 Weixiang Wang1†Zhou Lu2†Ge Teng3Zikang Yan1Lan Huang4
Weixiang Wang1†Zhou Lu2†Ge Teng3Zikang Yan1Lan Huang4 Zhongming Tan2
Zhongming Tan2 Zhiguo Liu5*Songning Ding1*
Zhiguo Liu5*Songning Ding1* Zhenjun Li5*
Zhenjun Li5*- 1Infectious Disease Prevention and Control Department, Nanjing Municipal Center for Disease Control and Prevention, Nanjing, Jiangsu, China
- 2National Health Commission (NHC) Key Laboratory of Enteric Pathogenic Microbiology, Jiangsu Provincial Center for Disease Control and Prevention, Nanjing, Jiangsu, China
- 3Microbiology Laboratory, Nanjing Municipal Center for Disease Control and Prevention, Nanjing, Jiangsu, China
- 4School of Public Health, Southeast University, Nanjing, Jiangsu, China
- 5National Key Laboratory of Intelligent Tracking and Forecasting for Infectious Diseases, National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing, China
Objective: Human brucellosis has re-emerged as a major public health threat in Jiangsu Province, but the sources and transmission dynamics of circulating strains remain poorly understood.
Methods: In this study, we integrated conventional biotyping, whole-genome sequencing single-nucleotide polymorphism (WGS-SNP) analysis, and core genome multilocus sequence typing (cgMLST) to investigate the phylogenetic relationships of Brucella melitensis in the region.
Results: Among 89 isolates analyzed, all were confirmed as B. melitensis (16 as biovar 1 and 73 as biovar 3), with a widespread geographic distribution across 15 cities in Jiangsu and adjacent areas, indicating extensive regional dissemination. All strains belonged to sequence type 8 (ST8) and genotype group II, clustering within the East Mediterranean lineage. Genomic resolution classified these strains into five SNP clades (C-I to C-V) and 17 SNP-based genotypes (STs), revealing a ladder-like phylogenetic structure. The lack of distinct geographic clustering suggests frequent cross-regional transmission, likely facilitated by the movement of infected sheep and goats. Phylogenomic analysis through cgMLST revealed distinct clustering of the 17 STs into two major groups (G-I and G-II), with 15 STs (88.2%) showing high genetic concordance between Jiangsu isolates and strains from China’s northeastern and northwestern. This compelling genomic evidence establishes that the current human brucellosis epidemic in Jiangsu is being driven by the nationwide expansion of dominant B. melitensis lineages.
Conclusion: The findings provide crucial insights into the infection sources and interregional transmission dynamics of brucellosis in southern China, highlighting the significant role of domestic animal movement in pathogen dissemination, demanding coordinated cross-regional interventions including strict implementing intervention strategies and enhance disease surveillance.
Introduction
Brucellosis is a common zoonotic disease caused by Brucella spp., which are Gram-negative, non-motile, non-spore-forming, slow-growing, and facultative intracellular bacteria (Di Bonaventura et al., 2021). Brucellosis, also known as undulant fever or Malta fever, was first reported in the 1850s in Malta from a patient who was said to have acquired the infection through consumption of infected goat’s milk (Hull and Schumaker, 2018). In 1886, David Bruce first isolated and identified Brucella melitensis from the spleens of four soldiers as the causative agent of the disease (Aleixo et al., 1999). Currently, 12 Brucella species have been identified, including six classical species and six novel species (Bricker et al., 2000). Among, four strains exhibit pathogenicity in humans and animals, B. melitensis is the most infectious, the other strains are Brucella suis and Brucella abortus and more recently human cases being infected with Brucella cetaceae have been reported (Gorvel, 2008; Orsini et al., 2022). Direct or indirect contact with infected animals and consumption of unpasteurized dairy products are the main transmission routes of brucellosis to humans, and people may also be infected by inhalation of contaminated dust or aerosols (Al Dahouk et al., 2003; Khalafalla, 2023). Domestic animals and wildlife, including goats, sheep, cattle, deer, and camels, are natural hosts of Brucella spp (Liu et al., 2020). The persistent worldwide prevalence of human brucellosis has caused serious public health concerns and vast economic losses in the farming industry (Singh et al., 2018).
Human brucellosis is prevalent worldwide, and an evidence-based report suggests that the global incidence of human brucellosis is 2.1 million new cases every year, with the highest incidence of human brucellosis in the Middle East and Central Asia (Laine et al., 2023). Furthermore, human brucellosis has become a serious public health challenge, and the cases reported in all 31 provinces in mainland China and the affected regions have expanded from northern to southern, including Jiangsu province (Lai et al., 2017). During the years 2006-2021, 1,347 brucellosis cases were reported in Jiangsu Province, with an average annual incidence of 0.1036 per 100,000 individuals (Zhang et al., 2023). Four cluster cases of brucellosis from one family were reported in Shuyang County, Jiangsu Province, China, and isolates from the four patients were indistinguishable by MLVA profiling, displaying a unique type in Jiangsu Province (Tan et al., 2015). Whole-genome sequencing single-nucleotide polymorphism (WGS-SNP) analysis is a widely used genotyping tool for identifying the transmission pattern and phylogenetic inferences of Brucella strains (Janowicz et al., 2018; Xue et al., 2023). And, cgMLST scheme that is applicable in Brucella molecular epidemiology and helps in accurately tracking and thus controlling the sources of infection (Abdel-Glil et al., 2022). In this study, we performed WGS-SNP and cgMLST analyses on Brucella melitensis strains isolated from human patients in Jiangsu Province to elucidate their transmission patterns and phylogenetic relationships, thereby providing critical insights for developing targeted surveillance and control strategies.
Materials and methods
Brucella strain isolation and convention identification
A total of 89 Brucella spp. was isolated from blood samples collected from patients with suspected brucellosis in Nanjing City. Brucella strains were isolated according to a standard bacteriology protocol (Yagupsky et al., 2019). Briefly, five (~10) fresh blood samples were collected from suspected patients, injected into a diphasic medium in a biosafety cabinet, and then incubated in a fully automated blood culture system (BACTECTM9000, BD) for at least two weeks. The culture-positive samples were screened by real-time PCR (Al Dahouk et al., 2007), transferred to new medium for purification and isolation, and subjected to AMOS-PCR and agglutination with anti-A and M monospecific sera tests to determine the species/biovars (Eltigani et al., 1991; Zhou et al., 2021).
DNA prepare, whole-genome sequencing and pangenome analysis
DNA preparation, quality assessment, and Whole-Genome Sequencing were based on a previous study (Xue et al., 2023). Genomic DNA was prepared from heat-inactivated (80°C, 10 min) pure cultures using the Fast Pure Bacterial DNA Isolation Mini Kit [Nanjing Novozymes Biotechnology Co., Ltd. (Nanjing, China)] according to the manufacturer’s instructions. After DNA preparation, all DNA samples were fragmented by sonication to a size of 350bp, sequencing library preparation was performed using the Nextera XT library preparation kit (Illumina Inc., San Diego, CA, USA), and whole-genome sequencing of the DNA sample was performed on the MGISEQ-2000 platform. Briefly, raw data were filtered and assembled using the CLC Genomics Workbench V23.0.1 software (QIAGEN, Hilden, Germany). A summary table of genome statistics (size, GC content, N50, contig counts, and gene counts) is provided in Supplementary Table S1, highlighting assembly quality and uniformity among isolates. Based on the Virulence Factor Database (VFDB) sequences with >98% coverage and identity, Virulence-associated genes were identified. Genomic mobile elements including IS711-family insertion sequences, plasmids, and integrative conjugative elements (ICEs) were comprehensively analyzed to evaluate horizontal gene transfer capacity. The IS elements were annotated using ISfinder (e-value cutoff 1e-5), plasmid contigs were identified through alignment with PlasmidFinder (95% identity threshold), and ICEs were predicted using ICEberg 2.0 with manual curation of conserved features (integrases, conjugation modules). Antimicrobial resistance (AMR) genes were systematically screened across all isolates using a AMRFinderPlus (v3.10.30) with default parameters. Only hits meeting ≥98% sequence identity and ≥98% coverage thresholds were considered valid. Panaroo tools (Tonkin-Hill et al., 2020) was used to identify core, and unique genes across the isolates, and interpreted their functional relevance.
Genome epidemiology investigation and phylogeny analysis of B. melitensis strains
Genome epidemiology investigation and phylogeny analysis of 89 Brucella melitensis strains was performed based on the Microbial Genomics Module in the CLC Genomics Workbench V23.0.1 software (QIAGEN, Hilden, Germany), including MLST and WGS-SNP analysis. Briefly, raw sequence reads were imported into CLC Genomics Workbench (version 22.0). Quality trimming was performed using the built-in Trim Sequences tool with fault parameters. Processed reads were aligned to the B. melitensis 16M reference genome (GCA_000007125.1) using Map Reads to Reference tool (default parameters). Duplicate reads were removed using the Remove Duplicate Mapped Reads tool (Strict Mode). High-confidence SNPs were identified using the Basic Variant Detection tool, and a core-genome SNP alignment was generated by extracting all high-quality SNPs and concatenating them into a single FASTA file (Create Alignment from the Variants tool). Sites with missing data (>10% isolates) or low confidence were removed, and the final alignment was exported in the PHYLIP format. Maximum likelihood (ML) phylogeny was inferred using the RAxML plugin (v8.2.12) integrated into the CLC with the GTR+G (selected via ModelTest based on BIC) substitution model. Maximum-likelihood trees of the 89 B. melitensis strains were constructed using a maximum-likelihood phylogenetic algorithm with 1,000 bootstrap replicates (Minh et al., 2020). As described previously (Janowicz et al., 2018), a global scale core-genome MLST (cgMLST) approach was employed to construct a minimum spanning tree, revealing the regional transmission patterns. The resulting Newick tree file was imported into the iTOL v6.5.7 for visualization (Letunic and Bork, 2021).
Results
The species/biovars and distribution of 89 Brucella strains
Based on AMOS-PCR amplification, a special 731 bp band was observed in all samples, implying that the strains were all Brucella melitensis strains. Furthermore, agglutination with anti-A and M monospecific sera tests showed that 16 strains of B. melitensis bv. 1 and 73 strains were identified as B. melitensis bv. 3 (Figure 1; Table 1). Based on the isolated location, 89 strains were isolated from three provinces and 15 cities (counties), one from Shandong, 23 from Anhui province, and 58 from Jiangsu provinces. The two provinces are adjacent to Naning, Jiangsu province, and patients were located near Nanjing for convenient medical treatment (Figure 1). These data indicate that B. melitensis bv. 3 was widely prevalent in Nanjing and bordering regions.
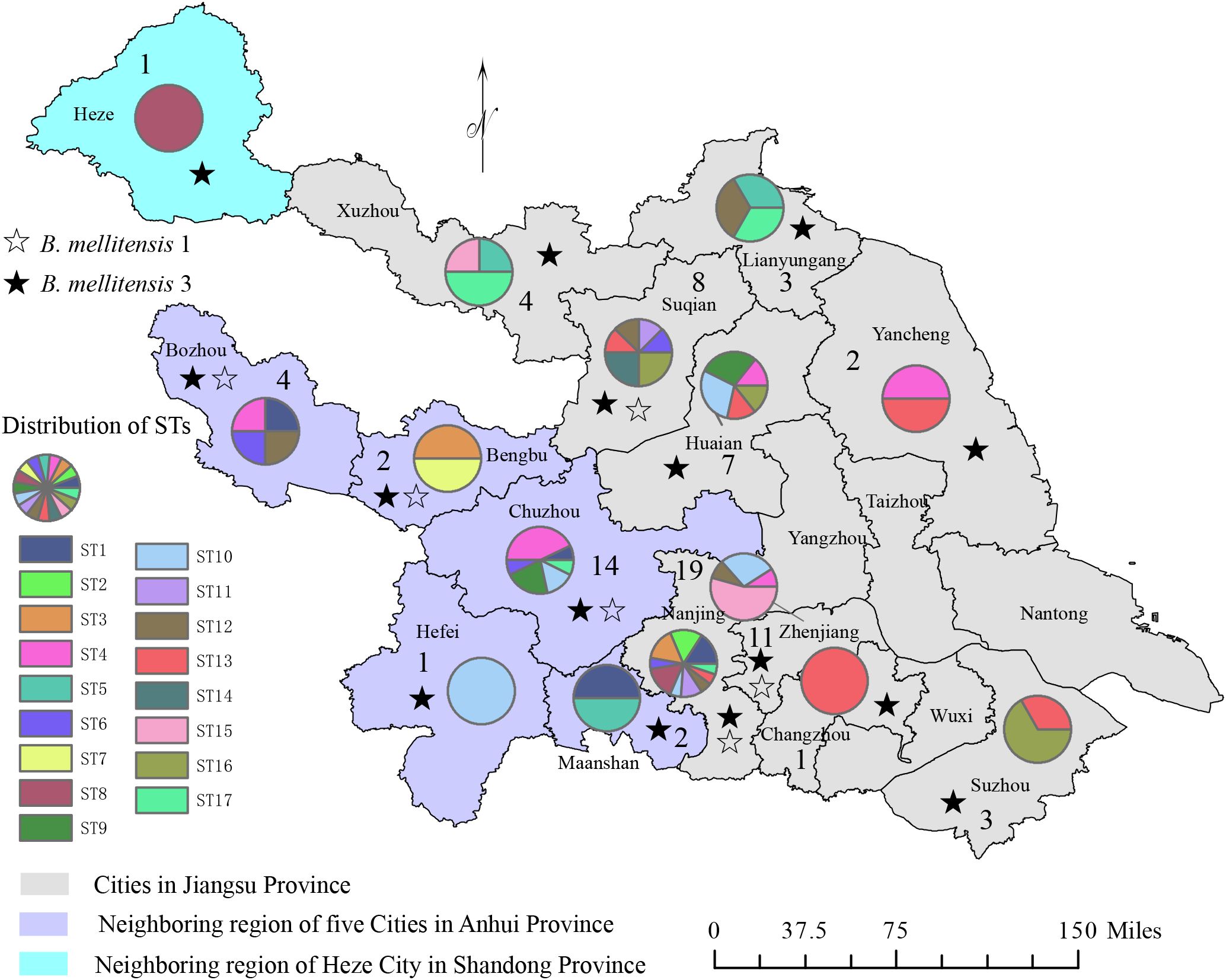
Figure 1. The distribution pattern of the number of strains, species/biovars, and STs of 89 B. melitensis from Jiangsu Province. The figure annotations are as follows: numerical values represent strain counts, ☆ symbols denote B. mellitensis bv. 1 strain, ★ symbols indicate B. mellitensis bv. 3 strains, and the color-coded pie chart illustrates the distribution of the 17 STs.
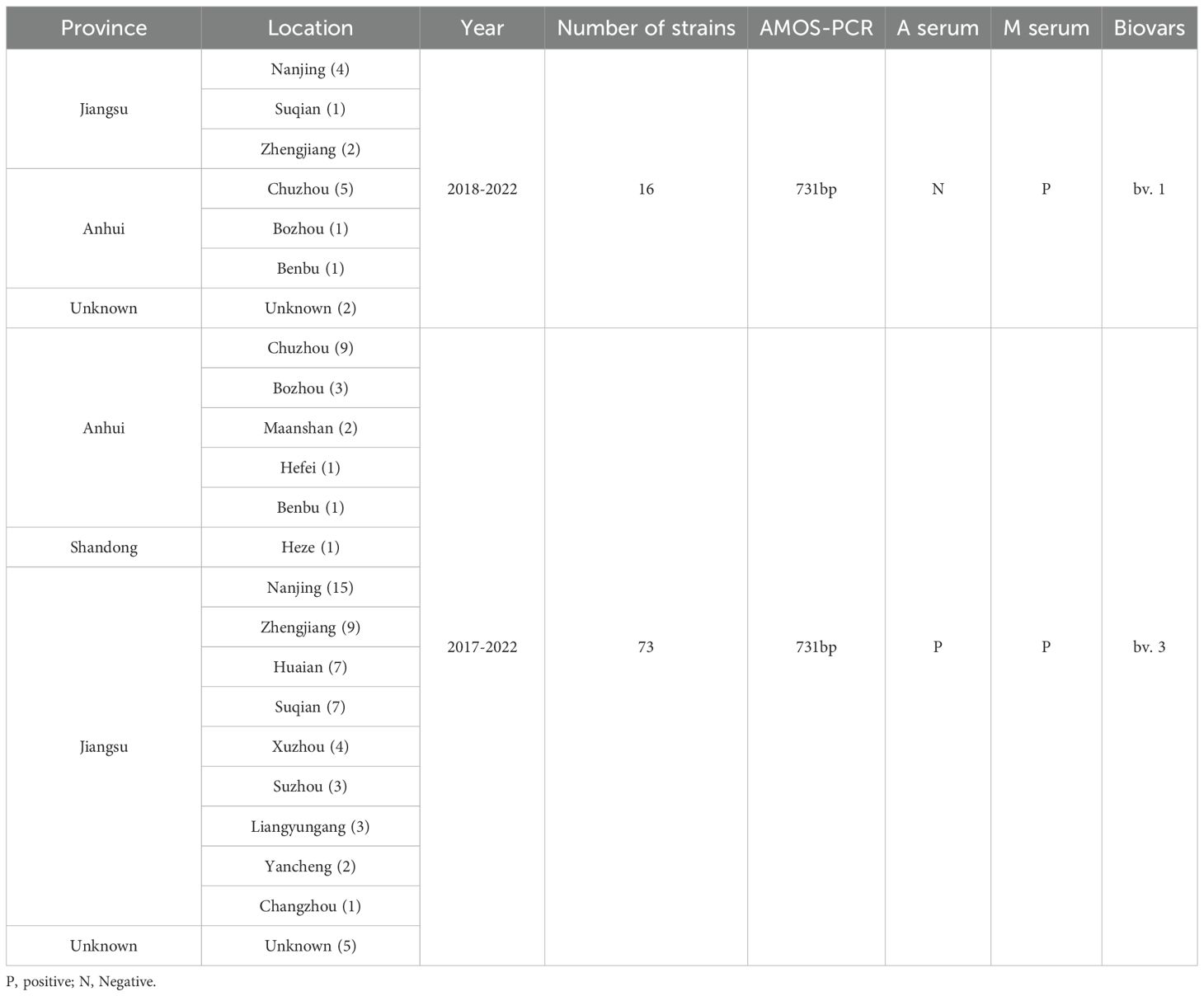
Table 1. The species/biovars, geographic distribution of 89 B. melitensis strains in this study.
Genome summary, virulence associated genes analysis and pangenome of B. melitensis
The analyzed strains demonstrated high genomic conservation with B. melitensis 16M, sharing an Average Nucleotide Identity (ANI) of 99.86–99.89%, confirming their close phylogenetic relationship. Genome assemblies showed moderate to high continuity, with scaffold counts ranging from 20 to 49, N50 values of 202,239–297,774 bp, and N70 values of 104,781–189,498 bp. All strains maintained a conserved genome size (~3.2 Mb; range: 3,238,490–3,389,613 bp) and encoded 1,412 protein-coding genes, consistent with the characteristic genomic architecture of Brucella spp. (Supplementary Table S1). Virulence-associated gene analysis revealed remarkable stability across the 89 strains, with 71–73 virulence genes retained per isolate. Notably, 14 genes exhibited partial absences, most frequently virB10 (missing in 84.3% of strains), followed by bspL (30.3%), wbkA (7.9%), and lpxE (6.7%) (Figure 2A). The remaining ten genes showed even lower absence frequencies (<5%). Functional annotation identified these variable genes as critical components of the VirB type IV secretion system (T4SS), T4SS-secreted effectors, and lipopolysaccharide (LPS) biosynthesis pathways—key determinants of bacterial virulence and host immune evasion. Antimicrobial resistance (AMR) screening detected only mprF (defensin resistance) across all strains, with no other AMR genes identified. Pangenome analysis delineated 2,884 core genes shared among all isolates, while strain-specific unique genes ranged from 1 to 141 (Figure 2B), reflecting both genomic stability and niche-specific adaptations.
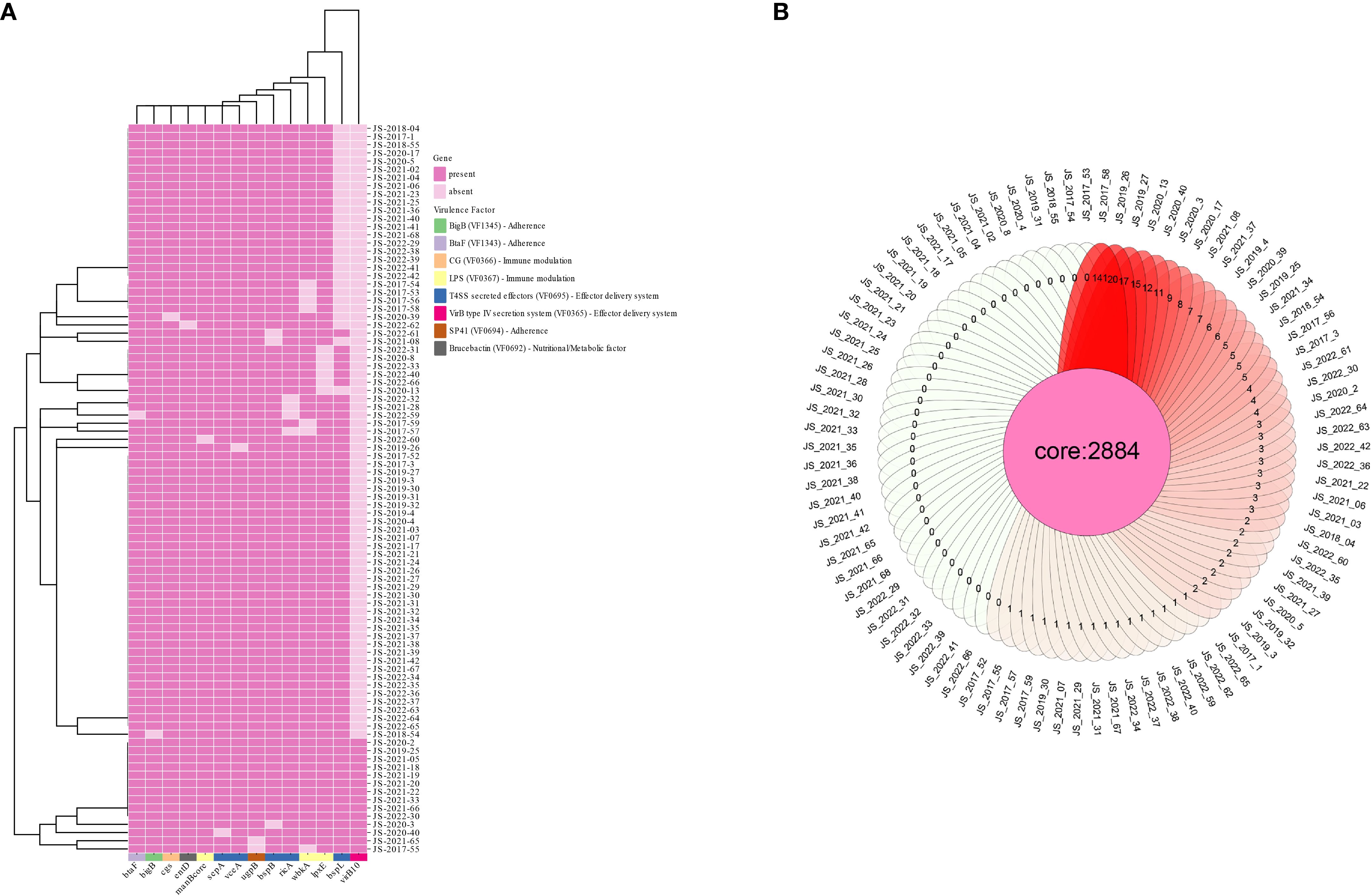
Figure 2. Distribution profile of virulence-associated genes (A) and core genes (B) among B. melitensis strains.
The epidemiology correlation analysis of 89 Brucella strains
Using both MLST-9 schemes, all strains were consistently classified as sequence type 8 (ST8). cgSNP analysis delineated the 89 B. melitensis into five phylogenetically distinct SNP clades (C-I to C-V) containing 17 SNP genotypes (STs), with C-I represented by ST1, C-II encompassing ST2-ST4, C-III comprising ST5-ST10 as the most diverse clade, C-IV containing ST11-ST12, and C-V including ST13-ST17. Notably, strains within each clade exhibited broad geographical distributions across multiple provinces, with the C-III clade demonstrating particularly extensive spread across Anhui, Shandong, and Jiangsu provinces. At finer spatial resolution, only ST2 showed restriction to a single region, while the remaining 16 STs contained strains from ≥2 regions collected across different time periods. The observed ladder-like phylogenetic topology and absence of geographical clustering strongly suggest recurrent cross-regional transmission events originating from multiple common infection sources, highlighting the complex dissemination patterns of B. melitensis in the study area (Figure 3).
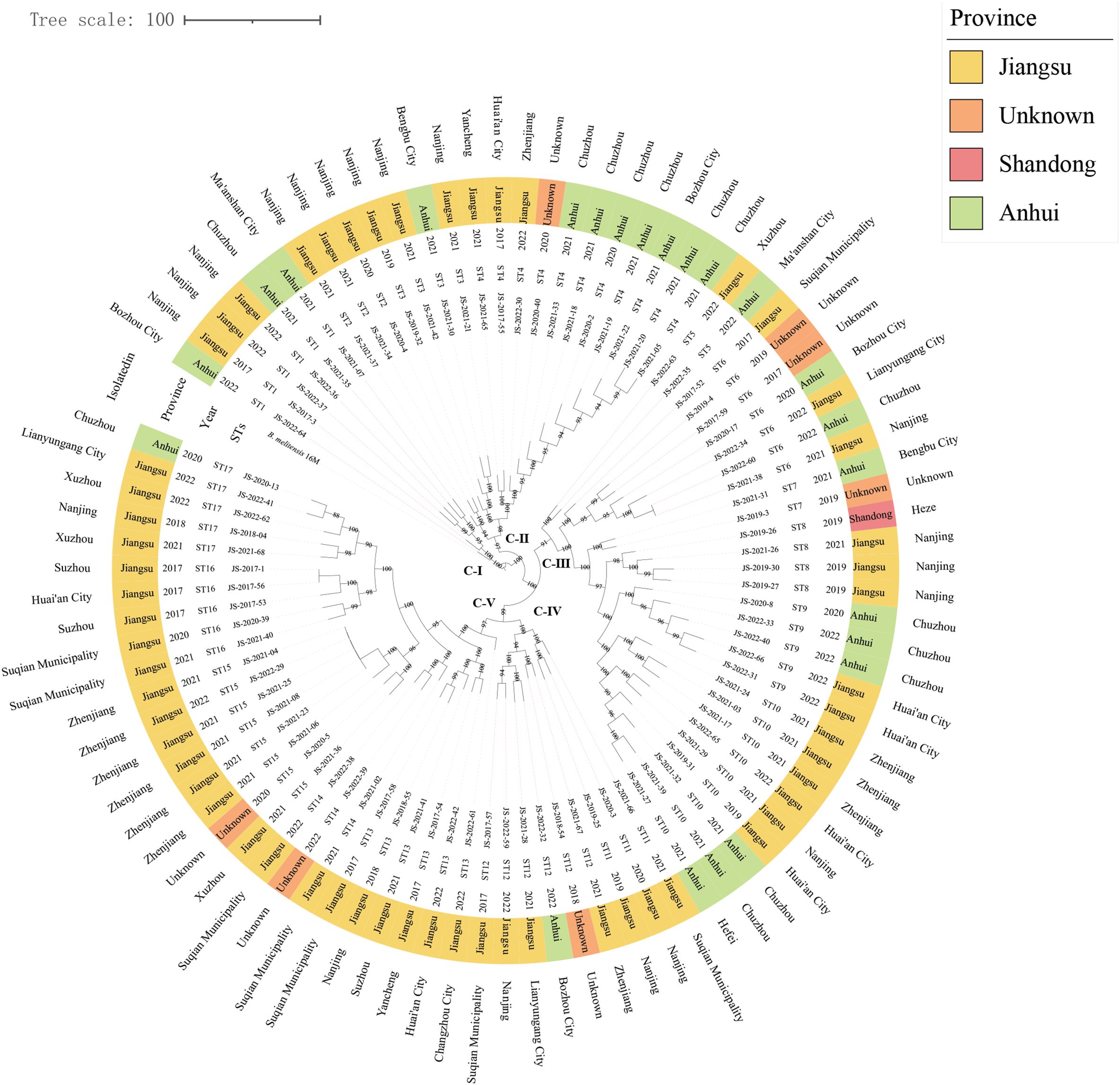
Figure 3. The maximum-likelihood tree generated by cgSNP matrix of 89 B. melitensis on county scale. The phylogeny trees of 89 B. melitensis strains were calculated using TreeBeST based on the Maximum-Likelihood Phylogenies (PHYML) algorithm with 1,000 bootstrap replicates. The maximum-likelihood tree depicts five major cgSNP-based clades (CI–CV), with bootstrap values (>85%) shown at branch nodes. The concentric circles (inner to outer) represent: Strain names, cgSNP genotypes (ST), Isolation year, Province (color-coded), and Geographic origin (prefecture/city).
Global cgMLST phylogenetic analysis of Brucella melitensis strains
Global cgMLST-based phylogenomic analysis demonstrated that all 89 strains consistently clustered within Genotype II of the East Mediterranean lineage, with the 17 STs segregating into two primary groups: ST1 and the remaining 16 STs. Strikingly, 15 of these STs exhibited genotype identities matching strains from multiple northern Chinese regions (Figure 4). Phylogenetic analysis revealed ST1’s close relatedness to Heilongjiang, Inner Mongolia, and Henan strains, while ST2-4 (C-II) formed a distinct clade with Heilongjiang, Liaoning, Hebei, and Inner Mongolia isolates; ST5-10 (C-III) showed tight clustering with Heilongjiang, Shandong, and Inner Mongolia strains; ST11-12 (C-IV) demonstrated high genetic concordance with Heilongjiang and Inner Mongolia isolates; and ST13-17 (C-V) exhibited clear phylogenetic linkages to strains from Heilongjiang, Xinjiang, Jilin, and Gansu (Figure 4). These results collectively indicate remarkable genetic homogeneity between the studied strains and northern Chinese (northeast/northwest) Brucella populations, strongly suggesting that the Nanjing-area human brucellosis epidemic likely originated through introduction of northern infection sources rather than representing an endemic focus.
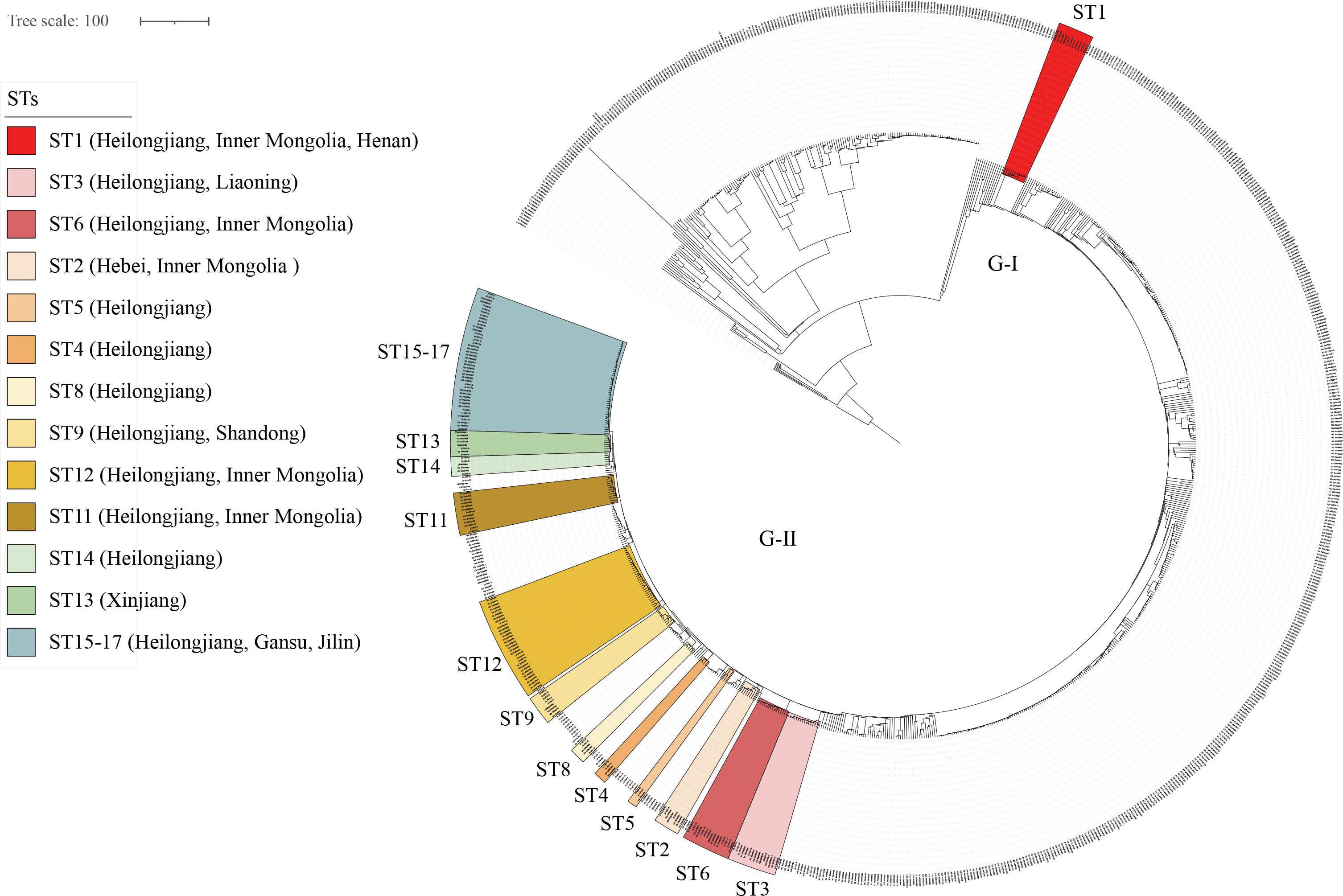
Figure 4. Global phylogenetic analysis of Brucella melitensis strains based on core genome multilocus sequence typing (cgMLST). Strains in each STs from study (highlighted) exhibit close phylogenetic relationships with isolates from the indicated regions (labeled by province in brackets), the tree topology reveals population structure and evolutionary divergence among global B. melitensis lineages, color-coded bars demonstrate complete cgSNP genotype matching between some or all strains of these sequence types (STs) and geographically distinct isolates from other provinces. The remaining unlabeled strains represent publicly available sequences sourced from GenBank.
Discussion
Human brucellosis is a re-emerging disease in Jiangsu province. Sporadic cases were reported after 2006, and the number of reported cases has significantly increased since 2011 (Zhang et al., 2023). However, the species/biovars, origin, and transmission patterns of circulating Brucella strains remain unclear. Bacteriology, bio-typing, and WGS-SNP analyses were applied to uncover the original and transmission patterns of circulating Brucella strains. Our investigation showed that B. meitensis strains were prevalent in Jiangsu province and neighboring regions, including Anhui and Shandong provinces, dominated by B. melitensis bv. 3. Previous studies have shown that human brucellosis has changed significantly over the past decades, and B. melitensis strains have become a dominant circulating species in China, and it has expanded from north to south regions (Zhu et al., 2020). An outbreak caused by B. melitensis at Jinchi Biotechnological Company engaged in collecting and disposing of kitchen waste from catering units in Lianyungang has been reported (Zhang et al., 2020). Brucellosis has become an important public health issue in Anhui Province, serum and milk samples obtained from goats in different regions of Anhui province were tested, the investigation frequency of brucellosis using RBPT, SAT, MRT, and PCR methods was 3.9% (n=7), 4.45% (n=8), 11.67% (n=7), and 86.67% (n=156), respectively (Rahman et al., 2019). These data show that ruminant brucellosis has been a serious public health concern in Jiangsu and Anhui provinces.
MLST analysis showed that all strains belonged to ST8, which is the dominant ST in the northern region (Sun et al., 2016). ST8 is the prevalent sequence type and the transmission of osteoarthritis-associated B. melitensis among different geographical areas in Inner Mongolia (Zhu et al., 2024). In Guangxi province, the population structure of Brucella strains had changed considerably; ST17 and ST21, two previously predominant populations, appeared to have been replaced by the recently emerging ST8 group (Liu et al., 2019). A previous report demonstrated that the cause of an outbreak was the plentiful influx of unchecked sheep from the northern part of China, and the employees in the process of sheep slaughtering or trading lacked effective prevention programs (Xiang et al., 2014).
WGS-SNP analysis revealed that 89 strains were divided into five SNP clades and 17 SNP genotypes (STs); each clade formed a ladder topology, with strains lacking apparent territory ownership, implying that cross-region cases occurred due to common sources of infection. Whole-genome SNP analysis showed that four B. melitensis strains were closely related to strains from China’s northern provinces, and the source of infection was partly human brucellosis in this province, which may have been from these regions (Li et al., 2020). In Shaanxi, multiple cross-county brucellosis outbreak events are driven by multiple indigenous circulating B. melitensis lineages (An et al., 2024).
The global phylogeny analysis showed that strains displayed a high genetic homogeneity with strains from multiple northern provinces, implying that multiple B. melitensis strain lineages were imported due to animal trade that co-drove the human brucellosis epidemic in Jiangsu. According to their territorial affiliation between the five clade strains, the absence of a clear differentiation suggests that strains continuously expanded and spread in Jiangsu province and neighboring regions. Similarly, 110 Chinese animal B. melitensis strains were grouped into five clusters, reflecting the existence of multiple lineages, and Chinese lineages were more closely related to strains collected from East Mediterranean and Middle East countries, such as Turkey, Kuwait, and Iraq (Sun et al., 2017). The active transfer and trade of animals (sheep and goats) between these regions is a reasonable explanation. A relevant survey showed that this province mainly breeds and raises its own cattle and sheep, the imported sheep come from Inner Mongolia, Shandong, Anhui and other provinces (Xu and Wang, 2018). The lack of geographic clustering among Jiangsu strains provides strong evidence for frequent cross-regional livestock movements, which significantly compromises local disease control efforts. The observed high genetic concordance with northern Chinese strains particularly highlights the need for an integrated One Health approach to effectively manage transmission risks at the human-animal interface. Key interventions should include: (1) implementing mandatory testing and quarantine protocols for livestock imported from endemic regions, (2) establishing real-time genomic surveillance systems to accurately trace outbreak origins, and (3) enhancing farmer education programs to mitigate high-risk behaviors such as direct contact with infected animals and consumption of unpasteurized dairy products (Liu et al., 2023). Finally, to effectively curb the southward spread of brucellosis, it is essential to strengthen inter-sectoral collaboration between agriculture and public health authorities while enhancing inspection and quarantine measures for introduced livestock.
Conclusion
At present, bacteriology and WGS-SNP have been applied to uncover the origin and transmission of Brucella strains, underscoring the fact that B. melitensis strains have become a severe public health issue in Jiangsu province and neighboring regions. Due to livestock trade and transfer, multiple B. melitensis lineages eventually trigger a local human brucellosis epidemic and spread. This investigation provides a vital cluse for devising and exerting a tailor control measure; however, further genomic epidemiological analysis on a regional and national scale will provide more detailed information to understand the transmission pattern of B. melitensis from north to south.
Data availability statement
The datasets generated during the current study are available in the National Microbiology Data Center repository (nmdc.cn), with accession number SUB1738305701652.
Ethics statement
The studies involving humans were approved by the ethics committee of the Jiangsu Provincial Center for Disease Control and Prevention. The study design was approved by the ethics committee of Jiangsu Provincial Center for Disease Control and Prevention. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
WW: Formal analysis, Investigation, Writing – original draft, Writing – review & editing, Resources, Software. ZuL: Investigation, Resources, Software, Writing – review & editing, Conceptualization, Methodology. TG: Formal analysis, Software, Writing – review & editing, Methodology, Resources. ZY: Data curation, Investigation, Project administration, Supervision, Writing – review & editing. LH: Conceptualization, Software, Writing – review & editing, Formal analysis, Supervision, Visualization. ZT: Investigation, Methodology, Project administration, Resources, Writing – review & editing. ZoL: Conceptualization, Formal analysis, Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing. SD: Conceptualization, Funding acquisition, Investigation, Methodology, Resources, Validation, Writing – review & editing. ZnL: Investigation, Methodology, Project administration, Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study was supported by the Medicine Key Science and Technology Development Project of Nanjing City (No. ZKX24060). The funders played no role in the study design, data collection and analysis, decision to publish, or manuscript preparation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2025.1603234/full#supplementary-material
Abbreviations
DNA, deoxyribonucleic acid; AMOS-PCR, abortus- melitensis-ovis-suis polymerase chain reaction; MLST, multi-locus sequence typing; WGS-SNP, whole-genome sequencing single-nucleotide polymorphism; cgMLST, core genome multilocus sequence typing.
References
Abdel-Glil, M. Y., Thomas, P., Brandt, C., Melzer, F., Subbaiyan, A., Chaudhuri, P., et al. (2022). Core genome multilocus sequence typing scheme for improved characterization and epidemiological surveillance of pathogenic brucella. J. Clin. Microbiol. 60, e0031122. doi: 10.1128/jcm.00311-22
Al Dahouk, S., Nöckler, K., Scholz, H. C., Pfeffer, M., Neubauer, H., and Tomaso, H. (2007). Evaluation of genus-specific and species-specific real-time PCR assays for the identification of Brucella spp. Clin. Chem. Lab. Med. 45, 1464–1470. doi: 10.1515/cclm.2007.305
Al Dahouk, S., Tomaso, H., Nöckler, K., Neubauer, H., and Frangoulidis, D. (2003). Laboratory-based diagnosis of brucellosis–a review of the literature. Part I: Techniques direct detection identification Brucella spp. Clin. Lab. 49, 487–505.
An, C., Nie, S., Luo, B., Zhou, D., Wang, W., Sun, Y., et al. (2024). Multiple Brucella melitensis lineages are driving the human brucellosis epidemic in Shaanxi Province, China: evidence from whole genome sequencing-based analysis. Front. Cell Infect. Microbiol. 14. doi: 10.3389/fcimb.2024.1452143
Bricker, B. J., Ewalt, D. R., MacMillan, A. P., Foster, G., and Brew, S. (2000). Molecular characterization of Brucella strains isolated from marine mammals. J. Clin. Microbiol. 38, 1258–1262. doi: 10.1128/jcm.38.3.1258-1262.2000
Di Bonaventura, G., Angeletti, S., Ianni, A., Petitti, T., and Gherardi, G. (2021). Microbiological laboratory diagnosis of human brucellosis: an overview. Pathogens 10(12):1623. doi: 10.3390/pathogens10121623
Eltigani, M. M., Al-Orainey, I. O., Saeed el, N. S., and Kambal, A. M. (1991). Biotyping of brucella isolates. Ann. Saudi Med. 11, 238–239. doi: 10.5144/0256-4947.1991.238
Gorvel, J. P. (2008). Brucella: a Mr "Hide" converted into Dr Jekyll. Microbes Infect. 10, 1010–1013. doi: 10.1016/j.micinf.2008.07.007
Hull, N. C. and Schumaker, B. A. (2018). Comparisons of brucellosis between human and veterinary medicine. Infect. Ecol. Epidemiol. 8, 1500846. doi: 10.1080/20008686.2018.1500846
Janowicz, A., De Massis, F., Ancora, M., Cammà, C., Patavino, C., Battisti, A., et al. (2018). Core genome multilocus sequence typing and single nucleotide polymorphism analysis in the epidemiology of brucella melitensis infections. J. Clin. Microbiol. 56(9):e00517-18. doi: 10.1128/jcm.00517-18
Khalafalla, A. I. (2023). Zoonotic diseases transmitted from the camels. Front. Vet. Sci. 10. doi: 10.3389/fvets.2023.1244833
Lai, S., Zhou, H., Xiong, W., Gilbert, M., Huang, Z., Yu, J., et al. (2017). Changing epidemiology of human brucellosis, China, 1955-2014. Emerg. Infect. Dis. 23, 184–194. doi: 10.3201/eid2302.151710
Laine, C. G., Johnson, V. E., Scott, H. M., and Arenas-Gamboa, A. M. (2023). Global estimate of human brucellosis incidence. Emerg. Infect. Dis. 29, 1789–1797. doi: 10.3201/eid2909.230052
Letunic, I. and Bork, P. (2021). Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W2w6. doi: 10.1093/nar/gkab301
Li, Z., Wang, X. M., Zhu, X., Wang, M., Cheng, H., Li, D., et al. (2020). Molecular characteristics of brucella isolates collected from humans in hainan province, China. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.00452
Liu, B., Liu, G., Ma, X., Wang, F., Zhang, R., Zhou, P., et al. (2023). Epidemiology, clinical manifestations, and laboratory findings of 1,590 human brucellosis cases in Ningxia, China. Front. Microbiol. 14. doi: 10.3389/fmicb.2023.1259479
Liu, Z., Wang, C., Wei, K., Zhao, Z., Wang, M., Li, D., et al. (2020). Investigation of genetic relatedness of brucella strains in countries along the silk road. Front. Vet. Sci. 7. doi: 10.3389/fvets.2020.539444
Liu, Z. G., Wang, M., Zhao, H. Y., Piao, D. R., Jiang, H., and Li, Z. J. (2019). Investigation of the molecular characteristics of Brucella isolates from Guangxi Province, China. BMC Microbiol. 19, 292. doi: 10.1186/s12866-019-1665-6
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., von Haeseler, A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. doi: 10.1093/molbev/msaa015
Orsini, M., Ianni, A., and Zinzula, L. (2022). Brucella ceti and Brucella pinnipedialis genome characterization unveils genetic features that highlight their zoonotic potential. Microbiologyopen. 11, e1329. doi: 10.1002/mbo3.1329
Rahman, S. U., Zhu, L., Cao, L., Zhang, Y., Chu, X., Feng, S., et al. (2019). Prevalence of Caprine brucellosis in Anhui province, China. Vet. World. 12, 558–564. doi: 10.14202/vetworld.2019.558-564
Singh, B. B., Khatkar, M. S., Aulakh, R. S., Gill, J. P. S., and Dhand, N. K. (2018). Estimation of the health and economic burden of human brucellosis in India. Prev. Vet. Med. 154, 148–155. doi: 10.1016/j.prevetmed.2018.03.023
Sun, M. J., Di, D. D., Li, Y., Zhang, Z. C., Yan, H., Tian, L. L., et al. (2016). Genotyping of Brucella melitensis and Brucella abortus strains currently circulating in Xinjiang, China. Infect. Genet. Evol. 44, 522–529. doi: 10.1016/j.meegid.2016.07.025
Sun, M., Jing, Z., Di, D., Yan, H., Zhang, Z., Xu, Q., et al. (2017). Multiple locus variable-number tandem-repeat and single-nucleotide polymorphism-based brucella typing reveals multiple lineages in brucella melitensis currently endemic in China. Front. Vet. Sci. 4. doi: 10.3389/fvets.2017.00215
Tan, Z., Huang, Y., Liu, G., Zhou, W., Xu, X., Zhang, Z., et al. (2015). A familial cluster of human brucellosis attributable to contact with imported infected goats in shuyang, Jiangsu province, China, 2013. Am. J. Trop. Med. Hyg. 93, 757–760. doi: 10.4269/ajtmh.15-0149
Tonkin-Hill, G., MacAlasdair, N., Ruis, C., Weimann, A., Horesh, G., Lees, J. A., et al. (2020). Producing polished prokaryotic pangenomes with the Panaroo pipeline. Genome Biol. 21, 180. doi: 10.1186/s13059-020-02090-4
Xiang, L., Zhou, W., Tang, F., Zhu, Y., Tan, Z., Liu, X., et al. (2014). An outbreak of brucellosis in a village in Jiangsu province. Zhonghua Liu Xing Bing Xue Za Zhi. 35, 1135–1137. doi: 10.3760/cma.j.issn.0254-6450.2014.10.013
Xu, Z. J. and Wang, X. Z. (2018). Preliminary investigation on brucellosis of cattle and sheep in Jiangsu province. China Anim. Quarantine. 35, 1–5. doi: 10.3969/j.issn.1005-944X.2018.03.001. XY X.
Xue, H., Zhao, Z., Wang, J., Ma, L., Li, J., Yang, X., et al. (2023). Native circulating Brucella melitensis lineages causing a brucellosis epidemic in Qinghai, China. Front. Microbiol. 14. doi: 10.3389/fmicb.2023.1233686
Yagupsky, P., Morata, P., and Colmenero, J. D. (2019). Laboratory diagnosis of human brucellosis. Clin. Microbiol. Rev. 33(1):e00073-19. doi: 10.1128/cmr.00073-19
Zhang, N., Fang, X. Y., Zhou, W. Z., Tan, Z. M., Liang, S. Y., Wang, X. C., et al. (2023). Epidemiological characteristics and temporal-spatial clustering analysis on human brucellosis in Jiangsu Province, 2006-2021. Sci. Rep. 13, 20024. doi: 10.1038/s41598-023-46690-z
Zhang, T., Liang, X., Zhu, X., Sun, H., and Zhang, S. (2020). An outbreak of Brucellosis via air-born transmission in a kitchen wastes disposing company in Lianyungang, China. Int. J. Infect. Dis. 96, 39–41. doi: 10.1016/j.ijid.2020.03.008
Zhou, Y., Meng, Y., Ren, Y., Liu, Z., and Li, Z. (2021). A retrospective survey of the abortion outbreak event caused by brucellosis at a blue fox breeding farm in Heilongjiang Province, China. Front. Vet. Sci. 8. doi: 10.3389/fvets.2021.666254
Zhu, L., Zhang, C., Liang, C., Peng, L., Yan, H., Liang, X., et al. (2024). Molecular epidemiological characteristics of osteoarthritis-associated Brucella melitensis in China: evidence from whole-genome sequencing-based analysis. Ann. Clin. Microbiol. Antimicrob. 23, 18. doi: 10.1186/s12941-024-00671-w
Keywords: human brucellosis, Brucella melitensis, WGS-SNP, phylogeny analysis, cgMLST
Citation: Wang W, Lu Z, Teng G, Yan Z, Huang L, Tan Z, Liu Z, Ding S and Li Z (2025) Phylogenetic evidence for nationwide expansion Brucella melitensis lineages drives the re-emerging and epidemic of human brucellosis in Jiangsu, China. Front. Cell. Infect. Microbiol. 15:1603234. doi: 10.3389/fcimb.2025.1603234
Received: 31 March 2025; Accepted: 01 August 2025;
Published: 02 September 2025.
Edited by:
Georgia Vrioni, National and Kapodistrian University of Athens, GreeceReviewed by:
Subbaiyan Anbazhagan, Indian Council of Medical Research (ICMR), IndiaDezhi Yang, Inner Mongolia International Hospital, China
Copyright © 2025 Wang, Lu, Teng, Yan, Huang, Tan, Liu, Ding and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiguo Liu, bGl1emhpZ3VvQGljZGMuY24=; Songning Ding, c29uZ25pbmcxMTI3QDE2My5jb20=; Zhenjun Li, bGl6aGVuanVuQGljZGMuY24=
†These authors have contributed equally to this work