 Ikram Khan
Ikram Khan Muhammad Irfan2
Muhammad Irfan2 Xiaodong Xie
Xiaodong Xie Zhiqiang Li
Zhiqiang Li- 1Department of Genetics, School of Basic Medical Sciences, Lanzhou University, Lanzhou, Gansu, China
- 2Department of Medical Laboratory Technology, Xcito School of Nursing and Allied Health Sciences, Chakdara, Khyber Pakhtunkhwa, Pakistan
- 3Department of Microecology, School of Basic Medical Sciences, Dalian Medical University, Dalian, Liaoning, China
- 4Department of Physiology, College of Basic Medical Sciences, Dalian Medical University, Dalian, Liaoning, China
- 5School of Stomatology, Key Laboratory of Oral Disease, Northwest Minzu University, Lanzhou, Gansu, China
The human-associated microbiome, encompassing diverse microbial communities across body sites, plays a pivotal role in maintaining host homeostasis. Disruption of this balance, termed dysbiosis, has been implicated in a spectrum of pathophysiological conditions. Traditionally, blood was considered a sterile microenvironment. However, emerging insights into the blood microbiome challenge the paradigm of blood sterility, revealing microbial signatures, including cell-free DNA and viable taxa, with putative implications for host physiology and disease. The blood taxonomic profile at the phylum level is dominated by Proteobacteria, with Bacteroidetes, Actinobacteria, and Firmicutes following in abundance. Dysbiosis in blood microbiome composition may indicate or contribute to systemic dysregulation, pointing to its potential role in disease etiology. These findings highlight the blood microbiome as a possible driver in the pathogenesis of infectious and non-infectious diseases, neurodegenerative disorders, and immune-mediated conditions. The detection of specific microbial profiles in circulation holds promise for biomarker discovery, enhancing disease stratification, and informing precision therapeutic strategies. However, advancing this field requires overcoming methodological challenges, including contamination control, standardization, and reproducibility. This review aims to present blood microbiome biomarkers across infectious, non-infectious, neurodegenerative, and immune-mediated diseases, while critically examining methodological variations, controversies, limitations, and future research directions. Elucidating these factors is critical to advancing blood microbiome biomarker validation and therapeutic targeting, thereby refining mechanistic insights into systemic disease pathogenesis.
Introduction
The human microbiome, an intricate assemblage of microorganisms residing within and on the human body, constitutes a dynamic ecosystem that profoundly influences human health. The composition and diversity of this microbiome have been linked to numerous physiological processes and disease states (Heintz-Buschart and Wilmes, 2018). Dysbiosis, characterized by an imbalance or perturbation in the microbiome’s equilibrium, has garnered attention for its potential implications in a wide array of health disorders (Shukla et al., 2024). Dysbiosis in the gut microbiome plays a potential role in both human health and disease states (Chen et al., 2021); however, the association between gut microbiome and human diseases is overlooked (Velmurugan et al., 2020). Beyond gut microbiome dysbiosis, recent metagenomic analyses have renewed interest in the long-standing hypothesis of a blood-resident microbiome, underscoring its potential role in disease pathophysiology (Tedeschi et al., 1969; Khan et al., 2022a). These developments compel further scrutiny into the nature and significance of microbes in the bloodstream.
The circulation is a closed system, and the blood in healthy individuals was earlier believed to represent a sterile environment, which is the basis for safe blood transfusions (Damgaard et al., 2015). However, recent studies have challenged this concept, revealing the presence of resident microbiomes in both healthy individuals and those with diseases (Castillo et al., 2019; Whittle et al., 2019; Velmurugan et al., 2020). These studies confirmed the presence of live bacteria, bacterial DNA (and associated metabolites), viral DNA (e.g., Rhabdoviridae and Anelloviridae), archaeal DNA (e.g., Euryarchaeota), and fungi (e.g., Basidiomycota, Ascomycota) in the blood (Panaiotov et al., 2018). While these findings support the idea of a circulating microbiome, debate persists about whether these microbes represent a stable, endogenous community or are transient migrants from colonized body sites (Jagare et al., 2023). For example, a recent large-scale study reported no consistent core blood microbiome, reinforcing the hypothesis of peripheral origin through translocation (Tan et al., 2023). These conflicting results underscore the complexity and need for standardization in blood microbiome research.
Nonetheless, a mountain of evidence suggests that the blood microbiome plays a crucial role in the development of various human diseases, including diabetes mellitus (Qiu et al., 2019), allergies (Funkhouser and Bordenstein, 2013), asthma (Lee et al., 2020), irritable bowel syndrome (Jagare et al., 2023), cardiovascular diseases (CVDs) (Amar et al., 2019; Khan et al., 2022b, 2022), and cancer (Poore et al., 2020; Søby et al., 2020; Yang et al., 2021). The blood microbiomes have also been detected in animals such as cats (Vientoos-Plotts et al., 2017), dogs (Scarsella et al., 2020, 2023), cows (Scarsella et al., 2021), pigs (Hyun et al., 2021), goats (Tilahun et al., 2022), and camels (Mohamed et al., 2021) in both healthy and diseased states. However, it remains unclear whether blood constitutes a stable ecological niche for bacteria with functional roles in human physiology or merely serves as a transient conduit for microbial migration between colonized sites. The most likely source of blood-associated microbes is translocation from microbe-rich environments, particularly the gastrointestinal tract and oral cavity, often triggered by mucosal injury (e.g., tooth brushing) or increased intestinal permeability (Castillo et al., 2019; Velmurugan et al., 2020). These observations lay the foundation for examining how blood microbiome signatures intersect with systemic disease processes.
Thus, this review examines the emerging concept of the blood microbiome and its potential involvement in the pathogenesis of systemic diseases. Drawing on recent findings that challenge the long-standing notion of blood sterility, we explore microbial signatures, both cell-free and viable, detected in circulation and their associations with infectious, non-infectious, neurodegenerative, and immune-mediated conditions. We emphasize the diagnostic and prognostic promise of blood microbiome profiles while addressing critical methodological pitfalls, including contamination risks and lack of standardization in low-biomass microbiome studies. By identifying key controversies and outlining future research priorities, this review aims to advance the clinical and mechanistic understanding of the blood microbiome in systemic disease.
Search strategy and eligibility criteria
We performed a systematic search of English-language literature via PubMed, Web of Science, and Google Scholar using MeSH terms and keywords including blood bacteria, blood microbiome, blood microbiota, circulating bacteria, circulating microbiome, circulating microbiota, bacteremia, and transient bacteremia. The search targeted cohort studies employing blood, plasma, or serum microbiome analyses using methods such as 16S rRNA sequencing, high-throughput RNA sequencing, Illumina MiSeq, shotgun metagenomic sequencing of cell-free DNA, and pyrosequencing. Eligible studies were observational (cohort, case-control, or retrospective) in patients with infectious, noninfectious, neurodegenerative, or immune-mediated conditions, analyzing blood-derived samples. Exclusion criteria included animal studies, intervention trials involving prebiotics or probiotics, studies assessing skin or gut microbiota via blood, those without comparative data on systemic diseases, and studies focusing on cardiometabolic or unrelated conditions.
Blood microbiome
The human microbiome is a diverse and dynamic community of microorganisms, including bacteria, viruses, fungi, and archaea, that inhabit various sites within the body. These microbes form a complex ecosystem, interacting closely and symbiotically with the human host. Understanding the composition and dynamics of the blood microbiome is essential for uncovering its specific impact on human health and disease [17–20], as maintaining balance within these microbial communities is critical for overall host function and resilience. Table 1 summarizes the key components of the microbiome, highlighting the unique characteristics and predominant taxa of the blood microbial community.
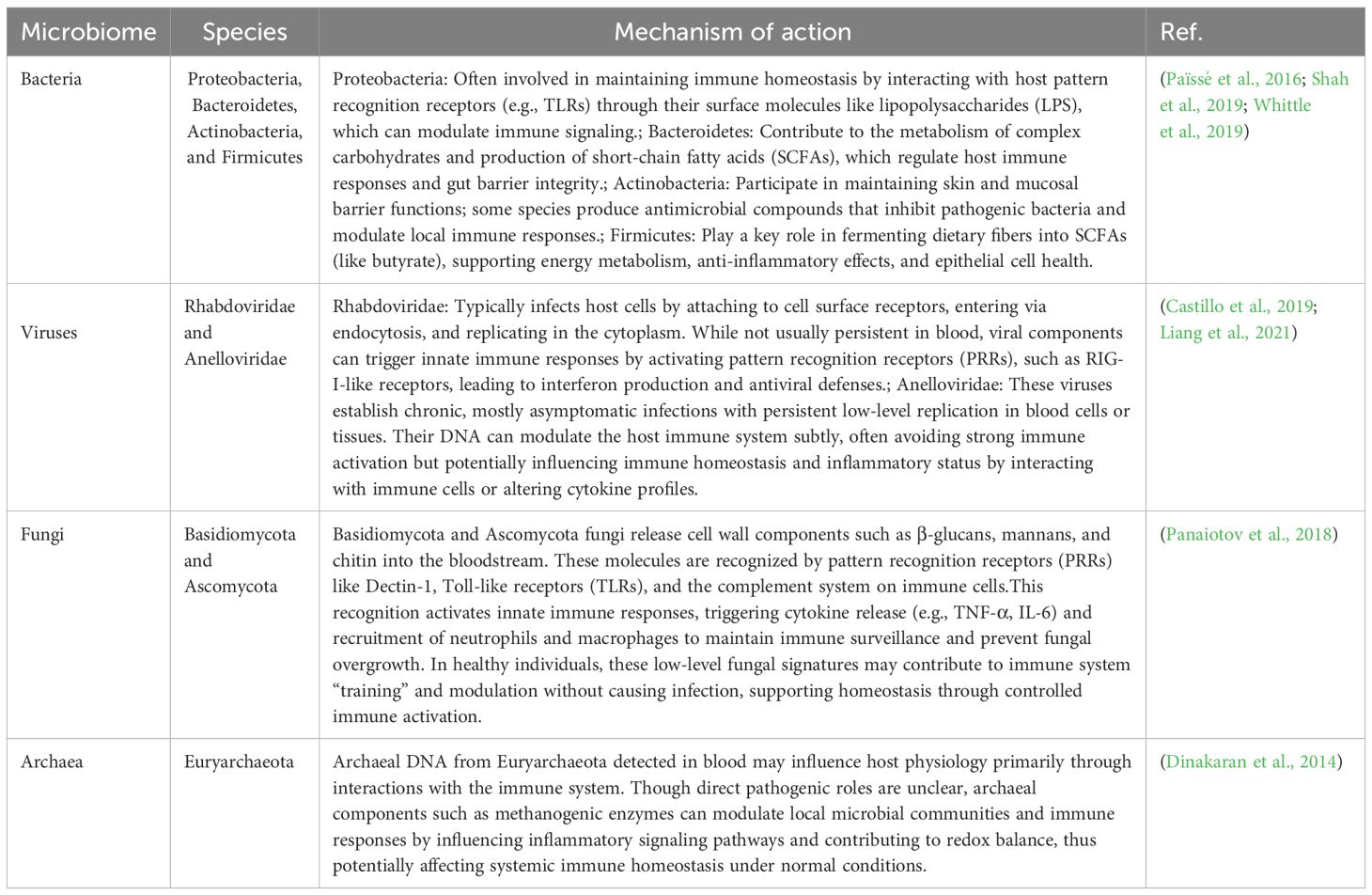
Table 1. Key blood microbial taxa and their possible roles in immune modulation, metabolism, and host homeostasis under normal conditions.
Alterations in the blood microbiome in systemic diseases
Although the presence of a blood microbiome was first noted over five decades ago, it has gained significant scientific interest since 2001 (Nikkari et al., 2001), with mounting evidence connecting it to various human diseases. Recent research has increasingly focused on the complex dynamics of the blood microbiome and its potential role in disease pathogenesis. This section synthesizes current knowledge on the composition and diversity of the blood microbiome across four key categories: infectious diseases, non-infectious diseases, neurological disorders, and immune-mediated conditions (Table 2).
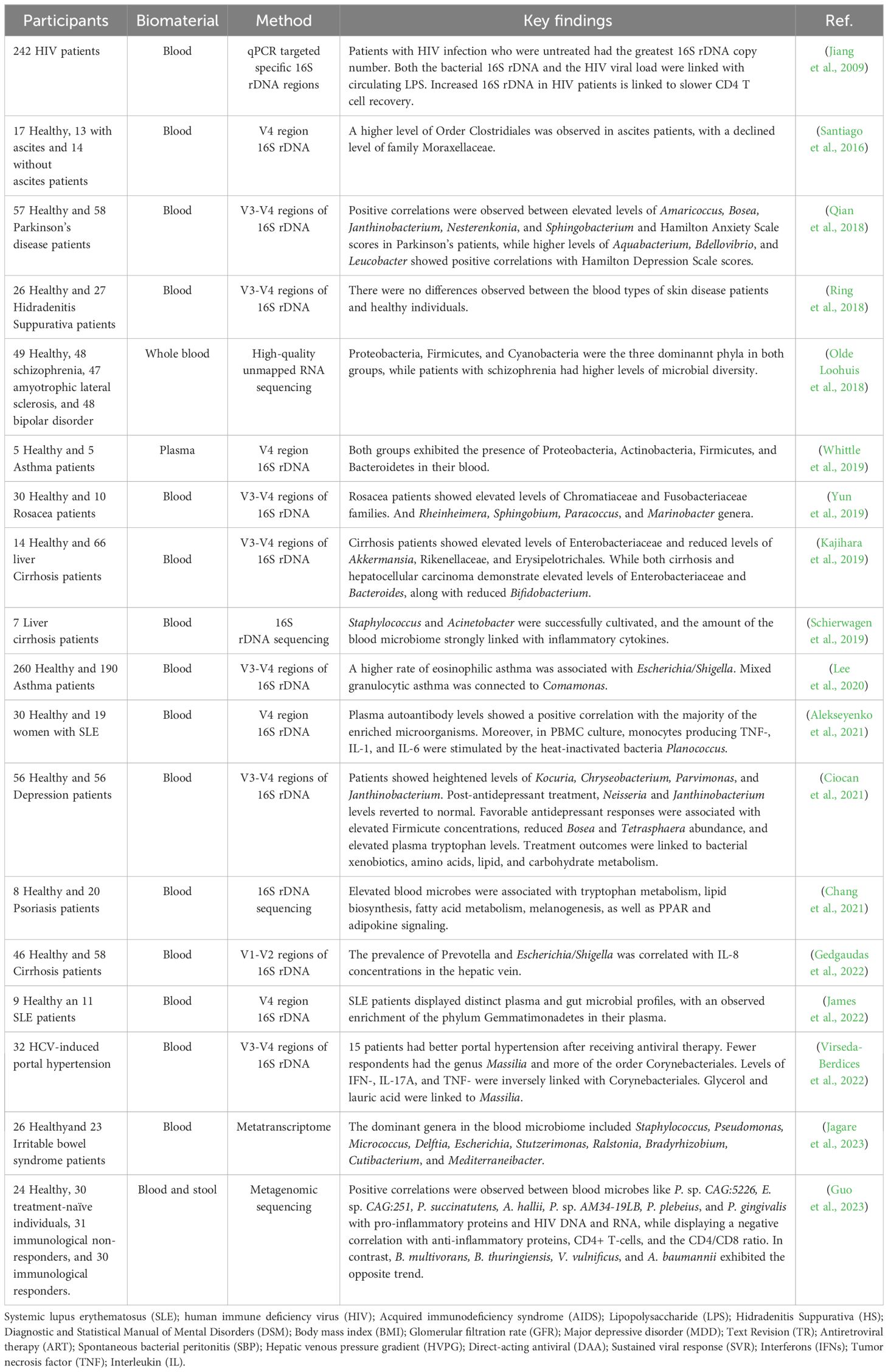
Table 2. Blood microbiome composition and diversity across infectious and non-infectious diseases, neurodegenerative disorders, and immune-mediated conditions.
Alterations in the blood microbiome in infectious diseases
Despite advancements in molecular biology, genetics, computation, and medicinal chemistry, infectious diseases remain a major and persistent threat to public health. Addressing the challenges of pathogen outbreaks, pandemics, and antimicrobial resistance requires collaborative, interdisciplinary efforts. Integrating systems and synthetic biology with blood microbiome research can accelerate progress in understanding human health and disease.
Recent studies observed alterations in the blood microbiome among patients with Human Immunodeficiency Virus (HIV) infection (Luo et al., 2019; Ancona et al., 2021). Libertucci et al. observed increased levels of Proteobacteria and decreased levels of Actinobacteria and Firmicutes phyla in the blood of HIV-positive individuals. They also found that elevated levels of Staphylococcaceae could alter the blood microbiome due to combination antiviral therapy (cART) (Libertucci and Young, 2019). This is clinically relevant, as cART-treated individuals may develop autoreactive B-cells and autoantibodies, suggesting a potential link between Staphylococcus and autoimmune manifestations in HIV. Blood microbiome disturbances may arise from gut bacterial translocation triggered by mucosal immune dysfunction and consequent epithelial barrier damage. Despite the effectiveness of cART, which may include treatments like non-nucleoside reverse transcriptase inhibitors or protease inhibitors, the compromise of gut epithelial barriers may persist in individuals with HIV infection (Lee et al., 2020). Nevertheless, these treatments may still contribute to ongoing gut bacterial translocation and sustained damage to the gut barrier (Søby et al., 2020). Luo et al. identified the presence of Massilia and Haemophilus in the blood of HIV patients undergoing effective cART. This finding suggests that these microbes may trigger the release of proinflammatory cytokines in the peripheral blood microbiome, potentially contributing to the progression of chronic systemic inflammation over time (Alekseyenko et al., 2021). A recent study by Guo et al. identified a specific blood microbiome signature, including P. sp. CAG:5226, E. sp. CAG:251, P. succinates, A. hallii, P. sp. AM34-19LB, P. plebeius, and P. gingivalis, which exhibited positive correlations with pro-inflammatory proteins, HIV DNA, and RNA. In contrast, these microbes showed negative correlations with anti-inflammatory proteins, CD4+ T-cells, and the CD4/CD8 ratio. Additionally, microbes such as B. multivorans, B. thuringiensis, V. vulnificus, and A. baumannii demonstrated an opposing pattern, suggesting the potential for identifying effective microbial and immunotherapeutic strategies for managing HIV infection (Guo et al., 2023). These findings imply that antiretroviral therapy may impair intestinal barrier integrity, while HIV infection itself could modulate the blood microbiome.
Evidence from the literature indicates that blood dysbiosis in septic patients is primarily associated with an overrepresentation of Proteobacteria or Bacteroidetes, while Actinobacteria are less commonly present. However, higher levels of Agrococcus within the Actinobacteria phylum have been suggested as a potential factor in the onset of sepsis (Païssé et al., 2016). Gosiewski et al. identified increased Proteobacteria and Bifidobacteriales in post-surgical sepsis patients, while Actinobacteria levels declined compared to healthy people (Gosiewski and Huminska, 2017). Several studies on lung diseases with suspected infections have identified significantly elevated levels of five bacterial genera in the blood microbiome, namely Veillonella, Prevotella, Cutibacterium, Corynebacterium, and Streptococcus, in patients with sarcoidosis (Hodzhev, 2023; Hodzhev et al., 2023). Investigating the blood microbiome in granulomatous lung diseases like sarcoidosis could provide insights into their origins and pathogenesis. The severity of COVID-19 has been associated with increased abundances of E. coli, Bacillus sp., Campylobacter hominis, Pseudomonas sp., Thermoanaerobacter pseudethanolicus, Thermoanaerobacterium thermosaccharolyticum, and Staphylococcus epidermidis (Dereschuk et al., 2021). These bacteria show that inflammation and the adaptive immune system are overactive (Figure 1). The findings provide insights into the blood microbiome profiles of smokers and COVID-19 patients, while also presenting a novel framework for investigating host–microbe interactions.
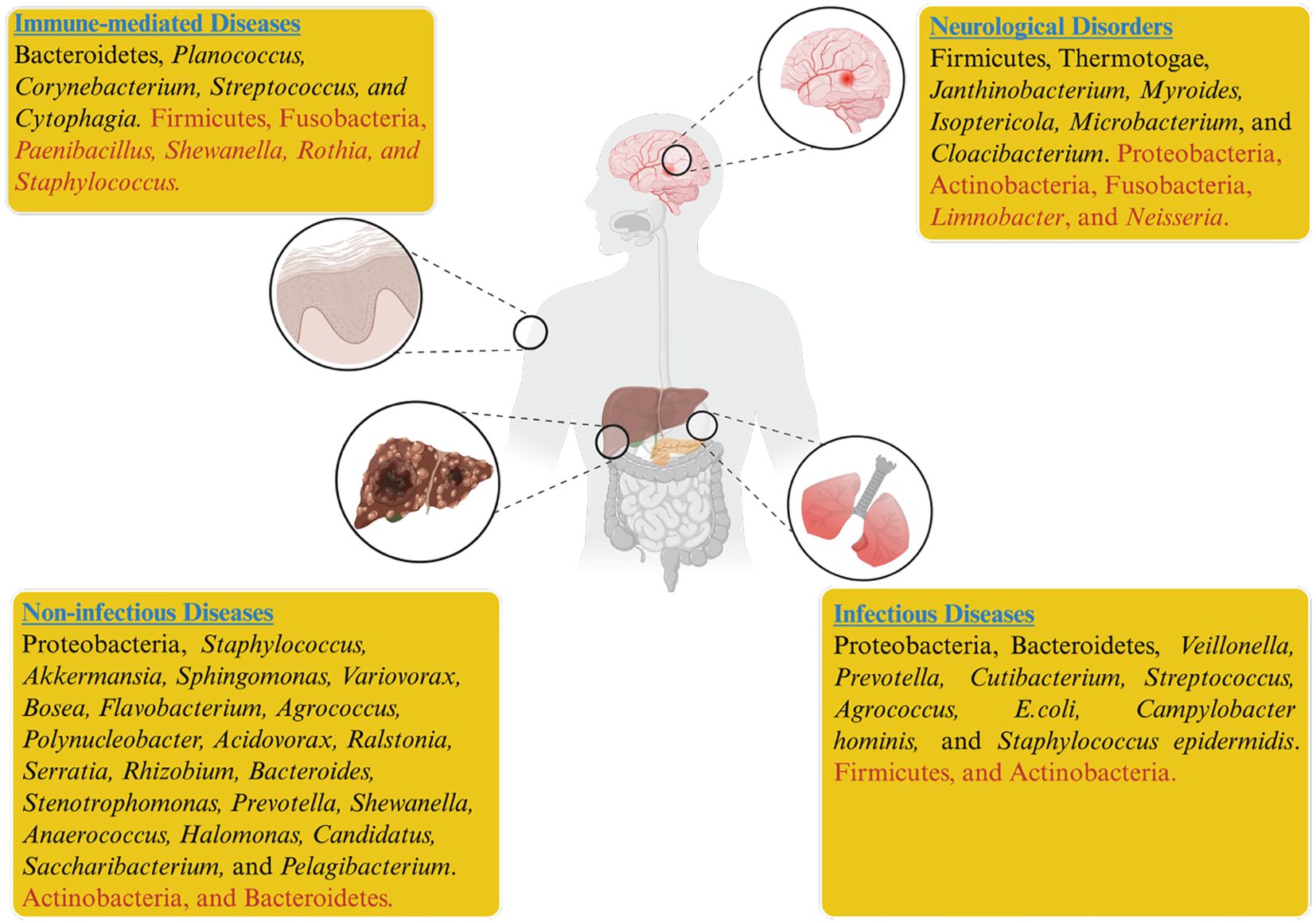
Figure 1. Dysbiosis of the blood microbiome has been associated with infectious, non-infectious, neurological, immune, and systemic diseases. In the figure, taxa shown in black indicate higher relative abundance, while those in red indicate reduced abundance across different disease states.
Alterations in the blood microbiome in non-infectious diseases
Non-infectious diseases are a leading cause of mortality and morbidity worldwide. The biological effects of conditions such as diabetes, elevated total cholesterol, obesity, and smoking, along with behavioral risk factors like sedentary behavior, harmful alcohol consumption, and poor diet, resemble those associated with other non-infectious diseases like cancer, CVDs, and chronic respiratory diseases. Since a healthy lifestyle can mitigate disease risk, these factors are modifiable. In contrast, age, sex, race, and genetic background are non-modifiable risk factors that are also known to contribute to various non-infectious disorders (Finland, 2021). Despite traditional risk factors, the role of the blood microbiome in human health and its association with disease is an emerging focus of current research. The Lelouvier team identified a distinct link between liver fibrosis and an enrichment of Proteobacteria, particularly elevated levels of the genera Sphingomonas, Variovorax, and Bosea in patients with non-alcoholic fatty liver disease (Lelouvier et al., 2016). This finding underscores the potential role of microbial dysbiosis in the progression of liver diseases, suggesting that alterations in specific bacterial taxa may influence the pathophysiology of liver fibrosis and related conditions (Figure 1). Similarly, increased microbial diversity of 16S rDNA was observed in patients diagnosed with pancreatitis and schizophrenia (Li et al., 2018; Olde Loohuis et al., 2018). Li and colleagues revealed that patients with pancreatitis exhibited elevated levels of Bacteroidetes and decreased Actinobacteria. At the same time, specific genera, including Serratia, Rhizobium, Bacteroides, Stenotrophomonas, Staphylococcus, and Prevotella, were found in the patient’s blood, regardless of disease severity (Li et al., 2018). Schierwagen et al. reported that liver cirrhosis patients exhibited elevated levels of Acinetobacteria and Staphylococcus, which correlated with inflammatory cytokines, while beneficial taxa such as Akkermansia, Rikenellaceae, and Erysipelotrichales were reduced; notably, Enterobacteriaceae dominated, indicating a disease-related microbial shift (Schierwagen et al., 2019).
Furthermore, elevated levels of Shewanella, Anaerococcus, Halomonas, Lachnospiraceae, Candidatus Saccharibacteria, Pelagibacterium, and Hyphomicrobiaceae, along with decreased levels of Bacteroidetes, may contribute to the pathogenesis of rheumatoid arthritis (RA) (Hammad et al., 2020; Mo et al., 2020). Multiple other disorders have been linked to blood microbiome dysbiosis, such as Rosacea and psoriasis, two chronic dermatological conditions exhibiting distinct blood microbiome signatures (Yun et al., 2019). Moreover, Chang et al. found that the elevated levels of Ralstonia, Staphylococcus, and Sphingomonas in psoriasis suggest potential involvement in adipocytokine signaling and lipid metabolic pathways contributing to chronic inflammation (Chang et al., 2021). Contrarily, patients with hidradenitis suppurativa, another chronic inflammatory skin condition, exhibit a blood microbiome akin to that of healthy people, hinting that its pathophysiology may not be associated with bacteremia (Ring et al., 2018). Markova et al. investigated blood samples from mothers and children with autism, which were used to isolate fungi and bacteria in their L-form. While they suggested pathophysiology remains a hypothesis and the study lacked statistical analysis, it hints at a potential vertical transfer of pathogens from mother to child that could influence autism development (Markova, 2020). Wang et al. identified increased levels of Flavobacterium, Agrococcus, Polynucleobacter, and Acidovorax in the blood samples of surgical patients who subsequently experienced postoperative septic shock. These genera were significantly correlated with disease severity and organ failure assessment scores (Wang, 2021). In contrast, catheter insertion appears to elevate the abundance of Burkholderiales in the blood of mice receiving enteral nutrition (Lucchinetti et al., 2022). However, total parenteral nutrition leads to substantial changes in gut bacterial composition while having a relatively minor effect on the blood microbiome. A study by Simões-Silva demonstrated that bloodstream bacteria primarily stem from the dysbiotic gut microbiome in end-stage renal disease (ESRD), and hemodialysis, to some extent, exacerbates microinflammation by promoting gut microbiota translocation due to impaired gut barrier function (Simoes-Silva et al., 2018). Simões-Silva and colleagues further compared the peritoneal bacterial profile with other body parts. Although the blood microbiome profile exhibited the closest match to peritoneal bacteria, their findings confirmed significant differences between the peritoneal and blood microbiomes (Simões-Silva et al., 2020). Larger cohort studies are crucial to validate the role of the blood microbiome in the progression of non-infectious diseases, providing deeper insights into its potential as a biomarker for disease development and progression.
Alterations in the blood microbiome in neurological diseases
The significant environmental and lifestyle changes in the modern era present a serious threat to human health, with the rise of various neurological disorders emerging as a major global challenge. Growing evidence suggests that the gut microbiota may influence brain function through the mediation of signaling pathways by microbial metabolites (Grochowska et al., 2019; Iannone et al., 2019). At the intersection of neuroscience and microbiology, groundbreaking studies from the past decade have revealed dynamic relationships between animals and their internal microbial populations. These interactions actively contribute to the development and functioning of neurological systems. The complex interplay of immunological, neural, and chemical signals plays a crucial role in maintaining health and advancing our understanding of neurological disorders (Morais et al., 2021). The gut-brain axis concept demonstrates how the gut microbiome can impact various brain-related health concerns (Mayer et al., 2022). Microbial components such as LPS and bacterial amyloid curli disseminate from the gut to the brain via circulation. These components tend to degrade the blood-brain barrier and cause aberrant accumulation of protein in the brain, which can lead to neuroinflammation (Suparan et al., 2022). Olde et al. analyzed the blood microbiome in patients with Amyotrophic Lateral Sclerosis (ALS), bipolar disorder, and schizophrenia. They observed elevated levels of Planctomycetes and Thermotogae in ALS patients compared to controls. However, patients with bipolar disorder and ALS displayed blood microbiomes that were similar to those of healthy individuals (Olde Loohuis et al., 2018). A previous study compared microbial DNA data derived from healthy individuals to human microbiome project (HMP) microbiome data. They demonstrated that, whereas the blood-microbiome closely resembles the skin and oral microbiomes, it differs substantially from the intestinal microbiome (Whittle et al., 2019). While most studies tend to consider the diffusion of bacteria into the blood-circulatory system as exceptional, this phenomenon may therefore occur rather frequently in healthy individuals (Moriyama et al., 2008; Païssé et al., 2016). These findings suggest that alterations in the blood microbiome observed in neurological disorders with gastrointestinal origins may reflect dysbiosis patterns similar to those seen in other systemic diseases (Figure 1).
Additionally, Ciocan et al. identified blood dysbiosis in patients with untreated major depressive episodes, marked by reduced Fusobacteria and Candidatus Saccharibacteria, enriched Janthinobacterium, and diminished Neisseria. Increased Firmicutes and decreased Proteobacteria and Actinobacteria were linked to positive treatment response (Ciocan et al., 2021). Liu et al. reported blood dysbiosis in Parkinson’s disease (PD), noting increased Myroides, Isoptericola, Microbacterium, Cloacibacterium, and Enhydrobacter, with reduced Limnobacter levels (Liu et al., 2021). Pérez-Soriano et al. found that the blood of Multiple System Atrophy (MSA) patients exhibited elevated microbiome levels, with distinct bacterial profiles for each subtype. For instance, cerebellar MSA showed increased Acinetobacter and decreased Blastococcus and Bacillus compared to PD (Pérez-soriano et al., 2020). However, these studies are observational, and more comprehensive cohort studies are needed to uncover new etiologies of neurological disorders associated with the blood microbiota. Such research could also aid in identifying diagnostic biomarkers and promising therapeutic strategies targeting blood microbiome dysbiosis in these conditions.
Alterations in the blood microbiome in immune-mediated diseases
Autoimmune diseases have a substantial impact on health, quality of life, healthcare usage, and the economy, resulting in increased mortality. Despite their rarity, they collectively affect 1 in 31 Americans and are a leading cause of death in young and middle-aged women (Jacobson et al., 1997; Walsh and Rau, 2000). Unlike diseases sharing common underlying causes, such as cancers or CVD, autoimmune disorders have typically been viewed as distinct entities, and their origins are largely unknown (Cooper and Stroehla, 2003). Global organizations have, therefore, underlined the necessity of population-based epidemiologic studies on autoimmune diseases (Committee, 2005).
Recent studies show that individuals with known or suspected autoimmune diseases have abnormal blood microbiome profiles that differ consistently from those of healthy individuals. Ogunrinde et al. reported that anti-double-stranded DNA antibodies and anti-nuclear factors play a role in systemic lupus erythematosus (SLE). Remarkably, depleted levels of Paenibacillus were detected in the blood of SLE patients and their first-degree relatives than those of healthy individuals (Ogunrinde et al., 2019). These findings suggest that genetic factors might be associated with this correlation. In contrast, Luo et al. observed elevated levels of Planococcus in patients with SLE. Exposure of peripheral blood mononuclear cells to Planococcus triggered the release of significant inflammatory cytokines, potentially contributing to the chronic inflammation characteristic of SLE (Alekseyenko et al., 2021). Jones et al. reported elevated levels of Cytophagia in the blood, indicating dysbiosis in patients with large vessel vasculitis, such as giant cell arteritis and Takayasu’s arteritis, compared to controls. In Takayasu’s arteritis, the presence of Staphylococcus in the blood may further exacerbate the condition (Jones et al., 2021). Cheng et al. reported that anti-rheumatic medications used to treat rheumatoid arthritis (RA) may potentially reverse blood dysbiosis by increasing the levels of Corynebacterium and Streptococcus while decreasing Shewanella (Cheng et al., 2023). Shewanella may contribute to RA pathogenesis through immune activation via its lipopolysaccharides, induction of pro-inflammatory cytokines, or molecular mimicry triggering autoimmunity, though direct causal evidence remains limited. Blood microbiome dysbiosis in RA may involve an elevated level of Lachnospiraceae, Halomonas, and Shewanella, while Corynebacterium1 and Streptococcus may decline (Chang et al., 2021). Lachnospiraceae appeared to continue elevation even after therapy, possibly indicating a compensatory response to blood dysbiosis. The elevation in Lachnospiraceae might have a positive impact on the course of the disease. In contrast, Puri et al. reported reduced levels of Firmicutes and Fusobacteria in the blood of psoriasis patients (Puri et al., 2018), while Han et al. stated that the pathophysiology of RA may involve a higher level of genus Pelagibacterium and PARP9 mRNA (Han and Lo, 2021). Despite common etiologies, blood microbiome configurations diverge across autoimmune diseases. In immune-mediated reversible obstructive airway disease and asthma, dysbiosis was characterized by a Bacteroidetes-enriched signature (Buford et al., 2018; Koliarakis et al., 2020). During airway inflammation, the lung microbiome, typically enriched in Bacteroidetes, may translocate into the bloodstream, reshaping the blood microbiome signature in these conditions (Zhang et al., 2019; Zhu et al., 2020). The long-term steroid treatment for airway constriction reduced Staphylococcus and Rothia, potentially altering the blood mirobiome in asthma (Figure 1). Systemic steroids were linked to increased levels of Prevotella 9, Intestinibacter, Lactobacillus, and Blautia (Buford et al., 2018). Large-scale, multi-cohort studies are needed to validate the contribution of identified taxa to disease progression and to uncover potential therapeutic targets.
Variability in blood microbiome study designs and their impact
Variability in blood microbiome study designs poses a significant challenge to establishing consistent microbial signatures linked to disease. Understanding these sources of variation is crucial for advancing the field and improving reproducibility.
● Methodological heterogeneity: Variations in blood sampling, DNA extraction, sequencing platforms, and bioinformatics workflows affect microbial detection and quantification (Regueira‐Iglesias et al., 2023), driving inconsistent results.
● Population diversity: Differences in cohort demographics, genetics, disease stages, and treatment history shape blood microbiome profiles (Gilbert et al., 2018), complicating cross-study comparisons.
● Temporal Dynamics: The blood microbiome fluctuates over time due to factors like diet, medication, and disease progression (Halfvarson et al., 2017), causing variability in findings from different sampling points.
● Environmental Influences: Geographic and ecological exposures modulate systemic microbiomes (Gacesa et al., 2022), adding variability across study populations.
● Analytical Focus: Targeted approaches emphasizing specific taxa may miss broader microbial shifts (Hanson et al., 2012), leading to divergent conclusions across studies.
Addressing these variables through standardized methodologies, comprehensive cohort characterization, longitudinal sampling, and unbiased analytical frameworks is essential to enhance reproducibility and accurately define the blood microbiome’s role in disease pathogenesis.
Vulnerability of low-biomass samples to contaminants
The extent of contaminant DNA and cross-contamination varies depending on the microbial biomass of each sample. Microbial biomass can be estimated by comparing microbial DNA quantities, such as 16S rRNA gene copies measured by qPCR, in samples versus DNA extraction blank controls (Lauder et al., 2016). High-biomass samples, like feces and soil, contain significantly more microbial DNA than blanks, whereas low-biomass samples, including blood, placenta, air, and built environments, often have DNA levels comparable to blanks. In low-biomass samples, the limited microbial DNA makes them highly susceptible to contamination from exogenous DNA or cross-contamination during processing, especially when handled alongside high-biomass samples, leading to false or misleading microbial profiles (Salter et al., 2014; Glassing et al., 2016; Lauder et al., 2016).
Many researchers question the blood microbiome’s existence because low-biomass samples are easily contaminated at multiple steps during processing (Chrisman et al., 2022). Several studies suggest that the blood microbiome in healthy individuals likely traces of microbial DNA from external sources or remnants of non-viable bacteria (Glassing et al., 2016; Martel et al., 2017). For similar reasons, claims of resident microbiomes in traditionally sterile, low-biomass niches, such as the prenatal womb, central nervous system, and tumor microenvironments, have been increasingly challenged and remain contentious (Kennedy et al., 2023). NGS is highly sensitive to trace microbial DNA contaminants originating from extraction kits, storage vessels, reagents, and even sequencing platforms. Additionally, skin-derived bacteria introduced during venepuncture and handling errors by clinical staff can further confound results. Most blood microbiome studies lack rigorous reporting on decontamination strategies, and even when addressed, efforts are often insufficient; comprehensive negative controls across all workflow stages are likely essential. Integrating bioinformatic and statistical tools into decontamination workflows has been proposed to enhance the accuracy of low-biomass microbiome profiling and reduce false-positive microbial signals (Davis et al., 2018). Therefore, establishing robust and contamination-resilient methodologies is fundamental for advancing microbiome research in low-biomass environments like the bloodstream.
Pitfalls in low-biomass metagenomics for blood microbiome research
Metagenomics enables culture-independent profiling of the blood microbiome (“hemobiome”) (Govender, 2024), but its application in low-biomass environments like blood is technically challenging. Microbial DNA comprises less than 0.1% of total nucleic acids and is easily masked by host DNA and environmental contaminants (Païssé et al., 2016; Selway et al., 2020). Artefacts introduced by reagents, lab surfaces, PCR biases, index hopping, and sequencing limitations can inflate false positives and compromise reproducibility (Païssé et al., 2016). Païssé et al. (2016) further reveal a highly diverse and quantitatively significant blood microbiome in healthy donors, varying across individuals and blood fractions. While molecular tools like qPCR and sequencing may help detect transfusion-transmitted bacterial infections, especially in immunocompromised recipients, their high sensitivity risks misclassifying clinically irrelevant DNA (Païssé et al., 2016). These findings underscore the need for cautious interpretation and standardized thresholds in low-biomass metagenomics to avoid false positives and guide safe, evidence-based blood screening practices. Furthermore, recent AI-based methods, particularly deep learning and ensemble classifiers (e.g., Random Forests, Gradient Boosting), offer a promising framework to overcome these limitations (Wani et al., 2022). These models can learn contamination patterns, filter noise, and extract biologically relevant features from noisy, sparse datasets. They also enable integration of heterogeneous inputs, such as metagenomic profiles, clinical metadata, and host response markers, to improve diagnostic precision. Some models have been trained to differentiate viable pathogens from remnant DNA fragments, further refining signal interpretation. As demonstrated by Wani et al. (2022), these approaches enhance the detection of clinically relevant taxa such as Staphylococcus spp. and position the hemobiome as a viable substrate for liquid biopsy and microbiome-guided precision diagnostics. For clinical application of metagenomic sequencing, rigorous interpretive thresholds must be established using metrics such as read counts, relative abundance, read quality scores, depth of coverage, and results from external and internal controls processed alongside clinical samples (Vijayvargiya et al., 2019). Given the inherent background noise and the difficulty of fully eliminating non-target microbial reads, metagenomic sequencing may be limited in reliably detecting low-abundance pathogens, particularly in low-biomass contexts like blood.
Unlike well-characterized niches such as the gut or skin, the blood microbiome remains poorly defined and highly contentious (Velmurugan et al., 2020; Chrisman et al., 2022). The technical pitfalls outlined above, especially contamination, low microbial load, and overreliance on DNA-based detection, contribute to inconsistent findings across studies. Discrepancies in reported microbial profiles raise fundamental questions about whether the signals reflect viable communities, transient translocation, or residual cell-free DNA (Glassing et al., 2016; Martel et al., 2017). Although rigorous controls and standardized workflows improve detection fidelity, they cannot fully eliminate the risk of artefactual signals (Jervis-Bardy et al., 2015; Davis et al., 2018). These challenges mirror debates in other low-biomass sites like the placenta, brain, and tumors (Kennedy et al., 2023), where microbial detection remains controversial. Crucially, next-generation sequencing lacks the resolution to confirm microbial viability or origin, limiting its interpretive value (Panaiotov et al., 2021). Emerging RNA-based methods, such as FISH, PETRI-seq, MATQ-seq, and BacDrop, enable single-cell resolution and functional assessment of microbial activity (Batani et al., 2019; Kuchina et al., 2021; Cheng et al., 2023; Homberger et al., 2023; Ma et al., 2023). These tools may help distinguish true microbial residents from technical artefacts or biological noise. Ultimately, realizing the potential of blood metagenomics will require methodological rigor, functional validation, and integration of multi-omic data to overcome the pitfalls inherent to low-biomass microbiome research.
Contamination in microbiome research: sources, dynamics, and impact on data integrity
Microbiome studies face two main contamination challenges: contaminant DNA and cross-contamination. Contaminant DNA can originate from various sources despite rigorous sample collection and processing protocols, including the sampling environment, laboratory settings (Llamas et al., 2017), personnel, plasticware (Motley et al., 2014), nucleic acid extraction kits (Glassing et al., 2016; Weyrich et al., 2017), and reagents such as PCR mastermixes (Shen et al., 2006; Eisenhofer et al., 2019). Additionally, contamination may arise from other samples or sequencing runs (Seitz et al., 2015; Ballenghien et al., 2017). More than 60 common contaminant taxa have been identified repeatedly in DNA extraction blanks and no-template controls across multiple studies. For instance, Salter et al. demonstrated that several contaminant taxa were consistently detected across different labs, extraction methods, and studies (Salter et al., 2014). These pervasive contaminants likely originate from sources such as kit and reagent manufacturing, human commensals on laboratory staff, and environmental exposure. However, contaminant profiles vary depending on extraction kits, laboratory environments (Glassing et al., 2016; Weyrich et al., 2017), and even fluctuate over time within the same lab (Weyrich et al., 2019).
Cross-contamination poses a further obstacle during microbiome sample processing and involves the unintended transfer of sample DNA, barcodes, or amplicons between adjacent wells or tubes, leading to batch effects (Nguyen et al., 2015). This can occur at various stages, including sample handling and tube or plate loading (Tamariz et al., 2006), as well as through aerosolization during pipetting or plate cover removal (Mainelis, 2020). Barcode contamination can arise when incorrect barcodes “jump” into neighboring samples, a process termed ‘tag switching’ (Carlsen et al., 2012). Additionally, cross-contamination may occur on sequencing platforms due to barcode sequencing errors, residual amplicons from previous runs, or ‘index hopping,’ where indexing reads are incorrectly assigned to sequencing reads (Larsson et al., 2018). Both contaminant DNA and cross-contamination are dynamic challenges that require continuous and careful monitoring throughout microbiome research workflows.
Blood microbiome research: unresolved challenges, controversies, and translational barriers
The study of the blood microbiome across various disease states has revealed microbial signatures linked to diagnosis, disease severity, and prognosis. Despite inconsistent findings, efforts to define blood dysbiosis are advancing, addressing key questions: “Who is there?” and “What do they do?” Current data suggest a predominance of bacteria, with a clear taxonomic structure in the blood microbiome, primarily composed of Proteobacteria, Bacteroidetes, Firmicutes, and Actinobacteria, which have been observed in blood across different health conditions (Amar et al., 2019; Shah et al., 2019; Scarsella et al., 2020; Khan et al., 2022b, 2022). However, the specific functions of distinct blood microbiome profiles and their roles in disease mechanisms remain largely unexplored. Given its potential public health impact, the blood microbiome warrants increased attention, as it has implications for both human and animal health. This perspective mirrors Tolstoy’s “Anna Karenina principle, “happy families are all alike; every unhappy family is unhappy in its own way.” Similarly, while a common foundation for health maintenance may exist within the blood microbiome, variations in microbial profiles may uniquely contribute to different pathological conditions, underscoring the need for targeted interventions and in-depth research (Zaneveld et al., 2017). Our understanding of the blood microbiome’s role in health and disease is still nascent, with significant gaps remaining in defining a “core blood microbiome,” understanding its health benefits, and elucidating its specific functions.
Despite growing interest in the blood microbiome, several studies report null or contradictory findings regarding microbial presence and associations with disease. Variations in sampling methods, contamination risks, and analytical techniques contribute to inconsistent results. Some studies fail to detect microbial signatures in blood, while others report conflicting microbial taxa linked to similar conditions. These discrepancies highlight the need for standardized protocols and rigorous controls to validate findings reliably. Future research on the blood microbiome should explore new areas while deepening our understanding of its mechanisms. Clarifying its role in disease progression, systemic inflammation, and comorbidities could revolutionize diagnostics and therapies. While current studies focus on microbial associations with systemic diseases, few have investigated interventions targeting the blood microbiome. Future studies should prioritize controlled trials, such as antimicrobial treatments, probiotics, or immune-modulating strategies, to assess causality and therapeutic potential. A better understanding of microbial-immune interactions is essential, especially in systemic inflammation and disease. Future research should focus on the molecular interactions between blood microbes and immune responses to develop targeted therapies. Personalized blood microbiome-based treatments hold promise, as tailored interventions may be more effective than generic ones. Identifying harmful microbial signatures linked to diseases could lead to targeted therapies, including bacteriophage treatment, which could reduce reliance on broad-spectrum antibiotics and combat antibiotic resistance. These precision strategies have the potential to transform disease prevention and treatment.
These insights pave the way for blood-based diagnostics and targeted microbiome-metabolome interventions. Biomarkers derived from circulating microbial taxa and metabolites hold promise for early disease detection. Therapeutically, metabolite supplementation (e.g., SCFAs, bile acids), antibiotics, probiotics, or vaccines (e.g., BCG) could modulate host–microbe interactions and inflammation. To unlock the full therapeutic potential of the blood microbiome, we must explore its “dark matter,” including the virome and uncultivated microbial taxa with functional significance that are not yet fully understood (Rodríguez del Río et al., 2024). However, challenges include distinguishing viable microbes from DNA fragments, understanding causal mechanisms, ensuring brain access, and improving omics integration. Longitudinal, multi-omic studies are essential to clarify whether these blood microbiome and metabolome changes are drivers or just bystanders in systemic disease.
Conclusion
The blood microbiome is increasingly recognized as a key player in the pathogenesis of diverse systemic diseases, including infectious, neurological, and immune-mediated conditions, through its influence on systemic inflammation, immune modulation, and metabolic disruption. Defining clear clinical outcomes and mechanistic parameters is essential to advance both research and therapeutic applications. However, current progress is hindered by methodological pitfalls such as inadequate contamination control, inconsistent sequencing protocols, and a lack of viability assessment. To overcome these barriers, rigorously designed, longitudinal multi-omic studies are needed to clarify causal relationships and enhance biomarker specificity. As we move beyond traditional approaches like probiotics and bacteriophage therapy, future translational success will rely on innovative, mechanistically guided microbiome-based interventions tailored to disease-specific blood microbial signatures, ultimately improving risk prediction, treatment, and patient outcomes.
Author contributions
IkK: Writing – review & editing, Writing – original draft. MI: Writing – review & editing. ImK: Investigation, Writing – review & editing. UN: Investigation, Writing – review & editing. XX: Visualization, Writing – review & editing, Resources, Validation, Supervision. ZL: Validation, Visualization, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We gratefully acknowledged all authors for their contribution and support.
Conflict of interest
The authors declare that this study was conducted without any commercial or financial relationships that could be construed as potential conflicts of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Alekseyenko, A. V., Ogunrinde, E., Li, M., Tsao, B. P., Kamen, D. L., Oates, J. C., et al. (2021). Rigorous plasma microbiome analysis method enables disease association discovery in clinic. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.613268
Amar, J., Lelouvier, B., Servant, F., Lluch, J., Burcelin, R., Bongard, V., et al. (2019). Blood microbiota modification after myocardial infarction depends upon low-density lipoprotein cholesterol levels. J. Am. Heart Assoc. 8, e011797. doi: 10.1161/JAHA.118.011797
Ancona, A., Petito, C., Iavarone, I., Petito, V., Galasso, L., Leonetti, A., et al. (2021). The gut–brain axis in irritable bowel syndrome and inflammatory bowel disease. Dig. Liver Dis. 53, 298–305. doi: 10.1016/j.dld.2020.11.026
Ballenghien, M., Faivre, N., and Galtier, N. (2017). Patterns of cross-contamination in a multispecies population genomic project: detection, quantification, impact, and solutions. BMC Biol. 15, 1–16. doi: 10.1186/s12915-017-0366-6
Batani, G., Bayer, K., Böge, J., Hentschel, U., and Thomas, T. (2019). Fluorescence in situ hybridization (FISH) and cell sorting of living bacteria. Sci. Rep. 9, 18618. doi: 10.1038/s41598-019-55049-2
Buford, T. W., Carter, C. S., VanDerPol, W. J., Chen, D., Lefkowitz, E. J., Eipers, P., et al. (2018). Composition and richness of the serum microbiome differ by age and link to systemic inflammation. Geroscience 40, 257–268. doi: 10.1007/s11357-018-0026-y
Carlsen, T., Aas, A. B., Lindner, D., Vrålstad, T., Schumacher, T., and Kauserud, H. (2012). Don’t make a mista (g) ke: is tag switching an overlooked source of error in amplicon pyrosequencing studies? Fungal Ecol. 5, 747–749. doi: 10.1016/j.funeco.2012.06.003
Castillo, D. J., Rifkin, R. F., Cowan, D. A., and Potgieter, M. (2019). The healthy human blood microbiome: Fact or fiction? Front. Cell. Infect. Microbiol. 9. doi: 10.3389/fcimb.2019.00148
Chang, C.-J., Zhang, J., Tsai, Y.-L., Chen, C.-B., Lu, C.-W., Huo, Y. P., et al. (2021). Compositional features of distinct microbiota base on serum extracellular vesicle metagenomics analysis in moderate to severe psoriasis patients. Cells 10, 2349. doi: 10.3390/cells10092349
Chen, Y., Zhou, J., and Wang, L. (2021). Role and mechanism of gut microbiota in human disease. Front. Cell. Infect. Microbiol. 11, 625913. doi: 10.3389/fcimb.2021.625913
Cheng, H. S., Tan, S. P., Meng, D., Wong, K., Ling, W., Koo, Y., et al. (2023). The blood microbiome and health : current evidence, controversies, and challenges. Intl. J. Mol. Sci. 24 (6), 5633. doi: 10.3390/ijms24065633
Chrisman, B., He, C., Jung, J.-Y., Stockham, N., Paskov, K., Washington, P., et al. (2022). The human “contaminome”: bacterial, viral, and computational contamination in whole genome sequences from 1000 families. Sci. Rep. 12, 9863. doi: 10.1038/s41598-022-13269-z
Ciocan, D., Cassard, A., Becquemont, L., Verstuyft, C., Voican, C. S., Asmar, K., et al. (2021). Blood microbiota and metabolomic signature of major depression before and after antidepressant treatment : a prospective case – control study. J. Psychiatry Neurosci. 46, 358–368. doi: 10.1503/jpn.200159
Committee, A. D. C. (2005). Progress in autoimmune diseases research: Report to Congress. US Dep. Heal. Hum. Serv.
Cooper, G. S. and Stroehla, B. C. (2003). The epidemiology of autoimmune diseases. Autoimmun. Rev. 2, 119–125. doi: 10.1016/S1568-9972(03)00006-5
Damgaard, C., Magnussen, K., Enevold, C., Nilsson, M., Tolker-Nielsen, T., Holmstrup, P., et al. (2015). Viable bacteria associated with red blood cells and plasma in freshly drawn blood donations. PloS One 10, e0120826. doi: 10.1371/journal.pone.0120826
Davis, N. M., Proctor, D. M., Holmes, S. P., Relman, D. A., and Callahan, B. J. (2018). Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 6, 1–14. doi: 10.1186/S40168-018-0605-2
Dereschuk, K., Apostol, L., Ranjan, I., Chakladar, J., Li, W. T., Rajasekaran, M., et al. (2021). Identification of lung and blood microbiota implicated in COVID-19 prognosis. Cells 10, 1452. doi: 10.3390/cells10061452
Dinakaran, V., Rathinavel, A., Pushpanathan, M., Sivakumar, R., Gunasekaran, P., and Rajendhran, J. (2014). Elevated levels of circulating DNA in cardiovascular disease patients: Metagenomic profiling of microbiome in the circulation. PloS One 9, e105221. doi: 10.1371/journal.pone.0105221
Eisenhofer, R., Minich, J. J., Marotz, C., Cooper, A., Knight, R., and Weyrich, L. S. (2019). Contamination in low microbial biomass microbiome studies: issues and recommendations. Trends Microbiol. 27, 105–117. doi: 10.1016/j.tim.2018.11.003
Finland, S. (2021). Official statistics of Finland (OSF): Causes of death. (Official Statistics of Finland (OSF))
Funkhouser, L. J. and Bordenstein, S. R. (2013). Mom knows best: the universality of maternal microbial transmission. PloS Biol. 11, e1001631. doi: 10.1371/journal.pbio.1001631
Gacesa, R., Kurilshikov, A., Vich Vila, A., Sinha, T., Klaassen, M. A. Y., Bolte, L. A., et al. (2022). Environmental factors shaping the gut microbiome in a Dutch population. Nature 604, 732–739. doi: 10.1038/s41586-022-04567-7
Gedgaudas, R., Bajaj, J. S., Skieceviciene, J., Varkalaite, G., Jurkeviciute, G., Gelman, S., et al. (2022). Circulating microbiome in patients with portal hypertension. Gut Microbes 14, 2029674. doi: 10.1080/19490976.2022.2029674/SUPPL_FILE/KGMI_A_2029674_SM1876.ZIP
Gilbert, J. A., Blaser, M. J., Caporaso, J. G., Jansson, J. K., Lynch, S. V., and Knight, R. (2018). Current understanding of the human microbiome. Nat. Med. 24, 392–400. doi: 10.1038/nm.4517
Glassing, A., Dowd, S. E., Galandiuk, S., Davis, B., and Chiodini, R. J. (2016). Inherent bacterial DNA contamination of extraction and sequencing reagents may affect interpretation of microbiota in low bacterial biomass samples. Gut Pathog. 8, 1–12. doi: 10.1186/S13099-016-0103-7
Gosiewski, T. and Huminska, K. (2017). Comprehensive detection and identification of bacterial DNA in the blood of patients with sepsis and healthy volunteers using next-generation sequencing method - the observation of DNAemia. Eur. J. Clin. Microbiol. Infect. Dis. 36, 329–336. doi: 10.1007/s10096-016-2805-7
Govender, K. N. (2024). Metagenomic sequencing directly from blood culture as a tool for clinical diagnosis [Doctoral dissertation]. University of Oxford.
Grochowska, M., Laskus, T., and Radkowski, M. (2019). Gut microbiota in neurological disorders. Arch. Immunol. Ther. Exp. (Warsz) 67, 375–383. doi: 10.1007/s00005-019-00561-6
Guo, X., Wang, Z., Qu, M., Guo, Y., Yu, M., Hong, W., et al. (2023). Abnormal blood microbiota profiles are associated with inflammation and immune restoration in HIV/AIDS individuals. Msystems 8 (5), e00467–23. doi: 10.1128/msystems.00467-23
Halfvarson, J., Brislawn, C. J., Lamendella, R., Vázquez-Baeza, Y., Walters, W. A., Bramer, L. M., et al. (2017). Dynamics of the human gut microbiome in inflammatory bowel disease. Nat. Microbiol. 2, 1–7. doi: 10.1038/nmicrobiol.2017.4
Hammad, D. B. M., Hider, S. L., Liyanapathirana, V. C., and Tonge, D. P. (2020). Molecular characterization of circulating microbiome signatures in rheumatoid arthritis. Front. Cell. Infect. Microbiol. 9. doi: 10.3389/fcimb.2019.00440
Han, D. S. C. and Lo, Y. M. D. (2021). The nexus of cfDNA and nuclease biology. Trends Genet. 37, 758–770. doi: 10.1016/j.tig.2021.04.005
Hanson, C. A., Fuhrman, J. A., Horner-Devine, M. C., and Martiny, J. B. H. (2012). Beyond biogeographic patterns: processes shaping the microbial landscape. Nat. Rev. Microbiol. 10, 497–506. doi: 10.1038/nrmicro2795
Heintz-Buschart, A. and Wilmes, P. (2018). Human gut microbiome: function matters. Trends Microbiol. 26, 563–574. doi: 10.1016/j.tim.2017.11.002
Hodzhev, Y. (2023). Analysis of blood microbiome dysbiosis in pulmonary sarcoidosis by decision tree model. Biotechnol. Biotechnol. Equip. 37, 2283133. doi: 10.1080/13102818.2023.2283133
Hodzhev, Y., Tsafarova, B., Tolchkov, V., Youroukova, V., Ivanova, S., Kostadinov, D., et al. (2023). Visualization of the individual blood microbiome to study the etiology of sarcoidosis. Comput. Struct. Biotechnol. J. 22, 50–57. doi: 10.1016/j.csbj.2023.10.027
Homberger, C., Hayward, R. J., Barquist, L., and Vogel, J. (2023). Improved bacterial single-cell RNA-seq through automated MATQ-seq and Cas9-based removal of rRNA reads. MBio 14, e03557–e03522. doi: 10.1128/MBIO.03557-22
Hyun, H., Lee, M. S., Park, I., Ko, H. S., Yun, S., Jang, D., et al. (2021). Analysis of porcine model of fecal- induced peritonitis reveals the tropism of blood microbiome. Front. Cell. Infect. Microbiol. 11, 1–14. doi: 10.3389/fcimb.2021.676650
Iannone, L. F., Preda, A., Blottière, H. M., Clarke, G., Albani, D., Belcastro, V., et al. (2019). Microbiota-gut brain axis involvement in neuropsychiatric disorders. Expert Rev. Neurother. 19, 1037–1050. doi: 10.1080/14737175.2019.1638763
Jacobson, D. L., Gange, S. J., Rose, N. R., and Graham, N. M. H. (1997). Epidemiology and estimated population burden of selected autoimmune diseases in the United States. Clin. Immunol. Immunopathol. 84, 223–243. doi: 10.1006/clin.1997.4412
Jagare, L., Rozenberga, M., Silamikelis, I., Ansone, L., Elbere, I., Briviba, M., et al. (2023). Metatranscriptome analysis of blood in healthy individuals and irritable bowel syndrome patients. J. Med. Microbiol. 72 (6), 1–12. doi: 10.1099/jmm.0.001719
James, W. A., Ogunrinde, E., Wan, Z., Kamen, D. L., Oates, J., Gilkeson, G. S., et al. (2022). A distinct plasma microbiome but not gut microbiome in patients with systemic lupus erythematosus compared to healthy individuals. J. Rheumatol. 49, 592–597. doi: 10.3899/jrheum.210952
Jervis-Bardy, J., Leong, L. E. X. X., Marri, S., Smith, R. J., Choo, J. M., Smith-Vaughan, H. C., et al. (2015). Deriving accurate microbiota profiles from human samples with low bacterial content through post-sequencing processing of Illumina MiSeq data. Microbiome 3, 1–11. doi: 10.1186/S40168-015-0083-8
Jiang, W., Lederman, M. M., Hunt, P., Sieg, S. F., Haley, K., Rodriguez, B., et al. (2009). Plasma levels of bacterial DNA correlate with immune activation and the magnitude of immune restoration in persons with antiretroviral-treated HIV infection. J. Infect. Dis. 199, 1177–1185. doi: 10.1086/597476
Jones, E., Stentz, R., Telatin, A., Savva, G. M., Booth, C., Baker, D., et al. (2021). The origin of plasma-derived bacterial extracellular vesicles in healthy individuals and patients with inflammatory bowel disease: A pilot study. Genes 12(10), 1636.
Kajihara, M., Koido, S., Kanai, T., Ito, Z., Matsumoto, Y., Takakura, K., et al. (2019). Characterisation of blood microbiota in patients with liver cirrhosis. Eur. J. Gastroenterol. Hepatol. 31, 1577–1583. doi: 10.1097/MEG.0000000000001494
Kennedy, K. M., de Goffau, M. C., Perez-Muñoz, M. E., Arrieta, M.-C., Bäckhed, F., Bork, P., et al. (2023). Questioning the fetal microbiome illustrates pitfalls of low-biomass microbial studies. Nature 613, 639–649. doi: 10.1038/s41586-022-05546-8
Khan, I. I. I. I. I., Khan, I. I. I. I. I., Jianye, Z., Xiaohua, Z., Khan, M., Gul, M., et al. (2022a). Exploring blood microbial communities and their influence on human cardiovascular disease. J. Clin. Lab. Anal. 36, 1–11. doi: 10.1002/jcla.24354
Khan, I., Khan, I., Kakakhel, M. A., Xiaowei, Z., Ting, M., Ali, I., et al. (2022). Comparison of microbial populations in the blood of patients with myocardial infarction and healthy individuals. Front. Microbiol. 13. doi: 10.3389/fmicb.2022.845038
Khan, I., Khan, I., Usman, M., Jianye, Z., Wei, Z. X., Ping, X., et al. (2022b). Analysis of the blood bacterial composition of patients with acute coronary syndrome and chronic coronary syndrome. Front. Cell. Infect. Microbiol. 12. doi: 10.3389/fcimb.2022.943808
Koliarakis, I., Athanasakis, E., Sgantzos, M., Mariolis-Sapsakos, T., Xynos, E., Chrysos, E., et al. (2020). Intestinal microbiota in colorectal cancer surgery. Cancers (Basel) 12, 3011. doi: 10.3390/cancers12103011
KuChina, A., Brettner, L. M., Paleologu, L., Roco, C. M., Rosenberg, A. B., Carignano, A., et al. (2021). Microbial single-cell RNA sequencing by split-pool barcoding. Sci. (80-.) 371, eaba5257. doi: 10.1126/SCIENCE.ABA5257
Larsson, A. J. M., Stanley, G., Sinha, R., Weissman, I. L., and Sandberg, R. (2018). Computational correction of index switching in multiplexed sequencing libraries. Nat. Methods 15, 305–307. doi: 10.1038/nmeth.4666
Lauder, A. P., Roche, A. M., Sherrill-Mix, S., Bailey, A., Laughlin, A. L., Bittinger, K., et al. (2016). Comparison of placenta samples with contamination controls does not provide evidence for a distinct placenta microbiota. Microbiome 4, 1–11. doi: 10.1186/s40168-016-0172-3
Lee, J.-H. H., Choi, J.-P. P., Yang, J., Won, H.-K. K., Park, C. S., Song, W.-J. J., et al. (2020). Metagenome analysis using serum extracellular vesicles identified distinct microbiota in asthmatics. Sci. Rep. 10, 1–9. doi: 10.1038/s41598-020-72242-w
Lelouvier, B., Servant, F., Païssé, S., Brunet, A. C., Benyahya, S., Serino, M., et al. (2016). Changes in blood microbiota profiles associated with liver fibrosis in obese patients: A pilot analysis. Hepatology 64, 2015–2027. doi: 10.1002/hep.28829
Li, Q., Wang, C., Tang, C., Zhao, X., He, Q., and Li, J. (2018). Identification and characterization of blood and neutrophil-associated microbiomes in patients with severe acute pancreatitis using next-generation sequencing. Front. Cell. Infect. Microbiol. 8. doi: 10.3389/fcimb.2018.00005
Liang, G., Bushman, F. D., and Liang, G. (2021). The human virome : assembly, composition and host interactions. Nat. Rev. Microbiol. 19, 514–527. doi: 10.1038/s41579-021-00536-5
Libertucci, J. and Young, V. B. (2019). The role of the microbiota in infectious diseases. Nat. Microbiol. 4, 35–45. doi: 10.1038/s41564-018-0278-4
Liu, X., Tang, S., Zhong, H., Tong, X., Jie, Z., Ding, Q., et al. (2021). A genome-wide association study for gut metagenome in Chinese adults illuminates complex diseases. Cell Discov. 7, 9. doi: 10.1038/s41421-020-00239-w
Llamas, B., Valverde, G., Fehren-Schmitz, L., Weyrich, L. S., Cooper, A., and Haak, W. (2017). From the field to the laboratory: Controlling DNA contamination in human ancient DNA research in the high-throughput sequencing era. STAR Sci. Technol. Archaeol. Res. 3, 1–14. doi: 10.1080/20548923.2016.1258824
Olde Loohuis, L. M, Mangul, S., Ori, A. P. S., Jospin, G., Koslicki, D., Yang, H. T., et al. (2018). Transcriptome analysis in whole blood reveals increased microbial diversity in schizophrenia. Transl. Psychiatry. 8, 1–9. doi: 10.1038/s41398-018-0107-9
Lucchinetti, E., Lou, P.-H., Lemal, P., Bestmann, L., Hersberger, M., Rogler, G., et al. (2022). Gut microbiome and circulating bacterial DNA (“blood microbiome”) in a mouse model of total parenteral nutrition: Evidence of two distinct separate microbiotic compartments. Clin. Nutr. ESPEN 49, 278–288. doi: 10.1016/j.clnesp.2022.03.038
Luo, Z., Li, M., Wu, Y., Meng, Z., Martin, L., Zhang, L., et al. (2019). Systemic translocation of Staphylococcus drives autoantibody production in HIV disease. Microbiome 7, 1–16. doi: 10.1186/s40168-019-0646-1
Ma, P., Amemiya, H. M., He, L. L., Gandhi, S. J., Nicol, R., Bhattacharyya, R. P., et al. (2023). Bacterial droplet-based single-cell RNA-seq reveals antibiotic-associated heterogeneous cellular states. Cell 186, 877–891. doi: 10.1016/j.cell.2023.01.002
Mainelis, G. (2020). Bioaerosol sampling: Classical approaches, advances, and perspectives. Aerosol Sci. Technol. 54, 496–519. doi: 10.1080/02786826.2019.1671950
Markova, N. (2020). Eubiotic vs. Dysbiotic Human Blood Microbiota: the Phenomenon of Cell Wall Deficiency and Disease-trigger Potential of Bacterial and Fungal L-forms. Discovery Med. 29 (156), 17–26.
Martel, J., Wu, C.-Y. C., Huang, P. P.-R., Cheng, W. W.-Y., Young, J. D., Reports, J. Y.-S., et al. (2017). Pleomorphic bacteria-like structures in human blood represent non-living membrane vesicles and protein particles. Sci. Rep. 7, 10650. doi: 10.1038/s41598-017-10479-8
Mayer, E. A., Nance, K., and Chen, S. (2022). The gut–brain axis. Annu. Rev. Med. 73, 439–453. doi: 10.1146/annurev-med-042320-014032
Mo, X., Dong, C., He, P., Wu, L., and Lu, X. (2020). Alteration of circulating microbiome and its associated regulation role in rheumatoid arthritis: Evidence from integration of multiomics data. Clinical and Translational Medicine 10 (7), e229. doi: 10.1002/ctm2.229
Mohamed, W., Mohamed, A., Ali, A. O., Mahmoud, H. Y. A. H., Omar, M. A., Chatanga, E., et al. (2021). Exploring prokaryotic and eukaryotic microbiomes helps in detecting tick-borne infectious agents in the blood of camels. Pathogens 10 (3), 351. doi: 10.3390/pathogens10030351
Morais, L. H., Schreiber, H. L., IV, and Mazmanian, S. K. (2021). The gut microbiota–brain axis in behaviour and brain disorders. Nat. Rev. Microbiol. 19, 241–255. doi: 10.1038/s41579-020-00460-0
Moriyama, K., Ando, C., Tashiro, K., Kuhara, S., Okamura, S., Nakano, S., et al. (2008). Polymerase chain reaction detection of bacterial 16S rRNA gene in human blood. Microbiol. Immunol. 52, 375–382. doi: 10.1111/j.1348-0421.2008.00048.x
Motley, S. T., Picuri, J. M., Crowder, C. D., Minich, J. J., Hofstadler, S. A., and Eshoo, M. W. (2014). Improved multiple displacement amplification (iMDA) and ultraclean reagents. BMC Genomics 15, 1–10. doi: 10.1186/1471-2164-15-443
Nguyen, N. H., Smith, D., Peay, K., and Kennedy, P. (2015). Parsing ecological signal from noise in next generation amplicon sequencing. New Phytol. 205, 1389–1393. doi: 10.1111/nph.12923
Nikkari, S., McLaughlin, I. J., Bi, W., Dodge, D. E., and Relman, D. A. (2001). Does blood of healthy subjects contain bacterial ribosomal DNA? J. Clin. Microbiol. 39, 1956–1959. doi: 10.1128/JCM.39.5.1956-1959.2001
Ogunrinde, E., Zhou, Z., Luo, Z., Alekseyenko, A., Li, Q., Macedo, D., et al. (2019). A link between plasma microbial translocation, microbiome, and autoantibody development in first- Degree relatives of systemic lupus erythematosus patients. Arthritis Rheumatol. 71, 1858–1868. doi: 10.1002/art.40935
Païssé, S., Valle, C., Servant, F., Courtney, M., Burcelin, R., Amar, J., et al. (2016). Comprehensive description of blood microbiome from healthy donors assessed by 16S targeted metagenomic sequencing. Transfusion 56, 1138–1147. doi: 10.1111/trf.13477
Panaiotov, S., Filevski, G., Equestre, M., Nikolova, E., Kalfin, R., Superiore, I., et al. (2018). Cultural isolation and characteristics of the blood microbiome of healthy individuals. Adv. Microbiol. 8, 406–421. doi: 10.4236/aim.2018.85027
Panaiotov, S., Hodzhev, Y., Tsafarova, B., Tolchkov, V., and Kalfin, R. (2021). Culturable and non-culturable blood microbiota of healthy individuals. Microorganisms 9, 1464. doi: 10.3390/microorganisms9071464
Pérez-soriano, A., Segura, M. A., and Botta-orfila, T. (2020). Transcriptomic differences in MSA clinical variants. Sci. Rep. 10 (1), 10310. 1–9. doi: 10.1038/s41598-020-66221-4
Poore, G. D., Kopylova, E., Zhu, Q., Carpenter, C., Fraraccio, S., Wandro, S., et al. (2020). Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature 579, 567–574. doi: 10.1038/s41586-020-2095-1
Puri, P., Liangpunsakul, S., Christensen, J. E., Shah, V. H., Kamath, P. S., Gores, G. J., et al. (2018). The circulating microbiome signature and inferred functional metagenomics in alcoholic hepatitis. Hepatology 67, 1284–1302. doi: 10.1002/hep.29623
Qian, Y., Yang, X., Xu, S., Wu, C., Qin, N., Chen, S.-D., et al. (2018). Detection of microbial 16S rRNA gene in the blood of patients with Parkinson’s disease. Front. Aging Neurosci. 10. doi: 10.3389/fnagi.2018.00156
Qiu, J., Zhou, H., Jing, Y., and Dong, C. (2019). Association between blood microbiome and type 2 diabetes mellitus : A nested case - control study. J. Clin. Lab. Anal. 33 (4), e22842. 1–7. doi: 10.1002/jcla.22842
Regueira-Iglesias, A., Balsa-Castro, C., Blanco-Pintos, T., and Tomás, I. (2023). Critical review of 16S rRNA gene sequencing workflow in microbiome studies: From primer selection to advanced data analysis. Mol. Oral. Microbiol. 38, 347–399. doi: 10.1111/omi.12434
Ring, H. C. C., Thorsen, J., Saunte, D. M. M., Lilje, B., Bay, L., Theut Riis, P., et al. (2018). Moderate to severe hidradenitis suppurativa patients do not have an altered bacterial composition in peripheral blood compared to healthy controls. J. Eur. Acad. Dermatol. Venereol. 32, 125–128. doi: 10.1111/jdv.14538
Rodríguez del Río, Á., Giner-Lamia, J., Cantalapiedra, C. P., Botas, J., Deng, Z., Hernández-Plaza, A., et al. (2024). Functional and evolutionary significance of unknown genes from uncultivated taxa. Nature 626, 377–384. doi: 10.1038/s41586-023-06955-z
Salter, S. J., Cox, M. J., Turek, E. M., Calus, S. T., Cookson, W. O., Moffatt, M. F., et al. (2014). Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12, 1–12. doi: 10.1186/s12915-014-0087-z
Santiago, A., Pozuelo, M., Poca, M., Gely, C., and Nieto, J. C. (2016). Alteration of the serum microbiome composition in cirrhotic patients with ascites. Sci. Rep. 6 (1), 25001. doi: 10.1038/srep25001
Scarsella, E., Meineri, G., Sandri, M., Ganz, H. H., and Stefanon, B. (2023). Characterization of the Blood Microbiome and Comparison with the Fecal Microbiome in Healthy Dogs and Dogs with Gastrointestinal Disease veterinary sciences Characterization of the Blood Microbiome and Comparison with the Fecal Microbiome in Healthy Dogs. Animals. (2021) 11(5), 1463. doi: 10.3390/vetsci10040277
Scarsella, E., Sandri, M., and Stefanon, B. (2020). Blood microbiome : A new marker of gut microbial population in dogs? Vet. Sci. 7 (4), 198. doi: 10.3390/vetsci7040198
Scarsella, E., Zecconi, A., and Cintio, M. (2021). Characterization of microbiome on feces, blood and milk in dairy cows with different milk leucocyte pattern. doi: 10.3390/ani11051463
Schierwagen, R., Alvarez-Silva, C., Madsen, M. S. A., Kolbe, C. C., Meyer, C., Thomas, D., et al. (2019). Circulating microbiome in blood of different circulatory compartments. Gut 68, 578–580. doi: 10.1136/gutjnl-2018-316227
Seitz, V., Schaper, S., Dröge, A., Lenze, D., Hummel, M., and Hennig, S. (2015). A new method to prevent carry-over contaminations in two-step PCR NGS library preparations. Nucleic Acids Res. 43, e135–e135. doi: 10.1093/nar/gkv694
Selway, C. A., Eisenhofer, R., and Weyrich, L. S. (2020). Microbiome applications for pathology: challenges of low microbial biomass samples during diagnostic testing. J. Pathol. Clin. Res. 6, 97–106. doi: 10.1002/cjp2.151
Shah, N. B., Allegretti, A. S., Nigwekar, S. U., Kalim, S., Zhao, S., Lelouvier, B., et al. (2019). Blood microbiome profile in CKD: A pilot study. Clin. J. Am. Soc Nephrol. 14, 692–701. doi: 10.2215/CJN.12161018
Shen, H., Rogelj, S., and Kieft, T. L. (2006). Sensitive, real-time PCR detects low-levels of contamination by Legionella pneumophila in commercial reagents. Mol. Cell. Probes 20, 147–153. doi: 10.1016/j.mcp.2005.09.007
Shukla, V., Singh, S., Verma, S., Verma, S., Rizvi, A. A., and Abbas, M. (2024). Targeting the microbiome to improve human health with the approach of personalized medicine: latest aspects and current updates. Clin. Nutr. ESPEN. 63, 813–820. doi: 10.1016/j.clnesp.2024.08.005
Simoes-Silva, L., Araujo, R., Pestana, M., Soares-Silva, I., and Sampaio-Maia, B. (2018). The microbiome in chronic kidney disease patients undergoing hemodialysis and peritoneal dialysis. Pharmacol. Res. 130, 143–151. doi: 10.1016/j.phrs.2018.02.011
Simões-Silva, L., Araujo, R., Pestana, M., Soares-Silva, I., and Sampaio-Maia, B. (2020). Peritoneal microbiome in end-stage renal disease patients and the impact of peritoneal dialysis therapy. Microorganisms 8, 173. doi: 10.3390/microorganisms8020173
Søby, J. H., Watt, S. K., Vogelsang, R. P., Servant, F., Lelouvier, B., Raskov, H., et al. (2020). Alterations in blood microbiota after colonic cancer surgery. BJS Open 4, 1227–1237. doi: 10.1002/bjs5.50357
Suparan, K., Sriwichaiin, S., Chattipakorn, N., and Chattipakorn, S. C. (2022). Human blood bacteriome: Eubiotic and dysbiotic states in health and diseases. Cells 11, 2015. doi: 10.3390/cells11132015
Tamariz, J., Voynarovska, K., Prinz, M., and Caragine, T. (2006). The application of ultraviolet irradiation to exogenous sources of DNA in plasticware and water for the amplification of low copy number DNA. J. Forensic Sci. 51, 790–794. doi: 10.1111/j.1556-4029.2006.00172.x
Tan, C. C. S., Ko, K. K. K., Chen, H., Liu, J., Loh, M., Consortium, S. G. K. H., et al. (2023). No evidence for a common blood microbiome based on a population study of. Nat. Microbiol. 8 (5), 973–985. doi: 10.1038/s41564-023-01350-w
Tedeschi, G. G., Amici, D., and Paparelli, M. (1969). Incorporation of nucleosides and amino-acids in human erythrocyte suspensions: possible relation with a diffuse infection of mycoplasms or bacteria in the L form. Nature 222, 1285–1286. doi: 10.1038/2221285a0
Tilahun, Y., Pinango, J. Q., Johnson, F., Lett, C., Smith, K., Gipson, T., et al. (2022). Transcript and blood − microbiome analysis towards a blood diagnostic tool for goats affected by Haemonchus contortus. Sci. Rep. 12 (1), 5362. doi: 10.1038/s41598-022-08939-x
Velmurugan, G., Dinakaran, V., Rajendhran, J., and Swaminathan, K. (2020). Blood microbiota and circulating microbial metabolites in diabetes and cardiovascular disease. Trends Endocrinol. Metab. 31, 835–847. doi: 10.1016/j.tem.2020.01.013
Vientoos-Plotts, A. I., Ericsson, A. C., Rindt, H., Grobman, M. E., Graham, A., Bishop, K., et al. (2017). Dynamic changes of the respiratory microbiota and its relationship to fecal and blood microbiota in healthy young cats. PloS One 12, 1–17. doi: 10.1371/journal.pone.0173818
Vijayvargiya, P., Jeraldo, P. R., Thoendel, M. J., Greenwood-Quaintance, K. E., Esquer Garrigos, Z., Sohail, M. R., et al. (2019). Application of metagenomic shotgun sequencing to detect vector-borne pathogens in clinical blood samples. PloS One 14, e0222915. doi: 10.1371/journal.pone.0222915
Virseda-Berdices, A., BroChado-Kith, O., Díez, C., Hontanon, V., Berenguer, J., González-García, J., et al. (2022). Blood microbiome is associated with changes in portal hypertension after successful direct-acting antiviral therapy in patients with HCV-related cirrhosis. J. Antimicrob. Chemother. 77, 719–726. doi: 10.1093/jac/dkab444
Walsh, S. J. and Rau, L. M. (2000). Autoimmune diseases: a leading cause of death among young and middle-aged women in the United States. Am. J. Public Health 90, 1463. doi: 10.2105/ajph.90.9.1463
Wang, C. (2021). Characterization of the blood and neutrophil - specific microbiomes and exploration of potential bacterial biomarkers for sepsis in surgical patients. Microbial Pathog. 152, 1343–1357. doi: 10.1002/iid3.483
Wani, A. K., Roy, P., and Kumar, V. (2022). Metagenomics and artificial intelligence in the context of human health. Infect. Genet. Evol. 100, 105267. doi: 10.1016/j.meegid.2022.105267
Weyrich, L. S., Duchene, S., Soubrier, J., Arriola, L., Llamas, B., Breen, J., et al. (2017). Neanderthal behaviour, diet, and disease inferred from ancient DNA in dental calculus. Nature 544, 357–361. doi: 10.1038/nature21674
Weyrich, L. S., Farrer, A. G., Eisenhofer, R., Arriola, L. A., Young, J., Selway, C. A., et al. (2019). Laboratory contamination over time during low-biomass sample analysis. Mol. Ecol. Resour. 19, 982–996. doi: 10.1111/1755-0998.13011
Whittle, E., Leonard, M. O., Harrison, R., Gant, T. W., and Tonge, D. P. (2019). Multi-method characterization of the human circulating microbiome. Front. Microbiol. 10. doi: 10.3389/fmicb.2018.03266
Yang, D., Wang, X., Zhou, X., Zhao, J., Yang, H., Wang, S., et al. (2021). Blood microbiota diversity determines response of advanced colorectal cancer to chemotherapy combined with adoptive T cell immunotherapy. Oncoimmunology 10, 1976953. doi: 10.1080/2162402X.2021.1976953
Yun, Y., Kim, H., Chang, Y., Lee, Y., Ryu, S., Shin, H., et al. (2019). Characterization of the blood microbiota in korean females with rosacea. Dermatology (Basel, Switzerland) 235, 255–259. doi: 10.1159/000496968
Zaneveld, J. R., McMinds, R., and Vega Thurber, R. (2017). Stress and stability: applying the Anna Karenina principle to animal microbiomes. Nat. Microbiol. 2, 1–8. doi: 10.1038/nmicrobiol.2017.121
Zhang, Y., Zhao, R., Shi, D., Sun, S., Ren, H., Zhao, H., et al. (2019). Characterization of the circulating microbiome in acute - on - chronic liver failure associated with hepatitis B. Liver Intl., 39(7), 1207–1216. doi: 10.1111/liv.14097
Keywords: systemic diseases, blood microbiome, controversies, challenges, future directions
Citation: Khan I, Irfan M, Khan I, Noor U, Xie X and Li Z (2025) Blood microbiome signatures in systemic diseases: current insights, methodological pitfalls, and future horizons. Front. Cell. Infect. Microbiol. 15:1616029. doi: 10.3389/fcimb.2025.1616029
Received: 23 April 2025; Accepted: 09 July 2025;
Published: 28 July 2025.
Edited by:
Keiji Nagano, Health Sciences University of Hokkaido, JapanReviewed by:
Atif Khurshid Wani, Lovely Professional University, IndiaMahaldeep Kaur, National Institutes of Health (NIH), United States
Copyright © 2025 Khan, Irfan, Khan, Noor, Xie and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaodong Xie, eGR4aWVAbHp1LmVkdS5jbg==; Zhiqiang Li, bGl6aGlxaWFuZzY3NjdAMTYzLmNvbQ==