 Liying Hu1,2†
Liying Hu1,2† Juane Lu1,2†Hongfei Fan3
Juane Lu1,2†Hongfei Fan3 Changcheng Niu1,2Yanping Han1,2
Changcheng Niu1,2Yanping Han1,2 Qinggele Caiyin1,4,5
Qinggele Caiyin1,4,5 Hao Wu2*
Hao Wu2* Jianjun Qiao1,2,4,5*
Jianjun Qiao1,2,4,5*- 1School of Chemical Engineering and Technology, Tianjin University, Tianjin, China
- 2Zhejiang Research Institute of Tianjin University (Shaoxing), Shaoxing, China
- 3Tianjin Key Laboratory of Food Science and Biotechnology, College of Biotechnology and Food Science, Tianjin University of Commerce, Tianjin, China
- 4Key Laboratory of Systems Bioengineering (Ministry of Education), Tianjin University, Tianjin, China
- 5State Key Laboratory of Synthetic Biology, Tianjin University, Tianjin, China
The FAS cell surface death receptor, a member of the tumor necrosis factor receptor family, activates both apoptotic and non-apoptotic signaling upon interaction with its ligand FASL. It is critical in cell migration, invasion, immune responses, and carcinogenesis. Pathogen infection can influence host cells’ behavior by modulating the FAS/FASL pathway, thereby influencing disease progression. Understanding the role of FAS signaling in the context of pathogen interactions is therefore crucial. This review examines FAS-mediated apoptotic and non-apoptotic signaling pathways, with particular emphasis on the mechanisms of apoptosis and inflammation induced by bacterial and viral infections. Additionally, it highlights therapeutic strategies, including drug, cytokine, antibody, and FASL recombinant protein therapies, providing new directions for treating pathogenic infections and cancers, as well as insights into developing novel therapeutic approaches.
1 Introduction
FAS (also known as CD95, APO-1), a member of the tumor necrosis factor receptor family, is ubiquitously expressed on the surface of most cells and tissues, particularly on immune cells such as activated macrophages and T cells (Nagata and Golstein, 1995; Park et al., 2003). Discovered by Trauth et al., 1989, FAS was identified as a cell surface protein that induces apoptosis upon interaction with the APO-1 monoclonal antibody (Trauth et al., 1989). In 1991, Nagata’s group cloned this receptor protein. Two years later, they cloned its homologous ligand, FASL (CD95L), which is primarily expressed on activated T cells and natural killer (NK) cells. T cells trigger apoptosis and exert cytotoxic effects on target cells by binding FASL to FAS (Itoh et al., 1991; Suda et al., 1993; Kägi et al., 1994; Oshimi et al., 1996; Malyshkina et al., 2017).
The FAS protein has a molecular weight of approximately 48 kDa and is composed of three primary regions: an extracellular domain containing three cysteine-rich subdomains essential for homotrimerization, a transmembrane structural domain, and a cytoplasmic region that includes a death domain and a potential negative regulatory region (Behrmann et al., 1994; Consonni et al., 2022). Activation of FAS triggers apoptosis, thereby inhibiting abnormal cell proliferation. Dysfunctional FAS signaling, however, can lead to autoimmune diseases or contribute to cancer progression (Nagata and Golstein, 1995). Impaired or mutated FAS signaling can activate pro-survival pathways, such as NF-κB and MAPK/ERK, enhancing cancer cell proliferation, migration, and inflammatory microenvironment formation (O’Brien et al., 2005). In advanced cancers, tumor cells may evade cytotoxic T-cell killing by upregulating FASL and downregulating FAS, creating an immune-privileged microenvironment that drives malignancy (O’Brien et al., 2005). FAS participates in both apoptotic and non-apoptotic signaling pathways and plays a pivotal role in cell migration, invasion, immune regulation, and cancer development.
Pathogens, including bacteria and viruses, can modulate host cell FAS-mediated signaling pathways through various strategies to facilitate their survival and replication. For example, Yersinia pestis degrades cell surface FASL via its protease Pla, thereby inhibiting FAS-mediated apoptosis and suppressing the release of inflammatory cytokine. This reduces immune cell recruitment, creating an immunosuppressive microenvironment that facilitates bacterial survival and replication (Caulfield et al., 2014). The novel pandemic influenza A virus (H1N1) upregulates FASL to enhance the extrinsic apoptotic pathway. This regulation likely maintains viral replicative niches within host cells either by promoting viral release during cell death or by exploiting the apoptotic environment to suppress host immune clearance (Wang et al., 2014). Additionally, other pathogens including parasites (such as Toxoplasma gondii, and Trypanosoma cruzi) manipulate the FAS/FASL signaling pathway to facilitate infection (Nakajima-Shimada et al., 2000; Vutova et al., 2007). However, pathogen manipulation of the FAS/FASL signaling pathway to resist host immune responses triggers persistent inflammation, organ damage, and tumor metastasis, thereby exacerbating disease progression. Recent advances in research have elucidated both the macro-regulation and micro-molecular mechanisms of pathogen interactions with FAS-mediated pathways, offering new therapeutic opportunities. Current treatment strategies targeting FAS include chemotherapy, radiotherapy, cytokine therapy, monoclonal antibody therapy, etc., which are primarily applied to cancers and autoimmune diseases associated with FAS gene mutations (Risso et al., 2022). In addition, natural small-molecule drugs have emerged as promising treatments for FAS-related diseases in recent years. These novel approaches offer valuable insights into managing pathogen-induced FAS-mediated inflammation and associated disorders.
This review discusses FAS-mediated apoptotic and non-apoptotic signaling pathways, focusing on the mechanisms of FAS-mediated apoptosis and inflammation triggered by bacterial and viral infections. It also highlights recent advancements in therapies targeting the FAS/FASL system, providing valuable insights into the pathogen-induced disease mechanisms and the development of innovative therapeutic strategies.
2 FAS-mediated signaling pathways
2.1 Apoptotic signaling pathway
The FAS-mediated apoptotic signaling pathway has been extensively studied. FAS is activated by its homologous ligand, FASL, located on the plasma membrane, which promotes aggregation and triggers conformational changes. FASL, a cell-surface glycoprotein, exists in two forms: transmembrane (mFASL) and soluble (sFASL). While mFASL induces cell death, sFASL primarily activates non-apoptotic signaling pathways (O’Reilly et al., 2009; Risso et al., 2023). Upon binding to mFASL, FAS recruits the adaptor protein FAS-associated death domain (FADD), which recruits procaspase-8, procaspase-10, and cellular FLICE inhibitory protein (c-FLIP) via their homotypic death effector domain (DED), forming a membrane-bound death-inducing signaling complex (DISC) (Lavrik and Krammer, 2012; Ranjan and Pathak, 2024). Within the DISC, procaspase-8 is cleaved into its active form, which subsequently activates downstream caspases, such as caspase-3 and caspase-7, thereby initiating apoptosis (Lavrik and Krammer, 2012; Sahoo et al., 2023) (Figure 1). Notably, c-FLIP exists in two common isoforms: the long form (c-FLIPL) and short form (c-FLIPS), which regulate the FAS apoptotic pathway through interactions with procaspase-8. As a bifunctional regulator, c-FLIPL can either promote or inhibit apoptosis. When binding to procaspase-8, it forms heterodimers that induce conformational changes, triggering partial autocleavage and allosteric activation of caspase-8 to initiate apoptosis (Chang et al., 2002; Yu et al., 2009). However, studies show that c-FLIPL may also block complete caspase-8 processing by retaining cleaved caspase-8 within the DISC, thereby terminating apoptotic signaling (Micheau et al., 2002). Its controversial pro- or anti-apoptotic roles appear context-dependent, influenced by concentration and cell type (Krueger et al., 2001). In contrast, c-FLIPS forms dysfunctional heterodimers with procaspase-8, and its truncated C-terminal domain fails to support procaspase-8 cleavage, thereby completely inhibiting DISC-mediated caspase-8 activation and apoptosis initiation (Schleich et al., 2016).
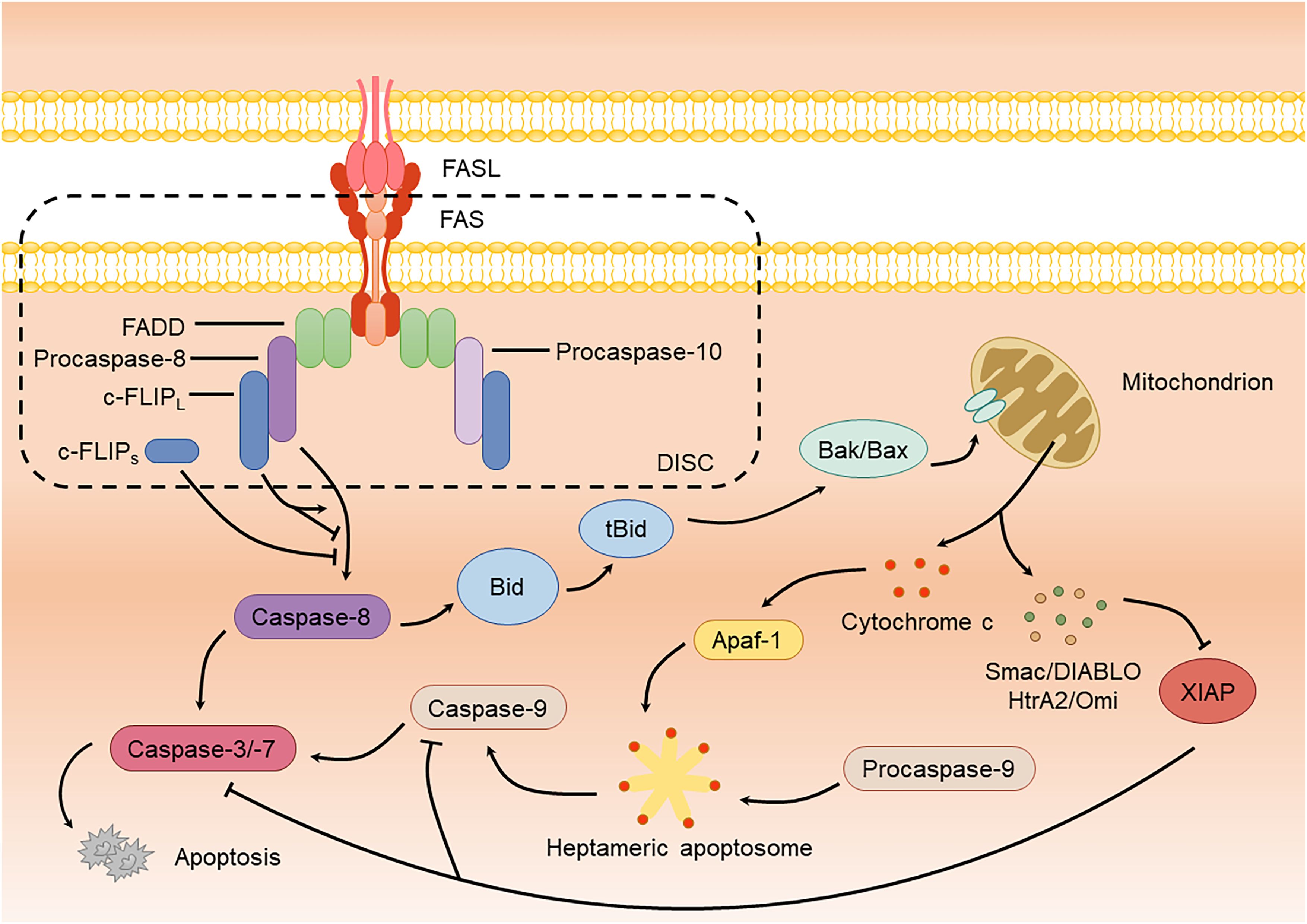
Figure 1. FAS-mediated apoptotic signaling pathway.
The aforementioned apoptotic pathway is categorized as extrinsic apoptosis with rapid DISC formation and significant caspase-8 production. In contrast, intrinsic apoptosis is more dependent on mitochondria and produces lower amounts of DISC and caspase-8 (Galluzzi et al., 2018). In this pathway, a small amount of caspase-8 cleaves Bid (BH3-interacting domain death agonist, belonging to the Bcl-2 family) (Kim et al., 2023). The truncated form of Bid (tBid) interacts with pro-apoptotic proteins Bak or Bax, also members of the Bcl-2 family, promoting their oligomerization and resulting in mitochondrial outer membrane permeabilization (MOMP), which releases cytochrome c (Flores-Romero et al., 2022). Once released into the cytoplasm, cytochrome c binds to Apaf-1 (apoptotic protease activating factor 1), forming a heptameric apoptosome. The apoptosome recruits caspase-9 monomers through their caspase recruitment domains (CARDs), catalyzing their activation (Li et al., 2017; Bock and Tait, 2020). Active caspase-9 subsequently activates downstream caspase-3 and caspase-7, inducing apoptosis (Srinivasula et al., 1998) (Figure 1). During this process, mitochondria also release Smac/DIABLO (diablo IAP-binding mitochondrial protein) and HtrA2/Omi (serine protease HTRA2), which block the inhibition of caspase-3/-7/-9 by XIAP (X-chromosome-linked inhibitor of apoptosis protein), thereby ensuring the efficient progression of apoptosis (Bock and Tait, 2020) (Figure 1).
In addition to caspase-mediated apoptosis, FAS can also mediate necroptosis via RIP kinase. Under FAS stimulation, RIPK1 is recruited to the DISC for activation, subsequently binding to RIPK3 via the RHIM (RIP homotypic interaction motif) to form the necrosome. RIPK3 then phosphorylates MLKL (mixed lineage kinase domain-like protein), inducing its oligomerization, membrane insertion, and disruption of membrane integrity, which triggers necroptosis (Holler et al., 2000; Sun et al., 2012; Cai et al., 2014; Liu et al., 2021).
2.2 Non-apoptotic signaling pathways
Upon activation, FAS triggers signaling pathways such as NF-κB, MAPK, and PI3K/AKT through different mechanisms. These pathways regulate immune responses and promote cell proliferation, migration, and invasion by inducing the production of inflammatory factors and chemokines (Figure 2).
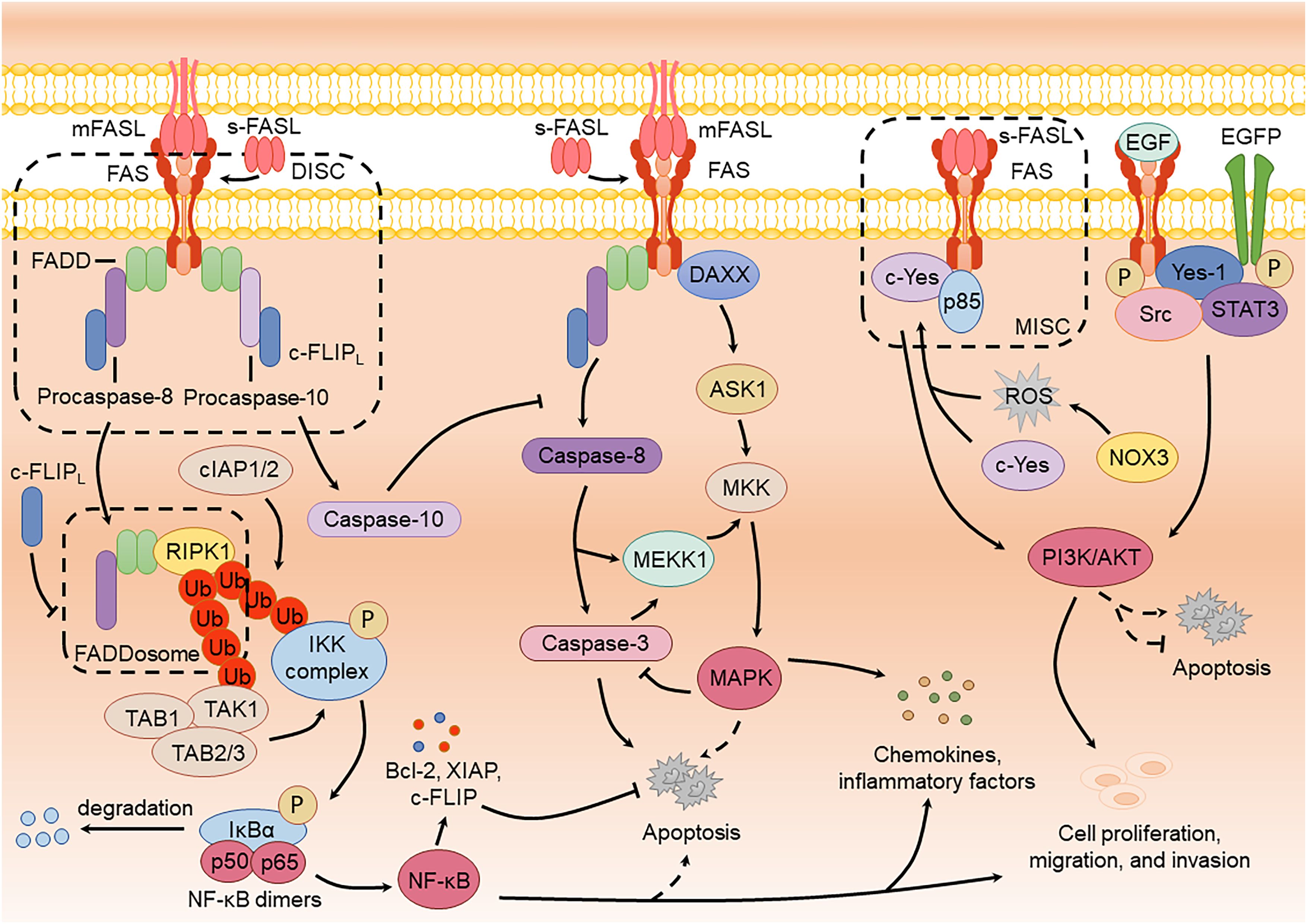
Figure 2. FAS-mediated non-apoptotic signaling pathways and the crosstalk with apoptotic signaling pathway.
2.2.1 NF-κB signaling pathway
Studies have shown that FAS activation can induce both apoptotic and NF-κB signaling within a single cell (Schmidt et al., 2015). However, the robust apoptotic response often obscures NF-κB activation. Upon FAS activation by FASL, the death domain recruits RIPK1 to FADD, forming a cytoplasmic procaspase-8-FADD-RIPK1 complex termed the “FADDosome” or “Complex II”. Within this complex, RIPK1 recruits the E3 ubiquitin ligases cIAP1/2 (cellular inhibitor of apoptosis protein 1/2), mediating its ubiquitination to generate ubiquitin chains. These chains recruit TAK1 (TGF-β-activated kinase 1) along with its adaptors, TAB1 and TAB2/3, as well as the IKK complex. Activated TAK1 phosphorylates and activates the IKK complex, which subsequently phosphorylates IκBα, leading to its degradation. This releases NF-κB dimers (such as p50/p65) for nuclear translocation, initiating the expression of pro-inflammatory cytokines such as IL-8 and driving inflammatory and immune regulatory responses (Kreuz et al., 2004; Ea et al., 2006; Henry and Martin, 2017). Activation of the NF-κB pathway through FAS can also lead to cell proliferation, migration, and invasion. For example, Zhang et al. showed that low-dose sFASL activates the FAS-mediated FADD-FLIP-TRAF-NF-κB pathway, resulting in elevated vascular endothelial growth factor (VEGF) levels and enhanced proliferation and migration of brain endothelial cells (Zhang et al., 2015). Notably, caspase-8 plays a crucial scaffold role in FADDosome, and its enzymatic activity is unrelated to the production of pro-inflammatory factors. The presence of enzymatic activity can cleave RIPK1, potentially explaining the masking of NF-κB activation (Kreuz et al., 2004; Henry and Martin, 2017).
Multiple factors are involved in regulating the activation of NF-κB in FAS, including caspase-10, TRAF2 (TNF receptor-associated factor 2), IAPs (inhibitor of apoptosis proteins), and A20 (deubiquitinating enzyme A20) (Davidovich et al., 2023). For instance, Horn et al. demonstrated that caspase-10 negatively regulates caspase-8-mediated cell death while facilitating NF-κB activation (Horn et al., 2017). In contrast, c-FLIP (mainly c-FLIPL) inhibits FADDosome formation, thereby suppressing FAS-induced NF-κB signaling and the production of inflammatory cytokines (Davidovich et al., 2023). Despite these insights, the precise mechanism underlying the formation of FADDosome remains to be fully elucidated. It is still unclear whether this process results from natural detachment from the receptor or is driven by other enzymatic activities that cleave the complex, necessitating further investigation.
2.2.2 MAPK signaling pathway
FAS is dependent on caspase activity to participate in inducing the activation of the MAPK signaling pathway. Ligand- or agonistic antibody-induced FAS activation generates active caspase-8 or caspase-3, which cleaves mitogen-activated protein kinase 1 (MEKK1), resulting in active MEKK1 (Deak et al., 1998; Park and Baek, 2022). Active MEKK1 interacts with mitogen-activated protein kinase kinases (MKKs), such as MKK6 and MKK7, leading to the phosphorylation of ERK, JNK, and p38 (Toyoshima et al., 1997; Park and Baek, 2022). These kinases, in turn, induce the production of inflammatory factors, enabling cells to resist external stimuli and enhance survival. Matsumoto et al. found that caspase-8 inhibition in HEK293 cells eliminates FAS-mediated JNK activation, thereby suppressing AP-1 and IL-8 production (Matsumoto et al., 2007). Similarly, Kober et al. reported that c-FLIP inhibits the MAPK pathway by preventing caspase-8 processing, resulting in a decrease in FAS-mediated activation of ERK and p38 (Kober et al., 2011). Activating the MAPK pathway through FAS can also lead to cell proliferation, migration, and invasion. Li et al. reported that oral cancer cell stemness and lung metastasis benefit from FAS and ERK phosphorylation (Li et al., 2022).
Alternatively, MAPK activation can occur independently of caspases. For example, FAS stimulation in primary T lymphocytes can induce ERK and p38 pathways even when caspases are inhibited (Scheller et al., 2002). This caspase-independent activation likely involves FAS interacting with the receptor-associated protein DAXX (death-domain associated protein 6), which subsequently interacts with apoptosis signal-regulating kinase 1 (ASK1), leading to MKK phosphorylation to induce JNK activation (Ichijo et al., 1997; Pleinis et al., 2017; Bogolyubova and Bogolyubov, 2021). Recent studies suggest that DAXX promotes the proliferation of ovarian cancer ascites cells by activating the ERK pathway and directly binding to CEBP-β (Liu et al., 2018). However, whether FAS plays a direct role in this pathway remains unclear and warrants further investigation.
2.2.3 PI3K/AKT signaling pathway
FAS-mediated PI3K/AKT signaling mainly promotes cell proliferation, migration, and invasion. During this process, sFASL binds to FAS, recruiting the Src family kinase c-Yes and the PI3K subunit p85 via NADPH oxidase 3 (NOX3) and ROS production, forming a motility-inducing signaling complex (MISC) that activates the PI3K/AKT pathway (Kleber et al., 2008; Tauzin et al., 2011; Akhiani and Martner, 2022). Activation of this pathway induces NF-κB activation and the cleavage of the extracellular matrix by MMPs (matrix metalloproteinases), including fibronectin, laminin, and type IV collagen, thereby enhancing cell migration and invasion. Furthermore, phosphorylated FAS interacts with epidermal growth factor receptor (EGFR) to form a complex comprising FAS, EGFR, Yes-1, Src, and STAT3 (signal transducer and activator of transcription 3) (Ta et al., 2018). Upon epidermal growth factor (EGF) stimulation, phosphorylated FAS accumulates in the nucleus, facilitating the nuclear localization of phosphorylated EGFR and STAT3. This process promotes cyclin D1 expression and STAT3-mediated activation of the AKT and MAPK pathways, thereby enhancing cell proliferation and migration (Ta et al., 2018; Seyrek and Lavrik, 2019).
2.3 Crosstalk between apoptotic and non-apoptotic pathways
FAS-mediated signaling pathways exhibit considerable crosstalk. FAS-mediated NF-κB activation induces anti-apoptotic proteins (including Bcl-2 family members, c-FLIP, XIAP) through its target genes, thereby suppressing FAS-driven apoptosis and establishing a negative feedback loop (Schram and Rothstein, 2003; Bernal-Mizrachi et al., 2006). Paradoxically, NF-κB can also promote apoptosis under specific contexts. As reported by Jennewein et al., NF-κB enhances DISC assembly to facilitate FAS-mediated apoptosis in glioblastoma cells (Jennewein et al., 2012). Additionally, MAPK activation suppresses FAS-mediated apoptosis by inhibiting caspase-8/-3 activity, while paradoxically, p38 MAPK has been reported to activate the intrinsic apoptotic pathway via FAS in CD8+ T cells (Holmström et al., 1999; Alvarado-Kristensson et al., 2004; Farley et al., 2006). The PI3K/AKT pathway also exhibits dual regulatory roles in FAS-mediated apoptosis (Lu et al., 2006; Liu et al., 2019b). These findings underscore the complexity of FAS signaling regulation, where pathway strength and outcomes depend on cell type and the surrounding microenvironment. Furthermore, inflammatory cytokines generated by non-apoptotic FAS signaling may reciprocally modulate apoptotic pathways, highlighting their therapeutic implications in FAS-driven diseases.
3 FAS-mediated apoptosis and inflammation induced by pathogen infection
Bacteria and viruses, two distinct classes of pathogens, differ significantly in infection mechanisms. Bacteria are prokaryotes with independent metabolic systems and reproductive capabilities. They invade hosts by secreting toxins to directly damage cells or injecting effector proteins via the type III secretion system to manipulate host cell signaling (Cossart and Sansonetti, 2004). In contrast, viruses are parasitic and rely on host cells for replication (König and Stertz, 2015). Hosts use apoptosis as a defense mechanism to limit infection and eliminate pathogens, while pathogens can exploit this process to enhance their spread. Bacteria like Salmonella induce apoptosis to escape host cells or evade immune clearance, whereas others such as Mycobacterium tuberculosis (M. tuberculosis) suppress apoptosis to prolong intracellular survival (Monack et al., 1996; Oddo et al., 1998). Similarly, many viruses inhibit apoptosis to sustain host cells for replication, as seen in human papilloma virus’s (HPV) E6 protein degrading p53, while some (such as adenoviruses) later trigger apoptosis to release progeny virions (Tollefson et al., 1996; Filippova et al., 2004). The differing reliance on apoptosis between two pathogen types stems from their survival strategies: bacteria depend on active dissemination, using apoptosis as an auxiliary mechanism to breach host barriers, while viruses require replication-release cycles, making apoptosis regulation critical to their life cycle. Host immunity plays a critical role in balancing the impact of infection, with chemotactic and inflammatory factors recruiting immune cells to eliminate infected cells via apoptosis, though excessive immune responses may occur. Here, we focus on FAS/FASL-mediated apoptosis or inflammation induced by bacterial and viral infections (Table 1).
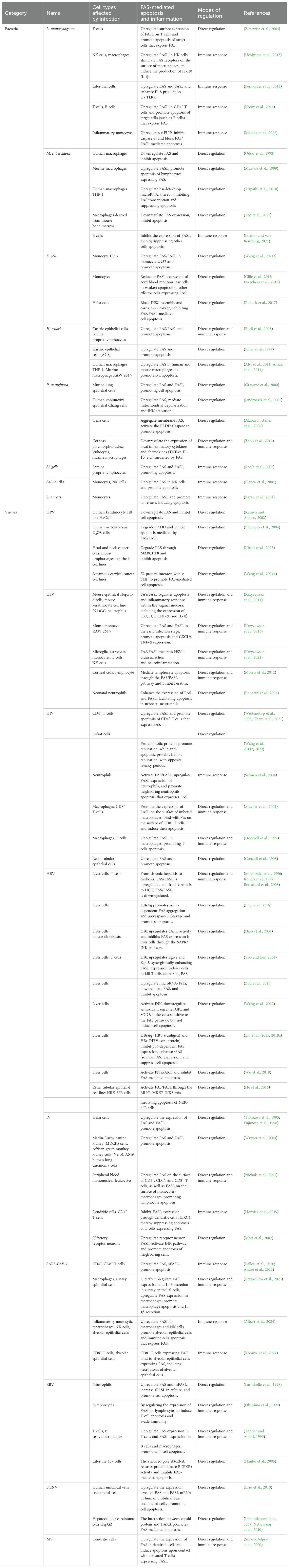
Table 1. Apoptosis and inflammation triggered by pathogen infection through FAS signaling.
3.1 Bacteria
3.1.1 Listeria monocytogenes
Listeria monocytogenes (L. monocytogenes), a foodborne pathogen, can cross the intestinal, blood-brain, and placental barriers, causing bacteremia and listeriosis (Kammoun et al., 2022). Early studies found that L. monocytogenes directly interacts with T cells through its virulence factors, listeriolysin O (LLO) and phosphatidylcholine-preferring phospholipase C (PC-PLC), promoting the surface expression of FASL. This FASL subsequently binds to FAS on neighboring cells, triggering apoptosis (Zenewicz et al., 2004). Later, Uchiyama et al. demonstrated that L. monocytogenes infection upregulates FASL expression in NK cells, which subsequently stimulates FAS receptors on the surface of macrophages, leading to IL-18/IL-1β production via a caspase-8 and ROS-dependent mechanism (Uchiyama et al., 2013). Fernandes et al. further reported that L. monocytogenes upregulates FAS, FASL and enhances IL-8 production via activating host Toll-like receptors (TLRs, TLR4 and TLR5) signaling (Fernandes et al., 2014). More recently, L. monocytogenes was shown to upregulate FASL expression in CD4+ T cells, inducing apoptosis in target cells that express FAS (Kotov et al., 2018). Additionally, the L. monocytogenes surface protein InlB upregulates c-FLIP through the c-Met-PI3Kα pathway, inhibiting caspase-8 activation and blocking FAS/FASL-mediated apoptosis, thereby facilitating bacterial invasion and spread (Maudet et al., 2022). These studies indicate that L. monocytogenes modulates FAS-mediated apoptosis and inflammatory responses through host immune mechanisms, rather than directly targeting FAS or FASL.
3.1.2 Mycobacterium tuberculosis
Mycobacterium tuberculosis (M. tuberculosis), the causative agent of tuberculosis (TB), is primarily transmitted through airborne droplets and is highly infectious, often existing in a latent state (Palittapongarnpim et al., 2024). M. tuberculosis can bind to macrophages via TLRs and activate intracellular signaling pathways leading to macrophage apoptosis or necroptosis (Yao et al., 2017). However, it can also affect the survival of host cells through FAS. M. tuberculosis evades immune clearance by differentially modulating FAS/FASL in host macrophages. In human macrophages, M. tuberculosis downregulates FAS directly to inhibit apoptosis and promote cell survival, while in mouse macrophages, it upregulates FASL directly to kill FAS-expressing lymphocytes, protecting infected cells (Oddo et al., 1998; Mustafa et al., 1999). Tripathi et al. further demonstrated that M. tuberculosis suppresses FAS transcription in THP-1 human macrophages by upregulating hsa-let-7b-5p, which binds to the FAS 3’ untranslated region (3’UTR), thereby blocking apoptosis and enhancing bacterial persistence (Tripathi et al., 2018). These mechanisms underlie M. tuberculosis’s chronic infection strategy.
Clinical studies indicate that FASL expression in B cells of TB patients is significantly reduced compared to healthy individuals, but it returns to normal levels after six months of anti-tuberculosis treatment, suggesting its potential as a biomarker for treatment response (Loxton and van Rensburg, 2021). This indicates that M. tuberculosis suppresses the host’s immune response through the FAS/FASL system, thereby promoting the survival of infected cells. However, some studies have shown that apoptosis-related genes, such as FADD and caspase-8, are significantly upregulated in T cell subsets of TB patients, potentially driving T cells apoptosis through FAS-independent mechanisms (Canaday et al., 2001; Wang et al., 2023). Based on recent studies, M. tuberculosis infection inhibits the expression of FAS/FASL, thereby suppressing apoptosis. It also induces the expression of various inflammatory factors, but there is no direct evidence to suggest that these effects are mediated by FAS.
3.1.3 Escherichia coli
Escherichia coli (E. coli), the most common gram-negative pathogen, is widely used as a model organism and is responsible for diseases such as colitis, diarrhea, and bacteremia (Gong et al., 2024). Studies investigating the interaction between E. coli and the FAS pathway primarily focus on monocytes as host cells. Wang et al. showed that E. coli infection directly upregulates FAS and FASL expression in U937 monocytes, triggering caspase-8-dependent apoptosis (Wang et al., 2011a). During bacterial infection, the apoptotic clearance of phagocytic monocytes suppresses the pro-inflammatory response in later stages (Dreschers et al., 2019). However, in neonatal monocytes, impaired apoptosis of phagocytic monocytes prolongs the pro-inflammatory phase, leading to persistent inflammation. Orlikowsky et al. showed that E. coli infection upregulates MMP-9 in cord blood mononuclear cells compared to peripheral blood mononuclear cells. MMP-9 cleaves mFASL, reducing cell surface FASL expression, which weakens apoptosis of FAS-expressing effector cells (such as T cells) and promotes persistent inflammation (Gille et al., 2013; Dreschers et al., 2019). Additionally, enteropathogenic Escherichia coli (EPEC) infection inhibits FAS-mediated apoptosis, primarily through the actions of the effector proteins NleB and NleF. In vitro infections, NleB glycosylates the death domain of FADD to block the formation of the DISC, thereby inhibiting caspase-8 activation. Conversely, NleF directly binds caspases-8, -4, and -9 to suppress their enzymatic activity (Pollock et al., 2017).
3.1.4 Helicobacter pylori
Helicobacter pylori (H. pylori), a gram-negative bacterium transmitted via the fecal-oral route, is associated with chronic gastritis, peptic ulcers, and even gastric cancer (Engelsberger et al., 2024). Research on FAS-mediated apoptosis and inflammation caused by H. pylori infection is still in its early stages. Early studies show that H. pylori infection promotes apoptosis in gastric epithelial cells by directly upregulating FAS/FASL, or mediates cell clearance via host immune responses by upregulating FAS/FASL in lamina propria lymphocytes (Rudi et al., 1998). Other co-culture experiments with H. pylori and gastric epithelial cells have also shown increased FAS expression, which induces apoptosis (Jones et al., 1999). This upregulation may be linked to elevated levels of the p73 protein, H. pylori stabilizes the p73 protein via the cag pathogenicity island (cag PAI) and c-Abl tyrosine kinase, prolonging its half-life and directly inducing FAS transcription to promote apoptosis (Wei et al., 2008). Further studies have demonstrated that H. pylori-induced apoptosis predominantly occurs through the mitochondrial pathway. Additionally, H. pylori activates NF-κB, which exerts anti-apoptotic effects (Maeda et al., 2002). However, Hasumi et al. observed that H. pylori infection induces apoptosis even in cells lacking the FAS receptor, suggesting that while FAS enhances apoptosis, it is not the sole mechanism (Hasumi et al., 2002). Furthermore, H. pylori infection increases FAS expression in human and mouse macrophages, activating caspase-8 and inducing apoptosis, a process driven by the virulence factor HP986 (Alvi et al., 2011; Ansari et al., 2014).
3.1.5 Pseudomonas aeruginosa
Pseudomonas aeruginosa (P. aeruginosa), a gram-negative bacterium, is associated with both acute and chronic infections, particularly in patients with cystic fibrosis (Jurado-Martín et al., 2021). Similar to H. pylori, research on the interaction between P. aeruginosa and the FAS pathway is still in its early stages. P. aeruginosa infection has been shown to directly upregulate FAS and FASL expression independently of the host immune response, thereby inducing apoptosis. The cystic fibrosis transmembrane conductance regulator (CFTR) also modulates FAS and FASL expression during infection (Grassmé et al., 2000; Cannon et al., 2003). Furthermore, Jendrossek et al. demonstrated that P. aeruginosa adheres to human conjunctiva epithelial Chang cells and directly upregulates FAS via its type III secretion system, leading to mitochondrial depolarization and JNK activation, thereby promoting apoptosis (Jendrossek et al., 2001). In addition, the ADP-ribosyltransferase activity of P. aeruginosa effector protein ExoS induces FAS receptor clustering in HeLa cells, activating FADD-caspase 8, but apoptosis remains unaffected by FASL-neutralizing antibody, suggesting alternative apoptotic mechanisms (Alaoui-El-Azher et al., 2006). P. aeruginosa infection also induces keratitis, a condition in which the regulation of the FAS/FASL pathway induces apoptosis and suppresses localized inflammatory expression of cytokines and chemokines, such as TNF-α and IL-1β. It also upregulates macrophage production of anti-inflammatory factor IL-10 to balance pro-inflammatory factor IL-12, demonstrating host immune responses against bacterial infection via the FAS/FASL system (Zhou et al., 2010).
3.1.6 Other bacteria
Other bacterial pathogens, including Shigella, Salmonella, and Staphylococcus aureus (S. aureus), also influence apoptosis and inflammation through the FAS pathway (Table 1). Shigella infection induces upregulation of FAS and FASL in intestinal lamina propria lymphocytes via host immune responses, leading to apoptosis (Raqib et al., 2002). Blanco et al. showed that Salmonella strains, including wild-type Ty2 and mutant TYT1231, upregulate FAS expression in NK cells following monocyte infection and co-culture with NK cells, this may be due to the stimulation of cytokines produced after monocyte infection (Blanco et al., 2001). Furthermore, after being phagocytosed by monocytes, S. aureus induces FASL release on the monocyte surface, activating caspase-3 and initiating apoptosis in itself and neighboring cells via the FAS/FASL pathway (Baran et al., 2001).
3.2 Viruses
3.2.1 Human papilloma virus
Human papilloma virus (HPV) is associated with several cancers, including cervical, head and neck, vaginal, and oropharyngeal cancer (Williamson, 2023). HPV is classified into low-risk and high-risk types, with high-risk types HPV16 and HPV18 receiving the most research attention (Williamson, 2023). The HPV genome encodes six early proteins (E1, E2, E4, E5, E6, E7) and two late proteins (L1, L2) (Nelson and Mirabello, 2023). Early studies indicate that the early proteins E5 and E6 play a critical role in inhibiting FAS-mediated apoptosis, thereby promoting HPV survival and replication within host cells (Nelson and Mirabello, 2023). The E5 protein, a membrane protein, suppresses the expression of the FAS receptor on the surface of the human keratinocyte cell line HaCaT, thereby preventing the binding of FASL to the receptor and inhibiting apoptosis (Kabsch and Alonso, 2002). In contrast, the E6 protein is widely recognized for its ability to rapidly ubiquitinate and degrade the tumor suppressor p53. Furthermore, E6 protects cells from FAS-mediated apoptosis by binding to the death domain of FADD and accelerating its degradation, which enhances viral survival and dissemination (Filippova et al., 2004). Recent studies have revealed that E6 upregulates the expression of the membrane-associated RING-CH-type finger ubiquitin ligase MARCHF8 via the MYC/MAX complex. MARCHF8 ubiquitinates FAS, leading to its degradation by the proteasome or lysosome and further inhibiting FAS-mediated apoptosis (Khalil et al., 2023). Additionally, the E2 protein interacts with c-FLIP, relieving the inhibition of c-FLIP on apoptosis signaling and promoting apoptosis. However, during HPV integration into the host genome, E2 protein expression is disrupted, impairing its anti-cancer effects (Wang et al., 2011b). In summary, HPV infection mainly promotes viral replication and spread in host cells by directly inhibiting the host cell FAS/FASL system and apoptosis.
3.2.2 Herpes simplex virus
Herpes simplex virus (HSV), a neurotropic pathogen responsible for chronic human infections, contains two types: HSV-1 and HSV-2 (Rathbun and Szpara, 2021; Wijesinghe et al., 2021). It primarily causes skin and mucosal disorders, with a predilection for the oral and genital regions (Rathbun and Szpara, 2021; Wijesinghe et al., 2021). Krzyzowska et al. found that in vitro HSV-2 infection of murine epithelial and keratinocyte cells upregulated FAS, FASL, anti-apoptotic proteins (Bcl-2, NF-κB, and AKT kinase), and pro-inflammatory/chemotactic cytokines (CXCL1/2, TNF-α, and IL-1β). The early induction of anti-apoptotic proteins delayed cell death, resulting in only moderate apoptosis at later stages. Additionally, cytokines secreted by keratinocytes facilitated neutrophil migration toward infected cells, exacerbating inflammation (Krzyzowska et al., 2011). These findings highlight the role of the FAS pathway in regulating apoptosis and inflammatory responses during HSV-2 infection.
Additionally, HSV-2 infection of monocytes induced early upregulation of FAS and FASL, rendering them susceptible to FAS-stimulated apoptosis. FAS/FASL signaling further enhanced CXCL9 and TNF-α expression in cells, thereby recruiting other immune cells and potentiating local immune responses (Krzyzowska et al., 2013). Recent research has also revealed that the FAS/FASL pathway is involved in HSV-1-mediated brain infection and neuroinflammation. HSV-1 infection upregulated FAS and FASL expression in brain microglia and astrocytes, along with pro-inflammatory/chemotactic cytokines (such as CXCL9, CXCL10, TNF-α, IL-6), recruiting FASL-expressing immune cells (such as T cells) to clear infected cells (Krzyzowska et al., 2021). Interestingly, exogenous FAS stimulation on infected cells suppressed inflammatory mediator expression. This indicates that immune cell recruitment to infection sites disrupts FAS-mediated apoptosis and pro-inflammatory responses, with this paradoxical regulation exacerbating local inflammation (Krzyzowska et al., 2021). Moreover, the FAS/FASL pathway also plays a role in HSV-1-induced corneal inflammation and neonatal neutrophil apoptosis (Ennaciri et al., 2006; Morris et al., 2012) (Table 1).
3.2.3 Human immunodeficiency virus
Human immunodeficiency virus (HIV) infection leads to acquired immunodeficiency syndrome (AIDS), severely impairing the immune system and increasing susceptibility to opportunistic infections and tumors (Kumah et al., 2023). A defining feature of HIV infection is the progressive depletion of CD4+ T cells, driven by multiple mechanisms including the FAS/FASL signaling pathway. Research indicates that exogenous FAS stimulation induces apoptosis in HIV-infected CD4+ T cells (Katsikis et al., 1995). The HIV-1 Tat and gp120 proteins upregulate FASL expression in T cells, which binds to FAS-expressing neighboring cells and the subsequent apoptosis of CD4+ T cells (Westendorp et al., 1995). Recent studies have elucidated the epigenetic mechanisms involved in this process. HIV-1 infection modifies histone H3 at the FASL promoter, increasing H3K4-trimethylation (H3K4Me3) and H3K9-acetylation (H3K9Ac) while reducing H3K9-trimethylation (H3K9Me3). These changes enhance FASL expression, making cells that express FAS more prone to apoptosis (Ghare et al., 2021). However, early studies also show that even with caspase inhibition or FAS deficiency, HIV-1 can still induce apoptosis in vitro, indicating that HIV-1 can directly kill T cells via FAS-independent pathways (Gandhi et al., 1998).
Wang et al. explored the effect of FAS/FASL-related apoptosis pathway molecules on HIV-1 replication. In Jurkat cells, the expression of pro-apoptotic proteins such as FASL, FADD, and p53 promoted viral replication, whereas anti-apoptotic factors like c-FLIP, Bcl-XL, and XIAP inhibited replication (Wang et al., 2011c). Notably, during HIV-1 latency, these proteins exhibited opposing effects, underscoring the intricate regulatory role of the FAS/FASL pathway in viral dynamics (Wang et al., 2022). HIV infection also increases the sensitivity of neutrophils, CD8+ T cells, macrophages, and renal tubular epithelial cells to FAS/FASL-mediated apoptosis, compounding the damage to the host’s immune and organ systems (Conaldi et al., 1998; Dockrell et al., 1998; Mueller et al., 2001; Salmen et al., 2004) (Table 1).
3.2.4 Hepatitis B virus
Hepatitis B virus (HBV) is a hepatotropic virus infecting approximately 30 million individuals annually. Chronic HBV infection can progress to severe liver conditions, including cirrhosis and hepatocellular carcinoma (HCC) (Asandem et al., 2024). The FAS signaling pathway plays distinct roles at various stages of liver disease. From chronic hepatitis to cirrhosis, the expression of FAS and FASL increases, promoting apoptosis, whereas their levels decline during the transition from cirrhosis to HCC, reducing cell death and facilitating tumor progression (Mochizuki et al., 1996; Kondo et al., 1997; Bortolami et al., 2008).
During chronic hepatitis, HBV surface antigen (HBsAg) induces endoplasmic reticulum (ER) stress, which inactivates upstream AKT signaling molecules (PDPK1 and mTORC2). This disruption reduces AKT phosphorylation, relieving its inhibitory effect on FAS aggregation and caspase-8 cleavage, ultimately promoting apoptosis (Jing et al., 2018).
Conversely, in HCC, HBV employs distinct mechanisms to inhibit apoptosis, supporting tumor development. Multiple HBV proteins contribute to this process (Table 1). For instance, HBx protein upregulates SAPK (stress-activated protein kinase) activity, inhibits FAS expression via the SAPK/JNK pathway, and protects hepatocytes from death (Diao et al., 2001). Additionally, HBx potently activates Egr-2 (early growth response factor-2) and Egr-3 transcription, synergistically enhancing FASL expression in hepatocytes to kill FAS-expressing T cells and evade immunity (Yoo and Lee, 2004). HBV infection also induces the promoter activity of microRNA-181a, leading to its upregulation. This microRNA subsequently targets and suppresses FAS expression, thereby facilitating tumor growth (Zou et al., 2015). Beyond its effects on liver cells, HBV infection contributes to extrahepatic diseases, as listed in Table 1.
3.2.5 Influenza virus
Influenza virus (IV) infects human and animal respiratory tracts, causing highly contagious seasonal epidemics (Liang, 2023). Early studies have shown that IV infection triggers low production of IFN-β while upregulating the serine/threonine kinase PKR, which is also activated by viral dsRNA. Activated PKR promotes FAS expression, which interacts with FASL on the surface of infected cells, leading to apoptosis (Takizawa et al., 1995; Fujimoto et al., 1998). Additionally, NF-κB promotes co-expression of both FAS and FASL on infected cell surfaces, a critical process for viral replication (Wurzer et al., 2004). Later, Nichols et al. demonstrated influenza A virus (IAV) infection increases FAS on CD3+/CD4+/CD8+ T cells and FASL on monocytes/macrophages in peripheral blood, driving lymphocyte apoptosis (Nichols et al., 2001). However, recent studies reveal that dendritic cell NLRC4 (an intracellular pattern recognition receptor) suppresses FASL expression, protecting FAS-expressing CD4+ T cells from apoptosis during IAV infection (Hornick et al., 2019). These findings provide valuable insights into the mechanisms underlying disease progression and T-cell immune responses.
3.2.6 Severe acute respiratory syndrome coronavirus 2
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a novel β coronavirus, is the causative agent of coronavirus disease 2019 (COVID-19). Its clinical manifestations include fever, cough, dyspnea, pneumonia, and, in severe cases, death (Mohamadian et al., 2021). Similar to HIV infection, severe COVID-19 exhibits lymphopenia, which is closely associated with the FAS/FASL-mediated pathway. Multiple studies have demonstrated that FAS expression on T cell surfaces is significantly upregulated in severe patients, accompanied by elevated serum sFASL levels and caspase-3 activation, leading to apoptosis of CD4+ and CD8+ T cells (Bellesi et al., 2020; André et al., 2022). As discussed by Leonardi et al., FAS activation may suppress FOXO1 (forkhead box protein O1) via AKT phosphorylation, inhibiting FOXP3 (forkhead box protein P3) expression and promoting T-cell differentiation into effector phenotypes. In the context of COVID-19, this leads to excessive T-cell activation, tissue infiltration, and apoptosis, contributing to immune dysregulation (Leonardi and Proenca, 2020).
The FAS/FASL system plays a dual role in COVID-19 pathogenesis. In mouse-adapted SARS-CoV-2 models, FASL upregulation on inflammatory monocytes/macrophages and NK cells triggers alveolar epithelial and immune cell apoptosis via FAS receptor activation, causing lung injury and acute respiratory distress syndrome (ARDS) (Albert et al., 2024). In severe COVID-19 patients, FASL-expressing CD8+ T cells induce alveolar epithelial necroptosis through FAS receptor engagement, exacerbating pulmonary damage (Komiya et al., 2024). Additionally, SARS-CoV-2-infected airway epithelial cells upregulate FASL, promoting FAS-mediated macrophage death while releasing IL-6, which stimulates the production of pro-inflammatory cytokines such as IL-1β (Fraga-Silva et al., 2023). These findings underscore the dual nature of FAS/FASL signaling, balancing antiviral defense with pathological hyperinflammation and tissue injury.
3.2.7 Other viruses
Other viruses, such as Epstein-Barr virus (EBV), dengue virus (DENV), and measles virus (MV) also mediate apoptosis via the FAS/FASL pathway (Table 1). EBV infection increases FAS and mFASL expression in neutrophils, with sFASL also elevated in culture supernatants. Direct cell-cell FAS/FASL crosslinking promotes apoptosis (Larochelle et al., 1998). DENV infection increases the expression of FAS and FASL mRNA in human umbilical vein endothelial cells, driving apoptotic cell death (Liao et al., 2010). Similarly, MV infection upregulates FAS expression in dendritic cells (DCs). Co-culture of DCs with activated T cells expressing FASL induces DC apoptosis, facilitating viral release (Servet-Delprat et al., 2000).
4 Treatment methods targeting the FAS/FASL system
Dysregulation of the FAS/FASL system during pathogen infections drives chronic inflammation or autoimmune damage, whereas targeted intervention strategies against this pathway demonstrate therapeutic potential.
4.1 Drug therapy
Certain chemical agents exhibit significant regulatory effects on the FAS/FASL pathway. For instance, anesthetics like chloral hydrate demonstrate anti-inflammatory and anti-infective properties by upregulating FAS expression in macrophages., This activation induces Caspase-8/-3-dependent apoptosis, thereby eliminating these cells (Cai et al., 2016). As a result, chloral hydrate suppresses lipopolysaccharide (LPS)-induced pro-inflammatory cytokine release, highlighting its potential to mitigate infection-driven inflammation through FAS signaling (Cai et al., 2016). During M. tuberculosis infection, glutathione-boosted NK cells upregulate FASL expression to bind FAS on infected monocytes, triggering apoptosis and suppressing intracellular bacterial proliferation (Guerra et al., 2012). Furthermore, in H. pylori-infected mouse models, prolonged rebamipide treatment downregulates Fas/FASL expression in gastric mucosal epithelial cells, reducing apoptosis and alleviating chronic inflammation. Concurrently, it suppresses NF-κB-mediated pro-inflammatory cytokine release, delaying the progression of precancerous lesion (Hahm et al., 2003).
Compared to synthetic drugs, natural small-molecule drugs offer notable advantages, including reduced toxicity and fewer adverse effects, making them compelling candidates for treating FAS-related diseases. For example, In HPV infection, luteolin markedly upregulates the expression of FAS, FASL, and FADD in HeLa cells, activating the caspase-8/-3 cascade to initiate the extrinsic apoptotic pathway. Concurrently, it synergizes with the mitochondrial pathway to trigger intrinsic apoptosis, establishing a dual pro-apoptotic mechanism that restores host-mediated clearance of infected cells (Ham et al., 2014). In contrast, during EV71 (Enterovirus 71) infection, baicalin suppresses viral 3D polymerase expression to block early replication, while concurrently inhibiting FASL and caspase-3 levels, thereby attenuating extrinsic apoptotic pathway activation. This dual action protects host cells from apoptosis and reduces viral dissemination and release (Li et al., 2015). Furthermore, recent studies have shown that ginsenoside Rb1 exhibits therapeutic potential in acute lung injury caused by S. aureus. Infection with S. aureus upregulates death receptor-related genes such as FAS in lung cells, while ginsenoside Rb1 reverses these changes, reducing apoptosis and achieving therapeutic benefits (Shaukat et al., 2021).
Collectively, these studies highlight the multifaceted effects of FAS/FASL-targeting agents, including the modulation of immune-mediated pathogen clearance, inhibition of excessive inflammation and tissue damage, and regulation of apoptosis in infected host cells. However, optimizing drug concentration is crucial, and thorough in vivo efficacy and safety assessments are necessary for the development of broad-spectrum anti-pathogen therapies.
4.2 Cytokine therapy
Both apoptotic and non-apoptotic FAS-mediated signaling pathways are critically regulated by cytokines, offering potential therapeutic avenues for FAS-related diseases. For example, In chronic hepatitis C induced by viral infection, interferon (IFN) therapy initially induces transient elevation of serum soluble FAS (sFAS) and sFASL, followed by increased membrane-bound FASL expression (Yoneyama et al., 2002). This dynamic modulation may preferentially amplify functional apoptotic signaling of membrane-bound FASL, thereby optimizing the balance between infected cell clearance and tissue damage. Additionally, IFN therapy regulates pathogen-infected cell clearance by immune cells. IFN-α directly stimulates peripheral blood mononuclear cells, selectively upregulating FASL expression on NK cells, thereby enhancing their cytotoxicity against FAS-sensitive targets and promoting NK cell-mediated pathogen elimination (Kirou et al., 2000). Additionally, IFN-γ activates FASL expression in CD8+ T cells, promoting the clearance of HCV-infected hepatocytes via the Fas/FASL pathway (Jirillo et al., 2000).
Similarly, interleukins (ILs) modulate immune cell-mediated clearance of infected cells via the FAS/FASL pathway. In mouse models of respiratory syncytial virus (RSV) infection, localized IL-4 overexpression upregulates FASL on CD4+ and CD8+ T cells, enhancing their cytotoxicity against FAS-expressing virus-infected cells. However, this nonspecific killing may induce collateral apoptosis in healthy tissues and exacerbate immune pathology (Aung and Graham, 2000). Therefore, FAS/FASL-targeting strategies involving ILs and IFNs must be carefully controlled in space and time, depending on the disease stage and immune context. Exploring combination therapies may offer a way to balance the competing demands of immune clearance and tissue damage.
4.3 Antibody therapy
The therapeutic potential of FAS/FASL-targeting antibodies was first revealed in 1989 with the identification of the APO-1 monoclonal antibody (Trauth et al., 1989). While FAS or FASL antibodies are primarily employed in treating autoimmune diseases and cancer, they also show promise against pathogen infections by either activating apoptosis in infected cells (via FAS antibodies) or blocking immune cell apoptosis (via FASL antibodies). For example, anti-FAS monoclonal antibodies eliminate HIV-infected cells without enhancing viral replication, effectively suppressing HIV transmission (Kobayashi et al., 1990). Similarly, FASL antibody treatment in simian immunodeficiency virus (SIV)-infected primates inhibits FAS-mediated apoptosis, preserving T/B lymphocyte survival and maintaining immune responses (Salvato et al., 2007; Poonia et al., 2009). However, certain FAS antibodies (such as Jo2) cause severe liver toxicity, necessitating the development of next-generation FAS antibodies (such as HFE7A) (Ichikawa et al., 2000). Additionally, combined therapy with IFN-γ and FAS antibody CH11 synergistically enhances apoptosis in human cytomegalovirus-infected cells by suppressing viral replication and inducing apoptosis, demonstrating dual therapeutic efficacy (Chaudhuri et al., 1999).
4.4 FASL recombinant proteins therapy
Advancements in genetic engineering have facilitated the development of recombinant FASL (rFASL) as a critical tool for dissecting apoptosis mechanisms and exploring therapeutic applications. Multiple rFASL expression systems, including baculoviral, adenoviral (AdFASL), and Pichia pastoris platforms, have been established to produce rFASL for both in vitro and in vivo apoptosis models (Mariani et al., 1996; Tanaka et al., 1997; Okuyama et al., 1998). Mountz et al. utilized FASL-deficient (gld/gld) mice infected with murine cytomegalovirus (MCMV) to demonstrate that rFASL exacerbates chronic inflammation by sustaining activated T-cell infiltration post-viral clearance (Zhang et al., 2000). Intravenous infusion of FASL-expressing antigen-presenting cells (APC-AdFASL) selectively targeted splenic marginal zones, inducing activated T-cell apoptosis and significantly reducing inflammation in the lungs, liver, and kidneys (Zhang et al., 2000). In a parallel study, localized AdFASL delivery to salivary glands attenuated MCMV-driven immune hyperactivity and alleviated chronic sialadenitis (Fleck et al., 2001). Importantly, this localized approach minimized systemic FASL activation-associated hepatotoxicity, supporting its clinical safety profile.
5 Perspectives
FAS cell surface death receptor is pivotal in both apoptotic and non-apoptotic signaling pathways, contributing to processes such as cell migration, invasion, immune regulation, and cancer progression. Although the role of FAS signaling in cancer and autoimmune disorders resulting from FAS mutations has been extensively studied, its function in pathogen-induced infections remains poorly understood. Pathogens, including bacteria and viruses, can cause a wide range of diseases with significant mortality. These pathogens can manipulate host cell immunity and survival by modulating the FAS signaling pathway. Consequently, exploring the mechanisms underlying FAS signaling in the context of pathogen infections is essential for enhancing our understanding of disease pathologies and targeted therapeutic strategies.
This article explores the mechanisms of FAS-mediated apoptosis and inflammation triggered by bacterial and viral infections, while also highlighting therapeutic strategies targeting the FAS/FASL system. Despite these advancements, the regulatory mechanisms governing the FAS signaling pathway remain highly intricate. Does pathogen infection directly or indirectly regulate FAS expression? What factors contribute to this regulation? How does host immunity dynamically interact with these processes? These questions require further investigation to uncover the precise dynamics of FAS signaling. In addition, although various therapeutic approaches targeting the FAS/FASL system have been developed, including drug therapy, cytokine therapy, antibody therapy, and FASL recombinant proteins therapy, most of these methods are still in the laboratory research stage and rarely enter clinical research, which is a significant challenge for researchers. Looking ahead, there is a pressing need to clarify additional FAS-regulated disease pathways and to develop innovative therapeutic strategies targeting the FAS/FASL system, ultimately paving the way for improved clinical applications.
Author contributions
LH: Conceptualization, Investigation, Visualization, Writing – original draft, Writing – review & editing. JL: Conceptualization, Resources, Visualization, Writing – original draft, Writing – review & editing. HF: Investigation, Visualization, Writing – review & editing. CN: Investigation, Visualization, Writing – review & editing. YH: Investigation, Visualization, Writing – review & editing. QC: Writing – review & editing. HW: Conceptualization, Resources, Supervision, Writing – review & editing. JQ: Conceptualization, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Key Research and Development Program of China (No. 2020YFA0907904) and the General Program of National Natural Science Foundation of China (No. 32070073).
Acknowledgments
We thank the current and former members of our laboratories and collaborators for their contributions to this review article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
FAS: tumor necrosis factor receptor superfamily member 6
FASL: FAS ligand
NK: natural killer
NF-κB: nuclear factor kappa-B
MAPK: mitogen-activated protein kinase
ERK: extracellular regulated protein kinases
mFASL: transmembrane FASL
sFASL: soluble FASL
FADD: FAS-associated death domain
c-FLIP: cellular FLICE inhibitory protein
DED: death effector domain
DISC: death-inducing signaling complex
Bid: BH3-interacting domain death agonist
tBid: truncated form of Bid
Bak: Bcl-2 homologous antagonist
Bax: apoptosis regulator Bax
MOMP: mitochondrial outer membrane permeabilization
Apaf-1: apoptotic protease activating factor 1
CARDs: caspase recruitment domains
Smac/DIABLO: diablo IAP-binding mitochondrial protein
HtrA2/Omi: serine protease HTRA2
XIAP: X-chromosome-linked inhibitor of apoptosis protein
RIPK1: receptor-interacting serine/threonine-protein kinase 1
RIPK3: receptor-interacting serine/threonine-protein kinase 3
RHIM: RIP homotypic interaction motif
MLKL: mixed lineage kinase domain-like protein
PI3K/AKT: phosphatidylinositol-3-kinase/protein kinase B
cIAP1/2: cellular inhibitor of apoptosis protein 1/2
TAK1: TGF-β-activated kinase 1
TAB: TAK1 binding protein
VEGF: vascular endothelial growth factor
TRAF2: TNF receptor-associated factor 2
IAPs: inhibitor of apoptosis proteins
A20: deubiquitinating enzyme A20
MEKK1: mitogen-activated protein kinase 1
MKKs: mitogen-activated protein kinase kinases
JNK: c-Jun N-terminal kinase
DAXX: death-domain associated protein 6
ASK1: apoptosis signal-regulating kinase 1
NOX3: NADPH oxidase 3
MISC: motility-inducing signaling complex
MMP: matrix metalloproteinase
EGFR: epidermal growth factor receptor
STAT3: signal transducer and activator of transcription 3
EGF: epidermal growth factor
LLO: listeriolysin O
PC-PLC: phosphatidylcholine-preferring phospholipase C
TLRs: Toll-like receptors
TB: tuberculosis
3’UTR: 3’ untranslated region
EPEC: enteropathogenic Escherichia coli
cag PAI: cag pathogenicity island
CFTR: cystic fibrosis transmembrane conductance regulator
HPV: Human papilloma virus
MARCHF8: membrane-associated RING-CH-type finger ubiquitin ligase
HSV: Herpes simplex virus
CXCL: C-X-C motif chemokine
HIV: Human immunodeficiency virus
AIDS: acquired immunodeficiency syndrome
HBV: Hepatitis B virus
HCC: hepatocellular carcinoma
HBsAg: HBV surface antigen
ER: endoplasmic reticulum
HBx: HBV x protein
SAPK: stress-activated protein kinase
Egr: early growth response factor
IV: influenza virus
IAV: influenza A virus
SARS-CoV-2: severe acute respiratory syndrome coronavirus 2
COVID-19: coronavirus disease 2019
FOXO1: forkhead box protein O1
FOXP3: forkhead box protein P3
ARDS: acute respiratory distress syndrome
EBV: Epstein-Barr virus
DENV: dengue virus
MV: measles virus
LPS: lipopolysaccharide
RSV: respiratory syncytial virus
SIV: simian immunodeficiency virus
MCMV: murine cytomegalovirus
HBeAg: HBV e antigen
sFAS: soluble FAS
HBc: HBV core protein
IFN: interferon
IL: interleukin
References
Akhiani, A. A., Martner, A. (2022). Role of phosphoinositide 3-kinase in regulation of NOX-derived reactive oxygen species in cancer. Antioxidants (Basel Switzerland) 12, 67. doi: 10.3390/antiox12010067
Alaoui-El-Azher, M., Jia, J., Lian, W., Jin, S. (2006). ExoS of Pseudomonas aeruginosa induces apoptosis through a Fas receptor/caspase 8-independent pathway in HeLa cells. Cell. Microbiol. 8, 326–338. doi: 10.1111/j.1462-5822.2005.00624.x
Albert, M. C., Uranga-Murillo, I., Arias, M., De Miguel, D., Peña, N., Montinaro, A., et al. (2024). Identification of FasL as a crucial host factor driving COVID-19 pathology and lethality. Cell Death Differ. 31, 544–557. doi: 10.1038/s41418-024-01278-6
Alvarado-Kristensson, M., Melander, F., Leandersson, K., Rönnstrand, L., Wernstedt, C., Andersson, T. (2004). p38-MAPK signals survival by phosphorylation of caspase-8 and caspase-3 in human neutrophils. J. Exp. Med. 199, 449–458. doi: 10.1084/jem.20031771
Alvi, A., Ansari, S. A., Ehtesham, N. Z., Rizwan, M., Devi, S., Sechi, L. A., et al. (2011). Concurrent proinflammatory and apoptotic activity of a Helicobacter pylori protein (HP986) points to its role in chronic persistence. PloS One 6, e22530. doi: 10.1371/journal.pone.0022530
André, S., Picard, M., Cezar, R., Roux-Dalvai, F., Alleaume-Butaux, A., Soundaramourty, C., et al. (2022). T cell apoptosis characterizes severe Covid-19 disease. Cell Death Differ. 29, 1486–1499. doi: 10.1038/s41418-022-00936-x
Ansari, S. A., Devi, S., Tenguria, S., Kumar, A., Ahmed, N. (2014). Helicobacter pylori protein HP0986 (TieA) interacts with mouse TNFR1 and triggers proinflammatory and proapoptotic signaling pathways in cultured macrophage cells (RAW 264.7). Cytokine 68, 110–117. doi: 10.1016/j.cyto.2014.03.006
Asandem, D. A., Segbefia, S. P., Kusi, K. A., Bonney, J. H. K. (2024). Hepatitis B virus infection: A mini review. Viruses 16, 724. doi: 10.3390/v16050724
Aung, S., Graham, B. S. (2000). IL-4 diminishes perforin-mediated and increases Fas ligand-mediated cytotoxicity In vivo. J. Immunol. 164, 3487–3493. doi: 10.4049/jimmunol.164.7.3487
Baran, J., Weglarczyk, K., Mysiak, M., Guzik, K., Ernst, M., Flad, H. D., et al. (2001). Fas (CD95)-Fas ligand interactions are responsible for monocyte apoptosis occurring as a result of phagocytosis and killing of Staphylococcus aureus. Infect. Immun. 69, 1287–1297. doi: 10.1128/IAI.69.3.1287-1297.2001
Behrmann, I., Walczak, H., Krammer, P. H. (1994). Structure of the human APO-1 gene. Eur. J. Immunol. 24, 3057–3062. doi: 10.1002/eji.1830241221
Bellesi, S., Metafuni, E., Hohaus, S., Maiolo, E., Marchionni, F., D’Innocenzo, S., et al. (2020). Increased CD95 (Fas) and PD-1 expression in peripheral blood T lymphocytes in COVID-19 patients. Br. J. Haematol. 191, 207–211. doi: 10.1111/bjh.17034
Bernal-Mizrachi, L., Lovly, C. M., Ratner, L. (2006). The role of NF-κB-1 and NF-κB-2-mediated resistance to apoptosis in lymphomas. Proc. Natl. Acad. Sci. U.S.A. 103, 9220–9225. doi: 10.1073/pnas.0507809103
Blanco, L., Puente, J., Carrasco, C., Miranda, D., Wolf, M. E., Mosnaim, A. D. (2001). Effect of Salmonella-infected human monocytes on natural killer cell cytotoxicity. In vitro studies. Int. Immunopharmacol. 1, 1285–1293. doi: 10.1016/S1567-5769(01)00060-1
Bock, F. J., Tait, S. W. G. (2020). Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 21, 85–100. doi: 10.1038/s41580-019-0173-8
Bogolyubova, I., Bogolyubov, D. (2021). DAXX is a crucial factor for proper development of mammalian oocytes and early embryos. Int. J. Mol. Sci. 22, 1313. doi: 10.3390/ijms22031313
Bortolami, M., Kotsafti, A., Cardin, R., Farinati, F. (2008). Fas/FasL system, IL-1beta expression and apoptosis in chronic HBV and HCV liver disease. J. Viral Hepat. 15, 515–522. doi: 10.1111/j.1365-2893.2008.00974.x
Cai, J., Peng, Y., Chen, T., Liao, H., Zhang, L., Chen, Q., et al. (2016). Chloral hydrate treatment induced apoptosis of macrophages via fas signaling pathway. Med. Sci. Monit. 22, 4836–4843. doi: 10.12659/msm.901772
Cai, Z., Jitkaew, S., Zhao, J., Chiang, H. C., Choksi, S., Liu, J., et al. (2014). Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell. Biol. 16, 55–65. doi: 10.1038/ncb2883
Canaday, D. H., Wilkinson, R. J., Li, Q., Harding, C. V., Silver, R. F., Boom, W. H. (2001). CD4+ and CD8+ T cells kill intracellular Mycobacterium tuberculosis by a perforin and Fas/Fas ligand-independent mechanism. J. Immunol. 167, 2734–2742. doi: 10.4049/jimmunol.167.5.2734
Cannon, C. L., Kowalski, M. P., Stopak, K. S., Pier, G. B. (2003). Pseudomonas aeruginosa -induced apoptosis is defective in respiratory epithelial cells expressing mutant cystic fibrosis transmembrane conductance regulator. Am. J. Respir. Cell Mol. Biol. 29, 188–197. doi: 10.1165/rcmb.4898
Caulfield, A. J., Walker, M. E., Gielda, L. M., Lathem, W. W. (2014). The Pla protease of Yersinia pestis degrades fas ligand to manipulate host cell death and inflammation. Cell Host Microbe 15, 424–434. doi: 10.1016/j.chom.2014.03.005
Chang, D. W., Xing, Z., Pan, Y., Algeciras-Schimnich, A., Barnhart, B. C., Yaish-Ohad, S., et al. (2002). c-FLIP(L) is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO. J. 21, 3704–3714. doi: 10.1093/emboj/cdf356
Chaudhuri, A. R., Jeor, S. S., Maciejewski, J. P. (1999). Apoptosis induced by human cytomegalovirus infection can be enhanced by cytokines to limit the spread of virus. Exp. Hematol. 27, 1194–1203. doi: 10.1016/S0301-472X(99)00044-2
Conaldi, P. G., Biancone, L., Bottelli, A., Wade-Evans, A., Racusen, L. C., Boccellino, M., et al. (1998). HIV-1 kills renal tubular epithelial cells in vitro by triggering an apoptotic pathway involving caspase activation and Fas upregulation. J. Clin. Invest. 102, 2041–2049. doi: 10.1172/JCI3480
Consonni, F., Gambineri, E., Favre, C. (2022). ALPS, FAS, and beyond: from inborn errors of immunity to acquired immunodeficiencies. Ann. Hematol. 101, 469–484. doi: 10.1007/s00277-022-04761-7
Cossart, P., Sansonetti, P. J. (2004). Bacterial invasion: the paradigms of enteroinvasive pathogens. Science 304, 242–248. doi: 10.1126/science.1090124
Davidovich, P., Higgins, C. A., Najda, Z., Longley, D. B., Martin, S. J. (2023). cFLIPL acts as a suppressor of TRAIL- and Fas-initiated inflammation by inhibiting assembly of caspase-8/FADD/RIPK1 NF-κB-activating complexes. Cell Rep. 42, 113476. doi: 10.1016/j.celrep.2023.113476
Deak, J. C., Cross, J. V., Lewis, M., Qian, Y., Parrott, L. A., Distelhorst, C. W., et al. (1998). Fas-induced proteolytic activation and intracellular redistribution of the stress-signaling kinase MEKK1. Proc. Natl. Acad. Sci. U.S.A. 95, 5595–5600. doi: 10.1073/pnas.95.10.5595
Diao, J., Khine, A. A., Sarangi, F., Hsu, E., Iorio, C., Tibbles, L. A., et al. (2001). X protein of hepatitis B virus inhibits Fas-mediated apoptosis and is associated with up-regulation of the SAPK/JNK pathway. J. Biol. Chem. 276, 8328–8340. doi: 10.1074/jbc.M006026200
Dockrell, D. H., Badley, A. D., Villacian, J. S., Heppelmann, C. J., Algeciras, A., Ziesmer, S., et al. (1998). The expression of Fas Ligand by macrophages and its upregulation by human immunodeficiency virus infection. J. Clin. Invest. 101, 2394–2405. doi: 10.1172/JCI1171
Dreschers, S., Platen, C., Ludwig, A., Gille, C., Köstlin, N., Orlikowsky, T. W. (2019). Metalloproteinases TACE and MMP-9 differentially regulate death factors on adult and neonatal monocytes after infection with Escherichia coli. Int. J. Mol. Sci. 20, 1399. doi: 10.3390/ijms20061399
Ea, C. K., Deng, L., Xia, Z. P., Pineda, G., Chen, Z. J. (2006). Activation of IKK by TNFα Requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol. Cell 22, 245–257. doi: 10.1016/j.molcel.2006.03.026
Engelsberger, V., Gerhard, M., Mejías-Luque, R. (2024). Effects of Helicobacter pylori infection on intestinal microbiota, immunity and colorectal cancer risk. Front. Cell. Infect. Microbiol. 14. doi: 10.3389/fcimb.2024.1339750
Ennaciri, J., Menezes, J., Proulx, F., Toledano, B. J. (2006). Induction of apoptosis by Herpes simplex virus-1 in neonatal, but not adult, neutrophils. Pediatr. Res. 59, 7–12. doi: 10.1203/01.pdr.0000191816.57544.b4
Farley, N., Pedraza-Alva, G., Serrano-Gomez, D., Nagaleekar, V., Aronshtam, A., Krahl, T., et al. (2006). p38 mitogen-activated protein kinase mediates the Fas-induced mitochondrial death pathway in CD8+ T cells. Mol. Cell. Biol. 26, 2118–2129. doi: 10.1128/MCB.26.6.2118-2129.2006
Fernandes, P., O’Donnell, C., Lyons, C., Keane, J., Regan, T., O’Brien, S., et al. (2014). Intestinal expression of Fas and Fas ligand is upregulated by bacterial signaling through TLR4 and TLR5, with activation of Fas modulating intestinal TLR-mediated inflammation. J. Immunol. 193, 6103–6113. doi: 10.4049/jimmunol.1303083
Filippova, M., Parkhurst, L., Duerksen-Hughes, P. J. (2004). The Human papillomavirus 16 E6 protein binds to Fas-associated death domain and protects cells from Fas-triggered apoptosis. J. Biol. Chem. 279, 25729–25744. doi: 10.1074/jbc.M401172200
Fleck, M., Zhang, H. G., Kern, E. R., Hsu, H. C., Muller-Ladner, U., Mountz, J. D. (2001). Treatment of chronic sialadenitis in a murine model of Sjögren’s syndrome by local fasL gene transfer. Arthritis. Rheumatol. 44, 964–973. doi: 10.1002/1529-0131(200104)44:4<964::AID-ANR154>3.0.CO;2-5
Flores-Romero, H., Hohorst, L., John, M., Albert, M., King, L. E., Beckmann, L., et al. (2022). BCL-2-family protein tBID can act as a BAX-like effector of apoptosis. EMBO J. 41, e108690. doi: 10.15252/embj.2021108690
Fraga-Silva, T. F. C., Cipriano, U. G., Fumagalli, M. J., Correa, G. F., Fuzo, C. A., Dos-Santos, D., et al. (2023). Airway epithelial cells and macrophages trigger IL-6-CD95/CD95L axis and mediate initial immunopathology of COVID-19. iScience 26, 108366. doi: 10.1016/j.isci.2023.108366
Fujimoto, I., Takizawa, T., Ohba, Y., Nakanishi, Y. (1998). Co-expression of Fas and Fas-ligand on the surface of influenza virus-infected cells. Cell Death. Differ. 5, 426–431. doi: 10.1038/sj.cdd.4400362
Galluzzi, L., Vitale, I., Aaronson, S. A., Abrams, J. M., Adam, D., Agostinis, P., et al. (2018). Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 25, 486–541. doi: 10.1038/s41418-017-0012-4
Gandhi, R. T., Chen, B. K., Straus, S. E., Dale, J. K., Lenardo, M. J., Baltimore, D. (1998). HIV-1 directly kills CD4+ T cells by a Fas-independent mechanism. J. Exp. Med. 187, 1113–1122. doi: 10.1084/jem.187.7.1113
Ghare, S. S., Chilton, P. M., Rao, A. V., Joshi-Barve, S., Peyrani, P., Reyes Vega, A., et al. (2021). Epigenetic mechanisms underlying HIV-infection induced susceptibility of CD4+ T cells to enhanced activation-induced FasL expression and cell death. J. Acquir. Immune Defic. Syndr. 86, 128–137. doi: 10.1097/QAI.0000000000002526
Gille, C., Dreschers, S., Leiber, A., Lepiorz, F., Krusch, M., Grosse-Opphoff, J., et al. (2013). The CD95/CD95L pathway is involved in phagocytosis-induced cell death of monocytes and may account for sustained inflammation in neonates. Pediatr. Res. 73, 402–408. doi: 10.1038/pr.2012.196
Gong, Z., Mao, W., Zhao, J., Ren, P., Yu, Z., Bai, Y., et al. (2024). TLR2 and NLRP3 orchestrate regulatory roles in Escherichia coli infection-induced septicemia in mouse models. J. Innate Immun. 16, 513–528. doi: 10.1159/000541819
Grassmé, H., Kirschnek, S., Riethmueller, J., Riehle, A., Von Kürthy, G., Lang, F., et al. (2000). CD95/CD95 ligand interactions on epithelial cells in host defense to Pseudomonas aeruginosa. Science 290, 527–530. doi: 10.1126/science.290.5491.527
Guerra, C., Johal, K., Morris, D., Moreno, S., Alvarado, O., Gray, D., et al. (2012). Control of Mycobacterium tuberculosis growth by activated natural killer cells. Clin. Exp. Immunol. 168, 142–152. doi: 10.1111/j.1365-2249.2011.04552.x
Hahm, K. B., Kim, D. H., Lee, K. M., Lee, J. S., Surh, Y. J., Kim, Y. B., et al. (2003). Effect of long-term administration of rebamipide on Helicobacter pylori infection in mice. Aliment Pharmacol. Ther. 18, 24–38. doi: 10.1046/j.1365-2036.18.s1.3.x
Ham, S., Kim, K. H., Kwon, T. H., Bak, Y., Lee, D. H., Song, Y. S., et al. (2014). Luteolin induces intrinsic apoptosis via inhibition of E6/E7 oncogenes and activation of extrinsic and intrinsic signaling pathways in HPV-18-associated cells. Oncol. Rep. 31, 2683–2691. doi: 10.3892/or.2014.3157
Hasumi, K., Tanaka, K., Saitoh, S., Takagi, A., Miwa, T., Mine, T., et al. (2002). Roles of tumor necrosis factor-alpha-receptor type 1 and Fas in the Helicobacter pylori-induced apoptosis of gastric epithelial cells. J. Gastroenterol. Hepatol. 17, 651–658. doi: 10.1046/j.1440-1746.2002.02757.x
He, P., Zhang, B., Liu, D., Bian, X., Li, D., Wang, Y., et al. (2016). Hepatitis B virus X protein modulates apoptosis in NRK-52E cells and activates Fas/FasL through the MLK3-MKK7-JNK3 signaling pathway. Cell. Physiol. Biochem. 39, 1433–1443. doi: 10.1159/000447846
Henry, C. M., Martin, S. J. (2017). Caspase-8 acts in a non-enzymatic role as a scaffold for assembly of a pro-inflammatory “FADDosome” complex upon TRAIL stimulation. Mol. Cell 65, 715–729.e5. doi: 10.1016/j.molcel.2017.01.022
Holler, N., Zaru, R., Micheau, O., Thome, M., Attinger, A., Valitutti, S., et al. (2000). Fas triggers an alternative, caspase-8–independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 1, 489–495. doi: 10.1038/82732
Holmström, T. H., Tran, S. E., Johnson, V. L., Ahn, N. G., Chow, S. C., Eriksson, J. E. (1999). Inhibition of mitogen-activated kinase signaling sensitizes HeLa cells to Fas receptor-mediated apoptosis. Mol. Cell. Biol. 19, 5991–6002. doi: 10.1128/MCB.19.9.5991
Horn, S., Hughes, M. A., Schilling, R., Sticht, C., Tenev, T., Ploesser, M., et al. (2017). Caspase-10 negatively regulates caspase-8-mediated cell death, switching the response to CD95L in favor of NF-κB activation and cell survival. Cell Rep. 19, 785–797. doi: 10.1016/j.celrep.2017.04.010
Hornick, E. E., Dagvadorj, J., Zacharias, Z. R., Miller, A. M., Langlois, R. A., Chen, P., et al. (2019). Dendritic cell NLRC4 regulates influenza A virus–specific CD4+ T cell responses through FasL expression. J. Clin. Invest. 129, 2888–2897. doi: 10.1172/JCI124937
Ichijo, H., Nishida, E., Irie, K., ten Dijke, P., Saitoh, M., Moriguchi, T., et al. (1997). Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 275, 90–94. doi: 10.1126/science.275.5296.90
Ichikawa, K., Yoshida-Kato, H., Ohtsuki, M., Ohsumi, J., Yamaguchi, J., Takahashi, S., et al. (2000). A novel murine anti-human Fas mAb which mitigates lymphadenopathy without hepatotoxicity. Int. Immunol. 12, 555–562. doi: 10.1093/intimm/12.4.555
Itoh, N., Yonehara, S., Ishii, A., Yonehara, M., Mizushima, S., Sameshima, M., et al. (1991). The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell 66, 233–243. doi: 10.1016/0092-8674(91)90614-5
Jendrossek, V., Grassmé, H., Mueller, I., Lang, F., Gulbins, E. (2001). Pseudomonas aeruginosa -induced apoptosis involves mitochondria and stress-activated protein kinases. Infect. Immun. 69, 2675–2683. doi: 10.1128/IAI.69.4.2675-2683.2001
Jennewein, C., Karl, S., Baumann, B., Micheau, O., Debatin, K. M., Fulda, S. (2012). Identification of a novel pro-apoptotic role of NF-κB in the regulation of TRAIL- and CD95-mediated apoptosis of glioblastoma cells. Oncogene 31, 1468–1474. doi: 10.1038/onc.2011.333
Jing, Z. T., Liu, W., Wu, S. X., He, Y., Lin, Y. T., Chen, W. N., et al. (2018). Hepatitis B virus surface antigen enhances the sensitivity of hepatocytes to Fas-mediated apoptosis via suppression of AKT phosphorylation. J. Immunol. 201, 2303–2314. doi: 10.4049/jimmunol.1800732
Jirillo, E., Pellegrino, N. M., Piazzolla, G., Caccavo, D., Antonaci, S. (2000). Hepatitis C virus infection: immune responsiveness and interferon-alpha treatment. Curr. Pharm. Des. 6, 169–180. doi: 10.2174/1381612003401271
Jones, N. L., Day, A. S., Jennings, H. A., Sherman, P. M. (1999). Helicobacter pylori induces gastric epithelial cell apoptosis in association with increased Fas receptor expression. Infect. Immun. 67, 4237–4242. doi: 10.1128/IAI.67.8.4237-4242.1999
Jurado-Martín, I., Sainz-Mejías, M., McClean, S. (2021). Pseudomonas aeruginosa: An audacious pathogen with an adaptable arsenal of virulence factors. Int. J. Mol. Sci. 22, 3128. doi: 10.3390/ijms22063128
Kabsch, K., Alonso, A. (2002). The Human papillomavirus type 16 E5 protein impairs TRAIL- and FasL-mediated apoptosis in HaCaT cells by different mechanisms. J. Virol. 76, 12162–12172. doi: 10.1128/JVI.76.23.12162-12172.2002
Kägi, D., Vignaux, F., Ledermann, B., Bürki, K., Depraetere, V., Nagata, S., et al. (1994). Fas and perforin pathways as major mechanisms of T cell-mediated cytotoxicity. Science 265, 528–530. doi: 10.1126/science.7518614
Kammoun, H., Kim, M., Hafner, L., Gaillard, J., Disson, O., Lecuit, M. (2022). Listeriosis, a model infection to study host-pathogen interactions in vivo. Curr. Opin. Microbiol. 66, 11–20. doi: 10.1016/j.mib.2021.11.015
Katsikis, P. D., Wunderlich, E. S., Smith, C. A., Herzenberg, L. A., Herzenberg, L. A. (1995). Fas antigen stimulation induces marked apoptosis of T lymphocytes in human immunodeficiency virus-infected individuals. J. Exp. Med. 181, 2029–2036. doi: 10.1084/jem.181.6.2029
Khalil, M. I., Yang, C., Vu, L., Chadha, S., Nabors, H., Welbon, C., et al. (2023). HPV upregulates MARCHF8 ubiquitin ligase and inhibits apoptosis by degrading the death receptors in head and neck cancer. PloS Pathog. 19, e1011171. doi: 10.1371/journal.ppat.1011171
Kim, J. H., Najy, A. J., Li, J., Luo, X., Kim, H.-R. C., Choudry, M. H. A., et al. (2023). Involvement of Bid in the crosstalk between ferroptotic agent-induced ER stress and TRAIL-induced apoptosis. J. Cell. Physiol. 237, 4180–4196. doi: 10.1002/jcp.30863
Kirou, K. A., Vakkalanka, R. K., Butler, M. J., Crow, M. K. (2000). Induction of Fas ligand-mediated apoptosis by interferon-alpha. Clin. Immunol. 95, 218–226. doi: 10.1006/clim.2000.4866
Kleber, S., Sancho-Martinez, I., Wiestler, B., Beisel, A., Gieffers, C., Hill, O., et al. (2008). Yes and PI3K bind CD95 to signal invasion of glioblastoma. Cancer Cell. 13, 235–248. doi: 10.1016/j.ccr.2008.02.003
Kobayashi, N., Hamamoto, Y., Yamamoto, N., Ishii, A., Yonehara, M., Yonehara, S. (1990). Anti-Fas monoclonal antibody is cytocidal to human immunodeficiency virus-infected cells without augmenting viral replication. Proc. Natl. Acad. Sci. U.S.A. 87, 9620–9624. doi: 10.1073/pnas.87.24.9620
Kober, A. M., Legewie, S., Pforr, C., Fricker, N., Eils, R., Krammer, P. H., et al. (2011). Caspase-8 activity has an essential role in CD95/Fas-mediated MAPK activation. Cell Death Dis. 2, e212. doi: 10.1038/cddis.2011.93
Komiya, Y., Kamiya, M., Oba, S., Kawata, D., Iwai, H., Shintaku, H., et al. (2024). Necroptosis in alveolar epithelial cells drives lung inflammation and injury caused by SARS-CoV-2 infection. Biochim. Biophys. Acta Mol. Basis. Dis. 1870, 167472. doi: 10.1016/j.bbadis.2024.167472
Kondo, T., Suda, T., Fukuyama, H., Adachi, M., Nagata, S. (1997). Essential roles of the Fas ligand in the development of hepatitis. Nat. Med. 3, 409–413. doi: 10.1038/nm0497-409
König, R., Stertz, S. (2015). Recent strategies and progress in identifying host factors involved in virus replication. Curr. Opin. Microbiol. 26, 79–88. doi: 10.1016/j.mib.2015.06.001
Kotov, D. I., Kotov, J. A., Goldberg, M. F., Jenkins, M. K. (2018). Many Th cell subsets have Fas ligand-dependent cytotoxic potential. J. Immunol. 200, 2004–2012. doi: 10.4049/jimmunol.1700420
Kreuz, S., Siegmund, D., Rumpf, J. J., Samel, D., Leverkus, M., Janssen, O., et al. (2004). NFκB activation by Fas is mediated through FADD, caspase-8, and RIP and is inhibited by FLIP. J. Cell Biol. 166, 369–380. doi: 10.1083/jcb.200401036
Krueger, A., Baumann, S., Krammer, P. H., Kirchhoff, S. (2001). FLICE-inhibitory proteins: regulators of death receptor-mediated apoptosis. Mol. Cell. Biol. 21, 8247–8254. doi: 10.1128/MCB.21.24.8247-8254.2001
Krzyzowska, M., Baska, P., Orlowski, P., Zdanowski, R., Winnicka, A., Eriksson, K., et al. (2013). HSV-2 regulates monocyte inflammatory response via the Fas/FasL pathway. PloS One 8, e70308. doi: 10.1371/journal.pone.0070308
Krzyzowska, M., Kowalczyk, A., Skulska, K., Thörn, K., Eriksson, K. (2021). Fas/FasL contributes to HSV-1 brain infection and neuroinflammation. Front. Immunol. 12. doi: 10.3389/fimmu.2021.714821
Krzyzowska, M., Shestakov, A., Eriksson, K., Chiodi, F. (2011). Role of Fas/FasL in regulation of inflammation in vaginal tissue during HSV-2 infection. Cell Death Dis. 2, e132. doi: 10.1038/cddis.2011.14
Kumah, E., Boakye, D. S., Boateng, R., Agyei, E. (2023). Advancing the global fight against HIV/Aids: strategies, barriers, and the road to eradication. Ann. Glob. Health 89, 83. doi: 10.5334/aogh.4277
Larochelle, B., Flamand, L., Gourde, P., Beauchamp, D., Gosselin, J. (1998). Epstein-Barr virus infects and induces apoptosis in human neutrophils. Blood 92, 291–299. doi: 10.1182/blood.V92.1.291.413k34_291_299
Lavrik, I. N., Krammer, P. H. (2012). Regulation of CD95/Fas signaling at the DISC. Cell Death Differ. 19, 36–41. doi: 10.1038/cdd.2011.155
Leonardi, A. J., Proenca, R. B. (2020). Akt-fas to quell aberrant T cell differentiation and apoptosis in covid-19. Front. Immunol. 11. doi: 10.3389/fimmu.2020.600405
Li, L. J., Chang, P. M., Li, C. H., Chang, Y. C., Lai, T. C., Su, C. Y., et al. (2022). FAS receptor regulates NOTCH activity through ERK-JAG1 axis activation and controls oral cancer stemness ability and pulmonary metastasis. Cell Death Discovery 8, 101. doi: 10.1038/s41420-022-00899-5
Li, X., Liu, Y., Wu, T., Jin, Y., Cheng, J., Wan, C., et al. (2015). The antiviral effect of baicalin on enterovirus 71 in vitro. Viruses 7, 4756–4771. doi: 10.3390/v7082841
Li, Y., Zhou, M., Hu, Q., Bai, X. C., Huang, W., Scheres, S. H. W., et al. (2017). Mechanistic insights into caspase-9 activation by the structure of the apoptosome holoenzyme. Proc. Natl. Acad. Sci. U.S.A. 114, 1542–1547. doi: 10.1073/pnas.1620626114
Liang, Y. (2023). Pathogenicity and virulence of influenza. Virulence 14, 2223057. doi: 10.1080/21505594.2023.2223057
Liao, H., Xu, J., Huang, J. (2010). FasL/Fas pathway is involved in dengue virus induced apoptosis of the vascular endothelial cells. J. Med. Virol. 82, 1392–1399. doi: 10.1002/jmv.21815
Limjindaporn, T., Netsawang, J., Noisakran, S., Thiemmeca, S., Wongwiwat, W., Sudsaward, S., et al. (2007). Sensitization to Fas-mediated apoptosis by dengue virus capsid protein. Biochem. Biophys. Res. Commun. 362, 334–339. doi: 10.1016/j.bbrc.2007.07.194
Liu, W., Guo, T. F., Jing, Z. T., Tong, Q. Y. (2019a). Repression of death receptor-mediated apoptosis of hepatocytes by Hepatitis B virus e antigen. Am. J. Pathol. 189, 2181–2195. doi: 10.1016/j.ajpath.2019.07.014
Liu, W., Jing, Z. T., Xue, C. R., Wu, S. X., Chen, W. N., Lin, X. J., et al. (2019b). PI3K/AKT inhibitors aggravate death receptor-mediated hepatocyte apoptosis and liver injury. Toxicol. Appl. Pharmacol. 381, 114729. doi: 10.1016/j.taap.2019.114729
Liu, S. B., Lin, X. P., Xu, Y., Shen, Z. F., Pan, W. W. (2018). DAXX promotes ovarian cancer ascites cell proliferation and migration by activating the ERK signaling pathway. J. Ovarian Res. 11, 90. doi: 10.1186/s13048-018-0462-4
Liu, W., Lin, Y. T., Yan, X. L., Ding, Y. L., Wu, Y. L., Chen, W. N., et al. (2015). Hepatitis B virus core protein inhibits Fas-mediated apoptosis of hepatoma cells via regulation of mFas/FasL and sFas expression. FASEB J. 29, 1113–1123. doi: 10.1096/fj.14-263822
Liu, X., Xie, X., Ren, Y., Shao, Z., Zhang, N., Li, L., et al. (2021). The role of necroptosis in disease and treatment. MedComm 2, 730–755. doi: 10.1002/mco2.108
Loxton, A. G., van Rensburg, I. C. (2021). FasL regulatory B-cells during Mycobacterium tuberculosis infection and TB disease. J. Mol. Biol. 433, 166984. doi: 10.1016/j.jmb.2021.166984
Lu, B., Wang, L., Stehlik, C., Medan, D., Huang, C., Hu, S., et al. (2006). Phosphatidylinositol 3-kinase/Akt positively regulates Fas (CD95)-mediated apoptosis in epidermal Cl41 cells. J. Immunol. 176, 6785–6793. doi: 10.4049/jimmunol.176.11.6785
Maeda, S., Yoshida, H., Mitsuno, Y., Hirata, Y., Ogura, K., Shiratori, Y., et al. (2002). Analysis of apoptotic and antiapoptotic signalling pathways induced by Helicobacter pylori. Gut. 50, 771–778. doi: 10.1136/gut.50.6.771
Malyshkina, A., Littwitz-Salomon, E., Sutter, K., Zelinskyy, G., Windmann, S., Schimmer, S., et al. (2017). Fas Ligand-mediated cytotoxicity of CD4+ T cells during chronic retrovirus infection. Sci. Rep. 7, 7785. doi: 10.1038/s41598-017-08578-7
Mariani, S. M., Matiba, B., Sparna, T., Krammer, P. H. (1996). Expression of biologically active mouse and human CD95/APO-1/Fas ligand in the baculovirus system. J. Immunol. Methods 193, 63–70. doi: 10.1016/0022-1759(96)00051-8
Matsumoto, N., Imamura, R., Suda, T. (2007). Caspase-8- and JNK-dependent AP-1 activation is required for Fas ligand-induced IL-8 production. FEBS J. 274, 2376–2384. doi: 10.1111/j.1742-4658.2007.05772.x
Maudet, C., Kheloufi, M., Levallois, S., Gaillard, J., Huang, L., Gaultier, C., et al. (2022). Bacterial inhibition of Fas-mediated killing promotes neuroinvasion and persistence. Nature 603, 900–906. doi: 10.1038/s41586-022-04505-7
Micheau, O., Thome, M., Schneider, P., Holler, N., Tschopp, J., Nicholson, D. W., et al. (2002). The long form of FLIP is an activator of caspase-8 at the Fas death-inducing signaling complex. J. Biol. Chem. 277, 45162–45171. doi: 10.1074/jbc.M206882200
Mochizuki, K., Hayashi, N., Hiramatsu, N., Katayama, K., Kawanishi, Y., Kasahara, A., et al. (1996). Fas antigen expression in liver tissues of patients with chronic hepatitis B. J. Hepatol. 24, 1–7. doi: 10.1016/S0168-8278(96)80178-4
Mohamadian, M., Chiti, H., Shoghli, A., Biglari, S., Parsamanesh, N., Esmaeilzadeh, A. (2021). COVID-19: Virology, biology and novel laboratory diagnosis. J. Gene Med. 23, e3303. doi: 10.1002/jgm.3303
Monack, D. M., Raupach, B., Hromockyj, A. E., Falkow, S. (1996). Salmonella typhimurium invasion induces apoptosis in infected macrophages. Proc. Natl. Acad. Sci. U.S.A. 93, 9833–9838. doi: 10.1073/pnas.93.18.9833
Mori, I., Goshima, F., Imai, Y., Kohsaka, S., Sugiyama, T., Yoshida, T., et al. (2002). Olfactory receptor neurons prevent dissemination of neurovirulent influenza A virus into the brain by undergoing virus-induced apoptosis. J. Gen. Virol. 83, 2109–2116. doi: 10.1099/0022-1317-83-9-2109
Morris, J. E., Zobell, S., Yin, X. T., Zakeri, H., Summers, B. C., Leib, D. A., et al. (2012). Mice with mutations in Fas and Fas ligand demonstrate increased herpetic stromal keratitis following corneal infection with HSV-1. J. Immunol. 188, 793–799. doi: 10.4049/jimmunol.1102251
Mueller, Y. M., De Rosa, S. C., Hutton, J. A., Witek, J., Roederer, M., Altman, J. D., et al. (2001). Increased CD95/Fas-induced apoptosis of HIV-specific CD8(+) T cells. Immunity 15, 871–882. doi: 10.1016/S1074-7613(01)00246-1
Mustafa, T., Phyu, S., Nilsen, R., Bjune, G., Jonsson, R. (1999). Increased expression of Fas ligand on Mycobacterium tuberculosis infected macrophages: a potential novel mechanism of immune evasion by Mycobacterium tuberculosis? Inflammation 23, 507–521. doi: 10.1023/a:1020286305950
Nagata, S., Golstein, P. (1995). The fas death factor. Science. 267, 1449–1456. doi: 10.1126/science.7533326
Nakajima-Shimada, J., Zou, C., Takagi, M., Umeda, M., Nara, T., Aoki, T. (2000). Inhibition of Fas-mediated apoptosis by Trypanosoma cruzi infection. Biochim. Biophys. Acta 1475, 175–183. doi: 10.1016/S0304-4165(00)00059-3
Nanbo, A., Yoshiyama, H., Takada, K. (2005). Epstein-Barr virus-encoded poly(A)- RNA confers resistance to apoptosis mediated through Fas by blocking the PKR pathway in human epithelial intestine 407 cells. JVirol. 79, 12280–12285. doi: 10.1128/JVI.79.19.12280-12285.2005
Nelson, C. W., Mirabello, L. (2023). Human papillomavirus genomics: Understanding carcinogenicity. Tumour Virus Res. 15, 200258. doi: 10.1016/j.tvr.2023.200258
Netsawang, J., Noisakran, S., Puttikhunt, C., Kasinrerk, W., Wongwiwat, W., Malasit, P., et al. (2010). Nuclear localization of dengue virus capsid protein is required for DAXX interaction and apoptosis. Virus Res. 147, 275–283. doi: 10.1016/j.virusres.2009.11.012
Nichols, J. E., Niles, J. A., Roberts, N. J. (2001). Human lymphocyte apoptosis after exposure to influenza A virus. J. Virol. 75, 5921–5929. doi: 10.1128/JVI.73.13.5921-5929.2001
O’Brien, D. I., Nally, K., Kelly, R. G., O’Connor, T. M., Shanahan, F., O’Connell, J. (2005). Targeting the Fas/Fas ligand pathway in cancer. Expert Opin. Ther. Targets. 9, 1031–1044. doi: 10.1517/14728222.9.5.1031
O’Reilly, L. A., Tai, L., Lee, L., Kruse, E. A., Grabow, S., Fairlie, W. D., et al. (2009). Membrane-bound Fas ligand only is essential for Fas-induced apoptosis. Nature 461, 659–663. doi: 10.1038/nature08402
Oddo, M., Renno, T., Attinger, A., Bakker, T., MacDonald, H. R., Meylan, P. R. (1998). Fas ligand-induced apoptosis of infected human macrophages reduces the viability of intracellular Mycobacterium tuberculosis. J. Immunol. 160, 5448–5454. doi: 10.4049/jimmunol.160.11.5448
Ohshima, K., Suzumiya, J., Sugihara, M., Nagafuchi, S., Ohga, S., Kikuchi, M. (1999). CD95 (Fas) ligand expression of Epstein-Barr virus (EBV)- infected lymphocytes: A possible mechanism of immune evasion in chronic active EBV infection. Pathol. Int. 49, 9–13. doi: 10.1046/j.1440-1827.1999.00816.x
Okuyama, T., Fujino, M., Li, X. K., Funeshima, N., Kosuga, M., Saito, I., et al. (1998). Efficient Fas-ligand gene expression in rodent liver after intravenous injection of a recombinant adenovirus by the use of a Cre-mediated switching system. Gene Ther. 5, 1047–1053. doi: 10.1038/sj.gt.3300704
Oshimi, Y., Oda, S., Honda, Y., Nagata, S., Miyazaki, S. (1996). Involvement of Fas ligand and Fas-mediated pathway in the cytotoxicity of human natural killer cells. J. Immunol. 157, 2909–2915. doi: 10.4049/jimmunol.157.7.2909
Palittapongarnpim, P., Tantivitayakul, P., Aiewsakun, P., Mahasirimongkol, S., Jaemsai, B. (2024). Genomic interactions between Mycobacterium tuberculosis and humans. Annu. Rev. Genomics Hum. Genet. 25, 183–209. doi: 10.1146/annurev-genom-021623-101844
Park, H. B., Baek, K. H. (2022). E3 ligases and deubiquitinating enzymes regulating the MAPK signaling pathway in cancers. Biochim. Biophys. Acta Rev. Cancer. 1877, 188736. doi: 10.1016/j.bbcan.2022.188736
Park, D. R., Thomsen, A. R., Frevert, C. W., Pham, U., Skerrett, S. J., Kiener, P. A., et al. (2003). Fas (CD95) induces proinflammatory cytokine responses by human monocytes and monocyte-derived macrophages. J. Immunol. 170, 6209–6216. doi: 10.4049/jimmunol.170.12.6209
Pleinis, J. M., Davis, C. W., Cantrell, C. B., Qiu, D. Y., Zhan, X. (2017). Purification, auto-activation and kinetic characterization of apoptosis signal-regulating kinase I. Protein Expr. Purif. 132, 34–43. doi: 10.1016/j.pep.2017.01.002
Pollock, G. L., Oates, C. V. L., Giogha, C., Wong Fok Lung, T., Ong, S. Y., Pearson, J. S., et al. (2017). Distinct roles of the antiapoptotic effectors NleB and NleF from Enteropathogenic Escherichia coli. Infect. Immun. 85, e01071–e01016. doi: 10.1128/IAI.01071-16
Poonia, B., Salvato, M. S., Yagita, H., Maeda, T., Okumura, K., Pauza, C. D. (2009). Treatment with anti-FasL antibody preserves memory lymphocytes and virus-specific cellular immunity in macaques challenged with simian immunodeficiency virus. Blood 114, 1196–1204. doi: 10.1182/blood-2009-02-202655
Ranjan, K., Pathak, C. (2024). Cellular dynamics of Fas-associated death domain in the regulation of cancer and inflammation. Int. J. Mol. Sci. 25, 3228. doi: 10.3390/ijms25063228
Raqib, R., Ekberg, C., Sharkar, P., Bardhan, P. K., Zychlinsky, A., Sansonetti, P. J., et al. (2002). Apoptosis in acute shigellosis is associated with increased production of Fas/Fas ligand, perforin, caspase-1, and caspase-3 but reduced production of Bcl-2 and interleukin-2. Infect. Immun. 70, 3199–3207. doi: 10.1128/IAI.70.6.3199-3207.2002
Rathbun, M. M., Szpara, M. L. (2021). A holistic perspective on herpes simplex virus (HSV) ecology and evolution. Adv. Virus Res. 110, 27–57. doi: 10.1016/bs.aivir.2021.05.001
Risso, V., Lafont, E., Le Gallo, M. (2022). Therapeutic approaches targeting CD95L/CD95 signaling in cancer and autoimmune diseases. Cell Death Dis. 13, 248. doi: 10.1038/s41419-022-04688-x
Risso, V., Thomas, M., Guével, B., Lavigne, R., Com, E., Martin, S., et al. (2023). Metalloprotease-mediated cleavage of CD95 ligand. FEBS J. 290, 3145–3164. doi: 10.1111/febs.16737
Rudi, J., Kuck, D., Strand, S., von Herbay, A., Mariani, S. M., Krammer, P. H., et al. (1998). Involvement of the CD95 (APO-1/Fas) receptor and ligand system in Helicobacter pylori-induced gastric epithelial apoptosis. J. Clin. Invest. 102, 1506–1514. doi: 10.1172/JCI2808
Sahoo, G., Samal, D., Khandayataray, P., Murthy, M. K. (2023). A review on caspases: Key regulators of biological activities and apoptosis. Mol. Neurobiol. 60, 5805–5837. doi: 10.1007/s12035-023-03433-5
Salmen, S., Terán, G., Borges, L., Goncalves, L., Albarrán, B., Urdaneta, H., et al. (2004). Increased Fas-mediated apoptosis in polymorphonuclear cells from HIV-infected patients. Clin. Exp. Immunol. 137, 166–172. doi: 10.1111/j.1365-2249.2004.02503.x
Salvato, M. S., Yin, C. C., Yagita, H., Maeda, T., Okumura, K., Tikhonov, I., et al. (2007). Attenuated disease in SIV-infected macaques treated with a monoclonal antibody against FasL. Clin. Dev. Immunol. 2007, 93462. doi: 10.1155/2007/93462
Scheller, C., Sopper, S., Ehrhardt, C., Flory, E., Chen, P., Koutsilieri, E., et al. (2002). Caspase inhibitors induce a switch from apoptotic to proinflammatory signaling in CD95-stimulated T lymphocytes. Eur. J. Immunol. 32, 2471–2480. doi: 10.1002/1521-4141(200209)32:9<2471::AID-IMMU2471>3.0.CO;2-E
Schleich, K., Buchbinder, J. H., Pietkiewicz, S., Kähne, T., Warnken, U., Öztürk, S., et al. (2016). Molecular architecture of the DED chains at the DISC: regulation of procaspase-8 activation by short DED proteins c-FLIP and procaspase-8 prodomain. Cell Death Differ. 23, 681–694. doi: 10.1038/cdd.2015.137
Schmidt, J. H., Pietkiewicz, S., Naumann, M., Lavrik, I. N. (2015). Quantification of CD95-induced apoptosis and NF-κB activation at the single cell level. J. Immunol. Methods 423, 12–17. doi: 10.1016/j.jim.2015.04.026
Schram, B. R., Rothstein, T. L. (2003). NF-kappa B is required for surface Ig-induced Fas resistance in B cells. J. Immunol. 170, 3118–3124. doi: 10.4049/jimmunol.170.6.3118
Servet-Delprat, C., Vidalain, P.-O., Azocar, O., Le Deist, F., Fischer, A., Rabourdin-Combe, C. (2000). Consequences of Fas-mediated human dendritic cell apoptosis induced by measles virus. J. Virol. 74, 4387–4393. doi: 10.1128/JVI.74.9.4387-4393.2000
Seyrek, K., Lavrik, I. N. (2019). Modulation of CD95-mediated signaling by post-translational modifications: towards understanding CD95 signaling networks. Apoptosis 24, 385–394. doi: 10.1007/s10495-019-01540-0
Shaukat, A., Shaukat, I., Rajput, S. A., Shukat, R., Hanif, S., Jiang, K., et al. (2021). Ginsenoside Rb1 protects from Staphylococcus aureus-induced oxidative damage and apoptosis through endoplasmic reticulum-stress and death receptor-mediated pathways. Ecotoxicol. Environ. Saf. 219, 112353. doi: 10.1016/j.ecoenv.2021.112353
Srinivasula, S. M., Ahmad, M., Fernandes-Alnemri, T., Alnemri, E. S. (1998). Autoactivation of procaspase-9 by apaf-1-mediated oligomerization. Mol. Cell. 1, 949–957. doi: 10.1016/S1097-2765(00)80095-7
Suda, T., Takahashi, T., Golstein, P., Nagata, S. (1993). Molecular cloning and expression of the fas ligand, a novel member of the tumor necrosis factor family. Cell 75, 1169–1178. doi: 10.1016/0092-8674(93)90326-l
Sun, L., Wang, H., Wang, Z., He, S., Chen, S., Liao, D., et al. (2012). Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227. doi: 10.1016/j.cell.2011.11.031
Ta, N. L., Chakrabandhu, K., Huault, S., Hueber, A. O. (2018). The tyrosine phosphorylated pro-survival form of Fas intensifies the EGF-induced signal in colorectal cancer cells through the nuclear EGFR/STAT3-mediated pathway. Sci. Rep. 8, 12424. doi: 10.1038/s41598-018-30804-z
Takizawa, T., Fukuda, R., Miyawaki, T., Ohashi, K., Nakanishi, Y. (1995). Activation of the apoptotic Fas antigen-encoding gene upon influenza virus infection involving spontaneously produced beta-interferon. Virology 209, 288–296. doi: 10.1006/viro.1995.1260
Tanaka, M., Suda, T., Yatomi, T., Nakamura, N., Nagata, S. (1997). Lethal effect of recombinant human Fas ligand in mice pretreated with Propionibacterium acnes. J. Immunol. 158, 2303–2309. doi: 10.4049/jimmunol.159.5.2303
Tanner, J. E., Alfieri, C. (1999). Epstein-Barr virus induces Fas (CD95) in T cells and Fas ligand in B cells leading to T-cell apoptosis. Blood 94, 3439–3447. doi: 10.1182/blood.V94.10.3439.422k23_3439_3447
Tauzin, S., Chaigne-Delalande, B., Selva, E., Khadra, N., Daburon, S., Contin-Bordes, C., et al. (2011). The naturally processed CD95L elicits a c-yes/calcium/PI3K-driven cell migration pathway. PloS Biol. 9, e1001090. doi: 10.1371/journal.pbio.1001090
Tollefson, A. E., Scaria, A., Hermiston, T. W., Ryerse, J. S., Wold, L. J., Wold, W. S. (1996). The adenovirus death protein (E3-11.6K) is required at very late stages of infection for efficient cell lysis and release of adenovirus from infected cells. J. Virol. 70, 2296–2306. doi: 10.1128/JVI.70.4.2296-2306.1996
Toyoshima, F., Moriguchi, T., Nishida, E. (1997). Fas induces cytoplasmic apoptotic responses and activation of the MKK7-JNK/SAPK and MKK6-p38 pathways independent of CPP32-like proteases. J. Cell Biol. 139, 1005–1015. doi: 10.1083/jcb.139.4.1005
Trauth, B. C., Klas, C., Peters, A. M., Matzku, S., Möller, P., Falk, W., et al. (1989). Monoclonal antibody-mediated tumor regression by induction of apoptosis. Science. 245, 301–305. doi: 10.1126/science.2787530
Tripathi, A., Srivastava, V., Singh, B. N. (2018). hsa-let-7b-5p facilitates Mycobacterium tuberculosis survival in THP-1 human macrophages by Fas downregulation.. FEMS Microbiol Lett. 365, fny040. doi: 10.1093/femsle/fny040
Uchiyama, R., Yonehara, S., Tsutsui, H. (2013). Fas-mediated inflammatory response in Listeria monocytogenes infection. J. Immunol. 190, 4245–4254. doi: 10.4049/jimmunol.1203059
Vutova, P., Wirth, M., Hippe, D., Gross, U., Schulze-Osthoff, K., Schmitz, I., et al. (2007). Toxoplasma gondii inhibits Fas/CD95-triggered cell death by inducing aberrant processing and degradation of caspase 8. Cell Microbiol. 9, 1556–1570. doi: 10.1111/j.1462-5822.2007.00893.x
Wang, W., Fang, Y., Sima, N., Li, Y., Li, W., Li, L., et al. (2011b). Triggering of death receptor apoptotic signaling by human papillomavirus 16 E2 protein in cervical cancer cell lines is mediated by interaction with c-FLIP. Apoptosis 16, 55–66. doi: 10.1007/s10495-010-0543-3
Wang, Q., Na, B., Ou, J. J., Pulliam, L., Yen, T. S. B. (2012). Hepatitis B virus alters the antioxidant system in transgenic mice and sensitizes hepatocytes to Fas signaling. PloS One 7, e36818. doi: 10.1371/journal.pone.0036818
Wang, J. H., Peng, Y., Yang, L. L., Wang, Y. B., Wu, B. G., Zhang, Y., et al. (2011a). Escherichia coli induces apoptosis in human monocytic U937 cells through the Fas/FasL signaling pathway. Mol. Cell. Biochem. 358, 95–104. doi: 10.1007/s11010-011-0925-z
Wang, X., Ragupathy, V., Zhao, J., Hewlett, I. (2011c). Molecules from apoptotic pathways modulate HIV-1 replication in Jurkat cells. Biochem. Biophys. Res. Commun. 414, 20–24. doi: 10.1016/j.bbrc.2011.09.007
Wang, Y., Sun, Q., Zhang, Y., Li, X., Liang, Q., Guo, R., et al. (2023). Systemic immune dysregulation in severe tuberculosis patients revealed by a single-cell transcriptome atlas. J. Infect. 86, 421–438. doi: 10.1016/j.jinf.2023.03.020
Wang, X., Tan, J., Zoueva, O., Zhao, J., Ye, Z., Hewlett, I. (2014). Novel pandemic influenza A (H1N1) virus infection modulates apoptotic pathways that impact its replication in A549 cells. Microbes Infect. 16, 178–186. doi: 10.1016/j.micinf.2013.11.003
Wang, X., Zhao, J., Biswas, S., Devadas, K., Hewlett, I. (2022). Components of apoptotic pathways modulate HIV-1 latency in Jurkat cells. Microbes Infect. 24, 104912. doi: 10.1016/j.micinf.2021.104912
Wei, J., O’Brien, D., Vilgelm, A., Piazuelo, M. B., Correa, P., Washington, M. K., et al. (2008). Interaction of Helicobacter pylori with gastric epithelial cells is mediated by the p53 protein family. Gastroenterology 134, 1412–1423. doi: 10.1053/j.gastro.2008.01.072
Westendorp, M. O., Frank, R., Ochsenbauer, C., Stricker, K., Dhein, J., Walczak, H., et al. (1995). Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature 375, 497–500. doi: 10.1038/375497a0
Wijesinghe, V. N., Farouk, I. A., Zabidi, N. Z., Puniyamurti, A., Choo, W. S., Lal, S. K. (2021). Current vaccine approaches and emerging strategies against herpes simplex virus (HSV). Expert Rev. Vaccines 20, 1077–1096. doi: 10.1080/14760584.2021.1960162
Williamson, A.-L. (2023). Recent developments in Human papillomavirus (HPV) vaccinology. Viruses 15, 1440. doi: 10.3390/v15071440
Wu, S. X., Chen, W. N., Jing, Z. T., Liu, W., Lin, X. J., Lin, X. (2018). Hepatitis B spliced protein (HBSP) suppresses Fas-mediated hepatocyte apoptosis via activation of PI3K/Akt signaling. J. Virol. 92, e01273–e01218. doi: 10.1128/JVI.01273-18
Wurzer, W. J., Ehrhardt, C., Pleschka, S., Berberich-Siebelt, F., Wolff, T., Walczak, H., et al. (2004). NF-kappaB-dependent induction of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and Fas/FasL is crucial for efficient influenza virus propagation. J. Biol. Chem. 279, 30931–30937. doi: 10.1074/jbc.M403258200
Yao, K., Chen, Q., Wu, Y., Liu, F., Chen, X., Zhang, Y. (2017). Unphosphorylated STAT1 represses apoptosis in macrophages during Mycobacteriumtuberculosis infection. J. Cell Sci. 130, 1740–1751. doi: 10.1242/jcs.200659
Yoneyama, K., Goto, T., Miura, K., Mikami, K., Ohshima, S., Nakane, K., et al. (2002). The expression of Fas and Fas ligand, and the effects of interferon in chronic liver diseases with hepatitis C virus. Hepatol. Res. 24, 327–337. doi: 10.1016/s1386-6346(02)00137-7
Yoo, Y. G., Lee, M. O. (2004). Hepatitis B virus X protein induces expression of Fas ligand gene through enhancing transcriptional activity of early growth response factor. J. Biol. Chem. 279, 36242–36249. doi: 10.1074/jbc.M401290200
Yu, J. W., Jeffrey, P. D., Shi, Y. (2009). Mechanism of procaspase-8 activation by c-FLIPL. Proc. Natl. Acad. Sci. U.S.A. 106, 8169–8174. doi: 10.1073/pnas.0812453106
Zenewicz, L. A., Skinner, J. A., Goldfine, H., Shen, H. (2004). Listeria monocytogenes virulence proteins induce surface expression of Fas ligand on T lymphocytes. Mol. Microbiol. 51, 1483–1492. doi: 10.1111/j.1365-2958.2003.03931.x
Zhang, H. G., Fleck, M., Kern, E. R., Liu, D., Wang, Y., Yang, P., et al. (2000). Antigen presenting cells expressing Fas ligand down-modulate chronic inflammatory disease in Fas ligand-deficient mice. J. Clin. Invest. 105, 813–821. doi: 10.1172/JCI8236
Zhang, C., Gao, F., Teng, F., Zhang, M. (2015). Fas/FasL complex promotes proliferation and migration of brain endothelial cells via FADD-FLIP-TRAF-NF-κB pathway. Cell Biochem. Biophys. 71, 1319–1323. doi: 10.1007/s12013-014-0351-4
Zhou, Z., Wu, M., Barrett, R. P., McClellan, S. A., Zhang, Y., Hazlett, L. D. (2010). Role of the Fas pathway in Pseudomonas aeruginosa keratitis. Invest. Ophthalmol. Vis. Sci. 51, 2537. doi: 10.1167/iovs.09-4152
Keywords: FAS, bacteria, virus, apoptosis, inflammation, treatment
Citation: Hu L, Lu J, Fan H, Niu C, Han Y, Caiyin Q, Wu H and Qiao J (2025) FAS mediates apoptosis, inflammation, and treatment of pathogen infection. Front. Cell. Infect. Microbiol. 15:1561102. doi: 10.3389/fcimb.2025.1561102
Received: 15 January 2025; Accepted: 25 March 2025;
Published: 22 April 2025.
Edited by:
Marcel Doerflinger, The University of Melbourne, AustraliaReviewed by:
Toru Okamoto, Juntendo University, JapanGregor Ebert, Technical University of Munich, Germany
Sven Engel, The University of Melbourne, Australia
Copyright © 2025 Hu, Lu, Fan, Niu, Han, Caiyin, Wu and Qiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hao Wu, ZHJlYW03MndoQHRqdS5lZHUuY24=; Jianjun Qiao, amlhbmp1bnFAdGp1LmVkdS5jbg==
†These authors have contributed equally to this work and share first authorship