 Xiaochun Xie1
Xiaochun Xie1 Jianxiong Zeng1,2,3*
Jianxiong Zeng1,2,3*- 1Key Laboratory of Animal Models and Human Disease Mechanisms of the Chinese Academy of Sciences, Kunming Institute of Zoology-The Chinese University of Hong Kong (KIZ-CUHK) Joint Laboratory of Bioresources and Molecular Research in Common Diseases, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, China
- 2Kunming National High-level Biosafety Research Center for Non-Human Primates, Center for Biosafety Mega-Science, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, China
- 3National Resource Center for Non-Human Primates, National Research Facility for Phenotypic and Genetic Analysis of Model Animals (Primate Facility), Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, China
Zika virus (ZIKV), which preferentially targets neural stem and progenitor cells (NSCs) especially in developing brain, is causally associated with fetal microcephaly, intrauterine retardation, and other congenital malformations in humans. However, there are, so far, no effective drugs and vaccines against ZIKV epidemics, warranting an enhanced understanding of ZIKV biology. Immune response is essential for neuronal cells to combat viral invasion. In turn, neurotropic ZIKV has developed a complex strategy of neuroimmune evasion to facilitate viral pathogenesis, especially developmental impairment in embryonic brain. Here, we review not only overall knowledge of ZIKV-related immune responses, but also current advances in our understanding of immune evasion in ZIKV infection. We also review several specific mechanisms underlying ZIKV protein-mediated immune evasion for viral pathogenesis.
Introduction
Zika virus (ZIKV) is a single-stranded positive-sense RNA virus that belongs to the genus Flavivirus of the family Flaviviridae. The RNA genome is translated directly into one large polyprotein, which produces 10 viral proteins (3 structural and 7 nonstructural proteins) via proteolytic cleavage by host and viral enzymes. The Flavivirus genus includes more than 50 arthropod-borne viruses with public health importance including Dengue virus (DENV), West Nile virus (WNV), Yellow fever virus (YFV), and Japanese encephalitis (JEV). ZIKV was originally isolated from a sentinel monkey in the Zika forest or Uganda in 1947 (Dick et al., 1952). ZIKV infection in human was just sporadically reported in a few African and Asian countries with mild symptoms (Weaver et al., 2016) and associated with Guillain–Barre disease (Cao-Lormeau et al., 2016). However, ZIKV unexpectedly emerged since 2015 and suddenly became a global public health threat due to its explosive outbreaks in the Americas. In a short time, ZIKV was considered as the etiological agent for fetal microcephaly and congenital Zika syndrome (CZS) (Rasmussen et al., 2016; Hoen et al., 2018). In the following years, ZIKV spread to 86 countries or territories worldwide, and it was estimated that ~3.6 billion people are living in areas at risk for transmission (Baud et al., 2017).
ZIKV is able to lead to devastating fetal microcephaly in infants born from infected pregnant mothers (Musso and Schot, 2016). Thousands of infants born from ZIKV-infected mothers in the Americas had impaired neurodevelopment with thinner cortical layers (Orioli et al., 2017). ZIKV is characterized by the intrinsic tropism for neural stem and progenitor cells (NSCs) in cell cultures, brain organoids, and fetal brain slices (Cugola et al., 2016; Dang et al., 2016; Garcez et al., 2016; Liang et al., 2016; Qian et al., 2016; Tang et al., 2016). ZIKV infection results in the impairment of NSC proliferation and differentiation, induces cell death, and ultimately causes cerebral developmental deficits (Shao et al., 2016; Nielsen-Saines et al., 2019; Zeng et al., 2020).
Immune response is critical for antagonizing neurotropism virus infection. The host utilizes multiple pattern recognition receptors (PRRs) to patrol diverse pathogen-associated molecular patterns (PAMPs), consequently activating antiviral responses including the production of interferons (IFNs) (Meylan et al., 2006). For example, retinoic acid-inducible gene-I (RIG-I) senses ZIKV-RNA and triggers MAVS-TBK1-IRF3 signaling and type I IFN production (Chazal et al., 2018). In order to evade the IFN-mediated surveillance, ZIKV evolved specific non-structural (NS) proteins for viral evasion, including NS1 (Xia et al., 2018; Zheng et al., 2018), NS3 (Riedl et al., 2019), and NS5 (Grant et al., 2016; Kumar et al., 2016). Notably, mammalian multipotent stem cells including NSCs produce little IFNs and respond poorly to IFN treatment compared to somatic cells (Hong and Carmichael, 2013; Wu et al., 2019). Instead, these cells rely on other antiviral machineries such as RNA interference (RNAi) (Ding and Voinnet, 2007). Therefore, this short review seeks to highlight recent advances in our understanding of ZIKV protein-mediated neuroimmune evasion, which contributes to virus pathogenicity.
ZIKV Capsid Targets Dicer to Inhibit miRNA Biogenesis in Developing Brain
In many eukaryotes, RNAi is a critical cellular mechanism by which short RNA oligos specifically pair with targeted mRNAs to regulate or inhibit their translation or gene expression, thereby maintaining homeostatic function in cells. Dicer as a critical microRNA (miRNA) biogenesis enzyme, is indispensable for the RNAi pathway, where it functions to cleave double-strand RNAs (dsRNAs) or stem-loop structure of pre-miRNAs into short dsRNAs, which are then loaded on the RNA-induced silencing complex (RISC) and processed into ~22-nucleotide (nt) mature miRNAs (Jinek and Doudna, 2009). Consequently, the mature miRNA pairs with the targeted complementary sequence in the 3’ untranslated region (UTR) of an mRNA molecule and causes a cleavage by Argonaute 2 (Ago2), the catalytic factor in the RISC, consequently resulting in the post-transcriptional gene silencing. Disruption of cellular miRNA homeostasis has been shown to be closely related to numerous diseases such as neurodevelopment deficits while miRNAs participate in almost every cellular process. Although type I IFN response provides a main protection against microbial invasion in most somatic cells, anti-viral RNAi especially miRNAs remains dominantly active in mammalian multipotent stem cells (Li et al., 2013; Maillard et al., 2013) since these stem cells have intrinsically less ability to respond to IFN treatment than other somatic cell types (Hong and Carmichael, 2013; Wu et al., 2018). ZIKV infection has been reported to disrupt the host miRNA profile in mammalian cells, and importantly neuronal Dicer deficient mice exhibit neurodevelopment disorders including microcephaly (Davis et al., 2008), strongly suggesting the potential correlation between Dicer dysfunction and ZIKV pathogenesis.
In a screening of the required host factors of ZIKV protein partners in NSCs to uncover the roles of the viral proteins in the context of ZIKV infection, ribonuclease III-like enzyme Dicer is the top hit within the resulted ZIKV–host interactome (Zeng et al., 2020). While Dicer is proven to be necessary for ZIKV replication by utilizing Dicer knock-outed mouse NSCs, it is intriguing that viral capsid-mediated interaction with Dicer exists only in ZIKV, but not in other flaviviruses including DENV2, WNV, JEV, YFV, JEV, HCV, OHFV, and TBEV. Such specific interaction also expectedly leads to the phenotype that only ZIKV capsid is able to suppress Dicer enzymatic activity by utilizing common Dicer substrates like shRNA, pre-miRNA, or double-strand RNA. In alignment comparison analysis of viral capsid between ZIKV, DENV2, WNV, and JEV, one capsid mutant (H41R) was successfully selected and completely abolished capsid–Dicer interaction, consequently losing the ability to inhibit Dicer enzymatic activity. Dicer is responsible for miRNA biogenesis in mammalian cells (Zeng et al., 2020). As a result, wild-type ZIKV (ZIKV-WT) infection led to significant reduction of total miRNA reads, and, however, such phenotype was absent in rescued ZIKV-H41R mutant virus-infected NSCs. Importantly, ZIKV-WT infection resulted in the downregulation of a panel of miRNAs including let-7a, miR-9, miR-17, and miR-19a, which has been shown to be important for neurogenesis and neurodevelopment. Similar miRNA biogenesis phenotype was also observed in an animal model of in utero ZIKV injection (Zeng et al., 2020). As expected, utilization of this mouse model also helps confirm that ZIKV capsid protein enabled to induce neurogenic deficits and corticogenesis impairment by directly targeting Dicer in developing brains. It has been known that the dysfunction to regulate miRNA homeostasis is closely linked to a large group of human diseases (Mendell and Olson, 2012). One example is that Dicer knockdown is associated with microcephaly-like phenotypes in an animal model (Davis et al., 2008). It has been proven that fetal brain development required the critical individual miRNAs like let-7a, miR-9, miR-17, and miR-19a (Rajman and Schratt, 2017). Accordingly, ZIKV infection-induced impairment of cellular miRNA homeostasis has been reported in human NSCs (Dang et al., 2019), astrocyte-like SVG-A cells (Kozak et al., 2017), and mosquitoes (Saldana et al., 2017). On the one hand, ZIKV-capsid specific association with Dicer represents a unique mechanism on why only ZIKV in flavivirus is related to microcephaly in clinic. On the other hand, miRNAs not only have antiviral function especially in neural stem cells but also are essential for normal neurogenesis, and thus ZIKV capsid achieves these two tasks by specifically targeting Dicer (Zeng et al., 2020).
ZIKV capsid hijacks host Dicer and suppresses its enzymatic activity to facilitate immune evasion and consequently disrupt cortical development (Figure 1A). As a viral structural protein, capsid is packaged into mature virion particle during virus assembly and is released right after virus entry. As a result, the capsid protein may not only wrap ZIKV RNA genome and protect it from outside the cells, but also initiate immune evasion by targeting Dicer-related RNAi pathway preceding the translation of viral genes. Therefore, ZIKV capsid targeting Dicer represents an elegant mechanism for ZIKV immune evasion in developing brain.
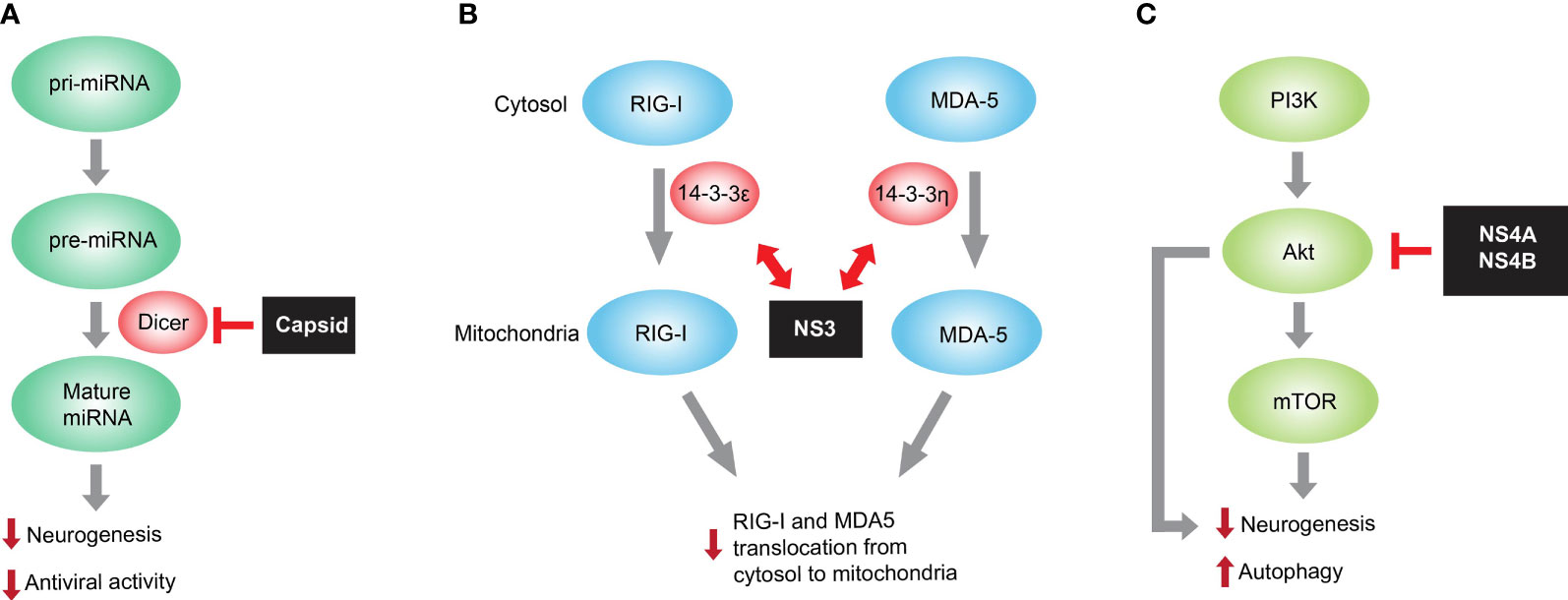
Figure 1 Representative ZIKV protein mediated immune evasion for facilitating viral pathogenesis. (A) ZIKV capsid interacts with Dicer, the host endoribonuclease responsible for producing mature miRNA from pre-miRNA, and dampens global miRNA production in neural stem cells. Because miRNA is necessary for both neurogenesis and antiviral function, the capsid-mediated miRNA inhibition causes neurogenesis impairment and reduced miRNA-mediated antiviral activity in developing brain. (B) ZIKV NS3 encodes a highly conserved 14-3-3-binding motif, which enables it to interact with 14-3-3ϵ/η required for translocation of RNA sensor RIG-I and MDA5, respectively, from cytosol to mitochondria. Such interaction prevents the translocation and thus RIG-I- and MDA-mediated antiviral interferon responses. (C) mTOR is a host factor necessary for neurogenesis and autophagy inhibition. ZIKV NS4A and NS4B inhibit Akt-mTOR signaling with unknown mechanisms and consequently impair neurogenesis and promote autophagy, thereby contributing to ZIKV pathogenesis in developing brain.
ZIKV Infection Induces Anti-Viral Small Interfering RNAs in Pluripotent Cells
RNAi is an evolutionarily conserved post-transcriptional gene silencing mechanism. Besides host microRNAs (miRNAs) regulating mRNA homeostasis and fine-tuning gene expression, virus-derived small interfering RNAs (vsiRNAs) can also be produced in infected host cells (Guo et al., 2019). In fact, vsiRNA production is an ancient innate immune response, defending the plants and animals from virus infection (Maillard et al., 2013; tenOever, 2016). In mammals, the physiological importance of vsiRNAs in somatic cells perhaps is still under investigation. It has been proposed that canonical vsiRNAs are generated by Dicer from double-stranded viral replicative intermediates, as evidenced by complementary pairs of vsiRNAs mapped successively to the viral RNA genome (Maillard et al., 2013). Once produced, vsiRNAs are loaded onto AGO family proteins of the RISCs to initiate the cleavage of host target genes and cognate viral RNAs (Guo et al., 2019). ZIKV infection has been demonstrated to induce abundant vsiRNAs in NSCs and hNPCs (Xu et al., 2019; Zeng et al., 2021). Zeng et al. reported not only that vsiRNA is a limiting factor for ZIKV infection in NSCS, but also that the total 29 vsiRNAs across the ZIKV genome through AGO-associated RNA-seq. More importantly, the production of these vsiRNAs is demonstrated to be dependent on Dicer. The vsiRNA-p18, the most abundant one within the identified 29 vsiRNAs, was detectable in ZIKV-infected NSCs while its physiologically antiviral effect remains unknown (Zeng et al., 2021). Similar evidence was also shown by Xu et al. that ZIKV infection induced abundant vsiRNA productions in human neural progenitors by direct deep RNA sequencing on ZIKV-infected cells. Similarly, the vsiRNAs’ physiological importance was uncovered by the fact that the ablation of key RNAi machinery components greatly facilitates ZIKV replication, and that increased anti-ZIKV activity in hNPCs was observed in the treatment of the enoxacin, a known RNAi enhancer (Xu et al., 2019). Although the advances described above have been achieved, further studies are needed to investigate how these vsiRNAs are produced and what are the physiological functions of these vsiRNAs.
ZIKV NS3 Antagonizes RIG-I-/MDA5-Mediated Innate Immunity
Innate immune sensors along with other critical transcription factors involved in interferon (IFN) signaling are necessary to restrict ZIKV pathogenesis (Serman and Gack, 2019). Upon viral RNA sensing, RIG-I and MDA5, both of the RIG-I-like receptor (RLR) family, translocate from the cytosol to the mitochondria and chemically activate kinase transcription factors including TBK1, IRF3, and IRF7. These transcription factors enter the nucleus to initiate IFN response, upregulate a large panel of IFN-stimulated genes (ISGs), and finally trigger to form antiviral status (Bowen et al., 2017; Chazal et al., 2018; Hertzog et al., 2018). Thereof, 14-3-3 family contains several immune-related protein members and translocates the RLR sensors to the targeted organelles in innate immunity. For example, 14-3-3ϵ facilitates the translocation from cytosol to mitochondria of the RIG-I, and 14-3-3η promotes the translocation of MDA5 to mitochondria and consequently enhances antiviral IFN response (Liu et al., 2012; Lin et al., 2019). ZIKV has been developed to inhibit or postpone IFN production and IFN-induced signaling such as ISG expression. One delicate study (Riedl et al., 2019) has shown that the ZIKV NS3 protein antagonized RIG-I- and MDA5-mediated anti-viral cascades via disguising itself to competitively associate with the 14-3-3 (Figure 1B). Interestingly, a NS3 mutation that fails to bind 14-3-3ϵ/η was defined and, as a result, the rescued NS3 mutation-containing recombinant ZIKV triggered enhanced anti-viral IFN response and consequently displayed the reduced replication capacity in SVGA cells. Interestingly, the viral protein-mediated mechanism targeting 14-3-3 is also present in other flaviviruses including DENV and WNV (Liu et al., 2012), and further delicate investigation of this immune evasion mechanism helps to look for molecule-targeting therapies or develop novel attenuated virus as a targeting strain for new vaccines.
ZIKV NS4A/4B Deregulates Akt-mTOR Signaling to Inhibit Neurogenesis
The PI3K-Akt-mTOR pathway is one of the cellular signaling pathways indispensable for neurogenesis and migration (Lee, 2015). Prenatal development is the most active stage regarding neurogenesis that populates the developing brain with neurons (Gotz and Huttner, 2005). Dysfunction in neurogenesis and differentiation could cause neurodevelopmental disorders such as microcephaly in humans (Ming and Song, 2011). Specifically, genetic mutations in the PI3K-Akt-mTOR pathway may be present in brain overdevelopment syndromes including megalencephaly-capillary malformation (MCAP), and megalencephaly-polydactyly-polymicrogyria-hydrocephalus (MPPH) (Mirzaa et al., 2013). Instead, mechanistic target of rapamycin (mTOR)-targeted suppression in developing brain caused microcephaly (Cloetta et al., 2013). Akt is an upstream molecule of mTOR and is the central player of the PI3K pathway. Interestingly, non-functional Akt mutation also caused microcephaly in humans, and instead, activating Akt mutation led to megalencephaly (Nellist et al., 2015). Viral pathogens have been reported to target the PI3K-Akt-mTOR pathway for their multiplication and pathogenicity in mammals (Buchkovich et al., 2008).
ZIKV, DENV, and HCV have been reported to hijack host cellular autophagy for viral multiplication (Heaton and Randall, 2010; Hamel et al., 2015). The mTOR kinase is the central player for autophagy induction, with activation of mTOR by Akt and MAPK inhibiting autophagy. Instead, AMPK and p53 signaling induced mTOR inactivation and facilitates autophagy (Jung et al., 2010). mTOR inactivation triggers downstream serine/threonine kinase UNC-51-like kinase-1 (ULK1), the mammalian homolog of yeast Atg1, which facilitates class III PI3K complex formation and finally induces autophagosome formation (Kim et al., 2011). Consistently, autophagy provides host protection against viral infection. Herpesviruses such as Kaposi’s sarcoma-associated herpesvirus (KSHV) and Epstein–Barr virus (EBV) inhibit cellular autophagy, utilizing viral proteins for their persistent infection establishment (Lee et al., 2009; Williams and Taylor, 2012; Liang et al., 2013).
Human neural stem cells and iPSC-derived neural organoids are known to be vulnerable to ZIKV infection (Garcez et al., 2016; Qian et al., 2016; Tang et al., 2016). This suggests the causal link between ZIKV infection and human microcephaly through neurogenesis suppression. Via two primary isolates of fetal neural stem cells (fNSCs), the study showed that the mechanism underlying ZIKV infection might destroy fetal brain development (Liang et al., 2016). Specifically, impairment of neurosphere growth and neural differentiation as well as aberrant autophagy were induced by ZIKV infection of human fNSCs. In the screening of individual ZIKV protein, it is interesting that both NS4A and NS4B synergistically inhibited host Akt-mTOR signaling, impaired the neurogenesis of human fNSCs, and upregulated autophagy (Figure 1C). The resulting promotion of viral replication in turn caused more impairment of neural development. Thus, ZIKV NS4A and NS4B are potential virulence determinants of viral neuroimmune evasion and viral pathogenesis, discovering the promising targets for anti-ZIKV therapeutic interventions.
ZIKV Inhibits the cGAS-STING Pathway
cGAS-STING as a critical innate immunity pathway has been shown to antagonize flavivirus infection (Schoggins et al., 2015; Aguirre et al., 2017; Sun et al., 2017) and accordingly flaviviruses also evolved multiple strategies to evade this pathway. cGAS-generated cGAMP-mediated IFN signaling in response to ZIKV infection in human fibroblasts was reduced while this reduction could not be observed in mouse fibroblasts. Mechanistically, ZIKV NS2b3 protein utilizes its enzymatic activity to cleave human but not mouse STING (Ding et al., 2018). However, although fibroblasts from rhesus macaque and chimpanzee are permissive to ZIKV infection, STING protein from these species can be cleaved by ZIKV NS2b3, suggesting the existence of another unknown mechanism. An interesting study reported that ZIKV NS1 not only triggered NLRP3 inflammasome activation but also contributed to caspase-1 stabilization, which facilitated caspase-1-mediated cleavage of cGAS in human THP-1 cells (Zheng et al., 2018). The further investigation on how NS2b3 or other ZIKV proteins modulate cGAS-STING signaling pathway is needed in future studies.
ZIKV Antagonizes the JAK-STAT Signaling Pathway
ZIKV infection has been demonstrated to regulate IFN response and its downstream signaling (Grant et al., 2016; Van der Hoek et al., 2017; Wu et al., 2017). JAK1, an important molecule in IFN signaling downstream, was decreased in ZIKV-infected A549 cells. A preliminary study showed that ZIKV NS2b3 protease was responsible for such decrease of JAK1 protein and JAK1 protein was stabilized by proteasome inhibitor MG132, suggesting the proteasome-mediated JAK1 degradation. However, details of relevant mechanisms are needed to uncover how ZIKV NS2b3 modulates JAK1 protein physiology. STAT1 and STAT2 phosphorylation is additionally a critical event in IFN downstream signaling. Strikingly, ZIKV infection prevented the phosphorylation of STAT1 at residue Tyr701 and STAT2 at residue Tyr689 in human A549 and dendritic cells. Specifically, ZIKV NS5 protein has been shown to not only inhibit endogenous STAT1 phosphorylation in HEK293T cells upon IFN-β stimulation but also degrade human STAT2 molecule in multiple cell lines. Further evidence showed that such degradation was mediated by the proteasome because the MG132 can inhibit STAT2 degradation (Grant et al., 2016). However, it is still unknown so far how ZIKV NS5 modulates the JAK-STAT signaling pathway for viral pathogenesis.
ZIKV Modulates TBK1-Mediated Immune Signaling
There are multiple lines of evidence supporting the modulation of immune molecule TBK1 by ZIKV infection (Wu et al., 2017; Xia et al., 2018; Lin et al., 2019). Endogenous TBK1 phosphorylation and thus IFN response was tremendously inhibited by ZIKV NS1 and NS4B expression in Sendai-infected A549 cells (Wu et al., 2017). ZIKV NS1 and NS4B were shown to associate with TBK1 to reduce TBK1 oligomerization; however, the consequence of such reduction remains unknown. ZIKV NS5 was associated with TBK1 and TRAF6, the latter being a TBK1-binding partner, and thus caused compromised TBK1–TRAF6 association. However, the detailed effect on such NS5-mediated modulation on physiological ZIKV infection remains to be investigated. Additionally, the residue 181 in ZIKV NS1 protein was critical for NS1-TBK1 binding and inhibited RIG-I mediated IFN signaling and production, because the mutation from valine to alanine at amino acid 181 in NS1 abolished TBK1- and IRF3-mediated phosphorylation of RIG-I-2CARD at Ser-172 and Ser-396 respectively, thereby resulting in the inability of RIG-I-mediated IFN response. It was reported that ZIKV NS5 not only decreased phosphorylation IRF3 at Ser-396 by TBK1 in HEK293 cells but also interacted with endogenous IRF3 through MTase domain (Xia et al., 2018), which together inhibited IRF3-mediated IFN response.
Concluding Remarks
ZIKV became a terrible pathogenic agent since 2015 due to its ability to cause neurogenesis impairment in developing brain and, consequently, microcephaly. Importantly, delayed childhood neurodevelopment and neurosensory alterations in ZIKV-exposed macaques or children have been documented (Nielsen-Saines et al., 2019; Valdes et al., 2019; Raper et al., 2020). Therefore, the existence of short-term and long-term effects of ZIKV infection is calling for further understanding ZIKV biology. Embryonic development, the stage in which ZIKV prefers to invade, is accompanied with complicated neuroimmune regulation involved interactively in mother, placenta, and embryo. The efficient evasion of innate immune defenses by ZIKV is indispensable to facilitate viral infection. Thus, investigation on how ZIKV could evade the host innate immune defenses is an essential topic to understand how ZIKV is able to invade fetal brain. More delicate studies on how ZIKV exerts neuroimmune evasion not only contribute to understanding ZIKV pathogenesis but also offer useful basis for anti-viral drug development.
Author Contributions
XX wrote the manuscript. JZ contributed to the framework and editing of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by start-up funding from Kunming Institute of Zoology in Chinese Academy of Sciences to JZ.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aguirre, S., Luthra, P., Sanchez-Aparicio, M. T., Maestre, A. M., Patel, J., Lamothe, F., et al. (2017). Dengue Virus NS2B Protein Targets cGAS for Degradation and Prevents Mitochondrial DNA Sensing During Infection. Nat. Microbiol. 2, 17037. doi: 10.1038/nmicrobiol.2017.37
Baud, D., Gubler, D. J., Schaub, B., Lanteri, M. C., Musso, D. (2017). An Update on Zika Virus Infection. Lancet 390, 2099–2109. doi: 10.1016/S0140-6736(17)31450-2
Bowen, J. R., Quicke, K. M., Maddur, M. S., O'Neal, J. T., McDonald, C. E., Fedorova, N. B., et al. (2017). Zika Virus Antagonizes Type I Interferon Responses During Infection of Human Dendritic Cells. PloS Pathog. 13, e1006164. doi: 10.1371/journal.ppat.1006164
Buchkovich, N. J., Yu, Y., Zampieri, C. A., Alwine, J. C. (2008). The TORrid Affairs of Viruses: Effects of Mammalian DNA Viruses on the PI3K-Akt-mTOR Signalling Pathway. Nat. Rev. Microbiol. 6, 265–275. doi: 10.1038/nrmicro1855
Cao-Lormeau, V. M., Blake, A., Mons, S., Lastere, S., Roche, C., Vanhomwegen, J., et al. (2016). Guillain-Barre Syndrome Outbreak Associated With Zika Virus Infection in French Polynesia: A Case-Control Study. Lancet 387, 1531–1539. doi: 10.1016/S0140-6736(16)00562-6
Chazal, M., Beauclair, G., Gracias, S., Najburg, V., Simon-Loriere, E., Tangy, F., et al. (2018). rig-I Recognizes the 5 ' Region of Dengue and Zika Virus Genomes. Cell Rep. 24, 320–328. doi: 10.1016/j.celrep.2018.06.047
Cloetta, D., Thomanetz, V., Baranek, C., Lustenberger, R. M., Lin, S., Oliveri, F., et al. (2013). Inactivation of Mtorc1 in the Developing Brain Causes Microcephaly and Affects Gliogenesis. J. Neurosci. 33, 7799–7810. doi: 10.1523/JNEUROSCI.3294-12.2013
Cugola, F. R., Fernandes, I. R., Russo, F. B., Freitas, B. C., Dias, J. L. M., Guimaraes, K. P., et al. (2016). The Brazilian Zika Virus Strain Causes Birth Defects in Experimental Models. Nature 534, 267–26+. doi: 10.1038/nature18296
Dang, J., Tiwari, S. K., Lichinchi, G., Qin, Y., Patil, V. S., Eroshkin, A. M., et al. (2016). Zika Virus Depletes Neural Progenitors in Human Cerebral Organoids Through Activation of the Innate Immune Receptor Tlr3. Cell Stem Cell 19, 258–265. doi: 10.1016/j.stem.2016.04.014
Dang, J. W., Tiwari, S. K., Qin, Y., Rana, T. M. (2019). Genome-Wide Integrative Analysis of Zika-Virus-Infected Neuronal Stem Cells Reveals Roles for MicroRNAs in Cell Cycle and Stemness. Cell Rep. 27, 3618–361+. doi: 10.1016/j.celrep.2019.05.059
Davis, T. H., Cuellar, T. L., Koch, S. M., Barker, A. J., Harfe, B. D., McManus, M. T., et al. (2008). Conditional Loss of Dicer Disrupts Cellular and Tissue Morphogenesis in the Cortex and Hippocampus. J. Neurosci. 28, 4322–4330. doi: 10.1523/JNEUROSCI.4815-07.2008
Dick, G. W., Kitchen, S. F., Haddow, A. J. (1952). Zika Virus. I. Isolations and Serological Specificity. Trans. R Soc. Trop. Med. Hyg 46, 509–520. doi: 10.1016/0035-9203(52)90042-4
Ding, Q., Gaska, J. M., Douam, F., Wei, L., Kim, D., Balev, M., et al. (2018). 'Species-Specific Disruption of STING-Dependent Antiviral Cellular Defenses by the Zika Virus NS2B3 Protease'. Proc. Natl. Acad. Sci. U. S. A. 115, E6310–E6E18. doi: 10.1073/pnas.1803406115
Ding, S. W., Voinnet, O. (2007). Antiviral Immunity Directed by Small Rnas. Cell 130, 413–426. doi: 10.1016/j.cell.2007.07.039
Garcez, P. P., Loiola, E. C., Da Costa, R. M., Higa, L. M., Trindade, P., Delvecchio, R., et al. (2016). Zika Virus Impairs Growth in Human Neurospheres and Brain Organoids. Science 352, 816–818. doi: 10.1126/science.aaf6116
Gotz, M., Huttner, W. B. (2005). The Cell Biology of Neurogenesis. Nat. Rev. Mol. Cell Biol. 6, 777–788. doi: 10.1038/nrm1739
Grant, A., Ponia, S. S., Tripathi, S., Balasubramaniam, V., Miorin, L., Sourisseau, M., et al. (2016). zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host Microbe 19, 882–890. doi: 10.1016/j.chom.2016.05.009
Guo, Z. X., Li, Y., Ding, S. W. (2019). Small RNA-Based Antimicrobial Immunity. Nat. Rev. Immunol. 19, 31–44. doi: 10.1038/s41577-018-0071-x
Hamel, R., Dejarnac, O., Wichit, S., Ekchariyawat, P., Neyret, A., Luplertlop, N., et al. (2015). Biology of Zika Virus Infection in Human Skin Cells. J. Virol. 89, 8880–8896. doi: 10.1128/JVI.00354-15
Heaton, N. S., Randall, G. (2010). dengue Virus-Induced Autophagy Regulates Lipid Metabolism. Cell Host Microbe 8, 422–432. doi: 10.1016/j.chom.2010.10.006
Hertzog, J., Dias, A. G., Rigby, R. E., Donald, C. L., Mayer, A., Sezgin, E., et al. (2018). Infection With a Brazilian Isolate of Zika Virus Generates RIG-I Stimulatory RNA and the Viral NS5 Protein Blocks Type I IFN Induction and Signaling. Eur. J. Immunol. 48, 1120–1136. doi: 10.1002/eji.201847483
Hoen, B., Schaub, B., Funk, A. L., Ardillon, V., Boullard, M., Cabie, A., et al. (2018). pregnancy Outcomes After ZIKV Infection in French Territories in the Americas. N Engl. J. Med. 378, 985–994. doi: 10.1056/NEJMoa1709481
Hong, X. X., Carmichael, G. G. (2013). innate Immunity in Pluripotent Human Cells ATTENUATED RESPONSE TO INTERFERON-Beta. J. Biol. Chem. 288, 16196–16205. doi: 10.1074/jbc.M112.435461
Jinek, M., Doudna, J. A. (2009). A Three-Dimensional View of the Molecular Machinery of RNA Interference. Nature 457, 405–412. doi: 10.1038/nature07755
Jung, C. H., Ro, S. H., Cao, J., Otto, N. M., Kim, D. H. (2010). mtor Regulation of Autophagy. FEBS Lett. 584, 1287–1295. doi: 10.1016/j.febslet.2010.01.017
Kim, J., Kundu, M., Viollet, B., Guan, K. L. (2011). AMPK and mTOR Regulate Autophagy Through Direct Phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–U71. doi: 10.1038/ncb2152
Kozak, R. A., Majer, A., Biondi, M. J., Medina, S. J., Goneau, L. W., Sajesh, B. V., et al. (2017). Microrna and mRNA Dysregulation in Astrocytes Infected With Zika Virus. Viruses-Basel 9, 297. doi: 10.3390/v9100297
Kumar, A., Hou, S., Airo, A. M., Limonta, D., Mancinelli, V., Branton, W., et al. (2016). Zika Virus Inhibits Type-I Interferon Production and Downstream Signaling. EMBO Rep. 17, 1766–1775. doi: 10.15252/embr.201642627
Lee, D. Y. (2015). Roles of mTOR Signaling in Brain Development. Exp. Neurobiol. 24, 177–185. doi: 10.5607/en.2015.24.3.177
Lee, J. S., Li, Q. L., Lee, J. Y., Lee, S. H., Jeong, J. H., Lee, H. R., et al. (2009). FLIP-Mediated Autophagy Regulation in Cell Death Control. Nat. Cell Biol. 11, 1355–U225. doi: 10.1038/ncb1980
Liang, Q. M., Chang, B., Brulois, K. F., Castro, K., Min, C. K., Rodgers, M. A., et al. (2013). Kaposi's Sarcoma-Associated Herpesvirus K7 Modulates Rubicon-Mediated Inhibition of Autophagosome Maturation. J. Virol. 87, 12499–12503. doi: 10.1128/JVI.01898-13
Liang, Q. M., Luo, Z. F., Zeng, J. X., Chen, W. Q., Foo, S. S., Lee, S. A., et al. (2016). Zika Virus NS4A and NS4B Proteins Deregulate Akt-mTOR Signaling in Human Fetal Neural Stem Cells to Inhibit Neurogenesis and Induce Autophagy. Cell Stem Cell 19, 663–671. doi: 10.1016/j.stem.2016.07.019
Li, Y., Lu, J. F., Han, Y. H., Fan, X. X., Ding, S. W. (2013). Rna Interference Functions as an Antiviral Immunity Mechanism in Mammals. Science 342, 231–234. doi: 10.1126/science.1241911
Lin, S., Yang, S., He, J., Guest, J. D., Ma, Z., Yang, L., et al. (2019). Zika Virus NS5 Protein Antagonizes Type I Interferon Production via Blocking TBK1 Activation. Virology 527, 180–187. doi: 10.1016/j.virol.2018.11.009
Liu, H. M., Loo, Y. M., Horner, S. M., Zornetzer, G. A., Katze, M. G., Gale, M. (2012). The Mitochondrial Targeting Chaperone 14-3-3 Epsilon Regulates a RIG-I Translocon That Mediates Membrane Association and Innate Antiviral Immunity. Cell Host Microbe 11, 528–537. doi: 10.1016/j.chom.2012.04.006
Maillard, P. V., Ciaudo, C., Marchais, A., Li, Y., Jay, F., Ding, S. W., et al. (2013). Antiviral RNA Interference in Mammalian Cells. Science 342, 235–238. doi: 10.1126/science.1241930
Mendell, J. T., Olson, E. N. (2012). Micrornas in Stress Signaling and Human Disease. Cell 148, 1172–1187. doi: 10.1016/j.cell.2012.02.005
Meylan, E., Tschopp, J., Karin, M. (2006). Intracellular Pattern Recognition Receptors in the Host Response. Nature 442, 39–44. doi: 10.1038/nature04946
Ming, G. L., Song, H. J. (2011). adult Neurogenesis in the Mammalian Brain: Significant Answers and Significant Questions. Neuron 70, 687–702. doi: 10.1016/j.neuron.2011.05.001
Mirzaa, G. M., Riviere, J. B., Dobyns, W. B. (2013). Megalencephaly Syndromes and Activating Mutations in the PI3K-AKT Pathway: MPPH and MCAP. Am. J. Med. Genet. Part C-Seminars Med. Genet. 163c, 122–130. doi: 10.1002/ajmg.c.31361
Musso, D., Schot, R. (2016). Zika Virus: What do We Know? Clin. Microbiol. Infection 11, 494–496. doi: 10.1016/j.cmi.2016.03.032
Nellist, M., Schot, R., Hoogeveen-Westerveld, M., Neuteboom, R. F., van der Louw, E. J., Lequin, M. H., et al. (2015). Germline Activating AKT3 Mutation Associated With Megalencephaly, Polymicrogyria, Epilepsy and Hypoglycemia. Mol. Genet. Metab. 114, 467–473. doi: 10.1016/j.ymgme.2014.11.018
Nielsen-Saines, K., Brasil, P., Kerin, T., Vasconcelos, Z., Gabaglia, C. R., Damasceno, L., et al. (2019). Delayed Childhood Neurodevelopment and Neurosensory Alterations in the Second Year of Life in a Prospective Cohort of ZIKV-Exposed Children. Nat. Med. 25, 1213–121+. doi: 10.1038/s41591-019-0496-1
Orioli, I. M., Dolk, H., Lopez-Camelo, J. S., Mattos, D., Poletta, F. A., Dutra, M. G., et al. (2017). Prevalence and Clinical Profile of Microcephaly in South America Pre-Zik-14: Prevalence and Case-Control Study. BMJ 359, j5018. doi: 10.1136/bmj.j5018
Qian, X. Y., Nguyen, H. N., Song, M. M., Hadiono, C., Ogden, S. C., Hammack, C., et al. (2016). Brain-Region-Specific Organoids Using Mini-Bioreactors for Modeling ZIKV Exposure. Cell 165, 1238–1254. doi: 10.1016/j.cell.2016.04.032
Rajman, M., Schratt, G. (2017). Micrornas in Neural Development: From Master Regulators to Fine-Tuners. Development 144, 2310–2322. doi: 10.1242/dev.144337
Raper, J., Kovacs-Balint, Z., Mavigner, M., Gumber, S., Burke, M. W., Habib, J., et al. (2020). Long-Term Alterations in Brain and Behavior After Postnatal Zika Virus Infection in Infant Macaques. Nat. Commun. 11, 2534. doi: 10.1038/s41467-020-16320-7
Rasmussen, S. A., Jamieson, D. J., Honein, M. A., Petersen, L. R. (2016). Zika Virus and Birth Defects–Reviewing the Evidence for Causality. N Engl. J. Med. 374, 1981–1987. doi: 10.1056/NEJMsr1604338
Riedl, W., Acharya, D., Lee, J. H., Liu, G. Q., Serman, T., Chiang, C., et al. (2019). Zika Virus NS3 Mimics a Cellular 14-3-3-Binding Motif to Antagonize RIG-I- and MDA5-Mediated Innate Immunity. Cell Host Microbe 26, 493–49+. doi: 10.1016/j.chom.2019.09.012
Saldana, M. A., Etebari, K., Hart, C. E., Widen, S. G., Wood, T. G., Thangamani, S., et al. (2017). Zika Virus Alters the microRNA Expression Profile and Elicits an RNAi Response in Aedes Aegypti Mosquitoes. PloS Neglected Trop. Dis. 11, e0005760. doi: 10.1371/journal.pntd.0005760
Schoggins, J. W., MacDuff, D. A., Imanaka, N., Gainey, M. D., Shrestha, B., Eitson, J. L., et al. (2015). Pan-Viral Specificity of IFN-Induced Genes Reveals New Roles for cGAS in Innate Immunity (Vol 505, Pg 691, 2014). Nature 525, 144–144. doi: 10.1038/nature12862
Serman, T. M., Gack, M. U. (2019). Evasion of Innate and Intrinsic Antiviral Pathways by the Zika Virus. Viruses-Basel 11, 970. doi: 10.3390/v11100970
Shao, Q., Herrlinger, S., Yang, S. L., Lai, F., Moore, J. M., Brindley, M. A., et al. (2016). Zika Virus Infection Disrupts Neurovascular Development and Results in Postnatal Microcephaly With Brain Damage. Development 143, 4127–4136. doi: 10.1242/dev.143768
Sun, B., Sundstrom, K. B., Chew, J. J., Bist, P., Gan, E. S., Tan, H. C., et al. (2017). Dengue Virus Activates cGAS Through the Release of Mitochondrial DNA. Sci. Rep. 7, 3594. doi: 10.1038/s41598-017-03932-1
Tang, H. L., Hammack, C., Ogden, S. C., Wen, Z. X., Qian, X. Y., Li, Y. J., et al. (2016). Zika Virus Infects Human Cortical Neural Progenitors and Attenuates Their Growth. Cell Stem Cell 18, 587–590. doi: 10.1016/j.stem.2016.02.016
tenOever, B. R. (2016). the Evolution of Antiviral Defense Systems. Cell Host Microbe 19, 142–149. doi: 10.1016/j.chom.2016.01.006
Valdes, V., Zorrilla, C. D., Gabard-Durnam, L., Muler-Mendez, N., Rahman, Z. I., Rivera, D., et al. (2019). Cognitive Development of Infants Exposed to the Zika Virus in Puerto Rico. JAMA Network Open 2, e1914061. doi: 10.1001/jamanetworkopen.2019.14061
Van der Hoek, K. H., Eyre, N. S., Shue, B., Khantisitthiporn, O., Glab-Ampi, K., Carr, J. M., et al. (2017). Viperin Is an Important Host Restriction Factor in Control of Zika Virus Infection. Sci. Rep. 7, 4475. doi: 10.1038/s41598-017-04138-1
Weaver, S. C., Costa, F., Garcia-Blanco, M. A., Ko, A. I., Ribeiro, G. S., Saade, G., et al. (2016). Zika Virus: History, Emergence, Biology, and Prospects for Control. Antiviral Res. 130, 69–80. doi: 10.1016/j.antiviral.2016.03.010
Williams, L. R., Taylor, G. S. (2012). Autophagy and Immunity - Insights From Human Herpesviruses. Front. Immunol. 3. doi: 10.3389/fimmu.2012.00170
Wu, X. F., Kwong, A. C., Rice, C. M. (2019). Antiviral Resistance of Stem Cells. Curr. Opin. Immunol. 56, 50–59. doi: 10.1016/j.coi.2018.10.004
Wu, Y. X., Liu, Q. X., Zhou, J., Xie, W. H., Chen, C., Wang, Z. F., et al. (2017). Zika Virus Evades Interferon-Mediated Antiviral Response Through the Co-Operation of Multiple Nonstructural Proteins In Vitro (Vol 3, 17006, 2017). Cell Discov. 3, 17006. doi: 10.1038/celldisc.2017.6
Wu, X. F., Thi, V. L. D., Huang, Y. M., Billerbeck, E., Saha, D., Hoffmann, H. H., et al. (2018). Intrinsic Immunity Shapes Viral Resistance of Stem Cells. Cell 172, 423–42+. doi: 10.1016/j.cell.2017.11.018
Xia, H., Luo, H., Shan, C., Muruato, A. E., Nunes, B. T. D., Medeiros, D. B. A., et al. (2018). An Evolutionary NS1 Mutation Enhances Zika Virus Evasion of Host Interferon Induction. Nat. Commun. 9, 414. doi: 10.1038/s41467-017-02816-2
Xu, Y. P., Qiu, Y., Zhang, B., Chen, G., Chen, Q., Wang, M., et al. (2019). Zika Virus Infection Induces RNAi-Mediated Antiviral Immunity in Human Neural Progenitors and Brain Organoids. Cell Res. 29, 265–273. doi: 10.1038/s41422-019-0152-9
Zeng, J. X., Dong, S. P., Luo, Z. F., Xie, X. C., Fu, B. S., Li, P., et al. (2020). The Zika Virus Capsid Disrupts Corticogenesis by Suppressing Dicer Activity and miRNA Biogenesis. Cell Stem Cell 27, 618–61+. doi: 10.1016/j.stem.2020.07.012
Zeng, J., Luo, Z., Dong, S., Xie, X., Liang, X., Yan, Y., et al. (2021). Functional Mapping of AGO-Associated Zika Virus-Derived Small Interfering RNAs in Neural Stem Cells. Front. Cell Infect. Microbiol. 11, 628887. doi: 10.3389/fcimb.2021.628887
Keywords: ZIKV, capsid, NS3, NS4A/4B, pathogenesis, NS5, immune evasion
Citation: Xie X and Zeng J (2021) Neuroimmune Evasion of Zika Virus to Facilitate Viral Pathogenesis. Front. Cell. Infect. Microbiol. 11:662447. doi: 10.3389/fcimb.2021.662447
Received: 01 February 2021; Accepted: 04 October 2021;
Published: 26 October 2021.
Edited by:
Daniel Adesse, Oswaldo Cruz Foundation (Fiocruz), BrazilReviewed by:
Patricia Garcez, Federal University of Rio de Janeiro, BrazilPedro Paulo Manso, Fundação Oswaldo Cruz, Brazil
Copyright © 2021 Xie and Zeng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianxiong Zeng, emVuZ2ppYW54aW9uZ0BtYWlsLmtpei5hYy5jbg==