 Ahlam Alsaadi1*
Ahlam Alsaadi1* Mohammed Imam2
Mohammed Imam2 Abdulrahman A. Alghamdi1
Abdulrahman A. Alghamdi1 Safia S. Aljedani1Amal Alsari1Haya Aljami1
Safia S. Aljedani1Amal Alsari1Haya Aljami1 Mohammad Bosaeed1,3
Mohammad Bosaeed1,3- 1King Abdullah International Medical Research Center, King Saud Bin Abdulaziz University for Health Sciences, Riyadh, Saudi Arabia
- 2Department of Microbiology and Parasitology, Qunfudah College of Medicine, Umm Al-Qura University, Al-Qunfudah, Makkah, Saudi Arabia
- 3Department of Medicine, King Abdulaziz Medical City, Ministry of National Guard Health Affairs, Riyadh, Saudi Arabia
Prophages are bacteriophages that integrate their genomes into the bacterial chromosome. This research aimed to analyze and characterize prophages integrated into 44 Pseudomonas aeruginosa strains isolated from tertiary hospitals in Saudi Arabia. A total of 97 intact prophages were identified among clinical strains, with 16 prophages found present in more than one strain simultaneously. All prophages were found to have lengths ranging from 7.7 kb to 74.1 kb, and their GC content was found to be between 49.91% and 64.9%. Our findings show that prophages are present in the majority of the isolated P. aeruginosa strains (41 out of 44). Additionally, several proteins related to viral defense (toxin/antitoxin modules and proteins against restriction-modification enzymes) were identified, supporting the idea that prophages influence bacterial pathogenesis and anti-phage defenses.
Backgrond
Pseudomonas aeruginosa is an opportunistic, Gram-negative pathogen and a member of the diverse and complex Pseudomonas genus (Pirnay et al., 2009). P. aeruginosa persists in both clinical and environmental settings and causes a wide spectrum of human infections, ranging from mild to life-threatening conditions (Diggle and Whiteley, 2020). The treatment of infections caused by P. aeruginosa presents one of the greatest therapeutic challenges (Pelegrin et al., 2021). Antibiotic resistance in P. aeruginosa can develop either through mutation that modify the expression or function of resistance-related genes or by acquiring resistance genes via mobile genetic elements (MGEs) (Silby et al., 2011). The accessory genome in P. aeruginosa consists mainly of mobile genetic elements such as plasmids, transposons, and temperate bacteriophages (prophages) (Silby et al., 2011). P. aeruginosa is listed in the ESKAPE group of pathogens, a designation for six bacteria recognized for critical clinical importance due to their increased antimicrobial-resistance rates (Rice, 2008).
Bacteriophages, or phages for short, are bacterial viruses considered obligate intracellular parasites. Phages are highly abundant on earth (Ackermann, 2007). During host infection, typically phages undergo one of two different life cycles: the lysogenic or the lytic cycle (Penadés et al., 2015; Williams et al., 2008). For phage therapy, strictly lytic phages, which directly undergo the lytic life cycle and lyse the target bacterial cells are preferable. Phages that follow the lysogenic life cycle integrate their genomes into the genome of their bacterial host, thereby contributing to bacterial fitness and bacterial genomic diversity, including the acquisition of antibiotic resistance genes and virulence factors (Gandon, 2016). Phages that integrate their genomes into the bacterial host are referred to as “prophages” or “temperate” phages (Gandon, 2016; Gordon et al., 2017). Recent studies have shown that prophages interact with their bacterial host’s regulatory cascade and interfere with the host’s immune system, in addition to encoding toxins, lytic proteins, and antimicrobial-resistance genes (Cieślik et al., 2021). For instance, in clinical P. aeruginosa prophage were found to code complete type II toxin-antitoxin module, exhibiting homology to the BrnT toxin and a CopG family antitoxin while other encode for complete type II TA system YoeB/YefM that is related to antibiotic resistance, biofilm formation, serum survival and host infection (Heaton et al., 2012; Ma et al., 2021). Moreover, the type II toxin YafO and a type IV antitoxin AbiEi were found in P. aeruginosa prophages all in which counteract bacterial defenses or to compete against external phages targeting the host (González de Aledo et al., 2023; Leroux and Laub, 2025). Therefore, the impact of prophages on human health and disease are largely unexplored and is still under investigation.
Early estimates found that prophages account for 10 to 20% of the host’s genome (Hatfull and Hendrix, 2011). The presence of prophages within the bacterial genome can provide the host with a selective advantage, for instance, the acquisition of prophages in clinical P. aeruginosa has been shown to reduce antibiotic susceptibility and enhance biofilm formation (Tariq et al., 2019). Among these, P. aeruginosa filamentous prophages Pf4 are highly upregulated and contributed to both biofilm formation and virulence (Hay and Lithgow, 2019; Platt et al., 2008; Rice et al., 2009). Additionally, prophages DMS3 and pp3 play distinct roles in host adaptation: DMS3 inhibits infection by phages that utilize the type IV pilus as a receptor, while pp3 promotes biofilm formation, facilitating bacterial persistence (Canchaya et al., 2004; Li et al., 2017; Little, 2005; Shah et al., 2021). Prophages can protect bacteria from environmental stressors and confer antibiotic resistance to bacterial cells by enhancing bacterial fitness through mechanisms such as, toxin-antitoxin systems, superinfection exclusion, biofilm formation and horizontal gene transfer (HGT) (Wang et al., 2010). These temperate phages significantly contribute to the extensive genomic diversity of bacteriophages through HGT and recombination (Dion et al., 2020).
Prophage-host interactions and dynamic adaptations over time have resulted in the acquisition of numerous defense mechanisms in response to selective pressures. These mechanisms including restriction-enzymes (RM) systems, clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated (cas) genes, the abortive infection (Abi) systems, as well as the accumulation of a variety of mutations in surface receptor proteins (Ambroa et al., 2022; Labrie et al., 2010). As the evolutionary race between bacteria and their viral predators continue, new anti-viral mechanisms have been discovered and describes, such as the use of cyclic nucleotides as signaling molecules, such as the cyclic oligonucleotide-based antiphage signaling system, the pyrimidine cyclase system for antiphage resistance, and restriction by an adenosine deaminase acting on RNA (Cohen et al., 2019; Duncan-Lowey et al., 2023; Tal et al., 2021). Additionally, NAD+ depletion is a widespread bacterial defense response to viral infection (Garb et al., 2022; Tal and Sorek, 2022). As phages evolve, they have developed mechanisms such as anti-CRISPR (Acr) proteins and viral DNA methyltransferases. Acr proteins, first discovered in prophages infecting P. aeruginosa strains (Bondy-Denomy et al., 2013), are small peptides known to inhibit CRISPR-Cas activity. They function by binding to the different elements of the CRISPR machinery, thereby preventing DNA recognition, or inhibiting Cas protein activity once the protein complex has assembled around the target DNA (Pawluk et al., 2018).
Most P. aeruginosa strains have been identified to contain at least one prophage-like element; some are poly-lysogens, harboring several prophages in their genome (Knezevic et al., 2015; Silby et al., 2011). Lysogenic phages in P. aeruginosa have been shown to confer selective beneficial traits, such as O antigen conversion, biofilm development, and virulence (Hayashi et al., 1990; Kuzio and Kropinski, 1983; Rice et al., 2009). Additionally, prophages can serve as viable candidates for phage therapy, as they can be genetically engineered to be strictly lytic. For instance, in 2019, a patient with disseminated drug-resistant Mycobacterium abscessus was successfully treated with a cocktail of mutant phages engineered to eliminate lysogeny associated genes (Dedrick et al., 2019). Thus, identifying P. aeruginosa prophages provides valuable insight into the role of phages in P. aeruginosa fitness and pathogenicity.
Here, we investigate publicly available P. aeruginosa genomes from Saudi Arabia, which currently include 44 published clinical bacterial genomes, with a focus on temperate P. aeruginosa phages. This study aims to expand understanding of the nature, composition, and role of prophages found within a multicenter hospital P. aeruginosa strain collection from Saudi Arabia. Additionally, we analyze the genes these prophages harbor to overcome bacterial defenses and disseminate antibiotic resistance genes. Identifying prophages with no AMR or virulence factor genes in their genome is beneficial as they can hold potential as phage therapy candidates.
Materials and methods
Origin of P. aeruginosa isolated genomes
A total of 44 complete genomes of P. aeruginosa isolates from 44 patients were obtained from the National Center for Biotechnology Information (NCBI) database (https://www.ncbi.nlm.nih.gov/) (Last accessed on April 17, 2023). Demographic data of the patients were extracted from the original study’s Supplementary Data and presented in the study (see Table 1; Supplementary Table 1), bacterial genomes were sequenced using the MiSeq Illumina platform with a 2 x 300 bp paired-end reads protocol, as previously described (Doumith et al., 2022). The whole genome extraction and sequencing methods are described (Doumith et al., 2022), and genomes were deposited in Genebank under the accession numbers PRJNA751257.
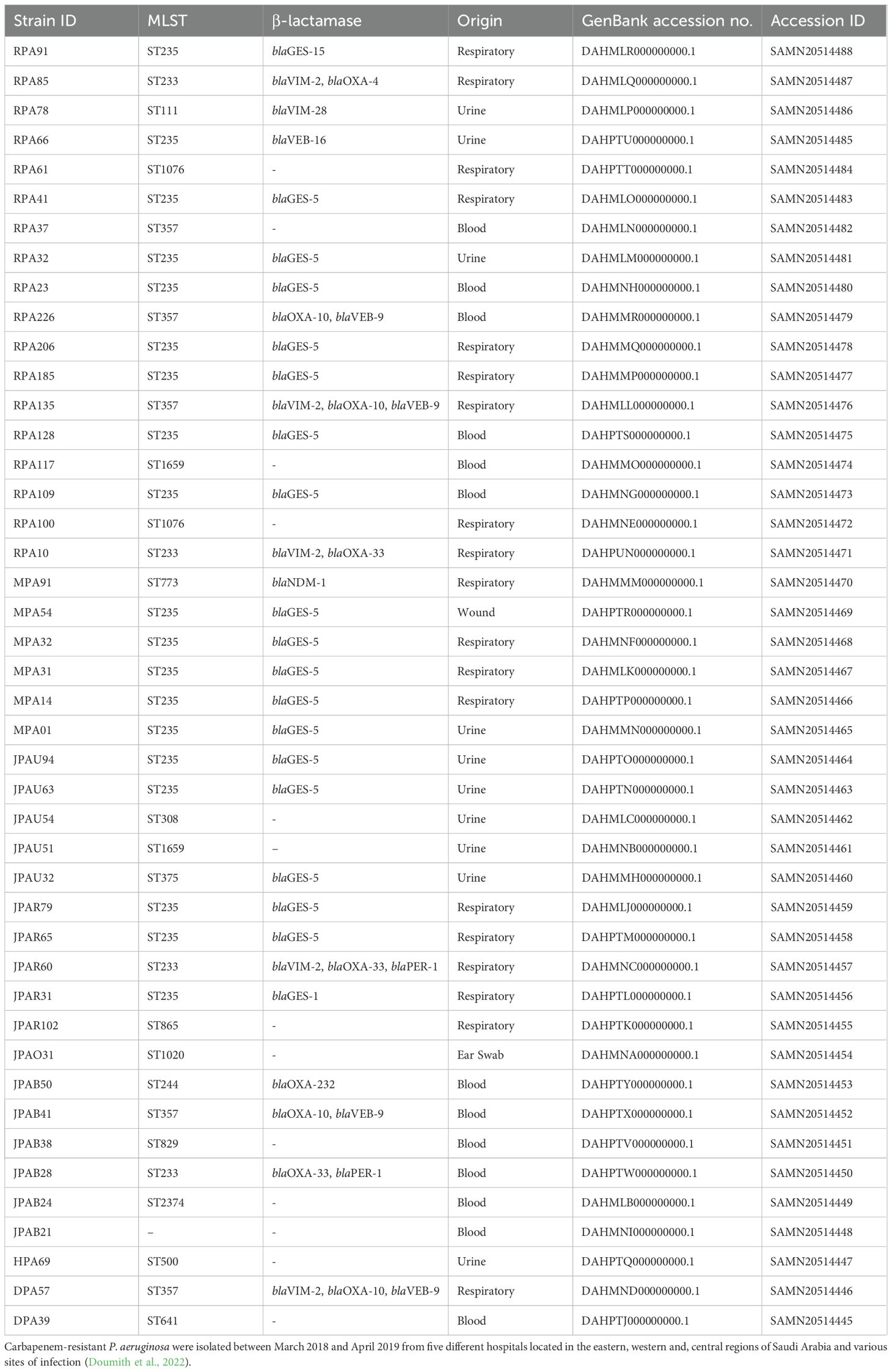
Table 1. Characteristics of the 44 P. aeruginosa clinical strains.
Identification of prophages in P. aeruginosa strains
Whole-genome sequences of P. aeruginosa clinical strains were used to identify and annotate prophages by submitting FASTA files via the web interface to PHASTER (PHAge Search Tool Enhanced Release) (https://phaster.ca/), a bioinformatics tool designed to rapidly identify and annotate putative prophage genomes sequences within the contigs of each bacterial genome. P. aeruginosa genomes were accessed on 23 04 2023. According to scoring criteria, PHASTER identifies prophages in three different categories: intact (score >90), questionable (score 70-90), and incomplete (score <70). Only intact prophages with scores of >90 were selected for this study. Additionally, PHASTER provides the location or insertion site of prophage within the bacterial genome, the number of coding DNA sequences (CDSs), and the GC% content of each prophage genome. Prophage genomes content were annotated using Prokka (https://github.com/tseemann/prokka) (last accessed on January 10, 2024) and the PHROGs database, which represents a library of families of different prokaryotic virus proteins generated using a new clustering approach based on remote homology detection, (https://phrogs.lmge.uca.fr/) (last accessed on January 10, 2024) (Terzian et al., 2021).
P. aeruginosa prophage classification based on genomic similarity
With the increasing amount of phage genomic data, phages are now classified based on their genome similarity, as defined by the International Committee on the Taxonomy of Viruses (ICTV). Taxonomic classification of dsDNA phage genomes was conducted using a recently published automated high-throughput computational tool (taxmyPHAGE) (https://github.com/amillard/tax_myPHAGE/tree/main) designed for genus- and species-level identification of bacteriophages (Millard et al., 2024). In this study, we searched for closely related phages based on their genome similarity to classify prophage genomes using taxmyPHAGE. Input phage genomes were prophages whole genome sequences as FASTA format files, and the output data included information on the phage genus and species, along with similarity scores. Based on the tool guidelines, dsDNA phage genomes with an average nucleotide identity (ANI) ≥95% are considered the same species, and bacteriophages with an ANI ≥ 70% over 100% of the genome are considered to belong to the same genus (Millard et al., 2024). Also, Genome-based phylogeny and tree construction was performed using the VICTOR web platform to compare Psedumonas prophage genomes from this study with three members of the Casadabanvirus genus, as currently recognized by the ICTV (https://ictv.global) (Meier-Kolthoff and Göker, 2017). Whole-genome amino acid sequences comparisons were conducted using the Genome-BLAST Distance Phylogeny method (GBDP) approach, applying the d0 distance formula (Meier-Kolthoff et al., 2013). The resulting intergenomic distances were used to construct a balanced minimum evolution phylogenetic tree using FASTME 2.0, incorporating subtree pruning and regrafting (SPR) optimization and supported by 100 pseudo-bootstrap replicates (Lefort et al., 2015). The final tree was midpoint-rooted and visualized using the ggtree package (Yu et al., 2017). Species, genus, and family demarcation thresholds were determined using the OPTSIL clustering algorithm (Göker et al., 2009). Applying standard parameters and an F value of 0.5, protein homology was assessed using HHPRED (Zimmermann et al., 2018) and PHYRE2 (Kelley et al., 2015), while domain architecture predictions were carried out using the SMART tool (Letunic et al., 2021).
Phylogenetic analysis of common Pseudomonas prophages
All identified complete prophage sequences were aligned using the MAFFT Version 7.0 program (https://maft.cbrc.jp/alignment/server/), using the strategy ‘auto’. A phylogenetic tree of the genomes was subsequently constructed using the phylogeny program (https://maft.cbrc.jp/alignment/server/phylogeny.html) using the neighbor-joining method using bootstrap values of 1000 replicates. The generated phylogenetic tree was visualized using the iTOL program (Baliga et al., 2021).
Identification of virulence factors in prophage genomes
Prophage genomes were used to screen for virulence factor (VF) genes against the Virulence Factors of Pathogenic Bacteria Database (VFDB) (http://www.mgc.ac.cn/cgi-bin/VFs/v5/main.cgi, last accessed on November 20, 2023) (Liu et al., 2019). The VFDB was established in 2004 to provide up-to-date information about VFs from various bacterial pathogens and serves as a comprehensive repository system of bacterial virulence factors. Furthermore, VF genes were identified using VirulanceFinder 2.0 (https://cge.cbs.dtu.dk/services/VirulenceFinder/, last accessed on November 20, 2023).
Identification of antibiotic resistance genes in prophage genomes
All prophage genomes were screened for the presence of antibiotic resistance genes in the Comprehensive Antibiotic Resistance Database (CARD) (https://card.mcmaster.ca/, last accessed on November 20, 2023) (Alcock et al., 2020). CARD is a biological database integrating molecular and sequence data while collecting resistance determinants and associated antibiotics. Resistance genes were also assessed by screening for AMR genes using the ResFinder 4.1 database (https://cge.cbs.dtu.dk/services/ResFinder-4.1/, last accessed on November 20, 2023).
Statistical analysis
The Pearson’s correlation coefficient (r) analysis was conducted using DATAtab: Online Statistics Calculator (DATAtab: DATAtabTeam, 2025) https://datatab.net. Pearson’s correlation was computed with a two-tailed test, and a threshold of p < 0.05 was considered statistically significant. Additionally, scatter plots were generated using DATAtab’s online tool, which provided visual representation of the relationships between variables. All statistical assumptions, including normality, were verified before analysis.
Results
Abundance of prophages in P. aeruginosa genomes
The genome sequences of 44 P. aeruginosa clinical isolates from 44 patients were screened for prophages. A total of 270 prophage-like elements were detected (Supplementary Table 2, 9), of which 97 were classified as intact, 96 as incomplete, and 77 as questionable (Figure 1). The number of intact bacteriophages found in each genome ranged from one to six, with a median of two, adding up to a final value of 97 prophages (Supplementary Table 2). The correlation between the prophage genome type and its distribution was investigated. The Pearson’s correlation coefficients (R-values) were Total & Incomplete (r = 0.79, p < 0.001), Total & Intact (r = 0.62, p < 0.001) and Total & Questionable (r = 0.4, p = 0.007). This correlation reveals a significant correlation between the total category and all three subcategories, as the strongest relationship is with Incomplete, followed by Intact, while the weakest is with Questionable. The R-values for all categories were greater than 0.5, indicating a positive correlation between increased the total prophages count with intact and incomplete prophage count increase. In total, 41 P. aeruginosa clinical (93.1%) were found to harbor at least one to six prophages. However, in strains JPAB41, JPAU51, and RPA226, no intact prophages were found. This indicates that prophages are abundant in the genomes of P. aeruginosa.
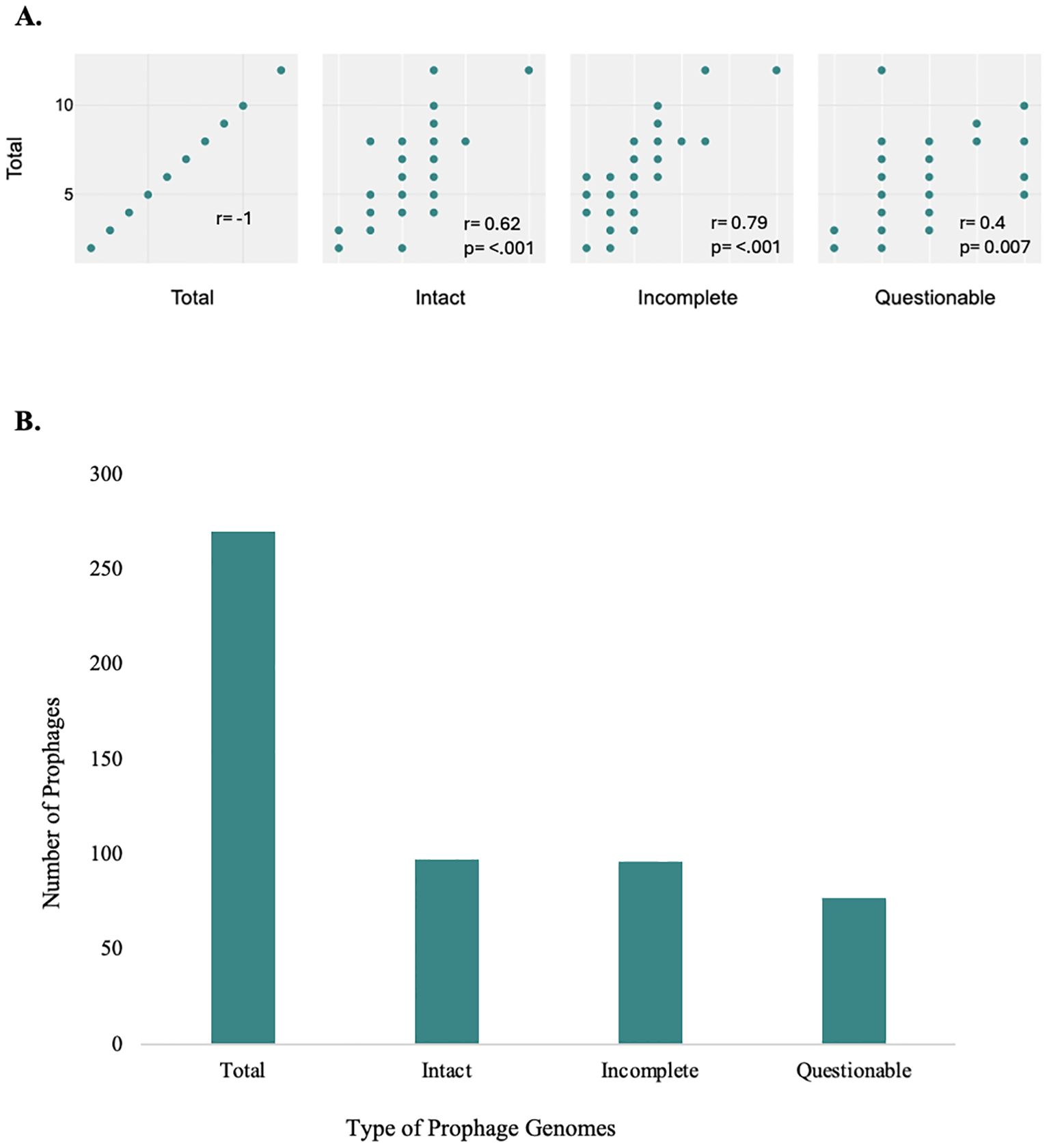
Figure 1. Prophage distribution in P. aeruginosa genomes. Illustration of the correlation coefficients between the total number of intact, incomplete, and questionable prophages (A), and total number of prophages in P. aeruginosa genomes, categorized as intact, incomplete, and questionable (B). Prophages were detected and classified using the PHASTER with default arguments and its scoring system. (A) Correlation and scatter plot of prophage genome types and their distribution. The analysis reveals a significant correlation between the total category and all three subcategories, as the strongest relationship is with Incomplete, followed by Intact, while the weakest is with Questionable, all with p-values <0.05; these correlations are statistically significant. (B) There were differences in the distribution of prophages of varying completeness on the chromosomes of P. aeruginosa. The integration of intact prophages in the bacterial genome indicates a recent infection with a temperate phage (Costa et al., 2018). There is a significantly high prevalence of defective phages (Figure 1, Supplementary Table 2).
AMR and virulence factors found in prophages
Prophages provide genomic plasticity and host adaptation for their bacteria (Shen et al., 2020), and act as important vehicles carrying virulence factor (VF) and antimicrobial resistance (AMR) genes (Colomer-Lluch et al., 2011). Among all prophages, only the prophage genome AA67 was found to carry a chloramphenicol resistance gene catB7_1 detected by ResFinder (Supplementary Table 10). There were no VF or AMR genes identified in other intact prophages in this study, this absence may be attributed to a complex decay process involving the accumulation of mutations, deletions or genetic rearrangements, leading to the loss of non-essential genes such as AMR genes. Additionally, carrying AMR genes can impose a metabolic burden on bacteria, especially in antibiotics-free environments, potentially reducing bacterial fitness. Thus, this selective pressure may favor retention of prophages lacking these genes (Kondo et al., 2021). However, VF genes were detected in the incomplete genomes of JPAO31R1in, JPAO31R7in, JPAR31R6in, JPAU63R6in, RPA10R10in, RPA41R7in, RPA109R8in, RPA128R6in, RPA135R4in, and MPA31R8in (Supplementary Table 3). In the clinical P. aeruginosa isolates, the presence of VF and/or AMR genes in incomplete prophages rather than intact prophages could be often associated with that intact prophages often retain genes essential for phage-related functions, whereas incomplete prophages may accumulate genes advantageous to bacterial survival. In the study by Kondo et al. (2021), they observed that AMR genes were frequently located near recombination-related genes, such as integrases and transposases, within prophage regions. In contrast, VF genes were less commonly associated with these recombination-related genes. The distinct distribution patterns imply that AMR and VF genes may be acquired and propagated through different evolutionary pathways within bacterial genomes (Kondo et al., 2021). In the genomes of nine different incomplete prophages, an annotated Escherichia coli iss gene was detected with 97.41% coverage (GenBank accession no. AF042279). The iss2 gene contributes to bacterial survival and is not usually found in Pseudomonas spp., suggesting acquisition through horizontal gene transfer. The incomplete prophage genome JPAO31R1in was found to carry eight different VF genes, including algL, algX, algG, algE, algK, alg44, alg8, and algD). Some of these alginates represent key exopolysaccharides involved in biofilm formation and alginate production by P. aeruginosa (Monday and Schiller, 1996).
Genetic diversity of P. aeruginosa prophages
Among the identified prophages, some prophages were found to be present in more than one clinical strain, with 13 P. aeruginosa genomes carrying the prophage AA18, 12 genomes harboring AA20 as the second most abundant prophage, and eight P. aeruginosa genomes carrying prophage AA19. The genomes of prophages AA13, AA08, and AA03 were found in six, five and four P. aeruginosa genomes, respectively (Figure 2). Four prophage genomes were present in three different genomes. In comparison, the other six common prophage genomes were only found twice in each bacterial genome (Supplementary Table 4; Figure 2). These common prophage genomes were distributed across a variety of strains with different sequence types and also had high percentages of integration on the chromosome. All prophage genome sizes ranged from 7.7 kb to 74.1 kb, and their GC% content was found to be between 49.91% and 64.9%, which is slightly lower than the host’s GC content of 65-67% for P. aeruginosa (Klockgether et al., 2011) (Supplementary Table 5). The differences in GC content among prophages are considered an evolutionary trait, indicative of the recent acquisition of prophage regions with exogenous origins (Fortier and Sekulovic, 2013). The most common prophage among P. aeruginosa strains, AA18, with a GC content of 62.67% indicates its adaptation to its host; the more similar the GC content of a prophage is to its host, the better adapted the prophage is. Additionally, prophages with shorter genome lengths present higher GC content, such as prophages AA29, AA47, AA67, and AA5 (Supplementary Table 5). It is important to note that the genomes were divided into contigs, which implies that PHASTER may have underestimated the correct number of intact prophages. Some prophages may have split into different contigs and thus identified as incomplete or questionable prophages.
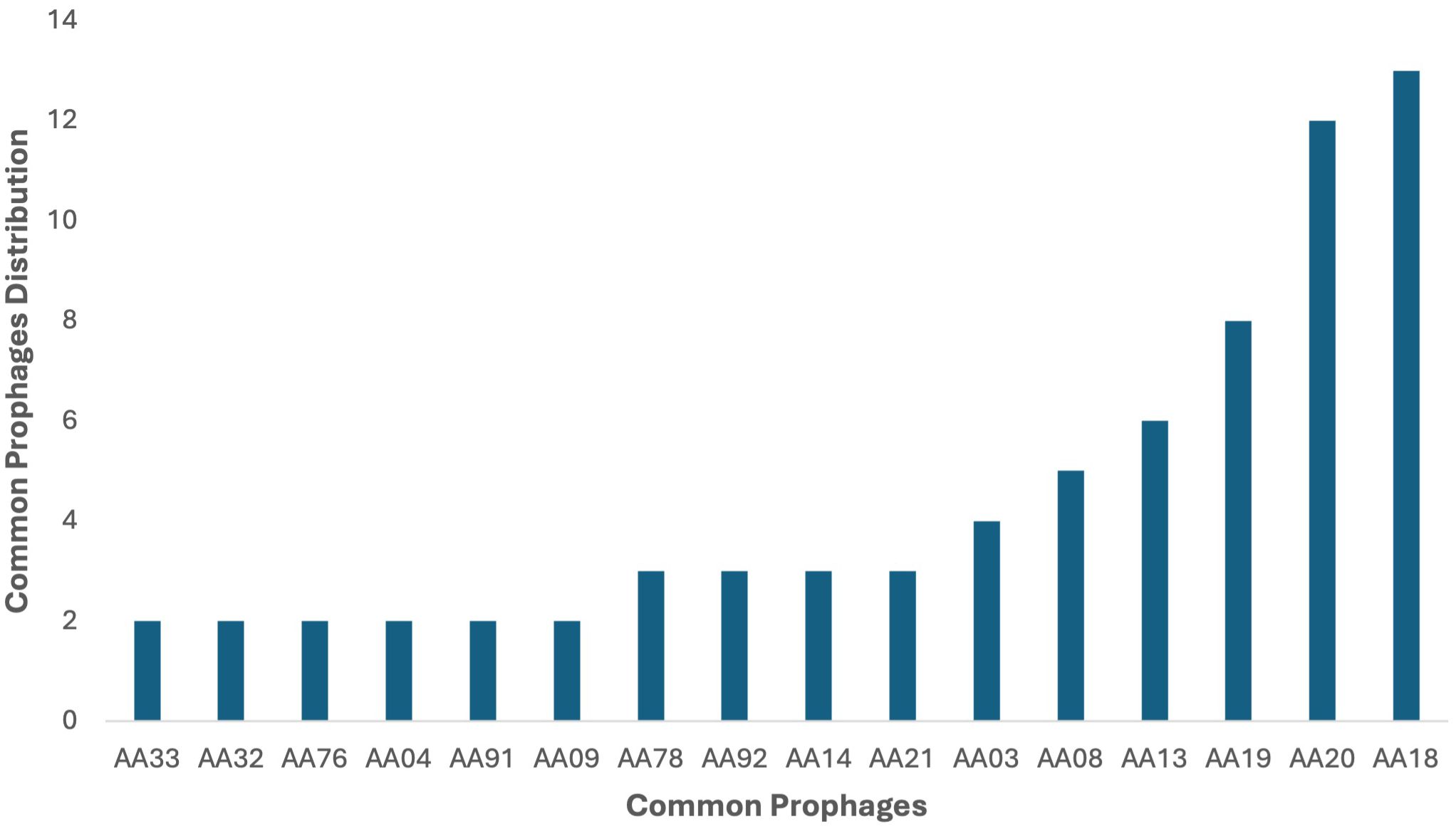
Figure 2. Distribution of intact prophages in the 44 P. aeruginosa strains. Among the identified prophages, AA18 was the dominant intact prophage in 13 P. aeruginosa genomes in the study, followed by AA20 in 12 genomes and AA19 in eight genomes. Prophages AA13, AA08, and AA03 were present in six, five, and four genomes, respectively. Four prophages were identified in three genomes each, while six other prophages were found in two genomes each.
Analysis of prophage distribution according to taxonomic classification and genome size
Using TaxmyPHAGE, 97 prophage genomes were classified into distinct taxonomic groups (Supplementary Table 6). TaxmyPHAGE identified the genera and species of the input prophage genome sequences; genomes were found to differ based on their genomic analysis (Figure 3). Genomic comparisons revealed that 28.1% of the genomes were from known genera, while 71.8% were classified to be from novel and unknown genera, 39.58% and 32.29% respectively. The majority of phages belong to novel or uncharacterized taxa at both the genus and species levels, suggesting the discovery of new phage groups. From the genera and species contributed significantly, like Casadabanvirus (7.2%), Lambdavirus (8.3%), and Citexvirus (6.25%). While genera and species such as Beetrevirus (2.08%) and Hollowayvirus (2.08%) were less common, suggesting they may be less widespread or specialized. Unexpectedly, the presence of Lambdavirus lambda was reported among Pseudomonas phages, in prophages genomes AA29, AA52, and AA62. These genome sequence annotations were verified using BLAST against the NCBI database to ensure accuracy, that the prophage genome annotations indicated Lambdavirus lambda. The results show a high degree of prophage diversity among the P. aeruginosa clinical strains. The average genome size of the prophages was 41.3kb, which is consistent with values reported from other studies working on different bacteria (Bobay et al., 2014; Canchaya et al., 2003).
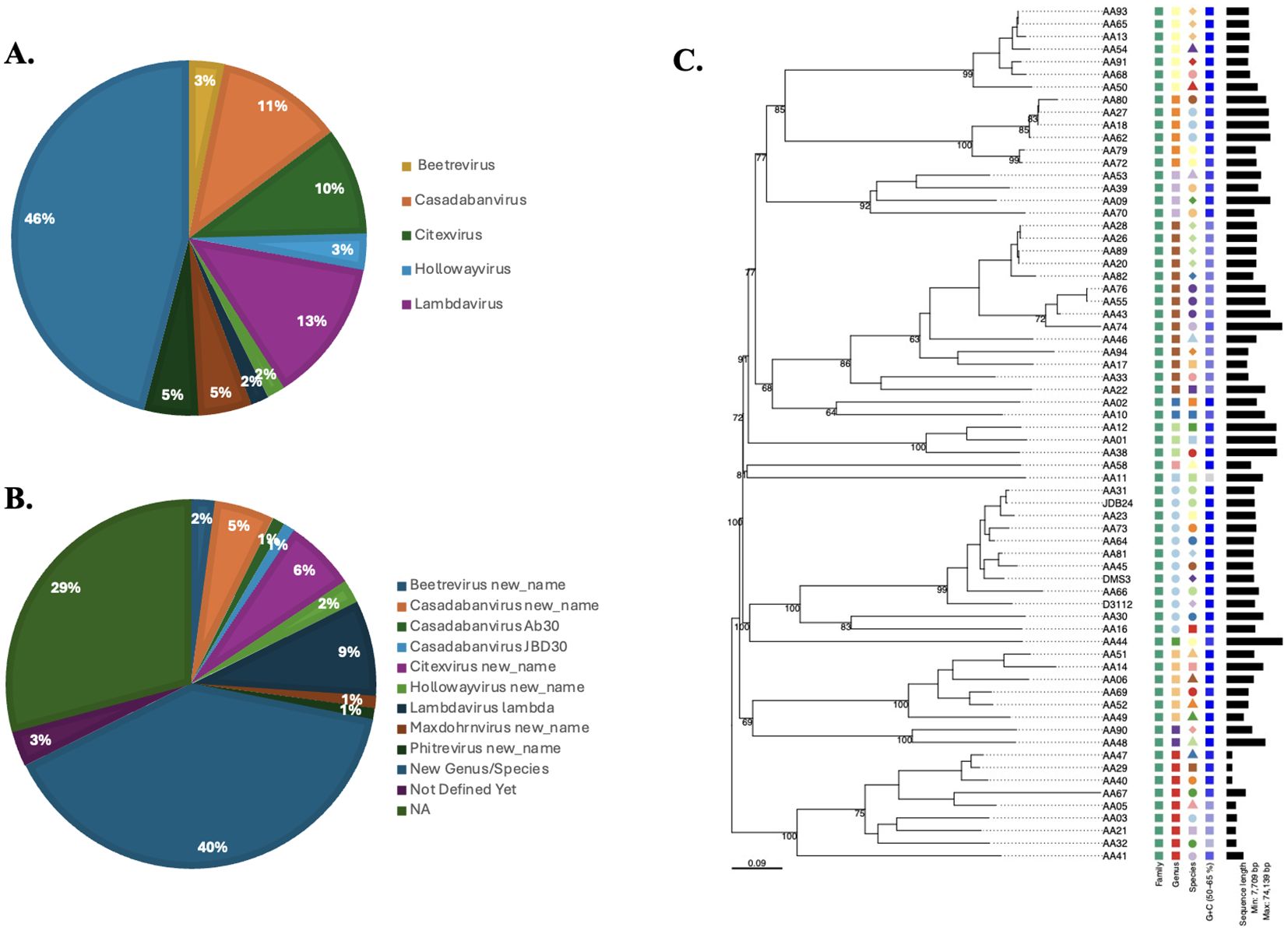
Figure 3. Taxonomic classification of prophage genomes. TaxmyPHAGE classified 97 prophage genomes, identifying 28.1% as belonging to known genera, and 71.8% as novel and unknown genera. Prominent taxa included Casadabanvirus (7.2%) and Lambdavirus (8.3%), while less frequent taxa, such as Beetrevirus (2.08%), were observed at both the genus (A) and species (B) levels. (C) Phylogeny of Pseudomonas prophages in current study and three different prophages (JDB24 (NC 020203), DMS3 (NC 008717), and D3112 (NC 005178) within the Casadabanvirus genus as listed by ICTV. The tree was inferred from the amino acid sequences using the VICTOR pipeline with the GBDP d0 formula.
Phylogenetic relationship between prophages
A phylogenetic tree was generated based on complete prophage sequences to evaluate diversity. The tree was constructed using the major capsid protein (MCP) as a reference (Figure 4). The phylogenetic analysis revealed that the prophages clustered according to their morphological family types, indicating that the predicted virion morphotypes correlate with the genomic phylogeny. This finding aligns with the new classification system of the International Committee on the Taxonomy of Viruses, which is based on genome and proteome data (Adriaenssens and Rodney Brister, 2017). Furthermore, phylogenetic analysis of the P. aeruginosa prophages investigated demonstrated a high diversity of evolutionary groupings and provided potential functional insights derived from the MCP sequences. Prophage genomes such as AA01, AA12, and AA09 are closely clustered, indicating lower genetic divergence and suggesting that their capsid structures may be highly conserved, which could imply a specific phage-host interaction. In contrast, prophages AA44, AA16, and AA73 share an evolutionary history, while the longer branches of AA45, AA09 with AA49, AA06, and AA92 suggest more distant relationships. These insights provide a framework for future studies on the diverse structural and functional roles of MCP in prophage adaptation. The tree was visualized using Interactive Tree of Life (iTOL) v6.
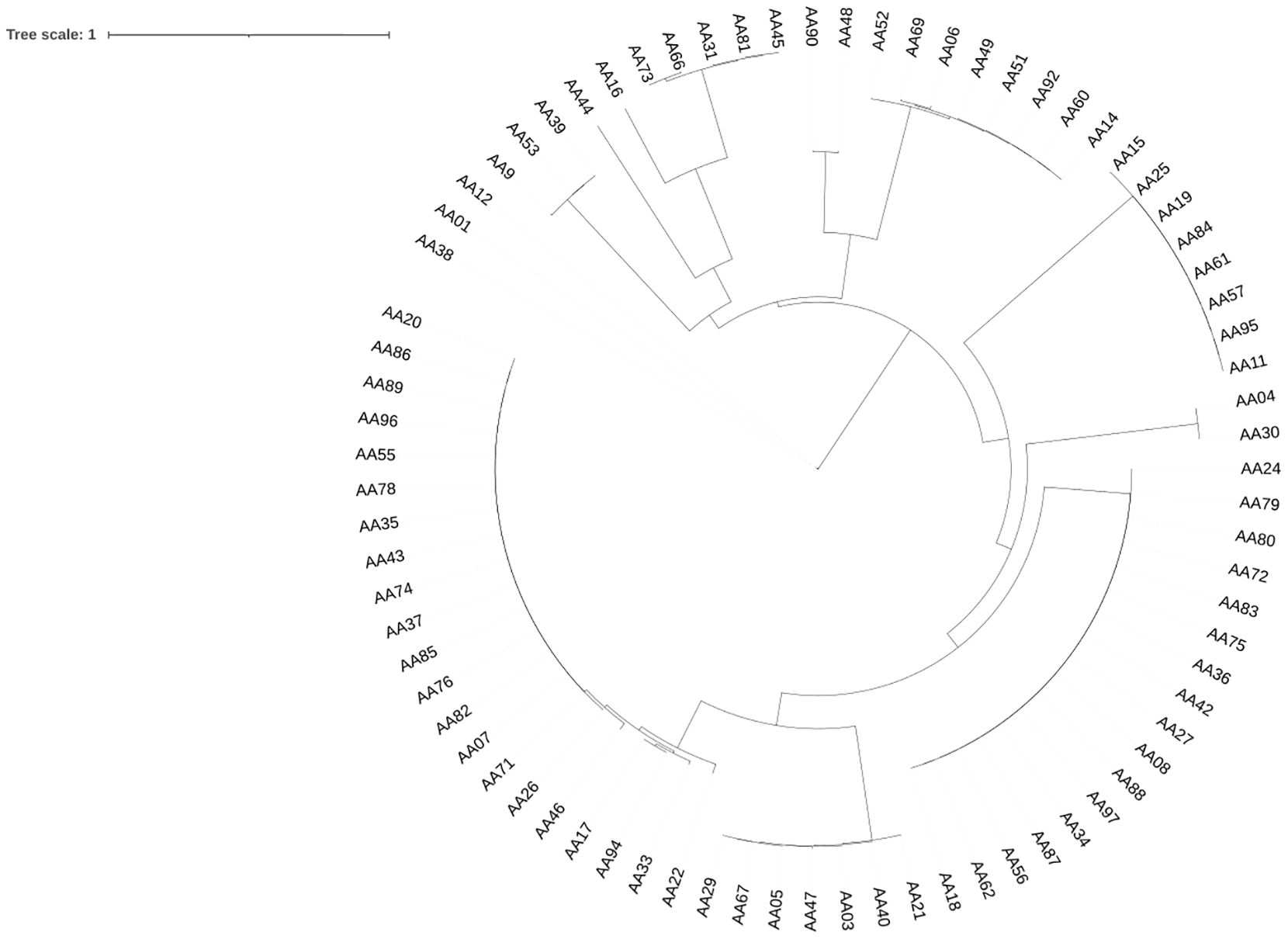
Figure 4. Phylogenetic Tree of P. aeruginosa-associated prophages. The phylogeny tree was constructed using the neighbor-joining method with the MAFFT program based on major capsid protein (MCP) sequences. Only prophage genomes containing MCP sequences were included in the analysis, while prophages without MCPs were excluded. The schematic representation of the tree was visualized using the iTOL v6 software (Letunic and Bork, 2021).
Sequence type distribution
In the sequence set consisting of 44 strains of P. aeruginosa, all strains had known sequence types (ST), except for one strain JPAB21 (Table 1), which were divided into 16 distinct types. The correlation between the P. aeruginosa ST and prophage harboring was studied (Supplementary Table 7). In the collection of 44 P. aeruginosa strains, ST235 was found to be the most prevalent type (n = 19), followed by ST233 (n = 5) and ST357 (n = 4), (Figure 5).
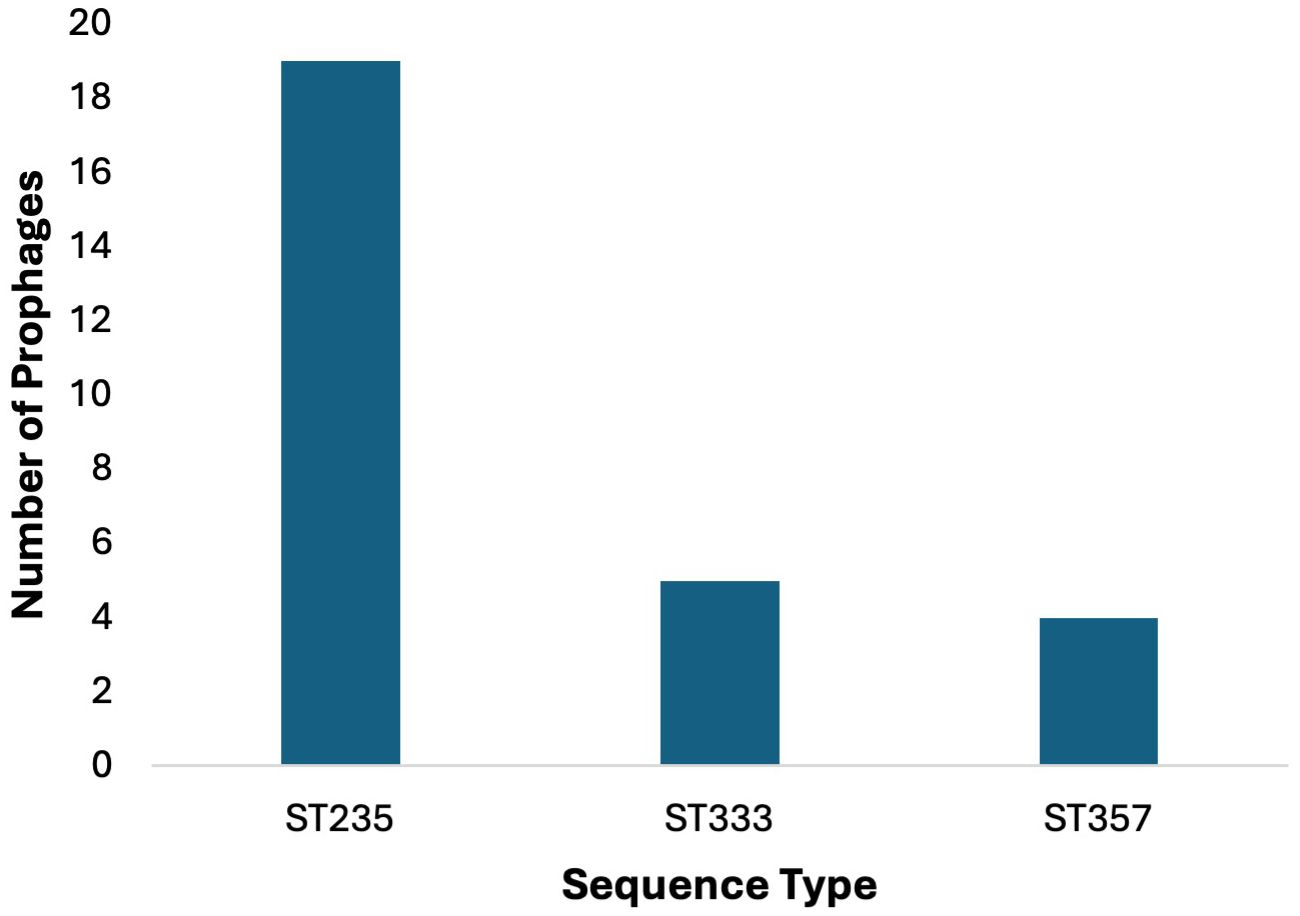
Figure 5. Prophage counts in most common sequence types (STs). Prophages were predominantly identified in the 44 P. aeruginosa strains belonging to ST235 (n=19), followed by ST233 (n = 5) and ST357 (n = 4).
In ST235, prophage AA18 was found in 13 out of 19 strains, prophage AA20 in 12 out of 19, and prophage AA19 in 5 out of 19 strains; in ST233 2 out of 4, and in ST111, 1 out of 1 strain. P. aeruginosa strains with ST235 harbor the three most common prophages, which carry the GES-5 and GES-15 β-lactamases. The other three P. aeruginosa strains JPAB41, JPAU51, and RPA226, which did not harbor any of the three most common prophages, carried the GES-1 and GES-5 β-lactamases. In other ST, the distribution of these prophages was rare or even absent. This suggests that ST may relate to the types of prophages integrated into the chromosome.
The strains belonging to the different STs were more diverse in their prophage arrangement. Although the remaining strains harbored at least one intact prophage, the distribution of these prophages was irregular and lacked a clear association.
Most common anti-phage defense systems identified in analyzed genomes
a. Defense against restriction-modification systems
In this study, 28 out of 97 prophages were found to encode DNA methyltransferases. Prophages use DNA methyltransferases to methylate their DNA to evade the host cell’s restriction-modification system, regulate viral gene expression, and facilitate DNA packaging into the preformed capsids (Burke et al., 2021; Loenen and Raleigh, 2014). Additionally, restriction alleviation proteins were found in 20 prophage genomes; these proteins are known to protect against the host cell’s restriction-modification systems (Supplementary Table 8) (Loenent and Murray1, 1986; Toothmant, 1981).
b. Toxin/antitoxin systems
Toxin-antitoxin (TA) modules play important roles in plasmid and prophage stability and are also key factors in bacterial physiology (Harms et al., 2018). A proposed system protects bacteria from phages, alongside CRISPR and restriction-modification systems. In this context, it is not surprising that prophages carry antitoxins to counteract bacterial defense mechanisms, or even toxins alone, to compete against external phages preying on their host (Leroux and Laub, 2022). In this study where the proposed system was detailed, prophages were found to encode TA modules belonging to the type II systems. The type II toxin YafO was found in 16 prophage genomes (Zhang et al., 2009). Additionally, the type II TA system putative toxin HigB2 was found in ten prophages. In bacteria, toxin HigB2 has been shown to reduce the expression of virulence-associated traits such as the production of pyochelin and pyocyanin, biofilm formation, and swarming motility. Thus, this system affects the pathogenicity of the strain in a manner not previously demonstrated for TA systems (Table 2; Supplementary Table 8) (Wood and Wood, 2016).
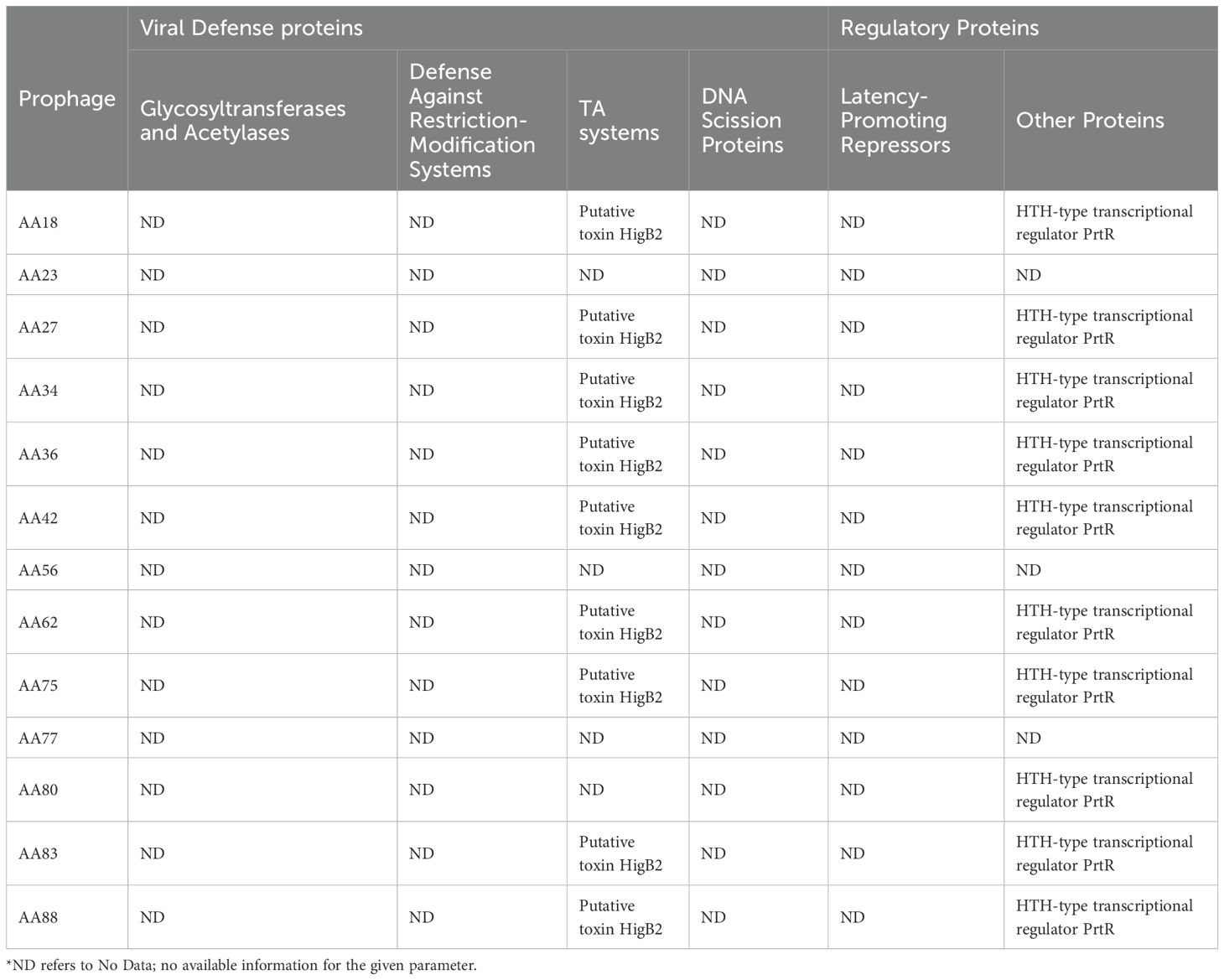
Table 2. Viral defense and regulatory proteins identified in 13 prophages common across all 44 P. aeruginosa genomes analyzed in this study.
DNA scission proteins
Prophages encode junction-resolving enzymes, such as Holliday junction resolvases. Specifically, prophages encode RusA-like Holliday junction resolvase, Lar-like restriction alleviation protein, and restriction alleviation Ral (Supplementary Table 8). These enzymes have been described as being involved in the degradation of the host’s DNA, self-DNA maturation, and cleavage prior to packaging (Wyatt and West, 2014).
Discussion
This study encompassed the search and analysis of prophages within a set of 44 multidrug-resistant (MDR) P. aeruginosa clinical strains isolated from different hospitals across different regions of Saudi Arabia (Doumith et al., 2022). Our findings demonstrate that these prophages are present in the majority of strains (Supplementary Tables 1 and 9). Many of the prophages were found in more than one strain, following a similar ST pattern. In only 6.8% of the strains (n = 3), no intact prophages as predicted by the PHASTER tool were identified, showing that prophage harboring is a very frequent trait among circulating P. aeruginosa strains in Saudi Arabia.
Prophages have been linked to bacterial diversification and evolution, exerting a strong selection pressure on bacterial fitness and virulence (Brüssow et al., 2004; Canchaya et al., 2004; Casjens, 2003; Desiere et al., 2001). A limited number of studies have characterized the prevalence of prophages in bacterial species and evaluated their role in virulence in the Arab region, specifically in clinical strains from Saudi Arabia. In this study, we report the analysis of prophage prevalence in clinical MDR P. aeruginosa and discuss their possible contribution to the evolution of the pathogenicity of P. aeruginosa.
We found that the 44 P. aeruginosa strains harbored a total of 270 prophages following genomic analysis. A significant number of these prophages (n = 97) were classified as intact prophages, while detective prophages (96 incomplete and 77 questionable) were also detected in large numbers. The subset of 97 intact prophages were classified into distinct taxonomic groups (Figure 3). The defective prophages were attributed to strong stress selection, which resulted in mutations or gene loss that inactivated the prophages (Bobay et al., 2014). Genomic diversification was observed among the sequences, revealing novel genera and species among the prophages. Using the TaxMYPHAGE tool, genomic comparisons indicated that 33.33% of the prophages within P. aeruginosa genomes were assigned to known genera (Figure 3), while 55% were classified as novel genera. This highlights the discovery of new phage groups and emphasizes the need for additional research to better understand phage diversity within clinically isolated P. aeruginosa strains. Interestingly, prophage genomes AA29, AA52, and AA62 were classified under the Lambdavirus genus, however, when compared against the NCBI database, they unexpectedly showed similarities to Pseudomonas associated phages. This could be attributed due to two key reasons; (i) complex evolutionary and taxonomic relationships, such as horizontal gene transfer (Hulin et al., 2023); (ii) mis-annotation due to sequence similarities, which may also explain the inclusion of Lambdaviruses among Pseudomonas phages as classification tools rely heavily on conserved genomic regions. Moreover, as genomic technologies advance even more rapidly, that there will be more discoveries in the near future. These findings suggest exercising caution when classifying phages, as reliance on specific conserved regions may lead to inaccuracies in taxonomic assignments. Overall, our findings reveal a significant degree of genomic diversity within the prophages within the clinically isolated P. aeruginosa strains.
The correlation between bacterial genome sizes and their GC content was studied by Chen et al. (2016). Applying their findings to prophages, shorter prophage genomes might have higher GC content due to selective pressures favoring energy-efficient nucleotide usage and the retention of stable, essential genes. However, it is important to note that this is a hypothesis derived from related studies and this relationship is influenced by various factors, including the specific bacterial host, environmental conditions, and evolutionary pressures.
Some genes expressed from prophage regions can alter the properties of the host, ranging from increased protection against further phage infection to increased virulence (Casjens, 2003). High-risk STs, such as ST235, were identified as carrying various prophages. ST235 is known for its ability to acquire mobile genetic elements, its elevated antimicrobial resistance rates, and its global distribution (Kabic et al., 2023; Treepong et al., 2018). In particular, ST235, the most prevalent ST among MDR P. aeruginosa clinical isolates (Table 1). has been shown to lack a functional CRISPR-Cas system, thus explaining its ability to acquire exogenous genetic elements, such as genomic matter from bacteriophage.
The prevalence of the putative toxin HigB2 was highlighted, as a part of a TA system in prophage genomes, which is known to stabilize the prophage within the bacterial cell. Under stressful conditions, the activation of HigB2 might initiate entering a state of dormancy, allowing bacterial cells to withstand adverse environments and delay prophages from entering the lytic cycle. The dormant state may contribute to the persistence of bacteria even in the presence of antibiotics (Leroux and Laub, 2022; Yang and Walsh, 2017). Collectively, these findings highlight the functional versatility of prophage-encoded toxins like HigB2, underscoring their roles in bacterial survival and adaptation in challenging environments.
The prophage-encoded HTH-type transcriptional regulator PrtR acts as a key modulator in controlling phage life cycle transitions, responding to host stress, and potentially influencing host virulence. Its regulatory function ensures that the prophage remains latent under stable conditions and activates lytic genes when the host environment becomes unfavorable, enhancing both phage survival and bacterial adaptability (Dodd et al., 2005; Oppenheim et al., 2005). In the presence of DNA damage, such as treatment with mitomycin C, the activation of RecA causes the PrtR repressor to self-cleave, which together with PrtN, regulates the production of pyocins (Matsui et al., 1993; Penterman et al., 2014; Walker, 1984; Wu and Jin, 2005). This highlights the importance of prophage-borne counter-defense mechanisms, which not only protect the prophage against the bacterial host’s immune system, but also protect the host from infection by other phages, enabling survival and transmission to bacterial progeny.
Prophages can be induced from their host and can contribute to phage therapy. Although prophages incorporate their genomes into the host and integrate within the host genome, they can also serve as effective candidates for phage therapy. Genetic engineering of prophages, such as the removal of the integrase protein, and genomes are cleared of virulence genes, resistance related genes, and generally undesirable genes can make engineered prophages safe for use it patients. In 2019, a patient with a disseminated multi-drug-resistant Mycobacterium abscessus was treated with a cocktail of mutant phages engineered to remove lysogeny associated genes (Dedrick et al., 2019). This case highlighted the feasibility of phage engineering for complex bacterial infections. Another form of phage engineering involves extracting specific lytic proteins, such as lysins and holin, and using them for therapeutic purposes instead of whole viral particles. Their effect of these phage derived enzymes on MDR P. aeruginosa has been investigated in vitro and reported in the literature (Cui et al., 2023). Building on the prophages identified in this study, these approaches could be adapted in the future to design tailored therapies for Pseudomonas-related infections.
On the other hand, the presence of prophages can have an impact on antibiotic and phage therapies. Certain antibiotics can induce prophages within bacterial genomes, leading to the release of phage particles and potential horizontal gene transfer. A study demonstrated that common oral medications, including antibiotics, can lead to prophage induction in gut bacterial isolates, suggesting that antibiotic therapy might inadvertently activate prophages, influencing bacterial behavior and resistance patterns (Sutcliffe et al., 2021). Additionally, Prophages can confer resistance to superinfection by other phages through mechanisms such as superinfection exclusion and the CRISPR-Cas system. This resistance poses challenges to phage therapy, as therapeutic phages may be rendered ineffective against lysogenic bacteria (Bondy-Denomy et al., 2016).
One limitation of our study is that some of the P. aeruginosa isolates were sequenced using short-read bridge amplification technology (Illumina, Oxford Genomics Centre, Oxford, UK), generating 150 bp fragments. After assembly, these sequences resulted in 206 to 3,252 contigs per genome (an average of 1,602 contigs per genome). The more fragmented the genomes are, the more difficult it is to identify intact prophages, meaning that some prophages would have not been identified based on the tools used as they would have been split across several contigs. To circumvent this issue, a combination of both short- and long-read sequencing could be performed to obtain high-quality, complete bacterial genomes. This study focused its analysis on intact prophages only. Overall, our results suggest a significant contribution of prophages to the evolution and adaptation of P. aeruginosa clinical strains.
In conclusion, this study demonstrates the pivotal role of prophages in shaping the genomic landscape and adaptive capabilities of MDR P. aeruginosa clinical strains. These findings not only advance our understanding of phage-bacteria interactions but also open new avenues for therapeutic applications and treatment of MDR infections. Future research should focus on integrating long-read sequencing technologies with functional studies to enhance the therapeutic potential of prophages. This will help refine prophage classification, uncover novel targets, and develop customized interventions against P. aeruginosa and other multidrug-resistant pathogens.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author contributions
AhA: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. AAA: Data curation, Formal Analysis, Software, Writing – review & editing. MI: Methodology, Validation, Writing – review & editing. SA: Validation, Visualization, Writing – review & editing. AmA: Data curation, Validation, Writing – review & editing. HA: Data curation, Validation, Writing – review & editing. MB: Validation, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The authors declare that financial support was received from KAIMRC for the research, authorship and/or publication of this article under the projects NRC22R-354-08.
Acknowledgments
The authors are grateful for the support from the management and staff of King Abdullah International Medical Research Center (KAIMRC).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2025.1563781/full#supplementary-material
References
Ackermann, H. W. (2007). 5500 Phages examined in the electron microscope. Arch. Virol. 152, 227–243. doi: 10.1007/s00705-006-0849-1
Adriaenssens, E. M., Rodney Brister, J. (2017). How to name and classify your phage: An informal guide. Viruses 9 (4), 1–9. doi: 10.3390/v9040070
Alcock, B. P., Raphenya, A. R., Lau, T. T. Y., Tsang, K. K., Bouchard, M., Edalatmand, A., et al. (2020). CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 48, D517–D525. doi: 10.1093/nar/gkz935
Ambroa, A., Blasco, L., López, M., Pacios, O., Bleriot, I., Fernández-García, L., et al. (2022). Genomic analysis of molecular bacterial mechanisms of resistance to phage infection. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.784949
Baliga, P., Shekar, M., Kallappa, G. S. (2021). Genome-wide identification and analysis of chromosomally integrated putative prophages associated with clinical klebsiella pneumoniae strains. Curr. Microbiol. 78, 2015–2024. doi: 10.1007/s00284-021-02472-2
Bobay, L. M., Touchon, M., Rocha, E. P. C. (2014). Pervasive domestication of defective prophages by bacteria. Proc. Natl. Acad. Sci. United. States America 111, 12127–12132. doi: 10.1073/pnas.1405336111
Bondy-Denomy, J., Pawluk, A., Maxwell, K. L., Davidson, A. R. (2013). Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 493, 429–432. doi: 10.1038/nature11723
Bondy-Denomy, J., Qian, J., Westra, E. R., Buckling, A., Guttman, D. S., Davidson, A. R., et al. (2016). Prophages mediate defense against phage infection through diverse mechanisms. ISME J. 10 (12), 2854–2866. doi: 10.1038/ismej.2016.79
Brüssow, H., Canchaya, C., Hardt, W.-D. (2004). Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 68, 560–602. doi: 10.1128/mmbr.68.3.560-602.2004
Burke, E. J., Rodda, S. S., Lund, S. R., Sun, Z., Zeroka, M. R., O’Toole, K. H., et al. (2021). Phage-encoded ten-eleven translocation dioxygenase (TET) is active in C5-cytosine hypermodification in DNA. PNAS 118, 2026742118. doi: 10.1073/pnas.2026742118/-/DCSupplemental
Canchaya, C., Fournous, G., Brüssow, H. (2004). The impact of prophages on bacterial chromosomes. Mol. Microbiol. 53, 9–18. doi: 10.1111/j.1365-2958.2004.04113.x
Canchaya, C., Proux, C., Fournous, G., Bruttin, A., Brüssow, H. (2003). Prophage genomics. Microbiol. Mol. Biol. Rev. 67, 473–473. doi: 10.1128/mmbr.67.3.473.2003
Casjens, S. (2003). Prophages and bacterial genomics: What have we learned so far? Mol. Microbiol. 49, 277–300. doi: 10.1046/j.1365-2958.2003.03580.x
Chen, W. H., Van Noort, V., Lluch-Senar, M., Hennrich, M. L., Wodke, J. A. H., Yus, E., et al. (2016). Integration of multi-omics data of a genome-reduced bacterium: Prevalence of post-transcriptional regulation and its correlation with protein abundances. Nucleic Acids Res. 44 (3), 1192–1202. doi: 10.1093/nar/gkw004
Cieślik, M., Bagińska, N., Jończyk-Matysiak, E., Węgrzyn, A., Węgrzyn, G., Górski, A. (2021). Temperate bacteriophages—the powerful indirect modulators of eukaryotic cells and immune functions. Viruses 13 (6), 1013. doi: 10.3390/v13061013
Cohen, D., Melamed, S., Millman, A., Shulman, G., Oppenheimer-Shaanan, Y., Kacen, A., et al. (2019). Cyclic GMP–AMP signalling protects bacteria against viral infection. Nature 574, 691–695. doi: 10.1038/s41586-019-1605-5
Colomer-Lluch, M., Imamovic, L., Jofre, J., Muniesa, M. (2011). Bacteriophages carrying antibiotic resistance genes in fecal waste from cattle, pigs, and poultry. Antimicrob. Agents Chemother. 55, 4908–4911. doi: 10.1128/AAC.00535-11
Costa, A. R., Monteiro, R., Azeredo, J. (2018). Genomic analysis of Acinetobacter baumannii prophages reveals remarkable diversity and suggests profound impact on bacterial virulence and fitness. Sci. Rep. 8 (1), 15346. doi: 10.1038/s41598-018-33800-5
Cui, J., Shi, X., Wang, X., Sun, H., Yan, Y., Zhao, F., et al. (2023). Characterization of a lytic Pseudomonas aeruginosa phage vB_PaeP_ASP23 and functional analysis of its lysin LysASP and holin HolASP. Front. Microbiol. 14. doi: 10.3389/fmicb.2023.1093668
DATAtab: DATAtab Team (2025). DATAtab: online Statistics Calculator (Graz, Austria: DATAtab e.U). Available at: https://datatab.net.
Dedrick, R. M., Guerrero-Bustamante, C. A., Garlena, R. A., Russell, D. A., Ford, K., Harris, K., et al. (2019). Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 25, 730–733. doi: 10.1038/s41591-019-0437-z
Desiere, F., McShan, W. M., Van Sinderen, D., Ferretti, J. J., Brüssow, H. (2001). Comparative genomics reveals close genetic relationships between phages from dairy bacteria and pathogenic streptococci: Evolutionary implications for prophage-host interactions. Virology 288, 325–341. doi: 10.1006/viro.2001.1085
Diggle, S. P., Whiteley, M. (2020). Microbe profile: Pseudomonas aeruginosa: Opportunistic pathogen and lab rat. Microbiol. (United Kingdom). 166, 30–33. doi: 10.1099/mic.0.000860
Dion, M. B., Oechslin, F., Moineau, S. (2020). Phage diversity, genomics and phylogeny. Nat. Rev. Microbiol. 18, 125–138. doi: 10.1038/s41579-019-0311-5
Dodd, I. B., Shearwin, K. E., Egan, J. B. (2005). Revisited gene regulation in bacteriophage λ. Curr. Opin. Genet. Dev. 15, 145–152. doi: 10.1016/j.gde.2005.02.001
Doumith, M., Alhassinah, S., Alswaji, A., Alzayer, M., Alrashidi, E., Okdah, L., et al. (2022). Genomic characterization of carbapenem-non-susceptible pseudomonas aeruginosa clinical isolates from Saudi Arabia revealed a global dissemination of GES-5-producing ST235 and VIM-2-producing ST233 sub-lineages. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.765113
Duncan-Lowey, B., Tal, N., Johnson, A. G., Rawson, S., Mayer, M. L., Doron, S., et al. (2023). Cryo-EM structure of the RADAR supramolecular anti-phage defense complex. Cell 186, 987–998.e15. doi: 10.1016/j.cell.2023.01.012
Fortier, L. C., Sekulovic, O. (2013). Importance of prophages to evolution and virulence of bacterial pathogens. Virulence 4, 354–365. doi: 10.4161/viru.24498
Gandon, S. (2016). Why be temperate: lessons from bacteriophage λ. Trends Microbiol. 24, 356–365. doi: 10.1016/j.tim.2016.02.008
Garb, J., Lopatina, A., Bernheim, A., Zaremba, M., Siksnys, V., Melamed, S., et al. (2022). Multiple phage resistance systems inhibit infection via SIR2-dependent NAD + depletion. Nat. Microbiol. 7 (11), 1849–1856. doi: 10.1101/2021.12.14.472415
Göker, M., García-Blázquez, G., Voglmayr, H., Tellería, M. T., Martín, M. P. (2009). Molecular taxonomy of phytopathogenic fungi: A case study in peronospora. PloS One 4 (7), e6319. doi: 10.1371/journal.pone.0006319
González de Aledo, M., Blasco, L., Lopez, M., Ortiz-Cartagena, C., Bleriot, I., Pacios, O., et al. (2023). Prophage identification and molecular analysis in the genomes of Pseudomonas aeruginosa strains isolated from critical care patients. MSphere 8 (4), e0012823. doi: 10.1128/msphere.00128-23
Gordon, M. A., Canals, R., Hammarlöf, D. L., Makumi, A., Aertsen, A., Feasey, N. A., et al. (2017). Characterization of the prophage repertoire of african salmonella typhimurium ST313 reveals high levels of spontaneous induction of novel phage BTP1. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.00235
Harms, A., Brodersen, D. E., Mitarai, N., Gerdes, K. (2018). Toxins, targets, and triggers: an overview of toxin-antitoxin biology. Mol. Cell 70, 768–784. doi: 10.1016/j.molcel.2018.01.003
Hatfull, G. F., Hendrix, R. W. (2011). Bacteriophages and their genomes. Curr. Opin. Virol. 1, 298–303. doi: 10.1016/j.coviro.2011.06.009
Hay, I. D., Lithgow, T. (2019). Filamentous phages: masters of a microbial sharing economy. EMBO Rep. 20 (6), e47427. doi: 10.15252/embr.201847427
Hayashi, T., Baba, T., Matsumoto, H., Terawaki, Y. (1990). Phage-conversion of cytotoxin production in Pseudomonas aerug’inosa. Mol. Microbiol. 4 (10), 1703–1709. doi: 10.1111/j.1365-2958.1990.tb00547.x
Heaton, B. E., Herrou, J., Blackwell, A. E., Wysocki, V. H., Crosson, S. (2012). Molecular structure and function of the novel BrnT/BrnA toxin-antitoxin system of Brucella abortus. J. Biol. Chem. 287, 12098–12110. doi: 10.1074/jbc.M111.332163
Hulin, M. T., Rabiey, M., Zeng, Z., Vadillo Dieguez, A., Bellamy, S., Swift, P., et al. (2023). Genomic and functional analysis of phage-mediated horizontal gene transfer in pseudomonas syringae on the plant surface. New Phytol. 237 (3), 959–973. doi: 10.1111/nph.18573
Kabic, J., Fortunato, G., Vaz-Moreira, I., Kekic, D., Jovicevic, M., Pesovic, J., et al. (2023). Dissemination of metallo-β-lactamase-producing pseudomonas aeruginosa in Serbian hospital settings: expansion of ST235 and ST654 clones. Int. J. Mol. Sci. 24 (2), 1519. doi: 10.3390/ijms24021519
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., Sternberg, M. J. E. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10 (6), 845–858. doi: 10.1038/nprot.2015.053
Klockgether, J., Cramer, N., Wiehlmann, L., Davenport, C. F., Tümmler, B. (2011). Pseudomonas aeruginosa genomic structure and diversity. Front. Microbiol. 2. doi: 10.3389/fmicb.2011.00150
Knezevic, P., Voet, M., Lavigne, R. (2015). Prevalence of Pf1-like (pro)phage genetic elements among Pseudomonas aeruginosa isolates. Virology 483, 64–71. doi: 10.1016/j.virol.2015.04.008
Kondo, K., Kawano, M., Sugai, M. (2021). Distribution of antimicrobial resistance and virulence genes within the prophage-associated regions in nosocomial pathogens. MSphere 6 (4), e0045221. doi: 10.1128/msphere.00452-21
Kuzio, J., Kropinski, A. M. (1983). O-Antigen Conversion in Pseudomonas aeruginosa PAO1 by Bacteriophage D3. J. Bacteriol. 155 (1), 203–212. doi: 10.1128/jb.155.1.203-212.1983
Labrie, S. J., Samson, J. E., Moineau, S. (2010). Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 8, 317–327). doi: 10.1038/nrmicro2315
Lefort, V., Desper, R., Gascuel, O. (2015). FastME 2.0: A comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 32 (10), 2798–2800. doi: 10.1093/molbev/msv150
Leroux, M., Laub, M. T. (2022). Toxin-antitoxin systems as phage defense elements. Annu. Rev. Microbiol. 76, 21–43. doi: 10.1146/annurev-micro-020722-013730
Leroux, M., Laub, M. T. (2025). Toxin-antitoxin systems as phage defense elements. Annu. Rev. Microbiol. 51, 56. doi: 10.1146/annurev-micro-020722
Letunic, I., Bork, P. (2021). Interactive tree of life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Li, G., Lu, S., Shen, M., Le, S., Shen, W., Tan, Y., et al. (2017). Characterization and interstrain transfer of prophage pp3 of Pseudomonas aeruginosa. PloS One 12 (3), e0174429. doi: 10.1371/journal.pone.0174429
Little, J. W. (2005). Lysogeny, Prophage Induction, and Lysogenic Conversion. Phages 37–54. doi: 10.1128/9781555816506.ch3
Liu, B., Zheng, D., Jin, Q., Chen, L., Yang, J. (2019). VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 47, D687–D692. doi: 10.1093/nar/gky1080
Loenen, W. A. M., Raleigh, E. A. (2014). The other face of restriction: Modification-dependent enzymes. Nucleic Acids Res. 42, 56–69. doi: 10.1093/nar/gkt747
Loenent, W. A. M., Murray1, N. E. (1986). Modification enhancement by the restriction alleviation protein (Ral) of bacteriophage lambda. J Mol Biol. 190 (1), 11–22. doi: 10.1016/0022-2836(86)90071-9
Ma, D., Gu, H., Shi, Y., Huang, H., Sun, D., Hu, Y. (2021). Edwardsiella piscicida yefM-yoeB: A type II toxin-antitoxin system that is related to antibiotic resistance, biofilm formation, serum survival, and host infection. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.646299
Matsui, H., Sano, Y., Ishihara, H., Shinomiya, T. (1993). Regulation of pyocin genes in pseudomonas aeruginosa by positive (prtN) and negative (prtR) regulatory genes. J. Bacteriol. 175 (5), 1257–1263. doi: 10.1128/jb.175.5.1257-1263.1993
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H.-P., Oker, M. G. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinf. 14, 60. doi: 10.1186/1471-2105-14-60
Meier-Kolthoff, J. P., Göker, M. (2017). VICTOR: genome-based phylogeny and classification of prokaryotic viruses. Bioinf. (Oxford England). 33, 3396–3404. doi: 10.1093/bioinformatics/btx440
Millard, A., Denise, R., Lestido, M., Thomas, M., Turner, D., Turner, D., et al. (2024). taxmyPHAGE: Automated taxonomy of dsDNA phage genomes at the genus and species level. PHAGE 6 (1), 5–11. doi: 10.1101/2024.08.09.606593
Monday, S. R., Schiller, N. L. (1996). Alginate synthesis in pseudomonas aeruginosa: the role of algL (Alginate lyase) and algX. J. Bacteriol. 178 (3), 625–632. doi: 10.1128/jb.178.3.625-632.1996
Oppenheim, A. B., Kobiler, O., Stavans, J., Court, D. L., Adhya, S. (2005). Switches in bacteriophage lambda development. Annu. Rev. Genet. 39, 409–429. doi: 10.1146/annurev.genet.39.073003.113656
Pawluk, A., Davidson, A. R., Maxwell, K. L. (2018). Anti-CRISPR: Discovery, mechanism and function. Nat. Rev. Microbiol. 16, 12–17. doi: 10.1038/nrmicro.2017.120
Pelegrin, A. C., Palmieri, M., Mirande, C., Oliver, A., Moons, P., Goossens, H., et al. (2021). Pseudomonas aeruginosa: A clinical and genomics update. FEMS Microbiol. Rev. 45 (6), fuab026. doi: 10.1093/femsre/fuab026
Penadés, J. R., Chen, J., Quiles-Puchalt, N., Carpena, N., Novick, R. P. (2015). Bacteriophage-mediated spread of bacterial virulence genes. Curr. Opin. Microbiol. 23, 171–178. doi: 10.1016/j.mib.2014.11.019
Penterman, J., Singh, P. K., Walker, G. C. (2014). Biological cost of pyocin production during the SOS response in Pseudomonas aeruginosa. J. Bacteriol. 196, 3351–3359. doi: 10.1128/JB.01889-14
Pirnay, J. P., Bilocq, F., Pot, B., Cornelis, P., Zizi, M., Van Eldere, J., et al. (2009). Pseudomonas aeruginosa population structure revisited. PloS One 4 (11), e7740. doi: 10.1371/journal.pone.0007740
Platt, M. D., Schurr, M. J., Sauer, K., Vazquez, G., Kukavica-Ibrulj, I., Potvin, E., et al. (2008). Proteomic, microarray, and signature-tagged mutagenesis analyses of anaerobic Pseudomonas aeruginosa at pH 6.5, likely representing chronic, late-stage cystic fibrosis airway conditions. J. Bacteriol. 190, 2739–2758. doi: 10.1128/JB.01683-07
Rice, L. B. (2008). Federal funding for the study of antimicrobial resistance in nosocomial pathogens: No ESKAPE. J. Infect. Dis. 197, 1079–1081. doi: 10.1086/533452
Rice, S. A., Tan, C. H., Mikkelsen, P. J., Kung, V., Woo, J., Tay, M., et al. (2009). The biofilm life cycle and virulence of Pseudomonas aeruginosa are dependent on a filamentous prophage. ISME. J. 3, 271–282. doi: 10.1038/ismej.2008.109
Shah, M., Taylor, V. L., Bona, D., Tsao, Y., Stanley, S. Y., Pimentel-Elardo, S. M., et al. (2021). A phage-encoded anti-activator inhibits quorum sensing in Pseudomonas aeruginosa. Mol. Cell 81, 571–583.e6. doi: 10.1016/j.molcel.2020.12.011
Shen, J., Zhou, J., Xu, Y., Xiu, Z. (2020). Prophages contribute to genome plasticity of Klebsiella pneumoniae and may involve the chromosomal integration of ARGs in CG258. Genomics 112, 998–1010. doi: 10.1016/j.ygeno.2019.06.016
Silby, M. W., Winstanley, C., Godfrey, S. A. C., Levy, S. B., Jackson, R. W. (2011). Pseudomonas genomes: Diverse and adaptable. FEMS Microbiol. Rev. 35, 652–680. doi: 10.1111/j.1574-6976.2011.00269.x
Sutcliffe, S. G., Shamash, M., Hynes, A. P., Maurice, C. F. (2021). Common oral medications lead to prophage induction in bacterial isolates from the human gut. Viruses 13 (3), 455. doi: 10.3390/v13030455
Tal, N., Morehouse, B. R., Millman, A., Stokar-Avihail, A., Avraham, C., Fedorenko, T., et al. (2021). Cyclic CMP and cyclic UMP mediate bacterial immunity against phages. Cell 184, 5728–5739.e16. doi: 10.1016/j.cell.2021.09.031
Tal, N., Sorek, R. (2022). SnapShot: bacterial immunity. Cell 185, 578–578.e1. doi: 10.1016/j.cell.2021.12.029
Tariq, M. A., Everest, F. L. C., Cowley, L. A., Wright, R., Holt, G. S., Ingram, H., et al. (2019). Temperate bacteriophages from chronic pseudomonas aeruginosa lung infections show disease-specific changes in host range and modulate antimicrobial susceptibility. MSystems 4 (4), e00191–18. doi: 10.1128/msystems.00191-18
Terzian, P., Olo Ndela, E., Galiez, C., Lossouarn, J., Pérez Bucio, R. E., Mom, R., et al. (2021). PHROG: Families of prokaryotic virus proteins clustered using remote homology. NAR. Genomics Bioinf. 3 (3), lqab067. doi: 10.1093/nargab/lqab067
Toothmant, P. (1981). Restriction alleviaion by bacteriophages lambda and lambda reverse. J. Virol. 38 (2), 621–631. doi: 10.1128/JVI.38.2.621-631.1981
Treepong, P., Kos, V. N., Guyeux, C., Blanc, D. S., Bertrand, X., Valot, B., et al. (2018). Global emergence of the widespread Pseudomonas aeruginosa ST235 clone. Clin. Microbiol. Infect. 24, 258–266. doi: 10.1016/j.cmi.2017.06.018
Walker, G. C. (1984). Mutagenesis and inducible responses to deoxyribonucleic acid damage in escherichia coli. Microbiol. Rev. 48 (1), 60–93. doi: 10.1128/mr.48.1.60-93.1984
Wang, X., Kim, Y., Ma, Q., Hong, S. H., Pokusaeva, K., Sturino, J. M., et al. (2010). Cryptic prophages help bacteria cope with adverse environments. Nat. Commun. 1 (9), 147. doi: 10.1038/ncomms1146
Williams, S. R., Gebhart, D., Martin, D. W., Scholl, D. (2008). Retargeting R-type pyocins to generate novel bactericidal protein complexes. Appl. Environ. Microbiol. 74, 3868–3876. doi: 10.1128/AEM.00141-08
Wood, T. L., Wood, T. K. (2016). The HigB/HigA toxin/antitoxin system of Pseudomonas aeruginosa influences the virulence factors pyochelin, pyocyanin, and biofilm formation. MicrobiologyOpen 5, 499–511. doi: 10.1002/mbo3.346
Wu, W., Jin, S. (2005). PtrB of Pseudomonas aeruginosa suppresses the type III secretion system under the stress of DNA damage. J. Bacteriol. 187, 6058–6068. doi: 10.1128/JB.187.17.6058-6068.2005
Wyatt, H. D. M., West, S. C. (2014). Holliday junction resolvases. Cold Spring Harbor Perspect. Biol. 6 (9), a023192. doi: 10.1101/cshperspect.a023192
Yang, Q. E., Walsh, T. R. (2017). Toxin-antitoxin systems and their role in disseminating and maintaining antimicrobial resistance. FEMS Microbiol. Rev. 41, 343–353. doi: 10.1093/femsre/fux006
Yu, G., Smith, D. K., Zhu, H., Guan, Y., Lam, T. T. Y. (2017). Ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 8 (1), 28–36. doi: 10.1111/2041-210X.12628
Zhang, Y., Yamaguchi, Y., Inouye, M. (2009). Characterization of YafO, an Escherichia coli toxin. J. Biol. Chem. 284, 25522–25531. doi: 10.1074/jbc.M109.036624
Keywords: Pseudomonas aeruginosa, prophages, multi-drug resistance, bacteriophages, Saudi Arabia
Citation: Alsaadi A, Imam M, Alghamdi AA, Aljedani SS, Alsari A, Aljami H and Bosaeed M (2025) Genomic analysis of prophages in 44 clinical strains of Pseudomonas aeruginosa isolated in Saudi Arabia. Front. Cell. Infect. Microbiol. 15:1563781. doi: 10.3389/fcimb.2025.1563781
Received: 20 January 2025; Accepted: 27 March 2025;
Published: 28 April 2025.
Edited by:
Mercedes Gonzalez Moreno, Leibniz Institute for Natural Product Research and Infection Biology, Hans Knoll Institute, GermanyReviewed by:
Jumpei Fujiki, Rakuno Gakuen University, JapanAhmed Elfadadny, Tokyo University of Agriculture and Technology, Japan
Copyright © 2025 Alsaadi, Imam, Alghamdi, Aljedani, Alsari, Aljami and Bosaeed. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ahlam Alsaadi, YWxzYWFkaS1haGxhbUBob3RtYWlsLmNvbQ==