 Chaitra Shankar1†
Chaitra Shankar1† Jobin John Jacob1†
Jobin John Jacob1† Karthick Vasudevan1
Karthick Vasudevan1 Rohit Biswas2Abi Manesh3
Rohit Biswas2Abi Manesh3 Dhiviya Prabaa Muthuirulandi Sethuvel1Santosh Varughese4
Dhiviya Prabaa Muthuirulandi Sethuvel1Santosh Varughese4 Indranil Biswas5
Indranil Biswas5 Balaji Veeraraghavan1*
Balaji Veeraraghavan1*- 1Department of Clinical Microbiology, Christian Medical College and Hospital, Vellore, India
- 2College of Biological Sciences, University of Minnesota, Saint Paul, MN, United States
- 3Department of Infectious Diseases, Christian Medical College and Hospital, Vellore, India
- 4Department of Nephrology, Christian Medical College and Hospital, Vellore, India
- 5Department of Microbiology, Molecular Genetics and Immunology, University of Kansas Medical Centre, Kansas City, KS, United States
Background: In recent years, the emergence of multidrug resistant hypervirulent K. pneumoniae (MDR hvKp) isolates poses severe therapeutic challenge to global public health. The present study used the complete genome sequence of two MDR hvKp isolates belonging to ST23 to characterize the phylogenetic background and plasmid diversity.
Methods: Two hvKp isolates from patients with bacteremia were sequenced using Ion Torrent PGM and Oxford Nanopore MinION platforms and assembled by hybrid genome assembly approach. Comparative genomics approaches were used to investigate the population structure, evolution, virulence, and antimicrobial resistance of MDR hvKp strains.
Results: The study isolates exhibited typical features of hvKp phenotypes associated with ST23. The convergence of multidrug resistance and hypervirulence were attributed by the presence of multiple plasmids including a 216 kb virulence plasmid and MDR plasmids belonging to IncA/C2, IncFIB, IncX3, and ColKP3 groups. The insertion of catA1 gene into virulence plasmid was observed along with genetic factors such as aerobactin, salmochelin, and rmpA2 that confer hvKp’s hypervirulent phenotype. The core genome single nucleotide polymorphism (SNP) phylogenetic analyses of the isolates showed the evolution of ST23 hvKp was predominantly driven by ICEKp acquisitions.
Conclusion: To the best of our knowledge, this is the first report of MDR hvKp isolates of ST23 with insertion of catA1 gene into the virulence plasmid which presents the possibility of hotspot integration sites on the plasmids to aid acquisition of AMR genes. ST23 is no longer confined to susceptible strains of hvKp. Our findings emphasize the need for more studies on recombinant events, plasmid transmission dynamics and evolutionary process involving hvKp.
Introduction
Klebsiella pneumoniae is a notorious nosocomial pathogen responsible for a wide range of healthcare associated infections and is commonly multidrug resistant (MDR). Thus, limited therapeutic options are available to control the infections caused by this pathogen (Martin and Bachman, 2018). However, hypermucoviscous K. pneumoniae, which mostly arises from the community associated infections, retains susceptibility to antimicrobials (Liu and Guo, 2019). The hypermucoviscous pathotype also exhibits hypervirulence (hv) and manifests invasive infections by causing pyogenic liver abscesses and subsequent bacteraemia, pneumonia, meningitis, or brain abscesses (Paczosa and Mecsas, 2016). Hypervirulent K. pneumoniae (hvKp) strains were initially reported from Taiwan, South Korea, and other South East Asian countries however these strains became increasingly prevalent worldwide (Siu et al., 2012).
In the past few years, several hvKp strains evolved into MDR-hvKp due to the acquisition of mobile genetic elements and MDR plasmids (Lee et al., 2017). The recent reports of carbapenem resistant hvKp (CR-hvKp) carrying blaKPC (Dong et al., 2018),blaNDM (Roulston et al., 2018; Yuan et al., 2019), and blaOXA-232 (Shu et al., 2019) is a matter of major public health concern. The convergence of virulence with antimicrobial resistance (AMR) in hvKp isolates represent a real threat to the treatment and management of K. pneumoniae infections (Zhang et al., 2020). Thus, understanding the genetic background and transmission of MDR-hvKp strains from different geographical locations is an urgent priority (Wyres et al., 2019).
HvKp isolates are frequently associated with K1 and K2 capsular serotypes that facilitate the pathogen to escape from phagocytosis and intracellular killing (Shon et al., 2013; Luo et al., 2014). A combination of iron acquisition systems such as, enterobactin (ent), yersiniabactin (ybt), and kfu in the chromosome together with other key virulence factors such as, aerobactin, salmochelin, rmpA, and rmpA2 encoded on a single large virulence plasmid (pLVPK) are associated with severe infections and a high mortality rate (Fu et al., 2019; Marr and Russo, 2019). Interestingly the ybt locus is mobilized by the chromosomally encoded integrative conjugative element ICEKp (Guo et al., 2017; Lam et al., 2018b).
To date, there remains only a few studies to understand the genetics and transmission of MDR hvKp in India. We recently reported ST23 hvKp isolates that were susceptible to all the antimicrobials tested (Shankar et al., 2018). Our ESBL and carbapenem resistant hvKp isolates did not belong to ST23 (Shankar et al., 2016a; Shankar et al., 2016b). The pan-susceptible ST23 hvKp carries a limited number of AMR plasmids in addition to a virulence plasmid. In contrast, the present study, describes ST23 MDR hvKp that carry up to seven AMR plasmids. To the best of our knowledge, this is the first study from India characterizing ST23 multidrug hypervirulent isolates carrying catA1, coding for chloramphenicol resistance, on the virulence plasmid and is studied using a hybrid genome assembly combining Ion Torrent and ONT MinION technologies.
Materials and Methods
Bacterial Isolates
The two K. pneumoniae isolates included in the study were obtained from the blood culture of two patients from different wards and were collected within a period of 4 months at Christian Medical College, Vellore, India. The isolate BA4656 was isolated from a patient who had been involved in a road traffic accident and suffered from acute kidney failure and sepsis. The patient had received antibiotic treatment elsewhere before being brought to the hospital. The second isolate BA34918 was isolated from a patient who was diagnosed with advanced cholangiocarcinoma type 3A and acute cholecystitis. Bacterial isolates were identified and confirmed by VITEK-MS (Database v2.0, bioMerieux, France). Screening for the hypermucoviscous phenotype was carried out using the string test as described previously (Shon et al., 2013).
Antimicrobial Susceptibility Testing
Antimicrobial susceptibility testing was performed for first and second line antibiotics using the Kirby-Bauer disc diffusion method (Bayer et al., 1966). The isolates were tested against ceftazidime (30 μg), cefepime (30 μg), piperacillin/tazobactam (100/10 μg), meropenem (10 μg), gentamicin (10 μg), amikacin (30 μg), ciprofloxacin (5 μg), and minocycline (30 μg). Multidrug resistance is defined as resistance to one or more antimicrobial agents in ≥ 3 classes (Magiorakos et al., 2012). Escherichia coli ATCC 25922, Enterococcus faecium ATCC 29212, and Pseudomonas aeruginosa ATCC 27853 were used as control strains for antimicrobial susceptibility testing. The minimum inhibitory concentration (MIC) of various antimicrobials was determined by VITEK2 using N281 card following the manufacturer’s protocol. Interpretation of the antimicrobial susceptibility results were done according to breakpoints defined by Clinical and Laboratory Standards Institute (CLSI) guidelines for K. pneumoniae (CLSI 2018). For tigecycline the MIC was determined using the broth microdilution method as per breakpoints defined by FDA.
Whole Genome Sequencing
Total genomic DNA from the isolates was extracted from an overnight culture (14–16 h) grown at 37°C on blood agar using the fully automated QIAsymphony instrument (Qiagen, Germany) according to the manufacturer’s instructions. The extracted DNA was quantified using NanoDrop One spectrophotometry (Thermo Fisher Scientific, MA, USA) and Qubit 3.0 Fluorometry (Life Technologies, CA, USA) and stored at −20°C until further use.
The genomic DNA samples were subjected to whole genome sequencing using the Ion Torrent PGM platform with Ion 316™ chip v2 for 400bp chemistry sequencing (Life Technologies, Carlsbad, CA). For this, DNA library was prepared using 1 g of the genomic DNA using Ion Xpress Plus Fragment Library Kit (Life Technologies) following the protocol recommended for 400 bp fragment library preparation. For long read sequencing, Oxford Nanopore MinION sequencing device was used with FLO-MIN106 R9 MinION flow cells. Long read DNA library was prepared using the SQK-LSK108 ligation sequencing kit (v.R9) along with ONT EXP-NBD103 Native Barcode Expansion kit following the manufacturer’s protocol (Oxford Nanopore Technologies, Oxford, UK). The library was loaded onto the flow cells, run for nearly 48 h using the standard MinKNOW software. The Fast5 files generated from MinION sequencing were subjected to base calling with Albacore software (v.2.0.1).
Hybrid Genome Assembly and Evaluation
Hybrid de novo assembly, quality check, and associated assembly statistics were performed using Ion Torrent and ONT MinION reads as described previously (Vasudevan et al., 2020). For accurate assembly the MinION long reads were error-corrected with the standalone Canu (v.1.7) using the “-correct -nanopore-raw” module (Koren et al., 2017). This was followed by hybrid de novo assembly using both Ion torrent and MinION reads using the Unicycler hybrid assembly pipeline (v 0.4.6) for prokaryotic genomes with the default settings (Wick et al., 2017). The complete circular genome was initially assembled and was polished with multiple rounds of Pilon (v.1.22) to reduce the base level errors as described previously (Walker et al., 2014). Genome assembly by combining the Ion Torrent short reads and the MinION long reads improved the contiguity and completeness of the genomes with fewer error rates. The quality measurements of the complete genome sequences after hybrid assembly were compared with the short reads assembled de novo using SPAdes v.3.12 algorithm (Bankevich et al., 2012).
To validate the quality of assembly after each polishing step the completeness, correctness and contiguity parameters were assessed using CheckM v1.0.5 (Parks et al., 2015) and Quast v4.5 (Gurevich et al., 2013). CheckM estimated the completeness and contiguity while Quast was used to detect mis-assemblies, mismatches, and indels by aligning the assemblies with the reference genome (AP006725). The genome sequences of the chromosomes and plasmids have been deposited in GenBank under the accession numbers CP035905-CP035912 and CP036190-CP036198 for BA4656 and BA34918 isolates, respectively.
Comparative Genome Analysis
Genomes were annotated using Prokaryotic Genome Annotation Pipeline (PGAP; v.4.1) from NCBI (Tatusova et al., 2016). Genome sequences of BA4656 (NZ_CP035905) and BA34918 (NZ_CP036190) were aligned with the genome sequences of two reference isolates, SGH10 (NZ_CP02580) and NTUH-K2044 (NC_012731) using the Geneious Prime software. The genomes were rotated and reverse complemented to set the start location for all the genomes as dnaA gene. The genomes were aligned using Geneious Prime software with progressive Mauve algorithm. We used default parameters to automatically calculate seed weight and minimum locally collinear blocks (LCBs) score. Gap alignment was performed using MUSCLE 3.6 program.
The resistance profile of the assembled genomes was identified using ResFinder (v.3.1) on web based server available at https://cge.cbs.dtu.dk/services/ResFinder. Similarly, the presence of plasmids in the genomes were identified and characterized using PlasmidFinder (v.1.3) available at https://cge.cbs.dtu.dk/services/PlasmidFinder. Further, sequence typing of the assembled genomes was determined by the seven gene multi-locus sequence typing (MLST) scheme available at Kleborate (Lam M. et al., 2018). The K and O antigen loci of BA4656 and BA34918 were also identified using Kleborate. The genomes were mapped and analyzed against the reference strains and were visualized using CGview server v.1.0 (Grant and Stothard, 2008).
Identification of virulence factors such as yersiniabactin, aerobactin, and other siderophore production systems were carried out using Kleborate. Mobile genetic elements (MGE) in the genome sequences were identified by IS finder (https://www-is.biotoul.fr/) for insertion sequences and INTEGRALL for integrons (http://integrall.bio.ua.pt/). Two transposons were identified in the isolates, for which the accession numbers were assigned by Transposon (Tn) Registry as Tn6691 and Tn6692 (https://transposon.lstmed.ac.uk/tn-registry). The ICEKp genomic island was identified using IslandViewer (Bertelli et al., 2017) and further confirmed through multiple alignment using Progressive Mauve (Darling et al., 2010), and NCBI BLAST (Johnson et al., 2008). The linear comparison of the ICEKp genomic island and the transposons were visualized using Easyfig (Sullivan et al., 2011). The presence TA systems in the strains were determine by using web-based TAfinder tool (https://db-mml.sjtu.edu.cn/TAfinder/index.php).
Phylogenetic Analysis
The genomic sequences from two K. pneumoniae isolates BA4656 and BA34918 were compared to the global collection of CG23 clones. The nucleotide sequences were obtained from GenBank (https://www.ncbi.nlm.nih.gov/genbank/). The sequencing reads for previously reported 192 hypervirulent K. pneumoniae global isolates (Lam et al., 2018a) were downloaded from ENA (https://www.ebi.ac.uk/ena) and subsequently assembled using SPAdes v.3.12 (Bankevich et al., 2012). Furthermore, the BacWGSTdb (http://bacdb.org/BacWGSTdb/) was used to investigate the relationship between the strains (Ruan and Feng, 2016).
The genomes were mapped to the reference genome ED23 (CP016814.1) using the BWA MEM (https://github.com/lh3/bwa) algorithm and Snippy v.4.5.1 (Seemann, 2015) was used to call the genomic variants. Additionally, the variants were then filtered using FreeBayes (https://github.com/ekg/freebayes). The core SNP genome alignment of all the genomes was generated with snippy-core. The recombination regions within the core genome alignment was further filtered and removed using the Gubbins (v. 2.4.1) algorithm (Croucher et al., 2015). The maximum likelihood (ML) phylogeny was constructed using FastTree v.2.1.8 (Price et al., 2009) using GTR model with 100 bootstrap replicates. The phylogenetic tree was rooted with the reference genome and labeled using the Interactive Tree of Life software (iTOL v.3) software (Letunic and Bork, 2016).
Results
Phenotypic Antibiotic Resistance Profiles of Multidrug Resistant Hypervirulent K. pneumoniae Isolates BA4656 and BA34918
We observed both BA4656 and BA34918 strains were phenotypically resistant to all the tested antimicrobials from disc diffusion assay. The MIC values for antimicrobials as determined by VITEK2 (bioMerieux) were as follows: piperacillin/tazobactam (≥128μg/ml), cefoperazone/sulbactam (≥ 64μg/ml), cefepime (≥ 64μg/ml), ceftazidime (≥ 64μg/ml), aztreonam (≥ 64μg/ml), imipenem (≥ 8μg/ml), meropenem (≥ 16µg/ml), doripenem (≥ 8µg/ml), amikacin (≥ 64μg/ml), gentamicin (≥ 16μg/ml), ciprofloxacin (≥4μg/ml), levofloxacin (≥8μg/ml), minocycline (≥16μg/ml), and trimethoprim/sulfamethoxazole (≥ 320µg/ml). The tigecycline MIC for BA4656 and BA34918 were 1 μg/ml and 0.06 μg/ml respectively. Based on the MIC values, we confirmed that both BA4656 and BA34918 isolates were MDR hypervirulent strains.
General Features of the BA4656 and BA34918 Genomes
The hybrid genomes of both the isolates consisted of a chromosome assembled into a single contig along with plasmids assembled into separate circular contigs. The genome of BA4656 comprised of a 5,432,984 bp chromosome with an average 57.51% GC content and seven circular plasmids (Supplementary Figure 1). The genome of BA34918 consisted of a 5,439,838 bp chromosome with an average 57.5% GC content and eight circular plasmids (Supplementary Figure 1). The N50 values for both the hybrid assemblies (5,432,984 and 5,439,838) were found to be higher than the short-read assemblies (59,132 and 52,852) suggesting highly contiguous assemblies. In addition, the higher BUSCO values validate the completeness of the hybrid genome assembly (100%) in comparison with the short-read assembly (97.5%). As mentioned before, both genomes belong to ST23 as determined by Kleborate.
From whole genome sequence analysis, it was found that both BA4656 and BA34918 isolates belong to ST23 with serotype K1 and O1v2. These are typical characteristics of hypervirulent K. pneumoniae. Comparison of the genomes of the clinical isolates characterized in this study with the two reference genomes SGH10 and NTUH2044 suggested the presence of several IS elements and other differences in the chromosomes (Figure 1). The two genomes in the present study were MDR and hence the presence of higher numbers of mobile genetic elements when compared to the reference genomes, which are susceptible to antimicrobials.
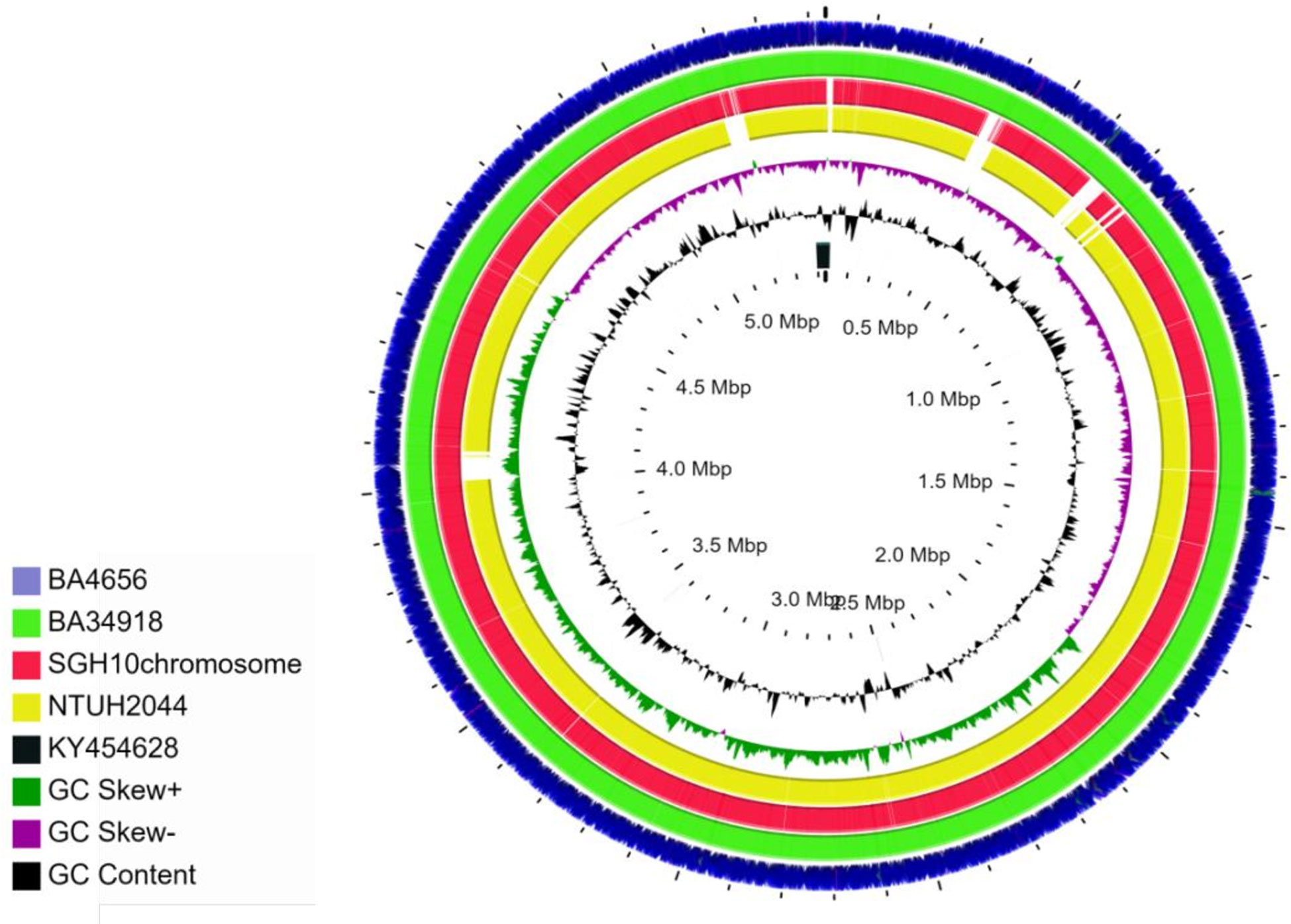
Figure 1 Circular representation of chromosomes of K. pneumoniae isolates BA4656 (NZ_CP035905) and BA34918 (NZ_CP036190) genomes displayed using CG view server with the reference genomes SGH10 (NZ_CP02580) and NTUH-K2044 (NC_012731). KY454628 is the reference used for ICEKp3. SGH10 and NTUH-2044 have ICEKp10 and ICEKp1, respectively. Genomes of BA4656 and BA34918 have numerous insertion sequences (IS) when compared to the reference genomes. These IS elements are represented as gaps in the two reference genomes.
Antimicrobial Resistance Among Multidrug Resistant Hypervirulent Klebsiella pneumoniae
The resistance phenotype of the two isolates was supported by the identification of extended spectrum β-lactamases blaSHV-190; quinolone efflux pumps oqxA and oqxB; and fosfomycin resistant gene fosA in the chromosome. The hybrid genome assembly also elucidated the presence of multiple MDR plasmids harboring resistance genes encoding for β-lactamases (blaTEM1B, blaCTX-M-15, blaCMY-4), aminoglycoside modifying enzymes [aac(6’)-Ib3, rmtF, aph(6)-Id, aph(3)-Ib], fluoroquinolone resistance (qnrB1), sulfonamide resistance (sul1, sul2), trimethoprim resistance (dfrA14), phenicols resistance (catA1, catB), and rifampicin resistance (ARR-2) (Table 1). Carbapenemase gene blaOXA-232 as well as macrolide resistance genes (mphE, msrE) were detected only in BA34918. The isolates carried 7–8 plasmids each and the distribution of resistance genes on the plasmids varied as mentioned in the Table 1.
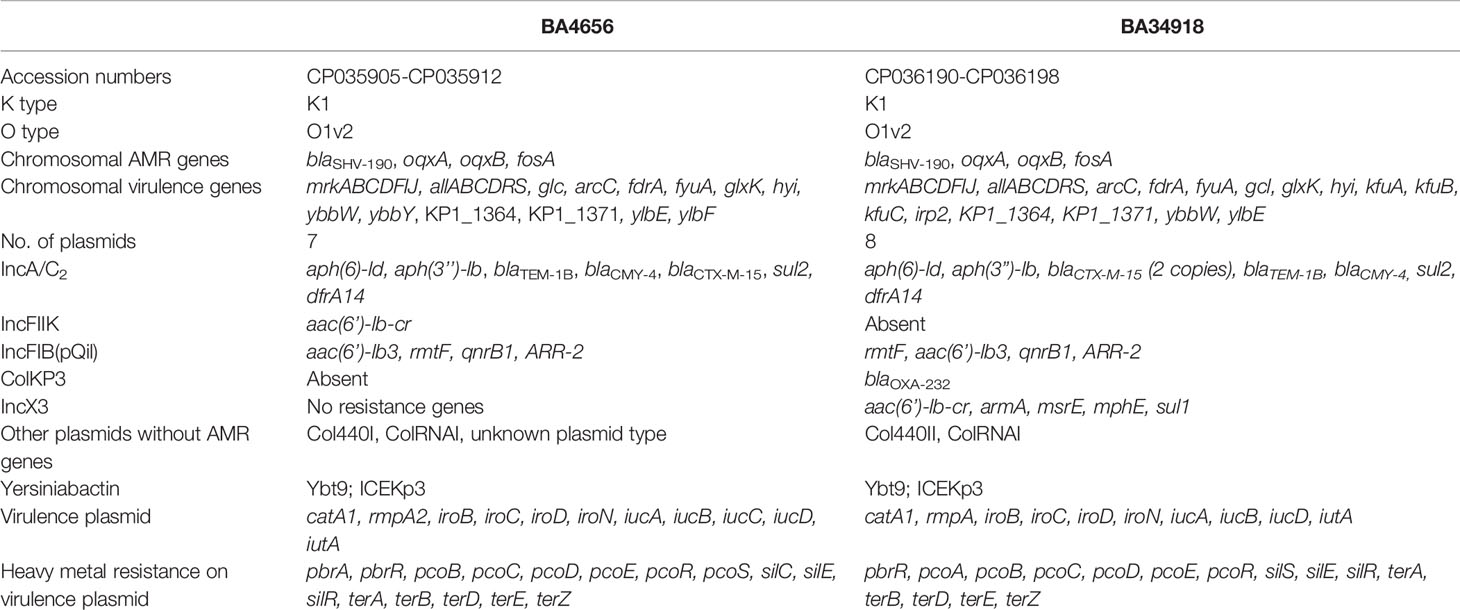
Table 1 Characteristics of the two multidrug resistant (MDR) hypervirulent Klebsiella pneumoniae isolates belonging to ST23.
Genomic Elements That Support Acquiring Foreign DNA
Genome alignment of the two isolates along with two reference genomes used in this study shows several areas in the genome that differ among the strains (Figures 1 and 2). The genome alignment has identified the presence of genes in the clinical isolates that are putatively part of restriction modification systems, which are absent in the two reference strains (Figures 2B, C). A total of 15 different TA systems in K. pneumoniae genomes were identified in all the four strains (including two references). Among the 15 TA systems, only 11 were common in all the strains that include all the major families. The study isolates BA34918 and BA4656 encode 11 and 14 TA systems respectively (Table 2).

Figure 2 Alignment of chromosomes of Klebsiella pneumoniae isolates BA4656 (NZ_CP035905) and BA34918 (NZ_CP036190) with reference genomes SGH10 (NZ_CP02580) and NTUH-K2044 (NC_012731). (A) 12 locally co-linear blocks (LCBs) of all four strains are shown. (B) Presence of restriction endonucleases (LCB 6) and exonucleases (LCB 2) in the clinical isolates and not in the reference strains. (C) The biosynthetic operon of the fim gene (LCB 11) shown here is flanked by the IS3 like insertion sequence in the clinical isolates and not in the reference genomes.
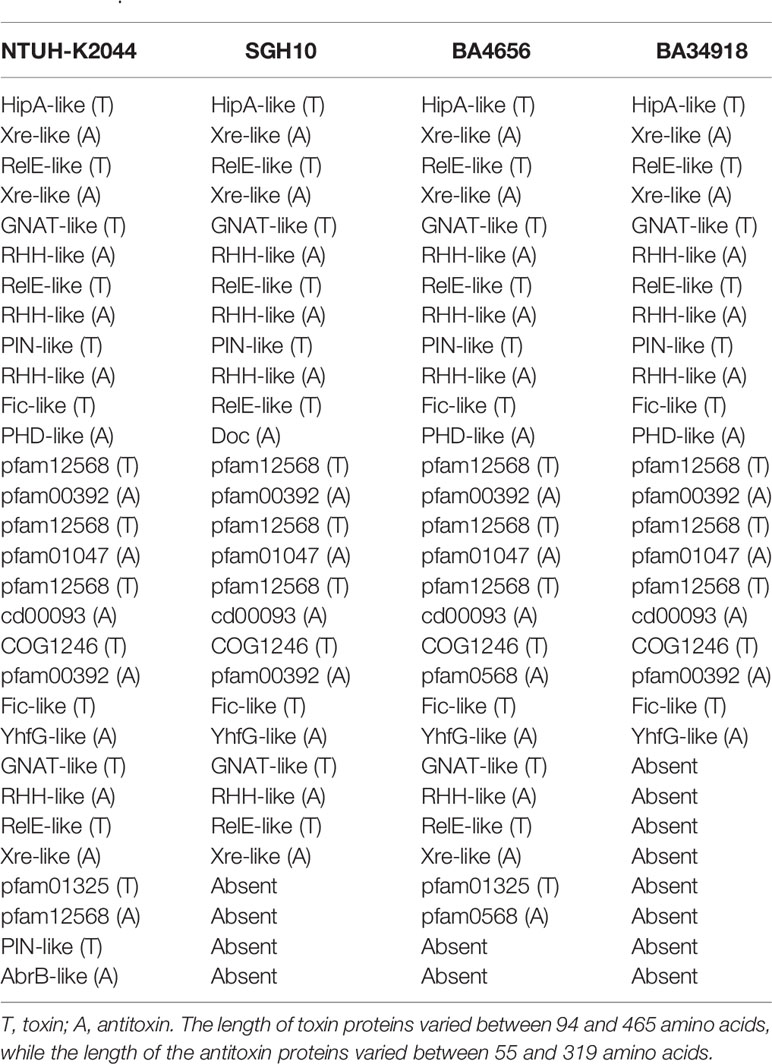
Table 2 Chromosomal toxin and antitoxin systems encoded by Klebsiella pneumoniae.
Mobile Genetic Elements
Plasmids and Transposons
Both the isolates harbored six common putative distinct plasmid replicons; namely IncA/C2, IncFIB (pQil), IncFIB, IncX3, ColRNAI, and Col440II along with a common virulence plasmid (Supplementary Figure 1). Surprisingly, the isolate BA34918 carried an additional ColKP3 non-conjugative plasmid that harbored the carbapenemase gene blaOXA-232 (CP036197). The presence of such a large number of plasmids among the ST23 sequence types has not been previously documented. Among the groups, the IncA/C2 plasmid is particularly important as it encodes multiple determinants (Table 1). These resistance genes were inserted within the transposon Tn6691, in the IncA/C2 plasmid (Figure 3). Sequence comparison with other IncA/C2 plasmid showed that the plasmid displayed 99% conservation with IncA/C2 plasmids from K. pneumoniae AR_0148 (CP021952), Citrobacter freundii (KX147633), E. coli (AP018143), and Salmonella Senftenberg (KP742988) (Figure 3). Considering the broad bacterial host range of IncA/C2 plasmid, acquisition of these plasmids contributed to the evolution of MDR hvKp from pan susceptible hvKp.
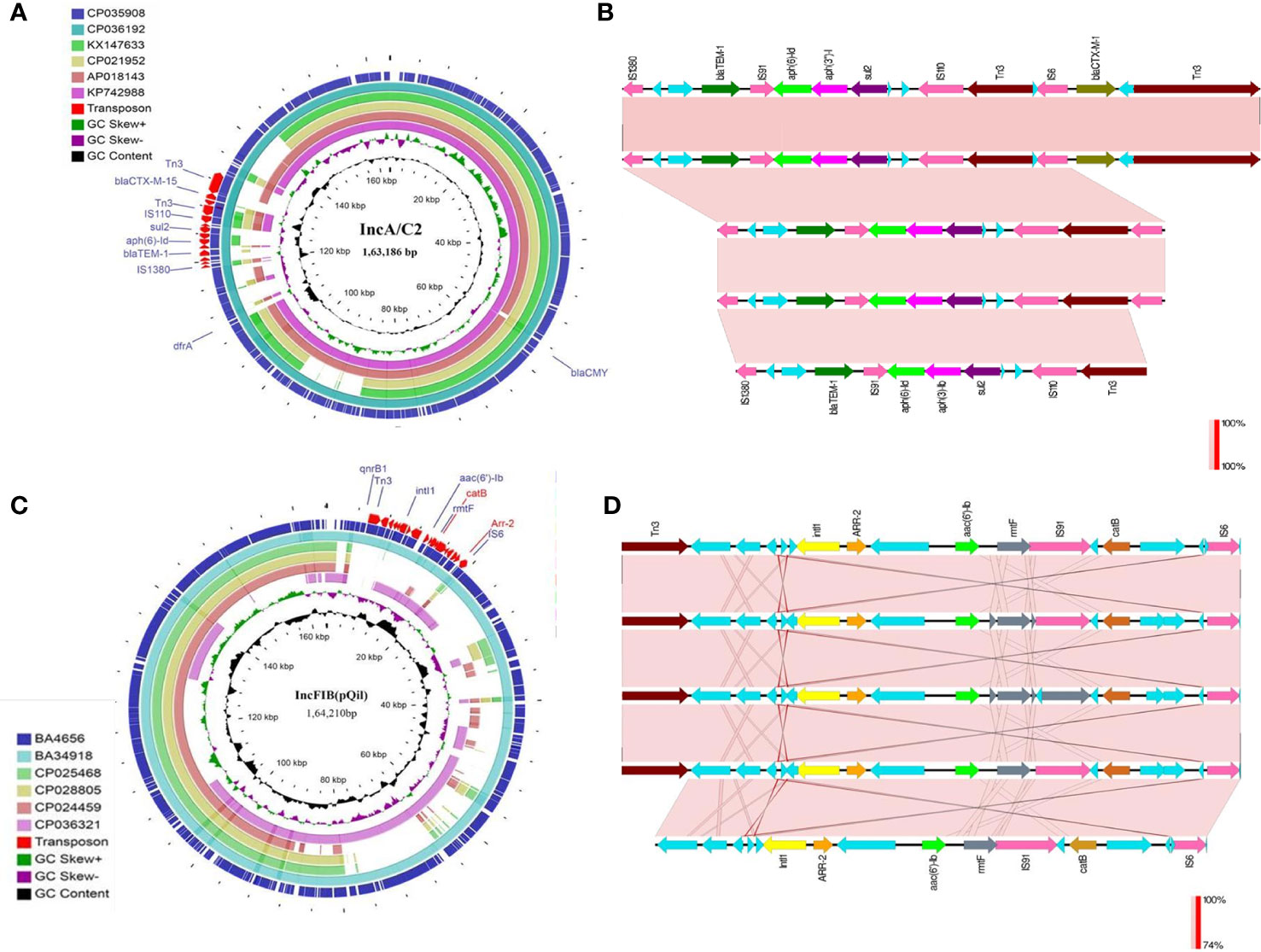
Figure 3 (A) The structures of the IncA/C2 plasmids identified in the present study (CP035908, 163,186bp and CP036192, 165,052bp). Sequence comparison with other IncA/C2 plasmids showed that the plasmid displayed 99% conservation with IncA/C2 plasmids from Klebsiella pneumoniae AR_0148 (CP021952), Citrobacter freundii (KX147633), Escherichia coli (AP018143), and Salmonella Senftenberg (KP742988). Tn6691 on IncA/C2 carried blaTEM-1B, blaCTX-M-15, aph(6)-ld and aph(3’’)-lb. (B) Comparison of the Tn6691 elements on IncA/C2 among B4656 (CP035908) and BA34918 (CP036192) with E. coli strain 109 (CP020523), K. pneumoniae strain INF249 (CP024489), and Salmonella enterica subsp. enterica serovar Typhimurium strain UGA14 (CP021463) in the above-mentioned order. (C) The structures of the IncFIB (pQil) plasmids identified in the present study (CP035907, 164,210bp and CP036194, 108,957bp). Tn6692 present on IncFIB(pQil) carried ARR-2, catB, rmtF, and aac(6’)-lb3. (D) Comparison of the transposon Tn6692 on IncFIB in the following order: K. pneumoniae strains AR0138 (CP021758), BA4656 (CP035907), BA34918 (CP036193), KPN1482 (CP020842), and SKGH01 (CP015500).
Similarly both the isolates harbored resistance genes qnrB1, catB, aac(6’)-Ib3, rmtF, and ARR-2 within a novel transposon Tn6692, on the IncFIB(pQil) plasmid (Figure 3). Although both BA4656 and BA34918 strains were found to harbor IncX3 plasmid, BA4656 lacked the resistance cassette aac(6’)-Ib-cr, armA, mphE, msrE, and sul-1 that is inserted into the IncX3 backbone of BA34918 (CP036195). The other two small ColRNAI (9.729 bp) and Col440II (4.166 bp) plasmid replicons does not carry any resistance genes or mobile genetic elements.
Virulence Plasmid
The large virulence plasmid (2,16,620 bp) of the two hvKp isolates were highly similar to the pLVPK-like virulence plasmid with < 50% sequence coverage. The BLAST based identity search for the homologous sequences showed similarity with well characterized virulence plasmids from other CC23 isolates such as K. pneumoniae strains NTUH-K2044 (AP006726; 99.63%), ED23 (CP016815; 99.63%), and SGH10 (CP025081; 99.62%). The comparison of the virulence plasmid of both the isolates with the reference virulence plasmid is shown in Figure 4. Surprisingly, we found that the virulence plasmid of both BA4656 and BA34918 isolates encode a chloramphenicol resistance gene (catA1), which is probably inserted through IS110 family transposase. The inserted region comprised of 2,744bp flanked by 10 bases of terminal repeat (TACCGGGAAG) and this was inserted between a hypothetical protein and IS5075 belonging to the IS110 family (Figure 4). The insertion of a resistance gene into the virulence plasmid indicates a potential hotspot for the further acquisition of other resistance genes into the virulence plasmid.
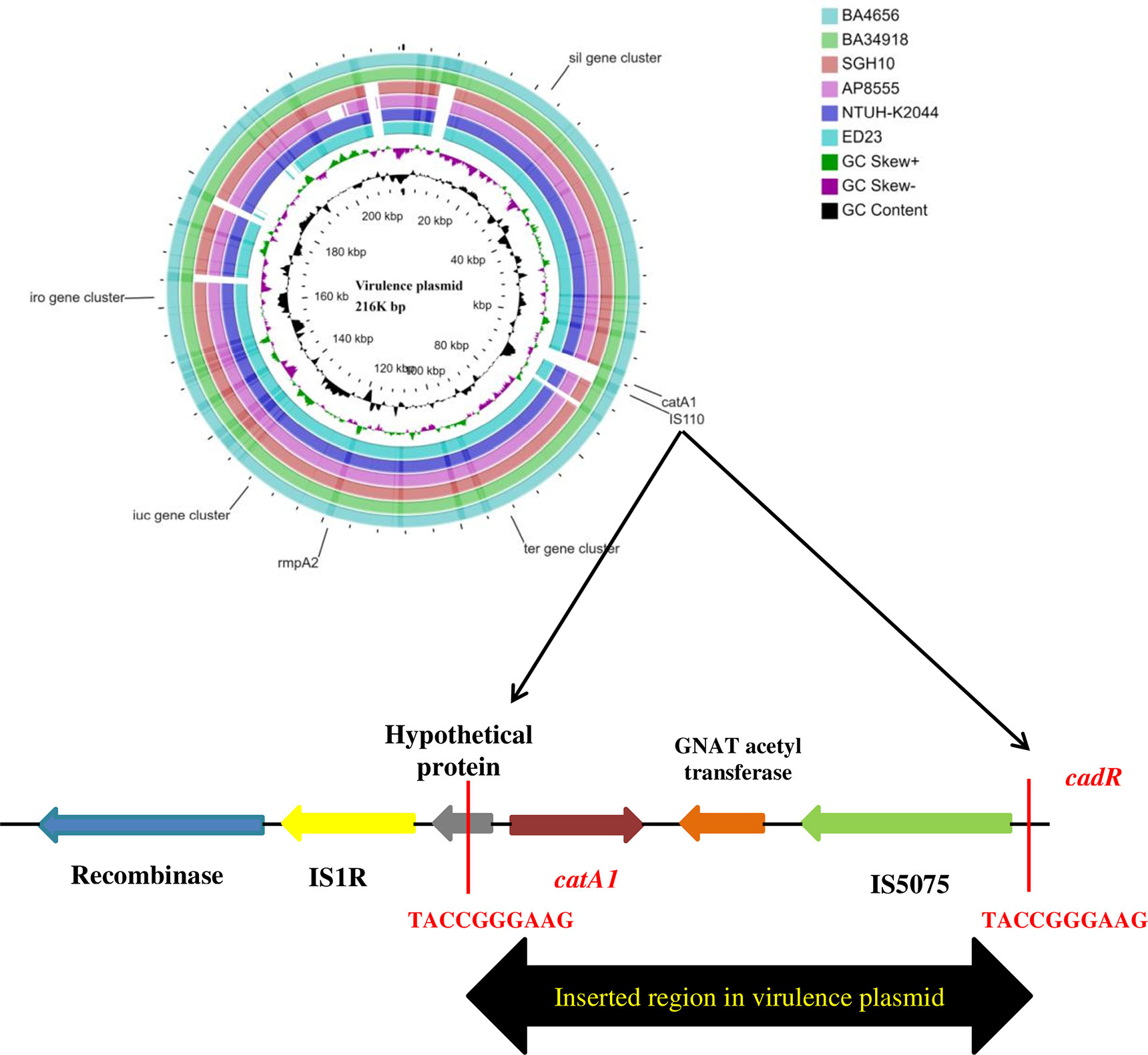
Figure 4 Comparison of virulence plasmid of hypervirulent (hv) Klebsiella pneumoniae isolates BA4656 and BA34918 with virulence plasmids from K. pneumoniae strains SGH10, NTUH-K2044, AP8555, and ED23. The plasmid carried virulence genes including rmpA/rmpA2, iro, and iuc gene clusters along with heavy metal resistance encoding ter, sil, and pco gene clusters. catA1, coding for chloramphenicol resistance with IS5075 (IS110 family) was inserted in plasmids of BA4656 and BA34918. Inserted region of 2,744 bp in the virulence plasmid carrying catA1 is shown below. Inserted region is flanked by a repeat region of 10bp (TACCGGGAAG) which could possibly be a hotspot for integration.
The phenotypic features encoded by the virulence plasmids (CP035906 and CP036191) included the salmochelin (iroBCDN) and aerobactin (iucABCDiutA) clusters which help in utilizing iron from the host (Shon et al., 2013). The presence of rmpA and rmpA2 along with K1 capsule type confirms a hypermucoid phenotype that is a characteristic feature of hypervirulent isolates (Shon et al., 2013). The virulence plasmids also code for resistance to heavy metals such as copper, lead, silver, and tellurite (Table 1). This contributes toward resistance to some disinfectants and helps hvKp thrive in hospital settings.
In addition to the virulence genes encoded by the plasmid, in K. pneumoniae the chromosome also codes for virulence factors. Chromosomal virulence factors include type3 fimbria coded by mrk operon (mrkABCDFIJ), allantoin regulation (allABCDRS), and kfu operon encoding iron uptake system (kfuABC) which contribute to the high virulence and invasiveness of the isolates. In addition, ybbW, ybbY glc, fdrA, glxK, arc, and hyi are other genes coding for iron uptake (Table 1).
Genomic Island
In hvKp, the yersiniabactin locus (ybtAPSTUX) was located within diverse Integrative congregative elements (ICEKp) present on the chromosome. Both the isolates carried the yersiniabactin locus ybt9 located in ICEKp3. The diversity of the detected ICEKp3 was analyzed by mapping against ICEKp regions from isolates ED23 (CP016814) and the K. pneumoniae strain 1670377 (KY454628) available in GenBank (Figure 5). Other than the yersiniabactin locus, the characteristic virulence factors of ICEKp including the siderophore genes irp1 and irp2, the ferric yersiniabactin uptake gene fyuA, the virB-type 4 secretion system (T4SS), and mobBC (mobilization) proteins were also identified. Another interesting observation is the fim operon, a genomic region encoding a major virulence factor, is flanked by insertion sequence elements only in the clinical isolates (Figure 2C). This indicates that the fim genes might have been recently acquired by the clinical strains as a selective advantage in infecting host tissues.
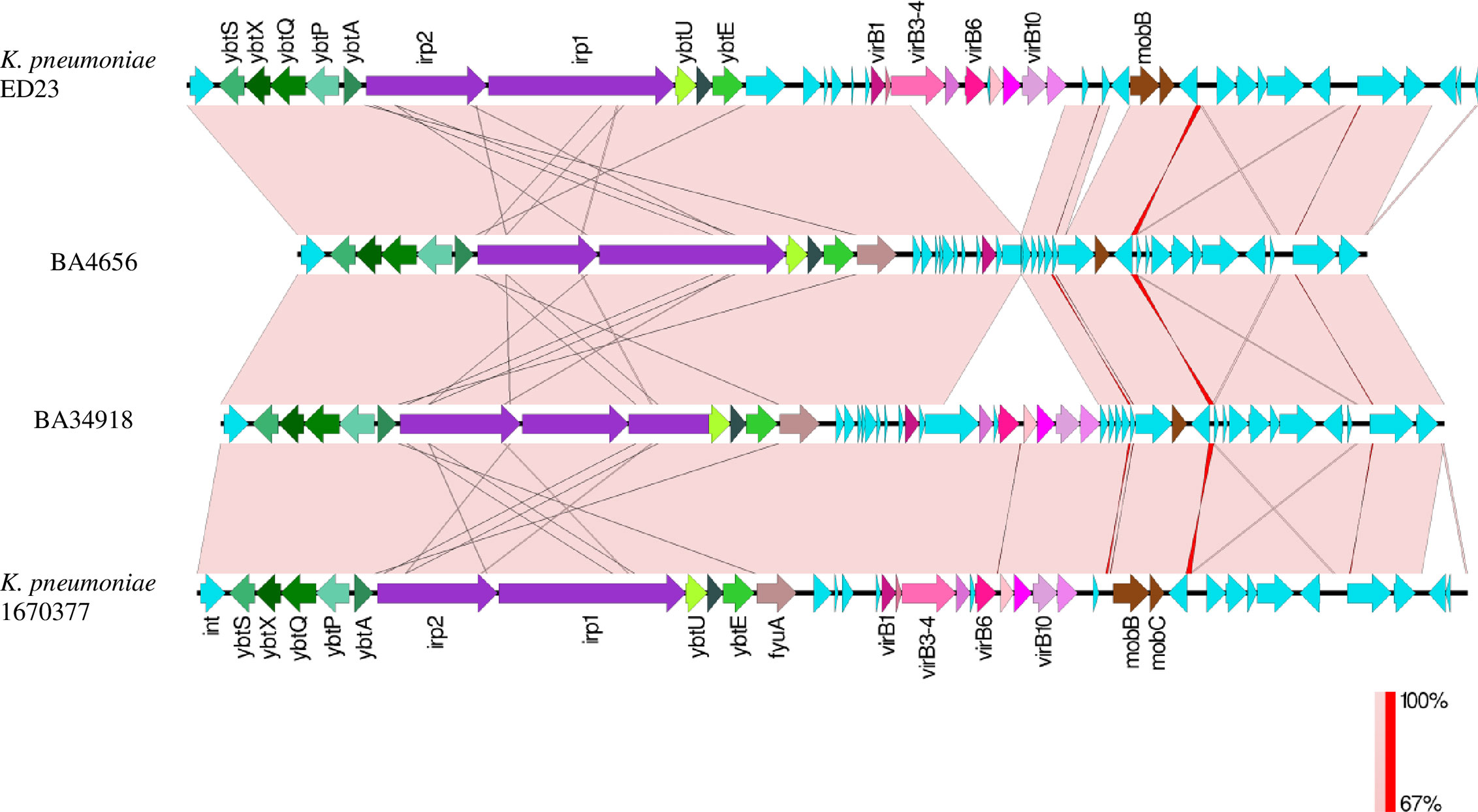
Figure 5 Comparison between ICEKp3 genomic islands (GI) of hypervirulent (hv) Klebsiella pneumoniae isolates BA4656 and BA34918 with ICEKp3 GI from other K. pneumoniae strains ED23 and 1670377. This island contains the yersiniabactin gene cluster and the type IV secretion system (T4SS) virB gene cluster. ICEKp3 of BA4656 was shorter than BA34918 since it lacked some of the T4SS genes.
Phylogenetic Analysis
The two hvKp isolates (BA4656 and BA34918), were compared with the genome sequences of 187 global CG23 isolates and 5 pan-susceptible Indian ST23 isolates. Phylogenetic analysis revealed a number of sub-lineages with the globally distributed CG23-I (CG23 sub-lineage I) comprising of 158 isolates. The study isolates BA4656 and BA34918 formed a separate sub-lineage (referred as sub-lineage II hereafter) with isolates from India, Southeast Asia, China, and Europe. An earlier pan-susceptible isolate from the same study center (BA253) as well as another isolate of Indian origin (VINI01) was also clustered with the sub-lineage II. The observed median pairwise SNP distance after removing the recombinant events was 191 SNPs (range 1–719 SNPs). Further the median pairwise SNP distance between CG23-I and sub-lineage II was identified as 307 SNPs (range 122–719 SNPs). The closest isolate (VINI01) differed from BA4656 and BA34918 by 311 and 328 SNPs respectively whereas the differences between the two study isolates were 453 SNPs.
The sub-lineage II either lacked yersiniabactin or carried ybt9 on ICEKp3 (Figure 6). Among the global collection, 153 (79.6%) isolates that belong to CG23-I were characteristically associated with ybt1 carried on ICEKp10 while other sub-lineages either lack ICEKp or carry ybt8/9 on ICEKp3. Further, isolates belonging to sub-lineage II produced aerobactin and salmochelin but lacked colibactin. Notably, all the 192 isolates belonged to K1 capsule type except for two European isolates. In contrast, six diverse types of O antigen were observed with O1v2 being the most common in 165 (86%) isolates. Also, 20 isolates predominantly from Asian countries, lacked rmpA and rmpA2.

Figure 6 Single nucleotide polymorphism (SNP) based phylogenetic tree based on the core genome alignment of 192 CG23 Klebsiella pneumoniae isolates from the global collection. The maximum likelihood phylogram is based on 9,300 SNPs after removing the recombinant regions. The phylogenetic tree was rooted by using the reference genome (ED23). The scale represents the evolutionary distances. The percentage of 100 bootstrap trials (100 replicates). The study isolates BA4656 and BA34918 formed a separate sub-lineage with 10 isolates distributed around the world in the phylogenetic tree (highlighted in orange color).
Discussion
Though our recent studies have indicated the characterization of hvKp from India (Shankar et al., 2016a; Shankar et al., 2018), the clinical, microbiological, and evolutionary features of MDR hvKp from India remain largely unknown. Both the isolates characterized in this study possessed the hypermucoviscous phenotype and the large virulence plasmid which has >90% similarity with the large virulence plasmid pLVPK of K. pneumoniae strain NTUH-K2044. Our results indicate the coexistence of multiple resistance plasmids such as IncA/C2, IncFIB, IncX3, and ColKP3 along with the virulence plasmid within the same hvKp isolate. Interestingly, these MDR plasmids are known to play an important role in the dissemination of antimicrobial resistance in K. pneumoniae worldwide (Martin and Bachman, 2018). These multiple acquisitions of MDR plasmids are surprising and unprecedented for hvKp as the clone is generally less likely to acquire multiple MDR plasmids (Wyres et al., 2019). However, due to antibiotic selection pressure hvKp appears to be acquiring multiple MDR plasmids leading to MDR-hvKp (Chen et al., 2020; Zhang et al., 2020).
The increasing genetic plasticity of hvKp has been reported from multiple geographical locations (Cejas et al., 2014; Zhang et al., 2016; Xie et al., 2018; Zhang et al., 2020). Moreover, several studies from other Asian countries have documented the predominant carbapenem resistance K. pneumoniae (CRKp) clones acquiring virulence plasmid from hvKp (Gu et al., 2018). Therefore, plasmid exchange appears to be happening both ways between hvKp and the prevalent sequence type of CRKp in study setting. We found that in Indian settings the possible exchange of hvKp and CRKp plasmids might have occurred between the hvKp clone ST23 and the CRKp clone ST231. Although the direct proof of exchange is absent, we speculate that ST231 K. pneumoniae is the most prevalent sequence type of CRKp in the same setting (Shankar et al., 2019). Given the high frequency and the number of MDR plasmids acquired, the MDR hvKp BA4656 and BA34918 can be considered as a “real superbug” and it might have serious implications to public health.
CC23 persisters showed higher survival rate at a dose of 10x MIC against a carbapenem class antibiotic meropenem (Lee et al., 2019). This indicates clinical isolates, belonging to CC23 complex, used in this study may be capable of forming persisters under high dose of carbapenem and thus can find opportunity to acquire resistance genes from the pool of dead bacteria. This, in turn, could allow for active growth in the presence of antibiotics from the once dormant persister bacteria. This is alarming because carbapenems are last resort to treat K. pneumoniae infection (Papp-Wallace et al., 2011). Since TA systems are the major players of persister formation, we investigated the presence of TA systems in the two study isolates (Table 2). Among the three additional TA systems present in the clinical isolates, one was uniquely present in the plasmid, while the other two were also present in the chromosome. In general, TA systems are also involved in diverse cellular processes. Plasmid maintenance, selfish alleles, gene regulation, growth control, persister formation, programmed cell arrest and death, anti-phage activity, biofilm formation, and general stress response can all be controlled by TAs (Ramage et al., 2009).
Among the multiple plasmids acquired by our isolates, IncA/C2, IncFIB, and ColKP3 are of particular importance. Notably the acquisition of the IncA/C2 and the IncFIB plasmids is considered to be responsible for the spread of resistance among Enterobacterales (Rozwandowicz et al., 2018). The nucleotide sequence similarity of plasmids in other Enterobacterales such as Citrobacter sp. (KX147633) E. coli (AP018143) and Salmonella sp. (KP742988) substantiates the horizontal dissemination of plasmid among different bacterial species. The Tn6691 transposon located on the IncA/C2 plasmid has been found to be responsible for the dissemination of AMR genes such as blaTEM-1B, blaCTX-M-15, aph(6)-ld, and aph(3’’)-lb. Similarly IncFIB, which was present in both the isolates, contributes to increased resistance to antimicrobials by carrying AMR genes such as aac(6’)-lb3, rmtF, qnrB1, ARR-2. The acquisition of the blaOXA-232-bearing ColKP3 plasmid by a single isolate of hvKp may mark a major evolutionary step toward further establishment of clonal complex 23 (CC23). Till now, the clonal dissemination of blaOXA-232-bearing K. pneumoniae majorly occurred by means of the ST231 carrying ColKP3 plasmid in India (Shankar et al., 2019). The emergence of carbapenemase producing hvKp in India can be hypothesized to be due to acquiring the ColKP3 plasmid from ST231 CRKp, as this sequence type is widely circulating in the same environment (Shankar et al., 2019). Thus, the emergence of blaOXA-232 producing hvKp is particularly concerning due to its combination of hypervirulence and carbapenem resistance.
Plasmids such as IncX3, ColKP3, IncA/C2 are common among members of Enterobacterales and are responsible of dissemination of antimicrobial resistance (AMR). Supplementary Figures 2–4 shows the comparison of these plasmids isolated from K. pneumoniae and E. coli from the study centre. Though similar AMR genes are mobilized by these plasmids in both the organisms, significant differences were observed among ColKP3. Two ColKP3 plasmids from E. coli carried blaOXA-181 and were 50% similar to K. pneumoniae ColKP3. IncX3 was highly similar among both the organisms. In the present study, IncFIB (pQil) and IncA/C2 that were present in K. pneumoniae were not identified among the E. coli in this study collection. Hence, from global database, representative IncA/C2 plasmids were used to construct a phylogenetic tree (Supplementary Figure 4). It was observed that IncA/C2 commonly coded for aminoglycoside and β-lactam resistance genes. blaNDM was the most common gene disseminated by this plasmid.
The characteristic feature of hvKp is the presence of the 216 kb large virulence plasmid with the mucoid regulators rmpA and rmpA2 being encoded in the plasmid. In addition, the virulence plasmid is characterized by the presence of the catA1 gene flanked by the insertion elements IS1 and IS110 suggesting that the region has been inserted into the virulence plasmid (Mahillon and Chandler, 1998). A similar recombination event in the same virulence plasmid was previously reported by Dong and colleagues (2018), where a blaKPC-2 carrying region was inserted into the virulence plasmid. Conversely there have been reports of the acquisition of a hvKp virulence plasmid by CRKp isolates (Gu et al., 2018). Notably, the emergence of a MDR hvKp carrying hybrid/fusion plasmid by the integration of a fragment of the hypervirulence plasmid into a MDR plasmid suggests the constant evolution of hvKp clonal lineages (Lam et al., 2019).
The SNP based phylogeny of the two isolates with respect to the representative CG23 global strains showed that the evolutionary events are not driven by geographical location. The phylogenetic distribution indicated distinct sub-lineages with a major sub-lineage (CG23-I) associated with liver abscess strains (Lam M. et al., 2018). This observation is in line with Lam and colleagues (2018) where this globally distributed sub-lineage is driven by ICEKp10 with ybt1. The sub-lineage II where two study isolates clustered were predominantly associated with sepsis and other invasive infection. However, the evolution of sub-lineage II is not consistent with ICEKp distribution.
In addition, when growth of the two study isolates was compared with ST231 K. pneumoniae from the same center, at 24 h, the isolate BA34918 and the ST231 Kp showed higher growth when compared to BA4656 (Supplementary Figure 5). However, after 25h, the ST231 Kp showed a rapid decline in growth while no decline in the growth of two hvKp was observed. The acquisition of antimicrobial resistance including colistin resistance in hvKp has been associated with increased fitness cost (Choi and Ko, 2015; Lee et al., 2017). Correspondingly, the ST23 hvKp with blaOXA-232 in the present study showed increased fitness when compared to the isolate without blaOXA-232.
Based on the genetic composition of the virulence plasmids, which had multiple insertion elements, we speculate further plasmid recombination events will occur leading to emergence of novel resistance-virulence encoding plasmids. Our findings are relevant in understanding the future risk of the emergence of individual K. pneumoniae strains carrying both the virulence and acquired resistance genes, capable of causing highly virulent infections, which will be extremely difficult to control. Specifically, our data indicate that MDR clones pose the greatest risk because they are more likely to acquire virulence genes than the hypervirulent clones acquiring resistance genes.
Data Availability Statement
The accession numbers of the genomes deposited in GenBank, NCBI, are mentioned in methods and Table 1.
Ethics statement
The study was reviewed and approved by Institutional Review Board, Christian Medical College and Hospital, Vellore with IRB min no. 9616 dated 1st September 2015. Written informed consent for participation was not required for this study since the bacterial isolates were used without patient identifier.
Author contributions
CS: Laboratory methods, data analysis and interpretation, manuscript writing. JJ: Data analysis, interpretation and manuscript writing. KV: Hybrid genome assembly and other bioinformatics methods. RB: Data analysis, manuscript writing. DPMS: Data analysis. AB: Clinical details and expert opinion on study design. SV: Clinical details and expert opinion on study design. IB: Manuscript correction. BV: Study design and supervising, manuscript writing, manuscript correction. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like the acknowledge Ms. Catherine Truman, Clinical Pharmacist, Christian Medical College and Hospital, India, for her valuable inputs in language editing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2020.575289/full#supplementary-material
References
Bankevich A., Nurk S., Antipov D., Gurevich A. A., Dvorkin M., Kulikov A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19 (5), 455–477. doi: 10.1089/cmb.2012.0021
Bertelli C., Laird M. R., Williams K. P., Simon Fraser University Research Computing Group, Lau B. Y., Hoad G., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45 (W1), W30–W35. doi: 10.1093/nar/gkx343
Bayer A. W., Kirby W. M. M., Sherris J. C., Turck M. (1966). Antibiotic susceptibility testing by a standardized single disc method. Am. J. Clin. Pathol. 45 (4), 493–496.
Cejas D., Canigia L. F., Cruz G. R., Elena A. X., Maldonado I., Gutkind G. O., et al. (2014). First isolate of KPC-2-producing Klebsiella pneumoniae sequence type 23 from the Americas. J. Clin. Microbiol. 52 (9), 3483–3485. doi: 10.1128/JCM.00726-14
Chen Y., Marimuthu K., Teo J., Venkatachalam I., Cherng B. P., De Wang L., et al. (2020). Acquisition of Plasmid with Carbapenem-Resistance Gene blaKPC2 in Hypervirulent Klebsiella pneumoniae, Singapore. Emerg. Infect. Dis. 26 (3), 549. doi: 10.3201/eid2603.191230
Choi M. J., Ko K. S. (2015). Loss of hypermucoviscosity and increased fitness cost in colistin-resistant Klebsiella pneumoniae sequence type 23 strains. Antimicrob. Agents Chemother. 59, 6763–6773. doi: 10.1128/AAC.00952-15
Croucher N. J., Page A. J., Connor T. R., Delaney A. J., Keane J. A., Bentley S. D., et al. (2015). Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43 (3), e15–. doi: 10.1093/nar/gku1196
Darling A. E., Mau B., Perna N. T. (2010). Progressive Mauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5 (6), e11147. doi: 10.1371/journal.pone.0011147
Dong N., Lin D., Zhang R., Chan E. W., Chen S. (2018). Carriage of blaKPC-2 by a virulence plasmid in hypervirulent Klebsiella pneumoniae. J. Antimicrob. Chemother. 73 (12), 3317–3321. doi: 10.1093/jac/dky358
Fu Y., Xu M., Liu Y., Li A., Zhou J. (2019). Virulence and genomic features of a blaCTX-M-3 and blaCTX-M-14 coharboring hypermucoviscous Klebsiella pneumoniae of serotype K2 and ST65. Infect. Drug Resist. 12, 145. doi: 10.2147/IDR.S187289
Grant J. R., Stothard P. (2008). The CGView Server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 36 (suppl_2), W181–W184. doi: 10.1093/nar/gkn179
Gu D., Dong N., Zheng Z., Lin D., Huang M., Wang L., et al. (2018). A fatal outbreak of ST11 carbapenem-resistant hypervirulent Klebsiella pneumoniae in a Chinese hospital: a molecular epidemiological study. Lancet Infect. Dis. 18 (1), 37–46. doi: 10.1016/S1473-3099(17)30489-9
Guo Y., Wang S., Zhan L., Jin Y., Duan J., Hao Z., et al. (2017). Microbiological and clinical characteristics of hypermucoviscous Klebsiella pneumoniae isolates associated with invasive infections in China. Front. Cell. Infect. Microbiol. 7, 24. doi: 10.3389/fcimb.2017.00024
Gurevich A., Saveliev V., Vyahhi N., Tesler G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29 (8), 1072–1075. doi: 10.1093/bioinformatics/btt086
Johnson M., Zaretskaya I., Raytselis Y., Merezhuk Y., McGinnis S., Madden T. L. (2008). NCBI BLAST: a better web interface. Nucleic Acids Res. 36 (suppl_2), W5–W9. doi: 10.1093/nar/gkn201
Koren S., Walenz B. P., Berlin K., Miller J. R., Bergman N. H., Phillippy A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27 (5), 722–736. doi: 10.1101/gr.215087.116
Lam M., Wick R. R., Wyres K. L., Holt K. E. (2018). Kleborate: comprehensive genotyping of Klebsiella pneumoniae genome assemblies. Available at: https://github.com/katholt/Kleborate.
Lam M. M., Wyres K. L., Duchêne S., Wick R. R., Judd L. M., Gan Y. H., et al. (2018a). Population genomics of hypervirulent Klebsiella pneumoniae clonal-group 23 reveals early emergence and rapid global dissemination. Nat. Commun. 9 (1), 1–0. doi: 10.1038/s41467-018-05114-7
Lam M. M., Wick R. R., Wyres K. L., Gorrie C. L., Judd L. M., Jenney A. W., et al. (2018b). Genetic diversity, mobilisation and spread of the yersiniabactin-encoding mobile element ICEKp in Klebsiella pneumoniae populations. Microbial. Genomics 4 (9). doi: 10.1099/mgen.0.000196
Lam M. M., Wyres K. L., Wick R. R., Judd L. M., Fostervold A., Holt K. E., et al. (2019). Convergence of virulence and MDR in a single plasmid vector in MDR Klebsiella pneumoniae ST15. J. Antimicrob. Chemother. 74 (5), 1218–1222. doi: 10.1093/jac/dkz028
Lee C. R., Lee J. H., Park K. S., Jeon J. H., Kim Y. B., Cha C. J., et al. (2017). Antimicrobial resistance of hypervirulent Klebsiella pneumoniae: epidemiology, hypervirulence-associated determinants, and resistance mechanisms. Front. Cell. Infect. Microbiol. 7, 483. doi: 10.3389/fcimb.2017.00483
Lee J. S., Choi J. Y., Chung E. S., Peck K. R., Ko K. S. (2019). Variation in the formation of persister cells against meropenem in Klebsiella pneumoniae bacteremia and analysis of its clinical features. Diagn. Microbiol. Infect. Dis. 95 (3), 114853. doi: 10.1016/j.diagmicrobio.2019.06.005
Letunic I., Bork P. (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44 (W1), W242–W245. doi: 10.1093/nar/gkw290
Liu C., Guo J. (2019). Hypervirulent Klebsiella pneumoniae (hypermucoviscous and aerobactin positive) infection over 6 years in the elderly in China: antimicrobial resistance patterns, molecular epidemiology and risk factor. Ann. Clin. Microbiol. Antimicrob. 18 (1), 4. doi: 10.1186/s12941-018-0302-9
Luo Y., Wang Y., Ye L., Yang J. (2014). Molecular epidemiology and virulence factors of pyogenic liver abscess causing Klebsiella pneumoniae in China. Clin. Microbiol. Infect. 20 (11), O818–O824. doi: 10.1111/1469-0691.12664
Magiorakos A. P., Srinivasan A., Carey R. B., Carmeli Y., Falagas M. E., Giske C. G., et al. (2012). Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 18 (3), 268–281. doi: 10.1111/j.1469-0691.2011.03570.x
Mahillon J., Chandler M. (1998). Insertion sequences. Microbiol. Mol. Biol. Rev. 62 (3), 725–774. doi: 10.1128/MMBR.62.3.725-774.1998
Marr C. M., Russo T. A. (2019). Hypervirulent Klebsiella pneumoniae: a new public health threat. Expert Rev. Anti-infect. Ther. 17, 2, 71–2, 73. doi: 10.1080/14787210.2019.1555470
Martin R. M., Bachman M. A. (2018). Colonization, infection, and the accessory genome of Klebsiella pneumoniae. Front. Cell. Infect. Microbiol. 8, 4. doi: 10.3389/fcimb.2018.00004
Paczosa M. K., Mecsas J. (2016). Klebsiella pneumoniae: going on the offense with a strong defense. Microbiol. Mol. Biol. Rev. 80 (3), 629–661. doi: 10.1128/MMBR.00078-15
Papp-Wallace K. M., Endimiani A., Taracila M. A., Bonomo R. A. (2011). Carbapenems: Past, Present, and Future. Antimicrob. Agents Chemother. 55 (11), 4943–4960. doi: 10.1128/AAC.00296-11
Parks D. H., Imelfort M., Skennerton C. T., Hugenholtz P., Tyson G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25 (7), 1043–1055. doi: 10.1101/gr.186072.114
Price M. N., Dehal P. S., Arkin A. P. (2009). FastTree: Computing Large Minimum-Evolution Trees with Profiles instead of a Distance Matrix. Mol. Biol. Evol. 26, 1641–1650. doi: 10.1093/molbev/msp077
Ramage H. R., Connolly L. E., Cox J. S. (2009). Comprehensive functional analysis of Mycobacterium tuberculosis toxin-antitoxin systems: implications for pathogenesis, stress responses, and evolution. PLoS Genet. 5 (12), e1000767. doi: 10.1371/journal.pgen.1000767
Roulston K. J., Bharucha T., Turton J. F., Hopkins K. L., Mack D. J. (2018). A case of NDM-carbapenemase-producing hypervirulent Klebsiella pneumoniae sequence type 23 from the UK. JMM Case Rep. 5 (9), e005130. doi: 10.1099/jmmcr.0.005130
Rozwandowicz M., Brouwer M. S., Fischer J., Wagenaar J. A., Gonzalez-Zorn B., Guerra B., et al. (2018). . Plasmids carrying antimicrobial resistance genes in Enterobacteriaceae. J. Antimicrob. Chemother. 73 (5), 1121–1137. doi: 10.1093/jac/dkx488
Ruan Z., Feng Y. (2016). BacWGSTdb, a database for genotyping and source tracking bacterial pathogens. Nucleic Acids Res. 44 (D1), D682–D687. doi: 10.1093/nar/gkv1004
Seemann T. (2015). Snippy: rapid haploid variant calling and core SNP phylogeny. Available at: https://github.com/tseemann/snippy.
Shankar C., Nabarro L. E., Ragupathi N. K., Sethuvel D. P., Daniel J. L., Veeraraghavan B. (2016a). Draft genome sequences of three hypervirulent carbapenem-resistant Klebsiella pneumoniae isolates from bacteremia. Genome Announc. 4 (6), e01081–e01016. doi: 10.1128/genomeA.01081-16
Shankar C., Santhanam S., Kumar M., Gupta V., Ragupathi N. K., Veeraraghavan B. (2016b). Draft genome sequence of an extended-Spectrum-β-Lactamase-positive Hypervirulent Klebsiella pneumoniae strain with novel sequence type 2318 isolated from a neonate. Genome Announc. 4 (6), e01273–e01216. doi: 10.1128/genomeA.01273-16
Shankar C., Veeraraghavan B., Nabarro L. E., Ravi R., Ragupathi N. K., Rupali P. (2018). Whole genome analysis of hypervirulent Klebsiella pneumoniae isolates from community and hospital acquired bloodstream infection. BMC Microbiol. 18 (1), 6. doi: 10.1186/s12866-017-1148-6
Shankar C., Mathur P., Venkatesan M., Pragasam A. K., Anandan S., Khurana S., et al. (2019). Rapidly disseminating blaOXA-232 carrying Klebsiella pneumoniae belonging to ST231 in India: multiple and varied mobile genetic elements. BMC Microbiol. 19 (1), 137. doi: 10.1186/s12866-019-1513-8
Shon A. S., Bajwa R. P., Russo T. A. (2013). Hypervirulent (hypermucoviscous) Klebsiella pneumoniae: a new and dangerous breed. Virulence 4 (2), 107–118. doi: 10.4161/viru.22718
Shu L., Dong N., Lu J., Zheng Z., Hu J., Zeng W., et al. (2019). Emergence of OXA-232 carbapenemase-producing Klebsiella pneumoniae that carries a pLVPK-like virulence plasmid among elderly patients in China. Antimicrob. Agents Chemother. 63 (3), e02246–e02218. doi: 10.1128/AAC.02246-18
Siu L. K., Yeh K. M., Lin J. C., Fung C. P., Chang F. Y. (2012). Klebsiella pneumoniae liver abscess: a new invasive syndrome. Lancet Infect. Dis. 12 (11), 881–887. doi: 10.1016/S1473-3099(12)70205-0
Sullivan M. J., Petty N. K., Beatson S. A. (2011). Easyfig: a genome comparison visualizer. Bioinformatics 27 (7), 1009–1010. doi: 10.1093/bioinformatics/btr039
Tatusova T., DiCuccio M., Badretdin A., Chetvernin V., Nawrocki E. P., Zaslavsky L., et al. (2016). J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44 (14), 6614–6624. doi: 10.1093/nar/gkw569
Vasudevan K., Ragupathi N. K., Jacob J. J., Veeraraghavan B. (2020). Highly accurate-single chromosomal complete genomes using IonTorrent and MinION sequencing of clinical pathogens. Genomics 112 (1), 545–551. doi: 10.1016/j.ygeno.2019.04.006
Walker B. J., Abeel T., Shea T., Priest M., Abouelliel A., Sakthikumar S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9 (11), p.e112963. doi: 10.1371/journal.pone.0112963
Wick R. R., Judd L. M., Gorrie C. L., Holt K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PloS Comput. Biol. 13 (6), e1005595. doi: 10.1371/journal.pcbi.1005595
Wyres K. L., Wick R. R., Judd L. M., Froumine R., Tokolyi A., Gorrie C. L., et al. (2019). Distinct evolutionary dynamics of horizontal gene transfer in drug resistant and virulent clones of Klebsiella pneumoniae. PloS Genet. 15 (4), e1008114. doi: 10.1371/journal.pgen.1008114
Xie Y., Tian L., Li G., Qu H., Sun J., Liang W., et al. (2018). Emergence of the third-generation cephalosporin-resistant hypervirulent Klebsiella pneumoniae due to the acquisition of a self-transferable blaDHA-1-carrying plasmid by an ST23 strain. Virulence 9 (1), 838–844. doi: 10.1080/21505594.2018.1456229
Yuan Y., Li Y., Wang G., Li C., Chang Y. F., Chen W., et al. (2019). blaNDM-5 carried by a hypervirulent Klebsiella pneumoniae with sequence type 29. Antimicrob. Resist. Infect. Control. 8 (1), 1–9. doi: 10.1186/s13756-019-0596-1
Zhang R., Lin D., Chan E. W., Gu D., Chen G. X., Chen S. (2016). Emergence of carbapenem-resistant serotype K1 hypervirulent Klebsiella pneumoniae strains in China. Antimicrob. Agents Chemother. 60 (1), 709–711. doi: 10.1128/AAC.02173-15
Keywords: Klebsiella pneumoniae, hypervirulence plasmid, ST23, hybrid genome, multidrug resistance, OXA-232
Citation: Shankar C, Jacob JJ, Vasudevan K, Biswas R, Manesh A, Sethuvel DPM, Varughese S, Biswas I and Veeraraghavan B (2020) Emergence of Multidrug Resistant Hypervirulent ST23 Klebsiella pneumoniae: Multidrug Resistant Plasmid Acquisition Drives Evolution. Front. Cell. Infect. Microbiol. 10:575289. doi: 10.3389/fcimb.2020.575289
Received: 23 June 2020; Accepted: 23 October 2020;
Published: 20 November 2020.
Edited by:
Dongsheng Zhou, Beijing Institute of Microbiology and Epidemiology, ChinaReviewed by:
Yunn Hwen Gan, National University of Singapore, SingaporeZhi Ruan, Zhejiang University, China
Copyright © 2020 Shankar, Jacob, Vasudevan, Biswas, Manesh, Sethuvel, Varughese, Biswas and Veeraraghavan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Balaji Veeraraghavan, dmJhbGFqaUBjbWN2ZWxsb3JlLmFjLmlu
†These authors have contributed equally to this work