 Fuka Kikuchi1,2,3
Fuka Kikuchi1,2,3 Keita Aoki2
Keita Aoki2 Satoshi D. Ohdachi4Kimiyuki Tsuchiya5†Masaharu Motokawa6Takamichi Jogahara7Nguyễn Trường Sơn8,9
Satoshi D. Ohdachi4Kimiyuki Tsuchiya5†Masaharu Motokawa6Takamichi Jogahara7Nguyễn Trường Sơn8,9 Saw Bawm10
Saw Bawm10 Kyaw San Lin11Thida Lay Thwe12
Kyaw San Lin11Thida Lay Thwe12 Chandika D. Gamage13Marie Claudine Ranorosoa14Hasmahzaiti Omar15Ibnu Maryanto16Hitoshi Suzuki17Keiko Tanaka-Taya1Shigeru Morikawa18Tetsuya Mizutani3Motoi Suzuki1
Chandika D. Gamage13Marie Claudine Ranorosoa14Hasmahzaiti Omar15Ibnu Maryanto16Hitoshi Suzuki17Keiko Tanaka-Taya1Shigeru Morikawa18Tetsuya Mizutani3Motoi Suzuki1 Richard Yanagihara19
Richard Yanagihara19 Satoru Arai1*
Satoru Arai1*- 1Infectious Disease Surveillance Center, National Institute of Infectious Diseases, Tokyo, Japan
- 2Department of Chemistry, Faculty of Science, Tokyo University of Science, Tokyo, Japan
- 3Research and Education Center for Prevention of Global Infectious Diseases of Animals, Tokyo University of Agriculture and Technology, Fuchu, Japan
- 4Institute of Low Temperature Science, Hokkaido University, Sapporo, Japan
- 5Laboratory of Bioresources, Applied Biology Co., Ltd., Tokyo, Japan
- 6The Kyoto University Museum, Kyoto University, Kyoto, Japan
- 7Faculty of Law, Economics and Management, Okinawa University, Naha, Japan
- 8Institute of Ecology and Biological Resources, Vietnam Academy of Science and Technology, Hanoi, Vietnam
- 9Graduate University of Science and Technology, Vietnam Academy of Science and Technology, Hanoi, Vietnam
- 10Department of Pharmacology and Parasitology, University of Veterinary Science, Nay Pyi Taw, Myanmar
- 11Department of Aquaculture and Aquatic Disease, University of Veterinary Science, Nay Pyi Taw, Myanmar
- 12Department of Zoology, Yangon University of Distance Education, Yangon, Myanmar
- 13Department of Microbiology, Faculty of Medicine, University of Peradeniya, Peradeniya, Sri Lanka
- 14Mention Foresterie et Environnement, Ecole Supérieur des Sciences Agronomiques, Université d'Antananarivo, Antananarivo, Madagascar
- 15Institute of Biological Sciences, Faculty of Science, University of Malaya, Kuala Lumpur, Malaysia
- 16Research Centre for Biology, Indonesian Institute of Sciences (LIPI), Bogor, Indonesia
- 17Laboratory of Ecology and Genetics, Graduate School of Environmental Science, Hokkaido University, Sapporo, Japan
- 18Department of Microbiology, Faculty of Veterinary Medicine, Okayama University of Science, Imabari, Japan
- 19Pacific Center for Emerging Infectious Diseases Research, John A. Burns School of Medicine, University of Hawaii at Manoa, Honolulu, HI, United States
Murid and cricetid rodents were previously believed to be the principal reservoir hosts of hantaviruses. Recently, however, multiple newfound hantaviruses have been discovered in shrews, moles, and bats, suggesting a complex evolutionary history. Little is known about the genetic diversity and geographic distribution of the prototype shrew-borne hantavirus, Thottapalayam thottimvirus (TPMV), carried by the Asian house shrew (Suncus murinus), which is widespread in Asia, Africa, and the Middle East. Comparison of TPMV genomic sequences from two Asian house shrews captured in Myanmar and Pakistan with TPMV strains in GenBank revealed that the Myanmar TPMV strain (H2763) was closely related to the prototype TPMV strain (VRC66412) from India. In the L-segment tree, on the other hand, the Pakistan TPMV strain (PK3629) appeared to be the most divergent, followed by TPMV strains from Nepal, then the Indian-Myanmar strains, and finally TPMV strains from China. The Myanmar strain of TPMV showed sequence similarity of 79.3–96.1% at the nucleotide level, but the deduced amino acid sequences showed a high degree of conservation of more than 94% with TPMV strains from Nepal, India, Pakistan, and China. Cophylogenetic analysis of host cytochrome b and TPMV strains suggested that the Pakistan TPMV strain was mismatched. Phylogenetic trees, based on host cytochrome b and cytochrome c oxidase subunit I genes of mitochondrial DNA, and on host recombination activating gene 1 of nuclear DNA, suggested that the Asian house shrew and Asian highland shrew (Suncus montanus) comprised a species complex. Overall, the geographic-specific clustering of TPMV strains in Asian countries suggested local host-specific adaptation. Additional in-depth studies are warranted to ascertain if TPMV originated in Asian house shrews on the Indian subcontinent.
Introduction
More than four decades following the original isolation of Thottapalayam thottimvirus (TPMV) from an Asian house shrew (Suncus murinus) in southern India (Carey et al., 1971), other genetically distinct hantaviruses (family Hantaviridae) have been detected in shrews (order Eulipotyphla, family Soricidae) of multiple species in Europe, Asia, Africa, and North America. To date, these include Imjin thottimvirus in the Ussuri white-toothed shrew (Crocidura lasiura) (Song et al., 2009), Kilimanjaro thottimvirus in the Kilimanjaro mouse shrew (Myosorex zinki) (Kang et al., 2014), Uluguru thottimvirus in the geata mouse shrew (Myosorex geata) (Kang et al., 2014), Tanganya orthohantavirus in the Therese's shrew (Crocidura theresae) (Klempa et al., 2007), Azagny orthohantavirus in the West African pygmy shrew (Crocidura obscurior) (Kang et al., 2011b), Jeju orthohantavirus in the Asian lesser white-toothed shrew (Crocidura shantungensis) (Arai et al., 2012), Bowé orthohantavirus in the Doucet's musk shrew (Crocidura douceti) (Gu et al., 2013), Cao Bǎng orthohantavirus in the Chinese mole shrew (Anourosorex squamipes) (Song et al., 2007c), Seewis orthohantavirus in the Eurasian shrew (Sorex araneus) (Song et al., 2007b), Ash River orthohantavirus in the masked shrew (Sorex cinereus) (Arai et al., 2008), Jemez Springs orthohantavirus in the dusky shrew (Sorex monticolus) (Arai et al., 2008), Kenkeme orthohantavirus in the flat-skulled shrew (Sorex roboratus) (Kang et al., 2010), Asikkala orthohantavirus in the Eurasian pygmy shrew (Sorex minutus) (Radosa et al., 2013), Yákèshí orthohantavirus in the taiga shrew (Sorex isodon) (Guo et al., 2013), Quan Hu Shan orthohantavirus in the greater stripe-backed shrew (Sorex cylindricauda) (Zuo et al., 2014), Boginia orthohantavirus in the Eurasian water shrew (Neomys fodiens) (Gu et al., 2013), and Camp Ripley orthohantavirus in the northern short-tailed shrew (Blarina brevicauda) (Arai et al., 2007). Data suggest that shrews, rather than rodents, may have served as the earlier mammalian hosts of hantaviruses (Song et al., 2007a; Kang et al., 2011a; Bennett et al., 2014; Yanagihara et al., 2014). That said, the evolutionary history of hantavirus expansion is still unclear. In particular, the genetic diversity and phylogeography of the prototype shrew-borne hantavirus (TPMV) warrants clarification.
Materials and Methods
Ethics Statement
The guidelines of the American Society of Mammalogists (Kirkland, 1998; Sikes and Animal Care and Use Committee of the American Society of Mammalogists, 2016) were followed for trapping and euthanasia of shrews and for tissue collection and processing. And approvals were obtained from the Ministry of Agriculture and Rural Development in Vietnam and the Institutional Animal Care and Use Committee of the National Institute of Infectious Diseases to conduct the study (permission numbers: 108074, 111126, 112152, 115162, 118180).
Animals
Asian house shrews were collected, using Sherman live traps, during biological distribution surveys from 2011 to 2016 in Asia (Indonesia, Japan, Malaysia, Myanmar, Pakistan, Sri Lanka, Vietnam, Yemen) and Africa (Comoros, Madagascar, Tanzania) (Ohdachi et al., 2016) (Figure 1). Lung tissues, preserved in RNAlater® Stabilization Solution, were analyzed for hantavirus RNA by reverse transcription polymerase chain reaction (RT-PCR) (Arai et al., 2016a). Asian house shrews from Bangladesh (early-onset diabetes in Suncus: EDS) and Nepal (Kathmandu: KAT), which were being maintained for experimentation, were also used for host genetic analysis (Jogahara, 2016) (Supplementary Data S1).
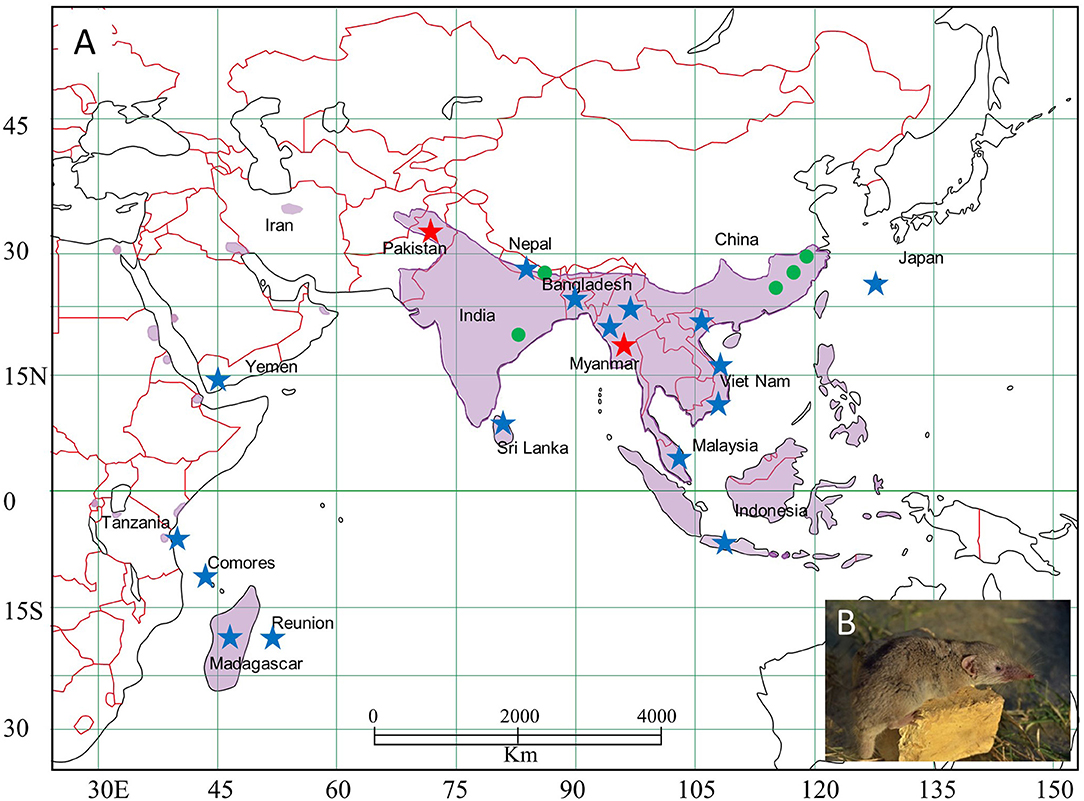
Figure 1. Map, showing the distribution of Asian house shrews (Suncus murinus) in Eurasia. (A) Asian house shrews were captured in multiple locations (indicated by star). TPMV-infected shrews were found in Pakistan (red star), Myanmar (red star), India (green circle), Nepal (green circle), and China (green circle). (B) Photo of Asian house shrew.
RNA Extraction and cDNA Synthesis
Total RNA was extracted from RNAlater®-preserved lung tissues, using the GC series Magtration®-MagaZorb® RNA Common N Kit or MagDEA RNA 100 Kit (Precision System Science, Matsudo, Japan), and then reverse transcribed, using PrimeScript™ II 1st strand cDNA Synthesis Kit (Takara Bio, Otsu, Japan) and oligonucleotide primer (OSM55F, 5′-TAGTAGTAGACTCC−3′), designed from the conserved 5′-ends of the S, M, and L segments of hantaviruses (Klempa et al., 2006, 2007; Song et al., 2007c; Arai et al., 2008, 2016a).
RT-PCR and DNA Sequencing
Nested primers for TPMV and other recently identified shrew-borne hantaviruses were used to initially screen tissues for hantavirus RNA (Song et al., 2009; Kang et al., 2011c; Gu et al., 2013). Thereafter, amplification of the full-length S-, M-, and L-genomic segments was attempted. Oligonucleotide primer sequences have been deposited as Supplementary Data S2. First- and second-round PCR was performed in 20-μL reaction mixtures, containing 250 μM dNTP, 2 mM MgCl2, and 0.25 μM of each primer. LA Taq hot start version (Takara Bio) and AmpliTaq gold 360 DNA polymerase (Applied Biosystems, Foster City, CA, USA) were used at 1 U each for the first- and second-round PCR, respectively (Arai et al., 2016b). Initial denaturation at 94°C for 2 min was followed by two cycles each of denaturation at 94°C for 30 s, two-degree step-down annealing from 48 to 38°C for 40 s, and elongation at 68°C for 1 min, then 32 cycles of denaturation at 94°C for 40 s, annealing at 42°C for 40 s, and elongation at 68°C for 1 min, in a Veriti thermal cycler (Applied Biosystems) and Mastercycler X50 (Eppendorf, Hamburg, Germany) (Arai et al., 2008, 2012). Amplicons were treated with Exonuclease I and Shrimp Alkaline Phosphatase (New England Biolabs, Ipswich, MA, USA) for 30 min. DNA was sequenced directly using an ABI Prism 377XL Genetic Analyzer (Applied Biosystems) (Arai et al., 2007; Kang et al., 2011c).
Genetic and Phylogenetic Analysis
Partial S-, M-, and L-segment nucleotide and amino acid sequences, amplified from Asian house shrews, were aligned with available hantavirus sequences, using the ClustalW in BioEdit (Thompson et al., 1994). The degree of sequence homology was assessed by pair-wise comparisons (Kang et al., 2009, 2011a). Phylogenetic trees were constructed using MrBayes 3.1.2 (Ronquist and Huelsenbeck, 2003), with the GTR+I+Γ model of evolution, as selected by using jModelTest version 2.1.7 (Darriba et al., 2012). Bayesian analysis consisted of 10 million Markov chain Monte Carlo generations to ensure convergence across two runs of six chains each, with average standard deviations of split frequencies <0.01 and effective sample sizes well over 100, resulting in consensus trees supported by posterior-node probabilities (Kang et al., 2009, 2011a). The co-evolutionary relationships between hantaviruses and their shrew and rodent reservoir hosts were analyzed by the comparative concordance between host and hantavirus cladograms in TreeMap 3b1243 (Charleston and Robertson, 2002; Kang et al., 2009; Arai et al., 2012).
mtDNA and Nuclear Genes Sequencing and Host Phylogeny
To verify the geographic diversity of Asian house shrews and to study their phylogenetic relationships, genomic DNA was extracted from lung tissue using the MagDEA® DNA 200 (GC) (Precision System Science). The entire 1,140-nucleotide cytochrome b (cytb) gene of mitochondrial DNA (mtDNA), the 1,545-nucleotide cytochrome c oxidase subunit I (COI) gene and the recombination activating gene 1 (RAG1) were amplified using the following primer sets: Cy-14724F (5′-GACYARTRRCATGAAAAAYCAYCGTTGT−3′)/Cy-15909R (5′-CYYCWTYIYTGGTTTA CAAGACYAG−3′) (Arai et al., 2016b) and KOD multi-enzyme (Toyobo, Osaka, Japan), MammMt-5533F (5′-CYCTGTSYTTRRATTTACAGTYYAA−3′)/MammMt-7159R (5′-GRGGTTCRAWW CCTYCCTYTCTT−3′) (Arai et al., 2019) and Phusion enzyme (New England Biolabs), and newly designed primers RAG1-61F (5′-TCTGCACCYGATGAAATTCARCACC−3′)/RAG1-3139R (5′-CTCCATTGAATCTTGGCTTTCC−3′) and KOD multi-enzyme, respectively. PCR was performed in 50-μL reaction mixtures, containing 200 μM dNTP and 1 U of KOD multi and Epi DNA polymerase or Phusion enzyme. Initial denaturation was at 95°C for 2 min, followed by two cycles each of denaturation at 95°C for 15 s, two-degree step-down annealing from 60 to 50°C for 30 s, and elongation at 68°C for 1 min 30 s, then 30 cycles of denaturation at 95°C for 15 s, annealing at 55°C for 30 s, and elongation at 68°C for 1 min 30 s, in a Veriti thermal cycler (Arai et al., 2019). PCR products were purified by Mobispin S-400 (Molecular Biotechnology, Lotzzestrasse, Germany) and were sequenced directly (Arai et al., 2012, 2019). The models of host nucleotide evolution were selected under jModeltest version 2.1.7, the GTR+I+Γ model for host phylogenetic sequence set, the TrN+G for Cytb, TIM3+ I for COI of and TPM2uf +I models for RAG1 of Suncus sequence sets. The results of modeltest were shown in Supplementary Data 3A–G. Host phylogenetic analysis also consisted of 10 million Markov chain Monte Carlo generations to ensure convergence across two runs of six chains each, with average standard deviations of split frequencies <0.01 and effective sample sizes well over 100, resulting in consensus trees supported by posterior-node probabilities.
Results
Hantavirus Detection
In all but two of the 198 shrew lung tissue samples, multiple attempts to detect hantavirus RNA were unsuccessful (Table 1). The exceptions were one of 11 and one of three Asian house shrews from Pakistan (captured in Karachi: 24.947802 N, 67.122999 E) and Myanmar (captured near a cattle farm in Taung gyi, Shan state: 20.804169 N, 97.060360 E, detail in Supplementary Table 1), respectively, collected in 2013. Sequence analysis of the amplicons revealed TPMV. Amplification of the full-coding region of the S segment and the partial M and L segments was achieved for TPMV strain H2763 (Myanmar), while only partial L-segment sequences were obtained for TPMV strain PK3629 (Pakistan).
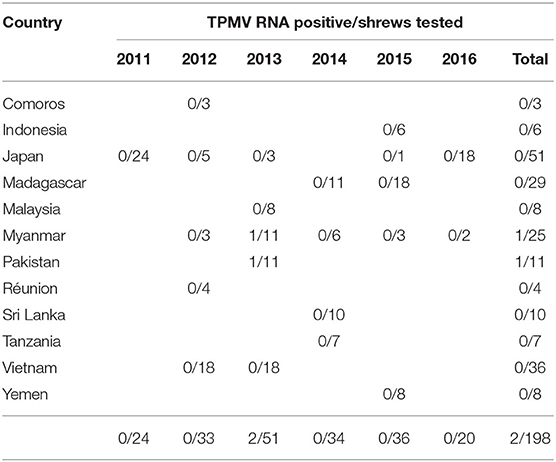
Table 1. RT-PCR detection of TPMV RNA in Asian house shrews.
Nucleotide and Amino Acid Sequence Analysis
Analysis of the S-, M-, and L-segment sequences of TPMV strain H2763 from Myanmar indicated an overall genomic organization similar to prototype TPMV strain VRC66412 from India. The 1,506-nucleotide S-genomic segment encoded a nucleocapsid (N) protein of 435 amino acids, possibly starting at nucleotide position 68, and a 130-nucleotide 3′-non-coding region. The TPMV S-genomic segment, like that of other recently described hantaviruses in shrews, did not contain the hypothetical NSs open reading frame, typically found in hantaviruses harbored by cricetid rodents.
Hantavirus Phylogeny
TPMV strain H2763 from Myanmar appeared as one cluster in phylogenetic trees, based on the S-, M-, and L-segment sequences, using the Bayesian methods (Figure 2). The TPMV strain PK3629 from Pakistan also constructed one cluster in a tree based on the L segment. The phylogenetic trees suggested that the primordial strain of TPMV originated in northern India and surrounding countries, including Pakistan or Nepal (Figure 2).
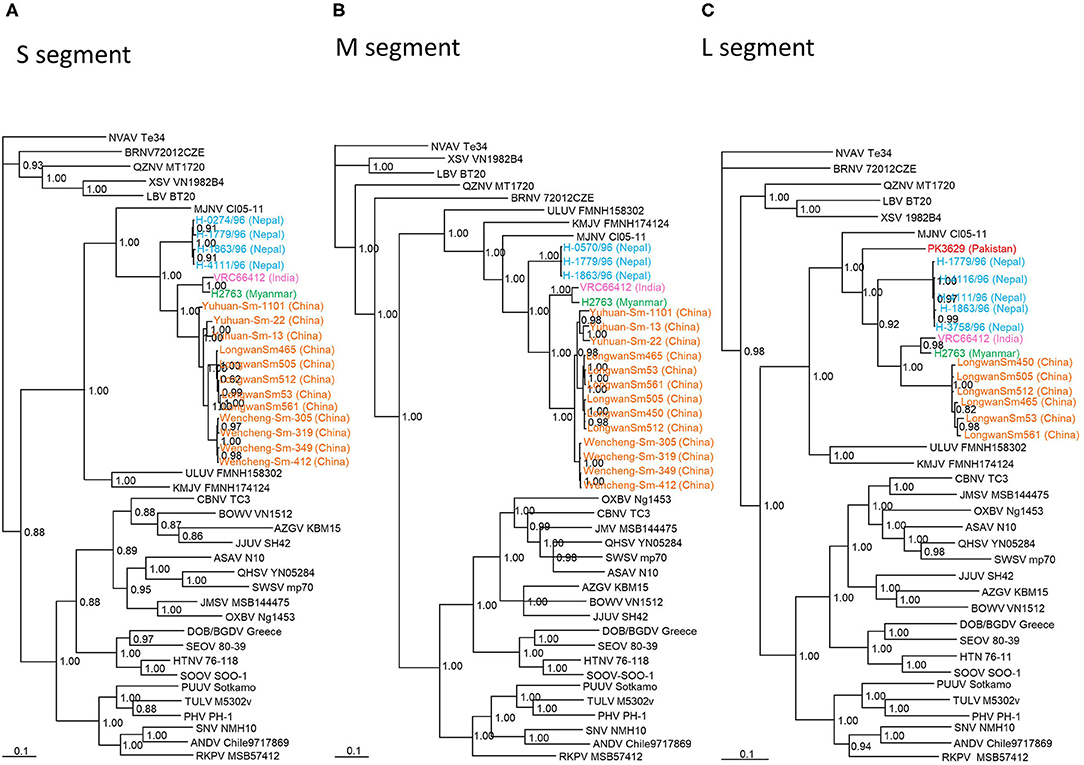
Figure 2. Phylogenetic trees generated by the Bayesian method, under the best-fit GTR+I+Γ model of evolution, based on the partial S-, M-, and L-genomic segments of TPMV strains H2763 (S: MT225396; M: MT225397; L: MT225398) in Myanmar and PK3629 in Pakistan (L: MT225399), and other representative hantaviruses. The phylogenetic positions of TPMV strains VRC66412 (S: AY526097, M: EU001329, L: EU001330) in India; TPMV strains LongwanSm53 (S: JF784172; M: JF784178; L: KJ420575), LongwanSm450 (S: JF784173, M: JF784179, L: KJ420576), LongwanSm465 (S: JF784174, M: JF784180), LongwanSm505 (S: JF784175, M: JF784181), LongwanSm512 (S: JF784176, M: JF784182), LongwanSm561 (S: JF784177, M: JF784183), Wencheng-Sm-305 (S: KJ420560, M: KJ420542), Wencheng-Sm-319 (S: KJ420561, M: KJ420543, L: KJ420569), Wencheng-Sm-349 (S: KJ420562, M: KJ420544, L: KJ420570), Wencheng-Sm-412 (S: KJ420563, M: KJ420545, L: KJ420571), Yuhuan-Sm-13 (S: KJ420564, M: KJ420546, L: KJ420572), Yuhuan-Sm-22 (S: KJ420565, M: KJ420547, L: KJ420573), and Yuhuan-Sm-1101 (S: KJ420566, M: KJ420548, L: KJ420574) in China; strains H-1091/96 (S: HQ831358), H-0274/96 (S: HQ831359), H-0570/96 (S: HQ831360, M: HQ831376), H-0898/96 (S: HQ831361), H-1779/96 (S: HQ831362, M: HQ831375, L: HQ831374), H-1863/96 (S: HQ831363, M: HQ831377, L: HQ831373), H-3753/96 (S: HQ831364), H-3758/96 (S: HQ831365, L: HQ831372), H-3858/96 (S: HQ831366), H-4066/96 (S: HQ831367), H-4111/96 (S: HQ831368, L: HQ831371), and H-4116/96 (S: HQ831369, L: HQ831370) in Nepal are shown in relationship to Imjin thottimvirus (MJNV Cl05-11, S: EF641804; M: EF641798; L: EF641806) from Crocidura lasiura, Uluguru thottimvirus (ULUV FMNH158302, S: JX193695; M: JX193696; L: JX193697) from Myosorex geata, Kilimanjaro thottimvirus (KMJV FMNH174124, S: JX193698; M: JX193699; L: JX193700) from Myosorex zinki, Jeju orthohantavirus (JJUV SH42, S: HQ663933; M: HQ663934; L: HQ663935) from Crocidura shantungensis, Oxbow orthohantavirus (OXBV Ng1453, S: FJ5339166; M: FJ539167; L: FJ593497) from Neurotrichus gibbsii, Cao Bǎng orthohantavirus (CBNV TC-3, S: EF543524; M: EF543526; L: EF543525) from Anourosorex squamipes, Azagny orthohantavirus (AZGV KBM15, S: JF276226; M: JF276227; L: JF276228) from Crocidura obscurior, Bowé orthohantavirus (BOWV VN1512, S: KC631782; M: KC631783; L: KC631784) from Crocidura douceti, Qian Hu Shan orthohantavirus (QHSV YN05-284, S: GU566023; M: GU566022; L:GU566021) from Sorex cylindricauda, Seewis orthohantavirus (SWSV mp70, S: EF636024; M: EF636025; L: EF636026) from Sorex araneus, Jemez Springs orthohantavirus from Sorex monticolus (JMSV MSB144475, S: FJ593499; M: FJ593500; L: FJ593501), Asama orthohantavirus (ASAV N10, S: EU929072; M: EU929075; L: EU929078) from Urotrichus talpoides, Nova mobatvirus (NVA Te34, S: KR072621, M: KR072622, L: KR072623) from Talpa europaea, and Rockport orthohantavirus (RKPV MSB57412, S: HM015223; M: HM015222; L: HM015221) from Scalopus aquaticus. Also shown are representative rodent-borne hantaviruses, including Sin Nombre orthohantavirus (SNV NMH10, S: NC_005216; M: NC_005215; L: NC_005217), Andes orthohantavirus (ANDV Chile9717869, S: AF291702; M: AF291703; L: AF291704), Prospect Hill orthohantavirus (PHV PH-1, S: Z49098; M: X55129; L: EF646763), Tula orthohantavirus (TULV M5302v, S: NC_005227; M: NC_005228; L: NC_005226), Puumala orthohantavirus (PUUV Sotkamo, S: NC_005224; M: NC_005223; L: NC_005225), Dobrava/Belgrade orthohantavirus (DOB/BGDV Greece, S: NC_005233; M: NC_005234; L: NC_005235), Hantaan orthohantavirus (HTNV 76-118, S: NC_005218; M: NC_005219; L: NC_005222) and Seoul orthohantavirus (SEOV 80-39, S: NC_005236; M: NC_005237; L: NC_005238), and bat-borne hantaviruses, Brno loanvirus (BRNV 7/2012/CZE, S: KX845678; M: KX845679; L: KX845680) from Nyctalus noctula, Láibin mobatvirus (LAIV BT20, S: KM102247; M: KM102248; L: KM102249) from Taphozous melanopogon, Xuân Son mobatvirus (XSV VN1982B4, S: KC688335; L: JX912953) from Hipposideros pomona and Quezon mobatvirus (QZNV MT1720/1657, S: KU950713; M: KU950714; L: KU950715) from Rousettus amplexicaudatus, respectively. The numbers at each node are posterior node probabilities based on 45,000 trees: two replicate Markov chain Monte Carlo runs consisting of six chains of two million generations each sampled every 100 generations with a burn-in of 7,500 (25%). The scale bar indicates nucleotide substitutions per site. (A) S-segment, (B) M-segment, (C) L-segment phylogenetic trees. Color of TPMV strains: red (Pakistan); orange (China); green (Myanmar); light blue (Nepal); pink (India).
Pair-Wise Alignment and Comparison
Pair-wise alignment and comparison of the S segment (1,506 nucleotides), M segment (2,382 nucleotides), and L segment (4,963nucleotides) revealed that TPMV strain H2763 from Myanmar exhibited high sequence similarity to prototype TPMV strain VRC66412 from southern India. The TPMV strain PK3629 from Pakistan showed low nucleotide sequence similarity (79.3%) in the L segment, but the encoded amino acid sequences were highly conserved (94.1–99.2%) with TPMV strains from Nepal, India, Pakistan, and China (Table 2). Compared with representative hantaviruses from rodents, shrews, and bats, the TPMV strain from Myanmar differed by ~20–60% at the nucleotide and amino acid levels for each segment.
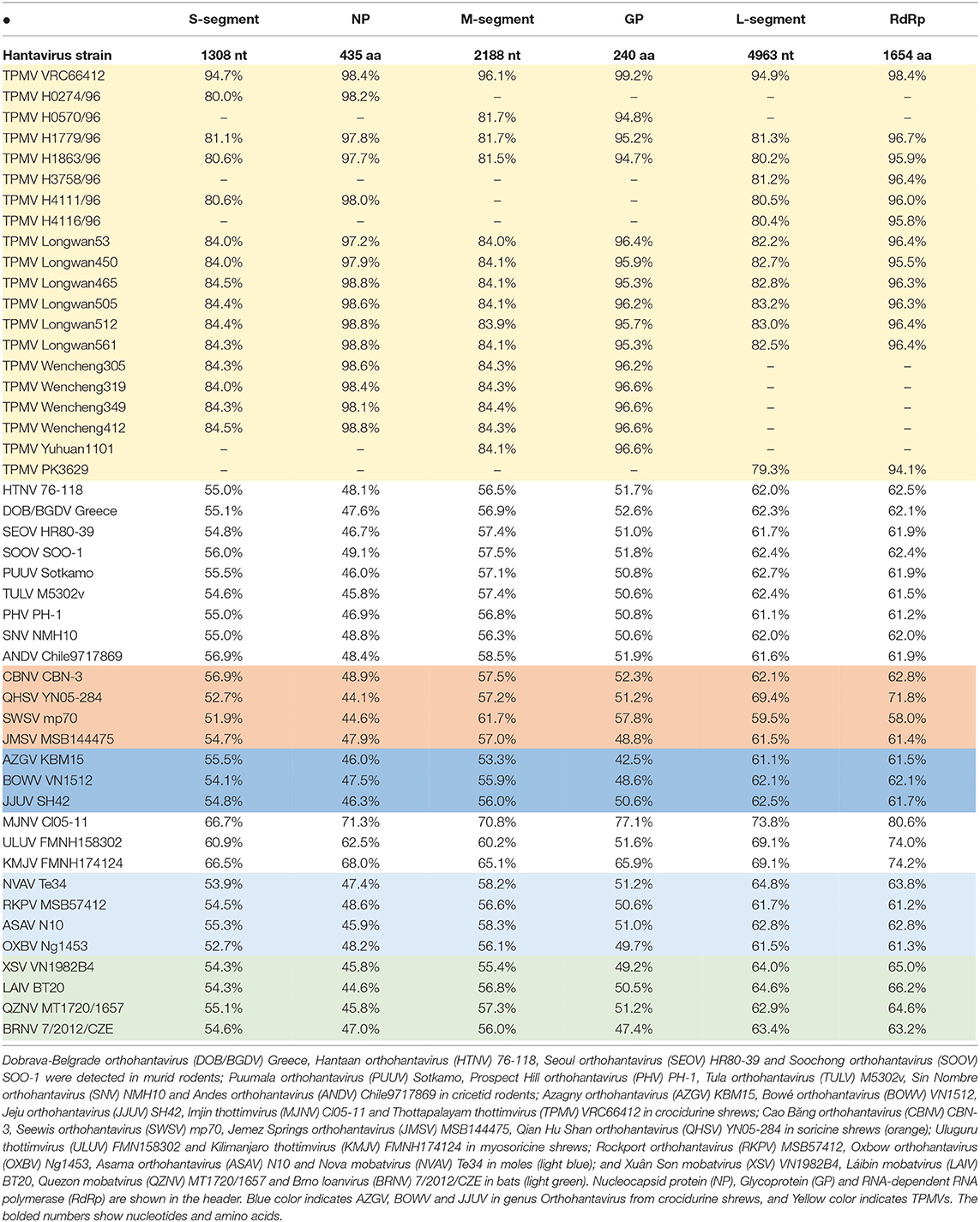
Table 2. Nucleotide and amino acid sequence similarity (%) between TPMV strain H2763 and other TPMV strains, as well as representative rodent-, shrew-, and bat-borne hantaviruses.
Co-phylogenetic Analysis of Asian House Shrew and TPMV
As evidenced by co-phylogeny mapping, using a consensus tree based on L-segment sequences, TPMV strains segregated according to the geographic locations of the Asian house shrews (Figures 3B,C). The phylogenetic positions of TPMV strains based on the S and M segments mirrored the phylogenetic relationships of their Asian house shrews, except for the Pakistan strain in the L-segment tree. The Pakistan strain was mismatched between virus and host phylogeography (Figure 3A).
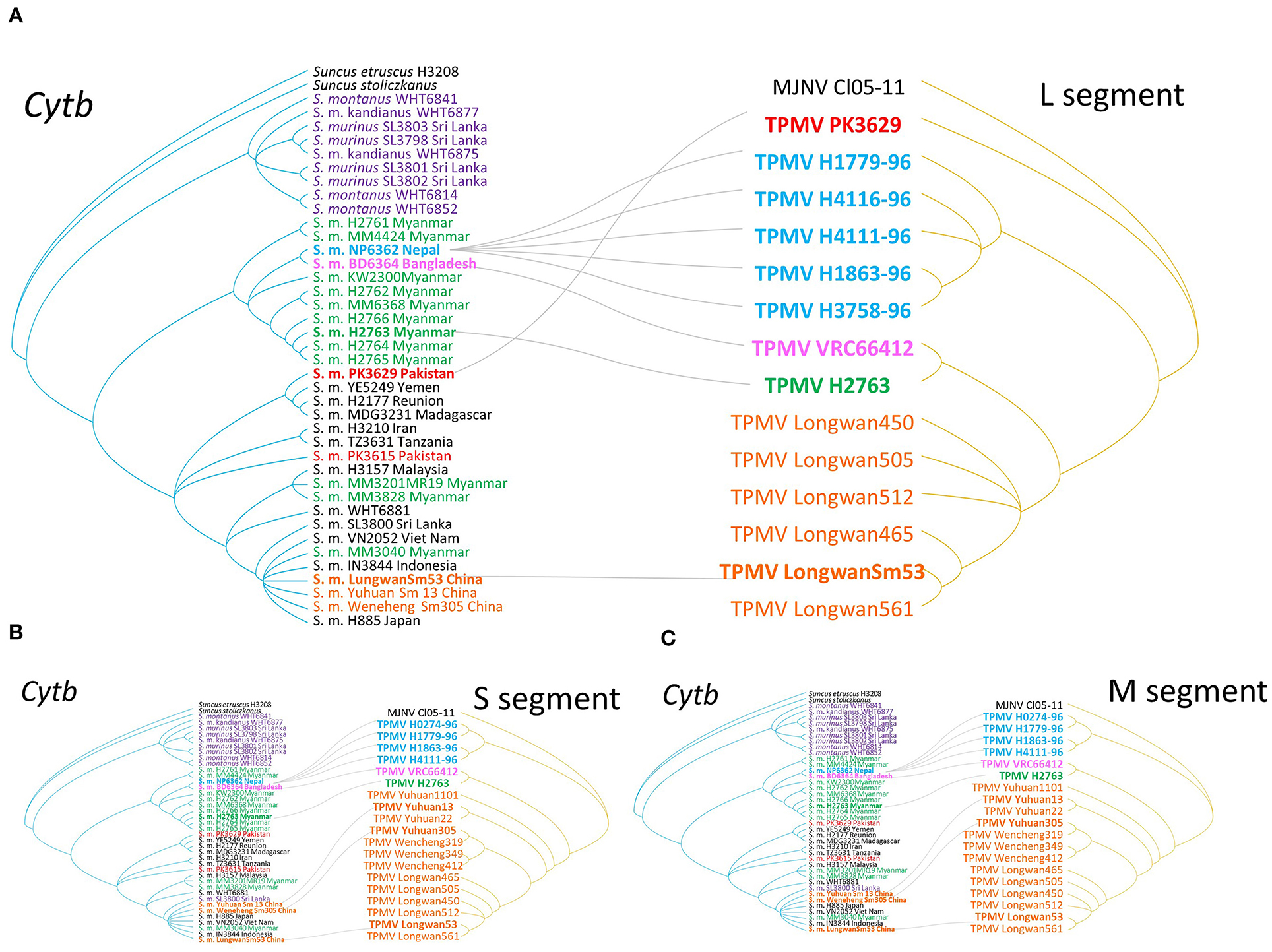
Figure 3. Comparisons of phylogenies of TPMV and their host species Asian house shrew. The host trees on the left were based on cytochrome b (cytb) gene, while the hantavirus tree on the right was based on the nucleotide sequences of L- (A), S- (B), and M-segments (C), respectively. Letterings for taxa are shown in purple for Sri Lanka, green for Myanmar, blue for Nepal, pink for Bangladesh and India, red for Pakistan, orange for China, black for the other countries, and out groups in both trees. Nepal Asian house shrew was adapted experimental animal strain NP6362. Bangladesh experimental strain BD6364 was used for alternative Indian strain.
Phylogenetic Analysis of Asian House Shrew
The molecular identification of TPMV-infected shrews was confirmed as S. murinus murinus by amplification and sequencing of the cytb and COI genes of mtDNA and RAG1 gene of nuclear DNA. Phylogenetic analysis based on the cytb gene indicated Asian house shrews and Etruscan shrews (Suncus etruscus) were clearly distinct (Figure 4). However, the relationships between Asian house shrews, Asian highland shrews (Suncus montanus) and, some S. murinus subspecies (such as S. m. murinus, S. m. kandianus, and S. m. caerulescens) (Meegaskumbura et al., 2010) were less clear.
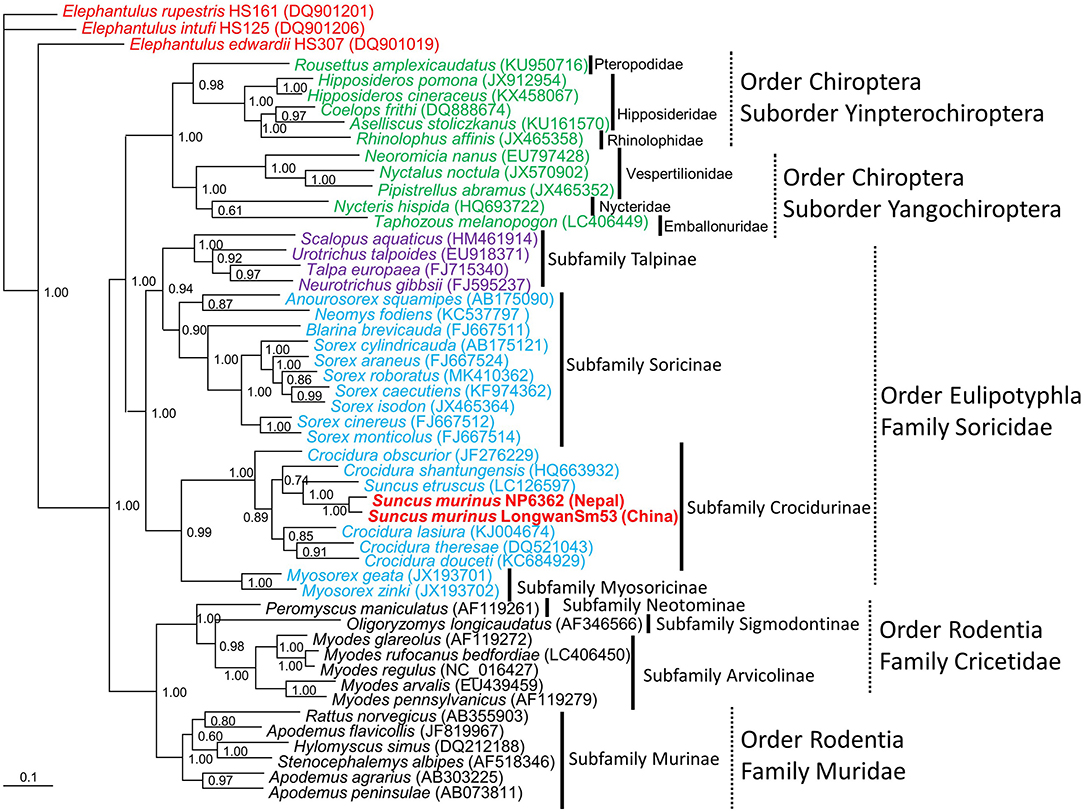
Figure 4. Bayesian phylogenetic tree, based on the 1,140-nucleotide cytochrome b gene of mtDNA of small mammals within the order Eulipotyphla (families Talpidae and Soricidae), order Rodentia (families Muridae and Cricetidae) and order Chiroptera, suborder Yinpterochiroptera (families Pteropodidae, Hipposideridae, Rhinolophidae), and suborder Yangochiroptera (families Nycteridae, Emballonuridae and Vespertilionidae). The tree was rooted using Elephantulus (order Macroscelidea, GenBank accession numbers DQ901019, DQ901206, and DQ901201) as the outgroup. Numbers at nodes indicate posterior probability values (>0.7) based on 150,000 trees: two replicate Markov chain Monte Carlo runs, consisting of six chains of 10 million generations each sampled every 100 generations with a burn-in of 25,000 (25%). Scale bars indicate nucleotide substitutions per site. Letterings for taxa are shown in green for bats, blue for shrews, purple for moles, black for rodents, red for Elephantulus, and red bold for Asian house shrew. The GenBank accession number for the cytb sequence for Asian house shrews are MT344840 in Nepal and JF784169 in China.
Segregation of Asian house shrews in Asia and Africa was demonstrated by co-phylogeny mapping, using consensus trees based on the cytb, COI and RAG1 genes (Figure 5). The phylogenetic positions of RAG1 in nuclear DNA and cytb and COI in mtDNA were not synchronized for each gene. These data suggest that S. murinus and S. montanus are hybrid species and comprise the S. murinus-S. montanus species complex.
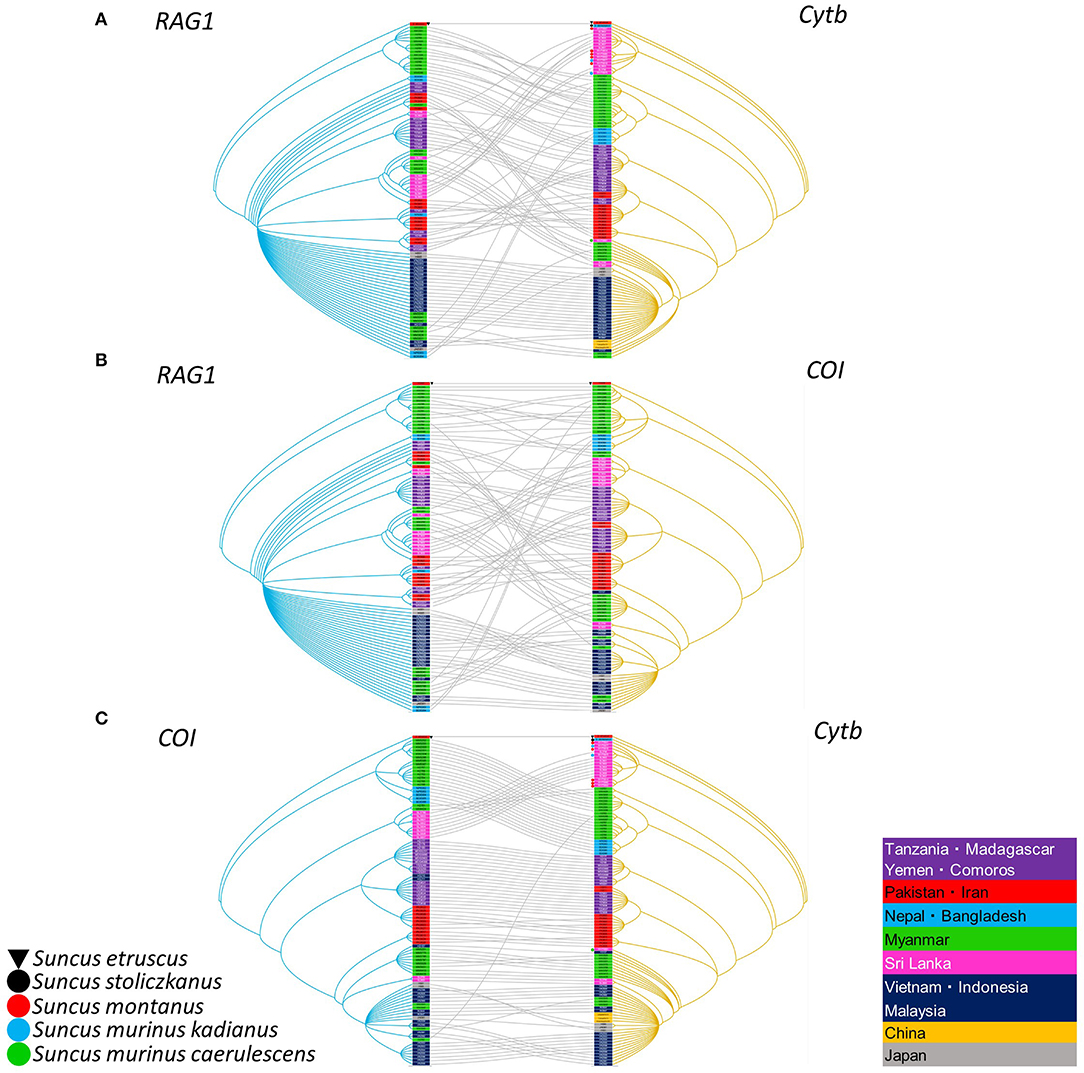
Figure 5. Comparisons of nuclear and mitochondria genes generated by TreeMap 3, using Bayesian method, based on the recombination activating gene 1 (RAG1), cytochrome b (cytb) gene, and cytochrome oxidase I (COI) gene of the Asian house shrew and related shrews. (A) Left tree on the RAG1 sequence, while the mitochondria tree on the right was based on the COI gene or (B) cytb gene, and (C) also comparison of cytb and COI genes, respectively. The nuclear (RAG-1) and mitochondria relationship (cytb and COI) were listed in Supplementary Table 1. Letterings for taxa are shown in right table. The deposited cytb sequences are attached dot. The tree was rooted using Suncus etruscus (order Eulipotyphla, family Soricidae, GenBank accession numbers: cytb, LC126597; COI, MK410384; and RAG1, MT344767). The RAG1 order in Figures 5A,B was the same. The order of COI and cytb in Figure 5C was adjusted for optimal.
Discussion
The Asian house shrew, one of 18 species in the genus Suncus, is widely distributed throughout Asia and the Pacific, Africa, and the Middle East (Figure 1). It is peridomestic, typically found within areas of human habitation, and has become dependent on discarded human food waste. Asian house shrews may have been intentionally introduced by humans, similar to Rattus rodents, into Africa (Egypt, Eritrea, Kenya, Republic of Djibouti, Rwanda, Sudan, and Tanzania), the Middle East (Iran, Iraq, Kingdom of Bahrain, Kuwait, Saudi Arabia, Sultanate of Oman, and Yemen), the islands within the Indian Ocean (Comoros, Republic of Madagascar, Republic of Mauritius, and Réunion), and Asia and the Pacific (Japan, Guam, and Philippines) (Kang et al., 2011c). Genetic analysis and treemap dendrograms of RAG1 and COI, RAG1 and cytb, and COI and cytb suggest that Asian house shrews may represent hybrids with the Asian highland shrew in Sri Lanka and some area of Eurasia (Figure 5).
The previously held conventional view that hantaviruses co-evolved with their reservoir hosts has been challenged recently by the conjecture that preferential host switching and local host-specific adaptation account for the congruent phylogenies of hantaviruses and their small mammal hosts (Ramsden et al., 2009). Multiple examples of host sharing are now known for hantaviruses hosted by rodents and shrews (Yanagihara et al., 2014). Whether or not TPMV exhibits such host sharing with evidence of carriage by other species of the genus Suncus requires future investigation.
Based on phylogenetic analysis of mtDNA and nuclear genes, as well as karyotype and morphological analysis, the taxonomy of the Asian house shrew is still unclear. Asian house shrews include at least two subspecies (S. murinus murinus, S. murinus kandianus, and S. murinus caerulescens), and the Asian highland shrew (S. montanus) is morphologically very similar. Our genetic analysis suggests that morphological based S. m. murinus, S. m. kadianus, S. m. caerulescens, and S. montanus represent hybrid species. Thus, a species complex has been proposed (Ohdachi et al., 2016).
Although genetically diverse strains of TPMV have been detected in Asian house shrews from Nepal (Kang et al., 2011c) and China (Guo et al., 2011), the geographic distribution and evolutionary origins of TPMV are still unclear. Our data suggest the possibility that TPMV expanded from the Indian subcontinent (Figure 6). The evolutionary time scale of TPMV is faster than that of its host and the host is older than the ancient trade routes between the Middle East and China.
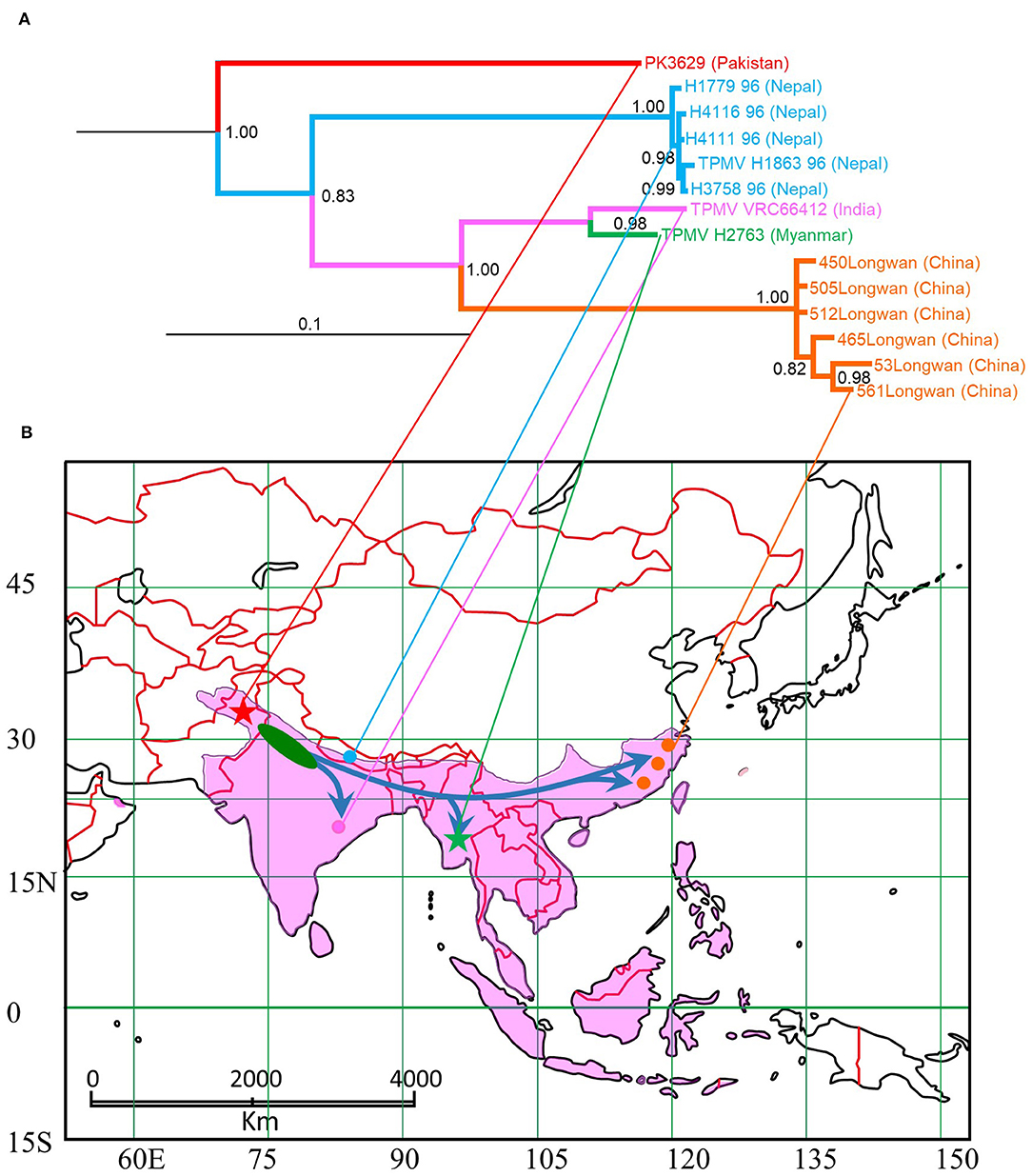
Figure 6. Map of distribution of Asian house shrew and phylogenetic tree based on L segment of TPMV. (A) TPMV cluster based on L-segment in phylogenetic analysis. (B) Blue arrows in map were estimated expansion root based on TPMV phylogeny. Pakistan strain was captured at red star, Nepal strains (Kang et al., 2011c) were captured at light blue circle, Indian strain (Carey et al., 1971) was captured at pink circle, Myanmar strain was captured at green star and Chinese strains (Guo et al., 2011) were captured at red circles. Pakistan and Myanmar strains were collected in this study (star symbols). Pink area is shown Asian house shrew distribution (Ohdachi et al., 2016).
Conclusion
Disappointingly, TPMV was detected in lung tissues of only two Asian house shrews, one from Myanmar and one from Pakistan. The reasons for this are not entirely clear, but it might be the result of the focal nature of TPMV infection, as is typical of other hantaviruses. Future studies on the phylogeography of TPMV and the Asian house shrew should provide valuable insights into the geographic radiation.
Data Availability Statement
The original contributions presented in the study are publicly available. This data can be found at: https://www.ncbi.nlm.nih.gov/, with the following accession numbers: MT225396—MT225399; MT344729—MT344941; MT363682—MT363701; MT364899.
Ethics Statement
The animal study was reviewed and approved by National Institute of Infectious Diseases (NIID), Institutional Animal Care and Use Committee.
Author Contributions
FK, KA, SM, RY, and SA conceived the study and designed the experiments. SO, KT-T, MM, TJ, NS, SB, KL, TT, CG, MR, HO, IM, HS, and SA conducted the trapping and field collections. FK, KA, and SA performed the experiments. FK, KA, RY, and SA analyzed the data. SO, KT-T, MM, TJ, and NS analyzed the host morphology. FK, KA, KT-T, SM, TM, MS, RY, and SA contributed reagents, materials, and analysis tools. FK, SM, TM, RY, and SA prepared the figures and draft manuscript. All authors contributed to the final manuscript.
Funding
This research was supported in part by a grant-in-aid from the Research Program on Emerging and Re-emerging Infectious Diseases, Japan Agency for Medical Research and Development (AMED) (JP15fk0108005, JP16fk0108117, JP17fk0108217, JP18fk0108017, JP19fk0108097, and JP20fk0108097); a grant-in-aid for scientific research from the Japan Society for the Promotion of Science (KAKENHI) (24405045 and JP18H03602); a grant-in-aid from the program of developing basic sciences in Chemistry, Life sciences, Earth sciences and Marine sciences, Vietnam (KHCBSS.01/20-22); as well as a grant from the U.S. National Institutes of Health (P30GM114737).
Conflict of Interest
KT, who is deceased, was previously employed by the Applied Biology Co., Ltd., in Tokyo, Japan.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the following individuals for supporting field investigations: Taher Ghadirian of the Persian Wildlife Heritage Foundation in Tehran, Iran; Razafindrakoto Todisoa and Mamisoa Colette Vincentine of the Botanical and Zoological Garden of Tsimbazaza in Antananarivo, Madagascar; Atushi Nakamoto of Okayama University of Science in Okayama, Japan; Chihiro Tanaka of the Yagiyama Zoological Park in Sendai, Japan; Shinichiro Kawada of the National Museum of Nature and Science in Tokyo, Japan; and Dai Fukui of the University of Tokyo Hokkaido Forests, the University of Tokyo in Furano, Japan. We also thank Shinichiro Kawada, Dai Fukui, and Takashiro Akitsu of the Tokyo University of Science in Tokyo, Japan, for helpful suggestions.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2020.00438/full#supplementary-material
References
Arai, S., Bennett, S. N., Sumibcay, L., Cook, J. A., Song, J.-W., Hope, A., et al. (2008). Phylogenetically distinct hantaviruses in the masked shrew (Sorex cinereus) and dusky shrew (Sorex monticolus) in the United States. Am. J. Trop. Med. Hyg. 78, 348–351. doi: 10.4269/ajtmh.2008.78.348
Arai, S., Gu, S. H., Baek, L. J., Tabara, K., Bennett, S. N., Oh, H. S., et al. (2012). Divergent ancestral lineages of newfound hantaviruses harbored by phylogenetically related crocidurine shrew species in Korea. Virology 424, 99–105. doi: 10.1016/j.virol.2011.11.013
Arai, S., Kang, H. J., Gu, S. H., Ohdachi, S. D., Cook, J. A., Yashina, L. N., et al. (2016a). Genetic diversity of Artybash virus in the Laxmann's shrew (Sorex caecutiens). Vector Borne Zoonotic Dis. 16, 468–475. doi: 10.1089/vbz.2015.1903
Arai, S., Kikuchi, F., Bawm, S., Son, N. T., Lin, K. S., Tu, V. T., et al. (2019). Molecular phylogeny of mobatviruses (Hantaviridae) in Myanmar and Vietnam. Viruses 11, 228. doi: 10.3390/v11030228
Arai, S., Song, J.-W., Sumibcay, L., Bennett, S. N., Nerurkar, V. R., Parmenter, C., et al. (2007). Hantavirus in northern short-tailed shrew, United States. Emerg. Infect. Dis. 13, 1420–1423. doi: 10.3201/eid1309.070484
Arai, S., Taniguchi, S., Aoki, K., Yoshikawa, Y., Kyuwa, S., Tanaka-Taya, K., et al. (2016b). Molecular phylogeny of a genetically divergent hantavirus harbored by the Geoffroy's rousette (Rousettus amplexicaudatus), a frugivorous bat species in the Philippines. Infect. Genet. Evol. 45, 26–32. doi: 10.1016/j.meegid.2016.08.008
Bennett, S. N., Gu, S. H., Kang, H. J., Arai, S., and Yanagihara, R. (2014). Reconstructing the evolutionary origins and phylogeography of hantaviruses. Trends Microbiol. 22, 473–482. doi: 10.1016/j.tim.2014.04.008
Carey, D. E., Reuben, R., Panicker, K. N., Shope, R. E., and Myers, R. M. (1971). Thottapalayam virus: a presumptive arbovirus isolated from a shrew in India. Indian J. Med. Res. 59, 1758–1760.
Charleston, M. A., and Robertson, D. L. (2002). Preferential host switching by primate lentiviruses can account for phylogenetic similarity with the primate phylogeny. Syst. Biol. 51, 528–535. doi: 10.1080/10635150290069940
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9:772. doi: 10.1038/nmeth.2109
Gu, S. H., Nicolas, V., Lalis, A., Sathirapongsasuti, N., and Yanagihara, R. (2013). Complete genome sequence and molecular phylogeny of a newfound hantavirus harbored by the Doucet's musk shrew (Crocidura douceti) in Guinea. Infect. Genet. Evol. 20, 118–123. doi: 10.1016/j.meegid.2013.08.016
Guo, W. P., Lin, X. D., Wang, W., Tian, J. H., Cong, M. L., Zhang, H. L., et al. (2013). Phylogeny and origins of hantaviruses harbored by bats, insectivores and rodents. PLoS Pathog. 9:e1003159. doi: 10.1371/journal.ppat.1003159
Guo, W. P., Lin, X. D., Wang, W., Zhang, X. H., Chen, Y., Cao, J. H., et al. (2011). A new subtype of Thottapalayam virus carried by the Asian house shrew (Suncus murinus) in China. Infect. Genet. Evol. 11, 1862–1867. doi: 10.1016/j.meegid.2011.07.013
Jogahara, T. (2016). Animal bio-resources in the Department of Zoology, Faculty of Science, Okayama University of Science. Proc. Okayama Assoc. Lab. Anim. Sci. 32, 12–18. Available online at: http://ousar.lib.okayama-u.ac.jp/files/public/5/54401/20160627151010751151/poalas_032_012_018.pdf
Kang, H. J., Arai, S., Hope, A. G., Cook, J. A., and Yanagihara, R. (2010). Novel hantavirus in the flat-skulled shrew (Sorex roboratus). Vector Borne Zoonotic Dis. 10, 593–597. doi: 10.1089/vbz.2009.0159
Kang, H. J., Bennett, S. N., Hope, A. G., Cook, J. A., and Yanagihara, R. (2011a). Shared ancestry between a newfound mole-borne hantavirus and hantaviruses harbored by cricetid rodents. J. Virol. 85, 7496–7503. doi: 10.1128/JVI.02450-10
Kang, H. J., Bennett, S. N., Sumibcay, L., Arai, S., Hope, A. G., Mocz, G., et al. (2009). Evolutionary insights from a genetically divergent hantavirus harbored by the European common mole (Talpa europaea). PLoS One 4:e6149. doi: 10.1371/journal.pone.0006149
Kang, H. J., Kadjo, B., Dubey, S., Jacquet, F., and Yanagihara, R. (2011b). Molecular evolution of Azagny virus, a newfound hantavirus harbored by the West African pygmy shrew (Crocidura obscurior) in Cote d'Ivoire. Virol. J. 8:373. doi: 10.1186/1743-422X-8-373
Kang, H. J., Kosoy, M. Y., Shrestha, S. K., Shrestha, M. P., Pavlin, J. A., Gibbons, R. V., et al. (2011c). Genetic diversity of Thottapalayam virus, a hantavirus harbored by the Asian house shrew (Suncus murinus) in Nepal. Am. J. Trop. Med. Hyg. 85, 540–545. doi: 10.4269/ajtmh.2011.11-0034
Kang, H. J., Stanley, W. T., Esselstyn, J. A., Gu, S. H., and Yanagihara, R. (2014). Expanded host diversity and geographic distribution of hantaviruses in sub-Saharan Africa. J. Virol. 88, 7663–7667. doi: 10.1128/JVI.00285-14
Kirkland, J. G. L. (1998). Guidelines for the capture, handling, and care of mammals as approved by the American Society of Mammalogists. J. Mammal. 79, 1416–1431. doi: 10.2307/1383033
Klempa, B., Fichet-Calvet, E., Lecompte, E., Auste, B., Aniskin, V., Meisel, H., et al. (2006). Hantavirus in African wood mouse, Guinea. Emerg. Infect. Dis. 12, 838–840. doi: 10.3201/eid1205.051487
Klempa, B., Fichet-Calvet, E., Lecompte, E., Auste, B., Aniskin, V., Meisel, H., et al. (2007). Novel hantavirus sequences in shrew, Guinea. Emerg. Infect. Dis. 13, 520–522. doi: 10.3201/eid1303.061198
Meegaskumbura, S., Meegaskumbura, M., and Schneider, C. J. (2010). Systematic relationships and taxonomy of Suncus montanus and S. murinus from Sri Lanka. Mol. Phylogenet. Evol. 55, 473–487. doi: 10.1016/j.ympev.2010.01.031
Ohdachi, S. D., Kinoshita, G., Oda, S.-I., Motokawa, M., Jogahara, T., Arai, S., et al. (2016). Intraspecific phylogeny of the house shrews, Suncus murinus-S. montanus species complex, based on the mitochondrial cytochrome b gene. Mammal Study 41, 229–238. doi: 10.3106/041.041.0408
Radosa, L., Schlegel, M., Gebauer, P., Ansorge, H., Heroldová, M., Jánová, E., et al. (2013). Detection of shrew-borne hantavirus in Eurasian pygmy shrew (Sorex minutus) in Central Europe. Infect. Genet. Evol. 19, 403–410. doi: 10.1016/j.meegid.2013.04.008
Ramsden, C., Holmes, E. C., and Charleston, M. A. (2009). Hantavirus evolution in relation to its rodent and insectivore hosts: no evidence for codivergence. Mol. Biol. Evol. 26, 143–153. doi: 10.1093/molbev/msn234
Ronquist, F., and Huelsenbeck, J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Sikes, R. S., and Animal Care and Use Committee of the American Society of Mammalogists (2016). Guidelines of the American Society of Mammalogists for the use of wild mammals in research and education. J. Mammal. 97, 663–688. doi: 10.1093/jmammal/gyw078
Song, J.-W., Baek, L. J., Schmaljohn, C. S., and Yanagihara, R. (2007a). Thottapalayam virus, a prototype shrewborne hantavirus. Emerg. Infect. Dis. 13, 980–985. doi: 10.3201/eid1307.070031
Song, J.-W., Gu, S. H., Bennett, S. N., Arai, S., Puorger, M., Hilbe, M., et al. (2007b). Seewis virus, a genetically distinct hantavirus in the Eurasian common shrew (Sorex araneus). Virol J. 4:114. doi: 10.1186/1743-422X-4-114
Song, J.-W., Kang, H. J., Gu, S. H., Moon, S. S., Bennett, S. N., Song, K. J., et al. (2009). Characterization of Imjin virus, a newly isolated hantavirus from the Ussuri white-toothed shrew (Crocidura lasiura). J. Virol. 83, 6184–6191. doi: 10.1128/JVI.00371-09
Song, J.-W., Kang, H. J., Song, K. J., Truong, T. T., Bennett, S. N., Arai, S., et al. (2007c). Newfound hantavirus in Chinese mole shrew, Vietnam. Emerg. Infect. Dis. 13, 1784–1787. doi: 10.3201/eid1311.070492
Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680. doi: 10.1093/nar/22.22.4673
Yanagihara, R., Gu, S. H., Arai, S., Kang, H. J., and Song, J. W. (2014). Hantaviruses: rediscovery and new beginnings. Virus Res. 187, 6–14. doi: 10.1016/j.virusres.2013.12.038
Keywords: Thottapalayam thottimvirus, Suncus murinus, genetic diversity, phylogeography, shrew-borne hantavirus
Citation: Kikuchi F, Aoki K, Ohdachi SD, Tsuchiya K, Motokawa M, Jogahara T, Sơn NT, Bawm S, Lin KS, Thwe TL, Gamage CD, Ranorosoa MC, Omar H, Maryanto I, Suzuki H, Tanaka-Taya K, Morikawa S, Mizutani T, Suzuki M, Yanagihara R and Arai S (2020) Genetic Diversity and Phylogeography of Thottapalayam thottimvirus (Hantaviridae) in Asian House Shrew (Suncus murinus) in Eurasia. Front. Cell. Infect. Microbiol. 10:438. doi: 10.3389/fcimb.2020.00438
Received: 22 April 2020; Accepted: 16 July 2020;
Published: 27 August 2020.
Edited by:
Connie S. Schmaljohn, Integrated Research Facility (NIAID), United StatesReviewed by:
Jiabo Ding, China Institute of Veterinary Drug Control, ChinaDavid Safronetz, Public Health Agency of Canada (PHAC), Canada
Copyright © 2020 Kikuchi, Aoki, Ohdachi, Tsuchiya, Motokawa, Jogahara, Sơn, Bawm, Lin, Thwe, Gamage, Ranorosoa, Omar, Maryanto, Suzuki, Tanaka-Taya, Morikawa, Mizutani, Suzuki, Yanagihara and Arai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Satoru Arai, YXJhaXNAbmloLmdvLmpw
†Deceased