012, 2015; Skovbakke et al., 2015; Molchanova et al., 2017b).
Antimicrobial Susceptibility Testing
Minimal inhibitory concentrations (MICs) of peptidomimetics, antibiotics and their combinations were determined by broth microdilution according to the Clinical Laboratory Standards Institute performance standards (Clinical and Laboratory Standards Institute, 2013). Based on previous studies indicating that among a range of tested antibiotics belonging to several classes azithromycin and rifampicin displayed a high propensity to synergize with peptides, and hence these antibiotics were chosen for the initial screening of peptidomimetics to identify possible antibiotic adjuvants (Jammal et al., 2015; Baker et al., 2016, 2018; Corbett et al., 2017; Lyu et al., 2017; Wu et al., 2017; Domalaon et al., 2018a,b; Yang et al., 2018). Here, a sub-MIC concentration of 0.5 μM peptidomimetic was tested in combination with 0.5 μg/mL rifampicin or 1 μg/mL azithromycin in 96-well plates, followed by inoculum addition according to CLSI guidelines. At these concentrations, control wells containing either peptidomimetic or antibiotic individually did not inhibit growth. Since rifampicin and azithromycin mainly are used to treat infections with Gram-positive pathogens only CLSI clinical breakpoints were available for such bacteria. Hence the clinically relevant test concentrations were selected based on the CLSI susceptibility breakpoints for Staphylococcus species (Clinical and Laboratory Standards Institute, 2017): rifampicin (1 μg/mL) and azithromycin (2 μg/mL). Potentiation of additional antibiotics by peptidomimetics was determined by using commercial 96-well plates containing varied concentrations of standard antibiotics (COMPAN1F; Trek Diagnostic Systems Inc.). The peptidomimetics were added immediately prior to inoculation with pathogen, and then MICs were determined as above. The MICs of peptidomimetic/antibiotic combinations were determined according to the method above, and the concentration ratios were selected based upon checkerboard results with 2:1 and 1:4 ratio of peptidomimetic with rifampicin or azithromycin, respectively. Minimal bactericidal concentrations (MBCs) were determined by using growth-curve assay plates. MIC plates were prepared as above, and then the plates were incubated at 37°C with continuous shaking. Growth was recorded at 10 min intervals as optical density (OD) at 600 nm during 24 h. At 24 h, for wells without growth, a sample was diluted 10-fold in sterile saline (0.9% sodium chloride) and plated onto MHA, following CLSI guidelines (Clinical and Laboratory Standards Institute, 1999).
Checkerboard Synergy Assay
The potential synergistic effects in peptidomimetic-antibiotic combinations were assessed by using a two-dimensional checkerboard assay (Garcia, 2010). Briefly, twofold dilutions of peptide and antibiotic were prepared in MBH II media along the X- and Y-axis, respectively, in 96-well plates. The inoculum was added to the plates, and subsequently these were incubated for 18–20 h. All steps for inoculum preparation and broth microdilution were in accordance with the CLSI guidelines (Clinical and Laboratory Standards Institute, 2013). The fractional inhibitory concentration index (FICI) was calculated as [MICA(A+B)/MICA(alone)] + [MICB(A+B)/MICB(alone)], where MICA(A+B) represents the MIC of antibiotic (A) in combination with peptidomimetic (B), while MICB(A+B) represents the MIC of peptidomimetic (B) in combination with antibiotic (A). MICA(alone) and MICB(alone) represent the MIC of each compound individually. FICI values of ≤0.5 were interpreted as synergy.
Determination of Cell Viability and Selectivity Indexes for Peptidomimetics
NIH 3T3 and HepG2 cells (both from ATCC, Manassas, VA, USA) were cultured in flat-bottomed 96-well MicroWell™ plates (NUNC, Roskilde, Denmark) at seeding densities of ~23,000 and ~22,000 cells/well, respectively, and then cultured for 22–24 h under standard culturing conditions (37°C, 5% CO2, humidified air) to reach 80–90% confluence. NIH 3T3 were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) newborn-calf serum (NCS) (Gibco, Paisly, UK), while HepG2 were cultured in Eagle's minimum essential medium (EMEM) supplemented with 10% (v/v) fetal bovine serum (FBS, Gibco, Paisly, UK), sodium pyruvate (1 mM), and non-essential amino acids (1%, v/v). Both culturing media were further supplemented with penicillin (100 U/mL), streptomycin (100 μg/mL), and L-glutamine (2 mM).
Effect on cell viability was determined in NIH 3T3 fibroblasts and HepG2 hepatocytes by using the MTS/PMS assay measuring metabolic activity. Briefly, the adhered cells were washed with 37°C Hanks' balanced salt solution (HBSS from Sigma-Aldrich, St. Louis, MO, USA), containing 10 mM HEPES (AppliChem, Darmstadt, Germany), and adjusted to pH 7.4, and were then exposed for 1 h to 100 μL of test compound(s) dissolved in the appropriate culturing medium without serum for each cell line. After exposure the cells were washed with HBSS containing 10 mM HEPES (pH 7.4) and 100 μL of an MTS/PMS solution, consisting of 240 μg/mL MTS (Promega, Madison, WI, USA) and 2.4 μg/mL PMS (SigmaAldrich, Buchs, Switzerland) in HBSS, was added to the cells, which then were incubated for 1 h at 37°C with horizontal shaking under light protection. Absorbance was measured at 492 nm by using a POLARstar OPTIMA plate reader (BMG Labtech, Offenburg, Germany). The relative viability was calculated according to Equation (1) with absorbance values obtained after incubation of cells with test compound; incubation with SDS (0.2%, w/v in medium) defined 100% cell death (Abspos), while the absorbance of cells incubated with medium defined 0% cell death (Absneg).
EC50 values were calculated by using GraphPad Prism 7 (GraphPad Software, La Jolla, CA, USA) via fitting of the relative cell viability to the concentration of the test compound by using Equation (2):
With top and bottom representing the mean highest and lowest observed values, respectively. The Hill slope represents a factor for the steepness of the linear part of the curve. In order to prevent erroneous calculation, the top and bottom values were constrained to 100 and 0%, respectively. Data were collected from technical triplicates. The selectivity index was calculated as the average effect on cell viability (EC50 values for HepG2 and NIH 3T3 cells) divided by the MIC.
Time-Kill Assay
Time-kill kinetic assays were performed in MDR E. coli ST131, MDR K. pneumoniae ST258, and P. aeruginosa ATCC 27853. Overnight cultures were subcultured in MHB II and grown to the logarithmic phase, after which ~106 CFU/mL cells were transferred to 15-mL round-bottom Falcon tubes. Bacterial cells were incubated at 37°C with aeration in the presence or absence of antibiotic, peptidomimetic or a combination of these at 2 × MIC (as observed in the growth curves; Table 6). At time points 0, 1, 2, 4, 8, and 24 h, 100 μl cells were serially diluted 10-fold in sterile 0.9% NaCl, and then 10 μl aliquots were plated on MHA in triplicate. The CFU/mL from each condition was calculated from counted colonies following 18–24 h incubation at 37°C. The detection limit was ~102 CFU/mL. The CFU/mL of each culture was graphed in GraphPad Prism 7 over time (h), and all graphs represent the average and standard error from two separate experiments (i.e., biological duplicates). Synergy was defined as a ≥2-log10 CFU/mL decrease for the cultures exposed to the antibiotic-peptidomimetic combination vs. either compound individually. Bactericidal activity was defined as a ≥3-log10 CFU/mL decrease after 24 h incubation.
Results
Identification of Two Subclasses of Peptidomimetics That Induce Susceptibility to Rifampicin and Azithromycin in Gram-Negative Pathogens
Screening of peptidomimetic-antibiotic combinations was performed in order to identify potentiators of rifampicin and azithromycin in MDR Escherichia coli sequence type 131 (ST131) and K. pneumoniae ST258 epidemic clones. This involved 42 selected diverse peptidomimetics that were combined at 0.5 μM with each antibiotic at concentrations approximating the clinical breakpoints in Gram-positive species (i.e., 0.5 μg/mL and 1 μg/mL, for rifampicin and azithromycin, respectively) (Clinical and Laboratory Standards Institute, 2017). Three peptidomimetics (i.e., 1, 15, and 26) were found to inhibit growth of both species in the presence of rifampicin, while one compound (i.e., 1) prevented growth in combination with azithromycin (Table 1). These peptidomimetics represent two subclasses of α-peptide/β-peptoid hybrids: subclass I (e.g., 1 and 15) comprising lysine-based compounds with achiral β-peptoid phenylalanine-like residues, and subclass II (e.g., 26) displaying both Lys and homoarginine (hArg) as cationic residues together with α-chiral β-peptoid hydrophobic Phe-like residues (Table 2; Figure 1). The effect on cell viability of these three peptidomimetics was tested toward the mammalian cells lines mouse fibroblasts (NIH 3T3) and human hepatocytes (HepG2), which revealed that 1 and 15 exhibited promising cell selectivity (i.e., EC50 >100 μM) while peptidomimetic 26 proved to affect cell viability the most with EC50 values in the range 50–60 μM (Table 2). These peptidomimetics were further screened for potentiation of additional 21 antibiotics (Table 3) at sub-MIC concentrations of ≤1 μM (Table 2). In presence of peptidomimetics 1, 15, or 26, the MIC of rifampicin (>2 μg/mL) was reduced to below 1 μg/mL in both MDR E. coli and K. pneumoniae, while the MIC of erythromycin was reduced from >4 μg/mL to ≤2 μg/mL in MDR E. coli, confirming the results from the primary screening. In addition, the MIC of clindamycin in MDR K. pneumoniae was reduced from >4 μg/mL to 2 μg/mL in presence of 1 μM of 26, while the MIC of ticarcilllin/clavulanic acid in E. coli was reduced from 64 μg/mL to 16 μg/mL in presence of 0.5 μM of 1. Since this E. coli strain was already susceptible to ticarcillin/clavulanic acid, this combination was not studied further. Growth inhibition occurred at concentrations ≥4-fold below the MICs of the peptidomimetic (except for 15 in E. coli) and antibiotic alone, indicating that the combinations exerted synergistic effects. Synergy was confirmed by the checkerboard assay, and fractional inhibitory concentration indexes (FICIs) ranged from 0.05 to 0.38 (Table 4). With FICIs ranging from 0.05 to 0.07, the synergistic peptidomimetic-rifampicin interactions in K. pneumoniae were especially potent. The peptidomimetic 26-clindamycin synergistic interaction in K. pneumoniae was also potent, however, due to the relatively high effect of 26 on the viability of eukaryotic cells, this combination was not studied further.

Table 1. Susceptibility of MDR E. coli ST131 and K. pneumoniae ST258 to rifampicin (RIF) or azithromycin (AZM) in combination with peptidomimeticsa.
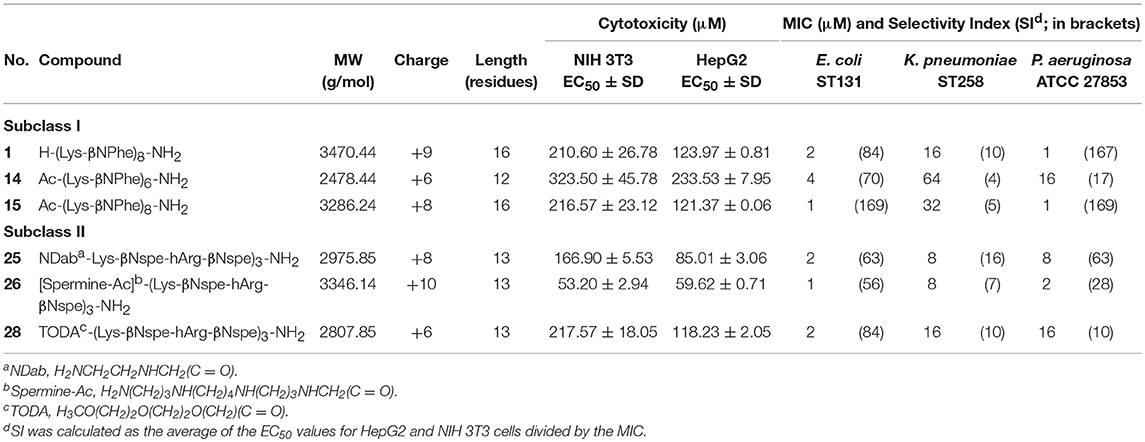
Table 2. Peptidomimetics studied: sequence, cytotoxicity, and MIC in clinical isolates of E. coli, K. pneumoniae and P. aeruginosa.
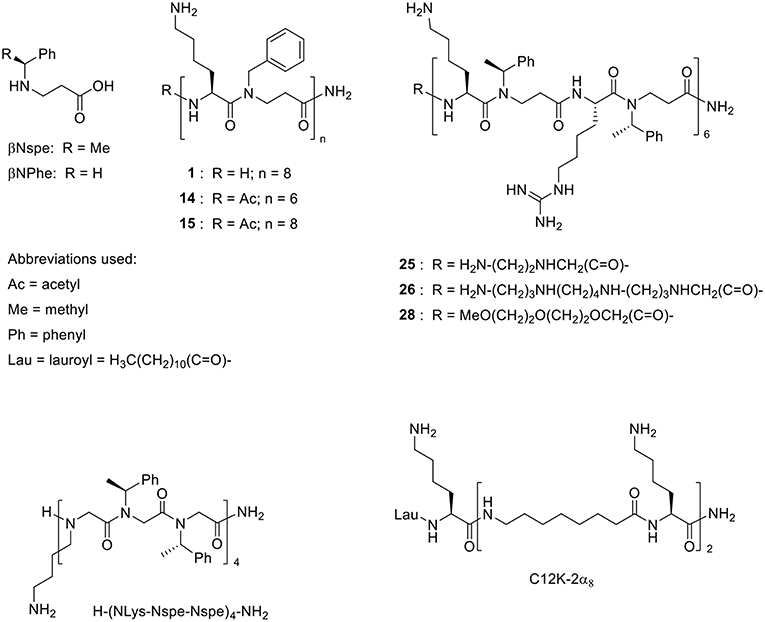
Figure 1. Structures of the α-peptide/β-peptoid hybrids listed in Table 2 and similar compounds that exhibit activity in vivo (Zaknoon et al., 2011; Czyzewski et al., 2016, respectively).
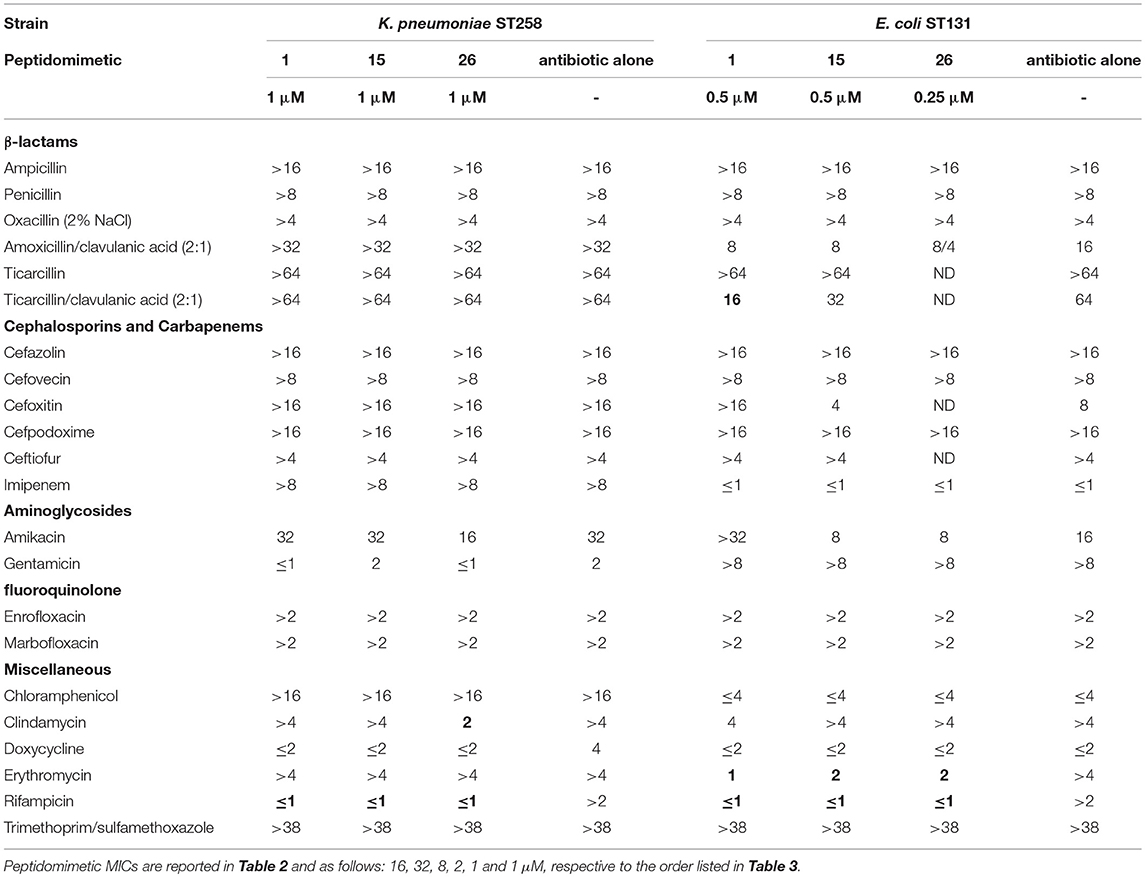
Table 3. Potentiation of antibiotics by peptidomimetics in MDR E. coli ST131 and K. pneumoniae ST258; MICs (μg/mL) of antibiotics in presence and absence of peptidomimetics; MICs reduced ≥4-fold in presence of a peptidomimetic are indicated in bold.
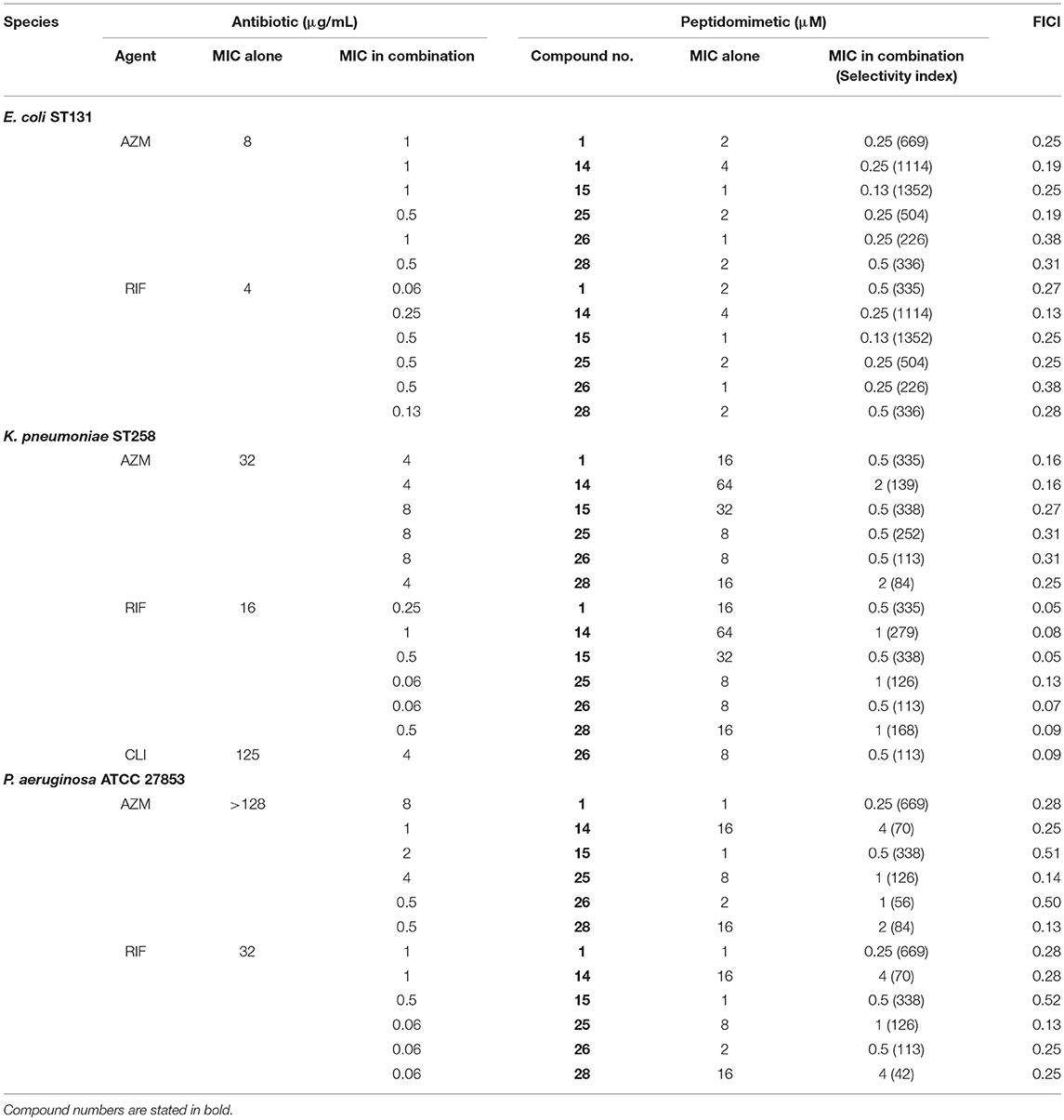
Table 4. Interaction of azithromycin (AZM) and rifampicin (RIF) with peptidomimetics in E. coli ST131, K. pneumoniae ST258, and P. aeruginosa as determined in the checkerboard assay.
To explore additional compounds within the two identified subclasses of peptidomimetics, found to enhance the antibacterial effect of rifampicin and azithromycin, the study was extended to include three additional peptidomimetics, 14 and 25, 28 (also belonging to subclasses I or II; Table 2). These three peptidomimetics also exhibited an ability to potentiate rifampicin and azithromycin in MDR E. coli and K. pneumoniae (Table 4). Overall, the degree of synergy was similar to that seen for combinations containing 1, 15, and 26, resulting in potentiation of rifampicin and azithromycin (to reach MICs of ≤1 and ≤8 μg/mL, respectively) in both species. Similar to the subclass I compounds (i.e., 1 and 15), peptidomimetic 14 displayed the lowest effect on cell viability with EC50 values of 323 μM and 234 μM in NIH 3T3 and HepG2, respectively (Table 2). In contrast, peptidomimetic 25 had a noticeably higher effect on cell viability with EC50 values in the range 80–170 μM, although somewhat less toxic than the highly cationic peptidomimetic 26. Among, the subclass II compounds, peptidomimetic 28 had the least effect on cell viability (EC50 >100 μM). Overall, the HepG2 liver cell line was more sensitive to the peptidomimetics than the fibroblast cells (i.e., NIH 3T3).
We extended our study to species outside Enterobacteriaceae by including a reference strain of P. aeruginosa, which displayed a susceptibility pattern similar to that found for E. coli (Table 2). Similarly, synergy was observed with FICIs ranging from 0.13 to 0.5, except for combinations with peptidomimetic 15, which only displayed a slight potentiation of antibiotic activity (i.e., FICIs >0.5; Table 4). In the presence of sub-MIC concentrations of peptidomimetics 1, 14, 15, 25, 26, or 28, the MICs of azithromycin were reduced 32- to 512-fold, corresponding to MIC values within the range 0.5-8 μg/mL in the synergistic combinations (Table 4). However, the most pronounced enhancement of antibiotic potency was observed when the peptidomimetics were used in combination treatment of K. pneumoniae, since the peptidomimetics in this case could be applied in 8- to 64-fold lower concentrations than their MICs as compared to only 2- to 16-fold lower than their MICs in E. coli and P. aeruginosa.
As a preliminary investigation of the therapeutic potential of the present subclasses of peptidomimetic antibiotic adjuvants, their cell selectivity was estimated as a measure that reflects both antibacterial activity and effect on viability of mammalian cells. Typically, a cell selectivity index (SI) is calculated as the ratio between the averaged EC50 values and the MIC of the peptidomimetic in each bacterial species. Peptidomimetics 1 and 15, belonging to subclass I, proved to be the most selective against E. coli and P. aeruginosa over mammalian cells with SIs within the range 84–169 (Table 2). When targeting MDR K. pneumoniae all compounds displayed poor selectivity, with 25 having a modest SI of 16. As the MICs of the peptidomimetics in combination with antibiotics were lower, the SIs of the combinations with azithromycin or rifampicin were improved, assuming a negligible contribution from the antibiotics to the negative effects on mammalian cell viability, since these were employed in clinically relevant concentrations (Baker et al., 2018). Toward E. coli, the most favorable combinations, containing peptidomimetics 14 and 15, would have approximated SIs >1,000 (Table 4). However, these two peptidomimetics were less selective against K. pneumoniae and P. aeruginosa with SIs within the range 70–338. While 14 had the highest EC50 (Table 2), the concentration of 14 required in synergistic combinations were ≥1 μM and 4 μM, in K. pneumoniae and P. aeruginosa, respectively, (Table 4). Likewise, compound 15 did not display synergy with any of the antibiotics in P. aeruginosa (Table 4). In contrast, peptidomimetic 1 exhibited synergy with both rifampicin and azithromycin at submicromolar concentrations with estimated SIs ≥335 against all three species (Table 4). With an average EC50 >150 μM, peptidomimetic 1 was also considered relatively non-toxic in itself. In addition, peptidomimetic 1 was the only compound that exhibited synergy at submicromolar concentrations in combination with antibiotics at clinically relevant concentrations in all three species, and thus this compound was selected for further studies.
Bacterial Growth and Killing in Presence of Peptidomimetic 1-Antibiotic Combinations
To ascertain that susceptibility of clinical isolates of Enterobacteriaceae and P. aeruginosa to combinations of peptidomimetic 1 with rifampicin or azithromycin could be induced, the combinations were tested with respect to MIC against several MDR isolates of each species. Overall, peptidomimetic 1 (at ≤1 μM) induced susceptibility to rifampicin and azithromycin in 83% of the isolates (i.e., in 30 of 36 isolates; Table 5). Notably, most E. coli isolates proved susceptible to 0.25–0.5 μg/mL rifampicin and 2–4 μg/mL azithromycin in combination with 0.5–1 μM peptidomimetic 1. While submicromolar concentrations of 1 in the majority of K. pneumoniae isolates were sufficient to induce susceptibility to rifampicin and azithromycin, three isolates required considerably higher concentrations of 1 (i.e., 2–8 μM). By contrast P. aeruginosa isolates typically required 1–8 μM of 1 to reduce the MIC of rifampicin and azithromycin to 0.5 μg/mL and 4–8 μg/mL, respectively.
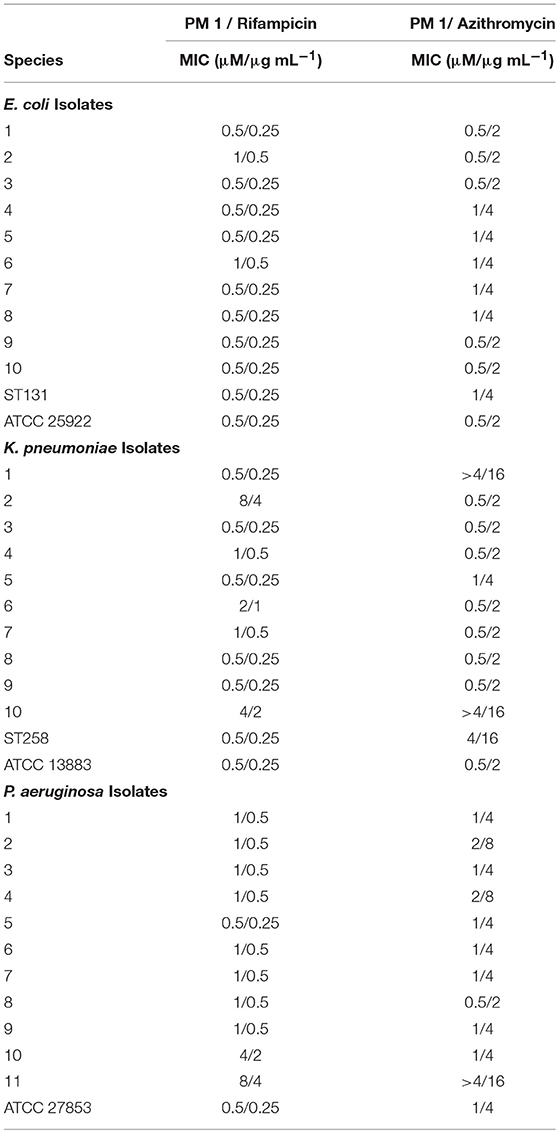
Table 5. MICs of Peptidomimetic (PM) 1-rifampicin and PM 1-azithromycin combinations in E. coli, K. pneumoniae, and P. aeruginosa clinical isolates and reference strains.
To understand the dynamics of the interaction of peptidomimetic 1 with antibiotics, the kinetics of bacterial growth and killing were investigated by growth-curve and time-kill assays. All combinations exhibited synergistic growth inhibition or bactericidal activity in growth-curve and time-kill assays. The growth curves of the combinations corresponded well to the MICs (Table 6) found in static growth assays (Tables 4, 5). In the time-kill assays, the concentrations of tested compounds were 2-fold above the MIC observed in the growth-curve assays, since a 2-fold higher bacterial concentration was used. The tested peptidomimetic 1-rifampicin combinations were bactericidal in all three species, as at least a 3-log reduction in CFU/mL was reached within the initial 4 h of exposure (Figures 3A–C). Similarly, peptidomimetic 1-azithromycin combinations (Figures 3D,F) were bactericidal in E. coli and P. aeruginosa. However, in K. pneumoniae (Figure 3E), the combination was bacteriostatic as it only reduced the initial inoculum CFU by ~1.5 log, also reflecting the high MBC, measured at the end of the growth-curve assay (Table 6). Although the MBCs of combinations in P. aeruginosa were only 2-fold higher than the MIC (Table 6), in the time-kill assay this species started to recover after 4 h of exposure to the rifampin- and azithromycin-peptidomimetic 1 combinations (Figures 3C,F). A similar, but less pronounced, trend was observed for the 1-rifampicin combination in E. coli, for which the CFU concentration had increased at the 24 h time point (Figure 3A). In addition, reduced growth was seen for E. coli and P. aeruginosa in the presence of 1 alone (at concentrations 2-fold below the MIC) within the first 4 h of exposure (Figures 3A,C,D).
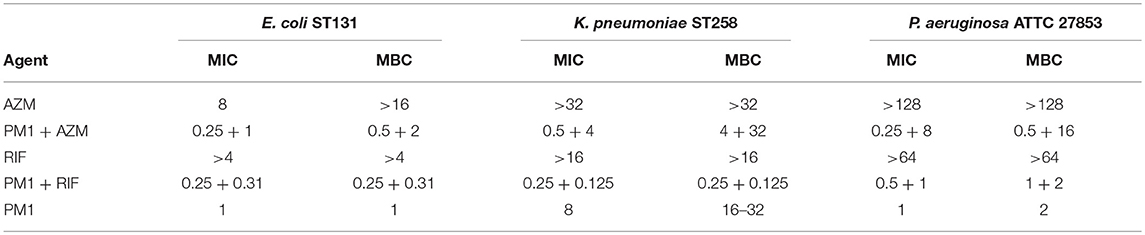
Table 6. MIC and MBC of peptidomimetic (PM) 1 (μM), azithromycin (AZM) and rifampicin (RIF) (μg/mL) and combinations (peptidomimetic + antibiotic) as determined in the growth-curve assay.
Discussion
In the present study an array of peptidomimetics was screened in order to identify possible potentiators of antibiotics against MDR Gram-negative pathogens. In particular, the aim was to repurpose antibiotics typically used against Gram-positive pathogens. This enabled identification of a lead compound, peptidomimetic 1, which exhibited high cell selectivity and displayed synergy with rifampicin and azithromycin at submicromolar, sub-MIC concentrations. Moreover, this compound was found capable of inducing susceptibility to antibiotics in MDR strains and clinical isolates of E. coli, K. pneumoniae and P. aeruginosa. Initial characterization of five additional peptidomimetics with similar structural composition also revealed potent antibacterial synergy with rifampicin and azithromycin, suggesting that the α-peptide/β-peptoid backbone constitutes a promising template for design of potentiators of antibiotics that Gram-negative pathogens are intrinsically resistant to.
The structural features of the peptidomimetics, identified as antibiotic adjuvants, have been examined in previous studies, focusing on the structure-activity relationships as potential directly antimicrobial agents (Jahnsen et al., 2014; Molchanova et al., 2017b); yet the present work constitutes the first report on their potential for synergy with traditional antibiotics. While the effect of human serum albumin and plasma on the antibiotic adjuvant activity was not addressed in this study, we have in previous work found that the antibacterial activity of similar peptidomimetics (belonging to the same peptidomimetic class) is independent of the presence of physiological concentration human serum albumin, while test in 25% plasma in fact increased their antimicrobial potency (Hein-Kristensen et al., 2013; Citterio et al., 2016). Likewise, the mechanism of synergy has not been investigated in the present study, but this type of peptidomimetics was previously found to exhibit bactericidal activity via membrane permeabilization (Hein-Kristensen et al., 2011), suggesting that at low concentrations these compounds may cause non-lethal membrane alterations that facilitate enhanced influx of antibiotics. The transient growth inhibition induced by these compounds at sub-MIC concentrations in growth-curve and time-kill assays (Figures 2, 3) supports this hypothesis. The six studied peptidomimetics range in length from 12 to 16 residues and display N-terminal end groups (Table 2; Figure 1). Subclass I peptidomimetics display a 1:1 ratio of alternating Lys residues and βNPhe peptoid units as well as either no end group or an acetyl group at the N-terminus (Table 1). Compound 1 had previously been found to be active against the Gram-negative E. coli, P. aeruginosa, Acinetobacter baumannii and Salmonella Typhimurium as well as the Gram-positive Staphylococcus pseudintermedius (Molchanova et al., 2017b), while 15 had proved antibacterial against E. coli (Jahnsen et al., 2012). Interestingly, compound 14 at 1 μM has been shown to block the pro-inflammatory effect of LPS (Skovbakke et al., 2015), thereby implying that LPS released from lysed bacteria might constitute a less critical issue when applying this compound as an antibiotic adjuvant.
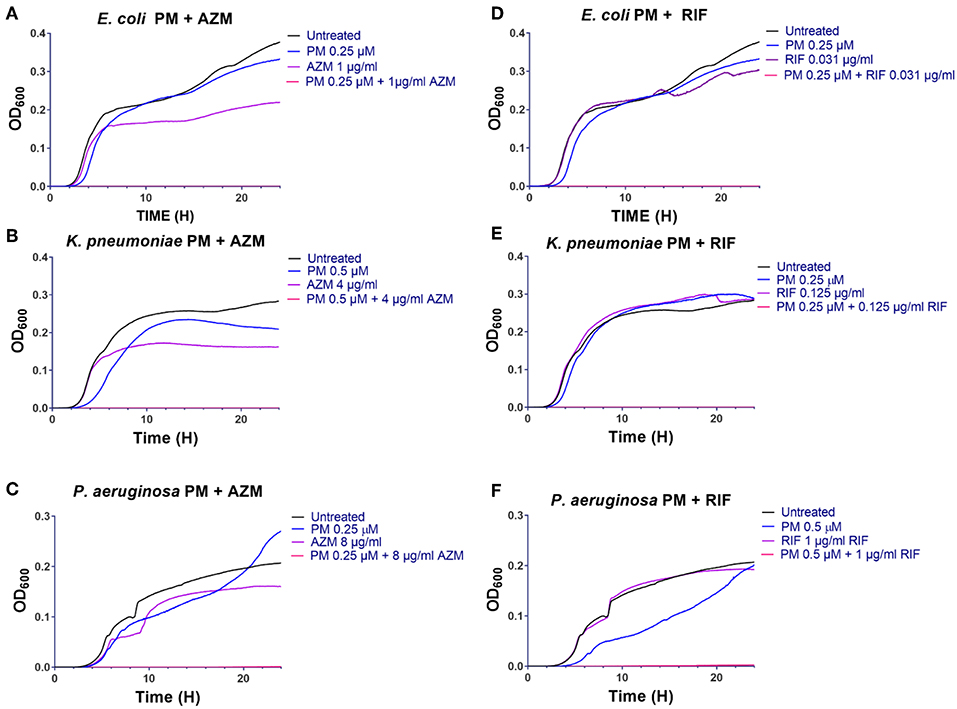
Figure 2. Peptidomimetic 1 potentiates activity of azithromycin and rifampicin in growth-curve assays (A–F). Growth of E. coli ST131 (A,D), K. pneumoniae ST258 (B,E) and P. aeruginosa ATCC 27853 (C,F) in absence or presence of antibiotic, peptidomimetic (PM) 1 or their combinations were recorded at regular intervals by measuring the optical density (OD) of each culture at 600 nm, and then these data were graphed over time (h). Initial bacterial concentration was ~5 ×105 CFU/mL. The MIC values of each compound are listed in Table 6.
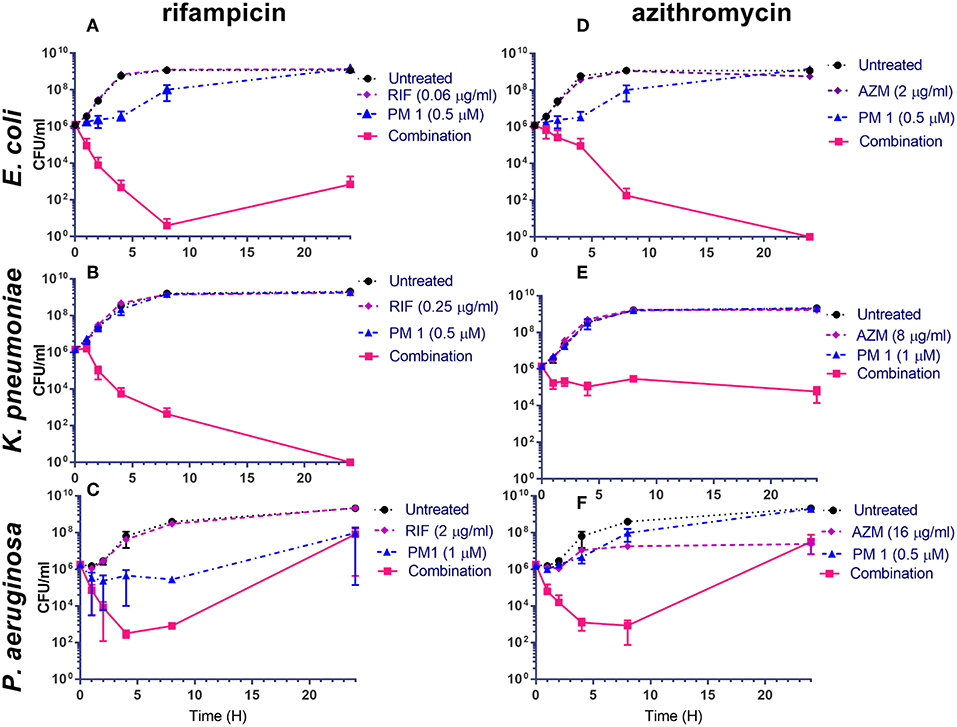
Figure 3. Peptidomimetic 1-combinations enhance bactericidal activity (A–F). Time-kill kinetics for E. coli ST131 (A,D), K. pneumoniae ST258 (B,E) and P. aeruginosa ATTC 27853 (C,F) exposed to peptidomimetic (PM) 1, azithromycin (AZM), rifampicin (RIF), peptidomimetic-antibiotic combinations or without treatment (controls); CFU/mL data are graphed at time points 0, 1, 2, 4, 8, and 24 h of exposure. Initial bacterial concentration was ~106 CFU/mL. The MBC values of each compound are listed in Table 6.
In accordance with the high structural similarity between 1 and 15, these analogs exhibited almost equipotent antibacterial, cytotoxic activity and potentiation of antibiotics (Tables 2, 4). Nonetheless, 1 appeared to exert slightly more efficient potentiation of antibiotics in P. aeruginosa. In contrast, 14 is four residues shorter than 15, and hence exhibited a less pronounced effect on the viability of mammalian cells, since this effect is strongly dependent on oligomer length (Jahnsen et al., 2014). Nonetheless, shortening of oligomer length gives rise to a concomitantly lower antibacterial activity, as also seen in the MDR strains of K. pneumoniae and P. aeruginosa (Table 2), yet 14 retained a similar degree of synergy with antibiotics (Table 4). Consequently, the selectivity indexes for 14 in combination with rifampicin or azithromycin in E. coli proved to be the most favorable among all of the peptidomimetic-antibiotic combinations tested. However, due to the typically 2- to 8-fold higher concentrations of 14 in synergistic combinations, these gave rise to less pronounced selectivity for K. pneumoniae and P. aeruginosa over mammalian cells (as reflected in its SIs; Table 2). While subclass I and II peptidomimetics share the same α-peptide/β-peptoid hybrid backbone structure, subclass II is characterized by a mixed Lys and hArg content (in a 1:1 ratio) of cationic residues (Table 2). The increased effect of subclass II peptidomimetics 25 and 26 on the viability of mammalian cell lines may be correlated to the presence of hArg residues displaying guanidinium functionalities, which confer an increased propensity to interact with, and thereby disrupt, mammalian membranes (Chan et al., 2006; Hein-Kristensen et al., 2011; Findlay et al., 2012; Jahnsen et al., 2014). However, even though 28 belongs to subclass II it had a similarly low effect on mammalian cell viability as that of compounds 1 and 15, which may be explained by its modification with a polar PEG-like moiety (i.e., 3,6,9-trioxadecanoyl; TODA) instead of a cationic moiety containing amine functionalities (i.e., NDab or spermine-acetyl moiety present in 25 and 26, respectively). Thus, the increased effect of 25 and 26 on mammalian cell viability may be due to their increased net cationic charge (Jahnsen et al., 2015). Moreover, while 28 retained antibacterial potency similar to its parent unmodified compound, it had a lower effect on mammalian cell viability than the parent compound (for NIH 3T3: EC50 ~218 μM vs. 117 μM) (Jahnsen et al., 2015), suggesting that introduction of the TODA moiety is beneficial with respect to therapeutic utility.
Interestingly, nine compounds in the peptidomimetic array contained the subclass II core structure, yet only four compounds were detected as potentiators of antibiotics. Another shorter peptidomimetic (i.e., 10) also potentiated rifampicin activity (but not that of azithromycin) in MDR E. coli. In contrast, the other five compounds (i.e., 9, 12, 13, 27, and 29) did not exhibit any potentiation activity, detectable at the low concentrations tested in the screening (Table 1). This suggests that addition of a palmitoyl moiety (in 9, 12, 13), a cinnamoyl moiety (in 27), a fluorinated phenylalanine (in 29), or shorter oligomers (i.e., 9 and 13) may result in lowered potency or abolish potentiation, while similar modifications (e.g., in 29) did not alter the antibacterial activity (Jahnsen et al., 2015). Based on these observations, there appears to be distinct physicochemical and/or overall structural requirements for achieving efficient potentiation of antibiotics and high direct antibacterial activity, respectively.
The present results infer that peptidomimetic 1 has a potential for repurposing rifampicin and azithromycin for treatment of MDR Gram-negative infections. While this peptidomimetic was considered to be the most promising due to its low toxicity (with ensuing favorable cell selectivity) and ability to exert synergy with antibiotics at submicromolar levels in all three species, the additional peptidomimetics with low effect on mammalian cell viability, characterized in this study, may also merit further investigation. Since a prerequisite for successful combination therapy is that the effective concentrations of both peptidomimetic and antibiotic can be achieved at the infection site, it is critical that future studies address in vivo pharmacokinetic/pharmacodynamics and acute toxicity to assess the clinical potential of such peptidomimetic-antibiotic combinations. In particular, co-formulation of antibiotic and peptidomimetics as nanoparticles would be preferable to ensure in vivo co-localization of the compounds in the appropriate ratios (Carmona-Ribeiro and de Melo Carrasco, 2014; Liu et al., 2016; Nordström and Malmsten, 2017). While in vivo data currently are lacking for the α-peptide/β-peptoid hybrids described in the present study, peptidomimetics (including the 12-mer, H-(NLys-Nspe-Nspe)4-NH2 and the pentamer C12K-2α8) displaying similar structural features such as a high content of both cationic and hydrophobic residues (aromatic or aliphatic) have previously been found to exhibit in vivo activity (antibacterial and antiparasitic, respectively) (Zaknoon et al., 2011; Czyzewski et al., 2016). Based on overall structural similarity, the peptidomimetics identified in the present study appear likely to possess physicochemical properties compatible with in vivo activity. Moreover, clinical breakpoints for rifampicin and azithromycin may be used to estimate the therapeutic potential of the antibiotics in these drug combinations. Although no clinical breakpoint points are available for these antibiotics in Enterobacteriaceae or P. aeruginosa, bactericidal efficacy of the 1-rifampicin combination occurred at concentrations ranging from 0.06 to 2 μg/mL in time-kill assays, which is below the resistance breakpoint for rifampicin alone in Staphylococcus species (R ≥4 μg/mL) (Clinical and Laboratory Standards Institute, 2017). Similarly, at concentrations below the azithromycin resistance breakpoint for Salmonella typhi (R ≥32 μg/mL), the 1-azithromycin combination exerted growth-inhibitory and bactericidal activity. Recent studies also indicate that antibacterial efficacy of azithromycin therapy against Gram-negative pathogens in vivo is considerably higher than implied by in vitro susceptibility testing in standard MHB media (Lin et al., 2015; Ersoy et al., 2017). Importantly, low concentrations (≤1 μM) of peptidomimetic 1 were able to induce susceptibility to azithromycin and rifampicin in most of the clinical isolates tested. Thus the low effect of 1 on viability of mammalian cells and its high potency in combination with antibiotics may facilitate circumvention of pharmacological obstacles frequently associated with antimicrobial peptides and peptidomimetics (Molchanova et al., 2017a).
Author Contributions
KB, HF, and LG conceived and designed the study. KB, BJ, HF, and LG contributed to experimental design. KB, BJ, and AH performed experiments. HN provided cytotoxicity experiments and data. KB wrote the first draft of the manuscript. HF, LG, AH, HN, and KB wrote sections of the manuscript. All authors edited, revised, and approved the manuscript for submission.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Laurent Poirel for providing the clinical isolates used in this study. The study was supported by the University of Copenhagen research centre for Control of Antibiotic Resistance (UC-CARE).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2019.00236/full#supplementary-material
References
Baker, K. R., Jana, B., Franzyk, H., and Guardabassi, L. (2016). A high-throughput approach to identify compounds that impair envelope integrity in Escherichia coli. Antimicrob. Agents Chemother. 60, 5995–6002. doi: 10.1128/AAC.00537-16
Baker, K. R., Jana, B., Hansen, A. M., Vissing, K. J., Nielsen, H. M., Franzyk, H., et al. (2018). Repurposing azithromycin and rifampicin against Gram-negative pathogens by combination with peptide potentiators. Int. J. Antimicrob. Agents. 53, 868–872. doi: 10.1016/j.ijantimicag.2018.10.025
Bonke, G., Vedel, L., Witt, M., Jaroszewski, J. W., Olsen, C. A., and Franzyk, H. (2008). Dimeric building blocks for solid-phase synthesis of α-peptide- β-peptoid chimeras. Synthesis. 2008, 2381–2390. doi: 10.1055/s-2008-1067171
Brown, D. (2015). Antibiotic resistance breakers: can repurposed drugs fill the antibiotic discovery void? Nat. Rev. Drug Discov. 14, 821–832. doi: 10.1038/nrd4675
Brown, E. D., and Wright, G. D. (2016). Antibacterial drug discovery in the resistance era. Nature 529, 336–343. doi: 10.1038/nature17042
Carmona-Ribeiro, A., and de Melo Carrasco, L. (2014). Novel formulations for antimicrobial peptides. Int. J. Mol. Sci. 15, 18040–18083. doi: 10.3390/ijms151018040
Cerquetti, M., Giufrè, M., García-Fernández, A., Accogli, M., Fortini, D., Luzzi, I., et al. (2010). Ciprofloxacin-resistant, CTX-M-15-producing Escherichia coli ST131 clone in extraintestinal infections in Italy. Clin. Microbiol. Infect. 16, 1555–1558. doi: 10.1111/j.1469-0691.2010.03162.x
Chan, D. I., Prenner, E. J., and Vogel, H. J. (2006). Tryptophan- and arginine-rich antimicrobial peptides: structures and mechanisms of action. Biochim. Biophys. Acta Biomembr. 1758, 1184–1202. doi: 10.1016/j.bbamem.2006.04.006
Citterio, L., Franzyk, H., Palarasah, Y., Emil, T., Valentina, R., and Gram, L. (2016). Improved in vitro evaluation of novel antimicrobials: potential synergy between human plasma and antibacterial peptidomimetics, AMPs and antibiotics against human pathogenic bacteria. Res. Microbiol. 167, 72–82. doi: 10.1016/j.resmic.2015.10.002
Clinical and Laboratory Standards Institute (1999). Methods for Determining Bactericidal Activity of Antimicrobial Agents; Approved Guidline, Wayne, PA: Vol 19. CLSI document M26-A, Clinical and Laboratory Standards Institute.
Clinical and Laboratory Standards Institute (2013). Performance Standards for Antimicrobial Disk and Dilution Susceptibility Tests for Bacteria Isolated From Animals; Approved Standard, 4th ed, Wayne, PA: vol 8. CLSI document VET01-A4, Clinical and Laboratory Standards Institute.
Clinical and Laboratory Standards Institute (2017). Performance Standards for Antimicrobial Susceptibility Testing, 27th ed, Wayne, PA: CLSI M100. Clinical and Laboratory Standards Institute.
Corbett, D., Wise, A., Langley, T., Skinner, K., Trimby, E., Birchall, S., et al. (2017). Potentiation of antibiotic activity by a novel cationic peptide: potency and spectrum of activity of SPR741. Antimicrob. Agents Chemother 22, 1–10. doi: 10.1128/AAC.00200-17
Czyzewski, A. M., Jenssen, H., Fjell, C. D., Waldbrook, M., Chongsiriwatana, N. P., Yuen, E., et al. (2016). In vivo, in vitro, and in silico characterization of peptoids as antimicrobial agents. PLoS ONE 11:e0135961. doi: 10.1371/journal.pone.0135961
Doi, Y., Bonomo, R. A., Hooper, D. C., Kaye, K. S., Johnson, J. R., Clancy, C. J., et al. (2017). Gram-negative bacterial infections: Research priorities, accomplishments, and future directions of the antibacterial resistance leadership group. Clin. Infect. Dis. 64, S30–S35. doi: 10.1093/cid/ciw829
Domalaon, R., Brizuela, M., Eisner, B., Findlay, B., Zhanel, G. G., and Schweizer, F. (2018a). Dilipid ultrashort cationic lipopeptides as adjuvants for chloramphenicol and other conventional antibiotics against Gram-negative bacteria. Amino Acids. 51, 383–393. doi: 10.1007/s00726-018-2673-9
Domalaon, R., Sanchak, Y., Koskei, L. C., Lyu, Y., Zhanel, G. G., Arthur, G., et al. (2018b). Short proline-rich lipopeptide potentiates minocycline and rifampin against multidrug- and extensively drug-resistant Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 62, e02374–e02317. doi: 10.1128/AAC.02374-17
Ersoy, S. C., Heithoff, D. M., Barnes, L., Tripp, G. K., House, J. K., Marth, J. D., et al. (2017). Correcting a fundamental flaw in the paradigm for antimicrobial susceptibility testing. EBioMedicine 20, 173–181. doi: 10.1016/j.ebiom.2017.05.026
Findlay, B., Szelemej, P., Zhanel, G. G., and Schweizer, F. (2012). Guanidylation and tail effects in cationic antimicrobial lipopeptoids. PLoS ONE 7:e41141. doi: 10.1371/journal.pone.0041141
Garcia, L. S. (ed.). (2010). “Synergism testing: broth microdilution checkerboard and broth macrodilution methods,” in Clinical Microbiology Procedures Handbook, 3rd Edition (Washington, DC: American Society of Microbiology Press, 140–162.
Ghosh, C., and Haldar, J. (2015). Membrane-Active Small Molecules: Designs Inspired by Antimicrobial Peptides. ChemMedChem 10, 1606–1624. doi: 10.1002/cmdc.201500299
Gill, E. E., Franco, O. L., and Hancock, R. E. (2015). Antibiotic adjuvants: diverse strategies for controlling drug-resistant pathogens. Chem. Biol. Drug Des. 85, 56–78. doi: 10.1111/cbdd.12478
Goldberg, K., Sarig, H., Zaknoon, F., Epand, R. F., Epand, R. M., and Mor, A. (2013). Sensitization of gram-negative bacteria by targeting the membrane potential. FASEB J. 27, 3818–3826. doi: 10.1096/fj.13-227942
Hein-Kristensen, L., Franzyk, H., Holch, A., and Gram, L. (2013). Adaptive evolution of Escherichia coli to an α-peptide/β-peptoid peptidomimetic induces stable resistance. PLoS ONE 8:e73620. doi: 10.1371/journal.pone.0073620
Hein-Kristensen, L., Knapp, K. M., Franzyk, H., and Gram, L. (2011). Bacterial membrane activity of α-peptide/β-peptoid chimeras: influence of amino acid composition and chain length on the activity against different bacterial strains. BMC Microbiol. 11:144. doi: 10.1186/1471-2180-11-144
Jahnsen, R. D., Frimodt-Møller, N., and Franzyk, H. (2012). Antimicrobial activity of peptidomimetics against multidrug-resistant Escherichia coli: a comparative study of different backbones. J. Med. Chem. 55, 7253–7261. doi: 10.1021/jm300820a
Jahnsen, R. D., Sandberg-Schaal, A., Vissing, K. J., Nielsen, H. M., Frimodt-Møller, N., and Franzyk, H. (2014). Tailoring cytotoxicity of antimicrobial peptidomimetics with high activity against multidrug-resistant Escherichia coli. J. Med. Chem. 57, 2864–2873. doi: 10.1021/jm401335p
Jahnsen, R. O., Sandberg-Schaal, A., Frimodt-Møller, N., Nielsen, H. M., and Franzyk, H. (2015). End group modification: efficient tool for improving activity of antimicrobial peptide analogues towards Gram-positive bacteria. Eur. J. Pharm. Biopharm. 95, 40–46. doi: 10.1016/j.ejpb.2015.01.013
Jammal, J., Zaknoon, F., Kaneti, G., Goldberg, K., and Mor, A. (2015). Sensitization of gram-negative bacteria to rifampin and OAK combinations. Sci. Rep. 5:9216. doi: 10.1038/srep09216
Jana, B., Cain, A. K., Doerrler, W. T., Boinett, C. J., Fookes, M. C., Parkhill, J., et al. (2017). The secondary resistome of pneumoniae. Sci. Rep. 7:42483. doi: 10.1038/srep42483
Kalan, L., and Wright, G. D. (2011). Antibiotic adjuvants: multicomponent anti-infective strategies. Expert Rev. Mol. Med. 13:e5. doi: 10.1017/S1462399410001766
Lainson, J. C., Daly, S. M., Triplett, K., Johnston, S. A., Hall, P. R., and Diehnelt, C. W. (2017). Synthetic antibacterial peptide exhibits synergy with oxacillin against MRSA. Med. Chem. Lett. 8, 853–857. doi: 10.1021/acsmedchemlett.7b00200
Lin, L., Nonejuie, P., Munguia, J., Hollands, A., Olson, J., Dam, Q., et al. (2015). Azithromycin synergizes with cationic antimicrobial peptides to exert bactericidal and therapeutic activity against highly multidrug-resistant gram-negative bacterial pathogens. EBioMedicine 2, 690–698. doi: 10.1016/j.ebiom.2015.05.021
Liu, X., Li, Z., Wang, X., Chen, Y., Wu, F., Men, K., et al. (2016). Novel antimicrobial peptide–modified azithromycin-loaded liposomes against methicillin-resistant Staphylococcus aureus. Int. J. Nanomed. 11, 6781–6794. doi: 10.2147/IJN.S107107
Liu, Y., Knapp, K. M., Yang, L., Molin, S., Franzyk, H., and Folkesson, A. (2013). High in vitro antimicrobial activity of β-peptoid-peptide hybrid oligomers against planktonic and biofilm cultures of Staphylococcus epidermidis. Int. J. Antimicrob. Agents 41, 20–27. doi: 10.1016/j.ijantimicag.2012.09.014
Lyu, Y., Yang, X., Goswami, S., Gorityala, B. K., Idowu, T., Domalaon, R., et al. (2017). Amphiphilic tobramycin–lysine conjugates sensitize multidrug resistant gram-negative bacteria to rifampicin and minocycline. J. Med. Chem. 60, 3684–3702. doi: 10.1021/acs.jmedchem.6b01742
Melander, R. J., and Melander, C. (2017). The challenge of overcoming antibiotic resistance: an adjuvant approach? ACS Infect. Dis. 3, 559–563 doi: 10.1021/acsinfecdis.7b00071
Méndez-Samperio, P. (2014). Peptidomimetics as a new generation of antimicrobial agents: current progress. Infect. Drug Resist. 7, 229–237. doi: 10.2147/IDR.S49229
Mojsoska, B., and Jenssen, H. (2015). Peptides and peptidomimetics for antimicrobial drug design. Pharmaceuticals 8, 366–415 doi: 10.3390/ph8030366
Molchanova, N., Hansen, P. R., Damborg, P., Nielsen, H. M., and Franzyk, H. (2017b). Lysine-based α-peptide/β-peptoid peptidomimetics: influence of hydrophobicity, fluorination, and distribution of cationic charge on antimicrobial activity and cytotoxicity. ChemMedChem 12, 312–318. doi: 10.1002/cmdc.201600553
Molchanova, N., Hansen, P. R., and Franzyk, H. (2017a). Advances in development of antimicrobial peptidomimetics as potential drugs. Molecules 22:1430. doi: 10.3390/molecules22091430
Moon, S. H., and Huang, E. (2018). Lipopeptide paenipeptin analogues potentiate clarithromycin and rifampin against carbapenem-resistant pathogens. Antimicrob. Agents Chemother. 62, e00329–e00318. doi: 10.1128/AAC.00329-18
Nordström, R., and Malmsten, M. (2017). Delivery systems for antimicrobial peptides. Adv. Colloid Interface Sci. 242, 17–34. doi: 10.1016/j.cis.2017.01.005
Renau, T. E., Léger, R., Yen, R., She, M. W., Flamme, E. M., Sangalang, J., et al. (2002). Peptidomimetics of efflux pump inhibitors potentiate the activity of levofloxacin in Pseudomonas aeruginosa. Bioorganic Med. Chem. Lett. 12, 763–766. doi: 10.1016/S0960-894X(02)00006-9
Ribeiro, S. M., Fuente-Núñez, C., Baquir, B., Faria-Junior, C., Franco, O. L., and Hancock, R. E. W. (2015). Antibiofilm peptides increase the susceptibility of carbapenemase-producing Klebsiella pneumoniae clinical isolates to β-lactam antibiotics. Antimicrob. Agents Chemother. 59, 3906–3912. doi: 10.1128/AAC.00092-15
Singh, S. B., Young, K., and Silver, L. L. (2017). What is an “ideal” antibiotic? discovery challenges and path forward. Biochem. Pharmacol. 133, 63–73. doi: 10.1016/j.bcp.2017.01.003
Skovbakke, S. L., Larsen, C. J., Heegaard, P. M. H., Moesby, L., and Franzyk, H. (2015). Lipidated α-peptide/β-peptoid hybrids with potent anti-inflammatory activity. J. Med. Chem. 58, 801–813. doi: 10.1021/jm501341h
World Health Organisation (2017). Global Priority List Of Antibiotic-Resistant Bacteria To Guide Research, Discovery, And Development Of New Antibiotics. World Health Organisation. Available online at: http://www.who.int/medicines/publications/global-priority-list-antibiotic-resistant-bacteria/en/ (accessed Jan 29, 2019)
Wu, X., Li, Z., Li, X., Tian, Y., Fan, Y., Yu, C., et al. (2017). Synergistic effects of antimicrobial peptide DP7 combined with antibiotics against multidrug-resistant bacteria. Drug Des. Devel. Ther. 11, 939–946. doi: 10.2147/DDDT.S107195
Yang, X., Domalaon, R., Lyu, Y., Zhanel, G., and Schweizer, F. (2018). Tobramycin-linked efflux pump inhibitor conjugates synergize fluoroquinolones, rifampicin and fosfomycin against multidrug-resistant Pseudomonas aeruginosa. J. Clin. Med. 7:158. doi: 10.3390/jcm7070158
Keywords: peptidomimetic, antibiotic potentiation, multidrug resistance, synergy, Gram-negative, antibiotic adjuvant
Citation: Baker KR, Jana B, Hansen AM, Nielsen HM, Franzyk H and Guardabassi L (2019) Repurposing Azithromycin and Rifampicin Against Gram-Negative Pathogens by Combination With Peptidomimetics. Front. Cell. Infect. Microbiol. 9:236. doi: 10.3389/fcimb.2019.00236
Received: 30 January 2019; Accepted: 14 June 2019;
Published: 02 July 2019.
Edited by:
Rodolfo García-Contreras, National Autonomous University of Mexico, MexicoReviewed by:
Frank Schweizer, University of Manitoba, CanadaCésar de la Fuente, Massachusetts Institute of Technology, United States
Copyright © 2019 Baker, Jana, Hansen, Nielsen, Franzyk and Guardabassi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kristin R. Baker, a3JpYmFrZUBzdW5kLmt1LmRr; Henrik Franzyk, aGVucmlrLmZyYW56eWtAc3VuZC5rdS5kaw==