 Yingjie Song1†
Yingjie Song1† Qiang Hu1†
Qiang Hu1† Yao Han1
Yao Han1 Hongbo Liu2Zhenyang Huang1
Hongbo Liu2Zhenyang Huang1 Mengwei Niu1Xue Dong1Kuocheng Yan1
Mengwei Niu1Xue Dong1Kuocheng Yan1 Li Jin1
Li Jin1 Hao Li1*Yansong Sun1*
Hao Li1*Yansong Sun1*- 1State Key Laboratory of Pathogen and Biosecurity, Academy of Military Medical Sciences, Beijing, China
- 2Chinese PLA Center for Disease Control and Prevention, Beijing, China
Introduction: Polymyxins are reserved as an ultimate defense against multidrug-resistant bacteria. The emergence of the polymyxin resistance gene mcr-1 poses a potential risk for the treatment of severe infections caused by Gram-negative bacteria. Timely detection and monitoring the mcr-1 gene are essential for guiding anti-infective therapy and controlling the spread of polymyxin resistance. Quantitative real-time PCR (qPCR) is one of the common methods for detecting resistance genes. However, qPCR has equipment dependency, and is not feasible in primary healthcare settings. Currently, there remains a lack of a highly sensitive and portable method for detecting the mcr-1 gene.
Methods: We established and optimized detection assays of the mcr-1 gene based on CRISPR/Cas13a system and lateral flow strips. The detection method was preliminarily evaluated using clinical isolates from Escherichia coli, compared with qPCR.
Results: The method for detecting the mcr-1 gene based on the CRISPR/Cas13a system and lateral flow strips was established, with a detection limit of 100 copies/mL. This method demonstrated high analytical specificity, with no cross-reactivity detected in non-mcr-1 and non-resistant strains. Among 36 clinical isolates, the method identified 31 strains as positive for the mcr-1 gene, and had a 100% concordance rate with the results of qPCR.
Conclusions: We established a detection method for the polymyxin resistance mcr-1 gene based on the CRISPR/Cas13a system. This method enables visual readouts without instruments, making it potentially applicable to primary healthcare settings and field surveillance.
1 Introduction
The overuse and misuse of antibiotics have significantly contributed to the rise of bacterial resistance, particularly the emergence of multidrug-resistant bacteria, which poses great challenges for the treatment of clinical infectious diseases (Willyard, 2017; Nanayakkara et al., 2021; Thompson, 2022; Martin-Mateos et al., 2024). Previous research indicated that 4.71 million deaths were associated with bacterial resistance, including 1.14 million deaths attributable to antibiotic resistance (Collaborators, G.B.D.A.R, 2024). Polymyxin is a peptide antibiotic derived from Paenibacillus polymyxa that inhibits the growth and reproduction of most Gram-negative bacteria, such as Escherichia coli and Pseudomonas aeruginosa (Chiu et al., 2022). In clinical settings, the polymyxin formulations mainly include polymyxin B and polymyxin E. These agents exhibit equivalent bactericidal potency and spectrum. The main difference is that polymyxin B is administered directly as the active drug, whereas polymyxin E, also known as colistin, is administered in the prodrug form of colistimethate sodium (Nation et al., 2014; Kelesidis and Falagas, 2015). Due to its bactericidal mechanism, which directly disrupts the integrity of the bacterial outer membrane, polymyxin has excellent efficacy against multidrug-resistant bacteria and is recognized as a last line of defense against multidrug-resistant bacteria (Andersson et al., 2016; Murray et al., 2022). The polymyxin resistance gene mcr-1 is generally located on bacterial plasmids and disseminates widely through horizontal gene transfer, enabling various bacterial strains to develop polymyxin resistance (Al-Tawfiq et al., 2017; Wise et al., 2018). Given the increasing public health threat posed by the emergence of the mcr-1 gene, timely detection and surveillance of the mcr-1 gene are critical for global antimicrobial resistance (AMR) control efforts.
Detection methods for the antibiotic-resistant phenotype of pathogenic bacteria include antimicrobial susceptibility testing (AST) and resistance genes detection. AST is the classic method for determining bacterial resistance phenotypes (Bartels et al., 2021; Tadesse et al., 2024), but it takes time to culture bacteria. It is a significant limitation for clinical decisions requiring rapid intervention. Quantitative real-time PCR (qPCR) is one of the common methods for detecting resistance genes, achieving rapid detection. Nevertheless, qPCR relies on specialized laboratories and sophisticated equipment, which limits its applicability in primary healthcare settings and hinders global AMR monitoring. Therefore, there is an urgent need for a sensitive and portable gene detection assay for antibiotic resistance.
The clustered regularly interspaced short palindromic repeats (CRISPR)/associated protein (Cas) system for nucleic acid detection has advanced rapidly (Gootenberg et al., 2017; Chen et al., 2018; Harrington et al., 2018; Kellner et al., 2019; Fozouni et al., 2021; Patchsung et al., 2023). The CRISPR/Cas13a system is characterized by targeted activation of additional cleavage activity. This system has been developed as a specific and high-sensitivity enzymatic reporter unlocking (SHERLOCK) assay, making it a prevalent choice for nucleic acid detection methods (Gootenberg et al., 2017; Kellner et al., 2019; Liu et al., 2017; Yin et al., 2022). In 2022, a study developed a highly sensitive CRISPR-based platform for mcr-1 gene detection but relied on a real-time fluorescence detector, limiting its applicability in primary healthcare settings (Honda et al., 2011; Gong et al., 2022). By combining recombinase aided amplification (RAA) and lateral flow strips, CRISPR assay can achieve 1 copy/μL sensitivity within 60 minutes rivaling qPCR while eliminating equipment dependency (Li et al., 2021). Besides, RAA assay eliminates the need for expensive thermal cyclers through isothermal nucleic acid amplification, reducing equipment costs compared to PCR thermocycler. CRISPR-based lateral flow strips further minimize operational expenses by replacing fluorescence readers with visual readouts. Therefore, CRISPR-based lateral flow strips assay is a highly sensitivity, fast, low-cost method for the detection of nucleic acid.
In a previous study, we have developed a CRISPR-ERASE detection method that is simple, rapid, sensitive, and specific for the detection of SARS-CoV-2, with a detection limit of 1 copies/μL (Li et al., 2021). This method combines CRISPR/Cas13a system and lateral flow strips, and has been approved as a medical device product (20203400919) by the National Medical Products Administration. In this study, we have adapted this method to detect the mcr-1 gene, which confers resistance to colistin. This novel application of CRISPR-ERASE enables rapid and cheap detection of the mcr-1 gene, with potential for global surveillance of colistin resistance mediated by the mcr-1 gene.
2 Materials and methods
2.1 Design and preparation of primers and crRNA
Sequences of the mcr-1 gene (NG_050417.1) were downloaded from GenBank database. Conserved sequences of the mcr-1 gene were identified by alignment and analysis using MEGA 7.0 software (Auckland, New Zealand). Four pairs of primers for isothermal amplification were then designed based on these conserved sequences. The T7 transcription promoter sequence was added at the 5’ end of the forward primer. Between the forward and reverse primers, we designed 3 crRNAs. The crRNA, probe, and primer sequences for the mcr-1 gene were shown in Supplementary Table 1. The DNA sequence of the mcr-1 gene was synthesized and cloned into pUC57 plasmid by Beijing Tianyihuiyuan Biotechnology Co., Ltd (Beijing, China), and the crRNA was prepared with reference to the literature (Li et al., 2020). The plasmid templates were absolutely quantified via digital PCR and then used as a template for analytical sensitivity and specificity detection experiments.
2.2 Samples preparation
In this study, 36 Escherichia coli strains were collected by the Chinese PLA Center for Disease Control and Prevention and shown in Supplementary Table 2. All strains were stored in 30% (w/v) glycerol broth at −80°C. The strains were incubated in Mueller-Hinton, followed by antimicrobial susceptibility test and plasmid extraction (TIANGEN, DP103). Plasmid templates were constructed by incorporating DNA fragment of the mcr-1 gene. The recombinant plasmids were absolutely quantified and diluted 10-fold serially to yield 103 copies/μL to 10-1 copies/μL.
2.3 Recombinase aided amplification
In accordance with the instructions provided with the DNA Isothermal Rapid Amplification Kit (Basic) (AMP Future Biotechnology Co., Ltd, WLB8201KIT), the amplification system consisted of 29.4 μL Buffer A, 2.5 μL Buffer B, 2 μL forward primer (10 nmol/L), 2 μL reverse primer (10 nmol/L), 9.1 μL non-enzymatic water, and 5 μL target DNA. The reaction system was incubated at a constant temperature of 39°C for 30 mins on a metal bath.
2.4 Fluorescence-based CRISPR assay
The crRNA mixed with the Cas13a enzyme to make the CRISPR/Cas13a-crRNA complex, which is used to incise the RAA product. The assay system consisted of 2 μL NTP Mix (2.5 mmol/L), 1 μL RNase Inhibitor (4 IU/μL), 0.5 μL T7 RNA polymerase (1 IU/μL), 0.5 μL HEPES (20 mmol/L), 0.25 μL MgCl2 (10 mmol/L), 10.75 μL enzyme-free water, 2.5 μL FAM-20U-BHQ1 (200 nmol/L), 1.5 μL crRNA (280 nmol/L), 1 μL Cas13a (45 nmol/L), and 5 μL RAA product. The fluorescence signals were collected every 2 min at 37°C to monitor the changes in fluorescence intensity. The threshold was determined by the mean fluorescence of the negative control group plus three times the standard deviation (Armbrecht et al., 2020).
2.5 CRISPR-ERASE assay
The total volume of the CRISPR-ERASE reaction system was 50 μL, and the mixture included 4 μL NTP Mix (2.5 mmol/L), 2 μL RNase Inhibitor (1.6 IU/μL), 1 μL T7 RNA polymerase (1 IU/μL), 1 μL HEPES (20 mmol/L), 0.5 μL MgCl2 (10 mmol/L), 26.5 μL DNase/RNase-Free Water, 5 μL FAM-20U-Biotin (2 nmol/L), 3 μL crRNA (2 nmol/L), 2 μL Cas13a (80 ng/L), and 5 μL RAA product. After incubation at 37°C for 30 min on a metal bath, the whole reaction system was transferred to ERASE lateral flow strip with a pipette gun. The reporter RNA molecule is labeled with FAM and biotin at its 5’ and 3’ ends, respectively. Colloidal gold particles coated with FAM antibodies are immobilized at the bottom of the test strip. When the target nucleic acid is present in the system, the activated Cas13a protein specifically cleaves the reporter RNA molecule. This cleavage releases the biotin moiety, which then binds to streptavidin encapsulated on the Test band. Simultaneously, the colloidal gold particles bind to the FAM moiety and the anti-FAM antibody encapsulated on the Control band, resulting in a visible signal only on the Control band. Conversely, when the target nucleic acid is absent, the reporter RNA molecule remains intact. In this case, the colloidal gold particles with FAM antibodies bind to streptavidin via the biotin molecule and also bind to the anti-FAM antibody, producing visible signals on both the Test and Control bands (Li et al., 2020). The results could be read with the naked eye after 5 mins.
2.6 Quantitative real-time PCR
A 10-fold gradient dilution of the mcr-1 plasmid template was detected using a quantitative Real-time PCR probe kit purchased from Thermo Fisher. The qPCR primer and probe are used as reported in the literature and shown in Supplementary Table 1 (Chabou et al., 2016). The reaction system was 10 μL TaqMan™ Fast Advanced Master Mix (2×), 0.5 μL forward primer (0.4 μmol/L), 0.5 μL reverse primer (0.4 μmol/L), 0.5 μL probe (0.4 μmol/L), 5.5 μL DNase/RNase-Free Water, and 3 μL target DNA. The qPCR conditions were as follows, the reaction mixtures were kept at 95°C for 30 s and subsequently put through 40 cycles of 95°C for 10 s and 60°C for 1 min, and the fluorescence signal was collected once at the end of each cycle. Real-time PCR was performed on serially diluted samples to measure cycle threshold (Ct) values. A standard curve was subsequently generated by plotting template concentrations (x-axis) against corresponding Ct values (y-axis). The limit of detection (LoD) was calculated as the minimum template concentration with Ct values statistically different from negative controls.
2.7 Antimicrobial susceptibility test
Polymyxin B susceptibility testing (Kangtai Biotechnology Co., Ltd, J1A079) was performed using the broth microdilution method, and results were interpreted according to European Committee on Antimicrobial Susceptibility Testing (EUCAST) breakpoints (EUCAST, 2025). Five colonies cultured for 18 h were taken by inoculation loop and put into sterile inoculum water, and the concentration was adjusted to 1.5*108 cfu/mL. Take 10 μL of the conditioned bacterial solution and add it to the liquid medium of antimicrobial susceptibility testing and mix well, and then add 100 μL per well to each concave control in the antimicrobial susceptibility plate. Finally, the plates were incubated in an incubator at 37°C for 18 h and then removed for interpretation of the results.
2.8 Statistical analysis
The data were analyzed by normality tests and then unpaired two-tailed Student’s t tests using GraphPad Prism 8.0.2 software (GraphPad, Inc., La Jolla, CA, USA).
3 Results
3.1 Schematic of CRISPR-ERASE detection assay for mcr-1 gene
In this study, we developed a detection assay for the mcr-1 resistance gene using CRISPR-ERASE technology. The schematic of the detection assay for the mcr-1 gene is shown in Figure 1. Plasmid DNA was extracted from Escherichia coli, and the mcr-1 gene fragment was amplified by recombinase-assisted amplification (RAA) and transcribed into an RNA sequence. When the CRISPR/Cas13a system recognizes the RNA target, it degrades the reporter RNA. The cleaved reporter RNA can be captured on a lateral flow strip and the results can be read with the naked eye using ERASE strips. A positive result is showed by the presence of only the Control band, while the appearance of both the Test and Control bands indicates a negative result.
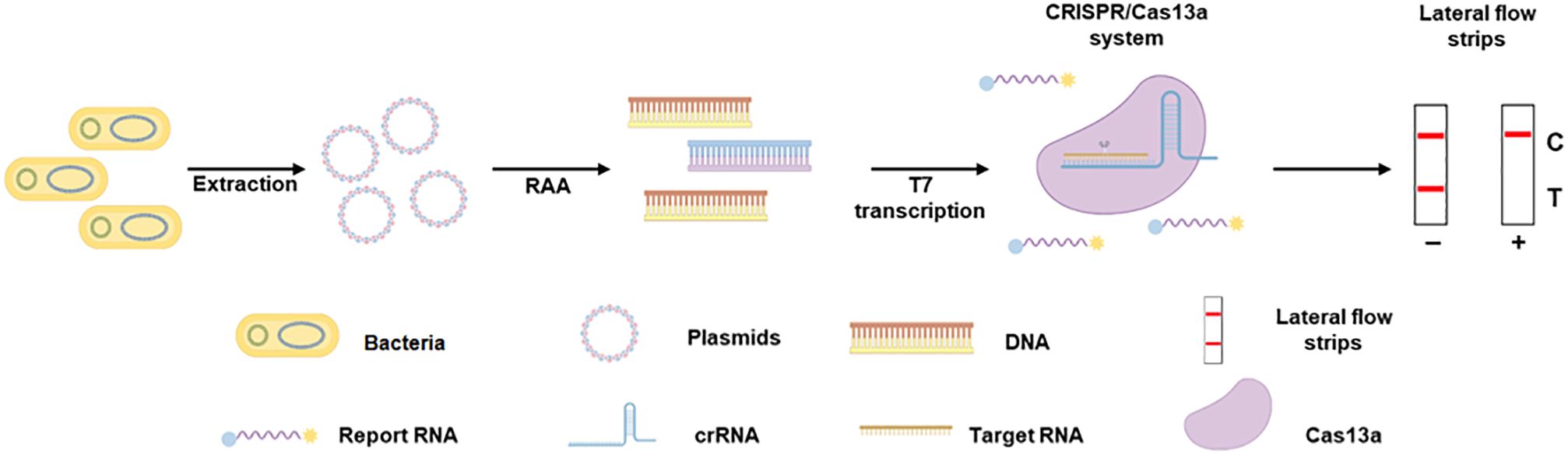
Figure 1. The schematic of CRISPR-ERASE assay for detecting the mcr-1 gene. Plasmid DNA was extracted from Escherichia coli, amplified the mcr-1 gene fragment by recombinase-assisted amplification (RAA) and transcribed into an RNA sequence. When CRISPR/Cas13a system recognizes the RNA target and degrades reporter RNA. The cleaved reporter RNA can be captured on a lateral flow strip and the results can be read with the naked eye using ERASE strips.
3.2 Design and screening of RAA primers and crRNAs
To screen efficient crRNA, we designed and screened 3 crRNAs and 4 pairs of RAA primers. The schematic representation of the positions of the crRNAs and RAA primers is shown in Figure 2A. Initially, we assessed the detection efficiency of the CRISPR/Cas13a system using three different crRNAs in conjunction with the same RAA product for the mcr-1 gene. The results indicated that the fluorescence profile of the CRISPR/Cas13a system increases after the initiation of the reaction. At 60 minutes, the average fluorescence value for the crRNA-1 group showed a 19.64 ± 1.24-fold increase, while the crRNA-2 group exhibited a 17.02 ± 2.58-fold increase, and the crRNA-3 group demonstrated a 12.38 ± 0.54-fold increase, all compared to the negative control group. The crRNA-1 group displayed significantly higher fluorescence values compared to the other two groups (Figures 2B, C). Consequently, crRNA-1 was selected for further mcr-1 gene detection.
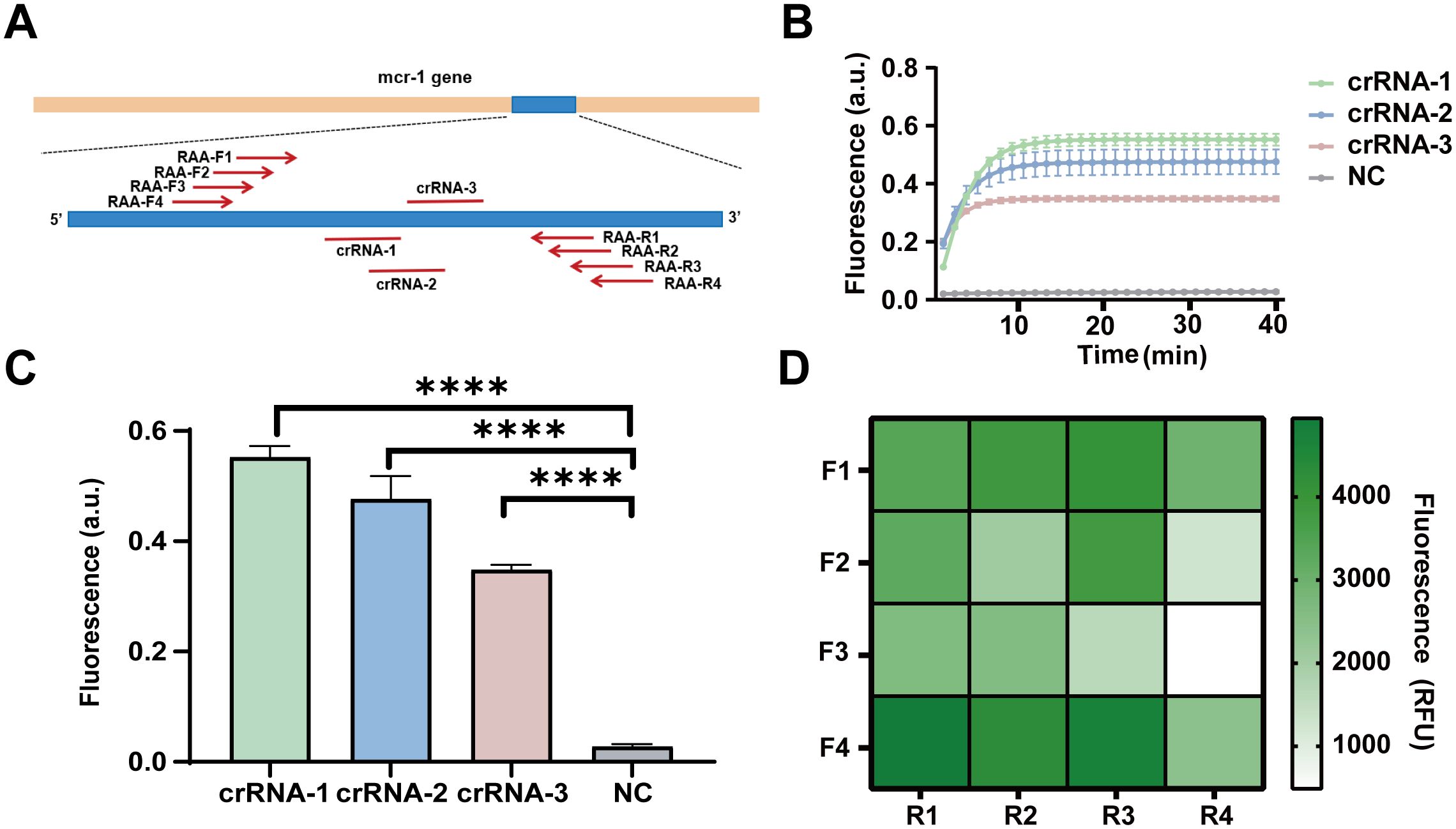
Figure 2. Design and screening of RAA primers and crRNAs for the detection of the mcr-1 gene. (A) The schematic diagram of the design of RAA primers and crRNA. (B) The dynamic fluorescence curves within 40 min of 3 different crRNAs for CRISPR-ERASE assay (n = 3). (C) The fluorescence values of different crRNAs designed for the mcr-1 gene were compared at the 40 min after detection reaction. ****p < 0.0001. The data were expressed as mean ± SEM (n = 3). (D) The fluorescence values produced at 40 min of CRSIPR fluorescence detection reaction with crRNA-1 and different RAA amplification primer combinations of mcr-1 gene. The data were expressed as mean ± SEM (n = 3).
To identify efficient RAA primers, we constructed 16 combinations by pairing four forward RAA primers with four reverse RAA primers targeting the mcr-1 gene. These combinations were tested to simultaneously amplify plasmids containing the mcr-1 gene fragment at a concentration of 10 copies/μL. The products were examined for fluorescence values using the CRISPR/Cas13a system with crRNA1. At 40 minutes, the primer combination F4R1 exhibited a fluorescence value of 4941 RFU, which was the highest among the 16 combinations (Figure 2D). Thus, the F4R1 primer was selected for subsequent mcr-1 gene detection.
3.3 Limit of detection of the CRISPR-ERASE assay using the plasmid containing the mcr-1 gene fragment
With the most efficient primer pairs and crRNA, we evaluated the analytical sensitivity of the fluorescence-based CRISPR assay. Initially, we determined the Limit of Detection of the CRISPR-ERASE assay. As illustrated in Figure 3A, the fluorescence values of the group of 103 to 10-1 copies/μL were significantly increased. To assess the experimental results quantitatively, we calculated the threshold using the mean fluorescence value of the negative control group plus three times the standard deviation. Fluorescence values above this threshold were classified as positive, while those below were classified as negative (Armbrecht et al., 2020; Ding et al., 2020; Sun et al., 2023). The fluorescence value of the CRISPR/Cas13a-based detection was calculated to be 4120.03 RFU. At a plasmid template concentration of 100 copies/mL, the fluorescence values for three replicate assays were 9765.63, 12903.47 and 12969.86 RFU, respectively, with all three replicates exceeding the threshold and thus classified as positive (Figure 3B). The LoD of the fluorescence-based CRISPR assay is 100 copies/mL.
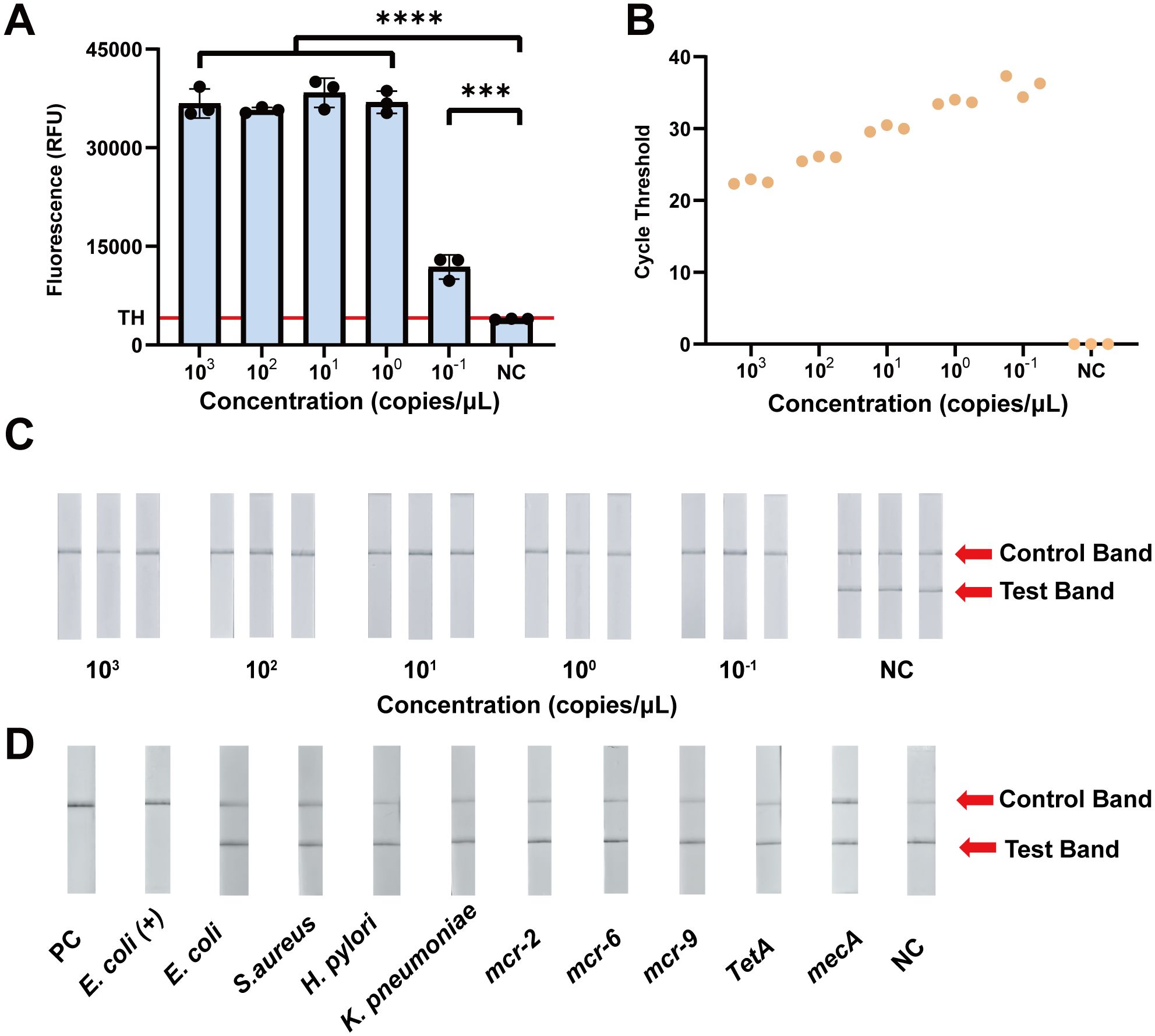
Figure 3. Analytical sensitivity and specificity evaluation of the CRISPR-ERASE assay for the detection of the mcr-1 gene. (A) Comparison of fluorescence values produced by CRISPR fluorescence detection of mcr-1 plasmid. TH means the threshold using the mean fluorescence value of the negative control group plus three times the standard deviation. ***p<0.001; ****p<0.0001. The data were expressed as mean ± SD (n = 3). (B) The results of qPCR at different mcr-1 plasmid concentrations (103 ~10-1 copies/μL) (n = 3). (C) The results of CRISPR-ERASE at different mcr-1 plasmid concentrations (103 ~10-1 copies/μL). Test band disappeared and control band was visible meaning that the test result is positive. Repeat each result three times. NC represents Negative Control. (D) CRISPR-ERASE assay was used to detect the mcr-1 gene of different strains. PC represents Positive Control; Escherichia coli (+) represents mcr-1-positive Escherichia coli; S.aureus represents Staphylococcus aureus; H. pylori represents Helicobacter pylori; K. pneumoniae represents Klebsiella pneumoniae (n = 3).
To further assess the analytical sensitivity of CRISPR/Cas13a-based detection, we compared the LoD of the CRISPR-based assay and qPCR. At a plasmid concentration of 100 copies/mL, all three replicates had cycle threshold (CT) values of 37.30, 34.38, and 36.28, showing a positive results. qPCR detected the plasmid template containing the mcr-1 gene at a concentration of 100 copies/mL (Figure 3B).
To avoid specialized instrumentation and achieve a portable detection method, we combined the CRISPR/Cas13a system with lateral flow strips. A plasmid containing the mcr-1 gene fragment was subjected to a 10-fold gradient dilution (103~10-1 copies/μL) and analyzed. At a template concentration of 100 copies/mL, only the Control band was visible on the ERASE strips across in all three replicate experiments, yielding consistently positive results (Figure 3C). These findings indicate that the LoD of the CRISPR-ERASE assay is 100 copies/mL.
3.4 Cross-reaction evaluation of the CRISPR-ERASE assay using the plasmid containing the mcr-1 gene fragment
To evaluate the cross-reaction of CRISPR-ERASE, we analyzed the DNA of five pathogens: mcr-1-positive Escherichia coli, Escherichia coli, Staphylococcus aureus, Helicobacter pylori, and Klebsiella pneumoniae. pUC57 plasmids containing the mcr-1 gene fragment were utilized as positive control templates, while pUC57 plasmids without the mcr-1 gene fragment were utilized as negative control templates. As illustrated in Figure 3D, genomic DNA from the mcr-1 carrying bacteria showed a disappearance of the Test band on the ERASE strips, while the Control band remained visible, indicating a positive result. In contrast, the ERASE strips for the remaining four bacterial pathogens displayed both Test and Control bands, confirming negative results. CRISPR-ERASE can specifically detect mcr-1 producing bacteria without cross-reacting with the genomic DNA of the other four pathogens.
To evaluate the performance of CRISPR-ERASE with non-mcr-1 genes, we compared the previously screened crRNA and RAA primers against the sequences of mcr-2 to mcr-11. Since mcr-2 and mcr-6 exhibit higher homology with the target sequences, while the other variants show no match (Rodríguez-Santiago et al., 2021), we selected mcr-2 and mcr-6 for analytical specificity experiments. Given the higher prevalence of mcr-9, it was also included in cross-reaction experiments. Additionally, we tested two other common resistance genes, tetA and mecA, which confer resistance to tetracycline and methicillin, respectively. As illustrated in Figure 3D, we assessed cross-reactivity with five non-mcr-1 genes and found no interference, thereby confirming the high analytical specificity of the assay.
3.5 Preliminary performance of CRISPR-ERASE assay in clinical isolates
For a preliminary evaluation of the CRISPR-ERASE assay, we tested 36 Escherichia coli strains isolated from clinical samples. We preformed antimicrobial susceptibility across a range of concentrations, determining the Minimum Inhibitory Concentration (MIC). For example, MIC values for clinical isolates No.30-33 were shown to be in the order of 4, 8, 1, and 1μg/mL (Figure 4A). According to EUCAST, an MIC of > 2 μg/mL is considered resistant, while an MIC of ≤ 2 μg/mL is classified as sensitive. As illustrated in Figure 4B, Supplementary Data 1, MIC values for clinical isolates No.1-19, 28, 29, and 31 were 4 μg/mL, while those for isolates No.20-27 and 30 were 8 μg/mL. Therefore, these 31 isolates were identified as polymyxin-resistant bacteria. In contrast, clinical isolates No.32, 33, 34, and 36 exhibited MIC values of 1 μg/mL, and isolate 35 had an MIC of 0.5 μg/mL, classifying these five isolates as polymyxin-sensitive bacteria.
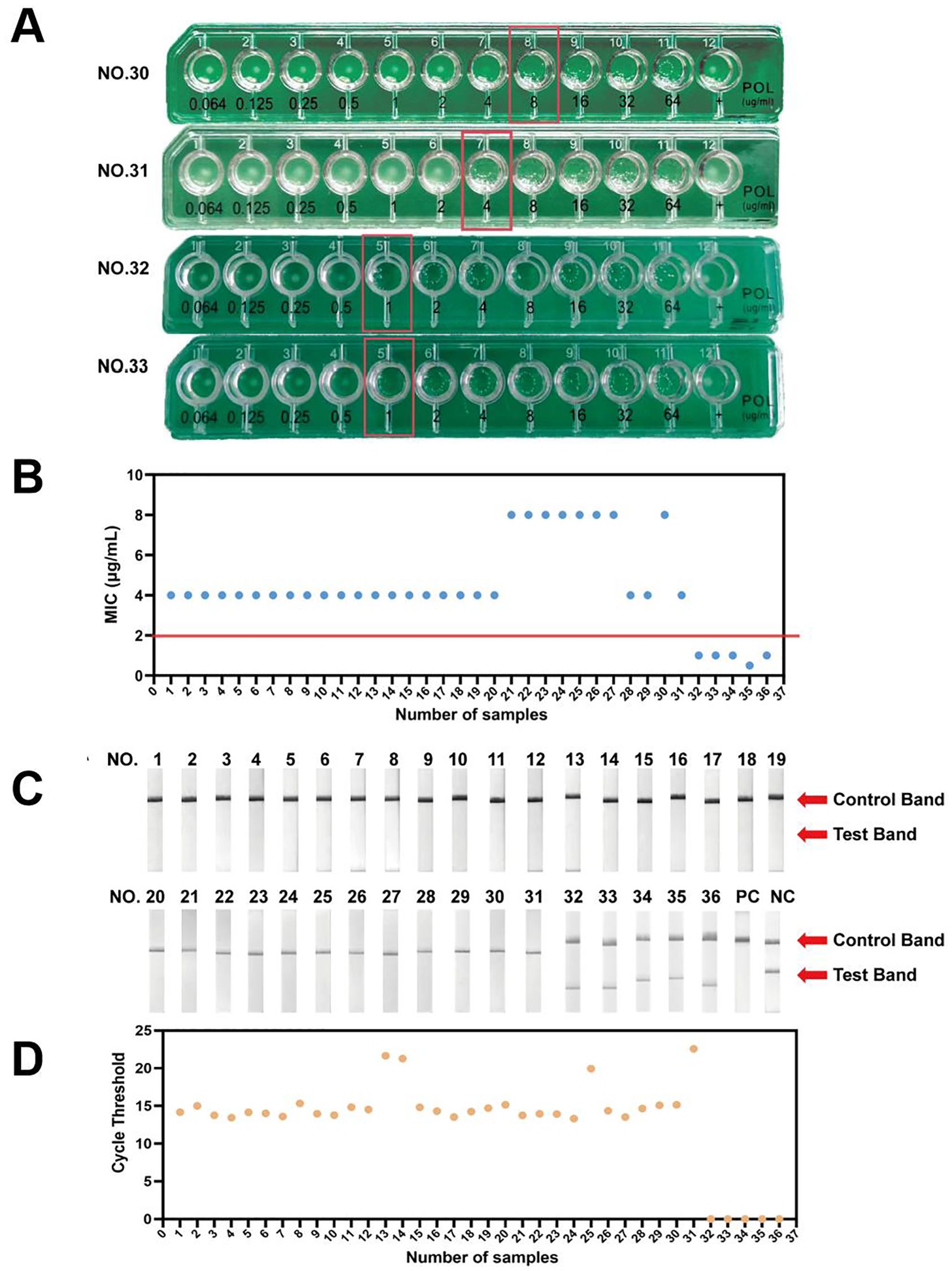
Figure 4. Preliminary validation of CRISPR-ERASE detection system to detect mcr-1 gene in isolated Escherichia coli samples. (A) The results of antimicrobial susceptibility testing for NO.31-33 isolated samples. (B) The MIC results of antimicrobial susceptibility testing for 36 isolated samples. (C) The results of CRISPR-ERASE detection assay for 36 isolated samples. PC represents Positive Control.NC represents Negative Control. (D) The results of qPCR for 36 isolated samples.
Subsequently, we preliminarily evaluated the assay on 31 polymyxin-resistant bacteria samples and 5 polymyxin-sensitive bacteria samples. Clinical isolates numbered 1-31 showed only the Control band and no Test band on the strips, and all of these samples tested positive for the mcr-1 gene. In contrast, clinical isolates numbered 32-36 displayed both Control band and Test bands on the strips and tested negative for the mcr-1 gene (Figure 4C). The CRISPR-ERASE method identified that these 31 polymyxin-resistant samples were all samples carrying the mcr-1 gene.
Furthermore, we performed qPCR on DNA samples extracted from 36 clinical isolates. Clinical isolates numbered 1-31 exhibited a maximum CT value of no more than 22.57, and all samples tested positive. In contrast, clinical isolates numbered 32-36 did not yield a readable CT value (Figure 4D). CRISPR-ERASE and qPCR results showed No.1-31 were tested positive for the mcr-1 gene, No.32-36 were tested negative for the mcr-1 gene. The results of quantitative real-time PCR were consistent with those of the CRISPR-ERASE method. Overall, the preliminary performance of CRISPR-ERASE assay in clinical isolates aligned with the findings from qPCR assay.
4 Discussion
Global efforts for the detection of antibiotic resistance genes have become increasingly urgent (Jiang et al., 2017; Wang et al., 2020; Kang et al., 2021; Zhang et al., 2022). In this study, we developed a rapid detection method for the mcr-1 gene based on the CRISPR-ERASE assay. This assay detects plasmid templates containing the mcr-1 gene at concentrations as low as 100 copies/mL, which exhibits a comparable LoD to qPCR using previously reported primers, suggesting a highly analytical sensitivity and accurate detection method for the mcr-1 gene. Lateral flow strips offer a portable and visual detection method that is not constrained by external experimental conditions, making it particularly suitable for decentralized settings such as rural clinics or field surveillance programs. The assay specifically targets the mcr-1 gene, with no cross-reactivity observed against non-resistant strains or other mcr variants, demonstrating high analytical specificity.
In our study, there are some limitations. First, while the CRISPR-ERASE assay avoids reliance on complex instruments, current nucleic acid extraction protocols still require centrifugation, which may hinder implementation in resource-limited regions. Future integration with direct lysis buffers could further streamline the workflow and save time. Second, validation was limited to Escherichia coli clinical isolates. Given the global prevalence of mcr-1 in other Gram-negative pathogens such as Klebsiella pneumoniae and Salmonella, extending testing to these species is critical to confirm broad applicability. Third, although the assay’s multiplex potential was preliminarily explored, simultaneous detection of multiple resistance genes will require rigorous optimization to prevent crRNA cross-reactivity and ensure signal fidelity. Forth, the sample size of 36 clinical isolates is relatively small, which may potentially limit the robustness of diagnostic sensitivity and specificity. Further validation in a larger cohort and diverse clinical settings is needed to confirm its clinical utility.
Polymyxin resistance mechanisms extend beyond plasmid-borne mcr-1 to include chromosomal mutations, with distinct pathways observed across bacterial species. While Escherichia coli predominantly acquires resistance through mcr-1-mediated plasmid transfer, Klebsiella pneumoniae primarily develops resistance via chromosomal mutations such as inactivation of the mgrB gene, a critical negative regulator of lipid A modification systems (Poirel et al., 2015). These species-specific resistance mechanisms highlight the complexity of polymyxin resistance in Gram-negative pathogens. From a clinical perspective, the CRISPR-ERASE assay has a potential impact on antimicrobial stewardship. The assay might be used to detect the inactivation of mgrB gene. Compared with traditional amplification and sequencing detection, this technology might detect inactivated genes more rapidly and at a lower cost. It is particularly relevant given that chromosomal-mediated resistance mechanisms like mgrB mutations are increasingly reported in clinical isolates worldwide (Poirel et al., 2015; Conceição-Neto et al., 2022). In veterinary or agricultural settings, this tool might support large-scale surveillance of mcr-1 transmission at lower costs and shorter turnaround times. However, widespread adoption would require validation in diverse matrices and integration into existing diagnostic pathways. For instance, a positive CRISPR-ERASE result could trigger reflex testing by sequencing, while negative results might expedite de-escalation of reserve antibiotics. With its high-resolution genotyping capacity, CRISPR-ERASE can track the spread of resistance genes across different bacterial strains and healthcare settings. This helps in understanding the transmission dynamics of resistance and supporting for antimicrobial resistance epidemiology studies.
In the future, the development of the assay may focus on expanding target pathogens to include multidrug-resistant Gram-negative bacteria, developing multiplexed CRISPR panels for co-detection of high-priority resistance genes, and conducting field trials in low-resource hospitals to evaluate real-world feasibility. This technology not only addresses an urgent diagnostic gap but also aligns with global priorities to combat antimicrobial resistance through precision surveillance and targeted therapy.
In summary, we established a CRISPR-ERASE assay for rapid detection of the mcr-1 resistance gene, achieving highly analytical sensitivity comparable to qPCR while eliminating reliance on specialized equipment. This platform enables timely identification of mcr-1-mediated colistin resistance, offering a potential tool to guide antimicrobial stewardship in clinical settings and support global surveillance efforts to curb the dissemination of plasmid-borne resistance.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Author contributions
YJS: Methodology, Validation, Writing – original draft, Writing – review & editing. QH: Formal Analysis, Methodology, Writing – original draft. YH: Data curation, Software, Writing – review & editing. HBL: Formal Analysis, Resources, Writing – review & editing. ZH: Methodology, Software, Writing – review & editing. MN: Visualization, Writing – review & editing. XD: Investigation, Visualization, Writing – review & editing. KY: Formal Analysis, Investigation, Writing – review & editing. LJ: Data curation, Investigation, Writing – review & editing. HL: Conceptualization, Funding acquisition, Project administration, Writing – original draft. YSS: Conceptualization, Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was funded by the National Key Research and Development Program of China (2023YFC2605100).
Acknowledgments
Figures of the schematic diagram were generated using Figdraw (https://www.home-for-researchers.com).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2025.1553681/full#supplementary-material.
References
Al-Tawfiq, J. A., Laxminarayan, R., and Mendelson, M. (2017). How should we respond to the emergence of plasmid-mediated colistin resistance in humans and animals? Int. J. Infect. Dis. 54, 77–84. doi: 10.1016/j.ijid.2016.11.415
Andersson, D. I., Hughes, D., and Kubicek-Sutherland, J. Z. (2016). Mechanisms and consequences of bacterial resistance to antimicrobial peptides. Drug Resist. Update 26, 43–57. doi: 10.1016/j.drup.2016.04.002
Armbrecht, L., Rutschmann, O., Szczerba, B. M., Nikoloff, J., Aceto, N., and Dittrich, P. S. (2020). Quantification of protein secretion from circulating tumor cells in microfluidic chambers. Adv. Sci. (Weinh) 7, 1903237. doi: 10.1002/advs.201903237
Bartels, M. D., Worning, P., Andersen, L. P., Bes, M., Enger, H., Ås, C. G., et al. (2021). Repeated introduction and spread of the MRSA clone t304/ST6 in northern Europe. Clin. Microbiol Infect. 27, 284.e281–284.e285. doi: 10.1016/j.cmi.2020.05.004
Chabou, S., Leangapichart, T., Okdah, L., Le Page, S., Hadjadj, L., and Rolain, J. M. (2016). Real-time quantitative PCR assay with Taqman(®) probe for rapid detection of MCR-1 plasmid-mediated colistin resistance. New Microbes New Infect. 13, 71–74. doi: 10.1016/j.nmni.2016.06.017
Chen, J. S., Ma, E., Harrington, L. B., Da Costa, M., Tian, X., Palefsky, J. M., et al. (2018). CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 360, 436–439. doi: 10.1126/science.aar6245
Chiu, S., Hancock, A. M., Schofner, B. W., Sniezek, K. J., Soto-Echevarria, N., Leon, G., et al. (2022). Causes of polymyxin treatment failure and new derivatives to fill the gap. J. Antibiot (Tokyo) 75, 593–609. doi: 10.1016/j.nmni.2016.06.017
Collaborators, G.B.D.A.R (2024). Global burden of bacterial antimicrobial resistance 1990-2021: a systematic analysis with forecasts to 2050. Lancet 404, 1199–1226. doi: 10.1016/S0140-6736(24)01867-1
Conceição-Neto, O. C., da Costa, B. S., Pontes, L., Silveira, M. C., Justo-da-Silva, L. H., de Oliveira Santos, I. C., et al. (2022). Polymyxin resistance in clinical isolates of K. pneumoniae in Brazil: update on molecular mechanisms, clonal dissemination and relationship with KPC-producing strains. Front. Cell. Infection Microbiol. 12, 898125. doi: 10.3389/fcimb.2022.898125
Ding, X., Yin, K., Li, Z., Lalla, R. V., Ballesteros, E., Sfeir, M. M., et al. (2020). Ultrasensitive and visual detection of SARS-CoV-2 using all-in-one dual CRISPR-Cas12a assay. Nat. Commun. 11, 4711. doi: 10.1038/s41467-020-18575-6
EUCAST (2025). Clinical breakpoints - breakpoints and guidance. Available online at: https://www.eucast.org/clinical_breakpoints (Accessed May 23, 2025).
Fozouni, P., Son, S., Díaz de León Derby, M., Knott, G. J., Gray, C. N., D’Ambrosio, M. V., et al. (2021). Amplification-free detection of SARS-CoV-2 with CRISPR-Cas13a and mobile phone microscopy. Cell 184, 323–333.e329. doi: 10.1016/j.cell.2020.12.001
Gong, L., Jin, Z., Liu, E., Tang, F., Yuan, F., Liang, J., et al. (2022). Highly sensitive and specific detection of mobilized colistin resistance gene mcr-1 by CRISPR-based platform. Microbiol Spectr. 10, e0188422. doi: 10.1128/spectrum.01884-22
Gootenberg, J. S., Abudayyeh, O. O., Lee, J. W., Essletzbichler, P., Dy, A. J., Joung, J., et al. (2017). Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 356, 438–442. doi: 10.1126/science.aam9321
Harrington, L. B., Burstein, D., Chen, J. S., Paez-Espino, D., Ma, E., Witte, I. P., et al. (2018). Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science 362, 839–842. doi: 10.1126/science.aav4294
Honda, M., Okuno, Y., Yoo, J., Ha, T., and Spies, M. (2011). Tyrosine phosphorylation enhances RAD52-mediated annealing by modulating its DNA binding. EMBO J. 30, 3368–3382. doi: 10.1038/emboj.2011.238
Jiang, X., Ellabaan, M. M. H., Charusanti, P., Munck, C., Blin, K., Tong, Y., et al. (2017). Dissemination of antibiotic resistance genes from antibiotic producers to pathogens. Nat. Commun. 8, 15784. doi: 10.1038/ncomms15784
Kang, K., Imamovic, L., Misiakou, M. A., Bornakke Sørensen, M., Heshiki, Y., Ni, Y., et al. (2021). Expansion and persistence of antibiotic-specific resistance genes following antibiotic treatment. Gut Microbes 13, 1–19. doi: 10.1080/19490976.2021.1900995
Kelesidis, T. and Falagas, M. E. (2015). The safety of polymyxin antibiotics. Expert Opin. Drug Saf. 14, 1687–1701. doi: 10.1517/14740338.2015.1088520
Kellner, M. J., Koob, J. G., Gootenberg, J. S., Abudayyeh, O. O., and Zhang, F. (2019). SHERLOCK: nucleic acid detection with CRISPR nucleases. Nat. Protoc. 14, 2986–3012. doi: 10.1038/s41596-019-0210-2
Li, H., Dong, X., Wang, Y., Yang, L., Cai, K., Zhang, X., et al. (2021). Sensitive and easy-read CRISPR strip for COVID-19 rapid point-of-care testing. Crispr J. 4, 392–399. doi: 10.1089/crispr.2020.0138
Li, H., Wang, S., Dong, X., Li, Q., Li, M., Li, J., et al. (2020). CRISPR-cas13a cleavage of dengue virus NS3 gene efficiently inhibits viral replication. Mol. Ther. Nucleic Acids 19, 1460–1469. doi: 10.1016/j.omtn.2020.01.028
Liu, Z., Banaei, N., and Ren, K. (2017). Microfluidics for combating antimicrobial resistance. Trends Biotechnol. 35, 1129–1139. doi: 10.1016/j.tibtech.2017.07.008
Martin-Mateos, R., Martínez-Arenas, L., Carvalho-Gomes, Á., Aceituno, L., Cadahía, V., Salcedo, M., et al. (2024). Multidrug-resistant bacterial infections after liver transplantation: Prevalence, impact, and risk factors. J. Hepatol 80, 904–912. doi: 10.1016/j.jhep.2024.02.023
Murray, C. J.L., Ikuta, K. S., Sharara, F., Swetschinski, L., Aguilar, G. R., Gray, A., et al. (2022). Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet J. 399, P629–P655. doi: 10.1016/S0140-6736(21)02724-0
Nanayakkara, A. K., Boucher, H. W., Fowler, V. G., Jr., Jezek, A., Outterson, K., and Greenberg, D. E. (2021). Antibiotic resistance in the patient with cancer: Escalating challenges and paths forward. CA Cancer J. Clin. 71, 488–504. doi: 10.3322/caac.21697
Nation, R. L., Velkov, T., and Li, J. (2014). Colistin and polymyxin B: peas in a pod, or chalk and cheese? Clin. Infect. Dis. 59, 88–94. doi: 10.1093/cid/ciu213
Patchsung, M., Homchan, A., Aphicho, K., Suraritdechachai, S., Wanitchanon, T., Pattama, A., et al. (2023). A multiplexed cas13-based assay with point-of-care attributes for simultaneous COVID-19 diagnosis and variant surveillance. Crispr J. 6, 99–115. doi: 10.1089/crispr.2022.0048
Poirel, L., Jayol, A., Bontron, S., Villegas, M. V., Ozdamar, M., Türkoglu, S., et al. (2015). The mgrB gene as a key target for acquired resistance to colistin in Klebsiella pneumoniae. J. Antimicrob Chemother. 70, 75–80. doi: 10.1093/jac/dku323
Rodríguez-Santiago, J., Cornejo-Juárez, P., Silva-Sánchez, J., and Garza-Ramos, U. (2021). Polymyxin resistance in Enterobacterales: overview and epidemiology in the Americas. Int. J. Antimicrobial Agents 58, 106426. doi: 10.1016/j.ijantimicag.2021.106426
Sun, Q., Oltra, E., Dijck-Brouwer, D. A. J., Chillon, T. S., Seemann, P., Asaad, S., et al. (2023). Autoantibodies to selenoprotein P in chronic fatigue syndrome suggest selenium transport impairment and acquired resistance to thyroid hormone. Redox Biol. 65, 102796. doi: 10.1016/j.redox.2023.102796
Tadesse, T., Alemayehu, H., Medhin, G., Akalu, A., Eguale, T., Chabou, S., et al. (2024). Antibiogram of Escherichia coli Isolated from Dairy Cattle and in-Contact Humans in Selected Areas of Central Ethiopia. Vet Med. (Auckl) 15, 117–127. doi: 10.2147/vmrr.S456247
Thompson, T. (2022). The staggering death toll of drug-resistant bacteria. Nature. doi: 10.1038/d41586-022-00228-x
Wang, Y., Lu, J., Engelstädter, J., Zhang, S., Ding, P., Mao, L., et al. (2020). Non-antibiotic pharmaceuticals enhance the transmission of exogenous antibiotic resistance genes through bacterial transformation. Isme J. 14, 2179–2196. doi: 10.1038/s41396-020-0679-2
Willyard, C. (2017). The drug-resistant bacteria that pose the greatest health threats. Nature 543, 15. doi: 10.1038/nature.2017.21550
Wise, M. G., Estabrook, M. A., Sahm, D. F., Stone, G. G., and Kazmierczak, K. M. (2018). Prevalence of mcr-type genes among colistin-resistant Enterobacteriaceae collected in 2014-2016 as part of the INFORM global surveillance program. PloS One 13, e0195281. doi: 10.1371/journal.pone.0195281
Yin, D., Yin, L., Wang, J., Shen, X., Pan, X., Hou, H., et al. (2022). Visual detection of duck tembusu virus with CRISPR/cas13: A sensitive and specific point-of-care detection. Front. Cell Infect. Microbiol 12, 848365. doi: 10.3389/fcimb.2022.848365
Keywords: mcr-1, polymyxin, antibiotic resistance, CRISPR/Cas13a, lateral flow strip
Citation: Song Y, Hu Q, Han Y, Liu H, Huang Z, Niu M, Dong X, Yan K, Jin L, Li H and Sun Y (2025) Detection assay of polymyxin resistance coding mcr-1 gene based on CRISPR/Cas13a system. Front. Cell. Infect. Microbiol. 15:1553681. doi: 10.3389/fcimb.2025.1553681
Received: 31 December 2024; Accepted: 23 May 2025;
Published: 05 June 2025.
Edited by:
Ghassan M Matar, American University of Beirut, LebanonReviewed by:
Mira El Chaar, University of Balamand, Beirut, LebanonJonathan Rodriguez-Santiago, Center for Research on Infectious Diseases (CISEI), Mexico
Tarek Alsanouri, The Eastern Mediterranean Public Health Network (EMPHNET), Jordan
Copyright © 2025 Song, Hu, Han, Liu, Huang, Niu, Dong, Yan, Jin, Li and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yansong Sun, c3VueXM2NDQzQDEyNi5jb20=; Hao Li, bGloYW84ODY2MzIzOUAxMjYuY29t
†These authors have contributed equally to this work and share first authorship