 Kiran Kumari1
Kiran Kumari1 Rajnish Prakash Singh
Rajnish Prakash Singh- 1Department of Bioengineering and Biotechnology, Birla Institute of Technology, Ranchi, Jharkhand, India
- 2Department of Biotechnology, Jaypee Institute of Information Technology, Noida, India
- 3Department of Plant Sciences and Landscape Architecture, University of Maryland, College Park, MD, United States
The present study aimed to identify the mechanisms underlying the survival of an environmental bacterium originally isolated from the waste-contaminated soil of Jhiri, Ranchi, India. Based on 16S rRNA, ANI (average nucleotide identity), and BLAST Ring Image Generator (BRIG) analysis, the isolated strain was identified as Pseudomonas aeruginosa. The present study extends the characterization of this bacterium through genomic and comparative genomic analysis to understand the genomic features pertaining to survival in stressed environments. The sequencing of the bacterium at Illumina HiSeq platform revealed that it possessed a 6.8 Mb circular chromosome with 65.9% GC content and 63 RNAs sequence. The genome also harbored several genes associated to plant growth promotion i.e. phytohormone and siderophore production, phosphate solubilization, motility, and biofilm formation, etc. The genomic analysis with online tools unraveled the various genes belonging to the bacterial secretion system, antibiotic resistance, virulence, and efflux pumps, etc. The presence of biosynthetic gene clusters (BCGs) indicated that large numbers of genes were associated to non-ribosomal synthesized peptide synthetase, polyketide synthetase, and other secondary metabolite production. Additionally, its genomes encode various CAZymes such as glycoside hydrolases and other genes associated with lignocellulose breakdown, suggesting that strain S-8 have strong biomass degradation potential. Furthermore, pan-genome analysis based on a comparison of whole genomes showed that core genome represented the largest part of the gene pools. Therefore, genome and comparative genome analysis of Pseudomonas strains is valuable for understanding the mechanism of resistance to metal stress, genome evolution, HGT events, and therefore, opens a new perspective to exploit a newly isolated bacterium for biotechnological applications.
Introduction
Pseudomonas genus is considered a major opportunistic pathogen, isolated from diverse ecological niches including, soil, water, and clinical specimens (Stover et al., 2000), and possess intrinsically advanced antibiotic resistance gene clusters (Lambert, 2002). Many strains have been associated with serious illness-causing nosocomial infections and various sepsis syndromes (Planquette et al., 2013; Philippart et al., 2015). The pathogenicity is attributed to the presence of virulence features like pili, flagella, exotoxin A, secretion system, and quorum-sensing proteins (Lyczak et al., 2000). Among various members, P. aeruginosa is a Gram-negative, aerobic, rod-shaped bacterium and environmental isolates of this bacterium easily adapt to a large variety of natural ecosystems (Selezska et al., 2012). As every P. aeruginosa has a certain pathogenic potential, they are all classified as risk group two and the degree of virulence varies substantially between strains (Hilker et al., 2015).
Besides, soil-dwelling Pseudomonas species form close relationships with plants, thereby positively affecting plant growth and nutrition (Cheng et al., 2017; Zamioudis et al., 2013), and also exhibits potent antagonistic activity toward pathogenic microorganisms (Biessy et al., 2019). However, the infection caused by various phytopathogens leads to a loss of ~25% crop production globally (Kim et al., 2017a, b). Additionally, the excessive use of chemical pesticides and fertilizers used to control phytopathogens imposes a serious effect on human and environmental health (Alvarez et al., 2012). Under these circumstances, microbial inoculants belonging to the genera Pseudomonas holds a promising substitute for conventional fertilizers and antibiotics (Stewart, 2001). Pseudomonas spp. influence plant health also by the production of diverse set of secondary metabolites (Nguyen et al., 2016; Stringlis et al., 2018), and through secreted proteins (Rangel et al., 2016). The production of phenazines and anthranilate by the P. aeruginosa contribute to the antagonism against plant pathogens (Anjaiah et al., 1998), and is thereby used as biocontrol agents and biofertilizers (Kwak and Weller, 2013). Moreover, Pseudomonas spp. also employ diverse mechanisms to colonize the plant rhizosphere (Little et al., 2019) and suppress a range of plant pathogens including bacteria (Arseneault et al., 2015), fungi (Michelsen et al., 2015), and insects (Flury et al., 2017), however, these attributes vary from strain to strain.
The genome of Pseudomonas spp. generally divided into a core genome containing sequences common to the species and an accessory genome, with the restriction of sequences present in some strains (Kung et al., 2010; Ozer et al., 2014). The virulence of Pseudomonas spp. is generally affected by the variation in both the core and accessory genome. However, many of the genomes investigation are still going, therefore, accessory genes associated with virulence in addition to mutation may be missed. This may limit the prediction of antibiotic resistance or virulence genes on mobile genetic elements (MGE) or standard chromosome regions as well as their prevalence in general (Beatson and Walker, 2014). It was suggested that the large genome size and complexity of several P. aeruginosa strains such as P. aeruginosa PAO1 (6.2 Mbp, 5683 genes), P. aeruginosa PA14 (6.5 Mbp, 5965 genes), and P. aeruginosa PA7 (6.5 Mbp, 6369 genes) reflects environmental adaptation with the highest proportion of regulatory genes, and a large number of genes involved in metabolism, transport and efflux systems allows the bacterium to survive in diverse environments (Stover et al., 2000; Winsor et al., 2011). The higher number of regulatory genes modulates the metabolic and genetic capacity of P. aeruginosa in varying environmental conditions. The increased availability of genes, genomics data, and further use of genomic tools suggests the potential applications of the strain for diverse biotechnological applications (Xia et al., 2018). Many of the Pseudomonas strains possess good industrial applications in-terms of production of highly stable enzymes (Grbavčić et al., 2011), biosurfactants (Rikalović, 2013), and polysaccharides (Dimitrijević et al., 2011). Additionally, many of the strains have been reported to tolerate high concentrations of heavy metals (Izrael-Zivkovic et al., 2018), which makes it industrial useful.
Due to the spectrum of ecological, biochemical and metabolic characteristics of the Pseudomonas genus, it is clearly evident that diversity among this bacterium extends to the genomic level. Klockgether et al. (2011) have suggested that sequencing of P. aeruginosa strains from environmental habitats provides an unbiased overview of the genetic repertoire. More than 200 P. aeruginosa genome sequences are available on the National Centre of Biotechnology Information (NCBI), however, less than 10% are of environmental strains. The increase in robust sequencing technologies has resulted in the economic cost of sequencing a bacterial genome. Next-generation sequencing provides valuable insight into the genome of organisms and allows the comprehensive analysis of genomic features (Roy et al., 2013). Furthermore, the functional annotation of genomic features can be utilized as a powerful tool for the development of genetically modified bacteria with improved functionality. Additionally, comparative genomics has emerged as a robust tool to identify and compare functionally important genomic elements (Wu et al., 2010). A comparison of genomes within the Pseudomonas group demonstrated evidence about the ecological and physiological diversity of these bacteria extends to the genomic level (Loper et al., 2012).
In the present study, we focused on the detailed genomic characterization of environmental isolate P. aeruginosa S-8 isolated from waste-dumping soil sample, which showed good antagonistic activity against tested bacterial and fungal pathogens. Therefore, the in-depth genome and comparative genome analysis will fill the gap in the genomic studies of environmental Pseudomonas strains. Additionally, the whole-genome analysis (WGS) of this strain will provide opportunities to identify genes involved in the biocontrol of pathogens, plant growth promotion (PGP), and genes involved in the production of secondary metabolites, etc. The available WGS data of many Pseudomonas strains in the public database (https://www.ncbi.nlm.nih.gov/datasets/genome/) have been used for the comparative genome analysis and pan-genome analysis with WGS data of P. aeruginosa S-8.
Materials and methods
Characterization of strain S-8
The bacterial strain S-8 was isolated from the metal-contaminated soil of Jhiri (23.40°N, 85.25°E), Ranchi, Jharkhand, India and its genome was submitted with accession number JARESC000000000. However, before genome sequencing, we performed the 16S rRNA gene sequencing following standard protocol (Singh et al., 2024). The strain was tested for its metal-stress tolerance against zinc sulfate (ZnSO4), copper sulfate (CuSO4), cadmium chloride (CdCl2), mercuric chloride (HgCl2), and nickel sulfate hexahydrate (NiSO4.6H2O), each with 5 mM concentration (Kumari et al., 2024). The isolate was tested for antagonistic activity against bacterial pathogens such as Bacillus subtilis, Salmonella typhi, Escherichia coli, and Staphylococcus aureus by using a well-diffusion method. The boiled culture was used as a control and the experiment was performed in triplicate. To test the antifungal activity, 100 µl fungal mycelia (in 0.85% saline) of Aspergillus niger, Microsporum gypseum, H. gypsium and Penicillium citrium was spread on the potato dextrose agar (PDA, Himedia, India) plate, and wells of 6 mm diameter were filled with 1×108 CFU/ml of S-8, and kept for incubation at 28°C for seven days. The activity was evaluated by measuring the ZOI (zone of inhibition) for which the parameter used was <10 mm = poor (+), 11 to 15 mm moderate (++), and between 16 to 20 mm = good (+++).
Isolate S-8 was tested for various motility behavior. To perform the swimming, S-8 was spot-inoculated on solidified tryptone swim plate composed of 1% tryptone, 0.5% NaCl, 0.3% agar, and incubated for 16 hr at 25°C. To evaluate the swarming, S-8 was spot inoculated on media containing 0.5% bacto-agar, 8g L-1 nutrient broth, 5g L-1 dextrose, and incubated for 24 hr at 30°C. For twitching, LB agar (1% agar) media was used for stab inoculation, and inoculated plates were incubated at 30°C for 24-48 hr. After incubation, the presence of a turbid circular zone indicated the swimming, movement of inoculation for swarming, and circular turbid zone for twitching activity (Connelly et al., 2004). The experiment was performed in duplicate.
Antibiotics sensitivity test
To check the antibiotics susceptibility, the disk diffusion method was used as recommended by the Clinical and Laboratory Standards Institute (CLSI). The isolate was grown in LB broth overnight for 14h with shaking at 180 rpm, spread in an agar plate, and then the paper disk containing the different antibiotics such as erythromycin (15 µg), ampicillin (10 µg), kanamycin (30 µg), tetracycline (30 µg), ciprofloxacin (5 µg), gentamicin (10 µg), fluconazole (25 µg), streptomycin (10 µg), vancomicin (30 µg), and voriconazole (10 µg) was placed in the agar plate for diffusion and incubated for 24-48 hr. The result was observed by measuring the diameter of the zone of inhibition created by the antibiotic disk against the tested isolates. These are the most common antibiotics used against both environmental and clinical strains (Singh et al., 2024).
Whole genome sequencing
The extracted genomic DNA was sequenced using an illumina HiSeq platform and the paired-end library was prepared by using the NEB Next Ultra DNA Library Prep Kit. Fast QC program was used for Quality control of Illumina reads (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). The Illumina reads were assembled using genome assembly tools SPAdes (Nurk et al., 2013). The transfer RNA (tRNA) and ribosomal RNA (rRNA) of the S-12 strain were identified using the tRNAscan-SE and RNAmmer (v1.2, http://www.cbs.dtu.dk/services/RNAmmer/) software, respectively. Further, the genome sequence was annotated by RAST (Rapid Annotation using Subsystem Technology) annotation server (Aziz et al., 2008) to annotate the open reading frames, and BLAT v2.0 to validate the predictions. COG functions of protein-coding sequences were determined using the RPS-BLAST algorithm for blast search against the COG database (https://ftp.ncbi.nih.gov/pub/wolf/COGs/) (Schaffer et al., 2001; Deb, 2022). The genes involved in metabolic pathways were annotated using KEGG and Blast2Go tools. Using Genome BLAST Distance Phylogeny (GBDP) method and tree builder service, the phylogeny tree of S-8 was created using its whole genome sequence.
Average nucleotide identity analysis
ANI analysis was done to explore the genetic distance and relatedness for the genome sets containing S-8, and publicly available P. aeruginosa genomes (Table 1) by MASH v2.2.2 (Ondov et al., 2016). To improve the speed for analyzing large amounts of sequence data, other methods have been developed including FastANIv1.32 which uses an alignment-free mapping algorithm (Mashmap) implemented to approximate ANI calculations in a range of 80–100% identity (Jain et al., 2018).
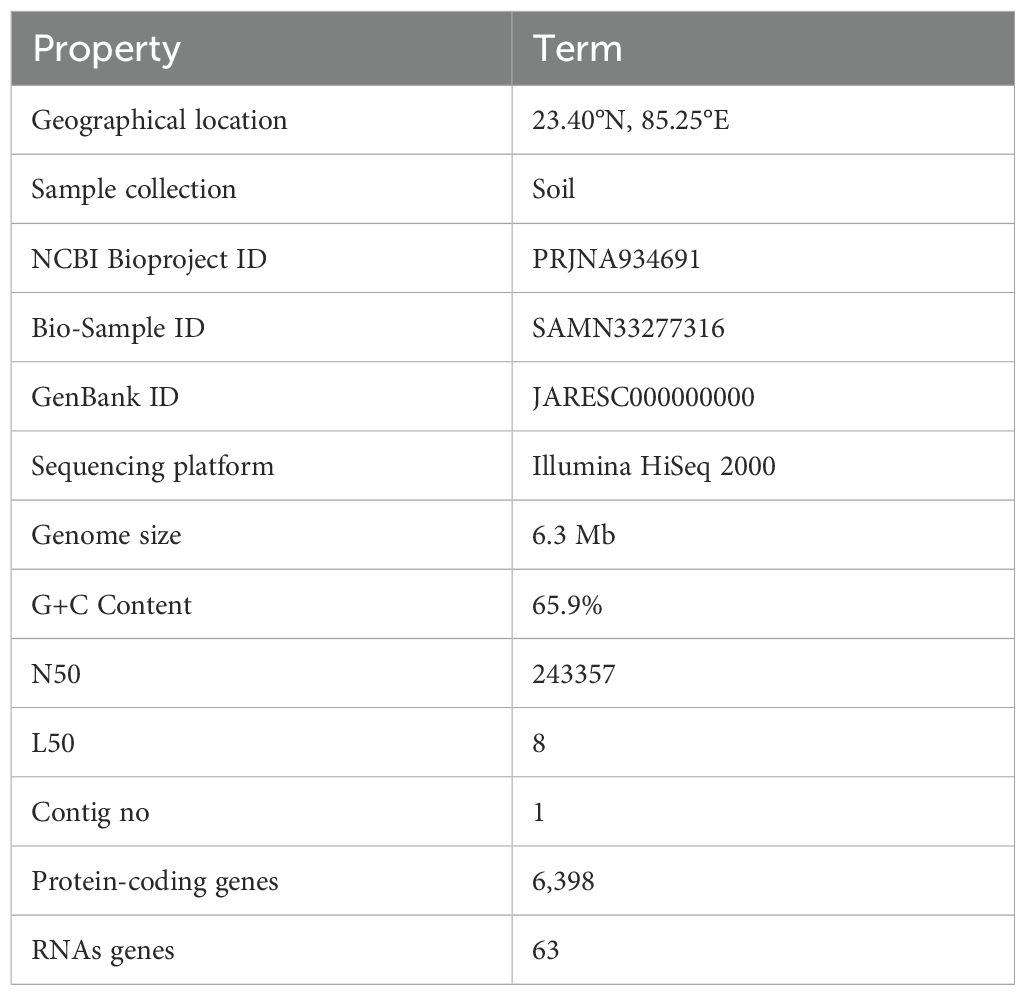
Table 1. General features of P. aeruginosa S-8 genome.
Antimicrobial and virulence analysis
The CARD database was used using a homology-based approach (BLASTX) against the genome sequence of S-8 to unravel the presence of AMR genes. For searching, BLAST output was filtered with a minimum of 80% identity and subject protein coverage. Similarly, the VFDB database was used against assembled genome with criteria of a minimum of 80% identity using a homology-based approach (BLASTX) to identify the virulence genes.
Prediction of biosynthetic gene clusters
The number and types of secondary metabolite BGCs in the genome sequence of P. aeruginosa S-8 were identified by antiSMASH version 5.1.2 in combination with Hidden Markov Model (HMM) to detect the BGCs-like region (Blin et al., 2021). Various unknown and characterized BGCs were identified and genetic similarities in gene clusters were predicted using antiSMASH 5.1.2.
Prediction of carbohydrate−active enzyme (CAZymes)
To unravel the presence of various CAZymes including glycosyltransferases (GTs), glycoside hydrolases (GHs), polysaccharide lyases (PLs), carbohydrate esterases (CEs), auxiliary activities (AAs) and carbohydrate-binding modules (CBMs), the protein sequences of S-8 was annotated using the dbCAN2 server, and BLAST-driven DIAMOND against the CAZy database. The diversity of CAZymes in the closest relatives of P. aeruginosa strains, P. aeruginosa NCTC9433, P. aeruginosa SCV20265, P. aeruginosa PA_D2, P. aeruginosa W36662, P. aeruginosa Paer4_119, P. aeruginosa F30658, P. aeruginosa PASGNDM345, P. aeruginosa PA1R, P. aeruginosa X78812, and P. aeruginosa W16407 was also performed to evaluate the comparative distribution.
Comparative genome analysis
The distribution of genes in the selected genome (Supplementary Table 1) under different functional category like virulence, metabolism, carbohydrate, stress responses, protein metabolism, amino acids & derivatives, and membrane transport was performed by RAST analysis. The analysis of orthologous gene clusters was analyzed using the Orthovenn2 program (Xu et al., 2019) with default parameters using the protein sequences of P. aeruginosa S-8, P. aeruginosa NCTC9433, P. aeruginosa SCV20265, P. aeruginosa PA_D2, P. aeruginosa W36662, and P. aeruginosa Paer4_119. The genomes were selected based on the high similarity to S-8 strains. The circular genome comparison of the assembly genome of S-8 was performed against the reference genomes (Supplementary Table 1) using the BRIG (Blast Ring Image Generator) Tool (Alikhan et al., 2011). BLAST was performed on five characteristic Pseudomonas genomes which were constructed using NCBI local BLAST-2.10.1.
Pan-genome analysis
Core and accessory genes in P. aeruginosa S-8 and its closest related strains were identified by using Roary 3.11.2 with default settings. The GFF3 files of all selected strains including P. aeruginosa S-8 genomes were generated by PROKKA 1.14.5 (Seemann, 2014). The maximum likelihood (ML) phylogenetic tree of P. aeruginosa S-8 and its closest related strains based on core-genome single nucleotide polymorphism was plotted after filtering the core genome alignment using the SNP-sites 2.5.1 (https://github.com/sanger-pathogens/snp-sites) (Page et al., 2016). The evolutionary history was calculated by the ML method based on the Tamura and Nei method (Tamura and Nei, 1993). To visualize the matrix showing the presence and absence of core genes in the used strains, Phandango was used. The summary file generated by Roary software was used to assess the proportions of the pan-genome.
HGT and SNP analysis
HGTector2 bioinformatics tool was used for the detection and analysis of horizontal gene transfer (HGT) events in microbial genomes. It utilizes computational methods to identify genes or genomic regions that have likely been acquired through horizontal transfer from distantly related species. Here, we used all the genomes as input for HGT analysis. Parsnp v.1.2 was used for whole-genome alignment and phylogenetic analysis of microbial genomes (Treangen et al., 2014). It compares genomes to a reference (using MUMmer) to identify core genome SNPs and build a phylogeny. It is part of the Harvest suite of tools developed by the University of Maryland, School of Medicine. The main purpose of Parsnp is to align and compare multiple bacterial genomes to identify genetic variations and to infer the phylogenetic relationships. It employs a progressive Mauve algorithm, which is a multiple genome alignment method that takes into account genome rearrangements, inversions, and horizontal gene transfer events.
Data deposition
The genome sequence of S-8 is available at NCBI with the BioProject PRJNA934691, Biosample SAMN33277316, and genome accession no. JARESC000000000.
Results
Characterization of strain S-8
The 16S rRNA sequencing confirmed that isolated strain S-8 belongs to P. aeruginosa (Supplementary Figure 1). The growth pattern of S-8 in metal enriched medium showed that strain S-8 growth behavior was higher in CuSO4-amended medium as compared to other heavy metals. The isolate showed a higher sensitivity (20 to 25 mm) against ampicillin, kanamycin, tetracycline, gentamicin, ciprofloxacine, vancomycin, and moderate sensitivity (12 to 18 mm) to streptomycin, and fluconazole. The strain was found to be resistant to voriconazole and erythromycin. The test isolate showed good antagonistic activity against S. typhi, E. coli, and moderate against B. subtilis and S. aureus. Against the tested fungal strains, S-8 showed good activity against A. niger, M. gypsium, and moderate against H. gypsium, P. citrium (Supplementary Figure 2). The test isolate S-8 showed the swimming, swarming, and twitching motility (Figure 1).
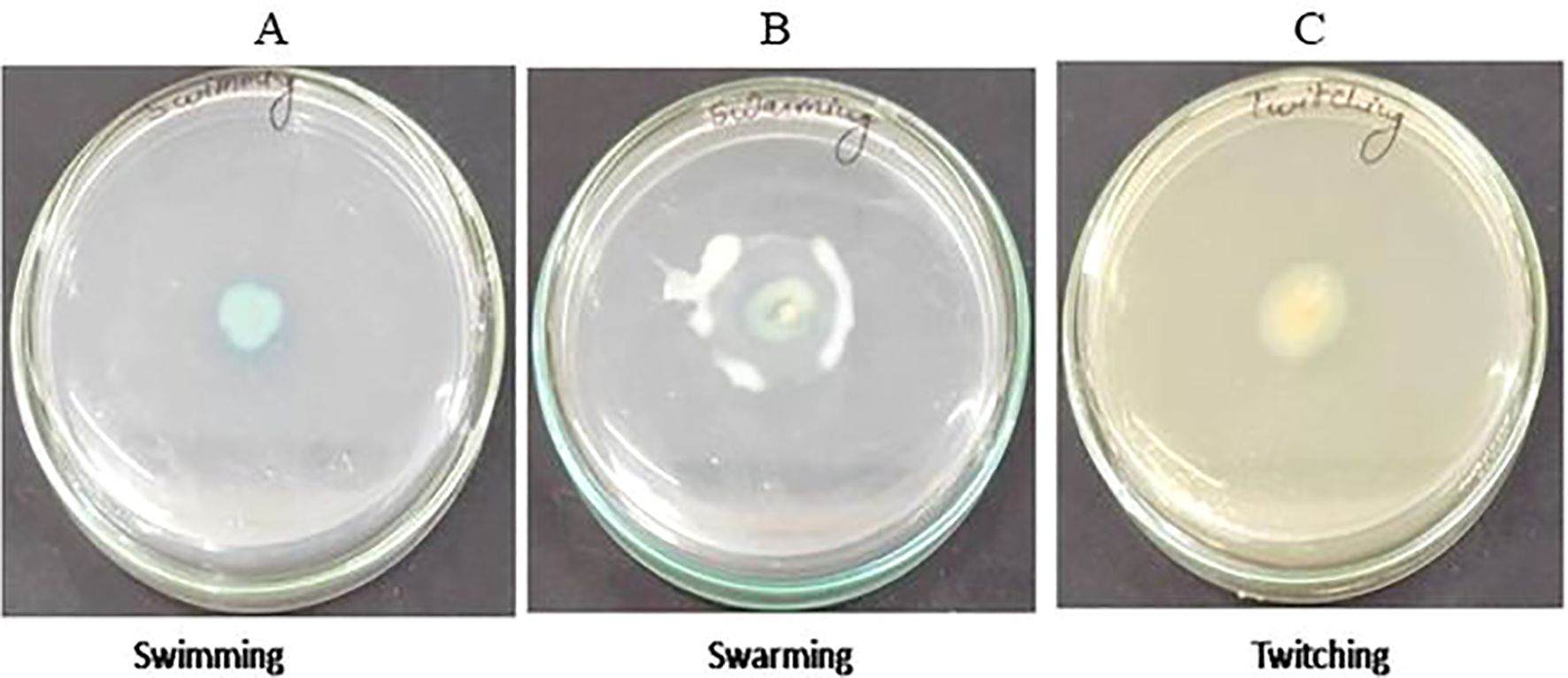
Figure 1. Test of motility swimming (A), swarming (B), and twitching (C) shown by P. aeruginosa S-8 on motility specific medium plate.
Genome analysis
A total of 6,908,234 sequencing reads were generated for strain S-8. The assembly of the genome sequence was performed using the Unicycler v-0.48 tool and a single contig of 6.8 Mb size was obtained (Figure 2). The assembly was validated using the NCBI-NR Blast program which showed the maximum homology with other Pseudomonas spp. The overall average G+C content of P. aeruginosa S-8 is 65.9% (Table 1) and therefore considered as G+C-rich, whereas genes acquired through horizontal gene transfer usually have a lower G+C content. A total of 63 RNAs were annotated in the S-8 genome. Further, gene/protein prediction from the draft genome using the Prokkav1.14 tool identified a total of 6,398 protein-coding genes.

Figure 2. The circular genome map (6.8 Mbp) of P. aeruginosa S-8. From the outer circle to the inner circle: coding DNA sequences (CDS) on the forward and reverse strand, RNA (tRNA/rRNA), GC content and GC skew, Pie chart representing the RAST subsystems categories in the P. aeruginosa S-8 genome. The most abundant systems on the category level are shown in the left piet chart, whereas, the right column showing the counts of features.
RAST functional annotation
The genome sequence of S-8 was further annotated by RAST, which showed that the top three subsystem features of S-8 are amino acids derivatives (729 genes), carbohydrates (467 genes), followed by cofactors/vitamins (371 genes) (Figure 3). The other subsystem includes the membrane transporter (322 genes), protein metabolism (305 genes), cell wall & capsule (228 genes), RNA metabolism (227 genes), and genes related to fatty acids metabolism (221). The gene annotation was performed with the closest relatives of S-8 including P. aeruginosa NCTC9433, P. aeruginosa SCV20265, P. aeruginosa PA_D2, P. aeruginosa W36662, P. aeruginosa Paer4_119, P. aeruginosa F30658, P. aeruginosa PASGNDM345, P. aeruginosa PA1R, P. aeruginosa X78812, and P. aeruginosa W16407 (Figure 4; Supplementary Table 1). The phylogenomic relationships of S-8 and other P. agglomerans strains was established based on the core genome analysis, which showed the closeness of S-8 to P. aeruginosa FRD1 and P. aeruginosa NCTC 10332 (Figure 5).
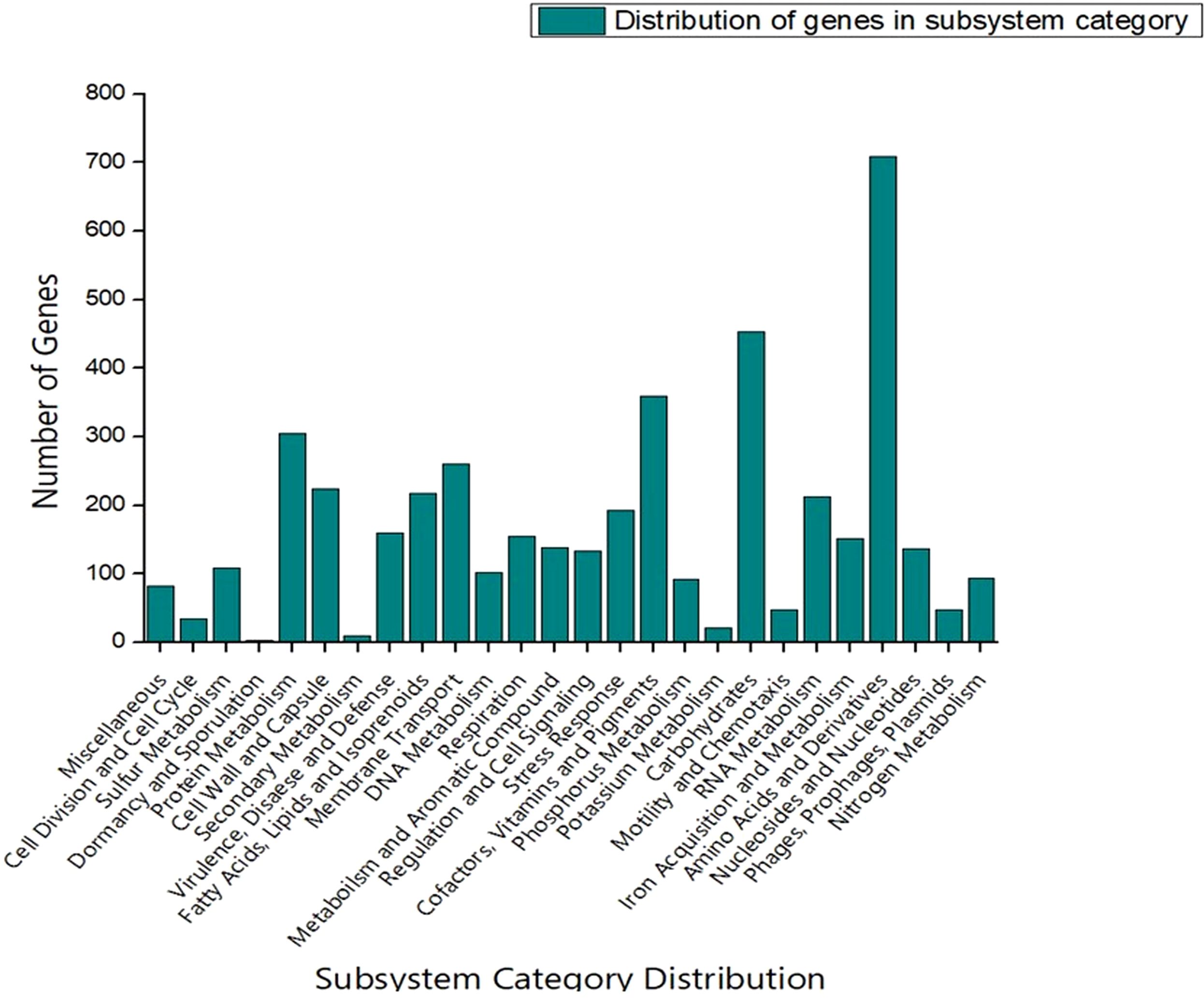
Figure 3. RAST-based comparative analysis of the distribution of genes which showed that S-8 possess a higher number of genes involved in amino acid synthesis, carbohydrate synthesis, cofactors, vitamin & pigments, and protein metabolism etc.
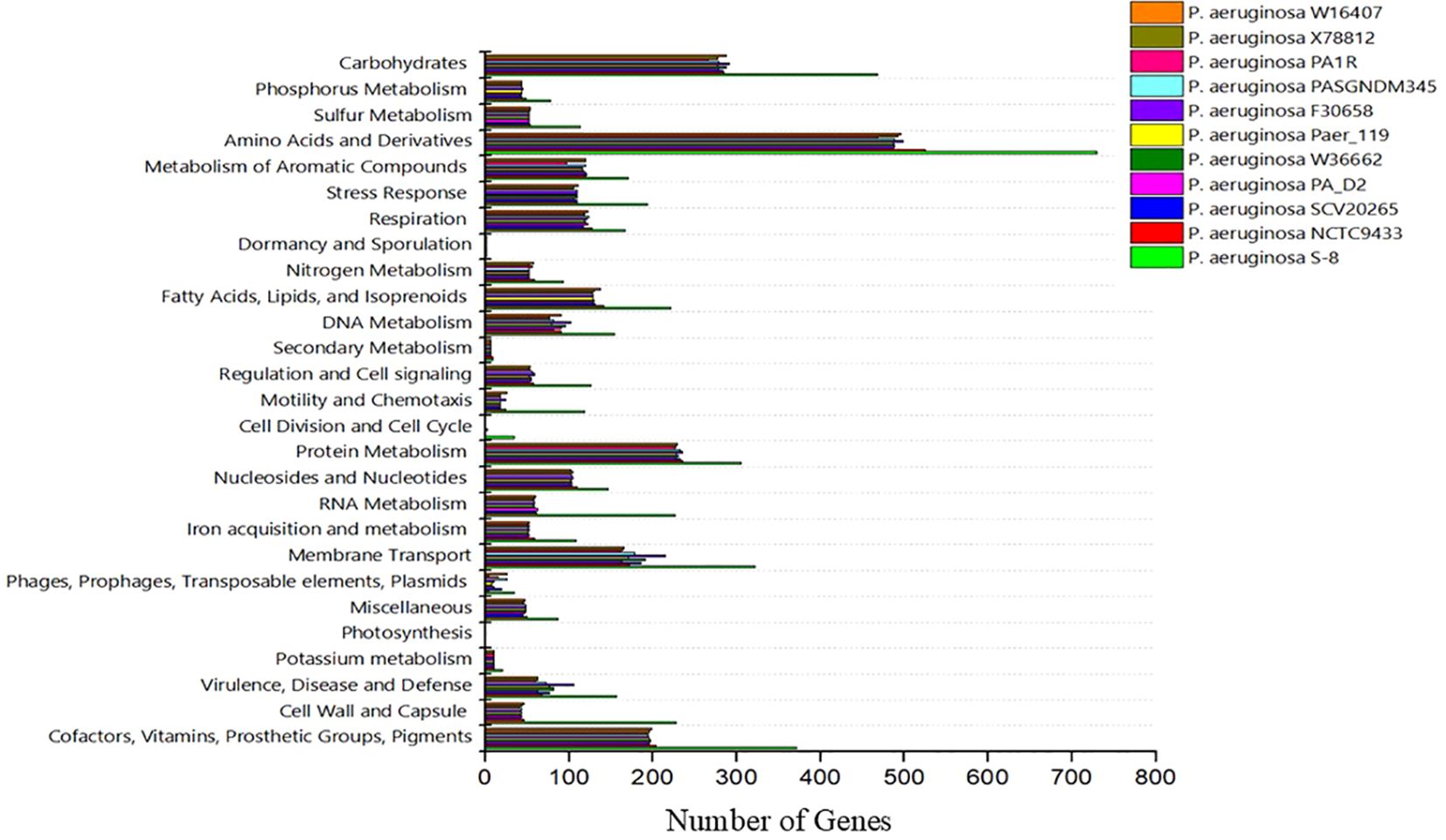
Figure 4. A compassion of gene distribution in the P. aeruginosa S-8 genome and its closest relatives genomes including P. aeruginosa NCTC9433, P. aeruginosa SCV20265, P. aeruginosa PA_D2, P. aeruginosa W36662, P. aeruginosa Paer4_119, P. aeruginosa F30658, P. aeruginosa PASGNDM345, P. aeruginosa PA1R, P. aeruginosa X78812, and P. aeruginosa W16407.
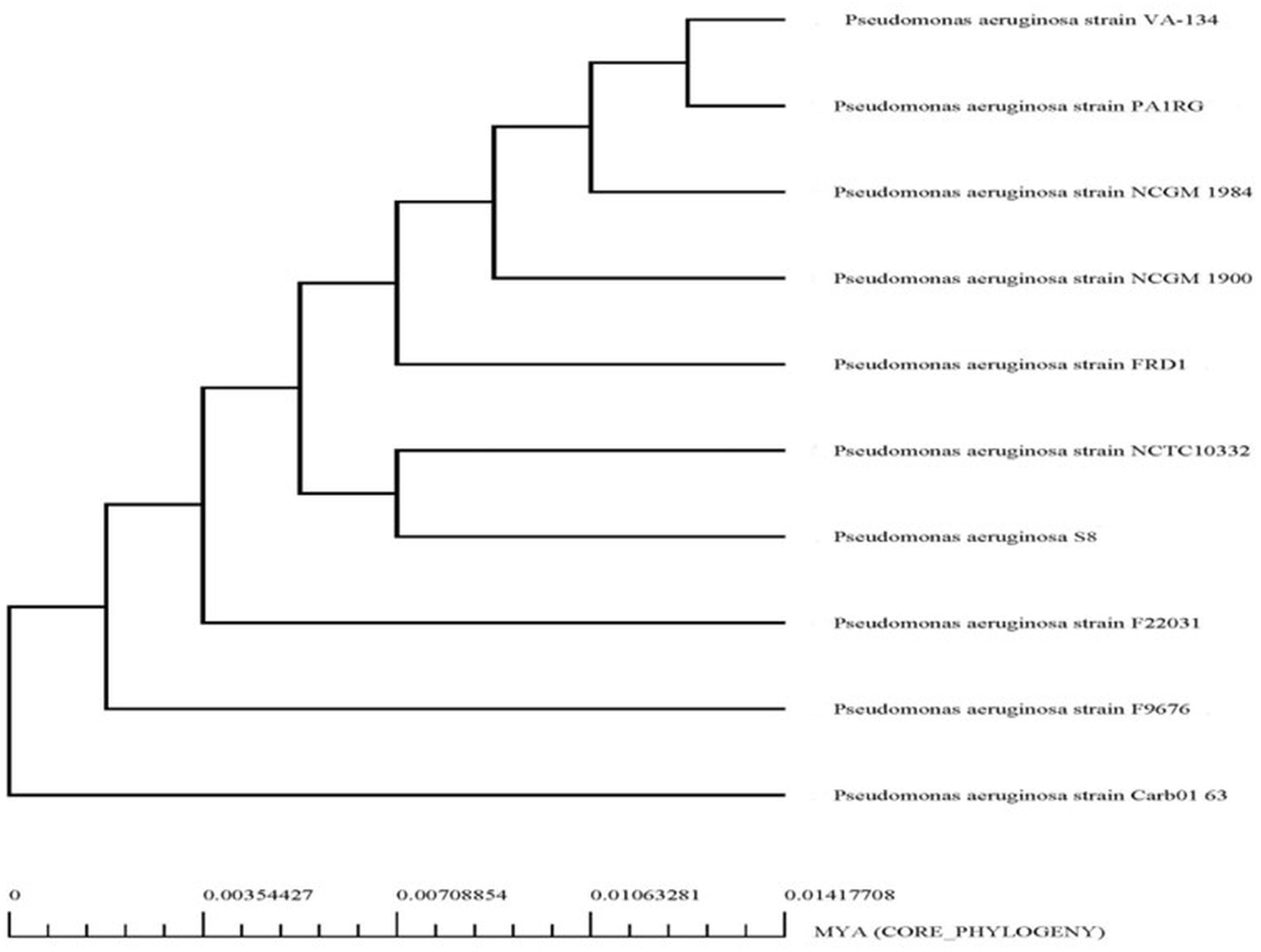
Figure 5. Phylogenetic analysis of P. aeruginosa S-8 based on WGS (whole genome sequences). All the closest related reference sequences were represented to PATRIC for phylogenetic analysis. Following MUSCLE alignment, RaxML was used to visualize the matrix.
Gene ontology
To assign the functionality of the classified proteins, gene ontology was investigated. A sum total of 66% of genes were related to biological processes, 18% to molecular functions, and 16% to cellular components (Figure 6). In the molecular functions, 23.95% genes were associated with different molecular functions, 15.19% related to catalytic activity, 6.40% to binding processes, 4.96% transferase activity and 3.92% to hydrolase activity (Supplementary File S1). In the cellular component, 24.50% of genes were related different cellular component, 23.22% to cellular anatomical entity, 12.23% to intracellular anatomical structure, and 11.67% to cytoplasm (Supplementary File S1). In the biological processes, the highest number of genes (8.06%) was observed for different biological processes. The other genes associated with different biological functionality have been summarized in Supplementary File S1.
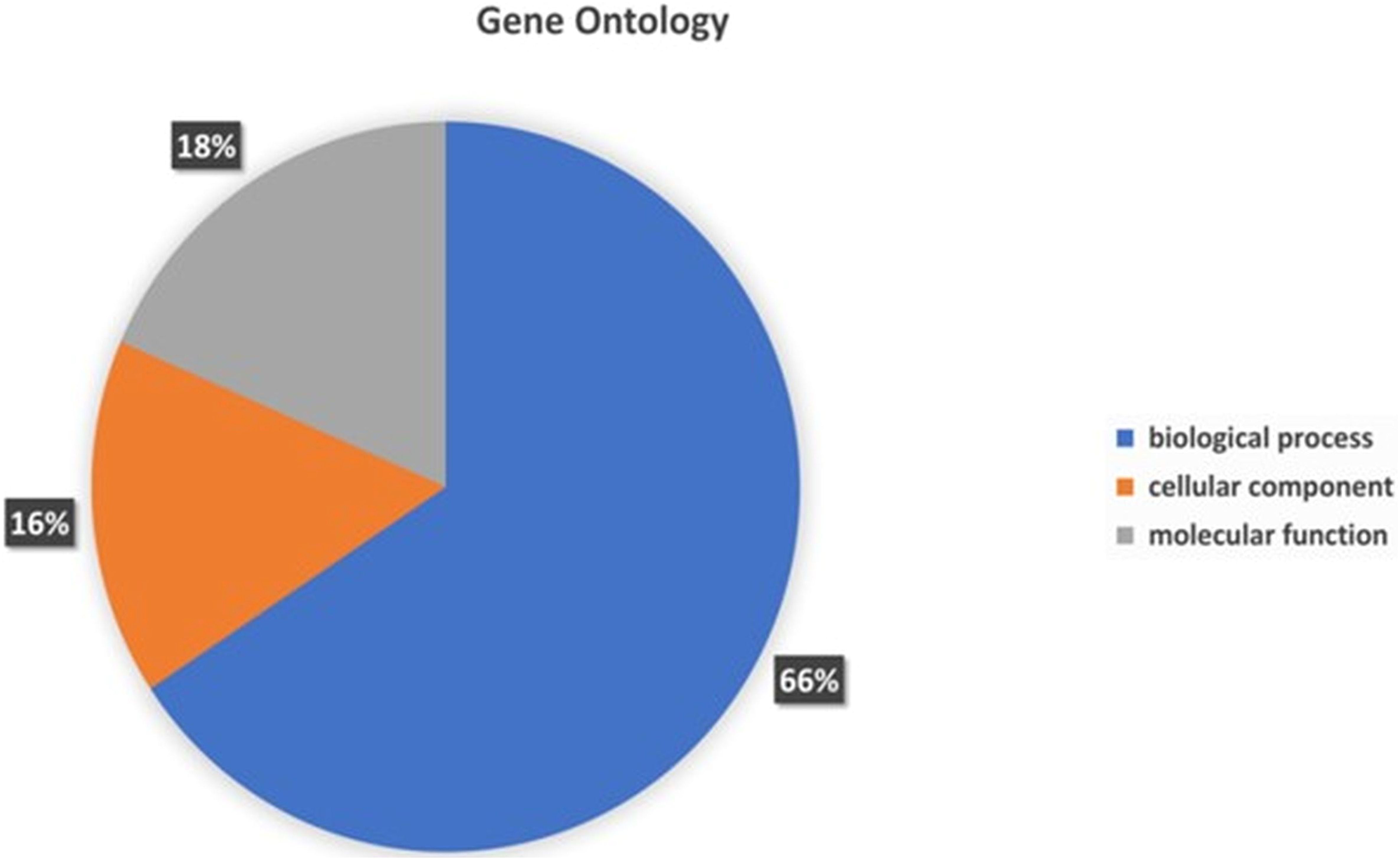
Figure 6. Gene Ontology functional annotations of the P. aeruginosa S-8 genome into 3 functional groups; biological processes, cellular component and molecular functions.
The COG database was used for the functional classification of predicted genes, whose distribution within the COG categories is provided in Figure 7. The COG prediction showed a higher number of genes (167), involved in amino acid transport and metabolism, followed by ribosomal structure and biogenesis (124), energy production (97), lipid transport and metabolism (61), coenzyme transport and metabolism (54) and nucleotide metabolism and metabolism (52). A total of 143 genes were identified with unknown functions. KEGG analysis identified the genes belonging to the various metabolic pathways (Figure 8). The highest number 383 was recorded for different metabolic pathways, 250 to the biosynthesis of secondary metabolites, 220 to the biosynthesis of amino acids, 170 to amino acid metabolism, 150 to nucleic acid metabolism, 110 to degradation of aromatic compounds, and 50 in the ribosome biosynthesis.
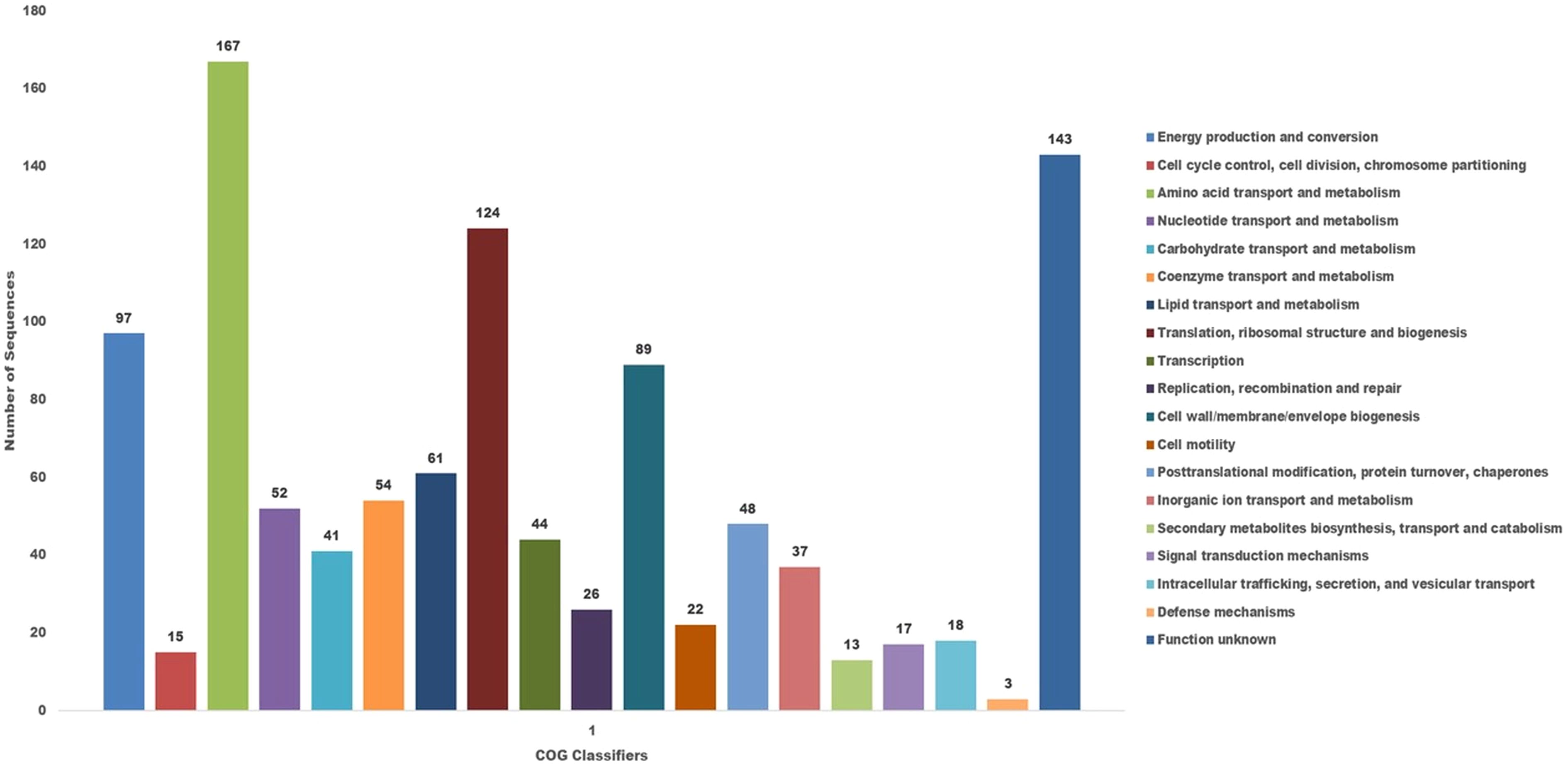
Figure 7. The cluster of orthologus analysis in P. aeruginosa S-8 using the COGs database based on orthologous groups. The identified genes in S-8 were divided into several functional subcategories based on the COG annotation (http://www.ncbi.nlm.nih.gov/COG/).
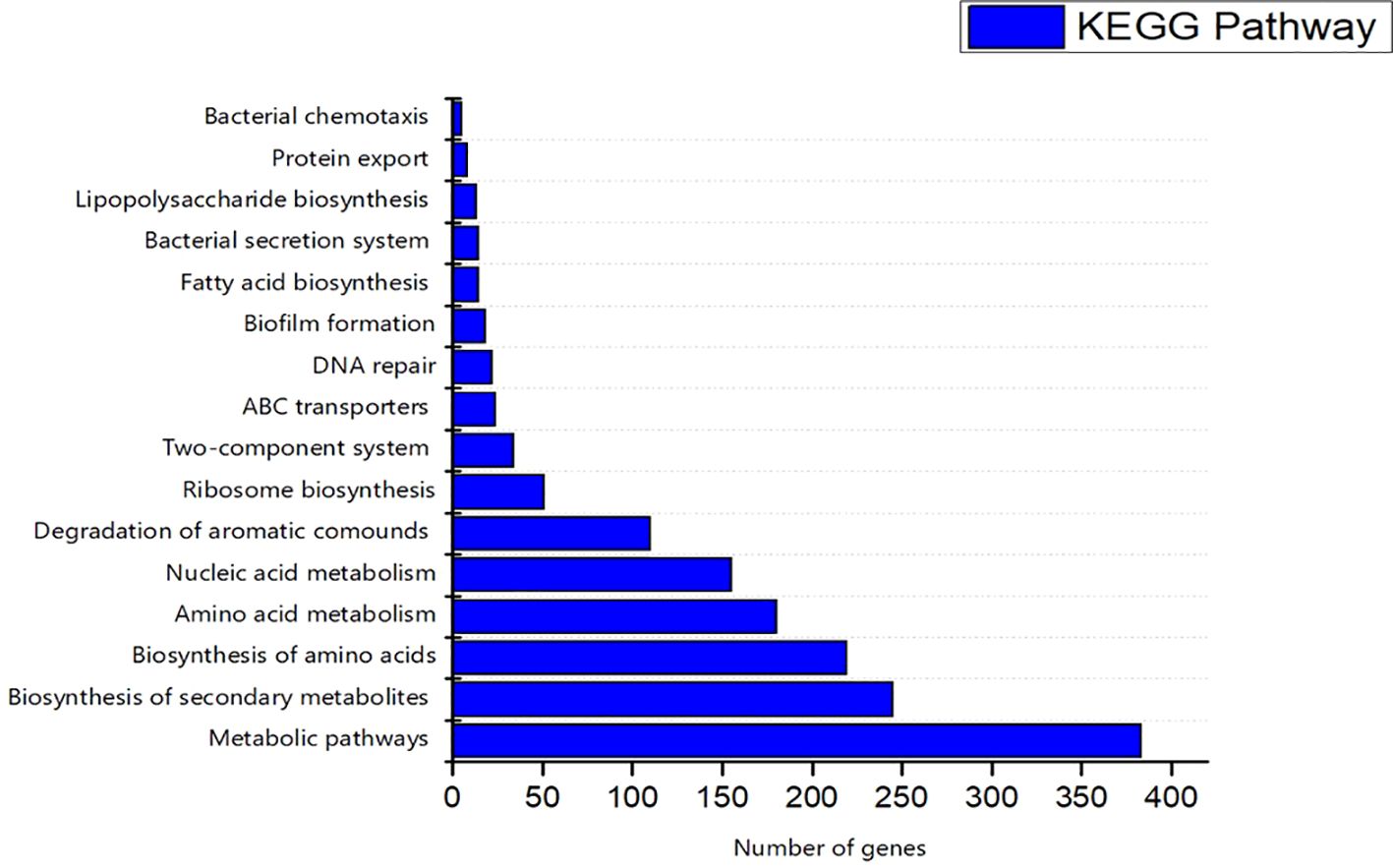
Figure 8. Kyoto Encyclopedia of Genes and Genomes (KEGG) were utilized for the retrieval of top ten (10) metabolic pathways of S-8 genes. KAAS server at KEGG database used for genes functional annotation by BLAST comparison with the manually curated database of KEGG- GENES.
Average nucleotide identity
To re-evaluate the phylogenetic relationship of S-8 within the Pseudomonas genus, ANI percentage of the S-8 genome was calculated with respect to other sequenced P. aeruginosa strains (Supplementary Table 1). We observed that our genome shows maximum genetic similarity with CP008869.2 (P. aeruginosa strain W16407), NC_023149.1 (P. aeruginosa SCV20265), CP008872.2 (P. aeruginosa strain X78812), and CP013113.1 (P. aeruginosa strain PAER4_119) with more than 99.2% similarity (Supplementary Figure 3). Similarly, the fastANI also showed the similar trend observed in our primary clustering dendrogram (Supplementary Figure 4).
AMR and VFDB analysis
CARD analysis identified the different classes of AMRs which have been summarized in Supplementary File S2. A number of resistance genes harbored within the genomes of the S-8 strain were identified and gene families associated to the resistance-nodulation-cell division (RND) antibiotic efflux pump, major facilitator superfamily (MFS) antibiotic efflux pump, beta-lactamase, ATP-binding cassette (ABC) antibiotic efflux pump, multidrug and toxic compound extrusion (MATE) transporter, and pmr phosphoethanolamine transferase were identified, which contributes to antibiotic resistance (Supplementary File S2). VFDB analysis identified the genes related to adherence, type IV pili and twitching motility genes, iron uptake related system related to achromobactin, pyoverdine, yersiniabactin biosynthesis, quorum sensing, and GacS/GacA two-component system. Enzymes related to hemolytic phospholipase, biosurfactant, and protease production were noted in S-8 genome. Moreover, genes related to Type VI secretion system (T6SS) like clpV, hcp, icmF, and vgrG were also annotated (Supplementary File S3). A comparison of virulence genes in the selected Pseudomonas genome was performed which showed that S-8 has the minimum virulence genes in all categories including adherence, antimicrobial activity, anti-phagocytosis, secretion system, protease activity, and toxin production, etc (Figure 9).
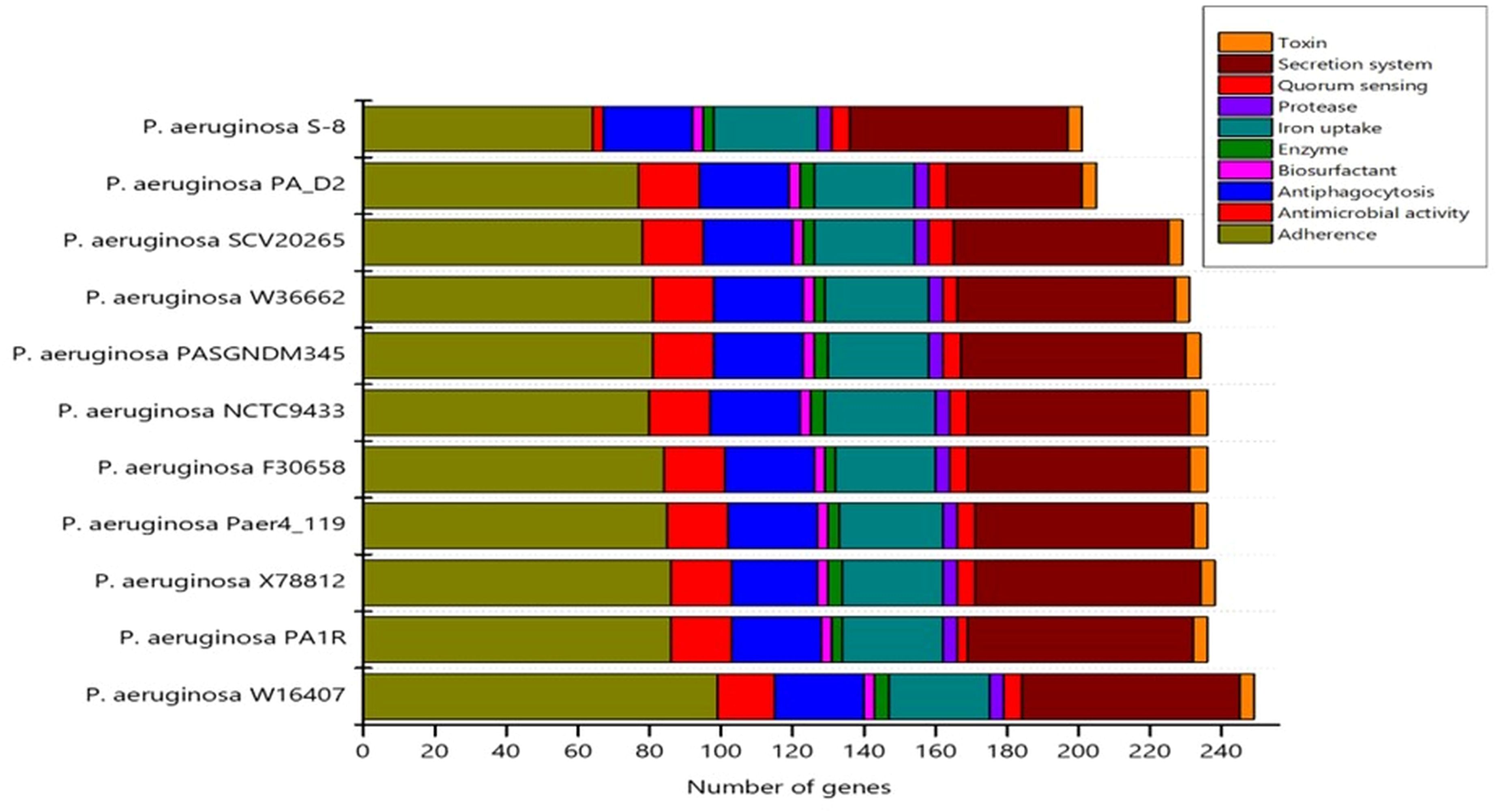
Figure 9. The distribution of virulence genes in the selected genomes was determined by virulence factor data base (VFDB) search tool.
BGCs analysis
In order to mine the secondary metabolite pathways, the various BGCs in the strain S-8 genome were annotated using the antiSMASH database, and revealed thirteen different secondary metabolites regions in the genome (Figure 10). In terms of biosynthetic paradigms, BGCs were comprised of non-ribosomal polypeptides (NRPS) like betalactone cluster, thiopeptide, hserlactone, redox-cofactor, and NRP- metallophore. The four NRPS clusters found in S-8 showed 100% similarity, to 2-amino-4-methoxy-trans-3-butenoic acid, pyoluterin, azetidomonamide, and pseudopaline, respectively (Table 2).
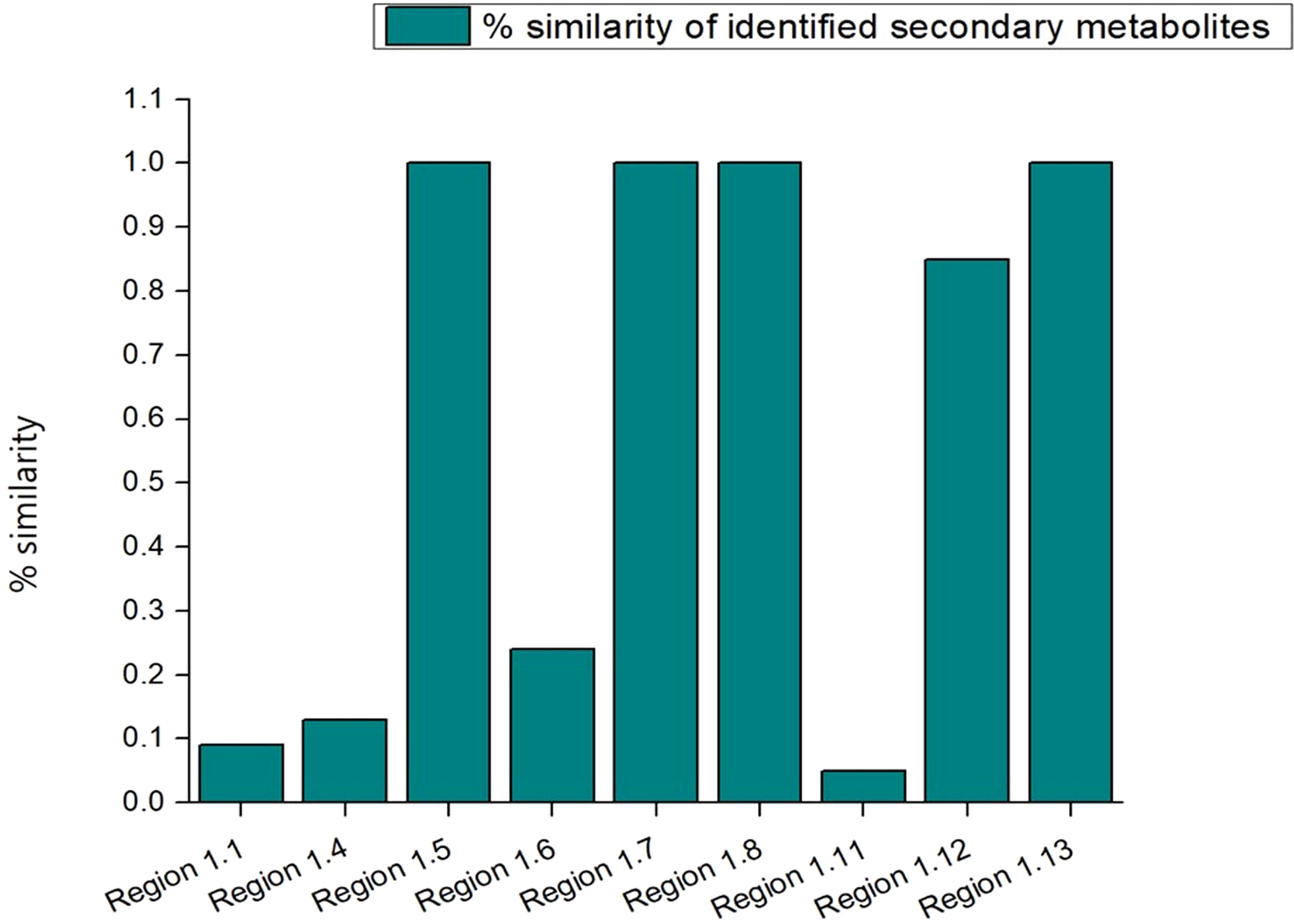
Figure 10. A total of 13 BGCs detected by antiSMASH in P. aeruginosa S-8 genome. The identified BGCs were belonging to different category like NRPS- (non-ribosomal peptides synthase) like betalactone, thiopeptide, NRP-metallophore, and PKs (polyketides synthase).

Table 2. Annotated BGCs in the P. aeruginosa S-8 genome.
CAZymes analysis
To investigate the industrial-relevant enzymes involved in the breakdown of complex carbohydrates, the S-8 genome was analyzed by the dbCAN2 server. As a result, 82 CAZymes genes were identified in the S-8 genome, which was classified into glycoside hydrolases (GHs), glycosyltransferases (GTs), carbohydrate-binding molecules (CBMs), carbohydrate esterases (CEs), and auxillary activities (AAs). Of these, the most abundant CAZymes were GHs and GTs with 30 and 29 genes, respectively, followed by AAs (10 genes), CE (6 genes), CBMs (4 genes), and PLs (3 genes) (Figure 11). The different groups of CAZymes were also compared to other ten genomes of P. aeruginosa strains (Supplementary Table 1) and a comparison of different CAZymes has been demonstrated in Figure 11. Among the compared genome, a higher number of CAZymes was observed for P. aeruginosa PA_D2 and P. aeruginosa PASGNDM345, followed by P. aeruginosa PA1R. The lowest diversity of CAZymes was noted for P. aeruginosa NCTC9433.
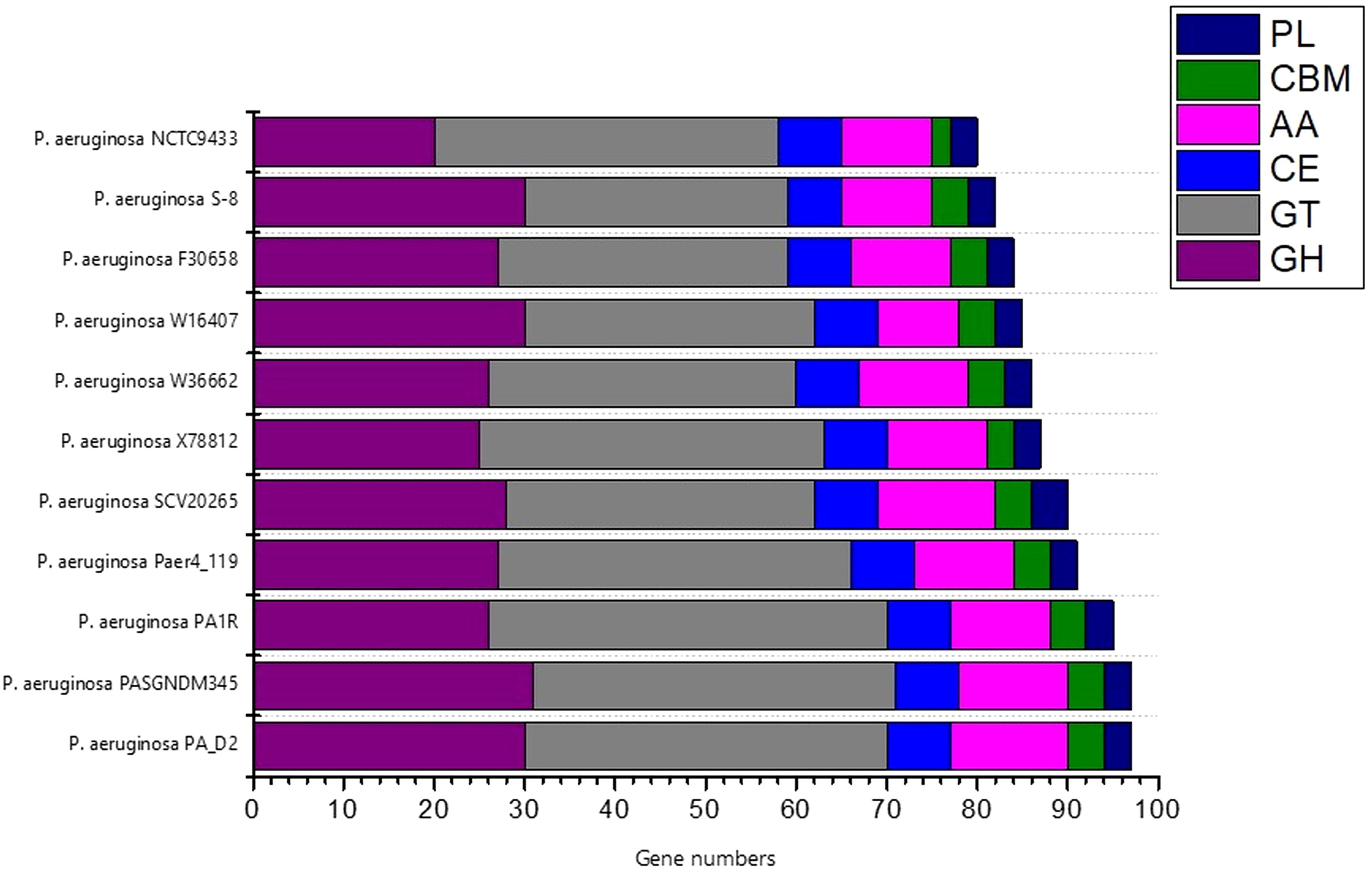
Figure 11. The distribution of various CAZymes like carbohydrate-binding modules (CBMs), glycoside hydrolases (GHs), glycosyl transferases (GTs), polysaccharide lyases (PLs), carbohydrate esterases (CEs), and auxiliary activities (AAs) in various P. aeruginosa species.
Comparative genome analysis
The protein coding gene comparison was performed between S-8 and the other closely related strains. The Venn diagram and the bar plot (Figure 12) showed that the numbers of core ortholog clusters shared by all the six species were 5,293, that suggests their conservation in the lineage after speciation events. The cumulative number of ortholog clusters shared between any two genomes, including the OS-1 was 30. A total of 59 gene clusters were unique to only a single genome. These clusters are probably gene clusters within multiple genes or in-paralog clusters which suggest that a lineage-specific gene expansion has occurred in these gene families. Additionally, the bar plot below the Venn diagram showed that the number of ortholog clusters for each species varied; P. aeruginosa S-8 (5,880), P. aeruginosa NCTC9433 (5,756), P. aeruginosa SCV20265 (6,090), P. aeruginosa PA_D2 (5,858), P. aeruginosa W36662 (5,990), and P. aeruginosa Paer4_119 (5,944). The formation of gene clusters was the first pattern, the second pattern shows the cluster counts and the third pattern represented in the form of stacker bar shows the total protein counts (Supplementary Figure 5A). The pairwise heatmap was performed for S-8 and other strains to highlight the overlapping number of gene clusters (Supplementary Figure 5B). A red color gradient showing the highest overlapping gene cluster thresholds was noted between P. aeruginosa S-8 and P. aeruginosa W16407 (Supplementary Figure 5B). A circular comparison performed by BRIG revealed the overall genome of P. aeruginosa S-8 has a high degree of sequence similarities (>99%) with other compared genomes (Figure 13).
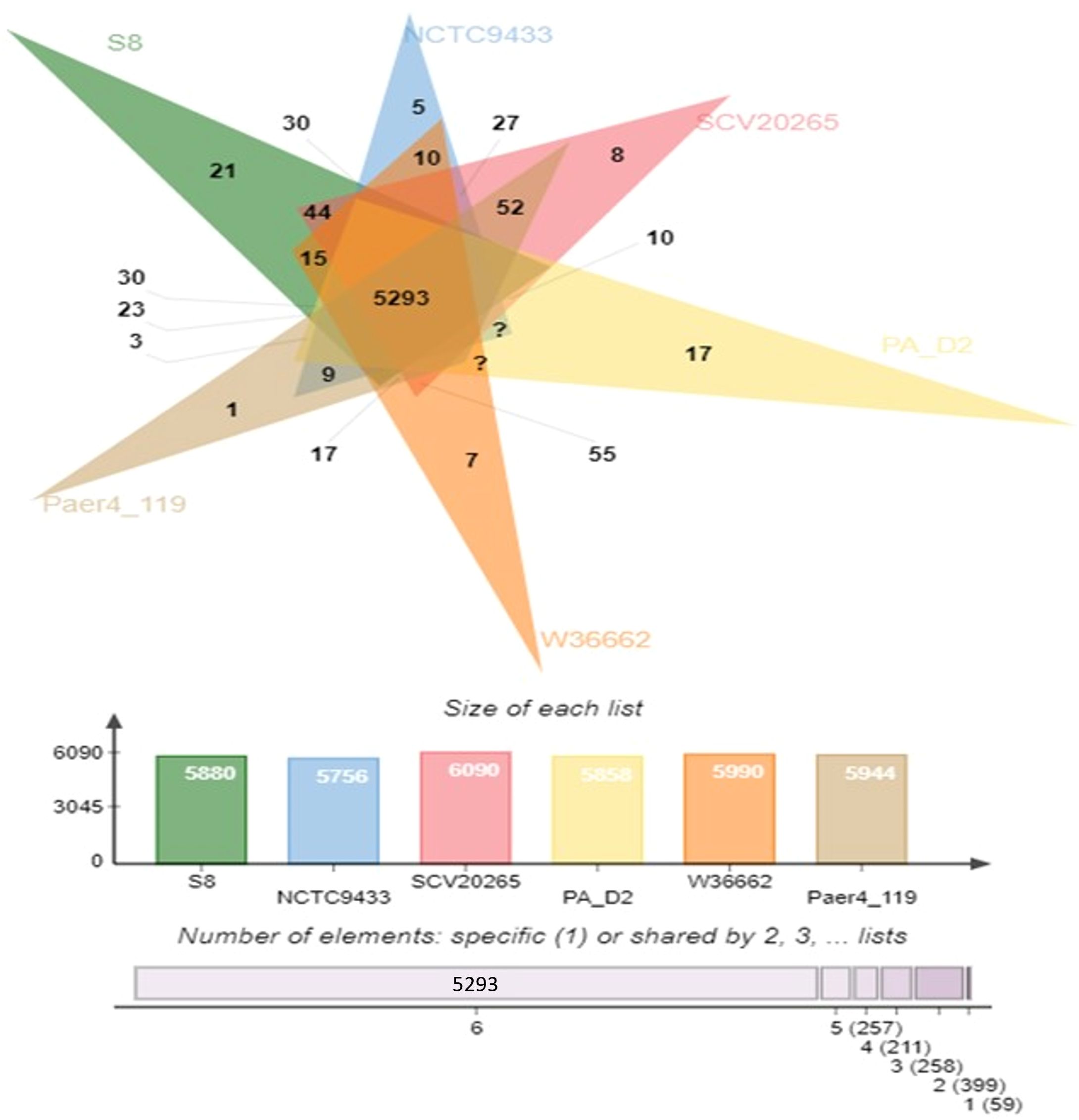
Figure 12. Venn diagram generated by Orthovenn2 represents the distribution of shared and unique gene clusters among selected Pseudomonas genomes. Compared to six Pseudomonas strains, the S-8 strain had twenty-one unique genes, however, a total of 5293 gene were shared among all six strains.
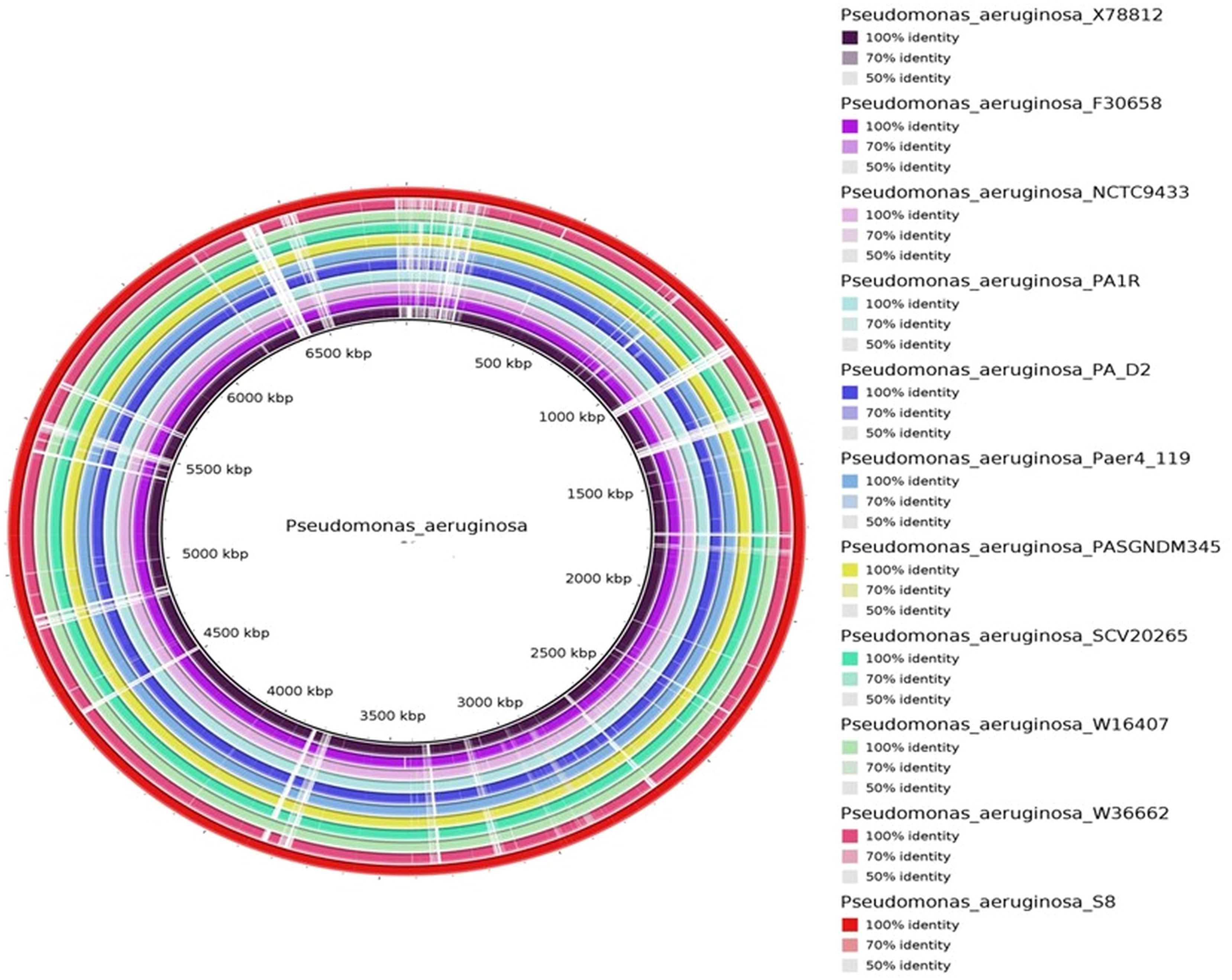
Figure 13. The circular genome comparison of assembled genome of S-8 was performed against the reference genome of P. aeruginosa X78812, P. aeruginosa F30658, P. aeruginosa NCTC9433, P. aeruginosa PA1R, P. aeruginosa PA_D2, P. aeruginosa Paer4_119, P. aeruginosa PASGNDM 345, P. aeruginosa SCV20265, P. aeruginosa W16407, and P. aeruginosa W3662 by using Blast Ring Image Generator (BRIG) (v 0.95) Tool.
Pan-genome analysis
The pan-genome and core-genome analysis was performed using the 10 genomes of the closest strains and the constructed tree showed two clusters based on presence/absence gene profiles (Figure 14). The resulting core genome SNPs-based phylogeny could be a good alternative for a better resolution than ANI-based analysis. For all 11 strains including P. aeruginosa S-8, the pan-genome contains 10,581 genes, of which the core genome represents 4,841 genes (approximately 40%), whereas the shell and cloud genome represents 2,201 (approximately 21%), and 3,539 genes (approximately 34%), respectively (Figures 14A-C). The core genome represented the largest part of the gene pools, this richness confirms the diversity and multiplicity of diverse traits. On average each strain contained 310 unique genes which correspond to approximately 3.0% of each genome. Box-plot analysis showed that among the selected strains, S-8 showed the less conserved genes (Supplementary Figure 6A), and a high number of unique genes (Supplementary Figure 6B).
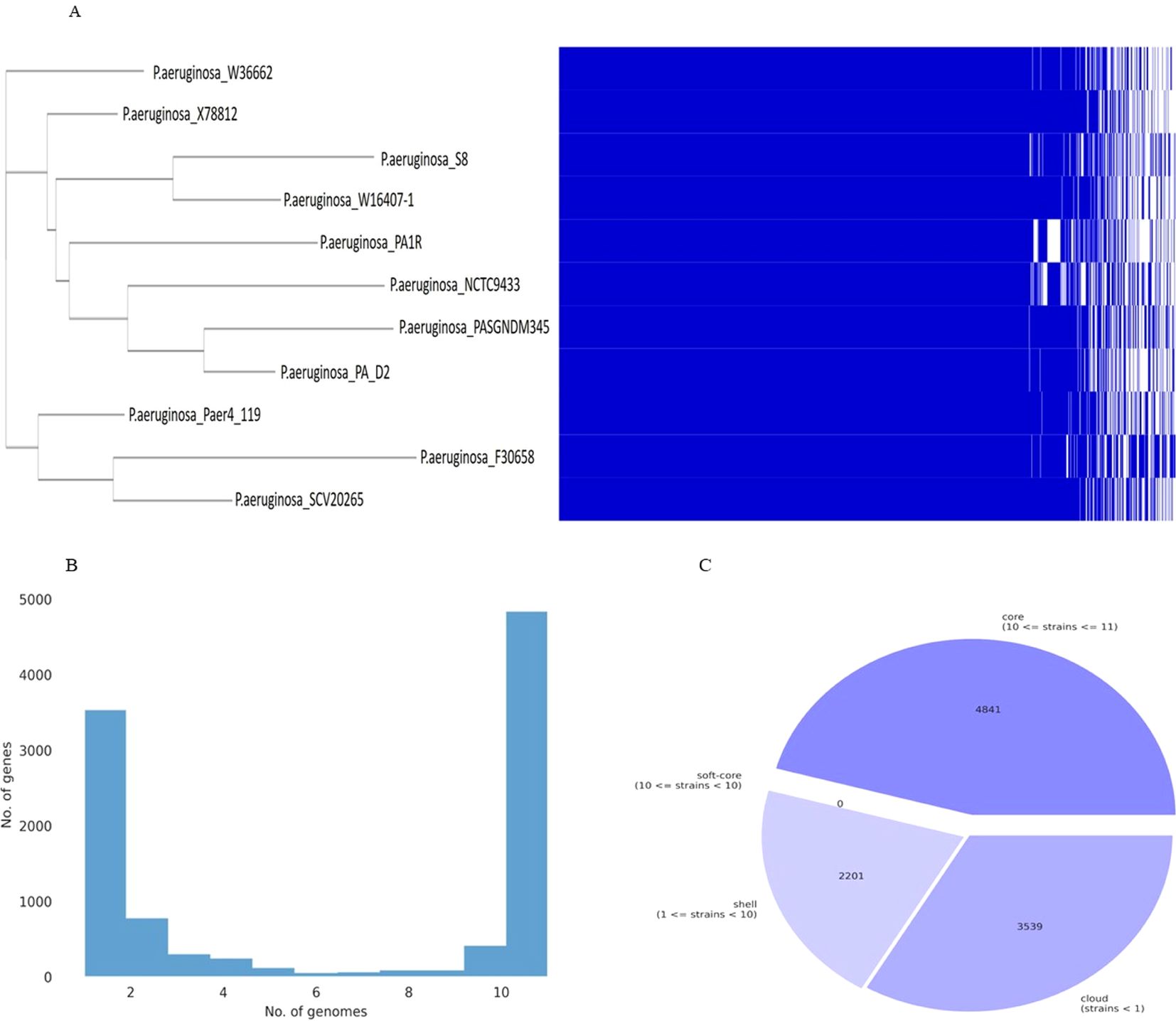
Figure 14. Graph of the pan-genome distribution of P. aeruginosa S-8 and other closely related genomes; (A) Gene distribution of P. aeruginosa strains based on the gene presence–absence matrix generated from Roary. A purple box, a green box, and an orange box to represent the core gene, accessory genes and specific genes, respectively; (B) The gene frequency plot within a whole genome set, demonstrating the distribution of genes per genome (C) Pie-chart representation of the gene distribution in the pan-genome with respect to presence of genes in proportion of strains out of ten strains selected.
Horizontal gene transfer analysis
HGTector2 analysis resulted in 911 HGTs form our S8 genome. Considering all genomes, we found a total of 10,547 genes. After identifying the horizontal gene transfers (HGTs) within the genome of interest, which in this case is S8, we conducted a KEGG pathway analysis specifically for these HGTs. The results showed that higher number (25%) was observed for the Quorum sensing and biofilm formation followed by phenazine biosynthesis (11%), amino acids and nucleotide metabolism (11%), and nucleotide sugar biosynthesis (11%) (Figure 15). The Parsnp based SNP tree showed a visual representation of the genetic clustering and divergence patterns among the analyzed strains of Pseudomonas. It represents a genome alignment approach to determine the precision of mapping, but with a reduced sensitivity. Analysis (Figure 16) showed that there was less variation between S-8 and P. aeruginosa W16407, whereas a high variation was observed between S-8 and P. aeruginosa PA1R.
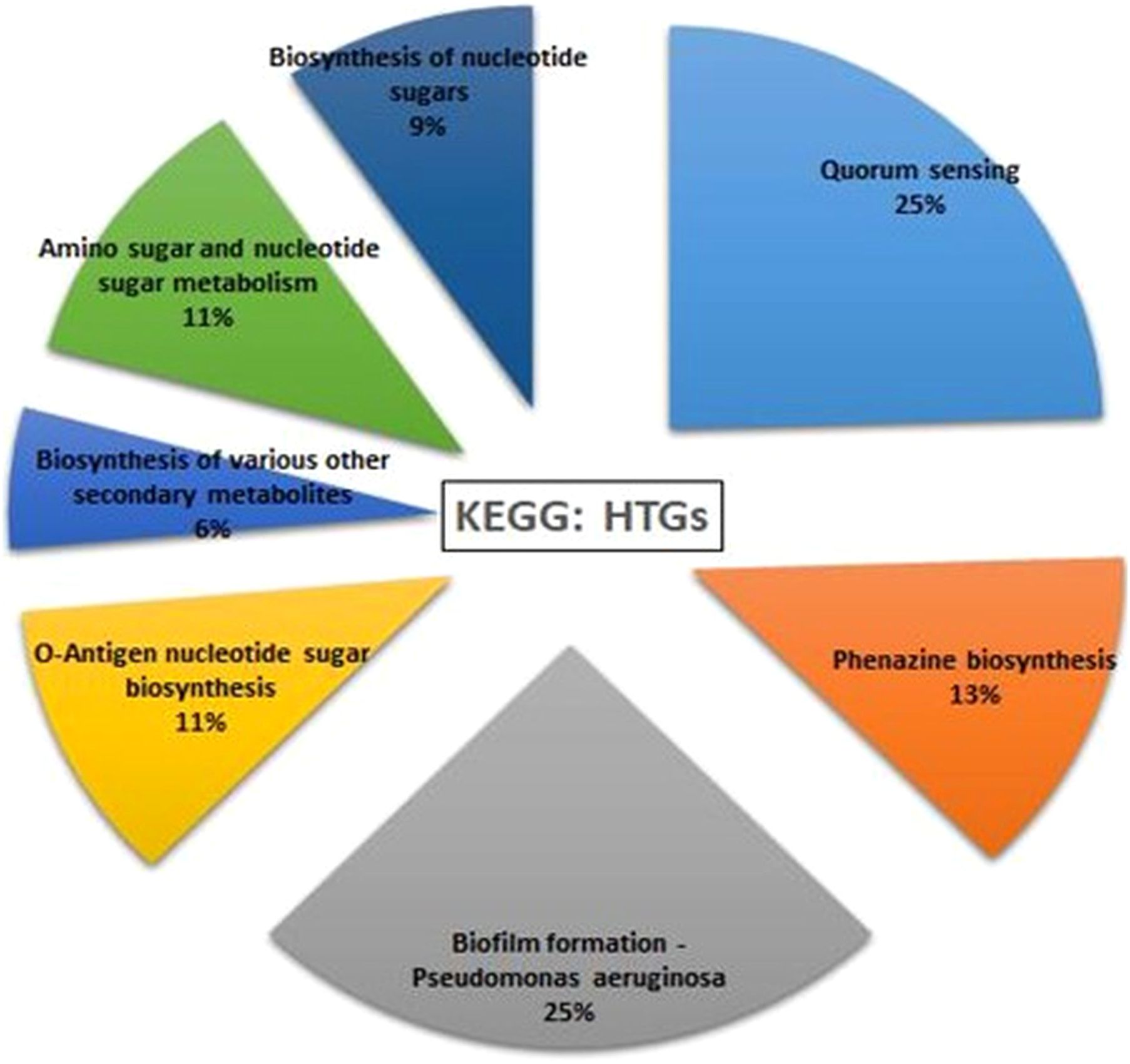
Figure 15. Horizontal gene transfer event was analyzed by HGTector2 which showed a total of 911 HGTs form our S8 genome. After identifying the horizontal gene transfers (HGTs) within the genome of interest, we conducted a KEGG pathway analysis specifically for these HGTs.
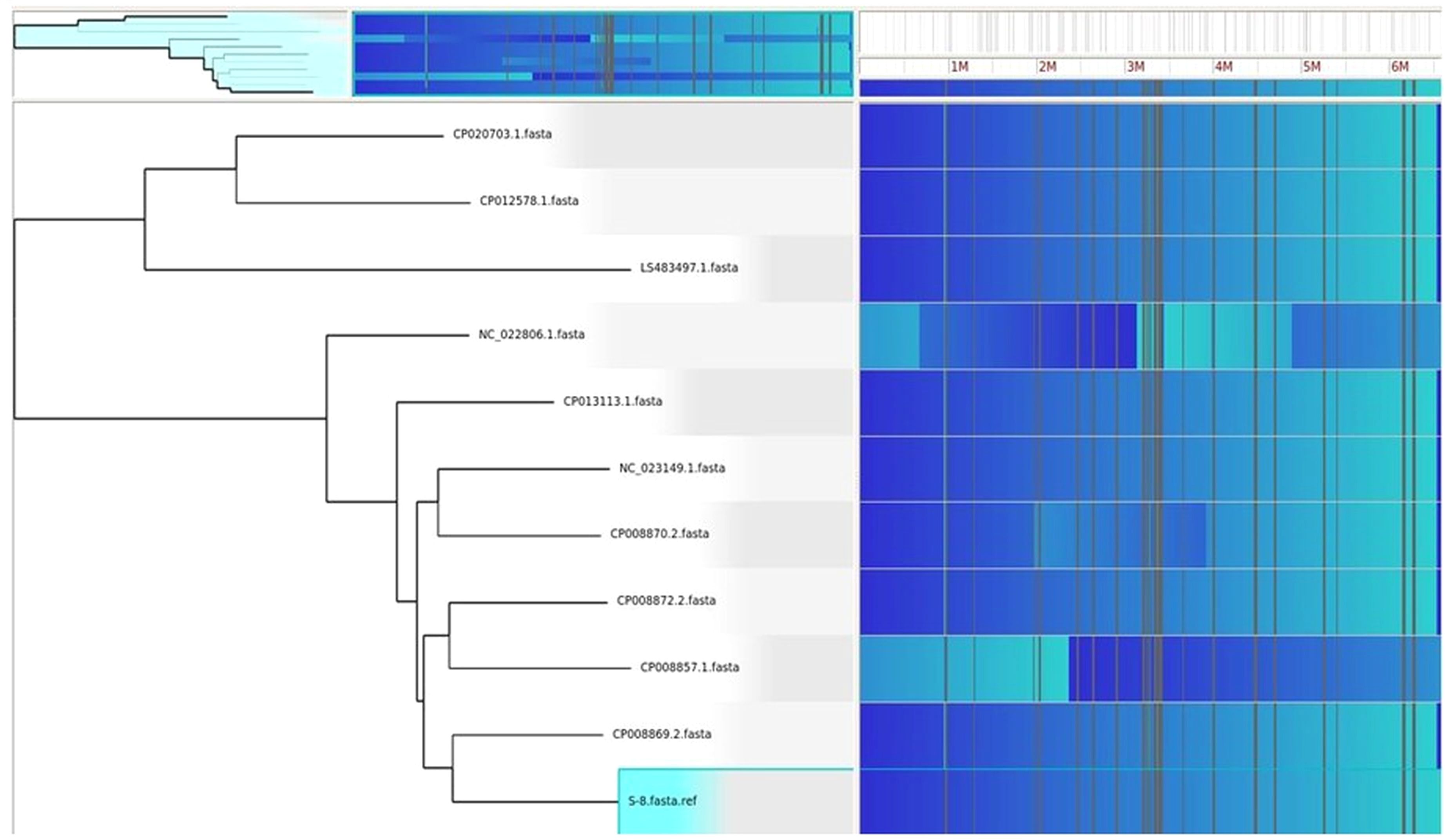
Figure 16. The Gingr generated tree based on parsnp analysis. The tree provides a visual representation of the genetic clustering and divergence patterns among the analyzed strains of Pseudomonas. The proximity of branches indicates a more recent common ancestry, while greater distances reflect greater genetic divergence.
Discussion
Utilizing the sequencing data of newly isolated strains, it is possible to make in silico-based predictions about the gene repository and virulence potential of a newly isolated strain P. aeruginosa S-8. This robust method decreases the need for a laborious trial, error-type wet lab experiments, and animal testing. In the present work, we investigated the in-depth genome, pan-genome, and comparative genome analysis of P. aeruginosa to assess the genomic differences and similarities between closely related strains. This type of study will provide insights into the adaption process of an environmental isolate to a particular environmental habitat. Moreover, the environmental isolates due to adaption to different habitats show more closeness among themselves than the clinical isolates (Sánchez et al., 2014). The genomes of several Pseudomonas-type strains have been deciphered, which has contributed to an improved understanding of the evolution and diversity of the Pseudomonas genus (Silby et al., 2011).
In particular, more P. aeruginosa isolates are being sequenced and showed the presence of various pathogenicity and virulence features (Klockgether et al., 2011; Marcelletti et al., 2011). Among the Pseudomonas genus, P. syringae comprise plant pathogens secreting effector’s proteins into plant cells by the type III secretion system (Lindeberg et al., 2012), whereas, several P. fluorescens are known for their biocontrol properties (Haas and Defago, 2005). In contrast, P. putida strains are mostly non-pathogenic and show robust metabolic capacities under stress conditions (Rojas et al., 2001). Flagella are used by bacteria for motility and many genes for flagella formation were annotated in the S-8 strain. The presence of flagella improves bacterial motility and consequently pathogenicity, and also provides the bacterium resistance to surfactant protein A (SP-A) (Zhang et al., 2007), a potent lung innate immune protein that kills microbial pathogens through opsonization (Tan et al., 2014a). Additionally, specific pili like type IV pili enhance the motility and antimicrobial resistance in P. aeruginosa (Tan et al., 2014b), and have been reported in severe cases of pneumonia, bacteremia, and increased mortality (Tang et al., 1995).
The KEGG analysis revealed that strain S-8 harbored the genes for the gluconeogenesis pathway, pentose phosphate pathway, purine and pyrimidine synthesis, fatty acid and peptidoglycan synthesis pathway. In terms of nitrogen metabolism, S-8 could take up ammonia using the ammonium family transporters. Besides sulfur metabolism, strain S-8 harbored the gene set for sulfate, phosphate, alaknesulfate and lipopolysaccharide, which enable S-8 to obtain and utilize nutrients such as nitrogen, carbon, phosphorus and sulfur from the environment to facilitate its survival in different environments. The genome analysis showed that S-8 could synthesize various amino acids including alanine, glycine, glutamate, serine, threonine, cysteine, valine, glutamine, proline, arginine, and phenylalanine. Moreover, genes for vitamin metabolism, peroxidase, superoxide dismutase, lipopolysaccharide biosynthesis, β-lactam resistance, and cationic antimicrobial peptide were also noted, which could enable the Pseudomonas strain S-8 to adapt to a complex and changeable environment.
Genome annotation identified the various metal resistance genes in S-8 genome. Genes coding for copper-translocating P-type ATPases (opA, copB, cusR, cusS, cueR) were identified which are a large family of transmembrane transporters and their role in ion homeostasis and tolerance of heavy-metal ions is well established. A previous study showed that P-type ATPases pumped out the metal ions from the cytoplasm to the periplasm. The cusRS is a two-component signal transduction system that activates the expression of cusCFBA operon and is involved in copper detoxification (Gilles-Gonzalez and Gonzalez, 2005). Copper-translocating P-type ATPases acts as an efflux pump in order to pump out excess Cu2+ from the cell (Ladomersky and Petris, 2015). Genes involved in lead resistance zntA and cadC, arsenic resistance arsC, arsR, genes involved in cobalt resistance czcD, and chromium resistance chrA were detected (Supplementary Table 2). Nickel-responsive regulator protein nikR and copper chaperone protein copZ were noted which belong to drug/metabolite transporter superfamily, and are presumed to expel the toxic metabolites out of the cell (Gudhka et al., 2015). P. aeruginosa S-8 was motile and we identified chemotaxis regulator proteins encoded by CheA, CheB, CheB1, CheR, CheV, CheY, CheW, and CheZ gene. Additionally, chemotaxis-associated proteins encoded by McpC, McpH, McpP, McpQ, PctA, PctB and CtpH genes along with flagellar proteins (FlhABF, FliCDEFGOP, MotAB) were also identified. Genes involved in the regulation of Fe uptake e.g. Fur and PchhR were detected in the S-8 genome (Supplementary Table 3).
S-8 genome also showed the presence of osmotic stress genes e.g. mdoB (Phosphoglycerol transferase I), mdoH (Glucans biosynthesis glucosyltransferase H), mdoG (Glucans biosynthesis protein G precursor), and mdoD (Glucans biosynthesis protein D precursor). Production of cold-shock and heat-shock proteins by microorganisms can help their survival in harsh environments and facilitate environmental adaption. The S-8 genome carries the various heat-shock and the cold-shock protein (Supplementary Table 4). Moreover, the S-8 genome encodes numerous proteins to protect the cell from oxidative stress including five peroxidases, three catalases, four superoxide dismutases, two hydroperoxide reductases and 11 glutathione S-transferases (GST). GST is a detoxification enzyme involved in cell protection against stress-induced reactive oxygen species (Oakley, 2011). Furthermore, many dehydrogenases/oxidoreductases were found in the S-8 genomes which play an important role as an oxidative protection in bacteria in response to heavy metals (Kaur et al., 2006; Williams et al., 2007). S-8 genome also encoded ions scavenging systems, such as thioredoxin (1 gene), thioredoxin reductase (2 genes), ferredoxin (3 genes), and glutaredoxin (2 gene) which act as defense proteins to protect microorganisms from oxidative damage (Gorriti et al., 2014).
AntiSMASH informatics predicted that S-8 genome has many secondary metabolite synthesis gene clusters with gene cluster types such as NRPS, PKS, T3PKs, and was able to predict pyoverdine, lankacidin, pyochelin, and pseudopaline etc. These gene clusters may all be involved in the antimicrobial process. It has been shown in the literature that pyoverdine, as a broad-spectrum antibacterial substance present in many Pseudomonas spp., exhibits bactericidal activity mainly by destabilizing the phospholipid membrane of the target pathogen, leading to cell lysis (Liu et al., 2021). The biosynthetic gene clusters of pyochelin has demonstrated other biological activity recently other than being only a chelating compound. This compound can particularly inhibit bacterial pathogens in a study conducted by Adler et al. (2012) and Ong et al. (2016). In this study, we found that S-8 contains a cluster of biosynthetic genes for many of the above-mentioned inhibitory substances based on genome sequencing, indicating that the bacterium not only has broad inhibitory activity against bacteria but also has the potential to inhibit pathogenic fungi.
Secondary metabolites are organic molecules that have diverse and powerful biological functions which facilitate the bacterial strain to adapt to the environment (Abegaz and Kinfe, 2019). The respective genes involved in the synthesis of these secondary metabolites are often clustered into biosynthetic gene clusters (Medema et al., 2015). In S-8 genome, we identified several gene clusters for secondary metabolites production which act as defensive molecules for the organism producing them (James, 2017). Non-ribosomal peptides (NRPs) are synthesized through enzyme-mediated condensation of amino acid residues where > 300 different precursor molecules help in the assemblage of NRPs (Saxena et al., 2020), and fall into the class of secondary metabolites with diverse properties as toxins, siderophores, antibiotics, immune-suppressants, and anticancer agents (Wang et al., 2014; Martínez-Núñez and López, 2016). All these BGCs comprised approximately 14.6% of the genome. Various gene clusters associated to NRPSs toxic for prokaryotes and eukaryotes were identified in S-8 genome (Rojas Murcia et al., 2015). Some of the NRPSs clusters showed similarity to the pyoverdine cluster with less than 100% similarity. Pyoverdine is a common siderophore found within P. aeruginosa species and could represent a novel drug or vaccine target (Fothergill et al., 2012).
Region 1.5 represented the clusters of gene for the biosynthesis of bicarbonate transport ATP-binding protein CmpD, nitrate import ATP-binding protein NrtD, cysteine/O-acetylserine efflux protein, purine ribonucleoside efflux pump NepI, alpha-ketoglutarate-dependent taurine dioxygenase, gamma-glutamyl putrescine oxidoreductase, and FMNH2-dependent monooxygenase SfnC (Supplementary Table 6). The nitrate import ATP-binding protein facilitates the nitrate uptake, nitrite transport, and responsible for energy coupling to the transport system (Watzer et al., 2019). The bicarbonate transport ATP-binding protein is involved in the specific and high affinity binding of nitrate and nitrite, and shows structural similarities to integral membrane subunits of ABC transporters (Poschenrieder et al., 2018).
The seventh cluster (Region 1.7) contained genes for the various transporters belonging to antibiotic efflux pump outer membrane protein (ArpC), multidrug ABC transporter permease (YbhR, YbhS, YbhF), gramicidin dehydrogenase (LgrE), vitamin B12 transporter (BtuB), L-lactate transporter, and helix-turn-helix (HTH)-type transcriptional regulator (MalT, YofA, DmlR) (Supplementary Table 7). The antibiotic efflux pump outer membrane protein form trimeric channels that facilitate the export of a variety of substrates in gram-negative bacteria, whereas, ABC transporter complex YbhFSR could be involved in efflux of cefoperazone (Feng et al., 2020). Vitamin B12 (or cobalamin) is an enzymatic cofactor essential for bacteria, however, it can be synthesized only by few microorganisms, so most bacteria need to take it up from the environment through the TonB-dependent transport system. Its import to bacterial cells occurs through the outer-membrane receptor called BtuB which forms a β-barrel with inner luminal domain and extracellular loops. Vitamin B12 binds with high affinity to the extracellular side of the BtuB protein (Pieńko and Trylska, 2021). The HTH family of transcription regulators is involved in the development of antibiotic resistance (Eckstein et al., 2021). Another enzymes, methane and alkanesulfonate monooxygenase belongs to the family of oxidoreductases and catalyzes the chemical reaction with O2 as oxidant and incorporation or reduction of oxygen (Liew et al., 2021).
The 8th cluster (Region 1.8) contained various genes for phosphoethanolamine transferase (EptA), phosphate-import ATP-binding protein (PhnC), acyl carrier protein, inner membrane transport protein (YdhP), ABC transporter phosphite binding protein (PhnD1), and transcriptional regulator (SlyA) (Supplementary Table 8). The phosphoethanolamine transferase (EptA) is an intramembrane enzyme that modifies the lipid-A portion of lipopolysaccharide (LPS) or lipooligosaccharide (LOS) by the addition of phosphoethanolamine, which resulted in reduction of overall net-negative charge of the outer membrane of some gram-negative bacteria, conferring resistance to polymyxin. This resistance mechanism has resulted in a global public health issue due to the increased use of polymyxin as last-resort antibiotic treatments against multi-drug-resistant pathogens (Samantha and Vrielink, 2020). The phosphate-import ATP-binding protein is involved in phosphonates, phosphate esters, phosphite and phosphate import, responsible for energy coupling to the transport system (Junhong et al., 2023). Another enzyme phosphate-import ATP-binding protein belongs to a larger family that includes phosphate, phosphite, and phosphonate transporters, which binds strongly to inorganic phosphate (Feingersch et al., 2012). The transcriptional regulators SlyA are often involved in the regulation of genes important for bacterial virulence and stress response. However, the slyA deletion mutant (ΔslyA) of Enterococcus faecalis showed more virulence in an insect infection model (Galleria mellonella), exhibited increased persistence in mouse kidneys and liver, and survives better inside peritoneal macrophages (Michaux et al., 2011).
The 12th cluster (Region 1.12) genes included efflux pump periplasmic linker (BepF) and membrane transporter (BepE), toluene efflux pump outer membrane protein (Ttg), Iron import ATP-binding/permease protein (IrtB & IrtA), D-alanine–D-alanyl carrier protein ligase, and regulatory protein (PchR) (Supplementary Table 9). Additionally, this region also possessed thioesterase (PikA5), isochorismate pyruvate lyase, salicylate biosynthesis isochorismate synthase, Inner membrane transport protein (YajR) and UvrABC system protein A. The UvrABC play critical role in DNA repair by nucleotide excision repair, replacing these aberrant nucleotides, involves the removal of twelve nucleotides where a genetic mutation has occurred followed by a DNA polymerase, and completing the DNA repair (Kraithong et al., 2021). Furthermore, this cluster possessed phenazine-1-carboxylate N-methyltransferase, which is involved in the biosynthesis of pyocyanin, a toxin produced and secreted by the P. aeruginosa, and plays a role in virulence (Mavrodi et al., 2001).
The 13th cluster (Region 1.13) genes included pseudopaline exporter and synthase, metal-pseudopaline receptor CntO, biosynthetic arginine decarboxylase and putative Nudix hydrolase YfcD (Supplementary Table 10). The pseudopaline genes are involved in biosynthesis of metallophores, their export in the extracellular medium, and the recovery of a metal-metallophore complex under metal scarce conditions (Lhospice et al., 2017). The Nudix hydrolase contribute to cellular ‘housekeeping’ through the breakdown of a wide range of nucleoside diphosphate derivatives (Tong et al., 2009).
A number of secretion systems were identified in S-8 genome such as type II secretion systems (T2SS) that secrete the folded proteins such as pseudolysin (lasB), phospholipase C (PlcH), or lipase (LipA) from the periplasm to extracellular milieu (Costa et al., 2015). The presence of type III secretion system (T3SS) allows the translocation of bacterial effectors proteins into the host cell (de Bentzmann and Plesiat, 2011; Girlich et al., 2004), and was associated with bacterial persistence in the lungs and increased mortality in patients suffering from acute respiratory infections (Anantharajah, 2017). Type VI (T6SS) plays crucial role in bacterial competition and pathogenesis (Wood et al., 2019). The presence of these diverse secretion systems also facilitates the survival of S-8 under different environment niche.
The VFDB and CARD based analysis revealed various genes associated with antibiotic resistance and virulence activity of strain S-8. Notably, the presence of gene encoded β-lactamases enzyme blaPDC-142 and blaPME-1 is consistent with carbapenem resistance. Additionally, the following cephalosporin-resistant genes, blaPDC-2, blaPDC-7, and blaPDC-9 was observed and one each for aminoglycoside (aph(3′)-IIb) fosfomycin (fosA) and chloramphenicol (catB7) resistance genes was present. The gene arnA modifies the lipid A with 4-amino-4-deoxy-l-arabinose (Ara4N), which allows Gram-negative bacteria to resist antimicrobial peptides and antibiotics such as polymyxin. A previous study (Udaondo et al., 2016) reported that P. putida strains that share 85% of the coding regions with P. aeruginosa bear the various genes encoded for transporters, enzymes and regulators for amino acid metabolism and reveals their environmental applications by the capacity to degrade pollutants and ability to promote plant growth. This research represents an effort to find the mechanisms underlying the ecology, pathogenicity and evolution history of Pseudomonas spp. that able to develop biotechnological advances. Cho et al., 2015 reported P. fluorescens PCL1751 genome and comparative genome study that reveals the integration of prophages that play an important role in genome rearrangements and this strain achieves biological control of pathogens through effective competition for nutrients including niches.
Exposure to metal stressors significantly inhibits the respiratory enzymatic activities as well as corresponding transcript level (Samanta et al., 2020). In S-8 genome, a large inventory of CAZymes was noted including sugar metabolizing enzymes, which confers respiratory metabolism under metal stressors, facilitating the high energy metabolism activity. P. aeruginosa shows a large degree of genomic heterogeneity both through variation in sequences found throughout the species (core genome) and through the presence or absence of sequences in different isolates (accessory genome) (Pincus et al., 2020). P. aeruginosa isolates also differ markedly in their ability to cause disease. In this study, we investigated the genomic features of a newly isolated environmental strain P. aeruginosa S-8. We showed that core genome, alone or in combination with the accessory genome are also predictive of virulence. Another interesting genomic trait, the presence of several GIs was observed in S-8 genome. In bacteria, these GIs are decorated with additional genes acquired via horizontal gene transfer (HGT) mechanism. The presence of these genes may render additional metabolic functions including adaptive traits and genome plasticity which may facilitate evolutionary survival (Pérez-Pantoja et al., 2013). Recent genomic investigation has revealed GIs encoded functional traits which are classified into PAIs (pathogenicity islands) encoding virulence genes, MIs (metabolic islands) encoding biosynthesis of secondary metabolites, RIs (resistance islands) encoding resistance genes to antibiotics, and SIs (symbiotic islands) encoding genes for symbiotic association of the host to microorganism. The observed results suggest a strong possibility that in S-8 strain GIs were recruited via HGT to facilitate the survival in different environmental conditions.
The microbial genome variation is essential for understanding microbial functions, microbe-host interactions, and to understand the effects of genetic variation on function or phenotype. The small genomic differences may influence the phenotype provides evidence about the functional consequence of sequence variation. Over decades, microbiologists have identified the function of numerous genes across multiple species mainly through investigating the effect of gene loss. The diversity of functional genetic features may greatly exceed the taxonomic diversity due to horizontal gene transfer (HGT) and rapid evolutionary adaptation (Wiedenbeck and Cohan, 2011). The BGCs clusters of S-8 strains showed both strain-specific and unidentified characteristics, which support the idea that bacteria perform metabolic activity exclusively for survival in a particular ecological environment and potentially construct alternative routes for new bioactive metabolite production. The test isolate S-8 showed a higher number of virulence genes as compared to other tested genomes, therefore, genome analysis of S-8 reveals the number of potential features that might be considered candidates for future studies to explore the virulence mechanism deployed by this bacterium. P. aeruginosa enriched with conserved core genome of low sequence diversity and variable genome components that communicate with other Pseudomonas by horizontal gene transfer (Klockgether et al., 2011).
Conclusion
In summary, several genomic features of P. aeruginosa S-8 were identified based on the whole-genome analysis. Phylogenetic based analysis showed that strain S-8 has a high similarity to P. aeruginosa strains. Comparative genomic analysis revealed that S-8 possesses genomic islands, prophages, etc. We identified some putative virulence factors and future studies should expand the number of isolates, so as to increase the confidence of results generated in the present study. The ability of the genome to predict antibiotic resistance genes opens the door for sequencing of new strains and it will further supplement or replace the traditional antimicrobial susceptibility testing. However, future studies are needed to provide detailed understandings of the role that genetic variation plays the ability of P. aeruginosa to cause disease by using suitable model system. By being aware of the potentially high virulence of the organisms, personal safety measurements can be increased to avoid an accidental exposition of the organism. The presence of various CAZymes illustrate its importance in industry regarding complex polysaccharide degradation and further energy production. Using newly sequenced data and their investigation can help to substantially speed up research in the future and to draw wider, more general conclusions.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
KK: Data curation, Investigation, Software, Writing – original draft. AS: Data curation, Formal analysis, Software, Writing – review & editing. PS: Data curation, Formal Analysis, Software, Investigation, Methodology, Visualization, Writing – review & editing. RS: Data curation, Formal analysis, Software, Writing – review & editing, Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The work was supported by the Ramalingswami Re-entry Fellowship, provided by Department of Biotechnology, Government of India.
Acknowledgments
The author acknowledges the Dept. of Bioengineering and Biotechnology, BIT Mesra for providing the infrastructure.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2025.1511507/full#supplementary-material
Supplementary Figure 1 | Phylogenetic analysis based on 16S rRNA confirmed that S-8 belongs to P. aeruginosa. The 16s rRNA gene was amplified and sequenced. The obtained sequence was aligned using CLUSTAL-X and tree was constructed using neighbor-joining (NJ) method with bootstraps of 1000 replicates.
Supplementary Figure 2 | Antagonistic activity of P. aeruginosa S-8 against bacterial pathogen E. coli (A), S. typhi (B), and fungal strain M. gypsium.
Supplementary Figure 3 | To re-evaluate the phylogenetic relationship of S-8, ANI analysis was calculated with respect to other sequenced P. aeruginosa strains.
Supplementary Figure 4 | The fastANI analyses confirmed the closest similarity of S-8 to P. aeruginosa F30658 and P. aeruginosa W16407.
Supplementary Figure 5 | (A) OrthoVenn diagram showing the number of common and separate protein clusters for S-8 and other closely related genomes, the occurrence table contains groups of gene clusters like cluster count and protein count. Row indicates the orthologous gene cluster for multiple species that summarized as a cell graph and column indicates different closely related bacterial species, (B) The pairwise protein sequence comparison for heatmap showing orthologous clusters between S-8 and other closely related strains.
Supplementary Figure 6 | (A) The pan-genome and core-genome analysis was performed using the Box-plot analysis which showed that among the selected strains, S-8 showed the less conserved genes, and a high number of unique genes (Supplementary Figure 6B).
References
Abegaz, B. M., Kinfe, H. H. (2019). Secondary metabolites, their structural diversity, bioactivity, and ecological functions: An overview. Phys. Sci. Rev. 4, 20180100. doi: 10.1515/psr-2018-0100
Adler, C., Corbalan, N. S., Seyedsayamdost, M. R., Pomares, M. F., de Creistobal, R. E., Clardy, J., et al. (2012). Catecholate siderophores protect bacteria from pyochelin toxicity. PloS One 7, e46754. doi: 10.1371/journal.pone.0046754
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L., Beatson, S. A. (2011). BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12, 402. doi: 10.1186/1471-2164-12-402
Alvarez, F., Castro, M., Príncipe, A., Borioli, G., Fischer, S., Mori, G., et al. (2012). The plant-associated Bacillus amyloliquefaciens strains MEP 218 and ARP 23 capable of producing the cyclic lipopeptides iturin or surfactin and fengycin are effective in biocontrol of sclerotinia stem rot disease. J. Appl. Microbiol. 112 (1), 159–174. doi: 10.1111/j.1365-2672.2011.05182.x
Anantharajah, A. (2017). Contribution to the understanding of Pseudomonas aeruginosa virulence in search of innovative therapeutic approaches: Focus on the type III secretion system, Secteur des sciences de lasante (Brussels, Belgium: Universite´ catholique de Louvain), 231.
Anjaiah, V., Koedam, N., Nowak-Thompson, B., Loper, J., Höfte, M., Tambong, J., et al. (1998). Involvement of phenazines and anthranilate in the antagonism of Pseudomonas aeruginosa PNA1 and Tn 5 derivatives Toward Fusarium spp. and Pythium spp. Mol. Plant Microbe Interact. 11 (9), 847–854. doi: 10.1094/MPMI.1998.11.9.847
Arseneault, T., Goyer, C., Filion, M. (2015). Pseudomonas fluorescens LBUM223 increases potato yield and reduces common scab symptoms in the field. Phytopathology 105, 1311–1317. doi: 10.1094/phyto-12-14-0358-r
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9, 75. doi: 10.1186/1471-2164-9-75
Beatson, S. A., Walker, M. J. (2014). Tracking antibiotic resistance. Science 345, 1454–1455. doi: 10.1126/science.1260471
Biessy, A., Novinscak, A., Blom, J., Léger, G., Thomashow, L., Cazorla, F. M., et al. (2019). Diversity of phytobeneficial traits revealed by whole-genome analysis of worldwide-isolated phenazine-producing Pseudomonas spp. Environ. Microbiol. 21, 437–455. doi: 10.1111/1462-2920.14476
Blin, K., Shaw, S., Kloosterman, A. M., Charlop-Powers, Z., van Wezel, G. P., Medema, M. H., et al. (2021). antiSMASH 6.0: improving cluster detection and comparison capabilities. Nucleic Acids Res. 49, W29–W35. doi: 10.1093/nar/gkab335
Cheng, X., van de Mortel, J. E., Dekkers, E., Nguyen, L., Medema, M. H., Raaijmakers, J. M. (2017). Genome-wide analysis of bacterial determinants of plant growth promotion and induced systemic resistance by Pseudomonas fluorescens. Environ. Microbiol. 19, 4638–4656. doi: 10.1111/1462-2920.13927
Cho, S. T., Chang, H. H., Egamberdieva, D., Kamilova, F., Lugtenberg, B., Kuo, C. H. (2015). Genome analysis of Pseudomonas fluorescens PCL1751: a rhizobacterium that controls root diseases and alleviates salt stress for its plant host. PloS One 10, e0140231. doi: 10.1371/journal.pone.0140231
Connelly, M. B., Young, G. M., Sloma, A. (2004). Extracellular proteolytic activity plays a central role in swarming motility in Bacillus subtilis. J. Bacteriol. 186, 4159–4167. doi: 10.1128/JB.186.13.4159-4167.2004
Costa, T. R., Felisberto-Rodrigues, C., Meir, A., Prevost, M. S., Redzej, A., Trokter, M., et al. (2015). Secretion systems in Gram-negative bacteria: structural and mechanistic insights. Nat. Rev. Microbiol. 13, 343–359. doi: 10.1038/nrmicro3456
Deb, K. (2022). [Review of cog verse, by v. selvaraj]. Indian Literature 66 (327), 189–191. Available online at: https://www.jstor.org/stable/27277222.
de Bentzmann, S., Ple´siat, P. (2011). Pseudomonas aeruginosa: use virulence complex. Rev. Francophonedes Laboratoires 2011, 73–81. doi: 10.1016/S1773-035X(11)71104-2
Dimitrijević, A., Veličković, D., Rikalović, M., Avramović, N., Milosavic, N., Jankov, R., et al. (2011). Simultaneous production of exopolysaccharide and lipase from extremophylic pseudomonas aeruginosa san-ai strain: A novel approach for lipase immobilization and purification. Carbohydr. Polymers 83 (3), 1397–1401. doi: 10.1016/j.carbpol.2010.10.005
Eckstein, S., Brehm, J., Seidel, M., Lechtenfeld, M., Heermann, R. (2021). Two novel XRE-like transcriptional regulators control phenotypic heterogeneity in Photorhabdus luminescens cell populations. BMC Microbiol. 21 (1), 63. doi: 10.1186/s12866-021-02116-2
Feingersch, R., Philosof, A., Mejuch, T., Glaser, F., Alalouf, O., Shoham, Y., et al. (2012). Potential for phosphite and phosphonate utilization by Prochlorococcus. ISME J. 6 (4), 827–834. doi: 10.1038/ismej.2011.149
Feng, Z., Liu, D., Wang, L., Wang, Y., Zang, Z., Liu, Z., et al. (2020). A putative efflux transporter of the ABC family, ybhFSR, in escherichia coli functions in tetracycline efflux and na+(Li+)/H+ Transport. Front. Microbiolr 11, 556. doi: 10.3389/fmicb.2020.00556
Flury, P., Vesga, P., Péchy-Tarr, M., Aellen, N., Dennert, F., Hofer, N., et al. (2017). Antimicrobial and insecticidal: Cyclic lipopeptides and hydrogen cyanide produced by plant-beneficial pseudomonas strains CHA0, CMR12a, and PCL1391 contribute to insect killing. Front. Microbiol. 8 (2). doi: 10.3389/fmicb.2017.00100
Fothergill, J. L., Winstanley, C., James, C. E. (2012). Novel therapeutic strategies to counter Pseudomonas aeruginosa infections. Expert Rev. anti-infect Ther. 10, 219–235. doi: 10.1586/eri.11.168
Gilles-Gonzalez, M. A., Gonzalez, G. (2005). Signal transduction by heme-containing PAS-domain proteins. J. Appl. Physiol. 96 (1), 67–73. doi: 10.1152/japplphysiol.00941.2003
Girlich, D., Naas, T., Nordmann, P. (2004). Biochemical characterization of the naturally occurring oxacillinaseOXA-50 of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 48, 2043–2048. doi: 10.1128/AAC.48.6.2043-2048.2004
Gorriti, M. F., Dias, G. M., Chimetto, L. A., Trindade-Silva, A. E., Silva, B. S., Mesquita, M. A. A., et al. (2014). Genomic and phenotypic attributes of novel salinivibrios from stromatolites, sediment and water from a high altitude lake. BMC Genomics 15, 473. doi: 10.1186/1471-2164-15-473
Grbavčić, S., Bezbradica, D., Izrael-Živković, L., Avramović, N., Milosavić, N., Karadžić, I., et al. (2011). Production of lipase and protease from an indigenous Pseudomonas aeruginosa strain and their evaluation as detergent additives: compatibility study with detergent ingredients and washing performance. Bioresource Technol. 102, 11226–11233. doi: 10.1016/j.biortech.2011.09.076
Gudhka, R. K., Neilan, B. A., Burns, B. P. (2015). Adaptation, ecology, and evolution of the halophilic stromatolite archaeon halococcus hamelinensis inferred through genome analyses. Archaea 2015, 241608. doi: 10.1155/2015/241608
Haas, D., Défago, G. (2005). Biological control of soil-borne pathogens by fluorescent pseudomonads. Nat. Rev. Microbiol. 3, 307–319. doi: 10.1038/nrmicro1129
Hilker, R., Munder, A., Klockgether, J., Losada, P. M., Chouvarine, P., Cramer, N., et al. (2015). Interclonal gradient of virulence in the Pseudomonas aeruginosa pangenome from disease and environment. Environ. Microbiol. 17 (1), 29–46. doi: 10.1111/1462-2920.12606
Izrael-Zivkovic, L., Rikalovic, M., Gojgic-Cvijovic, G., Kazazić, S., Vrvić, M., Brčeski, I., et al. (2018). Cadmium specific proteomic responses of highly resistant Pseudomonas aeruginosa san ai. RSC Adv. 8 (19), 10549–10560. doi: 10.1039/C8RA00371H
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9, 1–8. doi: 10.1038/s41467-018-07641-9
James, K. D. (2017). Animal metabolites: from amphibians, reptiles, aves/birds, and invertebrates. Pharmacognosy 20 (5), 401–411. doi: 10.1016/B978-0-12-802104-0.00019-6
Junhong, Li, Chan, Yu, Zeqin, L., Yan, W., Fei, W. (2023). Microplastic accelerate the phosphorus-related metabolism of bacteria to promote the decomposition of methyl phosphonate to methane. Sci. Environ. 858, 160020. doi: 10.1016/j.scitotenv.2022.160020
Kaur, A., Pan, M., Meislin, M., Facciotti, M. T., El-Gewely, R., Baliga, N. S. (2006). A systems view of haloarchaeal strategies to withstand stress from transition metals. Genome Res. 16, 841–854. doi: 10.1101/gr.5189606
Kim, Y., Koh, I., Lim, M. Y., Chung, W., Rho, M. (2017b). Pan-genome analysis of Bacillus for microbiome profiling. Sci. Rep. 7, 1–9. doi: 10.1038/s41598-017-11385-9
Kim, S. Y., Lee, S. Y., Weon, H. Y., Sang, M. K., Song, J. (2017a). Complete genome sequence of Bacillus velezensis M75, a biocontrol agent against fungal plant pathogens, isolated from cotton waste. J. Biotechnol. 241, 112–115. doi: 10.1016/j.jbiotec.2016.11.023
Klockgether, J., Cramer, N., Wiehlmann, L., Davenport, C. F., Tu¨mmler, B. (2011). Pseudomonas aeruginosa genomic structure and diversity. Front. Microbiol. 2. doi: 10.3389/fmicb.2011.00150
Kraithong, T., Hartley, S., Jeruzalmi, D., Pakotiprapha, D. A. (2021). Peek inside the machines of bacterial nucleotide excision repair. Int. J. Mol. Sci. 22, 952. doi: 10.3390/ijms22020952
Kumari, K., Dey, J., Mahapatra, S. R., Ma, Y., Sharma, P. K., Misra, N., et al. (2024). Protein profiling and immunoinformatic analysis of the secretome of a metal-resistant environmental isolate Pseudomonas aeruginosa S-8. Folia Microbiol. 69, 1095–1122. doi: 10.1007/s12223-024-01152-5
Kung, V. L., Ozer, E. A., Hauser, A. R. (2010). The accessory genome of Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 74, 621–641. doi: 10.1128/mmbr.00027-10
Kwak, Y. S., Weller, D. M. (2013). Take-all of wheat and natural disease suppression: A review. Plant Pathol. J. 29, 125–135. doi: 10.5423/PPJ.SI.07.2012.0112
Ladomersky, E., Petris, M. J. (2015). Copper tolerance and virulence in bacteria. Metallomics 7, 957–964. doi: 10.1039/C4MT00327F
Lambert, P. A. (2002). Mechanisms of antibiotic resistance in Pseudomonas aeruginosa. J. R Soc. Med. 95, 22–26.
Lhospice, S., Gomez, N. O., Ouerdane, L., Brutesco, C., Ghssein, G., Hajjar, C., et al. (2017). Pseudomonas aeruginosa zinc uptake in chelating environment is primarily mediated by the metallophore pseudopaline. Sci. Rep. 7 (1), 17132. doi: 10.1038/s41598-017-16765-9
Liew, J. J. M., El Saudi, I. M., Nguyen, S. V., Wicht, D. K., Dowling, D. P. (2021). Structures of the alkanesulfonate monooxygenase MsuD provide insight into C-S bond cleavage, substrate scope, and an unexpected role for the tetramer. J. Biologic Chem. 297. doi: 10.1016/j.jbc.2021.100823
Lindeberg, M., Cunnac, S., Collmer, A. (2012). Pseudomonas syringae type III effector repertoires: 28 last words in endless arguments. Trends Microbiol. 20, 199–208. doi: 10.1016/j.tim.2012.01.003
Little, R. H., Woodcock, S. D., Campilongo, R., Fung, R. K. Y., Heal, R., Humphries, L., et al. (2019). Differential regulation of genes for cyclic-di-GMP metabolism orchestrates adaptive changes during rhizosphere colonization by pseudomonas fluorescens. Front. Microbiol. 10, 1089. doi: 10.3389/fmicb.2019.01089
Liu, Y., Dai, C., Zhou, Y., Qiao, J., Tang, B., Yu, W., et al. (2021). Pyoverdines are essential for the antibacterial activity of Pseudomonas chlororaphis YL-1 under low-iron conditions. Appl. Environ. Microbiol. 87, e02840–e02820. doi: 10.1128/AEM.02840-20
Loper, J. E., Hassan, K. A., Mavrodi, D. V., Davis, E. W., 2nd, Lim, C. K., Shaffer, B. T., et al. (2012). Comparative genomics of plant-associated Pseudomonas spp.: Insight into diversity and inheritance of traits involved in multitrophic interactions. PloS Genet. 8 (7), 1–27. doi: 10.1371/journal.pgen.1002784
Lyczak, J. B., Cannon, C. L., Pier, G. B. (2000). Establishment of Pseudomonas aeruginosa infection, lessons from a versatile opportunist. Microbes Infect. 2, 1051–1060. doi: 10.1016/s1286-4579(00)01259-4
Marcelletti, S., Ferrante, P., Petriccione, M., Firrao, G., Scortichini, M. (2011). Pseudomonas syringae pv. actinidiae draft genomes comparison reveal strain specific features involved in adaptation and virulence to Actinidia species. PloS One 6, e27297. doi: 10.1371/journal.pone.0027297
Martínez-Núñez, M. A., López, V. E. L. Y. (2016). Non-ribosomal peptides synthetases and their applications in industry. Sustain Chem. Process 4, 13. doi: 10.1186/s40508-016-0057-6
Mavrodi, D. V., Bonsall, R. F., Delaney, S. M., Soule, M. J., Phillips, G., Thomashow, L. S. (2001). Functional analysis of genes for biosynthesis of pyocyanin and phenazine-1-carboxamide from Pseudomonas aeruginosa PAO1. J. Bacteriol. 183, 6454–6465. doi: 10.1128/jb.183.21.6454-6465.2001
Medema, M. H., Kottmann, R., Yilmaz, P., Cummings, M., Biggins, J. B., Blin, K., et al. (2015). Minimum information about a biosynthetic gene cluster. Nat. Chem. Biol. 11 (9), 625–631. doi: 10.1038/nchembio.1890
Michaux, C., Sanguinetti, M., Reffuveille, F., Auffray, Y., Posteraro, B., Gilmore, M. S., et al. (2011). SlyA is a transcriptional regulator involved in the virulence of Enterococcus faecalis. Infect. Immun. 79 (7), 2638–2645. doi: 10.1128/IAI.01132-10
Michelsen, C. F., Watrous, J., Glaring, M. A., Kersten, R., Koyama, N., Dorrestein, P. C., et al. (2015). Nonribosomal peptides, key biocontrol components for Pseudomonas fluorescens In5, isolated from a Green landic suppressive soil. MBio 6 (2), e00079. doi: 10.1128/mBio.00079-15
Nguyen, D. D., Melnik, A. V., Koyama, N., Lu, X., Schorn, M., Fang, J., et al. (2016). Indexing the Pseudomonas specialized metabolome enabled the discovery of poaeamide B and the bananamides. Nat. Microbiol. 2, 16197. doi: 10.1038/nmicrobiol.2016.197
Nurk, S., Bankevich, A., Antipov, D., Gurevich, A., Korobeynikov, A., Lapidus, A., et al. (2013). “Assembling genomes and mini-metagenomes from highly chimeric reads,” in Research in Computational Molecular Biology: 17th Annual International Conference, RECOMB 2013, Beijing, China, April 7-10, 2013. Berlin Heidelberg: Springer.
Oakley, A. (2011). Glutathione transferases: a structural perspective. Drug Metab. Rev. 43, 138–151. doi: 10.3109/03602532.2011.558093
Ondov, B. D., Treangen, T. J., Melsted, P., Mallonee, A. B., Bergman, N. H., Koren, S., et al. (2016). Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 17, 1–14. doi: 10.1186/s13059-016-0997-x
Ong, K. S., Aw, Y. K., Lee, L. H., Yule, C. M., Cheow, Y. L., Lee, S. M. (2016). Burkholderia paludis sp. nov., an antibiotic-siderophore producing novel Burkholderia cepacia complex species, isolated from Malaysian tropical peat swamp soil. Front. Microbiol. 7, 2046. doi: 10.3389/fmicb.2016.02046
Ozer, E. A., Allen, J. P., Hauser, A. R. (2014). Characterization of the core and accessory genomes of Pseudomonas aeruginosa using bioinformatic tools Spine and AGEnt. BMC Genomics 15, 737. doi: 10.1186/1471-2164-15-737
Page, A. J., Taylor, B., Delaney, A. J., Soares, J., Seemann, T., Keane, J. A., et al. (2016). SNP-sites: rapid efficient extraction of SNPs from multi-FASTA alignments. Microb. Genom. 2, e000056. doi: 10.1099/mgen.0.000056
Pérez-Pantoja, D., Nikel, P. I., Chavarría, M., de Lorenzo, V. (2013). Endogenous stress caused by faulty oxidation reactions fosters evolution of 2,4-dinitrotoluene-degrading bacteria. PloS Genet. 9, e1003764. doi: 10.1371/journal.pgen.1003764
Philippart, F., Gaudry, S., Quinquis, L., Lau, N., Ouanes, I., Touati, S., et al. (2015). Randomized intubation with polyurethane or conical cuffs to preventpneumonia in ventilated patients. Am. J. Respir. Crit. Care Med. 191 (6), 637–645. doi: 10.1164/rccm.201408-1398oc
Pieńko, T., Trylska, J. (2021). Correction: Extracellular loops of BtuB facilitate transport of vitamin B12 through the outer membrane of E. coli. PloS Comput. Biol. 17, e1009696. doi: 10.1371/journal.pcbi.1009696
Pincus, N. B., Ozer, E. A., Allen, J. P., Nguyen, M., Davis, J. J., Winter, D. R., et al. (2020). A genome-based model to predict the virulence of Pseudomonas aeruginosa isolates. mBio 11 (4), e01527–e01520. doi: 10.1128/mBio.01527-20
Planquette, B., Timsit, J. F., Misset, B. Y., Schwebel, C., Azoulay, E., Adrie, C., et al. (2013). Pseudomonas aeruginosa ventilator-associated pneumonia. Predictivefactors of treatment failure. Am. J. Respir. Crit. Care Med. 188 (1), 69–76. doi: 10.1164/rccm.201210-1897OC
Poschenrieder, C., Fernández, J. A., Rubio, L., Pérez, L., Terés, J., Barceló, J. (2018). Transport and use of bicarbonate in plants: current knowledge and challenges ahead. Int. J. Mol. Sci. 19, 1352. doi: 10.3390/ijms19051352
Rangel, L. I., Henkels, M. D., Shaffer, B. T., Walker, F. L., Davis, E. W., 2nd, Stockwell, V. O., et al. (2016). Characterization of toxin complex gene clusters and insect toxicity of bacteria representing four subgroups of Pseudomonas fluorescens. PloS One 11 (8), e0161120. doi: 10.1371/journal.pone.0161120
Rikalović, M. (2013). Ispitivanje ramnolipida dobijenih pomoću sojeva pseudomonas aeruginosa izolovanih iz sredina zagađenih naftom i naftnim derivatima (Doctoral dissertation, University of Belgrade, Faculty of Chemistry).
Rojas, A., Holguin, G., Glick and Y. Bashan, B. (2001). Synergism between Phyllobacterium sp. (N2-fixer) and Bacillus licheniformis (P-solubilizer), both from a semiarid mangrove rhizosphere. FEMS Microbiol. Ecol. 35, 181–187. doi: 10.1111/j.1574-6941.2001.tb00802.x
Rojas Murcia, N., Lee, X., Waridel, P., Maspoli, A., Imker, H. J., Chai, T., et al. (2015). The Pseudomonas aeruginosa antimetabolite L -2-amino-4-methoxy-trans-3-butenoic acid (AMB) is made from glutamate and two alanine residues via a thiotemplate-linked tripeptide precursor. Front. Microbiol. 6. doi: 10.3389/fmicb.2015.00170
Roy, A. S., Baruah, R., Gogoi, D., Borah, M., Singh, A. K., Boruah, H. P. D. (2013). Draft genome sequence of Pseudomonas aeruginosa strain N002, isolated from crude oil-contaminated soil from Geleky, Assam, India. Genome Announc. 1, e00104–e00112. doi: 10.1128/genomeA.00104-12
Samanta, S., Singh, A., Banerjee, A., Roychoudhury, A. (2020). Exogenous supplementation of melatonin alters representative organic acids and enzymes of respiratory cycle as well as sugar metabolism during arsenic stress in two contrasting indica rice cultivars. J. Biotechnol. 324, 220–232. doi: 10.1016/j.jbiotec.2020.10.013
Samantha, A., Vrielink, A. (2020). Lipid A phosphoethanolamine transferase: regulation, structure and immune response. J. Mol. Biol. 432, 5184–5196. doi: 10.1016/j.jmb.2020.04.022
Sánchez, D., Gomila, M., Bennasar, A., Lalucat, J., García-Valdés, E. (2014). Genome analysis of environmental and clinical P. aeruginosa isolates from sequence type-1146. PloS One 9, e107754. doi: 10.1371/journal.pone.0107754
Saxena, A. K., Kumar, M., Chakdar, H., Anuroopa, N., Bagyaraj, D. J. (2020). Bacillus species in soil as a natural resource for plant health and nutrition. J. Appl. Microbiol. 128, 1583–1594. doi: 10.1111/jam.14506
Schaffer, A. A., Aravind, L., Madden, T. L., Shavirin, S., Spouge, J. L., Wolf, Y. I., et al. (2001). Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res. 29, 2994–3005. doi: 10.1093/nar/29.14.2994
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Selezska, K., Kazmierzak, M., Mu¨sken, M., Garbe, J., Schobert, M., Häussler, S., et al. (2012). Pseudomonas aeruginosa population structure revisited under environmental focus: impact of water quality and phage pressure. Environ. Microbiol. 14, 1952–1967. doi: 10.1111/j.1462-2920.2012.02719.x
Silby, M. W., Winstanley, C., Godfrey, S. A., Levy, S. B., Jackson, R. W. (2011). Pseudomonas genomes: diverse and adaptable. FEMS Microbiol. Rev. 35, 652–680. doi: 10.1111/j.1574-6976.2011.00269.x
Singh, R. P., Sinha, A., Deb, S., Kumari, K. (2024). First report on in-depth genome and comparative genome analysis of a metal-resistant bacterium Acinetobacter pittii S-30, isolated from environmental sample. Front. Microbiol. 15. doi: 10.3389/fmicb.2024.1351161
Stewart, A. (2001). Commercial biocontrol — reality or fantasy? Australas Plant Pathol. 30, 127–131. doi: 10.1071/AP01011
Stover, C. K., Pham, X. Q., Erwin, A. L., Mizoguchi, S. D., Warrener, P., Hickey, M. J., et al. (2000). Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406 (6799), 959–964. doi: 10.1038/35023079
Stringlis, I. A., Zhang, H., Pieterse, C. M. J., Bolton, M. D., de Jonge, R. (2018). Microbial small molecules - weapons of plantsubversion. Natural Product Rep. 35, 410–433. doi: 10.1039/c7np00062f
Tamura, K., Nei, M. (1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 10 (3), 512–526. doi: 10.1093/oxfordjournals.molbev.a040023
Tan, R. M., Kuang, Z., Hao, Y., et al. (2014a). Type IV pilus glycosylation mediates resistance of Pseudomonas aeruginosa to opsonic activities of the pulmonary surfactant protein A. Infect. Immun. 83, 1339–1346. doi: 10.1128/IAI.02874-14
Tan, R. M., Kuang, Z., Hao, Y., Lau, G. W. (2014b). Type IV pilus of pseudomonas aeruginosa confers resistance to antimicrobial activities of the pulmonary surfactant protein-A. J. innate Immune 6 (2), 227–239. doi: 10.1159/000354304
Tang, H., Kays, M., Prince, A. (1995). Role of Pseudomonas aeruginosa pili in acute pulmonary infection. Infect. Immun. 63, 1278–1285. doi: 10.1128/IAI.63.4.1278-1285.1995
Tong, L., Lee, S., Denu, J. M. (2009). Hydrolase regulates NAD + metabolites and modulates cellular redox. J. biolog. Chem. 284, 11256–11266. doi: 10.1074/jbc.M809790200
Treangen, T. J., Ondov, B. D., Koren, S., Phillippy, A. M. (2014). The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 15, 524. doi: 10.1186/s13059-014-0524-x
Udaondo, Z., Molina, L., Segura, A., Duque, E., Ramos, J. L. (2016). Analysis of the core genome and pangenome of Pseudomonas putida. Environ. Microbiol. 18, 3268–3283. doi: 10.1111/emi.2016.18.issue-10
Wang, H., Fewer, D. P., Holm, L., Rouhiainen, L., Sivonen, K. (2014). Atlas of non-ribosomal peptide and polyketide biosynthetic pathways reveals common occurrence of nonmodular enzymes. Proc. Natl. Acad. Sci. U.S.A. 111, 9259–9264. doi: 10.1073/pnas.1401734111
Watzer, B., Spät, P., Neumann, N., Koch, M., Sobotka, R., Macek, B., et al. (2019). The signal transduction protein PII controls ammonium, nitrate and urea uptake in cyanobacteria. Front. Microbiol. 10, 1428. doi: 10.3389/fmicb.2019.01428
Wiedenbeck, J., Cohan, F. M. (2011). Origins of bacterial diversity through horizontal genetic transfer and adaptation to new ecological niches. FEMS Microbiol. Rev. 35, 957–976. doi: 10.1111/j.1574-6976.2011.00292.x
Williams, E., Lowe, T. M., Savas, J., DiRuggiero, J. (2007). Microarray analysis of the hyperthermophilic archaeon Pyrococcus furiosus exposed to gamma irradiation. Extremophiles 11, 19–29. doi: 10.1007/s00792-006-0002-9
Winsor, G. L., Lam, D., Fleming, L., Lo, R., Whiteside, M. D., Yu, N. Y., et al. (2011). Pseudomonas genome database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nuc. Acids Res. 39, D596–D600. doi: 10.1093/nar/gkq869
Wood, T. E., Howard, S. A., Forster, A., Nolan, L. M., Manoli, E., Bullen, N. P., et al. (2019). The pseudomonas aeruginosa T6SS delivers a periplasmic toxin that disrupts bacterial cell morphology. Cell Rep. 29 (1), 187–201. doi: 10.1016/j.celrep.2019.08.094
Wu, X., Monchy, S., Taghavi, S., Zhu, W., Ramos, J., van der Lelie, D. (2010). Comparative genomics and functional analysis of niche-specific adaptation in Pseudomonas putida. FEMS Microbiol. Rev. 35, 299–323. doi: 10.1111/j.1574-6976.2010.00249.x
Xia, X., Li, J., Zhou, Z., Wang, D., Huang, J., Wang, G. (2018). High-quality draft high genome sequence of the multiple heavy metal resistant bacterium Pseudamino bactermanganicus JH-7. Stand. Genomic Sci. 13, 1–8. doi: 10.1186/s40793-018-0330-2
Xu, L., Dong, Z., Fang, L., Luo, Y., Wei, Z., Guo, H., et al. (2019). OrthoVenn2: a web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 47, W52–W58. doi: 10.1093/nar/gkz333
Zamioudis, C., Mastranesti, P., Dhonukshe, P., Blilou, I., Pieterse, C. M. J. (2013). Unraveling root developmental programs initiated by beneficial Pseudomonas spp. bacteria Plant Physiol. 162, 304–318. doi: 10.1104/pp.112.212597
Keywords: bacteria, genome, pan-genome, AMR, virulence
Citation: Kumari K, Sinha A, Sharma PK and Singh RP (2025) In-depth genome and comparative genome analysis of a metal-resistant environmental isolate Pseudomonas aeruginosa S-8. Front. Cell. Infect. Microbiol. 15:1511507. doi: 10.3389/fcimb.2025.1511507
Received: 15 October 2024; Accepted: 05 February 2025;
Published: 27 February 2025.
Edited by:
Margherita Montalbano Di Filippo, National Institute of Health (ISS), ItalyReviewed by:
Vittoria Mattioni Marchetti, University of Pavia, ItalyCatherine Llanes, University of Franche-Comté, France
Mai Mahmoud Zafer, Ahram Canadian University, Egypt
Copyright © 2025 Kumari, Sinha, Sharma and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rajnish Prakash Singh, bWFuYXNyYWpuaXNoMjAwOEBnbWFpbC5jb20=