 Xiaofeng Hu1†
Xiaofeng Hu1† Boqian Wang2†Mingliang Chen3,4,5†Kexin Li1,6†Zhixi Peng1,6Lianqun Jin1
Boqian Wang2†Mingliang Chen3,4,5†Kexin Li1,6†Zhixi Peng1,6Lianqun Jin1 Junjie Yue2
Junjie Yue2 Hui Chen1Ling Zhang7
Hui Chen1Ling Zhang7 Shaofu Qiu1*
Shaofu Qiu1* Hongguang Ren2*
Hongguang Ren2* Hongbin Song1*
Hongbin Song1*- 1Chinese People’s Liberation Army of China (PLA) Center for Disease Control and Prevention, Beijing, China
- 2Beijing Institute of Biotechnology, State Key Laboratory of Pathogen and Biosecurity, Beijing, China
- 3Institute of Pathology and Southwest Cancer Center, Southwest Hospital, Army Medical University, Chongqing, China
- 4Institute of Toxicology, School of Military Preventive Medicine, Army Medical University, Chongqing, China
- 5State Key Laboratory of Trauma, Burn and Combined Injury, Third Military Medical University, Chongqing, China
- 6Institute of Pathology and Southwest Cancer Center, China Medical University, Shenyang, China
- 7Department of Laboratory Medicine, 73 Army Hospital, Xiamen, China
Background: BlaNDM, which encodes a metallo-β-lactamase that can hydrolyze most β-lactam antibiotics, has become a serious public health concern in China. It is crucial to investigate the evolution, dissemination, and genetic dynamics of blaNDM to develop potential strategies to control the proliferation of blaNDM.
Methods: In this study, we collected 1021 blaNDM-positive isolates, which features 67 new genomes from our laboratory and 954 genomes from NCBI. Through epidemiological big data analysis, phylogenetic tree-based geographic transmission analysis, and upstream-downstream genetic clustering evolution analysis, we systematically analyzed the evolution, dissemination, and genetic dynamics of blaNDM-positive bacteria.
Results: Analysis results indicate that blaNDM-5 is gradually supplanting blaNDM-1 in China and Acinetobacter has been replaced as the primary blaNDM-harboring genus by the Enterobacter, Escherichia, and Klebsiella, which are both within the Enterobacteriaceae family and more easily transmitted among humans. Furthermore, blaNDM-positive bacteria exhibit a distinct livestock-environment-human transmission cycle, while the phylogenetic diversity of blaNDM and tet(X)-co-carrying genera is progressively expanding with concomitantly enhanced resistance phenotypes. Currently, the predominant blaNDM-positive bacterial strains have likely disseminated from southwest China to coastal regions. We further identified multiple transposon structures beyond Tn125 that may facilitate blaNDM transfer.
Conclusions: The diversity of the blaNDM and its carrier bacterial strains is continuously increasing, and its transmission range is also expanding. Of greater concern, super-resistant strains co-harboring blaNDM and tet(X) genes exhibit high potential for imminent emergence in human populations. Considering that the blaNDM-carrier bacteria are increasingly adapted to inter-human spread, the analysis results above can provide methodological and data support for epidemiological surveillance, tracing, and early warning alerts.
Introduction
Antimicrobial resistance can result from point mutations of non-antimicrobial resistance genes or horizontal transfer of antimicrobial resistance genes from other strains (Boolchandani et al., 2019). New Delhi metallo-β-lactamase gene is an antimicrobial resistance gene that encodes a metallo-β-lactamase capable of hydrolyzing most β-lactam antibiotics, which are often utilized to manage critical infections of multidrug-resistant Gram-negative bacteria. The spread of blaNDM-positive bacteria has received widespread attention due to the potential impact on human health (Khan et al., 2017; Usmanqamar et al., 2021).
BlaNDM was initially identified in 2008 from a Klebsiella pneumoniae strain isolated from a urine sample from a patient in Sweden who had recently returned to the country from India (Yong et al., 2009). Although the worldwide spread the blaNDM can be traced to India in most cases, where blaNDM was first identified in 2005, an blaNDM-positive strain was isolated from a patient in Turkey with no history of travel outside of the country (Acman et al., 2022). Thus, accurate identification of the geographic origin of blaNDM is challenging (Poirel et al., 2011; Ignasi et al., 2014). BlaNDM predominantly occurs in the phylum Proteobacteria and has been confirmed in at least 11 different bacterial families (Fanny et al., 2020). blaNDM is frequently encoded by plasmids but can also be carried on the chromosomes of some strains (Baraniak et al., 2016; Rahman et al., 2018). Generally, the genome of blaNDM-positive strains codes for only one blaNDM, although rare strains can harbor multiple blaNDM genes, either within the same subtype or across different subtypes (Hu et al., 2017). The National Center for Biotechnology Information (NCBI) database has documented 32 blaNDM subtypes, which are primarily the result of point mutations, with blaNDM-1 being the most prevalent subtype (Basu, 2020). From a broader perspective, blaNDM is characterized by a range of genomic contexts based on the Tn125 backbone, which represents the most commonly occurring transposon structure (Wailan et al., 2015). Recently, several other transposon structures have been discovered involving various insertion sequence (IS) and transposase (Tn) elements related to blaNDM, including ISAba125, IS5, IS6, IS26, and Tn3, among others (Campos et al., 2015; Zhao et al., 2021; Zheng et al., 2021). Of these, ISCR27 places blaNDM downstream of ISAba125 using a rolling circle transposition mechanism, thereby providing potential evolutionary clues (Ilyina, 2012; Partridge and Iredell, 2012; Poirel et al., 2012).
Although antibiotic resistance via blaNDM poses a significant potential threat to patients with bacterial infections, most research into the prevalence of blaNDM in China has focused on individual strains, isolated events, or plasmid structures (Yang et al., 2017; Yang et al., 2018; Li et al., 2020; Li et al., 2020; Wang et al., 2020; Hu et al., 2022). Due to the lack of large-2scale data, the evolution, dissemination, and genetic dynamics of blaNDM in China remain unknown. Furthermore, the overuse of antibiotics in China emphasizes the necessity for relevant research of the unique characteristics and patterns of the evolution of blaNDM (Chang et al., 2019; Chen et al., 2019; Walley et al., 2021).
In this paper, the sequences of 1021 blaNDM-positive isolates were compiled, which included 954 bacterial genomes from NCBI and 67 newly collected bacterial genomes by our laboratory, effectively expanding early datasets of blaNDM-positive bacteria in NCBI and nearly doubling the number of isolates collected between 2010 and 2014.
Based on the collection, we conducted a comprehensive epidemiological analysis from multiple perspectives. Our key findings reveal that: (i) BlaNDM-positive bacteria exhibit a distinct livestock-environment-human transmission cycle, with significant clustered dissemination observed in economically developed and densely populated regions; (ii) The escalating misuse of antibiotics has led to a progressive increase in resistant strains, particularly those co-harboring blaNDM and tet(X) genes, which demonstrate both expanding host ranges and enhanced resistance profiles; and (iii) Although bacteria co-harboring blaNDM and tet(X) has currently only been detected in livestock and environmental samples, the established transmission patterns above strongly suggest imminent risks of human infection and potential community spread. Finally, referring to a previous study (Acman et al., 2022), we designed a more comprehensive analysis to intuitively show the differences and similarity of the blaNDM contexts in various bacterial isolates and investigate the diversity of potential blaNDM-carrier transposon structures, which could assist the transfer of blaNDM genes.
Results
Dataset of blaNDM-positive bacteria in China
A genomic dataset of 1021 blaNDM-positive isolates collected in China was assembled. On the one hand, 954 bacterial genomes are retrieved from the NCBI GenBank database (Benson et al., 2018) and the Reference Sequence database (O’Leary et al., 2016). The collection process is shown in the Methods section and their information is summarized in Supplementary File 1: SF1_NCBI_954.xlsx. Although sampling bias may be inherent, public genomic databases can provide the most comprehensive sequences (Wu et al., 2019). On the other hand, 67 bacterial genomes were collected by our laboratory, including 49 blaNDM-positive bacteria screened among 1403 carbapenem-resistant strains from 10 cities and 25 hospitals and 18 blaNDM-positive bacteria screened among 156 strains isolated from 16 hospital sewage samples. Genomic sequencing and assembly techniques are also shown in the Method section and their basic information is provided in Supplementary File 2: SF2_newly_collected_67.xlsx. The sequences of the 67 blaNDM-positive isolates were uploaded to the National Genomic Data Center.
The dataset of the 1021 blaNDM-positive isolates includes the province, year of isolation, strain, genus, and blaNDM subtype (Figure 1). The geographic distribution (provinces) of the isolates was collected and these with > 20 isolates are shown in red font and all others in green font (Figure 1a). The first number in the brackets denotes the dataset scale from the NCBI database, while the second number indicates the dataset scale of our collected bacterial genomes. The dataset is mainly distributed in the southwest, coastal, and central provinces of China. Among them, Shandong, Guangdong, Zhejiang, and Sichuan totally account for ~75%. The distribution of the bacterial isolates is categorized according to the taxonomy level of genera (Figure 1b). Escherichia, Klebsiella, and Enterobacter species account for >75% (PCT: percentage) of the dataset. Notably, the main genera worldwide are separately Klebsiella, Escherichia, and Acinetobacter (Acman et al., 2022), which slightly differs from the scenario in China. The main blaNDM subtypes are separately blaNDM-1, blaNDM-5, and blaNDM-9 and the other nine subtypes only accounted for ~2% of the dataset (Figure 1c). Finally, we show the distribution of the collection year for the blaNDM-positive bacteria (Figure 1d). The number of the collected isolates gradually increased from 2010 to 2015, and then decreased and fluctuated after 2015, which is similar to the global tendency, where the global peak occurred in 2016 (Acman et al., 2022). At the same time, the accumulative number of genera that carry blaNDM genes continuously increased from 2010 to 2019.
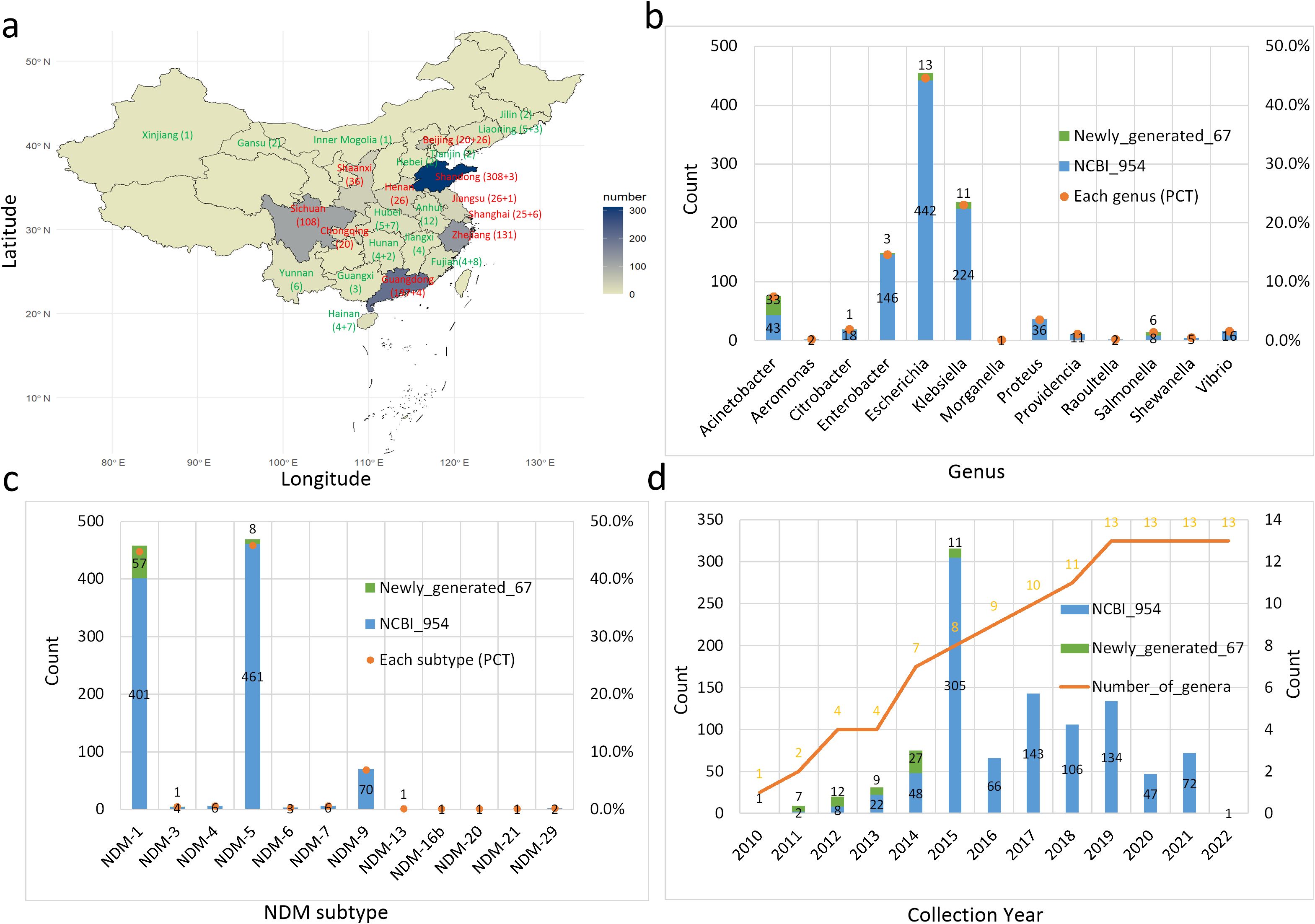
Figure 1. Composition of the China dataset of 1021 blaNDM-positive isolates. (a) Geographic distribution of blaNDM-positive assemblies. Provinces are colored by geographic region and the color reflects the number of isolates. The map was generated from coordinates using R software and the main provinces are highlighted. (b) Distribution of blaNDM-positive isolates at the genus level. (c) Distribution of blaNDM subtypes. (d) Distribution of isolate collection years and the accumulative number of genera carrying the blaNDM gene.
Distributions of blaNDM-positive bacteria in China
From the perspective of temporal distribution, we firstly focus on the main subtypes of blaNDM, which are separately blaNDM-1, blaNDM-5 and blaNDM-9. BlaNDM-1 was the only subtype identified in China from 2010 to 2012 (Figure 2a). However, the blaNDM-5 and blaNDM-9 subtypes surfaced after 2013, with blaNDM-5 gradually surpassing blaNDM-1 as the most predominant subtype. Then, we focus on the main blaNDM-positive bacteria genera, which are separately Acinetobacter, Enterobacter, Escherichia, Klebsiella, and Salmonella. BlaNDM initially occurred in Acinetobacter, which belongs to the Moraxellaceae family, before spreading to other genera, such as Enterobacter, Escherichia, and Klebsiella, which belong to the Enterobacteriaceae family (Figure 2b). After 2012, the ratios of Enterobacter, Escherichia, and Klebsiella gradually surpass the ratio of Acinetobacter among the blaNDM-positive genera. At the same time, the ratios of all Enterobacter, Escherichia, and Klebsiella isolates in NCBI (including both blaNDM-positive and blaNDM-negative isolates) are not increased compared to all Acinetobacter isolates (Supplementary Figure 1).
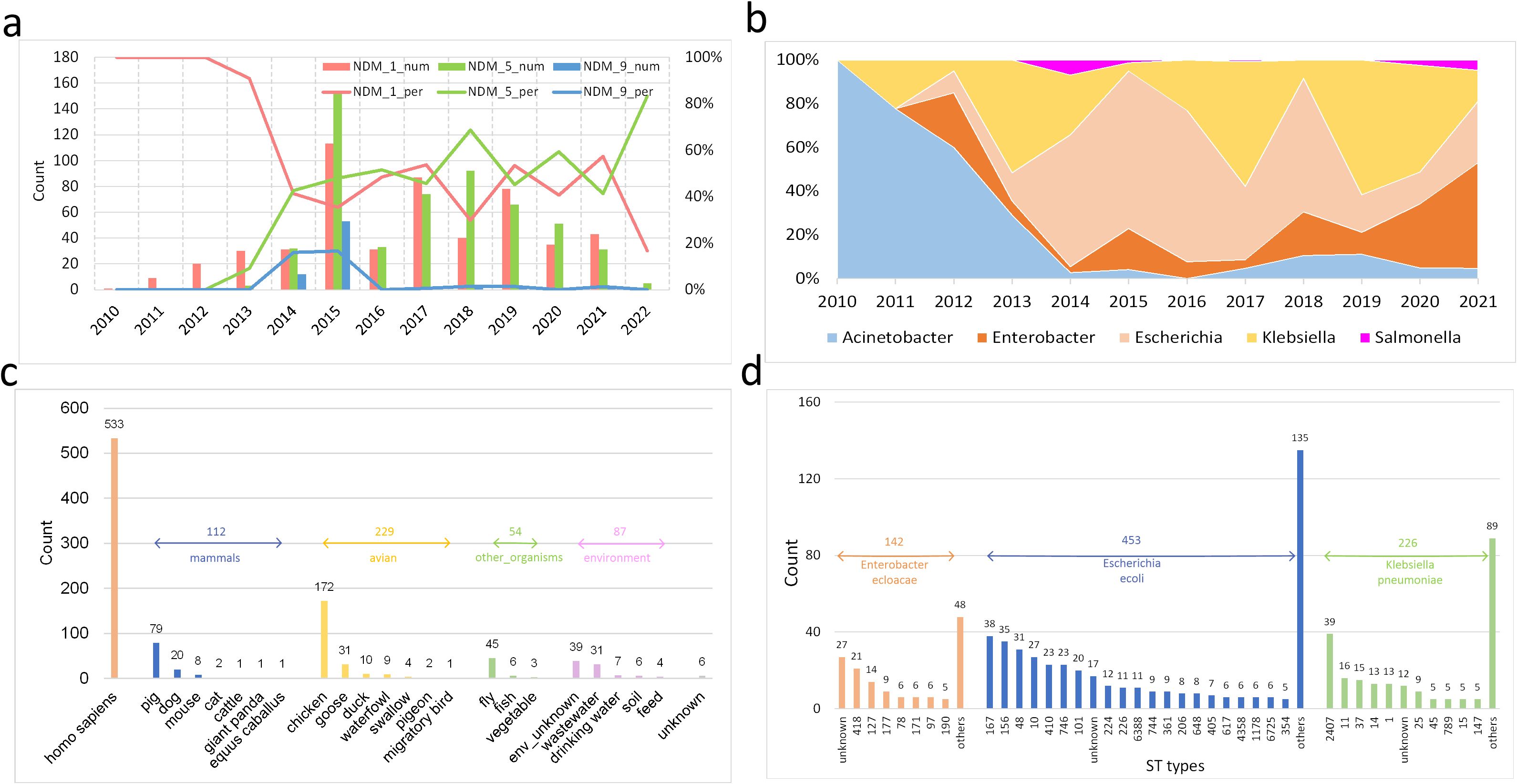
Figure 2. Distributions of the 1021 blaNDM-positive isolates in the China dataset (a) Temporal distribution of the three main blaNDM subtypes. For each year, the number and percentage of each blaNDM subtype are included. (b) Temporal distribution of the percentages of the five main bacterial genera carrying blaNDM-positive assemblies. (c) Collection sources distribution, which are categorized into five main groups with unknown source excluded. (d) ST types distribution of three main bacterial species by MLST.
From the perspective of geographic distribution, as shown in Supplementary Figure 2, blaNDM-1 was identified as the dominant subtype in six provinces, i.e., Beijing, Shanghai, Zhejiang, Henan, Shaanxi, and Chongqing provinces, whereas blaNDM-5 was the subtype dominant in the other four provinces, i.e., Shandong, Jiangsu, Guangdong, and Sichuan provinces. The predominant genera carrying blaNDM genes included Escherichia in Shandong, Jiangsu, and Guangdong provinces and Klebsiella in Beijing, Sichuan, and Shanghai provinces. Enterobacter emerged as the top genus in the central provinces of Shaanxi and Chongqing. Meanwhile, Enterobacter and Escherichia were the main carriers in Zhejiang province.
The temporal and geographic distributions of the other 9 blaNDM subtypes and 10 genera are recorded in Supplementary File 3: SF3_NDM_subtype_genus_year_province_distribution.xlsx.
Moreover, sample origins are crucial for epidemiological analysis of blaNDM-positive bacteria. We categorized the samples into five groups: Homo sapiens, other mammals, avian species, other organisms, and environmental sources (Figure 2c). The majority of specimens were derived from Homo sapiens (i.e., patients; 52.2%), with the remaining predominantly originating from avian species (22.4%), other mammals (11.0%), and environmental reservoirs (8.5%). This distribution strongly suggests significant transmission of blaNDM-positive bacteria along the livestock-environment-human pathway. Notably, flies (4.4%) appear to serve as important vectors in this transmission network.
Finally, clonal analysis of the predominant blaNDM-positive bacterial species is crucial for understanding the epidemiological patterns. We conducted multilocus sequence typing (MLST) analysis of the three major species: E. cloacae (n=142), E. coli (n=453), and K. pneumoniae (n=226) (Figure 2d). Among the characterized sequence types, both E. cloacae and E. coli subtypes exhibited gradually decreasing prevalence without dominant clones, whereas K. pneumoniae showed a predominant ST2407 clone. Although this study primarily focused on transmission tracing at the species level, more detailed investigations of clonal-specific evolutionary dynamics and geospatial transmission patterns within each pathogen will be essential for future epidemiological analysis and disease prevention strategies.
Geographic dissemination
Systematic phylogenetic analysis was conducted to investigate the dissemination of the main blaNDM-positive bacterial species throughout China. Four species with more than 15 isolates (i.e., Proteus mirabilis, K. pneumoniae, Escherichia coli, and Enterobacter hormaechei) were selected. The analysis process is described in the Methods section and the relevant configuration files are provided in availability of code (fasta_beast_setting).
To illustrate the dissemination process more clearly, different colors are utilized to separately mark the propagation paths (Figure 3). P. mirabilis was firstly discovered in Shandong and then spread to Beijing, Zhejiang, Sichuan, and Guangdong (Figure 3a). Meanwhile, both K. pneumoniae and E. coli were firstly detected in Sichuan and then spread to eastern China (Figures 3b, c). Similarly, E. hormaechei was also firstly discovered in Sichuan and then disseminated to southeast China and Xinjiang province (Figure 3d).
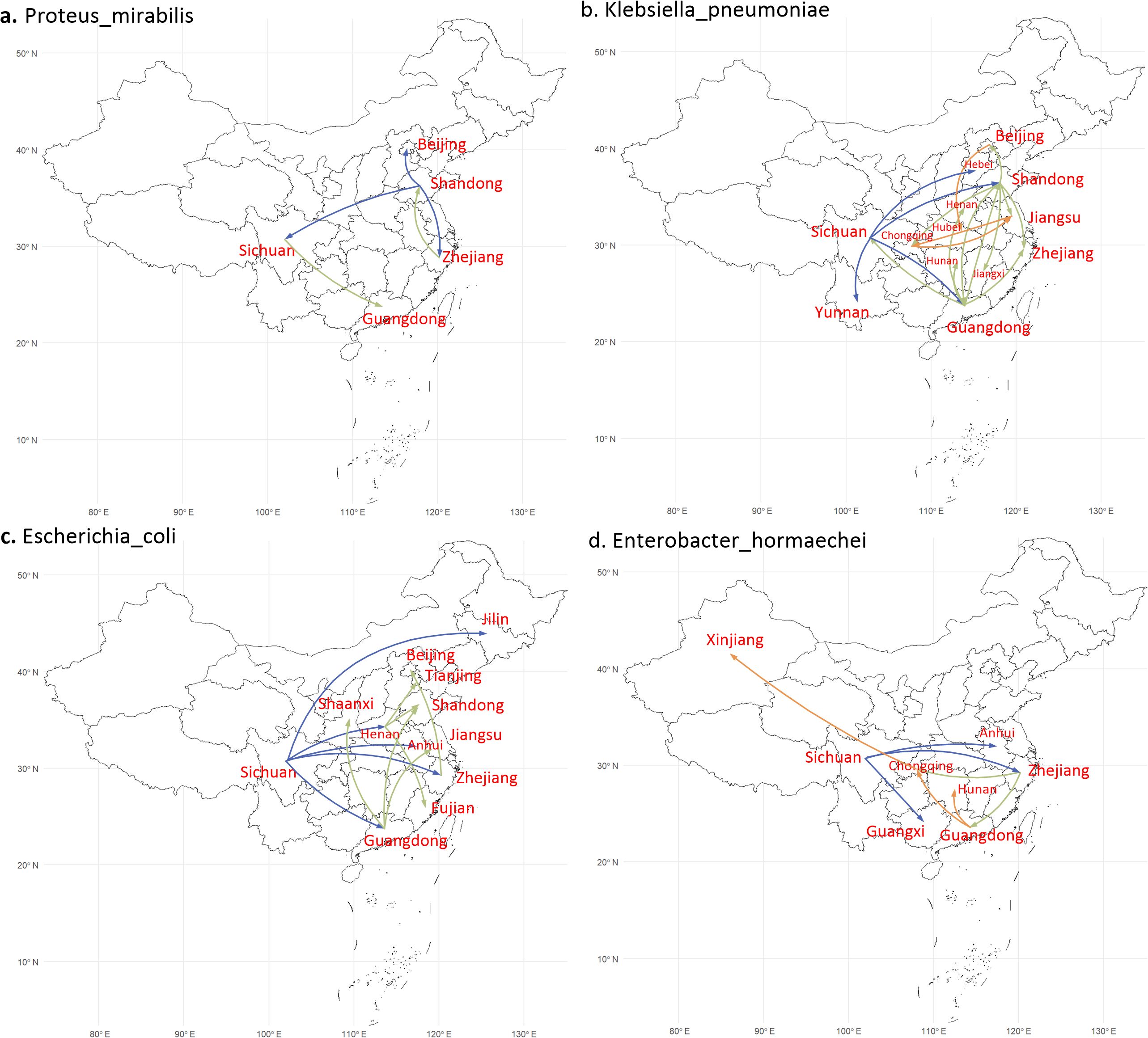
Figure 3. Geographic transfer of four bacterial species. The steps are indicated by different-colored arrows (blue, green and orange). (a) Geographic transfer of P. mirabilis (13 strains), which was first distributed from Shandong to Beijing, Zhejiang and Sichuan, and then diverted to Guangdong (from Sichuan) and back to Shandong (from Zhejiang). (b) Geographic transfer of K. pneumoniae (37 strains), which distributed from Sichuan to Shandong and Guangdong. (c) Geographic transfer of E coli (38 strains), which was distributed from Sichuan to Henan, Guangdong, and Zhejiang. (d) Geographic transfer of E hormaechei (24 strains), which was distributed from Sichuan to Zhejiang, Guangdong, and Chongqing.
blaNDM and tet(X) genes
The emergence of the plasmid-mediated high-level tigecycline resistance gene tet(X) has compromised the role of tigecycline as a “last-resort” antibiotic for treating carbapenem-resistant Gram-negative bacterial infections. Compared to the prototype tet(X), the enzyme activity of tet(X3) and tet(X4) variants is significantly enhanced. It is observed that tet(X3)/(X4) genes are often associated with blaNDM-1 in various Enterobacteriaceae spp. and Acinetobacter and confer just a slight fitness cost on the isolates (He et al., 2020). Other groups made similar observations with these pathogens with diverse tet(X) genes mainly but not exclusively in the veterinary sphere (Hirabayashi et al., 2021; Chen et al., 2024; Yao et al., 2025). For in-depth investigation, we identified 15 resistant isolates co-harboring both blaNDM and tet(X) genes from our collection of 1,021 strains (Figure 4). The tet(X) variants present in these samples were primarily three subtypes: tet(X3) and tet(X4) (MIC = 8 mg/L), along with tet(X5) (MIC = 4 mg/L) (Cui et al., 2021).
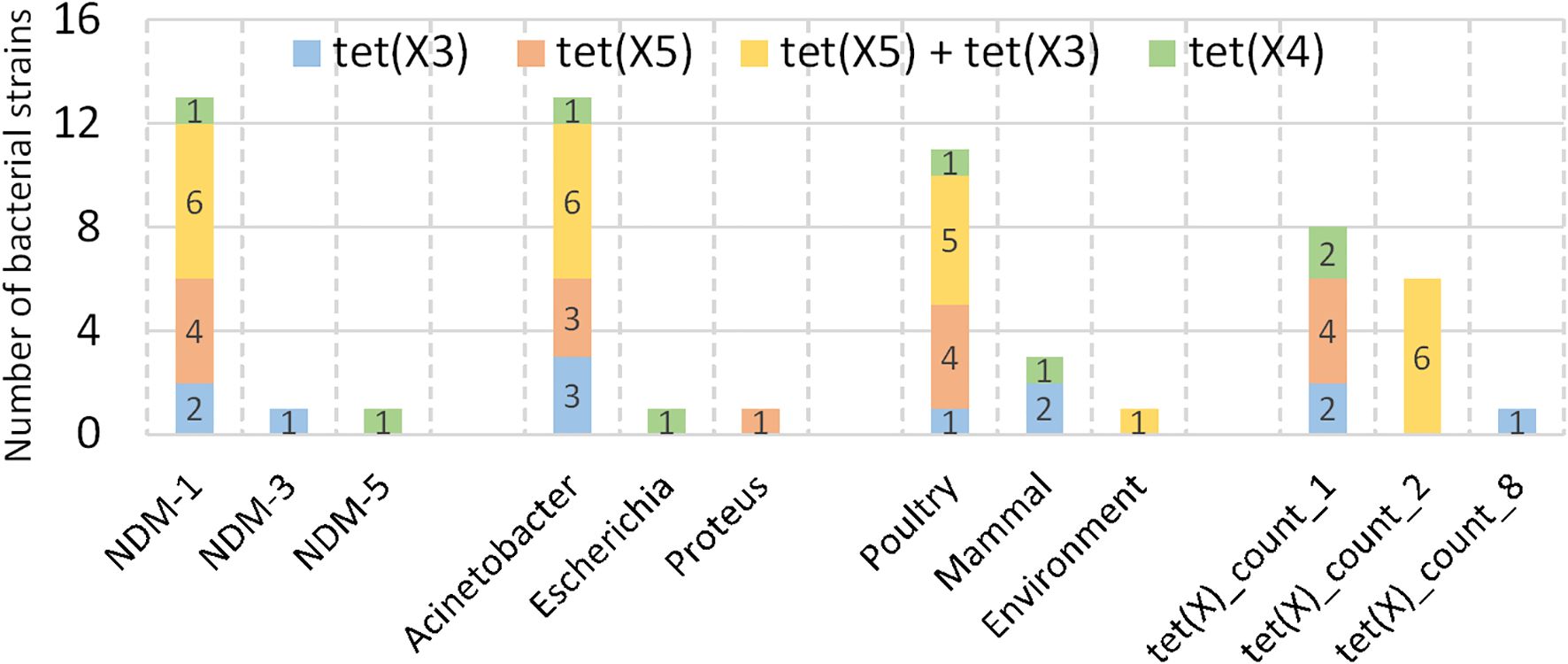
Figure 4. Co-occurrence of blaNDM and tet(X) genes. 15 strains co-harboring both blaNDM and tet(X) genes are identified. For each subtype of tet(X), various blaNDM subtypes, genera, sample sources and the count of tet(X) emergency in a strain were collected.
Notably, the strain GCA_015217905.1 harbored 8 tet(X3) genes and an unknown tet(X) variant, with all genes located in close genomic proximity. Additionally, six isolates carried two tet(X) variants, exclusively exhibiting the combination of tet(X3) and tet(X5). Regarding blaNDM variants, tet(X) genes predominantly co-occurred with blaNDM-1. There is only one isolate demonstrated coexistence of blaNDM-3 and tet(X3), and another blaNDM-5 and tet(X4). At the genus level, Acinetobacter represented the majority of isolates, while Escherichia and Proteus each accounted for only one isolate. Of particular note, the blaNDM-5 and tet(X4) co-occurrence was identified in the Escherichia isolate. These observations suggest potential genus-specific preferences for particular blaNDM and tet(X) variant combinations. However, further validation of these characteristics will require additional sample collection and comprehensive analytical verification in subsequent studies. Regarding host origins, poultry constituted the primary reservoir (n=11), with minimal environmental contamination detected (n=1).
Diversity of blaNDM context genes
The blaNDM context genes were extracted from the gbff files and automatically converted according to the Tn125 structure. More information of the blaNDM context genes is provided in Supplementary File 4: SF4_954_67_NDM_downstream_7.xlsx.
We utilize a clustering method similar to a previous study (Acman et al., 2022) to classify the blaNDM context genes based on their structural similarities (Figure 5). It firstly measures the structural similarity between two blaNDM downstream structures based on genes and positions (Figure 5a). Then, it generates a fully connected network where the node represents the bacterial isolate and the link represents similarity between two isolates (Figure 5b). By gradually eliminating links with increasing limitation, a clustering tree is established (Figure 5c). Specific process (Figure 5d) is described in the Methods section.
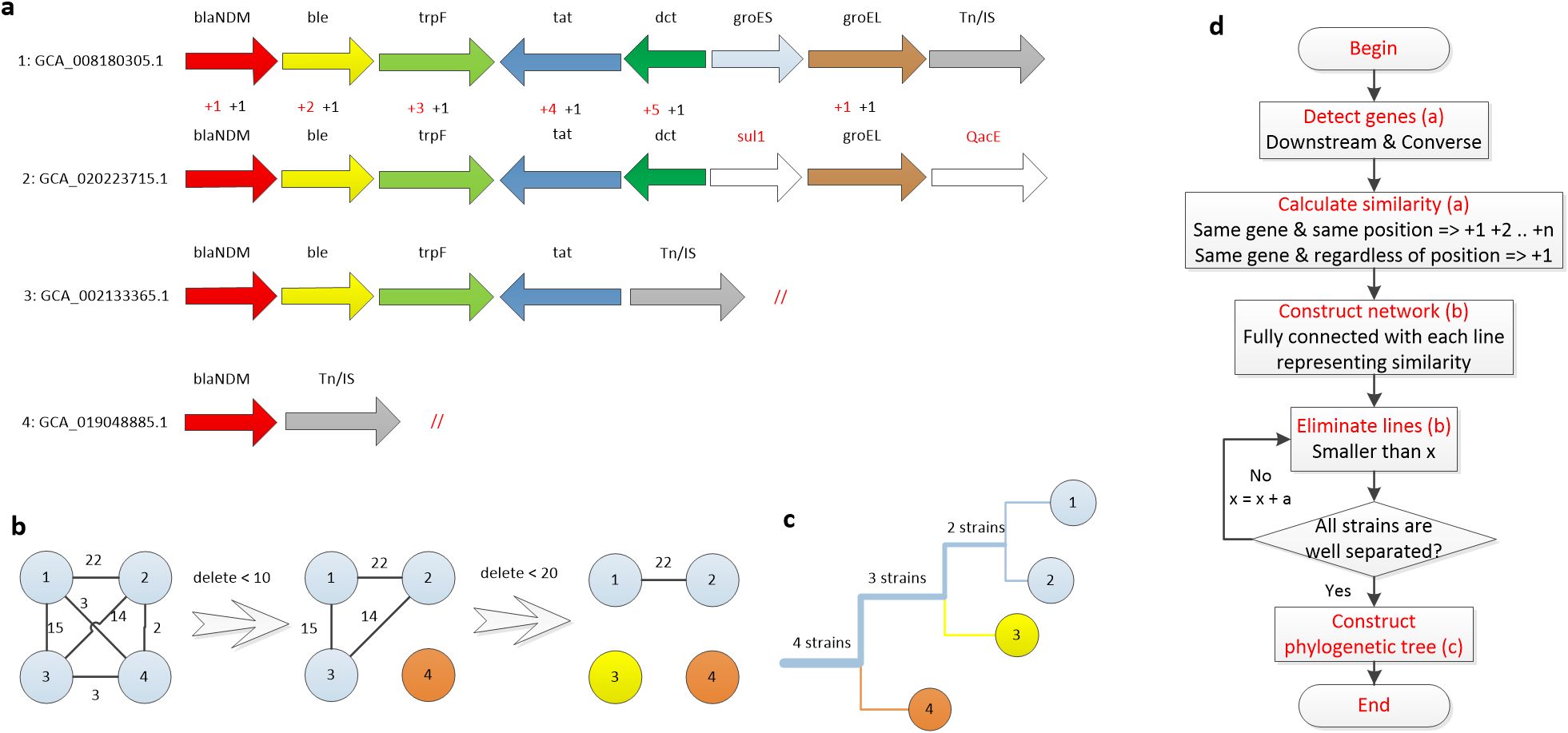
Figure 5. Similarity-based clustering of genes downstream of blaNDM (a) Genes downstream of blaNDM in four strains. (b) Fully connected network with each line representing the similarity of the connected two strains. Lines were gradually eliminated by a certain limitation until all strains were separated. (c) Clustering tree established according to results of step b where the thickness of each branch represents the number of strains. (d) The strain clustering method.
The clustering results revealed that most of the blaNDM context genes of the 1021 isolates were similar to the structure of Tn125 (Figure 6a). Among the 1021 isolates, 1010 (99%) carried a combination of the blaNDM, ble, and trpF genes; 88% (901) had a more extended group of genes, which included blaNDM, ble, trpF, and tat; 695 (68%) contained a gene structure consisting of blaNDM, ble, trpF, tat, and dct genes; 249 (24%) had a complete gene structure equivalent to Tn125, which excludes Tn/IS (Transposase/Insertion Sequence); 82 (8%) carried duplicated tat genes, while 95% of these also carried the functionally related genes groES and groEL. Other genes frequently found downstream of blaNDM included Resolvase, catB, dfrB, qacE, sul1, ANT, DHPS, ribosomal, NAD+, umuD, polV, RHS, and AraC, which were identified in >2% of the isolates.

Figure 6. Details of blaNDM context genes. (a) Clustering results of genes downstream of blaNDM. A tree was constructed in accordance with the method described in (Figure 5). The same gene in Tn125 context is marked with the same color. (b) A heat map of the relationships among different genes and the downstream positions from blaNDM.
A heatmap was constructed to visualize the diversity of genes located at different positions downstream of blaNDM (Figure 6b). Position P_4 was the primary point of gene diversity. Notably, from P_4 to P_8, the likelihood of gene diversity increased. The frequency of Tn/IS also increased from P_4 to P_8, with peak value occurring at P_6 (the position of groES in Tn125) and P_8 (the position of Tn/IS in Tn125).
Potential blaNDM-carrier transposons
To find the potential blaNDM-carrier transposons, further in-depth analysis of the characteristics of the Tn/IS elements upstream and downstream of blaNDM was conducted. Two upstream and nine downstream genes of blaNDM were extracted from the gbff files (Supplementary File 5). Especially, the positions of the Tn/IS elements were categorized based on the family and similarity in Supplementary File 6: SF6_IS_like.xlsx.
Totally, seven primary Tn/IS elements were identified, which included ISAba125, IS5, IS91, IS6, Tn3, ISL3, and IS1 and all infrequent Tn/IS elements were categorized into a single group called “others” (Supplementary Figure 3). Besides, the distribution of the seven main Tn/IS among different provinces, genera and collection years are also analyzed. Additional information is provided in Supplementary File 7: SF7_954_67_IS_distance_genus_province_year.xlsx.
A heat map was generated to clearly identify the possible positions of the Tn/IS elements upstream and downstream of blaNDM (Figure 7a). ISAba125 and IS5 were primarily situated upstream of blaNDM (up_1 and up_2), while IS6 and Tn3 occurred less frequently (IS6: up_1 and up_2; Tn3: up_2). Other Tn/IS genes rarely occurred upstream of blaNDM. In contrast, the downstream region of blaNDM was more complex. Specifically, IS91 was the most common gene and primarily located at four positions (down_3, down_4, down_5, and especially down_7). Meanwhile, IS6 frequently appeared downstream at positions down_5, down_6, and down_9. ISAba125 and ISL3 were less prevalent than IS91 and IS6 but more frequent than IS1 and IS5 downstream of blaNDM. These results demonstrate that the Tn/IS elements were asymmetrically distributed both upstream and downstream of blaNDM.
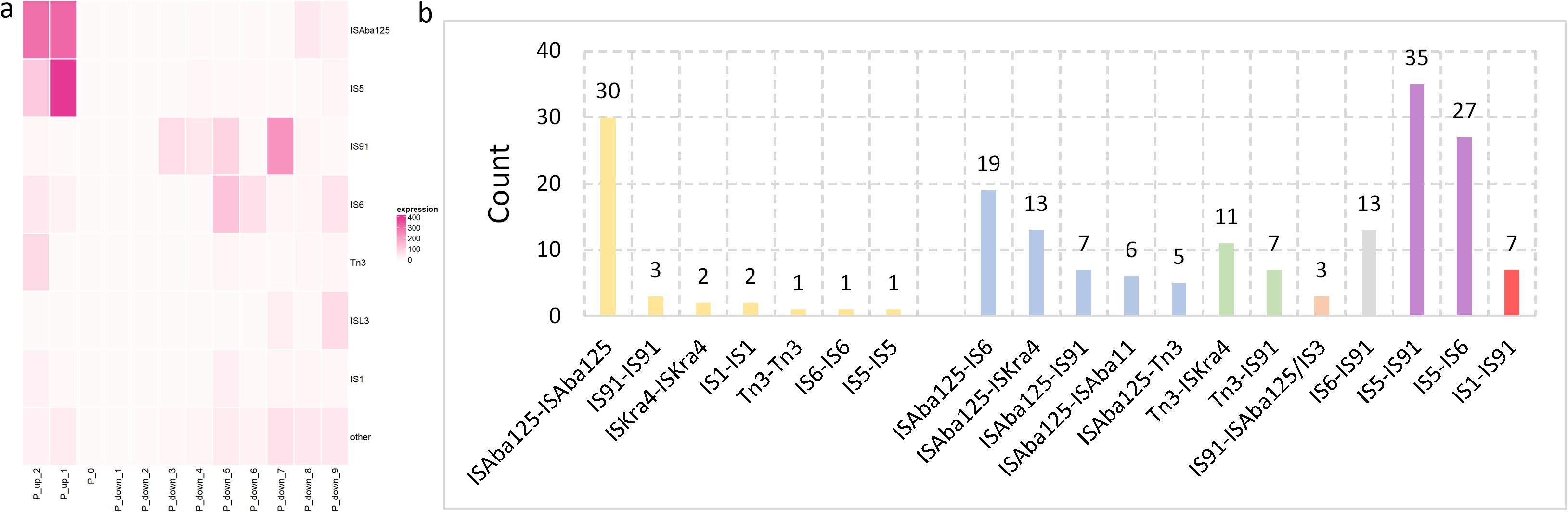
Figure 7. Positions and relationships of the Tn and IS elements downstream and upstream from blaNDM. (a) A heat map of the relationship among the 7 main Tn/IS types as well as other types, and corresponding positions from blaNDM. P_up_2 represents the second upstream gene from blaNDM, P_0 represents the position of blaNDM, and P_down_1 represents the first downstream gene from blaNDM. (b) Tn/IS pairs containing the blaNDM, ble, trpF, tat, dct groES and groEL genes were separated into two main groups: one including the same Tn or IS in the Tn/IS pair (yellow bars) and a second including different Tn or IS in the Tn/IS pair (blue, green, orange, grey, and purple bars with each color representing one upstream Tn/IS type).
Further comprehensive investigation into potential blaNDM-carrier transposons was conducted based on the distribution of the Tn/IS elements. The complete structure of Tn125 comprises seven genes between two ISAba125 elements: blaNDM, ble, trpF, tat, dct, groES, and groEL. There were 7 symmetrical combinations of Tn/IS pairs (yellow bars), which included ISAba125-ISAba125, IS91-IS91, ISKra4-ISKra4, IS1-IS1, Tn3-Tn3, IS6-IS6, and IS5-IS5 (Figure 7b). Notably, the ISAba125-ISAba125 combination (Tn125) was the most common and Tn3-Tn3 (IS3000-IS3000) and IS6-IS6 (IS26-IS26) were previously reported as novel blaNDM-harboring transposons (Campos et al., 2015; Zheng et al., 2021). There were also many other asymmetrical Tn/IS pairs with relatively high frequencies, especially the IS5-IS91 pair, which was observed 35 times, even higher than the frequency of ISAba125-ISAba125. More information is provided in Supplementary File 8: SF8_954_67_transposon_complete.xlsx.
Discussion
BlaNDM-positive bacteria are predominantly found in Asia, particularly mainland China (Acman et al., 2022). In this study, the basic characteristics, evolution, geographic dissemination, and genetic dynamics of blaNDM-positive bacteria in China were analyzed to help develop potential strategies to control the proliferation of blaNDM and prevent the development of drug-resistant bacteria. Although the analysis results could bias due to selection for sequencing, we strive to collect a sufficiently diverse range of data to ensure the validity of the results. Totally, 954 bacterial strains were retrieved from the NCBI database and 67 new blaNDM-positive bacteria genomes were identified by our laboratory, increasing the dataset by 7%. Especially, the 67 genomes effectively enhanced the NCBI dataset of early years (from 2010 to 2014) and early carrier of the blaNDM gene (Acinetobacter), offering insights into the complete evolution and dissemination process of blaNDM-positive bacteria in China.
From the perspective of geographic analysis, the majority of blaNDM-positive isolates were concentrated in economically developed and densely populated coastal, middle, and southwest provinces of China, including Guangdong, Zhejiang, Shanghai, Jiangsu, Shandong, Beijing, Sichuan, Chongqing, Shaanxi, and Henan (Figure 1a). The geographic dissemination analysis shows that the blaNDM-positive bacterial strains might be frequently transmitted from Sichuan to the coastal provinces (Figure 3). Although potential bias cannot be ruled out, the dissemination results reflect the trend of blaNDM spread to some extent. Compared with Beijing, Shanghai, and Guangdong, Sichuan is also a province with high population density. At the same time, it is near to South Asia, where the first blaNDM-positive bacterial isolate was discovered, which makes the dissemination results more reliable (Yong et al., 2009). The results of the geographic analysis indicate that blaNDM-positive bacteria are mainly distributed in economically developed and densely populated provinces in China, which will severely constrain people’s health and urban economic development. Therefore, we need to pay special attention to the prevention and control measures in these cities, as well as focus on preventing and controlling the main transmission route from west to east in China.
From the perspective of temporal analysis, there was a dramatic increase in blaNDM-positive bacteria from 2010 (first emergence in China) and the peak in 2015, which was followed by a steep decrease (Figure 1d). The consumption data of antibacterial drugs from 2012 to 2020 were obtained from the pharmaceutical database of China Pharmaceutical Industry Information Center and summarized in Supplementary Figure 4. The usage of carbapenems increased sharply from 2012 to 2015/2016, eventually stabilizing at a high level, suggesting a link between the increased prevalence of blaNDM-positive bacteria and continued overuse of antibiotics. Additionally, from 2010 to 2019, there was an increase in the cumulative number of genera carrying blaNDM genes, demonstrating that the blaNDM gene has continued dissemination among an increasing number of bacterial genera (Figure 1d). Among these genera, Escherichia, Klebsiella, and Enterobacter, members of the Enterobacteriaceae family, were identified as the most prevalent blaNDM-positive genera in China (Figure 1b), highlighting the importance of Enterobacteriaceae for the blaNDM transmission among humans (Perez and Bonomo, 2019). The temporal analysis shows that although the blaNDM resistance gene was initially detected in an Acinetobacter isolate, a shift has been observed from Acinetobacter to genera Escherichia, Klebsiella, and Enterobacter within the family Enterobacteriaceae (Figure 2b). Finally, the dominant blaNDM subtypes in China are separately blaNDM-1, blaNDM-5 and blaNDM-9 (Figure 1c). Nowadays, the prevalence of blaNDM-1 has decreased over time, gradually being replaced and possibly being surpassed by blaNDM-5 (Figure 2a). The results of the temporal analysis indicate that antibiotic abuse is one of the main causes of the growth of antibiotic-positive bacteria in China. Therefore, we need to avoid the overuse of antibiotics and focus on preventing the spread of blaNDM-positive genes especially caused by Enterobacteriaceae bacteria, as well as the two dominant subtypes genes blaNDM-1 and blaNDM-5.
The current study has limitations in spatiotemporal sampling heterogeneity, with significantly more samples collected in 2015 and from eastern coastal provinces compared to other time periods and regions (Figures 1a, b). Such sampling bias may potentially impact key conclusions in several aspects: (i) Distortion of transmission pathway inference - over-representation of developed areas could lead to overestimation of hospital/community-based transmission while underestimating cross-species transmission in agricultural regions; (ii) Miscalculation of evolutionary rates - disproportionate sampling during peak years (e.g., 2015) may introduce deviations in estimating genetic mutation accumulation rates; and (iii) Compromised strain representativeness - insufficient sampling in less-developed areas might miss locally prevalent clones and underestimate their importance in transmission networks. Nevertheless, several significant conclusions can still be drawn from the current dataset analysis. For example, epidemiological investigation of blaNDM-positive bacterial hosts (Figure 2c) revealed that the livestock-environment-human transmission chain likely serves as the intrinsic driver of the observed spatiotemporal dissemination patterns. However, to facilitate in-depth epidemiological analysis and effective infection control, future studies must prioritize detailed transmission tracing focusing on distinct clonal lineages (Figure 2d) of the predominant bacterial species.
Since tet(X) genes are well-established to confer a high-level resistance to tetracyclines (Sun et al., 2019) and were reported to be associated with low fitness cost, though certainly in a strain-specific fashion (He et al., 2019; Jiang et al., 2021; Xu et al., 2021; Tang et al., 2022), the carriage of the tet(X) genes should be beneficial to many multidrug resistance (MDR) pathogens in a high antibiotic environment. The screened samples spanned from 2015 to 2020 (Figure 4). Early isolates (2015–2018) exclusively belonged to the Acinetobacter genus and carried only tet(X3) and tet(X4) variants. However, in 2019 and 2020, we detected emergent Proteus and Escherichia genera harboring the novel tet(X4) variant. Notably, tet(X4) exhibits the highest enzymatic activity among all tet(X) subtypes, conferring the most significant resistance phenotype (Cui et al., 2021). Furthermore, its co-occurrence with blaNDM-5—a carbapenemase variant with enhanced resistance—amplifies clinical concerns. These findings collectively demonstrate: 1) Expanding host diversity: Gradual co-emergence of blaNDM and tet(X) across new bacterial genera (Proteus, Escherichia), indicating their accelerated interspecies dissemination; 2) Directed molecular evolution: Resistance genes are evolving toward higher enzymatic activity and stronger resistance profiles (e.g., tet(X4) and blaNDM-5 synergy).
While no human-derived co-resistant isolates have been identified yet, our prior research confirmed the livestock–environment–human transmission route for blaNDM-positive bacteria. Given the confirmed presence of blaNDM/tet(X) co-carriage in livestock and environmental reservoirs, potential zoonotic transmission remains plausible. This impending threat could severely compromise future clinical treatment options, necessitating urgent implementation of: 1) Genomic surveillance programs to track resistance gene flow; 2) Preemptive containment measures targeting agricultural and environmental reservoirs; 3) Stewardship protocols for last-resort antibiotics (e.g., tigecycline/carbapenems).
From the perspective of molecular analysis, the genetic dynamics of the blaNDM context genes were analyzed, with a particular focus on the IS/Tn elements that facilitate the transfer of blaNDM among bacteria. The genetic structure of 24% had the complete Tn125 structure and of >99% featured a combination of blaNDM, ble, and trpF, most of which are interrupted by Tn/IS (Figure 6a). The heat map also reflects that the gene diversity mainly emerged from P_4, and from P_4 to P_8, the gene diversity increased. Seven major Tn/IS, i.e., ISAba125, IS5, IS91, IS6, Tn3, ISL3, and IS1, are typically found either upstream or downstream of blaNDM (Figure 7a). Of these, Tn125, the combination of two ISAba125 elements, is the primary transposon (Figure 7b). The other six potential transposons (yellow bars) have a low occurrence rate, and among them, the Tn3-Tn3 and IS6-IS6 pairs have been confirmed as transposons in earlier reports (Campos et al., 2015; Zheng et al., 2021). The formation of a transposon necessitates symmetry of Tn/IS elements at both ends. However, asymmetrical distribution of Tn/IS elements both upstream and downstream of blaNDM is also very common, the emergency frequency of which are even higher than the reported transposons Tn3-Tn3 and IS6-IS6. The results of molecular analysis indicate that there are potential various structures of blaNDM-carrier transposon, which could facilitate the transmission and dissemination of blaNDM gene. At the same time, the upstream and downstream genes of blaNDM are primarily structured around Tn125. However, the further away from the blaNDM gene, the more diverse the genes tend to be, which is of high possibility due to the insertion of Tn/IS. Therefore, when preventing the spread of blaNDM genes, it is necessary to consider both the diversity of multiple transposon structures and blaNDM context genes.
Methods
Data collection
The NCBI database was comprehensively searched to identify all genera carrying blaNDM and all genomic sequences of sufficient assembly level (≥scaffold) were downloaded. Within these genera, strains carrying blaNDM were identified and further filtered based on the prevalence in China.
Point mutations can result in different, but similar, blaNDM subtypes. Therefore, the blaNDM-1 subtype was selected as a reference to search the NCBI database with the criteria of coverage >10% and identity >80%. The search results identified 2,894 potentially relevant species distributed across 34 genera (SF1: NCBI_collection), primarily in the family Enterobacteriaceae, order Enterobacterales, class Gammaproteobacteria, and phylum Proteobacteria. As of November 10, 2022, the NCBI database included 57,017 genomes (worldwide) for these species with 10,404 specifics to China. Among the 10,404 genomes specific to China (accession numbers are provided in SF1: 10404_China), 954 that fulfilled the following three rigorous criteria were selected as the NCBI dataset used in this study to make the analysis results more credible and reliable.
• Assembly level higher than contig and quality (=completeness–5*contamination) should be greater than 95%.
• Accurate collection year and location (province) data.
• Inclusion of the complete blaNDM sequence (813 bp).
Our collected bacterial genomes were extracted from cultured bacteria using the QIAamp DNA Mini Kit (Qiagen, Inc., Valencia, CA, USA) and sequenced by Novogene Co., Ltd. (Beijing, China) using the Illumina HiSeq 2500 platform (Illumina, Inc., San Diego, CA, USA) with an insert size of 350 bp. The genome was assembled de novo using the SOAPdenovo genome assembler (v2.04) (Li et al., 2010) with an average coverage of 110 fold. Scaffolding and gap filling were performed using SSPACE and GapFiller, respectively (Boetzer et al., 2011; Boetzer and Pirovano, 2012).
Geographic dissemination
To ensure adequate data for geospatial analysis, four species with >15 collected isolates were selected from the 1021 blaNDM-positive isolates. Ultimately, 13, 37, 38, and 24 isolates of P. mirabilis, K. pneumoniae, E. coli, and E. hormaechei, respectively, collected in different years and from different regions (provinces) were used for geospatial analysis (more information is provided on https://github.com/wr-sky/NDM:fasta_beast_setting/List_of_four_species.txt). BEAUti software in Beast (Bouckaert et al., 2014) was utilized to extract the region and year of collection of each species. The parameters included the substitution model (Blosum62), clock type (uncorrelated relaxed clock, relaxed distribution: lognormal), and other default options. Complete information on the fasta files and BEAUti settings are provided on https://github.com/wr-sky/NDM:fasta_beast_setting. Multiple runs were conducted using Beast until each parameter exceeded 200 when combined in Tracer log files to assure the analysis results more reliable and trustworthy. Multiple tree files were combined using LogCombiner software (https://beast.community/logcombiner) and then simplified by TreeAnnotator software (https://beast.community/treeannotator). The geographic dissemination of each species was simulated and calculated by SpreaD3 (Spatial Phylogenetics Reconstruction of Evolutionary Dynamics using Data-Driven Documents (D3)) (Bielejec et al., 2016) with the geographic data of the province in China as input (https://github.com/wr-sky/NDM:fasta_beast_setting/all_pro_pos.txt&china.json ).
Clustering of blaNDM context genes
The complete process of the clustering method is shown in Figure 5d. Initially, a set of seven downstream genes were be automatically extracted from the gbff file and formatted by the program. Then, manual inspection was applied for quality control. The final sequencing results of the downstream genes are provided in SF4: SF4_954_67_NDM_downstream_7.xlsx. Next, the similarity of each pair of gene sequences was measured based on the following principles:
• For the same gene in the same position, add 1, 2, and n scores in sequence. Once two genes in the same position are different, add score from 1 again.
• For the same gene, the score was increased by 1, regardless of the position.
A representative calculation of the similarity of the sequences of GCA_008180305.1 and GCA_020223715.1 is shown (Figure 5a). First, the score of each corresponding gene at the same position was calculated (red font). Since the five genes (blaNDM, ble, trpF, tat, and dct) are continuously aligned, scores of 1, 2, 3, 4, and 5 were added, respectively. Because groEL is not continuous with the other five genes, a score of 1 was added from the beginning. Next, the number of the same gene in both sequences was calculated and the corresponding numbers were added (black font). Finally, the similarity between these two gene sequences was assigned a score of 22.
After determining the similarity of each pair of sequences, a fully connected network was established where each node represents a sequence and each link represents the respective similarity score (Figure 5b). Subsequently, links were gradually eliminated with increasing similarity score limitations, resulting in a clustering tree. The first branch of the tree structure was formed by deleting links smaller than 10, which separated node 4 (Figure 5c). In the second branch, links smaller than 20 were deleted, resulting in the isolation of node 3. The remaining nodes 1 and 2 form the leaves of the last branch. The method generated a clustering tree of 1021 blaNDM-positive isolates (Figure 6a). Tn125 is marked in the same color, Tn/IS in brown, hypothetical proteins (hyp.ORF) in white, and all other 13 frequently emerged genes in purple.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://ngdc.cncb.ac.cn/, National Genomic Data Center PRJCA017599.
Author contributions
XH: Writing – review & editing, Supervision, Resources, Conceptualization. BW: Writing – review & editing, Software, Writing – original draft, Project administration, Validation, Data curation, Methodology, Resources. MC: Writing – review & editing, Formal analysis, Visualization, Investigation, Data curation. KL: Writing – original draft, Software, Resources, Methodology, Data curation, Formal analysis. ZP: Writing – review & editing, Data curation, Resources, Formal analysis. LJ: Data curation, Investigation, Resources, Formal analysis, Writing – review & editing. JY: Supervision, Writing – review & editing, Funding acquisition, Resources, Conceptualization. HC: Writing – review & editing, Methodology, Visualization. LZ: Methodology, Writing – review & editing, Investigation. SQ: Supervision, Conceptualization, Funding acquisition, Writing – review & editing. HR: Supervision, Writing – review & editing, Conceptualization, Project administration, Funding acquisition. HS: Supervision, Investigation, Conceptualization, Writing – review & editing, Project administration.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was financially supported the Beijing Municipal Natural Science Foundation of China (grant number: 7232229), and the National Natural Science Foundation of China (grant nos. 82003519, 32070025, and 62102439), and the Research Project of the State Key Laboratory of Pathogen and Biosecurity (grant nos. SKLPBS1807 and SKLPBS2214).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2025.1608826/full#supplementary-material
References
Acman, M., Wang, R., van Dorp, L., Shaw, L. P., Wang, Q., Luhmann, N., et al. (2022). Role of mobile genetic elements in the global dissemination of the carbapenem resistance gene bla NDM. Nat. Commun. 13, 1131. doi: 10.1038/s41467-022-28819-2
Baraniak, A., Izdebski, R., Fiett, J., Gawryszewska, I., Bojarska, K., Herda, M., et al. (2016). NDM-producing Enterobacteriaceae in Poland, 2012–14: inter-regional outbreak of Klebsiella pneumoniae ST11 and sporadic cases. J. Antimicrobial Chemotherapy 71, 85–91. doi: 10.1093/jac/dkv282
Basu, S. (2020). Variants of the New Delhi metallo-β-lactamase: new kids on the block. Future Med. 15, 465–467. doi: 10.2217/fmb-2020-0035
Benson, D. A., Cavanaugh, M., Clark, K., Karsch-Mizrachi, I., Ostell, J., Pruitt, K. D., et al. (2018). GenBank. Nucleic Acids Res. 46, :D41. doi: 10.1093/nar/gkx1094
Bielejec, F., Baele, G., Vrancken, B., Suchard, M. A., Rambaut, A., and Lemey, P. (2016). SpreaD3: interactive visualization of spatiotemporal history and trait evolutionary processes. Mol. Biol. Evol. 33, 2167–2169. doi: 10.1093/molbev/msw082
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D., and Pirovano, W. (2011). Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579. doi: 10.1093/bioinformatics/btq683
Boetzer, M. and Pirovano, W. (2012). Toward almost closed genomes with GapFiller. Genome Biol. 13, 1–9. doi: 10.1186/gb-2012-13-6-r56
Boolchandani, M., D’Souza, A. W., and Dantas, G. (2019). Sequencing-based methods and resources to study antimicrobial resistance. Nat. Rev. Genet. 20, 356–370. doi: 10.1038/s41576-019-0108-4
Bouckaert, R., Heled, J., Kühnert, D., Vaughan, T., Wu, C.-H., Xie, D., et al. (2014). BEAST 2: a software platform for Bayesian evolutionary analysis. PloS Comput. Biol. 10, e1003537. doi: 10.1371/journal.pcbi.1003537
Campos, J. C., Da Silva, M. J. F., Dos Santos, P. R. N., Barros, E. M., MdO, P., Seco, B. M. S., et al. (2015). Characterization of Tn 3000, a transposon responsible for bla NDM-1 dissemination among Enterobacteriaceae in Brazil, Nepal, Morocco, and India. Antimicrobial Agents chemotherapy 59, 7387–7395. doi: 10.1128/AAC.01458-15
Chang, Y., Chusri, S., Sangthong, R., Mcneil, E., and Tang, L. (2019). Clinical pattern of antibiotic overuse and misuse in primary healthcare hospitals in the southwest of China. PloS One 14, e0214779. doi: 10.1371/journal.pone.0214779
Chen, Y. L., Sng, W. J., Wang, D. Y., and Wang, X. Y. (2019). Antibiotic overuse and allergy-related diseases: an epidemiological cross-sectional study in the grasslands of Northern China. Ther. Clin. Risk Manage. 15, 783–789. doi: 10.2147/TCRM.S203719
Chen, H., Zhan, Y., Wang, L., Xiao, Z., Feng, D., Chen, Z., et al. (2024). Co-Occurrence of tet (X4) and bla NDM-5 in Escherichia coli Isolates of Inpatient Origin in Guangzhou, China. Microbial Drug Resistance 30, 153–163. doi: 10.1089/mdr.2023.0098
Cui, C.-Y., He, Q., Jia, Q.-L., Li, C., Chen, C., Wu, X.-T., et al. (2021). Evolutionary trajectory of the Tet (X) family: critical residue changes towards high-level tigecycline resistance. Msystems 6, e00050-21. doi: 10.1128/msystems.00050-21
Fanny, B., Anna, J., Joakim, L. D. G., and Erik, K. (2020). An updated phylogeny of the metallo-β-lactamases. J. Antimicrobial Chemotherapy. 76, 117–123. doi: 10.1093/jac/dkaa392
He, T., Li, R., Wei, R., Liu, D., Bai, L., Zhang, L., et al. (2020). Characterization of Acinetobacter indicus co-harbouring tet (X3) and bla NDM-1 of dairy cow origin. J. Antimicrobial Chemotherapy 75, 2693–2696. doi: 10.1093/jac/dkaa182
He, T., Wang, R., Liu, D., Walsh, T. R., Zhang, R., Lv, Y., et al. (2019). Emergence of plasmid-mediated high-level tigecycline resistance genes in animals and humans. Nat. Microbiol. 4, 1450–1456. doi: 10.1038/s41564-019-0445-2
Hirabayashi, A., Dao, T. D., Takemura, T., Hasebe, F., Trang, L. T., Thanh, N. H., et al. (2021). A transferable IncC-IncX3 hybrid plasmid cocarrying bla NDM-4, tet (X), and tmexCD3-toprJ3 confers resistance to carbapenem and tigecycline. Msphere 6, e0059221. doi: 10.1128/msphere.00592-21
Hu, X., Xu, X., Wang, X., Xue, W., Zhou, H., Zhang, L., et al. (2017). Diversity of New Delhi metallo-beta-lactamase-producing bacteria in China. Int. J. Infect. Dis. 55, 92–95. doi: 10.1016/j.ijid.2017.01.011
Hu, X., Yang, L., Dong, N., Lin, Y., Zhang, L., Wang, X., et al. (2022). Dissemination of bla NDM-5 in Escherichia coli through the IncX3 Plasmid from Different Regions in China. Microbial Drug Resistance 28, 453–460. doi: 10.1089/mdr.2021.0202
Ignasi, R., Noraida, M., Belgin, A., Espinal, P., and Murat, A. (2014). Molecular characterization of NDM-1-producing Acinetobacter pittii isolated from Turkey in 2006. J. Antimicrobial Chemotherapy. 69, 3437–8. doi: 10.1093/jac/dku306
Ilyina, T. S. (2012). Mobile ISCR elements: Structure, functions, and role in emergence, increase, and spread of blocks of bacterial multiple antibiotic resistance genes. Mol. Genet. Microbiol. Virol. 27, 135–146. doi: 10.3103/S0891416812040040
Jiang, L., Cai, W., Tang, F., Wang, Z., and Liu, Y. (2021). Characterization of fitness cost caused by tigecycline-resistance gene tet (X6) in different host bacteria. Antibiotics 10, 1172. doi: 10.3390/antibiotics10101172
Khan, A. U., Maryam, L., and Zarrilli, R. (2017). Structure, Genetics and Worldwide Spread of New Delhi Metallo-β-lactamase (NDM): a threat to public health. BMC Microbiol. 17, 101. doi: 10.1186/s12866-017-1012-8
Li, J., Hu, X., Yang, L., Lin, Y., Liu, Y., Li, P., et al. (2020). New Delhi Metallo-β-Lactamase 1-Producing Klebsiella pneumoniae ST719 isolated from a neonate in China. Microbial Drug Resistance 26, 492–496. doi: 10.1089/mdr.2019.0058
Li, P., Lin, Y., Hu, X., Liu, Y., Xue, M., Yang, L., et al. (2020). Characterization of blaNDM-1-and blaSHV-12-positive IncX3 plasmid in an Enterobacter hormaechei new sequence type 1000 from China. Infection Drug Resistance 13, 145–153. doi: 10.2147/IDR.S231366
Li, R., Zhu, H., Ruan, J., Qian, W., Fang, X., Shi, Z., et al. (2010). De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20, 265–272. doi: 10.1101/gr.097261.109
O’Leary, N. A., Wright, M. W., Brister, J. R., Ciufo, S., Haddad, D., McVeigh, R., et al. (2016). Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745. doi: 10.1093/nar/gkv1189
Partridge, S. R. and Iredell, J. R. (2012). Genetic contexts of bla NDM-1. Antimicrobial Agents Chemotherapy 56, 6065–6067. doi: 10.1128/AAC.00117-12
Perez, F. and Bonomo, R. A. (2019). Carbapenem-resistant Enterobacteriaceae: global action required. Lancet Infect. Dis. 19, 561–562. doi: 10.1016/S1473-3099(19)30210-5
Poirel, L., Bonnin, R. A., Boulanger, A., Schrenzel, J., Kaase, M., and Nordmann, P. (2012). Tn 125-related acquisition of bla NDM-like genes in Acinetobacter baumannii. Antimicrobial Agents chemotherapy 56, 1087–1089. doi: 10.1128/AAC.05620-11
Poirel, L., Bonnin, R. A., and Nordmann, P. (2011). Analysis of the resistome of a multidrug-resistant NDM-1-producing Escherichia coli strain by high-throughput genome sequencing. Antimicrob. Agents Chemother. 55, 4224–4229. doi: 10.1128/AAC.00165-11
Rahman, M., Prasad, K. N., Gupta, S., Singh, S., Singh, A., Pathak, A., et al. (2018). Prevalence and molecular characterization of New Delhi metallo-beta-lactamases in multidrug-resistant Pseudomonas aeruginosa and Acinetobacter baumannii from India. Microbial Drug Resistance 24, 792–798. doi: 10.1089/mdr.2017.0078
Sun, J., Chen, C., Cui, C.-Y., Zhang, Y., Liu, X., Cui, Z.-H., et al. (2019). Plasmid-encoded tet (X) genes that confer high-level tigecycline resistance in Escherichia coli. Nat. Microbiol. 4, 1457–1464. doi: 10.1038/s41564-019-0496-4
Tang, F., Cai, W., Jiang, L., Wang, Z., and Liu, Y. (2022). Large-scale analysis of fitness cost of tet (X4)-positive plasmids in Escherichia coli. Front. Cell. Infection Microbiol. 12, 798802. doi: 10.3389/fcimb.2022.798802
Usmanqamar, M., Slopes, B., Hassan, B., Khurshid, M., and Atoleman, M. (2021). The present danger of New Delhi metallo-β-lactamase: a threat to public health. Future Microbiol. 15, 1759–1778. doi: 10.2217/fmb-2020-0069
Wailan, A. M., Sartor, A. L., Zowawi, H. M., Perry, J. D., Paterson, D. L., and Sidjabat, H. E. (2015). Genetic contexts of bla NDM-1 in patients carrying multiple NDM-producing strains. Antimicrobial Agents chemotherapy 59, 7405–7410. doi: 10.1128/AAC.01319-15
Walley, J. D., Zhang, Z., and Wei, X. (2021). Antibiotic overuse in China: call for consolidated efforts to develop antibiotic stewardship programmes. Lancet Infect. Dis. 21, 597. doi: 10.1016/S1473-3099(21)00196-1
Wang, K., Li, P., Li, J., Hu, X., Lin, Y., Yang, L., et al. (2020). An NDM-1-producing Acinetobacter towneri isolate from hospital sewage in China. Infection Drug Resistance. 13, 1105–1110. doi: 10.2147/IDR.S246697
Wu, W., Feng, Y., Tang, G., Qiao, F., McNally, A., and Zong, Z. (2019). NDM metallo-β-lactamases and their bacterial producers in health care settings. Clin. Microbiol. Rev. 32, e00115–e00118. doi: 10.1128/CMR.00115-18
Xu, Y., Liu, L., Zhang, H., and Feng, Y. (2021). Co-production of Tet (X) and MCR-1, two resistance enzymes by a single plasmid. Environ. Microbiol. 23, 7445–7464. doi: 10.1111/1462-2920.15425
Yang, L., Hu, X., Xu, X., Yang, C., Xie, J., Hao, R., et al. (2017). Salmonella enterica serovar Typhimurium ST34 co-expressing bla NDM-5 and bla CTX-M-55 isolated in China. Emerging Microbes Infections 6, 1–3. doi: 10.1038/emi.2017.48
Yang, L., Li, P., Liang, B., Hu, X., Li, J., Xie, J., et al. (2018). Multidrug-resistant Citrobacter freundii ST139 co-producing NDM-1 and CMY-152 from China. Sci. Rep. 8, 10653. doi: 10.1038/s41598-018-28879-9
Yao, C., Jin, L., Wang, Q., Wang, M., Wang, R., Cai, M., et al. (2025). Unraveling the evolution and global transmission of high level tigecycline resistance gene tet (X). Environ. Int. 199, 109499. doi: 10.1016/j.envint.2025.109499
Yong, D., Toleman, M. A., Giske, C. G., Cho, H. S., Sundman, K., Lee, K., et al. (2009). Characterization of a New Metallo-β-Lactamase Gene, blaNDM-1, and a Novel Erythromycin Esterase Gene Carried on a Unique Genetic Structure in Klebsiella pneumoniae Sequence Type 14 from India. Antimicrob. Agents Chemother. 53, 5046–5054. doi: 10.1128/AAC.00774-09
Zhao, Q.-Y., Zhu, J.-H., Cai, R.-M., Zheng, X.-R., Zhang, L.-J., Chang, M.-X., et al. (2021). IS 26 is responsible for the evolution and transmission of bla NDM-harboring plasmids in Escherichia coli of poultry origin in China. Msystems 6, e00646–e00621. doi: 10.1128/msystems.00646-21
Zheng, X.-R., Sun, Y.-H., Zhu, J.-H., Wu, S.-L., Ping, C., Fang, L.-X., et al. (2021). Two novel blaNDM-1-harbouring transposons on pPrY2001-like plasmids coexisting with a novel cfr-encoding plasmid in food animal source Enterobacteriaceae. J Glob Antimicrob Resist. 26, 222–226. doi: 10.1016/j.jgar.2021.06.006
Keywords: blaNDM, evolution, dissemination, tet(X), genetic dynamics
Citation: Hu X, Wang B, Chen M, Li K, Peng Z, Jin L, Yue J, Chen H, Zhang L, Qiu S, Ren H and Song H (2025) Evolution, dissemination, and genetic dynamics of the carbapenem resistance gene blaNDM in China. Front. Cell. Infect. Microbiol. 15:1608826. doi: 10.3389/fcimb.2025.1608826
Received: 09 April 2025; Accepted: 24 July 2025;
Published: 11 August 2025.
Edited by:
Xiaoting Hua, Zhejiang University, ChinaReviewed by:
Miklos Fuzi, Independent Researcher, Seattle, WA, United StatesTian Jiang, Wenzhou Medical University, China
Copyright © 2025 Hu, Wang, Chen, Li, Peng, Jin, Yue, Chen, Zhang, Qiu, Ren and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongbin Song, aG9uZ2JpbnNvbmdAMjYzLm5ldA==; Hongguang Ren, YmlvcmVuQDE2My5jb20=; Shaofu Qiu, cWl1c2hmMDYxM0Bob3RtYWlsLmNvbQ==
†These authors have contributed equally to this work