 Cheng Zheng1†Jianmin Huang1†Shengming Xu1†Bin Lu1Hanxin Que1Tianhao Chen1Yubo Hou1,2Linlin He1,2Xia Fan1
Cheng Zheng1†Jianmin Huang1†Shengming Xu1†Bin Lu1Hanxin Que1Tianhao Chen1Yubo Hou1,2Linlin He1,2Xia Fan1 Fa-Ming Chen3*
Fa-Ming Chen3* Yi Wang1,4*
Yi Wang1,4* Hui Deng1,2*
Hui Deng1,2*- 1Institute of Stomatology, School and Hospital of Stomatology, Wenzhou Medical University, Wenzhou, Zhejiang, China
- 2Department of Periodontology, School and Hospital of Stomatology, Wenzhou Medical University, Wenzhou, Zhejiang, China
- 3State Key Laboratory of Oral & Maxillofacial Reconstruction and Regeneration, National Clinical Research Center for Oral Diseases, Shanxi International Joint Research Center for Oral Diseases, Department of Periodontology, School of Stomatology, The Fourth Military Medical University, Xi’ an, Shanxi, China
- 4Department of Orthodontics, School and Hospital of Stomatology, Wenzhou Medical University, Wenzhou, Zhejiang, China
Introduction: The direct infection of endothelial cells by Porphyromonas gingivalis (P. gingivalis), a keystone periodontal pathogen, has been implicated in the development of atherosclerosis. While non-selective autophagy facilitates its intracellular persistence in endothelial cells, the role of selective autophagy in this process remains unclear. This study investigated whether P. gingivalis hijacks mitophagy and lysosomes to persist in endothelial cells.
Methods: Human aortic endothelial cells (HAECs) were infected with P. gingivalis for 24 h. Mitophagy was detected by Western Blotting (WB), immunofluorescence, and transmission electron microscopy. Lysosomal function was assessed by acridine orange staining, lysosensor staining, and WB. The effects of mitophagy and lysosomes on P. gingivalis intracellular survival were evaluated by antibiotic protection assays and SYTO-9 staining.
Results: Our data demonstrated that P. gingivalis initiates PTENinduced putative kinase 1 (PINK1)-Parkin-mediated mitophagy in HAECs, leading to increased formation of autophagosomes and mitophagosomes, but disrupted autophagy/mitophagy flux. This blockage of autophagy/mitophagy flux was linked to lysosomal dysfunction, characterized by increased lysosome number, lysosomal membrane permeabilization, disruption of the lysosomal acidic environment, and decreased enzymatic activity. Additionally, antibiotic protection assays and SYTO-9 staining further revealed that P. gingivalis promotes its intracellular survival in endothelial cells by initiating mitophagy and impairing lysosomal function. Furthermore, the mitophagy activator decreased the co-localization of P. gingivalis with microtubule-associated protein 1 light chain 3 (LC3)-p62, LC3-NDP52, and lysosomal-associated membrane protein 1 (LAMP1), suggesting that P. gingivalis-initiated mitophagy inhibited xenophagosome formation and autophagosome/xenophagosome-lysosome fusion.
Conclusion: Our findings reveal that P. gingivalis may promote its intracellular survival in endothelial cells by initiating PINK1-Parkin-mediated mitophagy and impairing lysosomal function, thereby suppressing xenophagosome formation and xenophagic degradation. This study provides new insights into the mechanisms by which P. gingivalis persists in endothelial cells and its potential role in atherosclerosis progression.
1 Introduction
Porphyromonas gingivalis (P. gingivalis), a keystone pathogen in periodontal disease, is recognized for its ability to enter the bloodstream and colonize extra-oral tissues (Forner et al., 2006). Notably, the DNA of P. gingivalis has been identified in human atherosclerotic plaques, providing direct evidence of its association with atherosclerosis (Haraszthy et al., 2000). Animal studies further support this link, showing that P. gingivalis infection accelerates the progression of atherosclerotic lesions (Li et al., 2002; Brodala et al., 2005). Endothelial cells, which play a crucial role in atherosclerosis, are directly targeted by P. gingivalis (Gimbrone and García-Cardeña, 2016). This bacterium adheres to and invades endothelial cells, evades immune surveillance, and persists in a dormant state or replicating state, contributing to the pathology of atherosclerosis (Zheng et al., 2021). However, the mechanisms by which P. gingivalis survives and evades immune surveillance within endothelial cells remain largely unexplored.
Autophagy is the process by which cells eliminate intracellular pathogens, damaged organelles, and misfolded proteins. It begins when a phagophore, encapsulates cytoplasmic components to form autophagosomes. The autophagosomes then fuse with lysosomes to become autolysosomes, where their contents are degraded by lysosomal enzymes (Mizushima and Komatsu, 2011; Dikic and Elazar, 2018). Autophagy can be categorized into non-selective or selective. In non-selective autophagy, cells indiscriminately engulf and degrade parts of cytosolic components, such as intracellular bacteria and host cell proteins. Interestingly, upon invading endothelial cells and gingival epithelial cells, P. gingivalis triggers a non-selective autophagy pathway and directs itself into rich-nutrient autophagosomes to evade immune surveillance and promote its survival (Bélanger et al., 2006; Lee et al., 2018).
Selective autophagy targets specific components, such as damaged mitochondria (mitophagy) and intracellular bacteria (xenophagy), for degradation (Wang et al., 2023). Mitophagy is primarily regulated through a ubiquitin-dependent signaling pathway involving two major proteins: PTENinduced putative kinase 1 (PINK1) and E3 ubiquitin ligases Parkin. PINK1-Parkin-mediated mitophagy relies on ubiquitin-binding adaptor proteins such as p62, NDP52, and optineurin (Lazarou et al., 2015). Interestingly, mitophagy shares several molecular components, including those adaptor proteins, with antimicrobial selective autophagy (xenophagy), and they may compete for the same molecules (Stolz et al., 2014; Song et al., 2022; Rubio-Tomás et al., 2023).
In addition to the non-selective autophagy, certain bacteria have evolved mechanisms to exploit selective autophagy to support their survival. For instance, Burkholderia pseudomallei, Neisseria gonorrhoeae, and Listeria monocytogenes manipulate mitophagy to limit mitochondrial reactive oxygen species (mtROS) production, helping them evade host immune defenses (Zhang et al., 2019; Gao et al., 2024; Nan et al., 2024). Additionally, P. gingivalis has been shown to cleave NDP52 through its gingipains, enabling the bacterium to resist xenophagic degradation (Shiheido-Watanabe et al., 2023). Similarly, Mycobacterium bovis (M. bovis) inhibits host xenophagy by inducing mitophagy, thus enhancing its intracellular survival (Song et al., 2022).
Being the organelle responsible for the final degradation stage of autophagy, the lysosome is also vital in determining the fate of pathogens (Ballabio and Bonifacino, 2020). Some bacteria, such as Escherichia coli and Helicobacter pylori, survive within autophagosomes by reducing hydrolase activity through increasing lysosomal pH (Miao et al., 2015; Naskar et al., 2023). Recent studies suggested that P. gingivalis promotes lysosomal efflux in gingival epithelial cells, thereby preventing autophagosome-lysosome fusion and allowing the bacteria to survive (Liu et al., 2023). However, it remains uncertain whether P. gingivalis exploits mitophagy and lysosomal function to promote survival within endothelial cells.
This study aims to explore how P. gingivalis promotes its intracellular survival in endothelial cells by investigating the roles of mitophagy and lysosomal function. The findings of our study could provide new insights into the intracellular survival strategies of P. gingivalis.
2 Materials and methods
2.1 Reagents and primary antibodies
The reagents used in this study include carbonyl cyanide m-chlorophenylhydrazone (CCCP, MCE, Cat# HY-10094), ML-SA1 (MCE, Cat# HY-108462), Bafilomycin A1 (BafA1, MCE, Cat# HY-100558), and mitochondrial division inhibitor 1 (Mdivi-1, MCE, Cat# HY-15886). Other reagents included SYTO-9 (Invitrogen, Thermo Fisher Scientific, Cat# S34854), Actin-Tracker Red-Rhodamine (Beyotime Biotechnology, Cat# C2207S), acridine orange hydrochloride (AO, MCE, Cat# HY-101879), MitoSOX (Invitrogen, Thermo Fisher Scientific, Cat# M36007), and Mito-Tracker Red CMXRos (Beyotime Biotechnology, Cat# C1035). The details of primary antibodies for Western Blotting and immunofluorescence are described in Supplementary Tables 1, 2 respectively.
2.2 Cell culture
HAECs (iCell Bioscience, Cat# C1252, China) were cultured in endothelial cell medium (ECM) supplemented with 5% fetal bovine serum and 1% endothelial cell growth supplement. The cells were maintained in a humidified incubator at 37°C with 5% CO2.
2.3 Bacteria culture and fluorescent labeling
P. gingivalis W83 (ATCC, Manassas, VA, USA) was cultured on anaerobic blood agar plates within a chamber with 85% N2, 5% H2, and 10% CO2 at 37°C for 5–7 days. The bacteria were then inoculated into a liquid broth of brain heart infusion (BHI) supplemented with hemin (5 μg/mL) and vitamin K (5 μg/mL) and grown for over 24 h at 37°C. P. gingivalis was labeled with SYTO-9 according to the protocol provided by the manufacturer. HAECs were seeded onto coverslips in 24-well plates and co-cultured with SYTO-9-labeled P. gingivalis at a multiplicity of infection (MOI) of 1:100 for 2 h or 24 h. After incubation, the cells were stained with Actin-Tracker and DAPI for further analysis.
2.4 Immunofluorescence and imaging
HAECs were seeded onto coverslips in 24-well culture plates and pretreated with dimethyl sulfoxide (DMSO) (0.25%), BafA1 (20 nM), Mdivi-1 (25 μM), or CCCP (20 μM) for 2 h, or treated with ML-SA1 (10 μM) during P. gingivalis infection. Following pretreatments, cells were infected with P. gingivalis (MOI=100) for 24 h. Cells were sequentially fixed, permeabilized, and blocked to prepare samples for immunofluorescence. They were then incubated with the primary antibodies (Supplementary Table), followed by incubation with the secondary antibody. Mito-Tracker Red CMXRos was applied to stain mitochondria before fixation. Images were captured using a Leica Stellaris 5 confocal laser scanning microscope (CLSM, Leica Microsystems, Waltham, MA, USA) with a 63X oil immersion lens. Approximately 10–15 random images were acquired for each treatment group. The experiment was conducted in triplicate.
2.5 Antibiotic protection assays
HAECs were seeded at a density of 3 × 105 on 6-well plates. Cells were pretreated with DMSO (0.25%), BafA1 (20 nM), Mdivi-1 (25 μM), or CCCP (20 μM) for 2 h, or treated with ML-SA1 (10 μM) during P. gingivalis infection. Following pretreatments, cells were infected with P. gingivalis (MOI=100) for 2 h or 24 h. At each collection time point, HAECs were washed and then incubated in ECM containing gentamicin (300 μg/mL) and metronidazole (200 μg/mL) for 1 h to eliminate extracellular bacteria. The cells were subsequently lysed with iced double-distilled H2O (ddH2O). Lysates were serially diluted in ddH2O and plated on blood agar plates. Colony-forming units (CFUs) were determined after incubation to assess the number of intracellular bacteria. The experiment was conducted in triplicate.
2.6 Western blotting
Total protein extracts were prepared from cells using a modified RIPA buffer, containing freshly added protease and phosphatase inhibitors. Following the instructions provided by the manufacturer, protein concentration was determined using a BCA assay kit (Beyotime Biotechnology, Shanghai, China). The extracted proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Polyvinylidene fluoride (PVDF) membranes with transferred proteins were blocked in 5% non-fat dry milk and then incubated with the primary antibodies (Supplementary Table), followed by secondary antibody incubation. Protein expression levels were normalized to GAPDH and are expressed as fold changes relative to the control. The experiment was conducted in triplicate.
2.7 Transmission electron microscopy observation
Cells were fixed overnight at 4°C using 2.5% glutaraldehyde. After fixation, the cells were washed with sodium cacodylate buffer and fixed in 1% osmium tetroxide for 1 h, followed by dehydration with acetone. The cell pellet was infiltrated with resin and polymerized overnight at 60°C. Resin-embedded samples were sectioned into 70-nm ultrathin slices, and images were captured using a FEI Tecnai G2 F20 electron microscope (Thermo Fisher Scientific, USA). Each experiment was performed in triplicate.
2.8 LysoTracker green staining
LysoTracker Green staining (Beyotime Biotechnology, Shanghai, China) was used to label and quantify the lysosomes. After P. gingivalis treatment, HAECs were stained with LysoTracker Green (75 nM) at 37°C for 30 min, and then observed and imaged by CLSM.
2.9 siRNA transfection
Human Pink1-targeting siRNA plasmids were from TsingKe Biotech (China). The primers of each siRNA plasmid are as follows: siPink1–1 with the forward primer 5’ -GAGACCUGAAAUCCGACAA- 3’ and the reverse primer 5’-UUGUCGGAUUUCAGGUCUC-3’; siPink1–2 with the forward primer 5’ -GCCAUCUUGAACACAAUGA- 3’ and the reverse primer 5’-UCAUUGUGUUCAAGAUGGC-3’; siPink1–3 with the forward primer 5’ -GCAAAUGUGCUUCAUCUAA- 3’ and the reverse primer 5’-UUAGAUGAAGCACAUUUGC-3’.
Pink1 siRNA and negative control siRNA plasmids were transfected into HAECs at 20nM using Lipofectamine 3000 (Thermo Fisher Scientific, USA) following the manufacturer’s instructions. The transfection medium was replaced with fresh complete medium after 4 h, and the cells were transfected for 48 h. The efficiency of PINK1 silencing was detected by Western blot.
2.10 Acid phosphatase activity
After 24 h of exposure to P. gingivalis (MOI=100), the intracellular fluoride-resistant acid phosphatase activity was measured using a commercially available kit, according to the instructions provided by the manufacturer (ACP activity assay kit, Beyotime Biotechnology, Shanghai, China).
2.11 Lysosomal membrane permeability
Acridine orange (AO) staining was utilized to evaluate lysosomal membrane permeability (LMP). In brief, HAECs were exposed to P. gingivalis (MOI=100) for 24 h. As a positive control, cells pretreated with BafA1 (20 nM for 2 h) were included. Following this, the cells were stained with AO solution (5 μg/mL) for 20 min at 37°C and then observed under a fluorescence microscope.
2.12 Measurement of mtROS
HAECs were pretreated with DMSO (0.25%), Mdivi-1 (25 μM), or CCCP (20 μM) for 2 h. Following pretreatments, cells were infected with P. gingivalis (MOI=100) for 24 h. Intracellular mtROS level was detected using MitoSOX (5 μM) for 15–30 min at 37°C and then observed under a fluorescence microscope. Each experiment was performed in triplicate.
2.13 Statistical analysis
The normal distribution of data was checked by the Shapiro–Wilk test, and all data sets in the study passed the normality distribution test. Statistical significance was determined by using an Unpaired t-test (two-tailed). Comparisons among multiple groups were demonstrated utilizing one-way ANOVA along with Tukey’s post hoc test. Results were presented as mean ± SEM from a minimum of three biological replicates. P<0.05 was considered statistically significant, and all statistical analyses were performed with GraphPad Prism 9.0 (GraphPad Software, Boston, MA, USA).
3 Results
3.1 P. gingivalis initiated mitophagy and increased accumulation of mitophagosomes in endothelial cells
Following exposure to P. gingivalis (MOI=100) for 24 h in HAECs, we observed upregulation of proteins PINK1 and Parkin (Figures 1A, B), along with enhanced immunofluorescence co-localization of these proteins with mitochondria (Figures 1C–E), suggesting the activation of mitophagy-related signaling molecules. To validate these results, we assessed mitochondrial membrane potential (MMP) and mtDNA copy number. Our results showed a dose- and time-dependent decline in MMP (Supplementary Figures 1A–D) and a reduction in mtDNA copy number (Supplementary Figure 1E), suggesting that P. gingivalis induces mitochondrial dysfunction. These findings showed that P. gingivalis impaired mitochondrial function and initiated PINK1-Parkin-mediated mitophagy in endothelial cells.
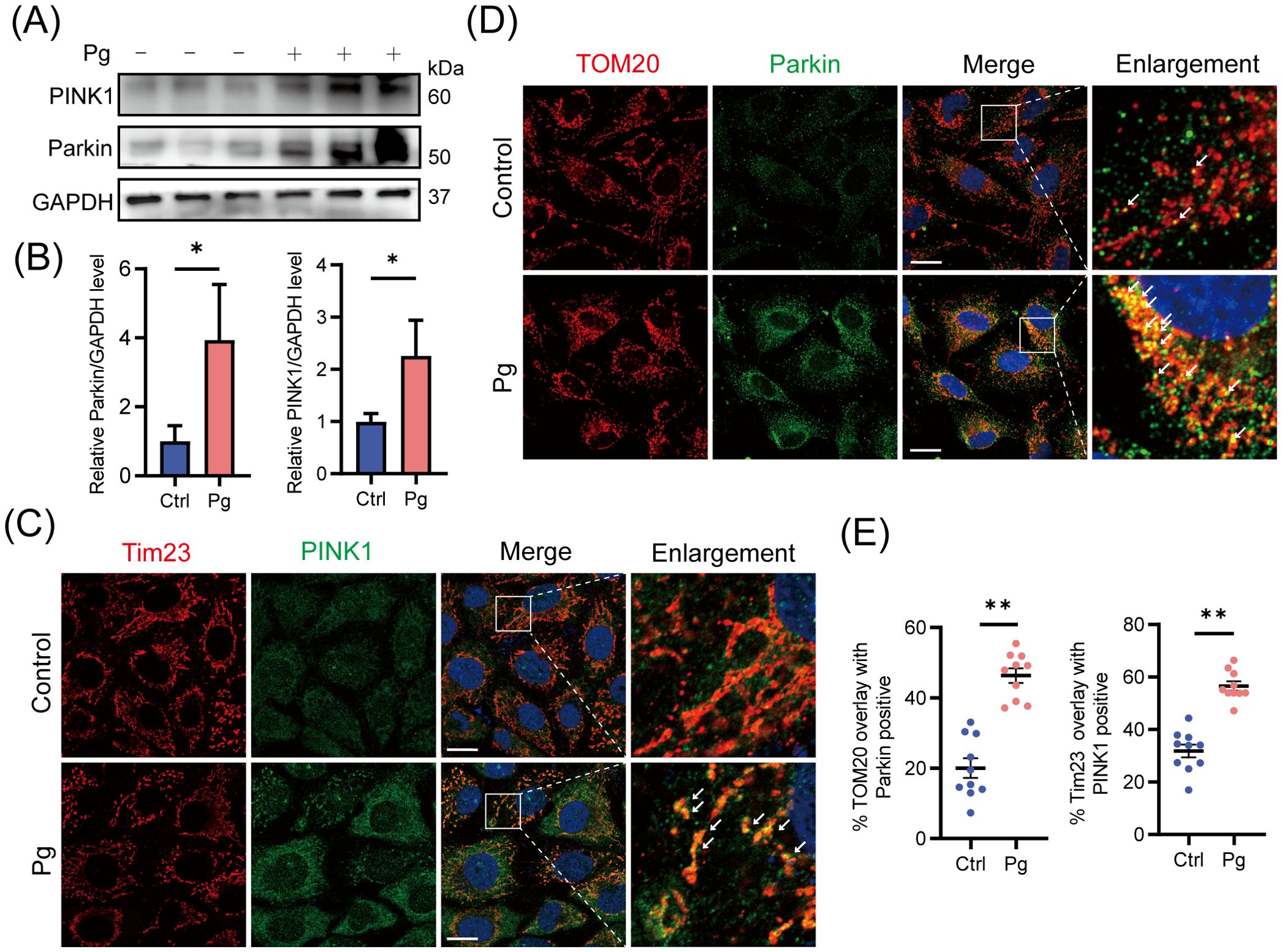
Figure 1. Initiation of PINK1-Parkin-mediated mitophagy upon Porphyromonas gingivalis stimulation at an MOI of 100 for 24 h in HAECs. (A) Representative western blot and (B) quantification of Parkin and PINK1 protein expression in control or Pg-infected HAECs (n=3 per group). (C, D) Representative images and (E) quantification of Tim23 (red) -PINK1 (green) and TOM20 (red) -Parkin (green) colocalization by confocal laser scanning microscope (CLSM). Scale bar: 15 μm. (n = 10–15 cells per group). Data are presented as the mean ± SEM. *P < 0.05 and **P< 0.01, compared with the control.
Mitophagy involves the formation of mitophagosomes, which are subsequently fused with lysosomes and degraded (Pickles et al., 2018). To investigate the development of autophagy/mitophagy, we examined the expression of lipidated microtubule-associated protein 1 light chain 3 proteins (LC3-II, autophagy formation marker) and p62 (autophagy degradation marker) (Klionsky et al., 2021) in HAECs exposed to P. gingivalis for 24 h, finding that P. gingivalis significantly enhanced the expression levels of LC3-II and p62 protein (Figure 2A). Given that p62 levels negatively correlate with autophagic activity (Klionsky et al., 2021), these results suggested that P. gingivalis may trigger the autophagy/mitophagy process but block lysosomal degradation.
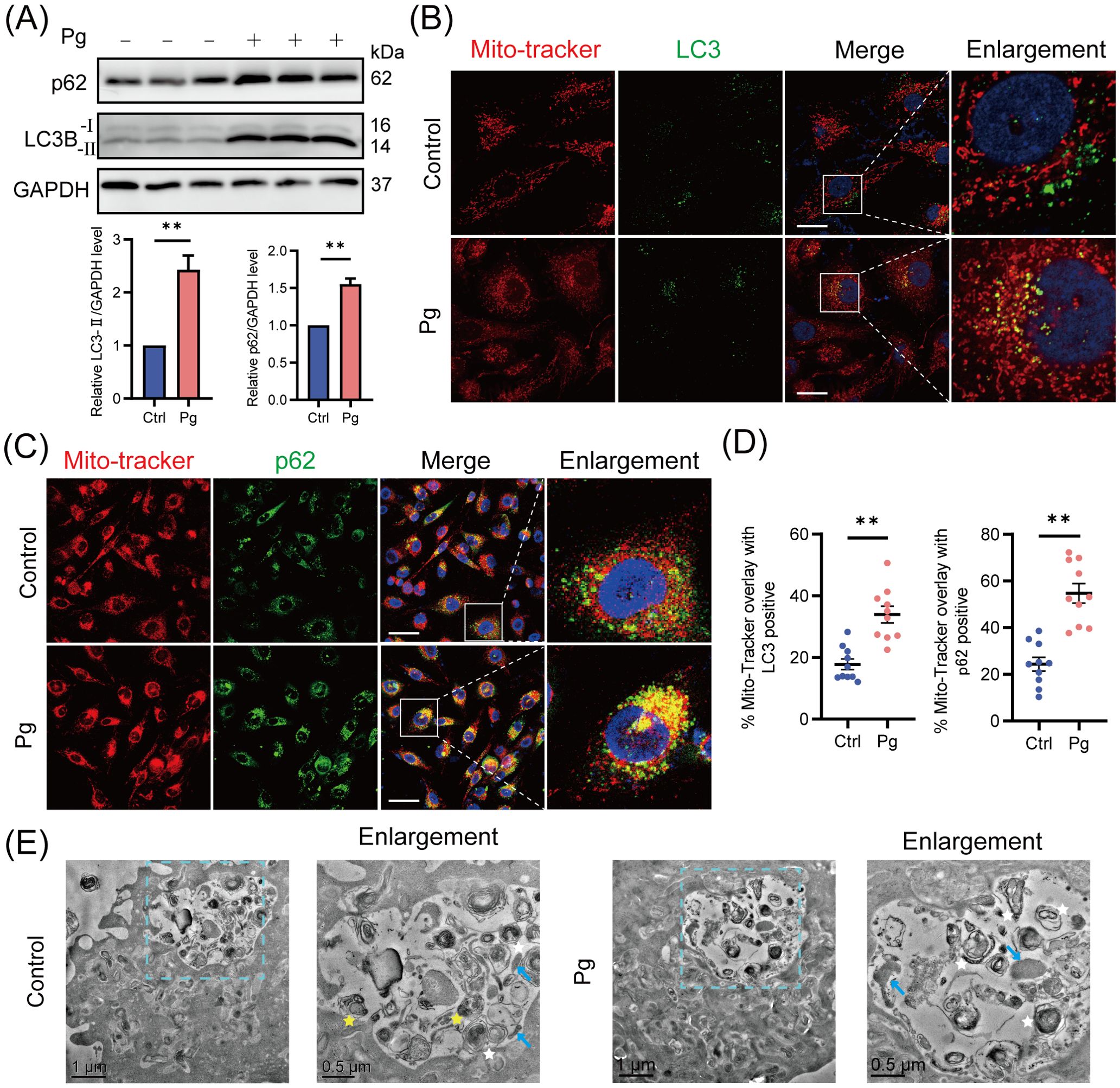
Figure 2. Mitophagosome formation accumulated upon Porphyromonas gingivalis infection. HAECs were infected with Pg at an MOI of 100 for 24 h. (A) Representative western blot and quantification of LC3-I/II and p62 protein expression in control or Pg-infected HAECs (n = 3 per group). (B) Representative images of Mito-Tracker (red) and LC3 (green) colocalization by CLSM. Scale bar: 15 μm. (C) Representative images of Mito-Tracker (red) and p62 (green) colocalization by CLSM. Scale bar: 30 μm. (D) The colocalization of MitoTracker-LC3 and MitoTracker-p62 was quantified (n = 10–15 cells per group). (E) Transmission electron microscopy (TEM) images revealed the mitophagosome-like vacuoles and mitophagolysosome-like vacuoles in control or Pg-infected HAECs (blue arrow: mitochondria, white star: mitophagosome, yellow star: mitophagolysosome, containing autophagosome). Data are presented as the mean ± SEM. **P< 0.01, compared with the control.
Furthermore, mitochondria co-localized with LC3 or cargo protein p62/NDP52 were analyzed to confirm the mitophagosome formation in HAECs. Upon infection with P. gingivalis, we observed an increase in the recruitment of p62/NDP52 to mitochondria and a significant rise in the percentage of mitophagosomes (yellow puncta/red puncta%) (Figures 2B–D; Supplementary Figures 2A, B), indicating enhanced formation of mitophagosomes. Next, transmission electron microscopy (TEM) was used to check subcellular structures within HAECs. We observed an increase in mitophagosome-like vacuoles with characteristic double-membrane structures, while mitophagolysosome-like vacuoles were rare in the P. gingivalis-treated group (Figure 2E). In summary, these results confirmed that P. gingivalis promotes an unhealthy accumulation of mitophagosomes in HAECs.
3.2 P. gingivalis increased the accumulation of mitophagosomes by reducing mitophagy/autophagy flux
To determine whether the accumulation of mitophagosomes is due to increased mitophagosome formation or impaired lysosomal clearance, HAECs were pretreated with the autophagosome-lysosome fusion inhibitor BafA1 for 2 h. After BafA1 treatment, we observed an increased LC3-II protein expression level and enhanced co-localization of LC3 with mitochondria in P. gingivalis-infected group compared with the uninfected ones (Figures 3A, B, E), suggesting that P. gingivalis promotes mitophagosome formation. Interestingly, the total accumulation of mitophagosomes (a comparison of LC3-II expression levels in control and P. gingivalis treatment groups) exceeded the level of mitophagosome formation, implying that P. gingivalis inhibited mitophagosome degradation in endothelial cells. Supporting the above observation on LC3 levels, there was no significant change in p62 protein levels between BafA1-treated and untreated HAECs exposed to P. gingivalis (Figure 3A), suggesting mitophagy/autophagy flux was reduced.
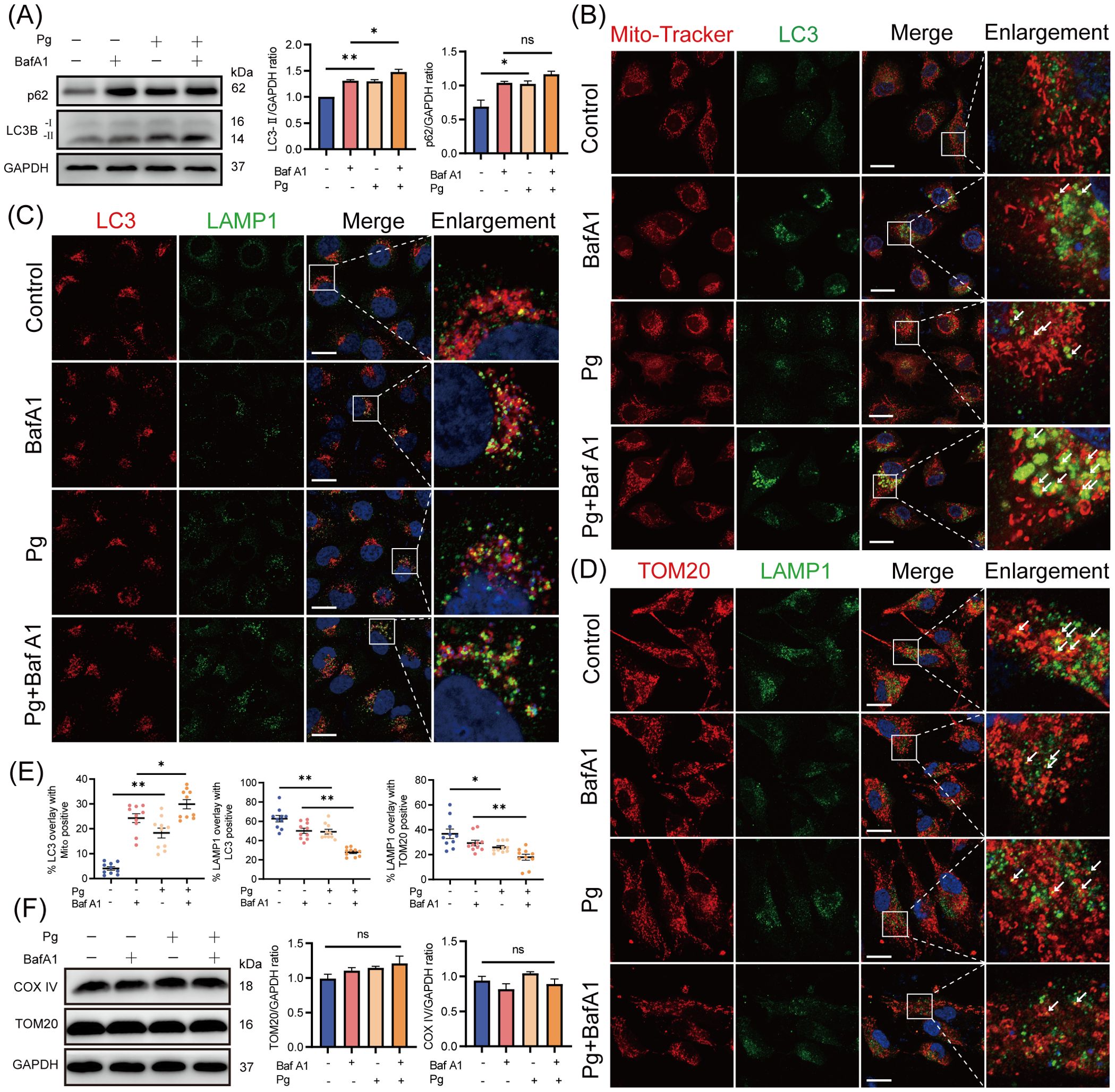
Figure 3. Porphyromonas gingivalis increased the accumulation of mitophagosomes by reducing mitophagy/autophagy flux. HAECs were pretreated with or without BafA1 (autophagosome-lysosome fusion inhibitor) for 2 h before Pg exposure (MOI=100, 24 h). (A) Representative western blot and quantification of LC3-I/II and p62 protein expression (n = 3 per group). (B-D) Representative images of Mito-Tracker (red)- LC3 (green), LC3 (red)- LAMP1 (green), and TOM20 (red)- LAMP1 (green) colocalization by CLSM. Scale bar: 15 μm. (E) The colocalization of MitoTracker-LC3, LC3-LAMP1, and TOM20-LAMP1 was quantified (n = 10–15 cells per group). (F) Representative western blot and quantification of mitochondrial marker COX IV and TOM20 protein expression (n = 3 per group). Data are presented as the mean ± SEM. *P < 0.05, and **P< 0.01, ns, not significant.
Additionally, we observed reduced co-localization of LC3 with lysosomal-associated membrane protein 1 (LAMP1, lysosomal marker) and translocase of outer mitochondrial membrane 20 (TOM20) with LAMP1 in P. gingivalis-infected HAECs (Figures 3C–E), suggesting that P. gingivalis inhibited mitophagosome-lysosome fusion. The expression levels of mitochondrial proteins TOM20 and COX IV did not decrease after P. gingivalis exposure (Figure 3F), indicating that damaged mitochondria were not effectively degraded. Overall, these results demonstrated that P. gingivalis increased the formation of mitophagosomes and reduced mitophagy/autophagy flux.
3.3 P. gingivalis reduced mitophagy/autophagy flux by impairing lysosomal function
A healthy lysosome is essential for the final degradation of autophagic substrates (Settembre et al., 2013). Therefore, we assessed the impact of P. gingivalis on lysosomal function in endothelial cells. Our results demonstrated a time- and dose-dependent increase in LysoTracker fluorescence intensity (Figures 4A, B) and LAMP1 protein expression (Supplementary Figures 3A, B) in the P. gingivalis treatment group, suggesting an elevation in lysosomal quantity due to P. gingivalis infection. These findings suggested that P. gingivalis is unlikely to reduce autophagy/mitophagy flux by promoting lysosomal efflux. Moreover, P. gingivalis exposure led to a decrease in the AO red/green ratio (Figures 4D, F), indicating increased lysosomal membrane permeability (LMP), which suggested that P. gingivalis may compromise lysosomal integrity. Additionally, the reduced LysoSensor fluorescence intensity suggested an elevated lysosomal pH value (Figures 4E, G). Given the observed lysosomal alkalization induced by P. gingivalis, it is necessary to further examine whether lysosomal hydrolases were affected, as hydrolase activity is responsible for waste disposal in an acidic environment. Our results revealed a reduction in the expression of mature cathepsin B (CTSB) and cathepsin D (CTSD) (Figure 4C), along with decreased acid phosphatase (ACP) activity (Figure 4H). Taking together, these findings indicated that P. gingivalis induces lysosomal dysfunction in endothelial cells, as evidenced by increased LMP, disruption of lysosomal acidity, and impaired degradative capacity.
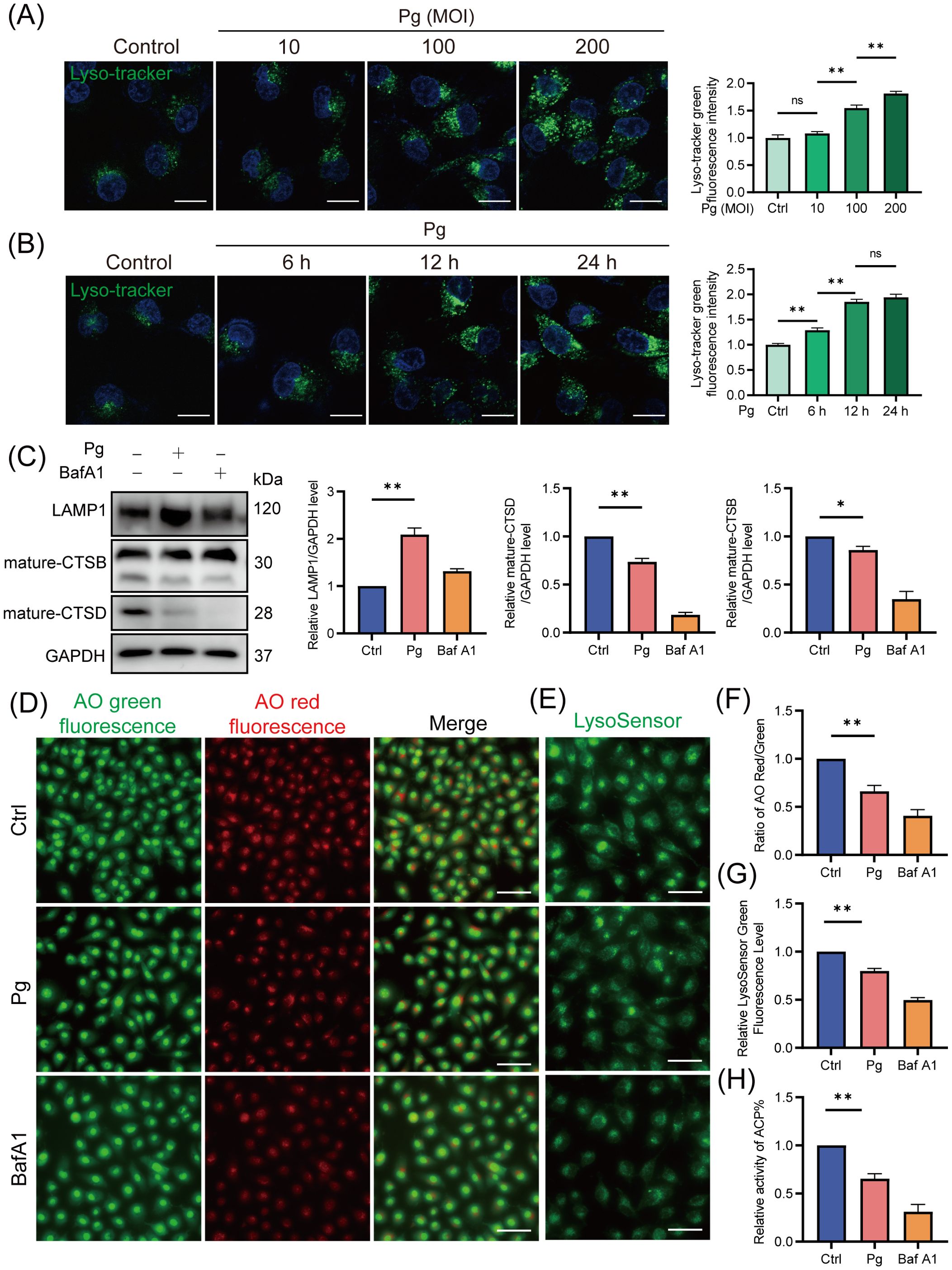
Figure 4. Porphyromonas gingivalis impaired lysosomal function in endothelial cells. HAECs were treated with Pg (MOI=100, 24 h) or not, and were pretreated with BafA1 for 2 h as a positive control. (A, B) Representative fluorescence images of Lyso-Tracker Green staining and the corresponding semi-quantification showed a dose- and time-dependent increase in lysosomal quantity in Pg-infected HAECs. Scale bar: 15 μm. (C) Representative western blot and quantification of mature-CTSB, mature-CTSD, and LAMP1 protein expression (n = 3 per group). (D) Representative fluorescence images of AO staining indicated the increase of LMP upon Pg stimulation. Scale bar: 200 μm. (E) Representative fluorescence images of LysoSensor green staining reflected changes in lysosomal pH value. Scale bar: 100 μm. (F) The relative LysoSensor green fluorescence intensity. (G) Ratio of AO red/green fluorescence intensity. (H) Acid phosphatase activity detection showed the activity of ACP. Data are represented from at least three independent experiments (mean ± SEM). *P < 0.05, and **P< 0.01, ns, not significant.
3.4 P. gingivalis initiated mitophagy and impaired lysosomal function to promote its intracellular survival
Given that certain bacteria can exploit host mitophagy to enhance their intracellular survival, we next examined whether P. gingivalis initiates mitophagy to support intracellular survival in endothelial cells. At 24 h post-infection, our results showed that treatment with the mitophagy inhibitor Mdivi-1 suppressed, while the mitophagy activator CCCP increased the intracellular titer of P. gingivalis in HAECs (Figure 5A). Furthermore, we analyzed the number of P. gingivalis in HAECs using a confocal laser scanning microscope (CLSM). The results showed that CCCP treatment enhanced, while Mdivi-1 treatment decreased, the number of SYTO9-stained P. gingivalis within cells. Besides, there was no significant difference in the number or intracellular titer of P. gingivalis at 2 h post-infection across the treatment groups, suggesting that neither CCCP nor Mdivi-1 affected bacterial invasion (Figures 5B, C). Moreover, HAECs were transfected with either Pink1 siRNA (20 nM) and negative control siRNA (20 nM) for 48 h, followed by P. gingivalis infection. We observed that PINK1 knockdown significantly reduced the intracellular survival of P. gingivalis (Supplementary Figures 4A–C; Figure 5D). These results indicated that P. gingivalis-initiated mitophagy promotes its intracellular survival.
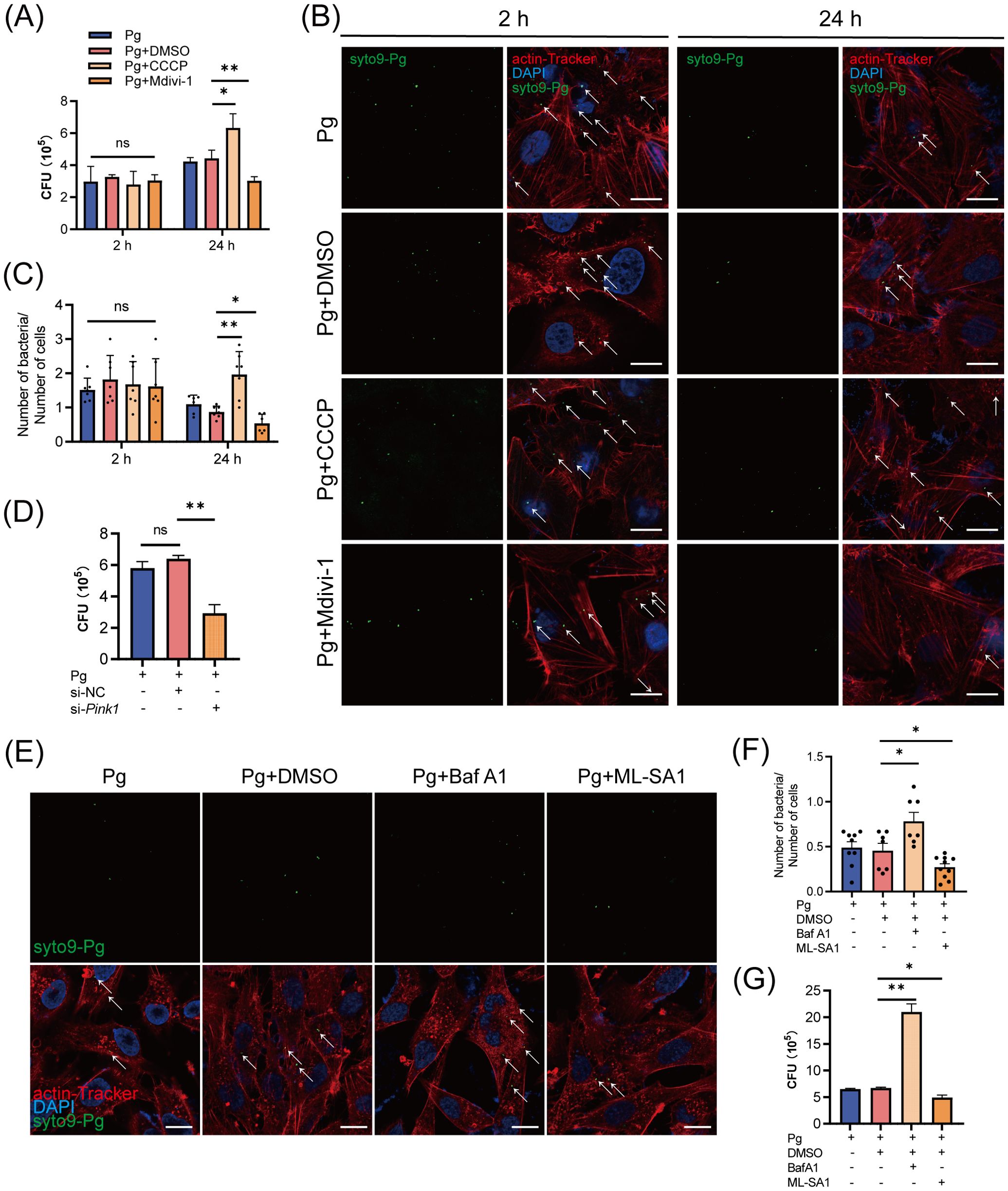
Figure 5. The initiation of mitophagy and lysosomal dysfunction increased the intracellular survival of Porphyromonas gingivalis in endothelial cells. (A) HAECs were treated with DMSO, CCCP (activation of mitophagy), or Mdivi-1 (inhibition of mitophagy), then infected with Pg (MOI = 100) for 2 h or 24 h. Pg intracellular survival was measured by antibiotic protection assays. (B) Representative fluorescence images of intracellular SYTO9-stained Pg load by CLSM. Scale bar: 15 μm. (C) The ratio of SYTO9-stained Pg to cell number. Cells in at least 6 fields were counted in each group. (D) HAECs were transfected with Pink1 siRNA (20 nM) or negative control siRNA (20 nM) for 48 h and then infected with Pg (MOI = 100). Intracellular Pg load was measured by antibiotic protection assays. (E) HAECs were treated with DMSO, BafA1 (inhibition of lysosomal function), or ML-SA1 (activation of lysosomal function), then infected with Pg (MOI = 100) for 24 h. Representative fluorescence images of intracellular SYTO9-stained Pg load by CLSM. Scale bar: 15 μm. (F) The ratio of SYTO9-stained Pg to cell number. Cells in at least 6 fields were counted in each group. (G) Pg intracellular survival was measured by antibiotic protection assays. Data are presented as the mean ± SEM. *P < 0.05, and **P< 0.01, ns, not significant.
To explore the role of lysosomal function in P. gingivalis survival, we further treated HAECs with BafA1 and the lysosomal agonist ML-SA1. ML-SA1 treatment suppressed while BafA1 treatment enhanced the intracellular titter and the number of SYTO9-stained P. gingivalis (Figures 5E–G). The results suggested that P. gingivalis impairs lysosomal function to prevent its xenophagic degradation, thereby promoting its intracellular survival.
3.5 P. gingivalis initiated mitophagy to suppress xenophagy in endothelial cells
Subsequently, we sought to understand the mechanisms by which mitophagy supports P. gingivalis survival. Mitophagy has been recognized to regulate bacterial intracellular survival by modulating mtROS production or influencing xenophagy (Zhang et al., 2019; Song et al., 2022). We observed that P. gingivalis infection increased the level of mtROS in an MOI- and time-dependent manner (Supplementary Figures 5A, B), with CCCP treatment further exacerbating the mtROS levels in HAECs (Figure 6A). The results suggested that P. gingivalis initiated mitophagy to promote its survival in HAECs, independently of the pathway involving mtROS generation.
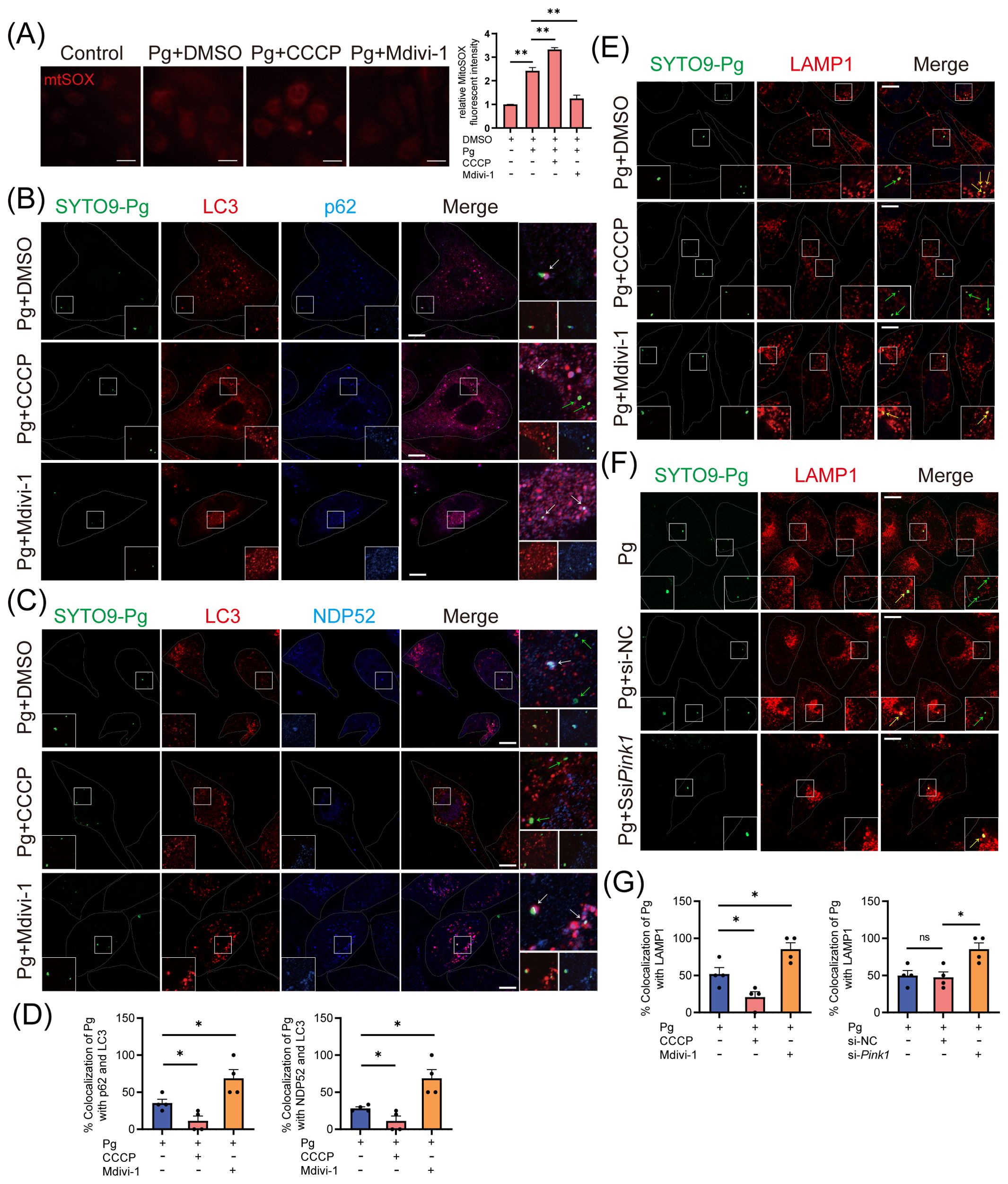
Figure 6. The initiation of mitophagy inhibits the host xenophagy, which targets Porphyromonas gingivalis to degrade. HAECs were treated with DMSO, CCCP (activation of mitophagy), or Mdivi-1 (inhibition of mitophagy), then infected with Pg (MOI = 100) for 24 h. (A) Representative fluorescence images and quantification of mitoSOX staining revealed the mitochondrial ROS production in each group. Scale bar: 65 μm. (B, C) Representative images and (D) quantification of LC3 (red)- Pg (green)- p62 (blue), LC3 (red)- Pg (green)- NPD52 (blue), and LAMP1 (red)- Pg (green) colocalization in DMSO, CCCP, or Mdivi-1 pretreated groups. Scale bar: 10 μm. (E) Representative images of SYTO9-stained Pg and LAMP1 colocalization in DMSO, CCCP, or Mdivi-1 pretreated groups. Scale bar: 10 μm. (F) HAECs were transfected with Pink1 siRNA (20 nM) or negative control siRNA (20 nM) for 48 h and then infected with Pg (MOI = 100). Representative images of SYTO9-stained Pg and LAMP1 colocalization. Scale bar: 10 μm. (G) The quantification of Pg and LAMP1 colocalization. Data are presented as the mean ± SEM. *P < 0.05, and **P< 0.01, ns, not significant.
Finally, we investigated how mitophagy initiation affects xenophagy during P. gingivalis infection. Our study examined LC3 (red fluorescence)- P. gingivalis (green fluorescence) and p62/NDP52 (blue fluorescence)- P. gingivalis colocalization to evaluate autophagosome formation engulfing P. gingivalis and cargo receptors’ involvement. CLSM analysis showed that Mdivi-1 enhanced, while CCCP inhibited, the colocalization between LC3 and p62/NDP52-localized P. gingivalis in HAECs (Figures 6B–D, F), suggesting that the recruitment of p62 or NDP52 to P. gingivalis and xenophagosome formation were inhibited due to the initiation of mitophagy. Next, CLSM analysis revealed that the colocalization of LAMP1 with P. gingivalis could be promoted by Mdivi-1 and suppressed by CCCP (Figures 6E, G), indicating that mitophagy initiation inhibited the xenophagic degradation of P. gingivalis. To further strengthen this result, we observed the colocalization of LAMP1 with P. gingivalis in HAECs transfected with Pink1 siRNA. The observation showed that PINK1 knockdown increased the colocalization of LAMP1 and P. gingivalis (Figures 6F, G). These results suggested that P. gingivalis initiates PINK1-mediated mitophagy to inhibit the formation of xenophagosomes, thereby enhancing its intracellular survival.
4 Discussion
Direct infection of P. gingivalis on endothelial cells has been linked to atherosclerosis. In this study, we found that P. gingivalis inhibited the formation of bacteria-containing xenophagosomes by initiating PINK1-Parkin-mediated mitophagy while simultaneously suppressing xenophagosome-lysosome fusion through lysosomal dysfunction. These findings contribute to our understanding of how P. gingivalis hijacks host mitophagy and lysosome function, shedding light on its role in pathogenesis and intracellular survival (Figure 7).
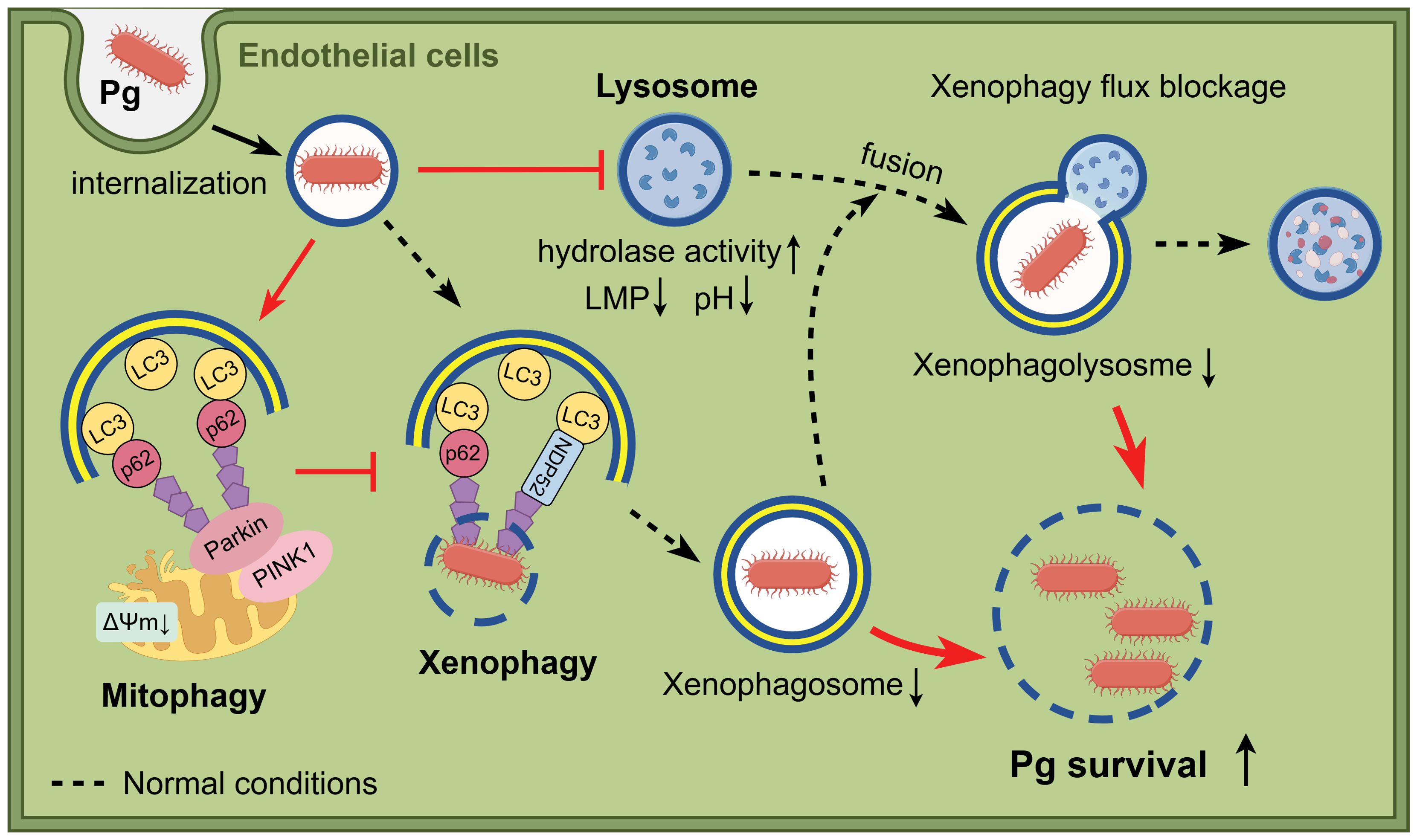
Figure 7. Schematic illustration of the study. When bacteria invade endothelial cells, P. gingivalis inhibits the formation of bacteria-containing xenophagosomes by initiating PINK1-Parkin-mediated mitophagy and simultaneously suppresses xenophagosome-lysosome fusion through lysosomal dysfunction, which results in enhanced intracellular survival. Figure created with cartoon components by Figdraw [www.figdraw.com].
During bacterial invasion, autophagy serves as an innate immune response to restrict bacterial growth, while bacteria have evolved strategies to combat autophagy (Huang and Brumell, 2014; Sudhakar et al., 2019). Upon internalization, P. gingivalis initially localizes within a phagosome in host cells (Yamatake et al., 2007). Some P. gingivalis then damage the phagosome membrane, exposing themselves to the host cytosol, where they are targeted for degradation by xenophagy (selective autophagy) (Mostowy, 2013; Lee et al., 2018). However, others evade degradation by trafficking to replicative vacuoles through non-selective autophagy, thereby promoting intracellular survival (Dorn et al., 2001; Bélanger et al., 2006). Additionally, P. gingivalis may manipulate host-selective autophagy pathways to support survival (Shiheido-Watanabe et al., 2023).
Mitophagy, a selective form of autophagy that eliminates damaged mitochondria, can also be exploited by intracellular pathogens to evade host defenses (Zhang et al., 2019; Song et al., 2022). However, whether P. gingivalis exploits mitophagy to survive within endothelial cells remains unclear. Previous studies have shown that P. gingivalis inhibits mitophagy in oral squamous cell carcinoma cells and bone marrow-derived macrophages (Jiang et al., 2023; Sheridan et al., 2024). In contrast, our findings indicated that P. gingivalis initiates mitophagy at an early stage, leading to increased mitophagosome formation. However, it inhibits mitophagosome-lysosome fusion and the final degradation stage, likely due to lysosomal dysfunction in endothelial cells. Interestingly, we found that the differential effects of P. gingivalis on early and late stages of mitophagy/autophagy contribute to its survival within endothelial cells through distinct mechanisms. Consistent with the role of mitophagy in promoting bacterial intracellular survival in previous studies, we confirmed that P. gingivalis initiates mitophagy to enhance its survival in endothelial cells, although it only triggers the early stage of the mitophagy process.
Mitophagy is well-documented for promoting bacterial survival by reducing mtROS production (Zhang et al., 2019; Gao et al., 2024; Nan et al., 2024). In our study, while P. gingivalis initiated mitophagy in HAECs, the mitophagy flux was ultimately blocked. As a result, damaged mitochondria accumulated, leading to increased mtROS levels, which couldn’t be reversed by the mitophagy activator. This suggested that P. gingivalis may not promote its survival by regulating mtROS levels in endothelial cells. Other bacteria can manipulate mitophagy to favor persistent infection through another strategy. Xenophagy, a form of selective autophagy closely linked to bacterial survival, shares several key molecules with mitophagy, including autophagy receptors like p62 and NDP52, and the E3 ubiquitin ligases like Parkin. This overlap implies potential crosstalk between mitophagy and xenophagy (Rubio-Tomás et al., 2023). A recent study demonstrated that M. bovis induces mitophagy to suppress host xenophagy, thereby facilitating its persistence (Vargas et al., 2023) Likewise, our findings revealed that P. gingivalis inhibited xenophagy by initiating mitophagy in HAECs. When mitophagy was inhibited using Mdivi-1, we observed increased recruitment of p62 and NDP52 to P. gingivalis, as indicated by enhanced colocalization. These receptors were directly bound to LC3, enabling the delivery of P. gingivalis to xenophagosomes and promoting its clearance when mitophagy was suppressed.
Lysosomes play a crucial role in the fusion and degradation of both mitophagosomes and xenophagosomes, which determine whether damaged mitochondrions are cleared and whether bacteria survive (Miao et al., 2015; Hu et al., 2019; Naskar et al., 2023). A study has shown that P. gingivalis blocks autophagosome-lysosome fusion by cleaving VAMP8 (a protein located on lysosomes) through its gingipains and by promoting lysosomal efflux (Shiheido-Watanabe et al., 2023; Liu et al., 2023). In contrast, we assessed lysosomal function and found that P. gingivalis induced lysosomal dysfunction. This dysfunction was characterized by increased lysosomal number, lysosomal membrane permeabilization, disruption of the lysosomal acidic environment, and reduced enzymatic activity (CTSB, CTSD, and ACP). Interestingly, the increased lysosomal number may reflect a compensatory response to these lysosomal function defects (Zhang et al., 2021). Additionally, we confirmed that activating lysosome function reduced the survival of P. gingivalis in HAECs, while inhibition of lysosomal function had the opposite effect, emphasizing lysosomal dysfunction as a potential mechanism by which P. gingivalis promotes its intracellular survival in endothelial cells.
Our study demonstrated that P. gingivalis initiates mitophagy and impairs lysosomal function in HAECs, inhibiting xenophagosome formation and its fusion with lysosomes. Both processes contribute to the intracellular survival of the bacterium. However, several limitations of our study should be noted. The specific virulence factors and mechanisms by which P. gingivalis initiates mitophagy to suppress xenophagy are yet to be identified, and the relevance of these mechanisms in vivo is unclear. Additionally, our analysis focused on a single strain of P. gingivalis, necessitating further studies to investigate strain-specific variations.
In conclusion, this study enhances our understanding of how P. gingivalis interacts with endothelial cells, particularly its manipulation of mitophagy and lysosomal dysfunction to promote survival. Our findings provide new insights for understanding the intracellular survival mechanism of P. gingivalis in endothelial cells.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics statement
Ethical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used.
Author contributions
CZ: Investigation, Methodology, Formal Analysis, Data curation, Writing – review & editing, Writing – original draft. JH: Writing – review & editing, Methodology, Formal Analysis, Writing – original draft, Investigation, Data curation. SX: Data curation, Formal Analysis, Methodology, Writing – review & editing, Investigation, Writing – original draft. BL: Writing – review & editing, Data curation, Investigation, Formal Analysis. HQ: Investigation, Formal Analysis, Writing – review & editing, Data curation. TC: Data curation, Formal Analysis, Investigation, Writing – review & editing. YH: Writing – review & editing, Investigation, Formal Analysis. LH: Writing – review & editing, Investigation, Formal Analysis. XF: Investigation, Formal Analysis, Writing – review & editing. F-MC: Writing – review & editing, Conceptualization, Supervision, Project administration, Methodology. YW: Methodology, Project administration, Supervision, Conceptualization, Writing – review & editing. HD: Methodology, Writing – review & editing, Supervision, Conceptualization, Writing – original draft, Project administration.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by grants from the National Natural Science Foundation of China (No.82470990 & 82101029), Zhejiang Provincial Medical and Health Science and Technology Plan (No.WKJ-ZJ-2544 & No. 2025KY107), Natural Science Foundation of Zhejiang Province (No. ZCLY24H1401), and Wenzhou Municipal Science and Technology Bureau (No. Y2023085).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2025.1613366/full#supplementary-material
References
Ballabio, A. and Bonifacino, J. S. (2020). Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 21, 101–118. doi: 10.1038/s41580-019-0185-4
Bélanger, M., Rodrigues, P. H., Dunn, W. A., Jr., and Progulske-Fox, A. (2006). Autophagy: A highway for porphyromonas gingivalis in endothelial cells. Autophagy 2, 165–170. doi: 10.4161/auto.2828
Brodala, N., Merricks, E. P., Bellinger, D. A., Damrongsri, D., Offenbacher, S., Beck, J., et al. (2005). Porphyromonas gingivalis bacteremia induces coronary and aortic atherosclerosis in normocholesterolemic and hypercholesterolemic pigs. Arterioscler. Thromb. Vasc. Biol. 25, 1446–1451. doi: 10.1161/01.ATV.0000167525.69400.9c
Dikic, I. and Elazar, Z. (2018). Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 19, 349–364. doi: 10.1038/s41580-018-0003-4
Dorn, B. R., Dunn, W. A., and Progulske-Fox, A. (2001). Porphyromonas gingivalis traffics to autophagosomes in human coronary artery endothelial cells. Infect. Immun. 69, 5698–5708. doi: 10.1128/IAI.69.9.5698-5708.2001
Forner, L., Larsen, T., Kilian, M., and Holmstrup, P. (2006). Incidence of bacteremia after chewing, tooth brushing and scaling in individuals with periodontal inflammation. J. Clin. Periodontol. 33, 401–407. doi: 10.1111/j.1600-051X.2006.00924.x
Gao, S., Gao, L., Yuan, D., Lin, X., and van der Veen, S. (2024). Gonococcal OMV-delivered PorB induces epithelial cell mitophagy. Nat. Commun. 15, 1669. doi: 10.1038/s41467-024-45961-1
Gimbrone, M. A. and García-Cardeña, G. (2016). Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ. Res. 118, 620–636. doi: 10.1161/CIRCRESAHA.115.306301
Haraszthy, V. I., Zambon, J. J., Trevisan, M., Zeid, M., and Genco, R. J. (2000). Identification of periodontal pathogens in atheromatous plaques. J. Periodontol. 71, 1554–1560. doi: 10.1902/jop.2000.71.10.1554
Hu, W., Zhang, L., Li, M. X., Shen, J., Liu, X. D., Xiao, Z. G., et al. (2019). Vitamin D3 activates the autolysosomal degradation function against Helicobacter pylori through the PDIA3 receptor in gastric epithelial cells. Autophagy 15, 707–725. doi: 10.1080/15548627.2018.1557835
Huang, J. and Brumell, J. H. (2014). Bacteria-autophagy interplay: a battle for survival. Nat. Rev. Microbiol. 12, 101–114. doi: 10.1038/nrmicro3160
Jiang, K., Li, J., Jiang, L., Li, H., and Lei, L. (2023). PINK1-mediated mitophagy reduced inflammatory responses to Porphyromonas gingivalis in macrophages. Oral. Dis. 29, 3665–3676. doi: 10.1111/odi.14286
Klionsky, D. J., Abdel-Aziz, A. K., Abdelfatah, S., Abdellatif, M., Abdoli, A., Abel, S., et al. (2021). Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)1. Autophagy 17, 1–382. doi: 10.1080/15548627.2020.1797280
Lazarou, M., Sliter, D. A., Kane, L. A., Sarraf, S. A., Wang, C., Burman, J. L., et al. (2015). The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. doi: 10.1038/nature14893
Lee, K., Roberts, J. S., Choi, C. H., Atanasova, K. R., and Yilmaz, Ö. (2018). Porphyromonas gingivalis traffics into endoplasmic reticulum-rich-autophagosomes for successful survival in human gingival epithelial cells. Virulence 9, 845–859. doi: 10.1080/21505594.2018.1454171
Li, L., Messas, E., Batista, E. L., Levine, R. A., and Amar, S. (2002). Identification of periodontal pathogens in atheromatous plaques. Circulation 105, 861–867. doi: 10.1161/hc0702.104178
Liu, M., Shao, J., Zhao, Y., Ma, B., and Ge, S. (2023). Porphyromonas gingivalis evades immune clearance by regulating lysosome efflux. J. Dent. Res. 102(5):555–64. doi: 10.1177/00220345221146097
Miao, Y., Li, G., Zhang, X., Xu, H., and Abraham, S. N. (2015). A TRP channel senses lysosome neutralization by pathogens to trigger their expulsion. Cell 161, 1306–1319. doi: 10.1016/j.cell.2015.05.009
Mizushima, N. and Komatsu, M. (2011). Autophagy: renovation of cells and tissues. Cell 147, 728–741. doi: 10.1016/j.cell.2011.10.026
Mostowy, S. (2013). Autophagy and bacterial clearance: a not so clear picture. Cell. Microbiol. 15, 395–402. doi: 10.1111/cmi.12063
Nan, D., Rao, C., Tang, Z., Yang, W., Wu, P., Chen, J., et al. (2024). Burkholderia pseudomallei BipD modulates host mitophagy to evade killing. Nat. Commun. 15, 4740. doi: 10.1038/s41467-024-48824-x
Naskar, M., Parekh, V. P., Abraham, M. A., Alibasic, Z., Kim, M. J., Suk, G., et al. (2023). α-Hemolysin promotes uropathogenic E. coli persistence in bladder epithelial cells via abrogating bacteria-harboring lysosome acidification. PloS Pathog. 19, e1011388. doi: 10.1371/journal.ppat.1011388
Pickles, S., Vigié, P., and Youle, R. J. (2018). Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. CB 28, R170–R185. doi: 10.1016/j.cub.2018.01.004
Rubio-Tomás, T., Sotiriou, A., and Tavernarakis, N. (2023). The interplay between selective types of (macro)autophagy: Mitophagy and xenophagy. Int. Rev. Cell Mol. Biol. 374, 129–157. doi: 10.1016/bs.ircmb.2022.10.003
Settembre, C., Fraldi, A., Medina, D. L., and Ballabio, A. (2013). Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 14, 283–296. doi: 10.1038/nrm3565
Sheridan, M., Chowdhury, N., Wellslager, B., Oleinik, N., Kassir, M. F., Lee, H. G., et al. (2024). Opportunistic pathogen Porphyromonas gingivalis targets the LC3B-ceramide complex and mediates lethal mitophagy resistance in oral tumors. iScience 27, 109860. doi: 10.1016/j.isci.2024.109860
Shiheido-Watanabe, Y., Maejima, Y., Nakagama, S., Fan, Q., Tamura, N., and Sasano, T. (2023). Porphyromonas gingivalis, a periodontal pathogen, impairs post-infarcted myocardium by inhibiting autophagosome-lysosome fusion. Int. J. Oral. Sci. 15, 42. doi: 10.1038/s41368-023-00251-2
Song, Y., Ge, X., Chen, Y., Hussain, T., Liang, Z., Dong, Y., et al. (2022). Mycobacterium bovis induces mitophagy to suppress host xenophagy for its intracellular survival. Autophagy 18, 1401–1415. doi: 10.1080/15548627.2021.1987671
Stolz, A., Ernst, A., and Dikic, I. (2014). Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 16, 495–501. doi: 10.1038/ncb2979
Sudhakar, P., Jacomin, A.-C., Hautefort, I., Samavedam, S., Fatemian, K., Ari, E., et al. (2019). Targeted interplay between bacterial pathogens and host autophagy. Autophagy 15, 1620–1633. doi: 10.1080/15548627.2019.1590519
Vargas, J. N. S., Hamasaki, M., Kawabata, T., Youle, R. J., and Yoshimori, T. (2023). The mechanisms and roles of selective autophagy in mammals. Nat. Rev. Mol. Cell Biol. 24, 167–185. doi: 10.1038/s41580-022-00542-2
Wang, S., Long, H., Hou, L., Feng, B., Ma, Z., Wu, Y., et al. (2023). The mitophagy pathway and its implications in human diseases. Signal Transduction Targeting Ther. 8, 304. doi: 10.1038/s41392-023-01503-7
Yamatake, K., Maeda, M., Kadowaki, T., Takii, R., Tsukuba, T., Ueno, T., et al. (2007). Role for gingipains in Porphyromonas gingivalis traffic to phagolysosomes and survival in human aortic endothelial cells. Infect. Immun. 75, 2090–2100. doi: 10.1128/IAI.01013-06
Zhang, X., Wei, M., Fan, J., Yan, W., Zha, X., Song, H., et al. (2021). Ischemia-induced upregulation of autophagy preludes dysfunctional lysosomal storage and associated synaptic impairments in neurons. Autophagy 17, 1519–1542. doi: 10.1080/15548627.2020.1840796
Zhang, Y., Yao, Y., Qiu, X., Wang, G., Hu, Z., Chen, S., et al. (2019). Listeria hijacks host mitophagy through a novel mitophagy receptor to evade killing. Nat. Immunol. 20, 433–446. doi: 10.1038/s41590-019-0324-2
Keywords: Porphyromonas gingivalis, endothelial cells, mitophagy, lysosomal function, xenophagy
Citation: Zheng C, Huang J, Xu S, Lu B, Que H, Chen T, Hou Y, He L, Fan X, Chen F-M, Wang Y and Deng H (2025) Porphyromonas gingivalis hijacks mitophagy and lysosomal function to persist in endothelial cells. Front. Cell. Infect. Microbiol. 15:1613366. doi: 10.3389/fcimb.2025.1613366
Received: 17 April 2025; Accepted: 21 July 2025;
Published: 08 August 2025.
Edited by:
Chun-Teh Lee, University of Texas Health Science Center at Houston, United StatesReviewed by:
Dongmei Zhang, China Medical University, ChinaXiaolin Tang, China Medical University, China
Copyright © 2025 Zheng, Huang, Xu, Lu, Que, Chen, Hou, He, Fan, Chen, Wang and Deng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Deng, aHVpZGVuZ0B3bXUuZWR1LmNu; ZGgwNzI2QDE2My5jb20=; Yi Wang, eS53YW5nQHdtdS5lZHUuY24=; Fa-Ming Chen, Y2Ztc3VuaGhAZm1tdS5lZHUuY24=
†These authors have contributed equally to this work