 Xin Wen
Xin Wen Xue Liu
Xue Liu Cheng Zhang
Cheng Zhang Xi Zhang
Xi Zhang- Medical Center of Hematology, Xinqiao Hospital of Army Medical University; Chongqing Key Laboratory of Hematology and Microenvironment, State Key Laboratory of Trauma and Chemical Poisoning, Army Medical University, Chongqing, China
Clostridioides difficile infection (CDI) is a significant cause of antibiotic-associated diarrhea and pseudomembranous colitis, manifesting as mild diarrhea, fulminant colitis, and even death. It is typically recognized as a healthcare-associated infection. Glucosyltransferase toxin A (TcdA) and toxin B (TcdB) are two major factors responsible for the pathogenicity of Clostridioides difficile (C. difficile). They bind to cell surface receptors and enter the cytoplasm via pH-dependent pore formation, causing cell death by inactivating GTPase. This review elucidates the pathogenic mechanisms of C. difficile glucosyltransferase toxins and discusses the interactions between the two toxins and host cells. It also summarizes current progresses in CDI therapies, providing a comprehensive understanding of the disease and laying the foundation for developing novel therapies and management strategies.
1 Introduction
Clostridioides difficile (C. difficile) is an obligate anaerobic, spore forming, Gram-positive bacillus, which was first isolated from the stool samples of healthy infants in 1935 and initially recognized as a kind of normal gut flora (Hall, 1935; Kelly et al., 1994b). It was not classified as an enteric pathogen until Bartlett and colleagues isolated toxin-producing C. difficile from the feces of patients with antibiotic-associated pseudomembranous colitis in 1978 (Bartlett et al., 1978). In an oxygen-rich environment, C. difficile forms spores to resist tough external conditions such as dryness, high temperatures, extreme pH levels, and even lethal effects of various chemicals and disinfectants (Paredes-Sabja et al., 2014). Due to the potent spreading capacity, the spores widely exist in medical environment and result in an inundate spread of C. difficile in the health system (Martin et al., 2016). The clinical manifestations of Clostridioides difficile infection (CDI) exhibit varied degrees of severity. These range from the mild forms such as asymptomatic colonization and mild diarrhea, to the severe conditions including pseudomembranous colitis, toxic megacolon, bowel perforation, and even death (Kociolek and Gerding, 2016). The occurrence of CDI is correlated with several high-risk factors, such as long-term use of antibiotics, weakened immune systems, severe underlying conditions, invasive procedures such as surgery, prolonged hospitalization, and advanced age (Bartlett, 2002; Leffler and Lamont, 2015; Wang et al., 2024).
The pathogenesis of CDI is driven by two types of toxins: large clostridial toxins (LCTs) TcdA and TcdB, and C. difficile transferases (CDT). TcdA and TcdB are considered the major virulence factors, each consisting of four functional domains: a glucosyltransferase domain (GTD), a cysteine proteinase domain (CPD), a transmembrane domain (TMD), and a C-terminal repetitive oligopeptide domain (CROP) (Figure 1) (Chandrasekaran and Lacy, 2017). TcdA and TcdB are internalized via receptor-mediated endocytosis. Glycoprotein 96 (gp96), sulfated glycosaminoglycans (sGAGs), and low-density lipoprotein receptor (LDLR) have been confirmed as the receptors for TcdA (Na et al., 2008; Tao et al., 2019; Schottelndreier et al., 2020), whereas chondroitin sulfate proteoglycan 4 (CSPG4), poliovirus receptor-like 3 (PVRL3), frizzled family (FZDs), and tissue factor pathway inhibitor (TFPI) are key cellular factors that mediate the binding and endocytosis of TcdB (Figure 2A) (Tao et al., 2016; Chen et al., 2021; Tian et al., 2022; Childress et al., 2023). Upon entering the cell, GTD is released into the cytoplasm through a pH-dependent autocleavage process. GTD inactivates GTPases through its glucosyltransferase (GT) activity, leading to disruption of the actin cytoskeleton and ultimately inducing cell death (Figure 2B) (Chandrasekaran and Lacy, 2017). Although both toxins ultimately result in cell death, TcdA is believed to induce apoptosis in a GT-dependent manner. In contrast, TcdB exhibits dose-dependent cytotoxicity, with apoptosis occurring at low doses in a GT-dependent manner and necrotic cell death at high doses in a GT-independent manner (Peritore-Galve et al., 2022).
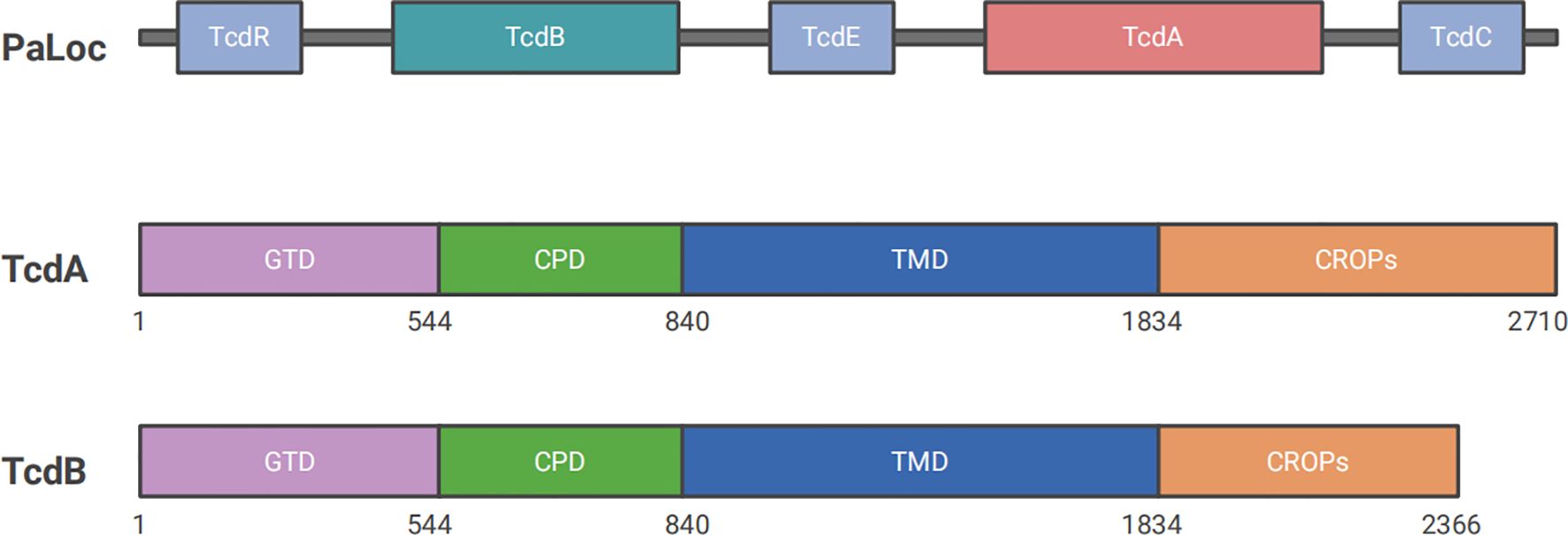
Figure 1. Structures of Clostridioides difficile PaLoc. PaLoc primarily encodes five genes: tcdR, tcdB, tcdE, tcdA, and tcdC. TcdA and tcdB encode two of the most important toxin proteins, TcdA and TcdB, which are responsible for CDI pathogenesis. Both toxins consist of four domains: CROP, which binds to target cell surface receptors; TMD, which is involved in the delivery process; CPD, a self-hydrolytic domain that cleaves and releases GTD into the cytoplasm to exert enzymatic functions; and GTD, which inactivates small GTPases to induce cell death.
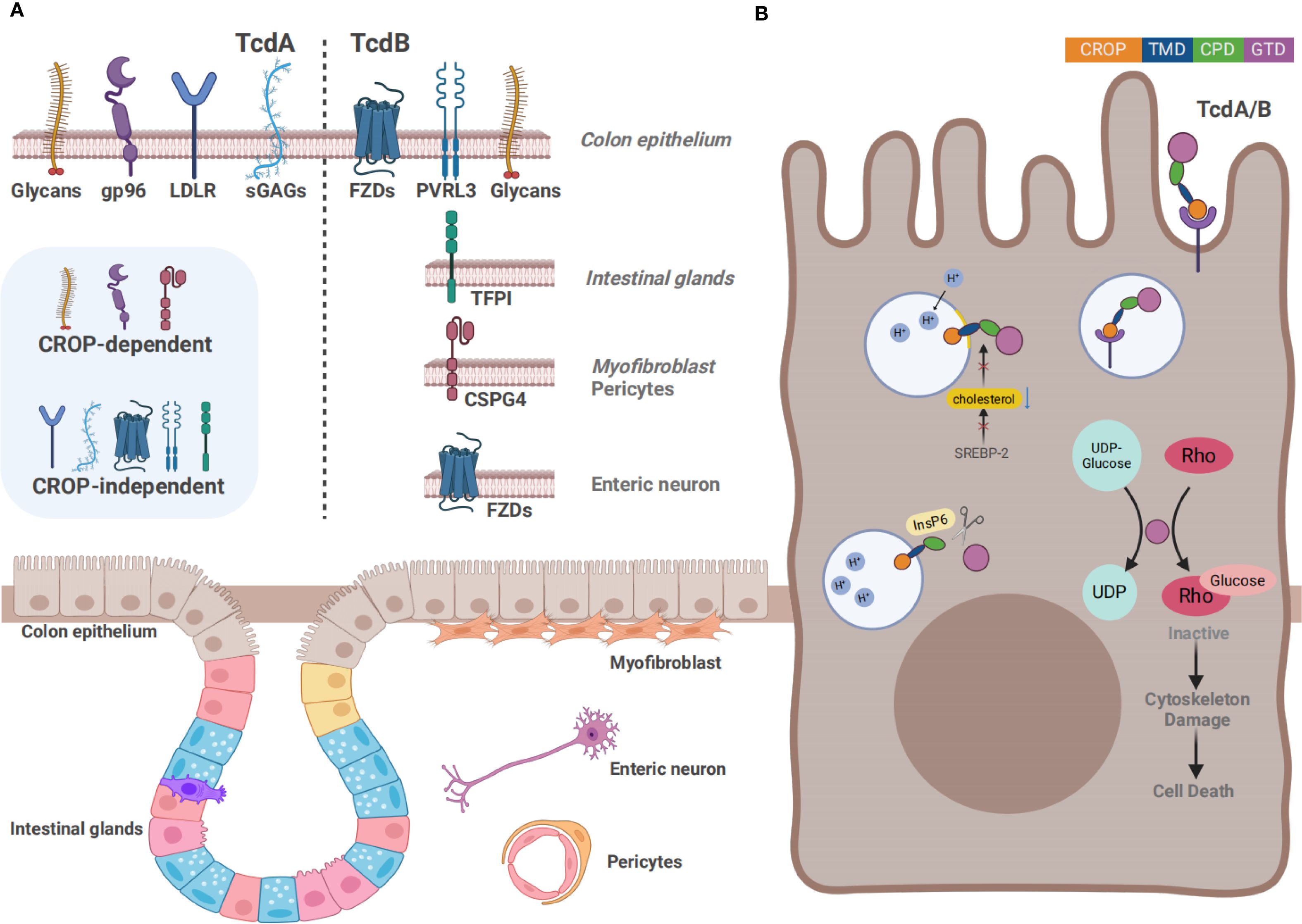
Figure 2. Overview of the action mechanism of TcdA and TcdB (A) The distinct cell surface receptors of TcdA and TcdB. Gp96, sGAGs, LDLR, and glycans such as Lewis X/Y/I have been confirmed as the receptors for TcdA, whereas CSPG4, PVRL3, FZDs, and TFPI are key receptors for TcdB. In addition to the CROP-dependent receptors, several receptors bind to the toxins independently of the CROP domain. Except for IECs, receptors are expressed on several other cell types, including intestinal glands, myofibroblasts, pericytes, and enteric neurons. (B) The action mechanism of TcdA and TcdB. Firstly, the toxins bind to the cell surface receptors through the CROP domain or other structure, followed by the internalization of toxins in acidic endosomes formed by endocytosis. Acidic endosomes subsequently trigger the pore formation and transport the CPD and GTD into the cytosol. Next, autocatalytic cleavage of the toxins is processed in the presence of InsP6, by which GTD is released into the cytosol. Rho GTPases are inactivated by transferring UDP-glucose to them, ultimately resulting in the induction of cytoskeletal damage.
Although treatments for CDI vary worldwide, antibiotics continue to be the first-line treatment option. However, due to its high mortality and recurrence rates, the prevention and treatment of CDI remain a great challenge in the field of healthcare (Maroo and Lamont, 2006; Surawicz, 2009). Except for antibiotics, scientists have been focusing on developing other therapeutic strategies, including monoclonal antibodies (mAbs), vaccines, gut microbiota restoration, and natural small molecular products. For example, fecal microbiota transplantation (FMT) was proposed as a treatment for recurrent Clostridioides difficile infection (rCDI) in 2013 (van Nood et al., 2013). Evidence from a phase III clinical trial indicated that the anti-TcdB mAb bezlotoxumab significantly reduced the recurrence rate of CDI in 2017 (Wilcox et al., 2017). In the same year, fidaxomicin replaced metronidazole as the first-line treatment for initial CDI episode (McDonald et al., 2018). With the deepening of the research on CDI therapies, the development of bioactive molecules and vaccines continues to provide new aspects for the disease prevention and treatment (Kordus et al., 2021; Bratkovic et al., 2024; Remich et al., 2024). To date, CDI remains an extremely complex issue that has garnered significant interest among researchers. This review focuses on elaborating the epidemiology of CDI, the mechanisms of toxin action, the toxin–host interaction pathways and recent advances in therapies. It aims to provide a comprehensive understanding of the disease and sketch a general view for developing novel therapies and management strategies for CDI.
2 Epidemiology and medical burden of Clostridioides difficile infection
CDI has become a concerning challenge worldwide, with rising trends in incidence and mortality rates. The epidemiological situation in medical institutions is even worse (Di Bella et al., 2024). Statistics indicate that in hospitals in North America and Europe, the annual incidence of CDI has reached approximately 4~10/1,000 cases of hospitalized patients (Rupnik et al., 2009). Compared to other healthcare-associated infections, CDI has a higher mortality rate. According to statistics from the Centers for Disease Control and Prevention, approximately 453,000 cases of CDI occur annually in the United States. These cases result in about 29,000 deaths and impose $1.5 billion in healthcare costs (Lessa et al., 2015). Moreover, CDI has a relatively high recurrence rate of about 20% to 30% (Wingen-Heimann et al., 2023). Numerous clinical trials conducted in Germany, France, Japan, and other countries have demonstrated that patients with rCDI experience longer hospital stays and incur higher medical costs than those with initial CDI, placing a heavy burden on the healthcare system (Heimann et al., 2015; Dinh et al., 2019; Kimura et al., 2020). Therefore, early diagnosis and treatment of CDI patients can reduce the risks of complications, recurrence, and infection-associated death.
The epidemiological characteristics of CDI vary by region and time, which have been evolving during the past three decades. It was initially prevalent in Western countries, then a sharp rise in CDI incidence and mortality rate occurred from 1991 to 2003, followed by its spread worldwide (Pepin, 2004). Later, McDonald et al. in the United States identified a new strain with greater virulence, namely NAP1/RT027. This strain is resistant to fluoroquinolones and rapidly spreads to Europe before prevailing worldwide, severely affecting human health and healthcare costs (McDonald et al., 2005; Arvand et al., 2014; Zhou et al., 2019). From 1999 to 2004, statistics showed a fourfold increase of CDI-related mortality in the United States (Redelings et al., 2007). Owing to the restrictions on the use of fluoroquinolone antibiotics, healthcare-associated events have decreased more or less since 2011. However, the prevalence of CDI remains widespread (Dingle et al., 2017). In recent years, another C. difficile strain, RT017, expressing TcdB only and exhibiting relatively strong virulence and spreading capacity, is prevailing rapidly from Asia to the whole world and attracting significant attention (Cairns et al., 2015; Imwattana et al., 2019). This aligns with our previous research on the epidemic and nosocomial transmission of C. difficile in China (Wen et al., 2022). The severity of this public health issue cannot be ignored, and it is of great significance to enhance the supervision over CDI and devote to clarifying its pathogenic mechanisms, so as to develop more effective clinical preventive and therapeutic strategies.
3 Overview of Clostridioides difficile toxins
The pathogenesis of C. difficile is mediated by two pathogenic islands: the pathogenicity locus (PaLoc) and the binary toxin encoding locus (CdtLoc). PaLoc is the primary pathogenic locus present in all toxic isolates, while CdtLoc is present only in a minority of PCR ribotypes (Awad et al., 2014).
TcdA and TcdB, two macromolecular proteins with molecular weights of 308 kDa and 270 kDa respectively, are encoded by specific genes on the PaLoc with a size of 19.6 kb. Another three genes are located on the PaLoc, including tcdR, tcdE, and tcdC, which primarily function in regulating the expression of the toxins (Dingle et al., 2014; Chandrasekaran and Lacy, 2017). TcdA and TcdB are two principal toxins responsible for CDI and belong to the LCT family (Orrell and Melnyk, 2021). Both of them comprise four functional domains: GTD, CPD, TMD, and CROP. Each domain plays an important role during the activation process of biological virulence. Firstly, the CROP domain binds to cell surface receptors (such as glycoproteins and glycolipids) to mediate cellular internalization by initiating endocytosis to form vesicles. Secondly, the conformational change of TMD under acidic environment facilitates the toxins to cross the cell membrane by the pore formation. Then, catalysed by inositol hexaphosphate (InsP6), CPD performs autocleavage by hydrolyzing specific protein substrates to release GTD into the cytoplasm. Finally, GTD, as a glycosylation enzyme, inactivates small GTPases of the Rho/Ras subfamily by adding glucose molecules in the cytoplasm (Jank and Aktories, 2008; Abt et al., 2016). Signaling proteins concluded in Rho/Ras subfamily regulate actin-dependent processes, such as cell migration, phagocytosis, and cell contraction. They also participate in various signaling pathways that control gene expression, cell cycle, and apoptosis (Jank et al., 2007a; Alam and Madan, 2024). As the first discovered toxin of C. difficile, TcdA has received extensive attention in various studies. TcdA was formerly known as an enterotoxin because it possessed intestinal cell adhesion properties. It mainly caused intestinal inflammation, exudation, and mucosal damage by disrupting intestinal barrier function (Triadafilopoulos et al., 1987; Smits et al., 2016). Evidence from previous studies has shown that TcdA is more effective in promoting secretion, causing mucosal damage, and initiating inflammation compared to TcdB (Triadafilopoulos et al., 1987). In contrast, TcdB was known as a cytotoxin that primarily functioned by disrupting the cytoskeleton and enhancing cell permeability, which led to cell rounding and death (Mileto et al., 2020). However, the specific roles and mechanisms of both toxins remain controversial due to conflicting evidence in the literature. An early study found that TcdA was shown to cause clinical symptoms independently, whereas TcdB toxicity depended on the presence of TcdA or pre-existing damage to the intestinal mucosa in animal models (Lyerly et al., 1985). Following, a report published in Nature in 2009 proposed that TcdB played a crucial role in pathogenesis, while strains expressing only TcdA were non-pathogenic in vitro and in a hamster disease model (Lyras et al., 2009). Shortly after, another study has claimed a different conclusion: both TcdA and TcdB are toxic (Kuehne et al., 2010). Researchers have speculated that the discrepancies arose from SNPs in toxin sequences, which is confirmed by the studies afterwards (Lanis et al., 2010; Rupnik and Janezic, 2015; Knight et al., 2015). The homology of amino acid sequences between TcdA and TcdB is 63% (von Eichel-Streiber et al., 1992). A study suggested that TcdB had a 100 to 1,000-fold higher efficacy than TcdA at the cellular level (Riegler et al., 1995). Building on this, another study using three different animal models demonstrated that TcdB played a more significant role in CDI, potentially leading to multiple organ dysfunction syndrome (Carter et al., 2015). Additionally, recent studies have shown that TcdA and TcdB target different receptors during the exertion of their toxic effects, leading to varying degrees of host immune and inflammatory responses (Liu et al., 2021a; Li and Saavedra, 2025). It is suggested that TcdB may play a more critical role in inducing cell death and promoting disease progression.
The CdtLoc, which is 6.2 kb in size, comprises three genes: cdtA, cdtB, and cdtR, among which cdtA and cdtB encode CDT to enhance the adhesion of C. difficile to target cells, while cdtR plays a regulatory role (Aktories et al., 2018). CDT, a binary ADP-ribosylation toxin, is detected in 5-30% of clinical C. difficile isolates, which tend to have higher virulence, such as RT027 and RT078 (Gerding et al., 2014). It consists of two parts: chain A and B, where chain A shows enzymatic activity and is responsible for ADP-ribosylation, and chain B is responsible for binding to host cell surface receptors, thereby mediating toxin entry. Chain A is activated after the toxin entry, which is able to transfer ADP-ribose groups to actin in the host cell, leading to changes in actin polymerization. The toxins affect cytoskeleton stability by modifying actin, thereby interfering with cell division and migration (Aktories et al., 2017).
These toxins work together to destroy the colon epithelium, causing the symptomatic manifestations of CDI, such as fluid secretion, inflammation, and tissue damage (Lyerly et al., 1988; Voth and Ballard, 2005; Kordus et al., 2021; Skinner et al., 2021; Pourliotopoulou et al., 2024). Although the enzymatic functions of the toxins have been majorly determined, the mechanism of toxin-host cell interaction remains unclear. Below, we will discuss the progress on the GT toxins, TcdA and TcdB.
3.1 The cellular receptors of TcdA and TcdB
The CROP domain, also known as the receptor-binding domain, is recognized as a necessary component for the interaction between toxins and host cell, which can mediate the initiation of toxin internalization. The CROP region of TcdA and TcdB binds to distinct surface receptors (Figure 2A) (Dingle et al., 2008). TcdA has been reported to bind to several protein receptor candidates, such as gp96, sGAGs, sucrase-isomaltase (SI), LDLR, Galα1-3Galβ1-4GlcNAc, and Lewis X/Y/I glycans. Nevertheless, there remain inconsistencies in related experimental data. Early studies indicated that TcdA bound to the trisaccharide Galα1-3Galβ1-4GlcNAc in vitro. However, this trisaccharide is not naturally expressed on human cells (Krivan et al., 1986; Clark et al., 1987). Another study indicated Lewis X/Y/I glycans as potential receptors for TcdA, which indeed exist on the human intestinal epithelial cells (IECs) (Tucker and Wilkins, 1991). TcdA can also bind to SI, a glycoprotein located on the brush-like edge of the rabbit small intestine (Pothoulakis et al., 1996). However, no such reports have been documented in human IECs. Gp96, a member of the heat shock protein family, is expressed on the endoplasmic reticulum as well as human colonocytes. Gp96 is regarded as one of the cell surface receptors for TcdA. However, gp96-deficient cells are only partially resistant to TcdA, suggesting that TcdA may also bind to other receptors (Na et al., 2008). Meanwhile, another study has indicated that gp96 is a binding receptor, whereas low-density lipoprotein receptor-associated protein 1 (LRP1) acts as an endocytic receptor for TcdA (Schottelndreier et al., 2020). Receptors LDLR and sGAGs are ubiquitously expressed on different mammalian cell surfaces (Jinno and Park, 2015). A recent study identified sGAGs and LDLR as CROP-independent host factors through genome-wide CRISPR-cas9 mediated screen technology, since both of them can mediate the binding and entry of the truncated TcdA which lacks the CROP domain (Tao et al., 2019). Subsequently, another in vitro study revealed that blocking the sGAGs could effectively inhibit the endocytosis of TcdA, further confirming that sGAGs played a crucial role in the endocytosis of TcdA (Zhang et al., 2025). Additionally, recent studies have found that the CROP domain exhibits a pH-dependent dynamic characteristic, which likely plays a crucial role in the cytotoxicity of TcdA (Chen et al., 2022a; Aminzadeh et al., 2022).
TcdB has been reported to bind to CSPG4, PVRL3, FZDs, TFPI, and a variety of glycans. CSPG4 is also known as neuron-glial antigen 2, which is highly expressed in sub-epithelial myofibroblast cells within the colonic tissues instead of the colonic epithelium (Tamburini et al., 2019). It was firstly confirmed as a CROP-dependent TcdB receptor through shRNA-mediated knock-down screen in 2014 (Yuan et al., 2015). There is a direct interaction between the N-terminus of CSPG4 and the C-terminus of TcdB, which can be promoted by extracellular Ca2+. The soluble peptide in the toxin-binding domain of CSPG4 can protect cells from TcdB (Yuan et al., 2015; Doyle et al., 2024). Meanwhile, another study showed that the cytotoxicity of TcdB was reduced when the expression of CSPG4 was down-regulated by inhibiting the Hippo signaling pathway (Larabee et al., 2023). However, TcdB was found to reduce IL-8 expression in CSPG4-knockout mice, but the mortality of CSPG4-knockout mice was not significantly different from that of the wild type mice, indicating that there may exist other receptors for TcdB (Yuan et al., 2015). Therefore, researchers have proposed the dual-receptor model for TcdB endocytosis, and the presence of alternative receptors was further confirmed by bezlotoxumab. It is an anti-TcdB antibody approved by the US Food and Drug Administration (FDA). The inhibition of TcdB binding to CSPG4 by the allosteric mechanism of bezlotoxumab did not show significant neutralization efficacy against numerous TcdB variants from prevalent hypervirulent strains (Chen et al., 2021). In the same year, another novel TcdB receptor, LRP1, was discovered by CRISPR-Cas9 screening in CSPG4-deficient HeLa cells by Shengjie Guo and his colleagues (Guo et al., 2022). However, previous research has indicated that LRP1 is not the endocytic receptor for TcdB in fibroblasts, suggesting that toxin receptors may exhibit cell type specificity (Schottelndreier et al., 2020). PVRL3, also known as Nectin-3, was identified as a cellular factor by a genetrap insertional mutagenesis screen, which was necessary for TcdB-mediated necrotic cell death. Additionally, it binds to TcdB independently of the CROP domain (LaFrance et al., 2015; Sakisaka and Takai, 2004). PVRL3 is highly expressed on the epithelial surface of the human colon. A study has revealed the unexpected localization of PVRL3 on the brush border of colonic epithelial cells by immunofluorescence microscopy, which is different from the localization of CSPG4 at epithelial cell junctions (Childress et al., 2023). In 2016, members of the Wnt receptor FZDs were identified as TcdB receptors by CRISPR/Cas9-mediated genome-wide screening by Liang Tao and his colleagues (Tao et al., 2016). Unlike CSPG4, FZDs are CROP-independent receptors. The FZDs family is a group of 7-pass transmembrane proteins. FZDs possess a unique extracellular domain termed as the cysteine-rich domain (CRD), which serves as the binding site of Wnt (Chen et al., 2018). It consists of 10 human genes (FZD1-10), among which FZD1/2/7 share sequence similarity of approximate 98% and are confirmed to facilitate TcdB entry into HeLa cells (MacDonald and He, 2012). Since both TcdB and Wnt bind to the FZD-CRD, it is theoretically feasible that the interaction between TcdB and FZDs may directly contribute to the disruption of the colon epithelium by blocking Wnt signaling (Chen et al., 2019). Another study has indicated that TcdB from epidemic NAP1/RT027 strains induced the dysfunctional stem cell state in both mice and human colonic organoids without binding to FZD1/2/7, instead it maintained the ability to interact with CSPG4 and Nectin-3 (Mileto et al., 2020). This suggested that different TcdB variants may adapt to various receptors. Recently, a number of studies have indicated that TcdB variants entered host cell by binding to distinct receptors, although they shared similar substrate profiles and cytotoxicity (Lopez-Urena et al., 2019; Henkel et al., 2020). In 2021, Liang Tao’s team has proposed that TcdB variants presented highly diversified receptor preferences: TcdB1 binds to two known receptors CSPG4 and FZDs, TcdB2 selectively binds to CSPG4, TcdB3 tends to interact with FZDs, and TcdB4 exerts toxic effects in a CSPG4/FZDs-independent manner (Pan et al., 2021). TFPI is highly expressed in the intestinal glands. Moreover, a recent study has identified TFPI as a colonic crypt receptor for TcdB from clade2 strain, a clinically prevalent and highly virulent strain. The severity of the toxic effects of clade2 strain may be related to the specific receptor of this TcdB variant (Tian et al., 2022; Luo et al., 2022).
With the development of the genome-wide CRISPR-Cas9 technology, researchers have identified a series of molecules as host receptors for TcdA and TcdB. While TcdA receptors remain conserved, TcdB subtypes are highly diversified in receptor specificity, translocation ability, inflammatory responses, and pathological outcomes. Several studies have shown that the family of Clostridioides GT toxins enter cells by binding to multiple receptors, with more than one host target (Manse and Baldwin, 2015; Lambert and Baldwin, 2016; Pan et al., 2021). With the proposal of the dual-receptor model, receptor binding sites for TcdA and TcdB are apparently not limited to the CROP domain. Recently, studies on toxin receptor revealed that TcdA and TcdB lacking the CROP domain could still interfere with host cell function, reflecting the presence of additional receptor binding regions (Schorch et al., 2014; Yuan et al., 2015; LaFrance et al., 2015; Tao et al., 2016; Doyle et al., 2024). Previous research has shown that TcdA and TcdB adopted distinct endocytic pathways. TcdB enters the cells via the clathrin-dependent endocytic pathway (Papatheodorou et al., 2019), while the internalization of TcdA is accomplished by a clathrin- and caveolae-independent mechanism mediated by PACSIN2 and dynamin (Chandrasekaran et al., 2016). However, the pathway by which toxins enter cells remains to be further clarified.
3.2 Pore-formation, translocation and autoprocessing
The TMD, also known as the delivery domain, plays the key role in forming pores to mediate the translocation of toxins. Endosomes form after toxins bind to cell surface receptors and are internalized into the cytoplasm through endocytosis. The acidic environment in endosomes is essential for the translocation of the toxins. Decreased pH in endosomes can induce a structural change of TMD, which enhances hydrophobicity and triggers the insertion of the TMD into the membrane, forming a pore-like α-helical structure (Qa’Dan et al., 2000; Orrell et al., 2017, Orrell et al., 2018). Then GTD and CPD translocate into the cytoplasm via the pores (Figure 2B). The formation of pores is usually accompanied by the formation of ion channels, which is a phenomenon historically reported for other translocating toxins (Barth et al., 2001). The optimal pH for the hydrophobic transition of TcdB ranges from 4.0 to 5.0, although fluctuations in the optimal pH have been observed among different TcdB subtypes (Lanis et al., 2013). An in vivo study has indicated that TcdB from hypervirulent C. difficile strains undergoes conformational changes at a higher pH to facilitate itself to translocate into cytoplasm more rapidly during the early stage of endocytosis (Lanis et al., 2010). A structurally related study has defined the minimal pore-forming region of TcdB, which is located at amino acid residues 830 and 990 (Genisyuerek et al., 2011). In an analysis covering over 8,000 tcdB genes, the sequence of the TMD was found to be the most evolutionarily conserved, indicating its potential in being an attractive target for broad-spectrum therapeutics (Mansfield et al., 2020). TcdA undergoes conformational changes and forms pores at a low pH level, and the process of pore formation by TcdA is cholesterol-dependent. Meanwhile, similar results were obtained for TcdB, suggesting that pore formation is dependent on the presence of cholesterol (Giesemann et al., 2006a). The sterol regulatory element–binding protein 2 (SREBP-2) pathway plays a crucial role in regulating the cholesterol content in cellular membranes. A recent in vitro study has shown that inhibiting the SREBP-2 pathway disrupted the cholesterol-dependent pore formation of TcdB in cell membranes, demonstrating that the SREBP-2 pathway may be a suitable target for antitoxin therapeutics against C. difficile toxins (Papatheodorou et al., 2019). Moreover, another in vitro study with cultured cells and human intestinal organoids, has found that amiodarone, a clinically common agent for treating cardiac arrhythmia, can inhibit cholesterol biosynthesis and subsequently interfere with the formation of translocation pores, suggesting its potential role in antitoxin therapy (Schumacher et al., 2023).
The CPD, also known as the autoprotease domain, facilitates the autoproteolytic cleavage and releases the GTD into the cytosol. With the pore formation, the GTD and CPD are unfolded and translocated to the host cytoplasm, where their biological activity was recovered through refolding under the assistance of the chaperonin TCP-1 ring complex/chaperonin containing TCP-1 (TRiC/CCT) (Giesemann et al., 2008; Steinemann et al., 2018). In order to initiate autoprocessing, CPD binds to the cellular host factor InsP6. Cytosolic InsP6, a molecule uniquely found within the cytosol of eukaryotic cells, acts as an allosteric activator to initiate the cysteine-protease activity of the CPD (Egerer et al., 2007; Reineke et al., 2007; Pruitt et al., 2009). At neutral pH, autocleavage occurs in the rear of a conserved leucine residue situated between the CPD and GTD, thereby GTD is released into the cytosol (Pfeifer et al., 2003; Rupnik et al., 2005). Though TcdA and TcdB operate by a similar mechanism, TcdB is more susceptible to autocleavage triggered by InsP6 than TcdA (Kreimeyer et al., 2011; Olling et al., 2014; Kordus et al., 2021). TcdB from epidemic NAP1/RT027 strains exhibited enhanced autoprocessing activity in vitro, suggesting that the sensitivity towards InsP6-mediated cleavage may be responsible for the varied toxicity of TcdB subtypes (Lanis et al., 2012). Research on crystal structures has claimed that zinc ions were essential for the autoprocessing activity of TcdA and TcdB in vitro (Chumbler et al., 2016). Another study proved that S-nitrosylation attenuated the toxicity of TcdA and TcdB in a CDI mouse model, which was further confirmed to be associated with the inhibition of toxin autocleavage (Savidge et al., 2011). Additionally, in mouse and human intestinal model, cysteine protease-mediated autoprocessing has been reported to be involved in the regulation of the proinflammatory activities of TcdA and TcdB (Zhang et al., 2018). To conclude, these studies provide new insights into the development of therapeutics against CDI, and further research on the mechanisms of autoprocessing is required to pave the way for new therapeutic approaches.
3.3 Glycosylation of the GTD and its enzymatic activity
Both GT and cysteine proteinase activities are essential for the virulence of C. difficile. A study has revealed that the GT activity of TcdB is a crucial factor for its cytotoxicity by screening a single-domain heavy-chain variable region (VHH) library, whereas cysteine proteinase activity merely plays a regulatory role in the release of GTD from the entire toxin molecule (Li et al., 2015). The release of GTD into the cytoplasm facilitated the selective transfer of UDP-glucose to the Rho and Ras GTPases, inactivating these GTPases by adding glucose molecules (Figure 2B). Rho GTPase serves as a molecular switch that regulates various processes, including the organization of the actin cytoskeleton, cell cycle progression, gene transcription, and the activity of numerous enzymes (Etienne-Manneville and Hall, 2002; Burridge and Wennerberg, 2004; Spiering and Hodgson, 2011; Chen et al., 2015). While Ras GTPase primarily controls cell differentiation and proliferation, angiogenesis, and cell adhesion (Macara et al., 1996; Colicelli, 2004). The inactivation of both proteins ultimately causes cell death. On the contrary, UDP-glucose deficiency renders the cells resistant to these toxins (Chaves-Olarte et al., 1996; Flores-Diaz et al., 1997). The GT activity is crucial for the toxic effects of both TcdA and TcdB. Although their sequences show similarity, TcdA and TcdB inactivate different GTPases in the host (Chaves-Olarte et al., 1997; Zeiser et al., 2013; Chen et al., 2022b). A study focusing on the differences in GT activity and substrate specificity between TcdA and TcdB has yielded an interesting result: TcdA is capable of modifying Rap2A in the Ras family, which is incapable for TcdB (Pruitt et al., 2012). Such difference has indicated that the ability to modify substrates from the Rho and Ras family is a prospective pointcut in understanding the pathogenic mechanisms of TcdA. Another study has proposed that TcdB can glycosylate members of the Rho protein family, including RhoA, Rac1, RhoG, TC10, and Cdc42, whereas TcdA cannot glycosylate RhoG and TC10 (Genth et al., 2008). These findings are significant for understanding the structural and functional distinctions between TcdA and TcdB. Meanwhile, several studies have proposed that toxins from various strains of C. difficile exhibit distinct preferences in GTPase substrates, which may contribute to the differential pathogenicity. A biomolecular structure study has further confirmed that TcdB variants selectively modify the structural basis of Rho and Ras GTPases through GTD, potentially causing diverse cytopathic effects in host cells (Liu et al., 2021b). Early in vitro studies have found that TcdB glycosylates Thr37 of RhoA, which leads to the degradation of the actin cytoskeleton and thereby causes cell death (Just et al., 1995). Subsequently, researchers have pinpointed Asp270, Arg273, Tyr284, Asn384, and Trp520 as essential amino acid residues for GT activity by alanine scanning techniques (Jank et al., 2007b). Mutations in these positions significantly reduce enzyme activity, indicating that these sites may serve as new therapeutic targets for C. difficile toxins.
Repairing the epithelium damaged by CDI and maintaining intestinal integrity are key steps in preventing rCDI. The Wnt/β-catenin pathway is a primary driver for epithelial cell proliferation in colonic crypts. An in vitro study has shown that TcdA inhibits Wnt/β-catenin signaling in a dose-dependent manner, which occurs primarily through inactivating the Rho GTPases rather than caspase-dependent β-catenin degradation (Bezerra et al., 2014). In 2020, another in vivo study further showed that TcdA inactivates Rac1 by glycosylation, by which the Wnt/β-catenin signaling pathway is inhibited and β-catenin is subsequently prevented from entering the nucleus, thereby inhibiting cell proliferation (Martins et al., 2020). Additionally, it has been reported that TcdA and TcdB affect the Hippo pathway, which is essential for tissue homeostasis and regeneration. YAP and TAZ, the downstream transcriptional co-activators of the Hippo pathway, are able to promote cell proliferation and intestinal regeneration (Gregorieff et al., 2015). The toxins have been demonstrated to inhibit YAP and TAZ by inactivating GTPases in IECs (Song et al., 2021). These findings suggest that these pathways may serve as therapeutic targets for CDI. Numerous studies have shown that TcdA and TcdB induce apoptosis in IECs by glycosylating Rho GTPases, which depends on the activation of caspase-3 (Qa’Dan et al., 2002; Nottrott et al., 2007). Caspase-6, -8, and -9 are also involved in toxins-induced apoptosis (Brito et al., 2002; Carneiro et al., 2006). TcdB has been found to induce apoptosis through both caspase-dependent and caspase-independent pathways. Caspase-dependent apoptosis involves the activation of caspase-3, while caspase-independent apoptosis may result from the GTD-induced inactivation of Rho, Rac, and Cdc42 (Qa’Dan et al., 2002). TcdA and TcdB can activate caspase-dependent apoptosis through a death receptor or mitochondria-dependent pathway (Elmore, 2007). In the mitochondria-dependent pathway, toxins can release cytochrome c and activate caspase-9 by altering mitochondrial outer membrane permeability, and this process is regulated by the anti-apoptotic members of the Bcl-2 family (Matarrese et al., 2007; Matte et al., 2009). Meanwhile, in the death receptor pathway, caspase-8 is activated by transmembrane death receptors, such as TNF-α, Fas, or IFN-γ, subsequently triggering IECs apoptosis (Gerhard et al., 2008). Recently, several molecules, such as the globular heads of C1q and junction plakoglobin, have been revealed in vitro and in vivo to play a crucial role in toxin-induced apoptosis of IECs in a mitochondria-dependent manner (Liang et al., 2020; Li et al., 2022). Moreover, it has been identified that the activation of caspase-3/7 by the intrinsic apoptotic pathway is crucial in triggering the apoptosis of IECs in vivo, and this activation does not rely on the pyrin inflammasome (Figure 3A) (Saavedra et al., 2018). Enteric glial cells (EGCs) are components of the enteric nervous system and contribute to maintaining normal intestinal function and the integrity of the intestinal barrier. Studies have indicated that TcdB can induce apoptosis in EGCs through a caspase-dependent but mitochondria-independent pathway which is not influenced by Bcl-2 family members (Macchioni et al., 2017; Fettucciari et al., 2017). Furthermore, stimulating EGCs with pro−inflammatory cytokines significantly enhanced the TcdB-induced apoptosis (Fettucciari et al., 2022). Recent studies have also proposed that adenosine receptors A2A and A2B, and the calcium-permeable channel TRPV4 can participate in modulating toxin-mediated apoptosis of EGCs and inflammatory responses in mice with CDI (Costa et al., 2022; Pacifico et al., 2025). Unlike IECs, myeloid cells like macrophages and dendritic cells show high expression of the cytosolic receptor pyrin (Sharma et al., 2018). In response to the GT activity of TcdA and TcdB, pyrin forms an inflammasome complex which functions as a sensor to activate inflammatory caspases, such as caspase-1 (Xu et al., 2014). Caspase-1 can initiate the maturation and the release of IL-1β and IL-18, and trigger an inflammatory programmed cell death known as pyroptosis (Bergsbaken et al., 2009; Jamilloux et al., 2018). Recent studies have proposed that C. difficile toxins can inactivate RhoA GTPases through the glycosylation of GTD, and subsequently activate the pyrin inflammasome to induce pyroptosis in mouse macrophages and human peripheral blood mononuclear cells (Xu et al., 2014; Gao et al., 2016; Van Gorp et al., 2016). Autophagy, as a pro-death mechanism under certain conditions, mediates degradation of cellular components via the lysosomal system (Kloft et al., 2010). Numerous studies have shown that autophagy plays a significant role in the pathogenicity of microorganisms (Wang et al., 2025b). In various autophagy-deficient cell lines, researchers have demonstrated that TcdB promotes the formation of the phosphoinositide 3-kinase complex and inhibits the mTOR signaling pathway through its GT activity. This, in turn, triggers autophagy and subsequently inhibits host cell proliferation (He et al., 2017). Interestingly, another study has demonstrated that non-toxigenic strains can also induce autophagy in Caco-2 cells, indicating that C. difficile induces autophagy through both toxin-dependent and toxin-independent mechanisms (Azimirad et al., 2023). Surface layer protein A of C. difficile has been shown to induce autophagy in human IECs, suggesting a potential role for other C. difficile virulence factors in regulating the autophagy process (Figure 3A) (Amirkamali et al., 2022).
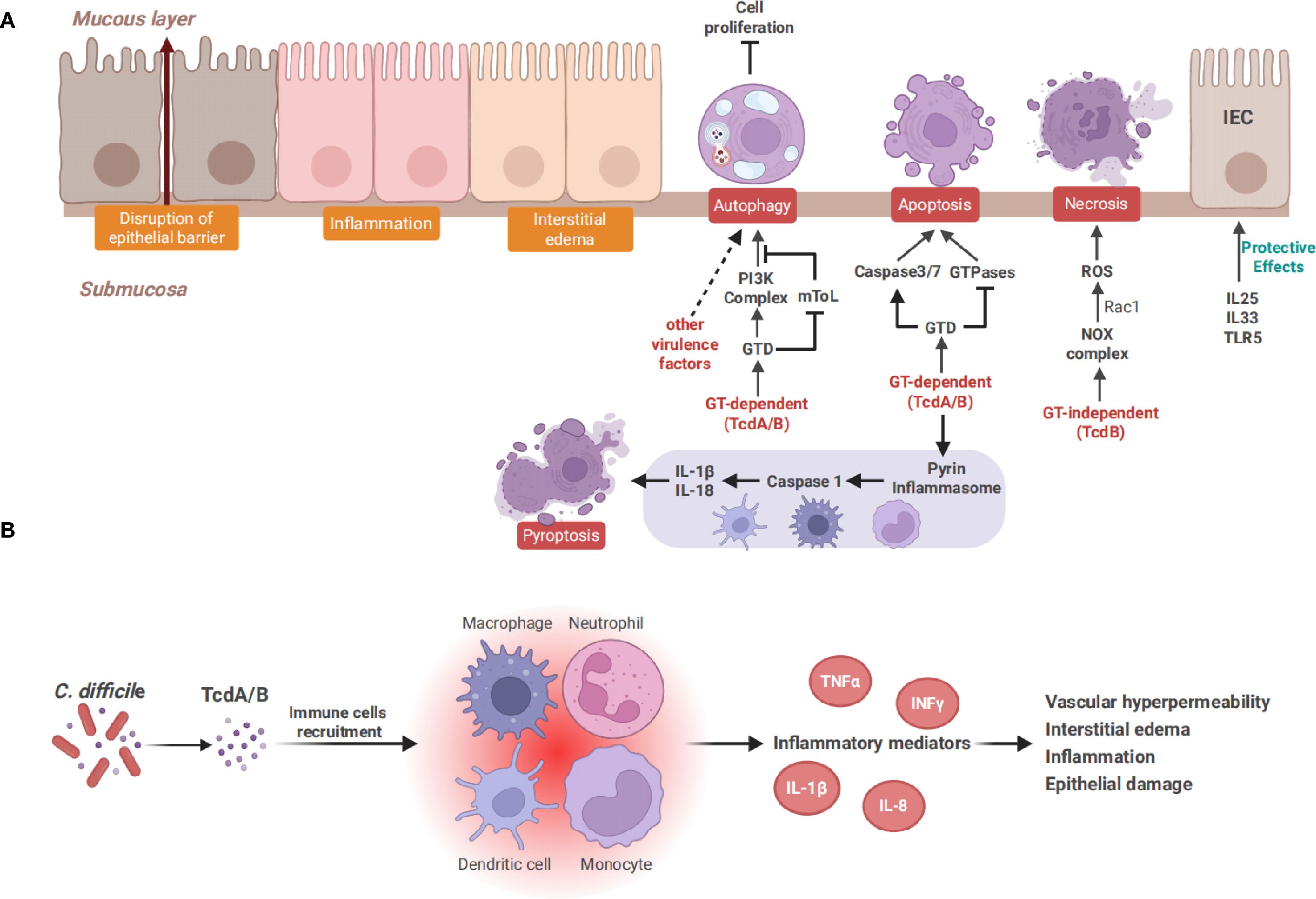
Figure 3. Toxin-mediated intestinal epithelial damage and the inflammatory response. (A) TcdA and TcdB cause cell death through distinct mechanisms. TcdA induces apoptosis in a GT-dependent manner, whereas TcdB exhibits dose-dependent cytotoxicity. At a low dose, TcdB induces GT-dependent apoptosis similar to TcdA, while a higher dose of TcdB triggers necrotic cell death. Both TcdA and TcdB can induce pyroptosis in immune cells, including monocytes, macrophages and dendritic cells. Pyroptosis is mediated by pyrin inflammasomes, which activate caspase-1 and subsequently release IL-1β and IL-18. Autophagy is also induced by TcdA and TcdB in a GT-dependent manner and contributes to the inhibition of cell proliferation. It can also be induced by other virulence factors of C. difficile, such as surface layer protein (A, B) TcdA and TcdB recruit immune cells such as neutrophils, monocytes, and macrophages, subsequently triggering the secretion of inflammatory mediators like TNF-α, IL-1β, and IL-8, leading to increased vascular permeability, interstitial edema, and intestinal epithelial damage, finally causing various forms of cell death.
The GT activity is essential for toxin-induced cell death. However, researchers have recently put forward a standpoint claiming that TcdB plays a more significant role in CDI due to its GT-dependent and GT-independent dual effects (Peritore-Galve et al., 2022). An acute intestinal infection mouse model established by GTD-deficient TcdB demonstrated that the GT activity of TcdB was essential for inducing disease symptoms (Yang et al., 2015b). Another mouse infection model established by GTD-deficient C. difficile showed that no significant alternation in the load of C. difficile, but a complete loss of pathogenicity to the mice was observed, which further confirmed the GT-dependent mechanism of TcdB (Bilverstone et al., 2020). However, under the condition of high concentrations of TcdB, GT-independent effects such as necrosis and pyknosis have been reported, suggesting that the GT-independent action may be concentration-dependent (Chumbler et al., 2012; Wohlan et al., 2014). TcdB-induced GT-independent necrosis depends on the assembly of the NADPH oxidase complex (NOX) in host epithelial cells and the production of reactive oxygen species (ROS) (Farrow et al., 2013). ROS generated by NOX is regulated by Rac1 (Hordijk, 2006). The GTD of TcdB rapidly accumulates to reach sufficient levels and drives early cell death in a Rac1-dependent manner. The loss of functional Rac1 can inhibit the early cell death induced by high concentrations of TcdB (Beer et al., 2018). Furthermore, TcdB can induce a Ras-dependent cell death termed pyknosis, in which Ras serves as a central upstream regulator of the GT-independent effect of TcdB (Figure 3A) (Stieglitz et al., 2022). The mechanism and significance of the cytotoxicity of C. difficile toxins remain to be further clarified.
3.4 Toxin-mediated host immune response
Multiple studies have shown that TcdA and TcdB cause tissue damage not only by direct cytotoxicity, but also by inducing inflammatory response. TcdA and TcdB can stimulate immune cells such as monocytes and macrophages, and trigger the secretion of inflammatory mediators, including IFNγ, TNF-α, IL-1β, IL-6, IL-8, IL-23, MIP-1α, and MIP-2 (Lee et al., 2007; Hasegawa et al., 2012; McDermott et al., 2017; Castagliuolo et al., 1998; Ishida et al., 2004). Neutrophils and other inflammatory cells were recruited by these inflammatory mediators to amplify the inflammatory cascade, promote vascular hyperpermeability and interstitial edema, and exacerbate intestinal tissue injury (Figure 3B) (Morteau et al., 2002; Madan and Petri, 2012; Saleh et al., 2019).
An early study has reported that TcdA interacted with specific surface receptors on rabbit neutrophils to activate a G protein-dependent signaling pathway, which induced neutrophil migration and tissue damage (Kelly et al., 1994a). Furthermore, TcdA has been found to induce monocyte necrosis, IL-1β release, and IL-8 production through the activation of ERK and p38 MAP kinase signaling pathways. It was also suggested that the activation of MAP kinase may not be related to the glycosylation of Rho proteins (Warny et al., 2000). Meanwhile, TcdA can also stimulate the upregulation of IL-8 and monocyte chemotactic protein 1 by activating the NF-κB signaling pathway, thereby inducing inflammatory responses in the intestinal mucosa (Kim et al., 2006). Another study has suggested that the endocytosis pathway of TcdA is necessary for the induction of TNF-α, and TcdA-induced secretion of TNF-α is dependent on its GT activity (Sun et al., 2009). Toll-like receptors (TLRs) are the primary components of the immune system, playing a crucial role in detecting pathogen-associated molecular patterns and in activating both innate and adaptive immune responses (Hsieh et al., 2025). A study revealed the important role of TLR9 in the pathogenesis of TcdA. TcdA binds to bacterial DNA to form a stable complex, which enters cells via a cell-penetrating peptide-like domain, and activates TLR9 pathway to trigger inflammatory responses (Chen et al., 2020a). Meanwhile, another study revealed that the activation of TLR5 signaling played a protective role against CDI, and it was presumed that TLR5 may protect IECs by inducing anti-apoptosis and cell proliferation (Jarchum et al., 2011).
TcdA and TcdB could induce the release of IL-1β by activating the inflammasome, which in turn triggers inflammation and intestinal damage, suggesting that the inhibition of the inflammasome or IL-1β signaling could be potential new strategies for treating CDI (Ng et al., 2010). Further research has indicated that the activation of inflammasomes and IL-1β signaling played a key role during the production of IL-23 stimulated by TcdA and TcdB (Cowardin et al., 2015). IL-23 is a key factor for driving neutrophil recruitment and the innate inflammatory response in C. difficile associated colitis, as confirmed in both human colon samples and animal experiments (Buonomo et al., 2013; McDermott et al., 2016). A retrospective study has shown that anti-IL-23 treatments significantly reduce the probability of all-cause death within 30 days, further confirming the inflammatory role of IL-23 in CDI patients (Madden et al., 2025). A transcriptome analysis revealed that although TcdA played a regulatory role in these responses, TcdB was actually the primary factor for inducing host innate immunity and pro-inflammatory responses (Carter et al., 2015). Notably, necrosis induced by TcdB is associated with ROS production mediated by NOX. Inhibition of ROS generation or abrogation of ROS can protect the colon from TcdB-induced damage (Farrow et al., 2013). Additionally, it has been reported that TcdB targets FZD1/2/7 in gut-innervating afferent neurons and CSPG4 in pericytes, releasing neuropeptide substances and inflammatory cytokines, which leads to neurogenic inflammation and subsequently causes CDI-associated histopathology in mouse models (Manion et al., 2023). This finding revealed a novel mechanism of TcdB inducing inflammation and offered a new approach for the targeted therapy of CDI.
However, a recent study suggested that toxin-induced host inflammation may offer potential benefits for C. difficile by altering the host’s nutritional environment and the structure of the gut microbiome (Fletcher et al., 2021). Another study has found that IL-33 stimulates the activation of colonic group 2 innate lymphoid cells, which in turn can prevent CDI. While the down-regulation of IL-33 results in severer illness and increased mortality. Moreover, the prevention of C. difficile associated mortality and epithelial cell damage by IL-33 is independent of bacterial load or toxin expression (Frisbee et al., 2019). Additionally, researchers have discovered that IL-25 maintained intestinal barrier integrity during CDI by inducing the increase of eosinophilia, and restoring the suppressed expression of IL-25 in CDI could decrease mortality and morbidity (Buonomo et al., 2016). These results indicate that both IL-25 and IL-33 may play protective roles in CDI. Further studies are needed to clarify the relationship between C. difficile toxins and host inflammation.
4 Strategies for treating and preventing CDI
The American College of Gastroenterology clinical guidelines in 2021 emphasized that vancomycin and fidaxomicin are the first-line treatments for CDI, in combination with parenteral metronidazole for fulminant CDI. Patients experiencing multiple relapses should be treated with FMT (Kelly et al., 2021). That same year, the clinical practice guideline issued by the Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America recommended fidaxomicin over a standard course of vancomycin for patients with either initial or recurrent CDI (Johnson et al., 2021). The European Society of Clinical Microbiology and Infectious Diseases guidelines emphasized the significance of discontinuing predisposing antibiotic therapy and recommended using fidaxomicin for the treatment of CDI when available and feasible (van Prehn et al., 2021). In 2025, updated guidelines from the Australasian Society of Infectious Diseases reaffirm that the cessation of antimicrobial therapies is crucial for CDI prevention and optimal management, while highlighting the significant role of FMT in rCDI (Longhitano et al., 2025).
Treatment for CDI varies worldwide according to these clinical guidelines. Antibiotics remain the first-line treatment option, however they can interfere with the normal gut microbiota composition, leading to the recurrence of infection (Sehgal et al., 2022; Kapandji et al., 2025). At present, several narrow-spectrum antibiotics specifically for C. difficile, such as ridinilazole (phase III) and ibezapolstat (phase II), are undergoing clinical trials (Okhuysen et al., 2024; Eubank et al., 2025). Given the remarkable results, these antibiotics are expected to be widely applied clinically in the future. In recent years, it has been recognized that a balanced gut microbiota plays a crucial role in maintaining the health of the host (Liu et al., 2023; Chen et al., 2023). FMT has emerged as a powerful therapeutic approach for managing patients with rCDI. However, the lack of standardization in preparation and administration of fecal material poses inherent risks and limits its large-scale application (Wilcox et al., 2020; Feuerstadt et al., 2022). Although SER-109 and REBYOTA have currently been approved by the FDA to prevent rCDI, more real-world clinical trials are needed to further confirm their safety and efficacy (Gonzales-Luna et al., 2023). Antitoxin-targeted therapies for C. difficile toxins, as well as the development of vaccines utilizing inactivated toxins, have also achieved promising results preclinically, and have gradually progressed to clinical trials (Bratkovic et al., 2024). Further studies are still needed to evaluate their long-term efficacy and safety. CDI remains a significant challenge in the health field, and it is urgent to develop new therapies to control the increasing incidence, rising severity and high recurrence rate of CDI. Table 1 summarizes the existing achievements, limitations, as well as the future direction of progress in CDI treatment.
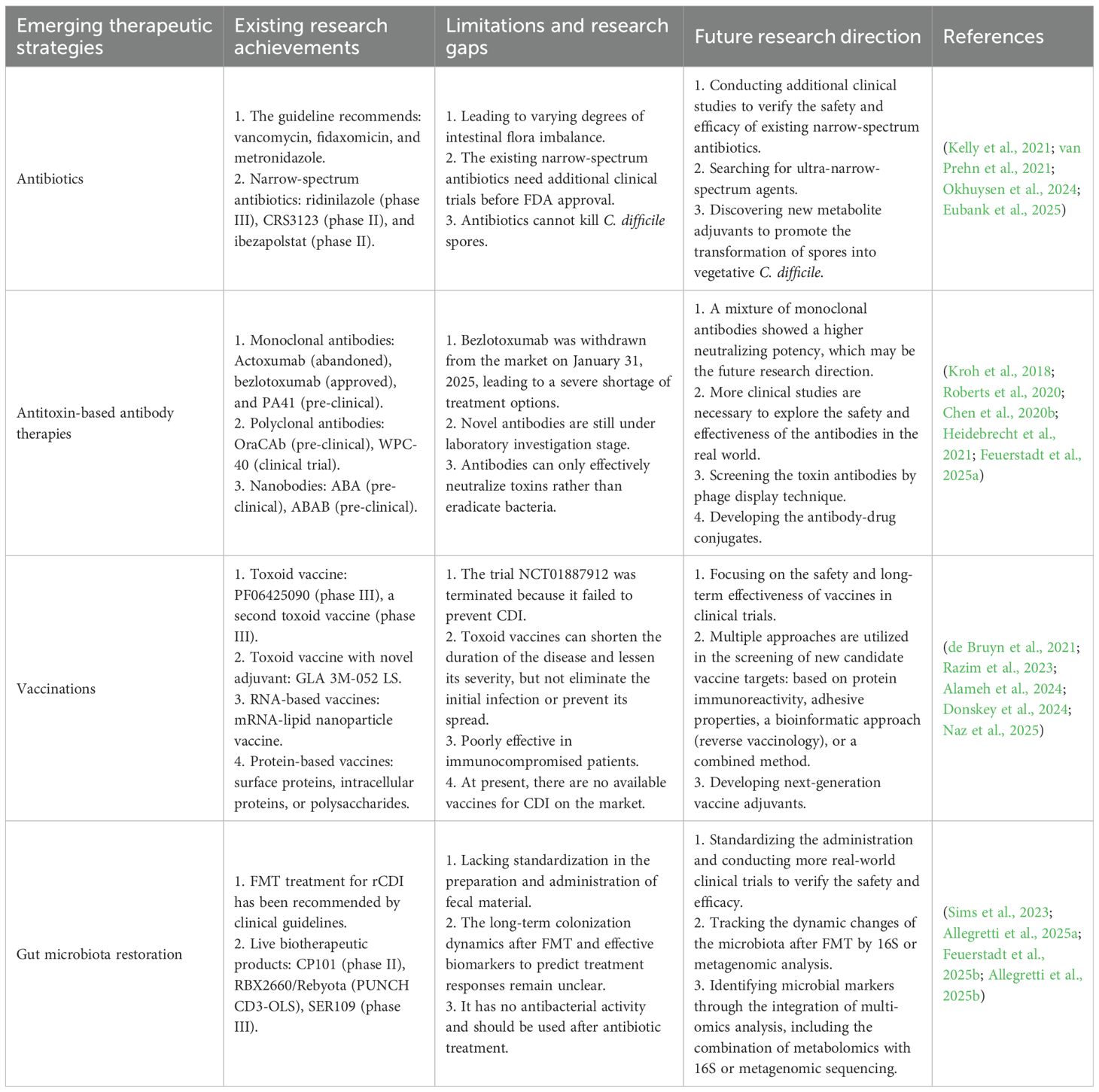
Table 1. Overview of the development of CDI treatment.
4.1 Antitoxin-based antibody therapies
The standard therapy for CDI primarily relies on antibiotics, yet it has a high recurrence rate. In recent years, researchers have sought to develop new strategies that target toxins instead of pathogens to reduce recurrence rates. Hence, antitoxin-based antibody therapies have attracted significant attention.
It is confirmed that passive immunization using antitoxin mAbs against TcdA and TcdB can achieve reductions in mortality, morbidity, and recurrence rates to different extents in various animal infection models (Kink and Williams, 1998; Giannasca et al., 1999; Babcock et al., 2006; Yang et al., 2015a). In 2012, novel antitoxin mAbs, anti-TcdA PA50 and anti-TcdB PA41, were successfully generated and humanized. The combination of PA50 and PA41 significantly improved the survival rates in a hamster model of CDI (Marozsan et al., 2012). Subsequently, the mechanism of PA41 was elucidated through a combination of structural, biochemical, and cellular functional studies. PA41 recognizes a highly conserved epitope on the GTD of TcdB and prevents its translocation into the cytosol (Kroh et al., 2018). In addition to studies on animal models, data from CDI patients also supported the feasibility of adopting TcdA and TcdB antibodies. Morever, the serum levels of anti-TcdA and anti-TcdB mAbs were correlated with CDI relapse (Kyne et al., 2001). In a phase II clinical study (NCT00350298), simultaneous administration of CDA1 and CDB1, human mAbs targeting TcdA and TcdB, during antibiotic treatment significantly reduced the recurrence rate of CDI (Lowy et al., 2010; Gupta et al., 2016). Actoxumab and bezlotoxumab are human mAbs against C. difficile TcdA and TcdB, respectively, which have been proven effective in several preclinical studies (Gupta et al., 2016). Two phase III clinical trials (NCT01241552 and NCT01513239) also confirmed that bezlotoxumab significantly reduced the recurrence rate and exhibited a safety profile similar to that of the placebo. However, actoxumab failed to exhibit such effect, and the therapeutic effect was not significantly enhanced when combining with bezlotoxumab (Wilcox et al., 2017). Both actoxumab and bezlotoxumab bind to the CROP domains to prevent the toxins from binding to mammalian cells. A study has shown that bezlotoxumab binds to two homologous but distinct epitopes on the CROP domain, thereby preventing TcdB from binding to the surface of host cells (Orth et al., 2014). However, actoxumab cannot bind to both epitopes simultaneously because they are situated on the opposite sides of the CROP domain of TcdA, which may be one reason for its insufficient efficacy (Hernandez et al., 2017). Therefore, exploring anti-TcdA mAbs that target different epitopes may offer improved protection against TcdA. The binding of bezlotoxumab can induce an allosteric change in TcdB, thereby disrupting the CSPG4-binding site (Chen et al., 2021). However, bezlotoxumab does not affect the interaction between TcdB and FZD1/2/7 or Nectin-3, since TcdB binding to these receptors is independent of the CROP domain. This indicates that neutralizing antibodies targeting other toxin domains provide comparable or enhanced protective effects (Manse and Baldwin, 2015). Several animal studies have confirmed that a mixture of mAbs targeting different toxin domains exhibits a higher neutralizing efficacy compared to a single mAb (Davies et al., 2013; Qiu et al., 2016), highlighting the potential value of hybrid mAbs in future research. As the direct neutralizing effect of antibodies is crucial in inactivating toxins, antibody fragments, such as nanobodies, are expected to serve as effective alternatives to full-length mAbs. Researchers have developed a tetravalent, bispecific antibody comprising two VHH binding domains against both TcdA and TcdB, namely ABA. It exhibited the ability to simultaneously neutralize TcdA and TcdB and the efficacy in preventing and treating CDI in mice (Yang et al., 2014). Subsequently, ABAB, a tetra-specific antibody consisting of four distinctive toxin-neutralizing VHHs, has shown a broad neutralizing capacity in mice and hamsters (Chen et al., 2020b). Several studies have indicated that although the neutralization of both toxins was necessary for the maximum protection of rodents, the neutralization of TcdB alone seemed to be adequate for mammals, which suggested that the neutralization effect may depend on host species (Leav et al., 2010; Steele et al., 2013).
Currently, bezlotoxumab remains the only anti-toxin antibody approved by FDA to prevent rCDI (Mullard, 2016; Wilcox et al., 2017). However, since Merck, the manufacturer, has announced that bezlotoxumab will be withdrawn from the market on January 31, 2025, many medical institutions will face a severe shortage of treatment options (Feuerstadt et al., 2025a). Additionally, due to the presence of multiple TcdB variants, current antibodies exhibit low neutralizing potency against the TcdB variants of various epidemic pathogenic strains. Recently, several new technologies, such as phage display technique, have emerged to facilitate the screening of new C. difficile toxin antibodies (Kordus et al., 2023; Raeisi et al., 2023). However, there is still a gap between laboratory achievements and clinical efficacy, and further research on C. difficile toxin antibodies is necessary. It is important to recognize that the antibodies mentioned above are effective in neutralizing toxins rather than eradicating the bacteria. Therefore, the development of antibody-drug conjugates is anticipated to be a novel research frontier (Wang et al., 2025a). Existing antibody therapies have been discontinued, and the development of new antibodies remains in preclinical studies. Despite the promising results, there are inherent limitations due to the lengthy clinical trial process.
4.2 Vaccination
CDI continues to be a significant and costly medical issue, primary prevention is greatly needed. The development of vaccines has prospective efficacy in protecting individuals with high risks of developing CDI. Although there is yet no available vaccine for C. difficile on the market, data from several clinical trials have indicated its potential feasibility. C. difficile vaccines primarily include inactivated toxins, recombinant toxins, and RNA vaccines, which are designed to induce systemic antibody responses against TcdA and TcdB (Knisely et al., 2016; Tang et al., 2025). A phase II clinical trial (NCT02561195) has demonstrated satisfactory tolerance, bio-safety as well as immunogenicity of the C. difficile vaccine in healthy US adults, which supported the further development of the vaccine (Kitchin et al., 2020). It further showed that immune responses to the C. difficile vaccine persisted for 48 months after the third dose, and a four-dose administration was found to prolong the immunogenicity up to 3 years with safety in an extension study involving adults aged 65 to 85 years (Remich et al., 2024). However, a phase III clinical trial (NCT01887912) demonstrated that although a bivalent C. difficile toxoid vaccine exhibited good immunogenicity and safety, it was ineffective in preventing CDI. Consequently, the study was ultimately terminated (de Bruyn et al., 2021). Another clinical trial (NCT03090191) evaluated PF-06425090, a detoxified toxin-A/B vaccine for primary CDI prevention. It demonstrated that while the vaccine significantly reduced the CDI events requiring treatment and effectively shortened the symptom duration, it also failed to lower the incidence of primary CDI events (Donskey et al., 2024). Various clinical studies have shown that toxoid vaccines can shorten the duration of the disease and lessen its severity, but do not eliminate the initial infection or prevent its spread. The development of next-generation vaccines against C. difficile faces numerous challenges.
Given the suboptimal performance of aluminum as an adjuvant in C. difficile vaccines for inducing immunity, researchers have shifted their focus to seeking novel adjuvants. A recent study has revealed that GLA 3M-052 LS, a dual Toll-like receptor ligand liposome, can enhance the immunogenicity of TcdB vaccines in mice (Naz et al., 2025). This breakthrough holds promise for advancing the development of next-generation vaccines against CDI. Recently, mRNA vaccines have shown significant potential in combating a variety of pathogens. Researchers have successfully developed an mRNA-lipid nanoparticle vaccine targeting C. difficile toxins and virulence factors, and have verified its effectiveness in preventing CDI and promoting bacterial clearance in multiple clinically relevant animal models (Alameh et al., 2024). Numerous studies have indicated that toxoid vaccines do not prevent the transmission of bacteria among patients or eradicate the pathogen during the initial stages of infection. Therefore, an ideal vaccine should incorporate additional antigenic components that provoke an immune response early in the infection process, such as surface proteins or polysaccharides (Razim et al., 2023). These studies remain at the stage of animal experiments and urgently require further clinical trials to verify their safety and efficacy. Although the development of C. difficile vaccines still faces multiple challenges, it is of great significance to provide better solutions for preventing CDI.
4.3 Gut microbiota restoration
Recently, FMT has garnered significant attention from researchers due to its potential in preventing rCDI. Studies suggest that microbial imbalance is closely associated with the occurrence of CDI, with the widespread use of antibiotics being one of the main causes of gut microbiota disruption (Surawicz, 2009). Antibiotic treatment-induced dysbiosis leads to the excessive proliferation of harmful bacteria such as C. difficile, resulting in subsequent infections (Leffler and Lamont, 2015). Research has shown that the abundance of Enterococci significantly increases, while beneficial bacteria such as Bifidobacterium and Ruminococcus significantly decrease in patients treated with β-lactam antibiotics, leading to high susceptibility to C. difficile (Berkell et al., 2021). Furthermore, a study found that patients with CDI have already shown lower microbial diversity before antibiotic treatment, further suggesting that changes in gut microbiota diversity are related to the occurrence and development of CDI (Chen et al., 2022). The prognosis of CDI may be improved by restoring the balance of intestinal microbiota. Recent studies have shown that probiotics such as Firmicutes can effectively inhibit C. difficile growth and simultaneously promote the recovery of beneficial intestinal flora in patients with CDI (Herrera et al., 2021). Additionally, results from various animal models have indicated that the microbial therapies can also inhibit bacterial growth and toxin secretion by altering intestinal metabolites, such as short-chain fatty acid, lactic acid, and bile acid (Buffie et al., 2015; Li et al., 2024, Li et al., 2025). Recently, caffeic acid phenethyl ester and equol have been identified to reduce intestinal damage from toxins by screening the natural compounds library. They inhibit bacterial growth and toxin secretion by regulating intestinal metabolism in mouse models while ensuring the integrity of the intestinal microbiota, revealing their potential as a therapeutic for the management of CDI (Guo et al., 2025a, Guo et al., 2025b). A healthy gut microbiota not only inhibits the growth of C. difficile but also activates the host’s immune response. A restored microbiota can regulate immune cells and promote the release of anti-inflammatory factors, thereby reducing infection-induced inflammatory responses (Chiu et al., 2021; Yang et al., 2024). Microbial metabolites, including succinate and citrulline, have been demonstrated in CDI mouse models to exert anti-inflammatory effects by activating immune cells, effectively protecting against CDI-induced damage (Xie et al., 2025; Kellogg et al., 2025). Therefore, the restoration of gut microbiota as an adjunctive strategy in treating CDI has demonstrated its importance and potential.
FMT is recommended by multiple clinical guidelines for preventing rCDI due to its benefits in restoring the balance of the gut microbiota (van Prehn et al., 2021; Johnson et al., 2021). A clinical trial (NCT03005379) enrolled a veteran population with rCDI to test the efficacy and safety of capsule-delivered FMT, which contains lyophilized microbiota isolated from fecal material from standardized donors. Unfortunately, the trial was terminated because there were no significant advantages of FMT in preventing rCDI or reducing mortality rates (Drekonja et al., 2025). Due to the lack of standardization in the compositions, dosages, and administrations of fecal material, the outcomes of FMT have been less than satisfactory. Recently, several live biotherapeutic products have shown encouraging results. CP101, a full-spectrum oral microbiome therapy, has been proven to be more effective than placebo in reducing the recurrence rate of CDI, with comparable safety profiles, in phase II clinical trials (NCT03110133, NCT03497806) (Allegretti et al., 2025b). The phase III clinical trial of CP101 (NCT05153499) is still in the recruitment stage for subjects (Gonzales-Luna et al., 2023). SER-109 is an oral capsule consisting of live, purified Firmicutes bacterial spores. It is designed to achieve therapeutic goals via a dual mechanism involving competitive metabolism and bile acid regulation. SER-109 has completed phase II (NCT02437487), phase III (NCT03183128) and open-label phase III (NCT03183141) trials (McGovern et al., 2021; Feuerstadt et al., 2022; Sims et al., 2023). The results have consistently shown that SER-109 has good tolerability and significantly reduces CDI recurrence rates. Additionally, the SER-109 group showed increased production of secondary bile acids, which inhibited the germination and growth of C. difficile spores (Feuerstadt et al., 2022). REBYOTA (formerly known as RBX2660), a single-dose broad consortia microbiota based live biotherapy, has achieved significant data in phase II and phase III clinical studies (Khanna et al., 2022; Orenstein et al., 2022; Dubberke et al., 2018). Recently, in a PUNCH CD3-OLS clinical trial (NCT03931941), REBYOTA has further demonstrated its safety and efficacy in preventing rCDI (Feuerstadt et al., 2025b). Meanwhile, the results of another PUNCH CD3-OLS clinical trial (NCT03244644) have shown that the efficacy of REBYOTA in preventing rCDI is not weakened in patients with inflammatory bowel disease (Allegretti et al., 2025a). Currently, SER-109 and REBYOTA have been approved by the FDA to prevent rCDI. However, live biotherapeutic products still need to be carried out on the basis of antibiotic treatment. More large-scale, real-world randomized controlled trials are needed to further verify the efficacy and safety of microbial therapy for rCDI. Moreover, exploring optimal intervention timing and methods across different clinical scenarios is necessary to achieve better management of rCDI.
4.4 Novel small molecule strategies
Antibiotics are the preferred small molecule agents recommended for the treatment of CDI. However, they often lead to recurrence by interfering with the normal intestinal flora (Leffler and Lamont, 2015). There has been a shift in focus towards small molecule strategies without direct bactericidal or bacteriostatic activity. Currently, screening libraries of medications approved for other diseases is a significant initiative for searching potential treatment agents for CDI.
Niclosamide, an anthelmintic drug, has recently been found to neutralize the cytotoxic effects of TcdA, TcdB, and CDT in infected mice. It functions by inhibiting the process of pore formation while maintaining the balance of the intestinal flora (Tam et al., 2018). Cholesterol in the cell membrane is crucial for the formation of pores by TcdA and TcdB (Giesemann et al., 2006). Statins, a group of cholesterol-lowering medications used in clinics, have been demonstrated to prevent the cytotoxic effects of these toxins in vitro, highlighting their potential therapeutic benefits in treating CDI (Papatheodorou et al., 2019). Additionally, the antiarrhythmic drug amiodarone has been confirmed to provide protection against both TcdA and TcdB by inhibiting cholesterol biosynthesis under in vitro conditions (Schumacher et al., 2023). Another study has revealed that calcium channel signaling acts as a key mediator of TcdB-induced necrosis through small molecule screening, further suggesting that the calcium channel blocker amiodarone may offer a general protective effect against the severe consequences of CDI (Farrow et al., 2020). Future studies focusing on drugs that inhibit cholesterol synthesis in the cell membrane and other molecules that block toxin action may facilitate the development of effective therapies against CDI. Auranofin, an FDA-approved oral anti-rheumatic drug, may reduce spore and toxin production by inhibiting selenium metabolism in C. difficile, or by interfering with its biosynthesis in vivo. This suggests that Auranofin has potential as a promising therapeutic option for CDI, especially in reducing disease recurrence and controlling nosocomial infections (Hutton et al., 2020). Misoprostol, an FDA-approved stable prostaglandin E1 analogue, has been demonstrated to protect against C. difficile-associated mortality by decreasing the intestinal mucosal permeability in mouse models of CDI. Additionally, it contributes to the recovery of gut microbiota following antibiotic perturbation (Zackular et al., 2019). Bile acid metabolites are crucial for influencing the life cycle of C. difficile (Buffie et al., 2015). Obeticholic acid, an antagonist of farnesoid X receptors, has been approved for the treatment of primary biliary cholangitis. It was found to decrease the bacterial load and improve the prognosis of CDI in mice by reducing the synthesis of primary bile acids (Jose et al., 2021). Ketotifen, an anti-allergy drug, was found to inhibit enteritis induced by TcdA in rats, primarily by suppressing the release of mediators derived from mast cells and neutrophils (Pothoulakis et al., 1993). The antidepressant amoxapine, the respiratory stimulant doxapram, and the antipsychotic trifluoperazine were recently found to effectively reduce the bacterial burden and toxin levels in mice with CDI. Moreover, these drugs have minimal impact on the composition of the microbiota as they have neither bacteriostatic nor bactericidal properties (Andersson et al., 2018). The team further explored the mechanism of action using RNA-seq technology and discovered that these drugs protect against CDI by modulating the host innate immune defenses (Andersson et al., 2020). These results emphasize the importance of immune regulation as a potential therapeutic option for CDI. Berberine, a natural compound found in traditional Chinese medicine, exhibits multiple biological functions, including anti-inflammatory and antioxidant properties. It is used clinically to treat intestinal infections. Recent studies have revealed its potential protective effects against rCDI in animal models, indicating the potential role of traditional Chinese medicine in the treatment of CDI (Wang et al., 2025).
Additionally, numerous new therapies are currently under development, including viruses, bacteriophages and their derivatives such as endolysins and tailocins, as well as microbial metabolites like antimicrobial peptides and α-defensins (Jank et al., 2008; Danis-Wlodarczyk et al., 2021; Phothichaisri et al., 2022; Mondal et al., 2020; Ghosh et al., 2024; Lietz et al., 2025). These novel treatments have demonstrated their potential in the preclinical stage. Despite promising results, a lengthy clinical trial process is the basis for the future clinical applications in the treatment and prevention of recurrent and refractory CDI.
5 Conclusion
In this review, we summarized the epidemiology of CDI, the action mechanism of TcdA and TcdB, the interaction between toxins and host cells, and the progress of treatments. As one of the most common healthcare-associated diseases, CDI still has many unsolved scientific questions (Di Bella et al., 2024). For instance, the cell surface receptor for toxins warrants further investigation. The role of GTPases as toxin targets in the cytoplasm has been established. However, when the toxin reaches a certain concentration, the GT-independent cell death pathway and the associated pathological damage require further investigation (Orrell and Melnyk, 2021). With the widespread transmission of various subtypes of virulent C. difficile globally, researchers have discovered that these strains display diverse mechanisms of toxin action. Research into the structure of toxin subtypes not only aids in addressing issues at the root of pathogenic mechanisms, but also offers robust support for the development of new targeted drugs. In the exploration of therapeutic strategies, small-molecule metabolites show promising potential. These compounds not only inhibit GT activity of toxins but also restore gut microbiota balance. This dual effect offers a new therapeutic strategy for CDI. Although research on anti-toxin antibodies and CDI vaccines is still very scarce at present (Donskey et al., 2024; Naz et al., 2025), progress in novel narrow-spectrum antibiotics and approval of various live biotherapeutic products have significantly advanced the clinical management of CDI (Sims et al., 2023; Okhuysen et al., 2024; Feuerstadt et al., 2025b).
In conclusion, to fully understand the pathogenic mechanism of CDI and develop new anti-CDI drugs, there is still a long way to go. It is hoped that this review will be beneficial to researchers who are seeking a comprehensive acknowledgement of the action mechanisms of C. difficile toxins and recent advances in the treatment of CDI.
Author contributions
XW: Writing – original draft. XL: Writing – original draft. KW: Writing – original draft. HL: Writing – original draft. CZ: Writing – review & editing. XZ: Writing – review & editing. QW: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the National Key Research and Development Program of China, under the project title Precision Molecular Typing and Development of New Detection Technologies for Patients with Refractory Acute Leukemia (Grant No. 2023YFC2508905), the Special Project for Talent Construction Program in Xinqiao Hospital (Grant No. 2023YQB062).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
C. difficile, Clostridioides difficile; CDI, Clostridioides difficile infection; rCDI, recurrent Clostridioides difficile infection; LCT, large clostridial toxin; TcdA, Clostridioides difficile toxin A; TcdB, Clostridioides difficile toxin B; GTD, glucosyltransferase domain; CPD, cysteine proteinase domain; TMD, transmembrane domain; CROP, C-terminal repetitive oligopeptide domain; InsP6, inositol hexaphosphate; mAbs, monoclonal antibodies; IECs, intestinal epithelial cells; FMT, fecal microbiota transplantation; PaLoc, pathogenicity locus; CdtLoc, the binary toxin encoding locus; CDT, Clostridioides difficile transferases; gp96, glycoprotein 96; sGAGs, sulfated glycosaminoglycans; SI, sucrase-isomaltase; LDLR, the low-density lipoprotein receptor family; LRP1, low-density lipoprotein receptor-associated protein-1; CSPG4, chondroitin sulfate proteoglycan 4; PVRL3, poliovirus receptor-like 3; FZDs, frizzled family; TFPI, tissue factor pathway inhibitor; FDA, Food and Drug Administration; CRD, cysteine-rich domain; SREBP-2, sterol regulatory element–binding protein 2; TRiC/CCT, chaperonin TCP-1 ring complex/chaperonin containing TCP-1; GT, glucosyltransferase; VHH, heavy-chain variable region; EGCs, enteric glial cells; NOX, NADPH oxidase complex; ROS, reactive oxygen species; TLRs, Toll-like receptor.
References
Abt, M. C., McKenney, P. T., and Pamer, E. G. (2016). Clostridium difficile colitis: pathogenesis and host defence. Nat. Rev. Microbiol. 14, 609–620. doi: 10.1038/nrmicro.2016.108
Aktories, K., Papatheodorou, P., and Schwan, C. (2018). Binary Clostridium difficile toxin (CDT) - A virulence factor disturbing the cytoskeleton. Anaerobe 53, 21–29. doi: 10.1016/j.anaerobe.2018.03.001
Aktories, K., Schwan, C., and Jank, T. (2017). Clostridium difficile toxin biology. Annu. Rev. Microbiol. 71, 281–307. doi: 10.1146/annurev-micro-090816-093458
Alam, M. Z. and Madan, R. (2024). Clostridioides difficile toxins: host cell interactions and their role in disease pathogenesis. Toxins (Basel) 16, 6. doi: 10.3390/toxins16060241
Alameh, M. G., Semon, A., Bayard, N. U., Pan, Y. G., Dwivedi, G., Knox, J., et al. (2024). A multivalent mRNA-LNP vaccine protects against Clostridioides difficile infection. Science 386, 69–75. doi: 10.1126/science.adn4955
Allegretti, J. R., Feuerstadt, P., Knapple, W. L., Orenstein, R., Pinton, P., Sheh, A., et al. (2025a). Safety and efficacy of fecal microbiota, live-jslm (REBYOTA(R)), for the prevention of recurrent clostridioides difficile infection in participants with inflammatory bowel disease in PUNCH CD3-OLS. Inflammation Bowel Dis. 31 (8), 2112–2122. doi: 10.1093/ibd/izae291
Allegretti, J. R., Kelly, C. R., Louie, T., Fischer, M., Hota, S., Misra, B., et al. (2025b). Safety and tolerability of CP101, a full-spectrum, oral microbiome therapeutic for the prevention of recurrent clostridioides difficile infection: A phase 2 randomized controlled trial. Gastroenterology 168, 357–366.e3. doi: 10.1053/j.gastro.2024.09.030
Aminzadeh, A., Larsen, C. E., Boesen, T., and Jorgensen, R. (2022). High-resolution structure of native toxin A from Clostridioides difficile, EMBO Rep, Vol. 23 No. 1 pp, e53597. doi: 10.15252/embr.202153597
Amirkamali, S., Azimirad, M., Nasiri, G., Goudarzi, H., Noori, M., Yadegar, A., et al. (2022). Surface layer protein A from hypervirulent Clostridioides difficile ribotype 001 can induce autophagy process in human intestinal epithelial cells. Microb. Pathog. 169, 105681. doi: 10.1016/j.micpath.2022.105681
Andersson, J. A., Peniche, A. G., Galindo, C. L., Boonma, P., Sha, J., Luna, R. A., et al. (2020). New host-directed therapeutics for the treatment of clostridioides difficile infection. mBio 11, 2. doi: 10.1128/mBio.00053-20
Andersson, J. A., Sha, J., Kirtley, M. L., Reyes, E., Fitts, E. C., Dann, S. M., et al. (2018). Combating multidrug-resistant pathogens with host-directed nonantibiotic therapeutics. Antimicrob. Agents Chemother. 62 (1), e01943-17. doi: 10.1128/AAC.01943-17
Arvand, M., Vollandt, D., Bettge-Weller, G., Harmanus, C., and Kuijper, E. J. (2014). Increased incidence of Clostridium difficile PCR ribotype 027 in Hesse, German to 2013. Euro Surveill 19, 10. doi: 10.2807/1560-7917.ES2014.19.10.20732
Awad, M. M., Johanesen, P. A., Carter, G. P., Rose, E., and Lyras, D. (2014). Clostridium difficile virulence factors: Insights into an anaerobic spore-forming pathogen. Gut Microbes 5, 579–593. doi: 10.4161/19490976.2014.969632
Azimirad, M., Noori, M., Amirkamali, S., Nasiri, G., Asadzadeh, A. H., Yadegar, A., et al. (2023). Clostridioides difficile PCR ribotypes 001 and 084 can trigger autophagy process in human intestinal Caco-2 cells. Microb. Pathog. 185, 106450. doi: 10.1016/j.micpath.2023.106450
Babcock, G. J., Broering, T. J., Hernandez, H. J., Mandell, R. B., Donahue, K., Boatright, N., et al. (2006). Human monoclonal antibodies directed against toxins A and B prevent Clostridium difficile-induced mortality in hamsters. Infect. Immun. 74, 6339–6347. doi: 10.1128/IAI.00982-06
Barth, H., Pfeifer, G., Hofmann, F., Maier, E., Benz, R., and Aktories, K. (2001). Low pH-induced formation of ion channels by clostridium difficile toxin B in target cells. J. Biol. Chem. 276, 10670–10676. doi: 10.1074/jbc.M009445200
Bartlett, J. G. (2002). Clinical practice. Antibiotic-associated diarrhea. N Engl. J. Med. 346, 334–339. doi: 10.1056/NEJMcp011603
Bartlett, J. G., Chang, T. W., Gurwith, M., Gorbach, S. L., and Onderdonk, A. B. (1978). Antibiotic-associated pseudomembranous colitis due to toxin-producing clostridia, new england journal of medicine, vol. 298 No. 10 pp, 531–534. doi: 10.1056/NEJM197803092981003
Beer, L. A., Tatge, H., Reich, N., Tenspolde, M., Olling, A., Goy, S., et al. (2018). Early cell death induced by Clostridium difficile TcdB: Uptake and Rac1-glucosylation kinetics are decisive for cell fate. Cell Microbiol. 20, e12865. doi: 10.1111/cmi.12865
Bergsbaken, T., Fink, S. L., and Cookson, B. T. (2009). Pyroptosis: host cell death and inflammation. Nat. Rev. Microbiol. 7, 99–109. doi: 10.1038/nrmicro2070
Berkell, M., Mysara, M., Xavier, B. B., van Werkhoven, C. H., Monsieurs, P., Lammens, C., et al. (2021). Microbiota-based markers predictive of development of Clostridioides difficile infection. Nat. Commun. 12, 2241. doi: 10.1038/s41467-021-22302-0
Bezerra, L. B., Faria, F. B., Da, G. A. N., Moreira, L. D., Albuquerque, R. R., Garcia, A. J., et al. (2014). Clostridium difficile toxin A attenuates Wnt/beta-catenin signaling in intestinal epithelial cells. Infect. Immun. 82, 2680–2687. doi: 10.1128/IAI.00567-13
Bilverstone, T. W., Garland, M., Cave, R. J., Kelly, M. L., Tholen, M., Bouley, D. M., et al. (2020). The glucosyltransferase activity of C. difficile Toxin B is required for disease pathogenesis. PloS Pathog. 16, e1008852. doi: 10.1371/journal.ppat.1008852
Bratkovic, T., Zahirovic, A., Bizjak, M., Rupnik, M., Strukelj, B., and Berlec, A. (2024). New treatment approaches for Clostridioides difficile infections: alternatives to antibiotics and fecal microbiota transplantation. Gut Microbes 16, 2337312. doi: 10.1080/19490976.2024.2337312
Brito, G. A., Fujji, J., Carneiro-Filho, B. A., Lima, A. A., Obrig, T., and Guerrant, R. L. (2002). Mechanism of Clostridium difficile toxin A-induced apoptosis in T84 cells. J. Infect. Dis. 186, 1438–1447. doi: 10.1086/344729
Buffie, C. G., Bucci, V., Stein, R. R., McKenney, P. T., Ling, L., Gobourne, A., et al. (2015). Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517, 205–208. doi: 10.1038/nature13828
Buonomo, E. L., Cowardin, C. A., Wilson, M. G., Saleh, M. M., Pramoonjago, P., and Petri, W. A. (2016). Microbiota-Regulated IL-25 Increases Eosinophil Number to Provide Protection during Clostridium difficile Infection. Cell Rep. 16, 432–443. doi: 10.1016/j.celrep.2016.06.007
Buonomo, E. L., Madan, R., Pramoonjago, P., Li, L., Okusa, M. D., and Petri, W. A. (2013). Role of interleukin 23 signaling in clostridium difficile colitis. J. Infect. Dis. 208, 917–920. doi: 10.1093/infdis/jit277
Burridge, K. and Wennerberg, K. (2004). Rho and Rac take center stage. Cell 116, 167–179. doi: 10.1016/S0092-8674(04)00003-0
Cairns, M. D., Preston, M. D., Lawley, T. D., Clark, T. G., Sta bler, R. A., and Wren, B. W. (2015). Genomic epidemiology of a protracted hospital outbreak caused by a toxin A-negative clostridium difficile sublineage PCR ribotype 017 strain in london, england. J. Clin. Microbiol. 53, 141–3147. doi: 10.1128/JCM.00648-15
Carneiro, B. A., Fujii, J., Brito, G. A., Alcantara, C., Oria, R. B., Lima, A. A., et al. (2006). Caspase and bid involvement in Clostridium difficile toxin A-induced apoptosis and modulation of toxin A effects by glutamine and alanyl-glutamine in vivo and in vitro. Infect. Immun. 74, 81–87. doi: 10.1128/IAI.74.1.81-87.2006
Carter, G. P., Chakravorty, A., Pham Nguyen, T. A., Mileto, S., Schreiber, F., Li, L., et al. (2015). Defining the Roles of TcdA and TcdB in Localized Gastrointestinal Disease, Systemic Organ Damage, and the Host Response during Clostridium difficile Infections. mBio 6, e00551. doi: 10.1128/mBio.00551-15
Castagliuolo, I., Keates, A. C., Wang, C. C., Pasha, A., Valenick, L., Kelly, C. P., et al. (1998). Clostridium difficile toxin A stimulates macrophage-inflammatory protein-2 production in rat intestinal epithelial cells. J. Immunol. 160, 6039–6045. doi: 10.4049/jimmunol.160.12.6039
Chandrasekaran, R., Kenworthy, A. K., and Lacy, D. B. (2016). Clostridium difficile toxin A undergoes clathrin-independent, PACSIN2-dependent endocytosis. PloS Pathog. 12, e1006070. doi: 10.1371/journal.ppat.1006070
Chandrasekaran, R. and Lacy, D. B. (2017). The role of toxins in Clostridium difficile infection. FEMS Microbiol. Rev. 41, 723–750. doi: 10.1093/femsre/fux048
Chaves-Olarte, E., Florin, I., Boquet, P., Popoff, M., von Eichel-Streiber, C., and Thelestam, M. (1996). UDP-glucose deficiency in a mutant cell line protects against glucosyltransferase toxins from Clostridium difficile and Clostridium sordellii. J. Biol. Chem. 271, 6925–6932. doi: 10.1074/jbc.271.12.6925
Chaves-Olarte, E., Weidmann, M., Eichel-Streiber, C., and Thelestam, M. (1997). Toxins A and B from Clostridium difficile differ with respect to enzymatic potencies, cellular substrate specificities, and surface binding to cultured cells. J. Clin. Invest. 100, 1734–1741. doi: 10.1172/JCI119698
Chen, B., Basak, S., Chen, P., Zhang, C., Perry, K., Tian, S., et al. (2022a). Structure and conformational dynamics of Clostridioides difficile toxin A. Life Sci. Alliance 5, 6. doi: 10.26508/lsa.202201383
Chen, B., Liu, Z., Perry, K., and Jin, R. (2022b). Structure of the glucosyltransferase domain of TcdA in complex with RhoA provides insights into substrate recognition. Sci. Rep. 12, 9028. doi: 10.1038/s41598-022-12909-8
Chen, Y., Lv, T., Yan, D., Zheng, L., Zheng, B., Wang, J., et al. (2022). Disordered intestinal microbial communities during clostridioides difficile colonization and subsequent infection of hepatic cirrhosis patients in a tertiary care hospital in China. Front. Cell Infect. Microbiol. 12, 825189. doi: 10.3389/fcimb.2022.825189
Chen, S., Sun, C., Wang, H., and Wang, J. (2015). The role of rho GTPases in toxicity of clostridium difficile toxins. Toxins (Basel) 7, 5254–5267. doi: 10.3390/toxins7124874
Chen, P., Tao, L., Wang, T., Zhang., J., He., A., Lam, K. H., et al. (2018). Structural basis for recognition of frizzled proteins by Clostridium difficile toxin B. Science. 360 (6389), 664–669. doi: 10.1126/science.aar1999
Chen, P., Tao, L., Liu, Z., Dong, M., and Jin, R. (2019). Structural insight into Wnt signaling inhibition by Clostridium difficile toxin B. FEBS J. 286, 874–881. doi: 10.1111/febs.14681
Chen, X., Yang, X., de Anda, J., Huang, J., Li, D., Xu, H., et al. (2020a). Clostridioides difficile toxin A remodels membranes and mediates DNA entry into cells to activate toll-like receptor 9 signaling. Gastroenterology 159, 2181–2192.e1. doi: 10.1053/j.gastro.2020.08.038
Chen, P., Zeng, J., Liu, Z., Thaker, H., Wang, S., Tian, S., et al. (2021). Structural basis for CSPG4 as a receptor for TcdB and a therapeutic target in Clostridioides difficile infection. Nat. Commun. 12, 3748. doi: 10.1038/s41467-021-23878-3
Chen, X., Zhang, H., Ren, S., Ding, Y., Remex, N. S., Bhuiyan, M. S., et al. (2023). Gut microbiota and microbiota-derived metabolites in cardiovascular diseases. Chin. Med. J. (Engl) 136, 2269–2284. doi: 10.1097/CM9.0000000000002206
Chen, K., Zhu, Y., Zhang, Y., Hamza, T., Yu, H., Saint, F. A., et al. (2020b). A probiotic yeast-based immunotherapy against Clostridioides difficile infection. Sci. Transl. Med. 12 (567), eaax4905. doi: 10.1126/scitranslmed.aax4905
Childress, K. O., Cencer, C. S., Tyska, M. J., and Lacy, D. B. (2023). Nectin-3 and shed forms of CSPG4 can serve as epithelial cell receptors for Clostridioides difficile TcdB. mBio 14, e0185723. doi: 10.1128/mbio.01857-23
Chiu, C. W., Tsai, P. J., Lee, C. C., Ko, W. C., and Hung, Y. P. (2021). Application of microbiome management in therapy for clostridioides difficile infections: from fecal microbiota transplantation to probiotics to microbiota-preserving antimicrobial agents. Pathogens 10, 6. doi: 10.3390/pathogens10060649
Chumbler, N. M., Farrow, M. A., Lapierre, L. A., Franklin, J. L., Haslam, D. B., Goldenring, J. R., et al. (2012). Clostridium difficile Toxin B causes epithelial cell necrosis through an autoprocessing-independent mechanism. PloS Pathog. 8, e1003072. doi: 10.1371/journal.ppat.1003072
Chumbler, N. M., Rutherford, S. A., Zhang, Z., Farrow, M. A., Lisher, J. P., Farquhar, E., et al. (2016). Crystal structure of Clostridium difficile toxin A. Nat. Microbiol. 1, 15002. doi: 10.1038/nmicrobiol.2015.2
Clark, G. F., Krivan, H. C., Wilkins, T. D., and Smith, D. F. (1987). Toxin A from Clostridium difficile binds to rabbit erythrocyte glycolipids with terminal Gal alpha 1-3Gal beta 1-4GlcNAc sequences. Arch. Biochem. Biophys. 257, 217–229. doi: 10.1016/0003-9861(87)90561-3
Colicelli, J. (2004). Human RAS superfamily proteins and related GTPases. Sci. STKE 2004, RE13. doi: 10.1126/stke.2502004re13
Costa, D., Shin, J. H., Goldbeck, S. M., Bolick, D. T., Mesquita, F. S., Loureiro, A. V., et al. (2022). Adenosine receptors differentially mediate enteric glial cell death induced by Clostridioides difficile Toxins A and B. Front. Immunol. 13, 956326. doi: 10.3389/fimmu.2022.956326
Cowardin, C. A., Kuehne, S. A., Buonomo, E. L., Marie, C. S., Minton, N. P., Petri, J. W. A., et al. (2015). Inflammasome activation contributes to interleukin-23 production in response to Clostridium difficile. mBio 6 (1), e02386-14. doi: 10.1128/mBio.02386-14
Danis-Wlodarczyk, K. M., Wozniak, D. J., and Abedon, S. T. (2021). Treating bacterial infections with bacteriophage-based enzybiotics: in vitro, in vivo and clinical application. Antibiotics (Basel) 10, 12. doi: 10.3390/antibiotics10121497
Davies, N. L., Compson, J. E., Mackenzie, B., O’Dowd, V. L., Oxbrow, A. K., Heads, J. T., et al. (2013). A mixture of functionally oligoclonal humanized monoclonal antibodies that neutralize Clostridium difficile TcdA and TcdB with high levels of in vitro potency shows in vivo protection in a hamster infection model. Clin. Vaccine Immunol. 20, 377–390. doi: 10.1128/CVI.00625-12
de Bruyn, G., Gordon, D. L., Steiner, T., Tambyah, P., Cosgrove, C., Martens, M., et al. (2021). Safety, immunogenicity, and efficacy of a Clostridioides difficile toxoid vaccine candidate: a phase 3 multicentre, observer-blind, randomised, controlled trial. Lancet Infect. Dis. 21, 252–262. doi: 10.1016/S1473-3099(20)30331-5
Di Bella, S., Sanson, G., Monticelli, J., Zerbato, V., Principe, L., Giuffre, M., et al. (2024). Clostridioides difficile infection: history, epidemiology, risk factors, prevention, clinical manifestations, treatment, and future options. Clin. Microbiol. Rev. 37, e0013523. doi: 10.1128/cmr.00135-23
Dingle, K. E., Didelot, X., Quan, T. P., Eyre, D. W., Stoesser, N., Golubchik, T., et al. (2017). Effects of control interventions on Clostridium difficile infection in England: an observational study. Lancet Infect. Dis. 17, 411–421. doi: 10.1016/S1473-3099(16)30514-X
Dingle, K. E., Elliott, B., Robinson, E., Griffiths, D., Eyre, D. W., Stoesser, N., et al. (2014). Evolutionary history of the clostridium difficile pathogenicity locus. Genome Biol. Evol. 6, 36–52. doi: 10.1093/gbe/evt204
Dingle, T., Wee, S., Mulvey, G. L., Greco, A., Kitova, E. N., Sun, J., et al. (2008). Functional properties of the carboxy-terminal host cell-binding domains of the two toxins, TcdA and TcdB, expressed by Clostridium difficile. Glycobiology 18, 698–706. doi: 10.1093/glycob/cwn048
Dinh, A., Le Monnier, A., Emery, C., Alami, S., Torreton, E., Duburcq, A., et al. (2019). Predictors and burden of hospital readmission with recurrent Clostridioides difficile infection: a French nation-wide inception cohort study. Eur. J. Clin. Microbiol. Infect. Dis. 38, 1297–1305. doi: 10.1007/s10096-019-03552-9
Donskey, C. J., Dubberke, E. R., Klein, N. P., Liles, E. G., Szymkowiak, K., Wilcox, M. H., et al. (2024). CLOVER (CLOstridium difficile Vaccine Efficacy tRial) Study: A Phase 3, Randomized Trial Investigating the Efficacy and Safety of a Detoxified Toxin A/B Vaccine in Adults 50 Years and Older at Increased Risk of Clostridioides difficile Infection. Clin. Infect. Dis. 79, 1503–1511. doi: 10.1093/cid/ciae410
Doyle, D. A., DeAngelis, P. L., and Ballard, J. D. (2024). CSPG4-dependent cytotoxicity for C. difficile TcdB is influenced by extracellular calcium and chondroitin sulfate. mSphere 9, e0009424. doi: 10.1128/msphere.00094-24
Drekonja, D. M., Shaukat, A., Huang, Y., Zhang, J. H., Reinink, A. R., Nugent, S., et al. (2025). A randomized controlled trial of efficacy and safety of fecal microbiota transplant for preventing recurrent clostridioides difficile infection. Clin. Infect. Dis. 80, 52–60. doi: 10.1093/cid/ciae467
Dubberke, E. R., Lee, C. H., Orenstein, R., Khanna, S., Hecht, G., and Gerding, D. N. (2018). Results from a randomized, placebo-controlled clinical trial of a RBX2660-A microbiota-based drug for the prevention of recurrent clostridium difficile infection. Clin. Infect. Dis. 67, 1198–1204. doi: 10.1093/cid/ciy259
Egerer, M., Giesemann, T., Jank, T., Satchell, K. J., and Aktories, K. (2007). Auto-catalytic cleavage of Clostridium difficile toxins A and B depends on cysteine protease activity. J. Biol. Chem. 282, 25314–25321. doi: 10.1074/jbc.M703062200
Elmore, S. (2007). Apoptosis: a review of programmed cell death. Toxicol. Pathol. 35, 495–516. doi: 10.1080/01926230701320337
Etienne-Manneville, S. and Hall, A. (2002). Rho GTPases in cell biology. Nature 420, 629–635. doi: 10.1038/nature01148
Eubank, T. A., Jo, J., Alam, M. J., Begum, K., McPherson, J. K., Le, T. M., et al. (2025). Efficacy, safety, pharmacokinetics, and associated microbiome changes of ibezapolstat compared with vancomycin in adults with Clostridioides difficile infection: a phase 2b, randomised, double-blind, active-controlled, multicentre study. Lancet Microbe 101126 (8), 101126. doi: 10.1016/j.lanmic.2025.101126
Farrow, M. A., Chumber, N. M., Bloch, S. C., King, M., Moton-Melancon, K., Shupe, J., et al. (2020). Small molecule inhibitor screen reveals calcium channel signaling as a mechanistic mediator of clostridium difficile tcdB-induced necrosis. ACS Chem. Biol. 15, 1212–1221. doi: 10.1021/acschembio.9b00906
Farrow, M. A., Chumbler, N. M., Lapierre, L. A., Franklin, J. L., Rutherford, S. A., Goldenring, J. R., et al. (2013). Clostridium difficile toxin B-induced necrosis is mediated by the host epithelial cell NADPH oxidase complex. Proc. Natl. Acad. Sci. U.S.A. 110, 18674–18679. doi: 10.1073/pnas.1313658110
Fettucciari, K., Marguerie, F., Fruganti, A., Marchegiani, A., Spaterna, A., Brancorsini, S., et al. (2022). Clostridioides difficile toxin B alone and with pro-inflammatory cytokines induces apoptosis in enteric glial cells by activating three different signalling pathways mediated by caspases, calpains and cathepsin B. Cell Mol. Life Sci. 79, 442. doi: 10.1007/s00018-022-04459-z
Fettucciari, K., Ponsini, P., Gioe, D., Macchioni, L., Palumbo, C., Antonelli, E., et al. (2017). Enteric glial cells are susceptible to Clostridium difficile toxin B. Cell Mol. Life Sci. 74, 1527–1551. doi: 10.1007/s00018-016-2426-4
Feuerstadt, P., Allegretti, J., and Khanna, S. (2025a). Treatment of clostridioides difficile: the times they are changing. Am. J. Gastroenterol doi: 10.14309/ajg.0000000000003445
Feuerstadt, P., Chopra, T., Knapple, W., Van Hise, N. W., Dubberke, E. R., Baggott, B., et al. (2025b). PUNCH CD3-OLS: A phase 3 prospective observational cohort study to evaluate the safety and efficacy of fecal microbiota, live-jslm (REBYOTA) in adults with recurrent clostridioides difficile infection. Clin. Infect. Dis. 80, 43–51. doi: 10.1093/cid/ciae437
Feuerstadt, P., Louie, T. J., Lashner, B., Wang, E., Diao, L., Bryant, J. A., et al. (2022). SER-109, an oral microbiome therapy for recurrent clostridioides difficile infection. N Engl. J. Med. 386, 220–229. doi: 10.1056/NEJMoa2106516
Fletcher, J. R., Pike, C. M., Parsons, R. J., Rivera, A. J., Foley, M. H., McLaren, M. R., et al. (2021). Clostridioides difficile exploits toxin-mediated inflammation to alter the host nutritional landscape and exclude competitors from the gut microbiota. Nat. Commun. 12, 462. doi: 10.1038/s41467-020-20746-4
Flores-Diaz, M., Alape-Giron, A., Persson, B., Pollesello, P., Moos, M., von Eichel-Streiber, C., et al. (1997). Cellular UDP-glucose deficiency caused by a single point mutation in the UDP-glucose pyrophosphorylase gene. J. Biol. Chem. 272, 23784–23791. doi: 10.1074/jbc.272.38.23784
Frisbee, A. L., Saleh, M. M., Young, M. K., Leslie, J. L., Simpson, M. E., Abhyankar, M. M., et al. (2019). IL-33 drives group 2 innate lymphoid cell-mediated protection during Clostridium difficile infection. Nat. Commun. 10, 2712. doi: 10.1038/s41467-019-10733-9
Gao, W., Yang, J., Liu, W., Wang, Y., and Shao, F. (2016). Site-specific phosphorylation and microtubule dynamics control Pyrin inflammasome activation. Proc. Natl. Acad. Sci. U.S.A. 113, E4857–E4866. doi: 10.1073/pnas.1601700113
Genisyuerek, S., Papatheodorou, P., Guttenberg, G., Schubert, R., Benz, R., and Aktories, K. (2011). Structural determinants for membrane insertion, pore formation and translocation of Clostridium difficile toxin B. Mol. Microbiol. 79, 1643–1654. doi: 10.1111/j.1365-2958.2011.07549.x
Genth, H., Dreger, S. C., Huelsenbeck, J., and Just, I. (2008). Clostridium difficile toxins: more than mere inhibitors of Rho proteins. Int. J. Biochem. Cell Biol. 40, 592–597. doi: 10.1016/j.biocel.2007.12.014
Gerding, D. N., Johnson, S., Rupnik, M., and Aktories, K. (2014). Clostridium difficile binary toxin CDT: mechanism, epidemiology, and potential clinical importance. Gut Microbes 5, 15–27. doi: 10.4161/gmic.26854
Gerhard, R., Nottrott, S., Schoentaube, J., Tatge, H., Olling, A., and Just, I. (2008). Glucosylation of Rho GTPases by Clostridium difficile toxin A triggers apoptosis in intestinal epithelial cells. J. Med. Microbiol. 57, 765–770. doi: 10.1099/jmm.0.47769-0
Ghosh, S., Erickson, D., Chua, M. J., Collins, J., and Jala, V. R. (2024). The microbial metabolite urolithin A reduces Clostridioides difficile toxin expression and toxin-induced epithelial damage. mSystems 9 (2), e0125523. doi: 10.1128/msystems.01255-23
Giannasca, P. J., Zhang, Z. X., Lei, W. D., Boden, J. A., Giel, M. A., Monath, T. P., et al. (1999). Serum antitoxin antibodies mediate systemic and mucosal protection from Clostridium difficile disease in hamsters. Infect. Immun. 67, 527–538. doi: 10.1128/IAI.67.2.527-538.1999
Giesemann, T., Egerer, M., Jank, T., and Aktories, K. (2008). Processing of Clostridium difficile toxins. J. Med. Microbiol. 57, 690–696. doi: 10.1099/jmm.0.47742-0
Giesemann, T., Jank, T., Gerhard, R., Maier, E., Just, I., Benz, R., et al. (2006). Cholesterol-dependent pore formation of Clostridium difficile toxin A. J. Biol. Chem. 281, 10808–10815. doi: 10.1074/jbc.M512720200
Gonzales-Luna, A. J., Carlson, T. J., and Garey, K. W. (2023). Emerging options for the prevention and management of clostridioides difficile infection. Drugs 83, 105–116. doi: 10.1007/s40265-022-01832-x
Gregorieff, A., Liu, Y., Inanlou, M. R., Khomchuk, Y., and Wrana, J. L. (2015). Yap-dependent reprogramming of Lgr5(+) stem cells drives intestinal regeneration and cancer. Nature 526, 715–718. doi: 10.1038/nature15382
Guo, S., Chen, Y., Liu, J., Zhang, X., Liu, Z., Zhou, Z., et al. (2022). Low-density lipoprotein receptor-related protein 1 is a CROPs-associated receptor for Clostridioides infection toxin B. Sci. China Life Sci. 65, 107–118. doi: 10.1007/s11427-021-1943-9
Guo, Y., Liu, Z., Wang, J., Deng, X., He, L., Zhang, Y., et al. (2025a). Equol neutralizes toxin B to combat Clostridioides difficile infection without disrupting the gut microbiota. Microbiol. Res. 298, 128-219. doi: 10.1016/j.micres.2025.128219
Guo, Y., Zhang, Y., Wang, G., Liu, H., Wang, J., Deng, X., et al. (2025b). Caffeic acid phenethyl ester protects Clostridioides difficile infection by toxin inhibition and microbiota modulation. Elife 13, RP101757. doi: 10.7554/eLife.101757.4
Gupta, S. B., Mehta, V., Dubberke, E. R., Zhao, X., Dorr, M. B., Guris, D., et al. (2016). Antibodies to toxin B are protective against clostridium difficile infection recurrence. Clin. Infect. Dis. 63, 730–734. doi: 10.1093/cid/ciw364
Hall, I. C. (1935). Intestinal flora in new-born infants with a description of a new pathogenic anaerobe, Bacillus Difficilis. Am. J. Dis. Child, 49390–49402.
Hasegawa, M., Kamada, N., Jiao, Y., Liu, M. Z., Nunez, G., and Inohara, N. (2012). Protective role of commensals against Clostridium difficile infection via an IL-1beta-mediated positive-feedback loop. J. Immunol. 189, 3085–3091. doi: 10.4049/jimmunol.1200821
He, R., Peng, J., Yuan, P., Yang, J., Wu, X., Wang, Y., et al. (2017). Glucosyltransferase activity of clostridium difficile toxin B triggers autophagy-mediated cell growth arrest. Sci. Rep. 7, 10532. doi: 10.1038/s41598-017-11336-4
Heidebrecht, H. J., Lagkouvardos, I., Reitmeier, S., Hengst, C., Kulozik, U., and Pfaffl, M. W. (2021). Alteration of Intestinal Microbiome of Clostridioides difficile-Infected Hamsters during the Treatment with Specific Cow Antibodies. Antibiotics (Basel) 10, 6. doi: 10.3390/antibiotics10060724
Heimann, S. M., Vehreschild, J. J., Cornely, O. A., Wisplinghoff, H., Hallek, M., Goldbrunner, R., et al. (2015). Economic burden of Clostridium difficile associated diarrhoea: a cost-of-illness study from a German tertiary care hospital. Infection 43, 707–714. doi: 10.1007/s15010-015-0810-x
Henkel, D., Tatge, H., Schottelndreier, D., Tao, L., Dong, M., and Gerhard, R. (2020). Receptor binding domains of tcdB from clostridioides difficile for chondroitin sulfate proteoglycan-4 and frizzled proteins are functionally independent and additive. Toxins (Basel) 12, 12. doi: 10.3390/toxins12120736
Hernandez, L. D., Kroh, H. K., Hsieh, E., Yang, X., Beaumont, M., Sheth, P. R., et al. (2017). Epitopes and mechanism of action of the clostridium difficile toxin A-neutralizing antibody actoxumab. J. Mol. Biol. 429, 1030–1044. doi: 10.1016/j.jmb.2017.02.010
Herrera, G., Paredes-Sabja, D., Patarroyo, M. A., Ramirez, J. D., and Munoz, M. (2021). Updating changes in human gut microbial communities associated with Clostridioides difficile infection. Gut Microbes 13, 1966277. doi: 10.1080/19490976.2021.1966277
Hordijk, P. L. (2006). Regulation of NADPH oxidases: the role of Rac proteins. Circ. Res. 98, 453–462. doi: 10.1161/01.RES.0000204727.46710.5e
Hsieh, M. L., Nishizaki, D., Adashek, J. J., Kato, S., and Kurzrock, R. (2025). Toll-like receptor 3: a double-edged sword. biomark. Res. 13, 32. doi: 10.1186/s40364-025-00739-5
Hutton, M. L., Pehlivanoglu, H., Vidor, C. J., James, M. L., Thomson, M. J., and Lyras, D. (2020). Repurposing auranofin as a Clostridioides difficile therapeutic. J. Antimicrob. Chemother. 75, 409–417. doi: 10.1093/jac/dkz430
Imwattana, K., Knight, D. R., Kullin, B., Collins, D. A., Putsathit, P., Kiratisin, P., et al. (2019). Clostridium difficile ribotype 017 - characterization, evolution and epidemiology of the dominant strain in Asia. Emerging Microbes Infections 8, 796–807. doi: 10.1080/22221751.2019.1621670
Ishida, Y., Maegawa, T., Kondo, T., Kimura, A., Iwakura, Y., Nakamura, S., et al. (2004). Essential involvement of IFN-gamma in Clostridium difficile toxin A-induced enteritis. J. Immunol. 172, 3018–3025. doi: 10.4049/jimmunol.172.5.3018
Jamilloux, Y., Magnotti, F., Belot, A., and Henry, T. (2018). The pyrin inflammasome: from sensing RhoA GTPases-inhibiting toxins to triggering autoinflammatory syndromes. Pathog. Dis. 76, 3. doi: 10.1093/femspd/fty020
Jank, T. and Aktories, K. (2008). Structure and mode of action of clostridial glucosylating toxins: the ABCD model. Trends Microbiol. 16, 222–229. doi: 10.1016/j.tim.2008.01.011
Jank, T., Giesemann, T., and Aktories, K. (2007a). Rho-glucosylating Clostridium difficile toxins A and B: new insights into structure and function. Glycobiology 17, 15R–22R. doi: 10.1093/glycob/cwm004
Jank, T., Giesemann, T., and Aktories, K. (2007b). Clostridium difficile glucosyltransferase toxin B-essential amino acids for substrate binding. J. Biol. Chem. 282, 35222–35231. doi: 10.1074/jbc.M703138200
Jank, T., Ziegler, M. O., Schulz, G. E., and Aktories, K. (2008). Inhibition of the glucosyltransferase activity of clostridial Rho/Ras-glucosylating toxins by castanospermine. FEBS Lett. 582, 2277–2282. doi: 10.1016/j.febslet.2008.05.025
Jarchum, I., Liu, M., Lipuma, L., and Pamer, E. G. (2011). Toll-Like Receptor 5 Stimulation Protects Mice from Acute Clostridium difficile Colitis. Immun 79 (4), 1495–1503. doi: 10.1128/IAI.01196-10
Jinno, A. and Park, P. W. (2015). Role of glycosaminoglycans in infectious disease. Methods Mol. Biol. 1229, 567–585. doi: 10.1007/978-1-4939-1714-3_45
Johnson, S., Lavergne, V., Skinner, A. M., Gonzales-Luna, A. J., Garey, K. W., Kelly, C. P., et al. (2021). Clinical practice guideline by the infectious diseases society of america (IDSA) and society for healthcare epidemiology of america (SHEA): 2021 focused update guidelines on management of clostridioides difficile infection in adults. Clin. Infect. Dis. 73, 755–757. doi: 10.1093/cid/ciab718
Jose, S., Mukherjee, A., Horrigan, O., Setchell, K., Zhang, W., Moreno-Fernandez, M. E., et al. (2021). Obeticholic acid ameliorates severity of Clostridioides difficile infection in high fat diet-induced obese mice. Mucosal Immunol. 14, 500–510. doi: 10.1038/s41385-020-00338-7
Just, I., Selzer, J., Wilm, M., von Eichel-Streiber, C., Mann, M., and Aktories, K. (1995). Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature 375, 500–503. doi: 10.1038/375500a0
Kapandji, N., Salmona, M., Lemoine, A., Ulmann, G., Calderaro, J., Roche, B., et al. (2025). Unravelling neutropenic enterocolitis: insights from gut microbiota, and intestinal barrier analyses. Exp. Hematol. Oncol. 14, 74. doi: 10.1186/s40164-025-00661-4
Kellogg, T. D., Ceglia, S., Mortzfeld, B. M., Tanna, T. M., Zeamer, A. L., Mancini, M. R., et al. (2025). Succinate-producing microbiota drives tuft cell hyperplasia to protect against Clostridioides difficile. J. Exp. Med. 222, 1. doi: 10.1084/jem.20232055
Kelly, C. P., Becker, S., Linevsky, J. K., Joshi, M. A., O’Keane, J. C., Dickey, B. F., et al. (1994a). Neutrophil recruitment in Clostridium difficile toxin A enteritis in the rabbit. J. Clin. Invest. 93, 1257–1265. doi: 10.1172/JCI117080
Kelly, C. R., Fischer, M., Allegretti, J. R., LaPlante, K., Stewart, D. B., Limketkai, B. N., et al. (2021). ACG clinical guidelines: prevention, diagnosis, and treatment of clostridioides difficile infections. Am. J. Gastroenterol. 116, 1124–1147. doi: 10.14309/ajg.0000000000001278
Kelly, C. P., Pothoulakis, C., and LaMont, J. T. (1994b). Clostridium difficile colitis. N Engl. J. Med. 330, 257–262. doi: 10.1056/NEJM199401273300406
Khanna, S., Assi, M., Lee, C., Yoho, D., Louie, T., Knapple, W., et al. (2022). Efficacy and safety of RBX2660 in PUNCH CD3, a phase III, randomized, double-blind, placebo-controlled trial with a bayesian primary analysis for the prevention of recurrent clostridioides difficile infection. Drugs 82, 1527–1538. doi: 10.1007/s40265-022-01797-x
Kim, J. M., Lee, J. Y., Yoon, Y. M., Oh, Y. K., Youn, J., and Kim, Y. J. (2006). NF-kappa B activation pathway is essential for the chemokine expression in intestinal epithelial cells stimulated with Clostridium difficile toxin A. Scand. J. Immunol. 63, 453–460. doi: 10.1111/j.1365-3083.2006.001756.x
Kimura, T., Stanhope, S., and Sugitani, T. (2020). Excess length of hospital stay, mortality and cost attributa ble to Clostridioides (Clostridium) difficile infection and recurrence: a nationwide analysis in Japan. Epidemiol. Infect. 148, e65. doi: 10.1017/S0950268820000606
Kink, J. A. and Williams, J. A. (1998). Antibodies to recombinant Clostridium difficile toxins A and B are an effective treatment and prevent relapse of C. difficile-associated disease in a hamster model of infection. Infect. Immun. 66, 2018–2025. doi: 10.1128/IAI.66.5.2018-2025.1998
Kitchin, N., Remich, S. A., Peterson, J., Peng, Y., Gruber, W. C., Jansen, K. U., et al. (2020). A phase 2 study evaluating the safety, tolerability, and immunogenicity of two 3-dose regimens of a clostridium difficile vaccine in healthy US adults aged 65 to 85 years. Clin. Infect. Dis. 70, 1–10. doi: 10.1093/cid/ciz153
Kloft, N., Neukirch, C., Bobkiewicz, W., Veerachato, G., Busch, T., von Hoven, G., et al. (2010). Pro-autophagic signal induction by bacterial pore-forming toxins. Med. Microbiol. Immunol. 199, 299–309. doi: 10.1007/s00430-010-0163-0
Knight, D. R., Elliott, B., Chang, B. J., Perkins, T. T., and Riley, T. V. (2015). Diversity and evolution in the genome of clostridium difficile. Clin. Microbiol. Rev. 28, 721–741. doi: 10.1128/CMR.00127-14
Knisely, J. M., Liu, B., Ranallo, R. T., and Zou, L. (2016). Vaccines for healthcare-associated infections: promise and challenge. Clin. Infect. Dis. 63, 657–662. doi: 10.1093/cid/ciw333
Kociolek, L. K. and Gerding, D. N. (2016). Breakthroughs in the treatment and prevention of Clostridium difficile infection. Nat. Rev. Gastroenterol. Hepatol. 13, 150–160. doi: 10.1038/nrgastro.2015.220
Kordus, S. L., Kroh, H. K., Rodriguez, R. C., Shrem, R. A., Peritore-Galve, F. C., Shupe, J. A., et al. (2023). Nanobodies against C. difficile TcdA and TcdB reveal unexpected neutralizing epitopes and provide a toolkit for toxin quantitation in vivo. PloS Pathog. 19 (10), e1011496. doi: 10.1371/journal.ppat.1011496
Kordus, S. L., Thomas, A. K., and Lacy, D. B. (2021). Clostridioides difficile toxins: mechanisms of action and antitoxin therapeutics. Nat. Rev. Microbiol 20 (5), 285–298. doi: 10.1038/s41579-021-00660-2
Kreimeyer, I., Euler, F., Marckscheffel, A., Tatge, H., Pich, A., Olling, A., et al. (2011). Autoproteolytic cleavage mediates cytotoxicity of Clostridium difficile toxin A, Naunyn Schmiedebergs. Arch. Pharmacol. 383, 253–262. doi: 10.1007/s00210-010-0574-x
Krivan, H. C., Clark, G. F., Smith, D. F., and Wilkins, T. D. (1986). Cell surface binding site for Clostridium difficile enterotoxin: evidence for a glycoconjugate containing the sequence Gal alpha 1-3Gal beta 1-4GlcNAc. Infect. Immun. 53, 573–581. doi: 10.1128/iai.53.3.573-581.1986
Kroh, H. K., Chandrasekaran, R., Zhang, Z., Rosenthal, K., Woods, R., Jin, X., et al. (2018). A neutralizing antibody that blocks delivery of the enzymatic cargo of Clostridium difficile toxin TcdB into host cells. J. Biol. Chem. 293, 941–952. doi: 10.1074/jbc.M117.813428
Kuehne, S. A., Cartman, S. T., Heap, J. T., Kelly, M. L., Cockayne, A., and Minton, N. P. (2010). The role of toxin A and toxin B in Clostridium difficile infection. Nature 467, 711–713. doi: 10.1038/nature09397
Kyne, L., Warny, M., Qamar, A., and Kelly, C. P. (2001). Association between antibody response to toxin A and protection against recurrent Clostridium difficile diarrhoea. Lancet 357, 189–193. doi: 10.1016/S0140-6736(00)03592-3
LaFrance, M. E., Farrow, M. A., Chandrasekaran, R., Sheng, J., Rubin, D. H., and Lacy, D. B. (2015). Identification of an epithelial cell receptor responsible for Clostridium difficile TcdB-induced cytotoxicity. Proc. Natl. Acad. Sci. U.S.A. 112, 7073–7078. doi: 10.1073/pnas.1500791112
Lambert, G. S. and Baldwin, M. R. (2016). Evidence for dual receptor-binding sites in Clostridium difficile toxin A. FEBS Lett. 590, 4550–4563. doi: 10.1002/1873-3468.12487
Lanis, J. M., Barua, S., and Ballard, J. D. (2010). Variations in TcdB activity and the hypervirulence of emerging strains of Clostridium difficile. PloS Pathog. 84 (1), e1001061. doi: 10.1371/journal.ppat.1001061
Lanis, J. M., Heinlen, L. D., James, J. A., and Ballard, J. D. (2013). Clostridium difficile 027/BI/NAP1 encodes a hypertoxic and antigenically variable form of TcdB. PloS Pathog. 9, e1003523. doi: 10.1371/journal.ppat.1003523
Lanis, J. M., Hightower, L. D., Shen, A., and Ballard, J. D. (2012). TcdB from hypervirulent Clostridium difficile exhibits increased efficiency of autoprocessing. Mol. Microbiol. 84, 66–76. doi: 10.1111/j.1365-2958.2012.08009.x
Larabee, J. L., Doyle, D. A., Ahmed, U., Shadid, T. M., Sharp, R. R., Jones, K. L., et al. (2023). Discovery of Hippo signaling as a regulator of CSPG4 expression and as a therapeutic target for Clostridioides difficile disease. PloS Pathog. 19, e1011272. doi: 10.1371/journal.ppat.1011272
Leav, B. A., Blair, B., Leney, M., Knauber, M., Reilly, C., Lowy, I., et al. (2010). Serum anti-toxin B antibody correlates with protection from recurrent Clostridium difficile infection (CDI). Vaccine 28, 965–969. doi: 10.1016/j.vaccine.2009.10.144
Lee, J. Y., Park, H. R., Oh, Y. K., Kim, Y. J., Youn, J., Han, J. S., et al. (2007). Effects of transcription factor activator protein-1 on interleukin-8 expression and enteritis in response to Clostridium difficile toxin A. J. Mol. Med. (Berl) 85, 1393–1404. doi: 10.1007/s00109-007-0237-7
Leffler, D. A. and Lamont, J. T. (2015). Clostridium difficile infection. N Engl. J. Med. 373, 287–288. doi: 10.1056/NEJMc1506004
Lessa, F. C., Mu, Y., Bamberg, W. M., Beldavs, Z. G., Dumyati, G. K., Dunn, J. R., et al. (2015). Burden of Clostridium difficile infection in the United States. N Engl. J. Med. 372, 825–834. doi: 10.1056/NEJMoa1408913
Li, Y., Rui, W., Sheng, X., Deng, X., Li, X., Meng, L., et al. (2025). Bifidobacterium breve synergizes with Akkermansia muciniphila and Bacteroides ovatus to antagonize Clostridioides difficile. ISME J. 19, 1. doi: 10.1093/ismejo/wraf086
Li, D. and Saavedra, P. (2025). The interplay between host immunity and Clostridioides difficile infection. mBio 16 (8), e0356224. doi: 10.1128/mbio.03562-24
Li, S., Shi, L., Yang, Z., Zhang, Y., Perez-Cordon, G., Huang, T., et al. (2015). Critical roles of Clostridium difficile toxin B enzymatic activities in pathogenesis. Infect. Immun. 83, 502–513. doi: 10.1128/IAI.02316-14
Li, Y., Wang, Z., Bai, L. L., Li, Y. Z., Jiang, Y. J., Xu, T. L., et al. (2024). Positive intervention of distinct peptides in clostridioides difficile infection in a mouse model. Commun. Biol. 7, 1172. doi: 10.1038/s42003-024-06850-x
Li, Y., Xu, W., Ren, Y., Cheung, H. C., Huang, P., Kaur, G., et al. (2022). Plakoglobin and high-mobility group box 1 mediate intestinal epithelial cell apoptosis induced by clostridioides difficile tcdB. mBio 13 (5), e0184922. doi: 10.1128/mbio.01849-22
Liang, J., Ning, Y., Dong, L., Ma, X., Li, S., Yang, H., et al. (2020). The role of the globular heads of the C1q receptor in TcdA-induced human colonic epithelial cell apoptosis via a mitochondria-dependent pathway. BMC Microbiol. 20, 274. doi: 10.1186/s12866-020-01958-6
Lietz, S., Sokolowski, L. M., Lindner, K., Rodriguez, A. A., Standker, L., Vogel, V., et al. (2025). The antimicrobial peptide Angie 5 inhibits TcdA and TcdB from Clostridioides difficile. Cell Mol. Life Sci. 82, 265. doi: 10.1007/s00018-025-05799-2
Liu, L., Wang, H., Chen, X., and Xie, P. (2023). Gut microbiota: a new insight into neurological diseases. Chin. Med. J. (Engl) 136, 1261–1277. doi: 10.1097/CM9.0000000000002212
Liu, Z., Zhang, S., Chen, P., Tian, S., Zeng, J., Perry, K., et al. (2021). Structural basis for selective modification of Rho and Ras GTPases by Clostridioides difficile toxin B. Sci. Adv. 7, eabi4582. doi: 10.1126/sciadv.abi4582
Longhitano, A., Roder, C., Blackmore, T., Campbell, A., May, M., and Athan, E. (2025). Australasian Society of Infectious Diseases updated guidelines for the management of Clostridioides difficile infection in adults and children in Australia and New Zealand. Intern. Med. J. 55, 503–513. doi: 10.1111/imj.16638
Lopez-Urena, D., Orozco-Aguilar, J., Chaves-Madrigal, Y., Ramirez-Mata, A., Villalobos-Jimenez, A., Ost, S., et al. (2019). Toxin B Variants from Clostridium difficile Strains VPI 10463 and NAP1/027 Share Similar Substrate Profile and Cellular Intoxication Kinetics but Use Different Host Cell Entry Factors. Toxins (Basel) 11, 6. doi: 10.3390/toxins11060348
Lowy, I., Molrine, D. C., Leav, B. A., Blair, B. M., Baxter, R., Gerding, D. N., et al. (2010). Treatment with monoclonal antibodies against Clostridium difficile toxins. N Engl. J. Med. 362, 197–205. doi: 10.1056/NEJMoa0907635
Luo, J., Yang, Q., Zhang, X., Zhang, Y., Wan, L., Zhan, X., et al. (2022). TFPI is a colonic crypt receptor for TcdB from hypervirulent clade 2 C. difficile. Cell 185, 980–994.e15. doi: 10.1016/j.cell.2022.02.010
Lyerly, D. M., Krivan, H. C., and Wilkins, T. D. (1988). Clostridium difficile: its disease and toxins. Clin. Microbiol. Rev. 1, 1–18. doi: 10.1128/CMR.1.1.1
Lyerly, D. M., Saum, K. E., MacDonald, D. K., and Wilkins, T. D. (1985). Effects of Clostridium difficile toxins given intragastrically to animals. Infect. Immun. 47, 349–352. doi: 10.1128/iai.47.2.349-352.1985
Lyras, D., Connor, O., Howarth, P. M., Sambol, S. P., Carter, G. P., Phumoonna, T., et al. (2009). Toxin B is essential for virulence of Clostridium difficile. Nature 458, 1176–1179. doi: 10.1038/nature07822
Macara, I. G., Lounsbury, K. M., Richards, S. A., McKiernan, C., and Bar-Sagi, D. (1996). The ras superfamily of GTPases, FASEB J, vol. 10 No. 5 pp, 625–630. doi: 10.1096/fasebj.10.5.8621061
Macchioni, L., Davidescu, M., Fettucciari, K., Petricciuolo, M., Gatticchi, L., Gioe, D., et al. (2017). Enteric glial cells counteract Clostridium difficile Toxin B through a NADPH oxidase/ROS/JNK/caspase-3 axis, without involving mitochondrial pathways. Sci. Rep. 7, 45569. doi: 10.1038/srep45569
MacDonald, B. T. and He, X. (2012). Frizzled and LRP5/6 receptors for Wnt/beta-catenin signaling. Cold Spring Harb. Perspect. Biol. 4 (12), a007880. doi: 10.1101/cshperspect.a007880
Madan, R. and Petri, W. J. (2012). Immune responses to Clostridium difficile infection. Trends Mol. Med. 18, 658–666. doi: 10.1016/j.molmed.2012.09.005
Madden, G. R., Preissner, R., Preissner, S., and Petri, W. A. (2025). Anti-interleukin-23 treatment linked to improved Clostridioides difficile infection survival. Gut Microbes 17, 2480195. doi: 10.1080/19490976.2025.2480195
Manion, J., Musser, M. A., Kuziel, G. A., Liu, M., Shepherd, A., Wang, S., et al. (2023). C. difficile intoxicates neurons and pericytes to drive neurogenic inflammation. Nature 622, 611–618. doi: 10.1038/s41586-023-06607-2
Manse, J. S. and Baldwin, M. R. (2015). Binding and entry of Clostridium difficile toxin B is mediated by multiple domains. FEBS Lett. 589, 3945–3951. doi: 10.1016/j.febslet.2015.11.017
Mansfield, M. J., Tremblay, B. J., Zeng, J., Wei, X., Hodgins, H., Worley, J., et al. (2020). Phylogenomics of 8,839 Clostridioides difficile genomes reveals recombination-driven evolution and diversification of toxin A and B. PloS Pathog. 16, e1009181. doi: 10.1371/journal.ppat.1009181
Maroo, S. and Lamont, J. T. (2006). Recurrent clostridium difficile. Gastroenterology 130, 1311–1316. doi: 10.1053/j.gastro.2006.02.044
Marozsan, A. J., Ma, D., Nagashima, K. A., Kennedy, B. J., Kang, Y. K., Arrigale, R. R., et al. (2012). Protection against Clostridium difficile infection with broadly neutralizing antitoxin monoclonal antibodies. J. Infect. Dis. 206, 706–713. doi: 10.1093/infdis/jis416
Martin, J. S., Monaghan, T. M., and Wilcox, M. H. (2016). Clostridium difficile infection: epidemiology, diagnosis and understanding transmission. Nat. Rev. Gastroenterol. Hepatol. 13, 206–216. doi: 10.1038/nrgastro.2016.25
Martins, C. S., Costa, D., Lima, B. B., Leitao, R., Freire, G. E., Silva, G., et al. (2020). Clostridioides difficile toxin A-induced wnt/beta-catenin pathway inhibition is mediated by rac1 glucosylation. Front. Microbiol. 11, 1998. doi: 10.3389/fmicb.2020.01998
Matarrese, P., Falzano, L., Fabbri, A., Gambardella, L., Frank, C., Geny, B., et al. (2007). Clostridium difficile toxin B causes apoptosis in epithelial cells by thrilling mitochondria. Involvement of ATP-sensitive mitochondrial potassium channels. J. Biol. Chem. 282, 9029–9041. doi: 10.1074/jbc.M607614200
Matte, I., Lane, D., Cote, E., Asselin, A. E., Fortier, L. C., Asselin, C., et al. (2009). Antiapoptotic proteins Bcl-2 and Bcl-XL inhibit Clostridium difficile toxin A-induced cell death in human epithelial cells. Infect. Immun. 77, 5400–5410. doi: 10.1128/IAI.00485-09
McDermott, A. J., Falkowski, N. R., McDonald, R. A., Frank, C. R., Pandit, C. R., Young, V. B., et al. (2017). Role of interferon-gamma and inflammatory monocytes in driving colonic inflammation during acute Clostridium difficile infection in mice. Immunology 150, 468–477. doi: 10.1111/imm.12700
McDermott, A. J., Falkowski, N. R., McDonald, R. A., Pandit, C. R., Young, V. B., and Huffnagle, G. B. (2016). Interleukin-23 (IL-23), independent of IL-17 and IL-22, drives neutrophil recruitment and innate inflammation during Clostridium difficile colitis in mice. Immunology 147, 114–124. doi: 10.1111/imm.12545
McDonald, L. C., Gerding, D. N., Johnson, S., Bakken, J. S., Carroll, K. C., Coffin, S. E., et al. (2018). Clinical practice guidelines for clostridium difficile infection in adults and children: 2017 update by the infectious diseases society of america (IDSA) and society for healthcare epidemiology of america (SHEA). Clin. Infect. Dis. 66, 987–994. doi: 10.1093/cid/ciy149
McDonald, L. C., Killgore, G. E., Thompson, A., Owens, R. J., Kazakova, S. V., Sambol, S. P., et al. (2005). An epidemic, toxin gene-variant strain of Clostridium difficile. N Engl. J. Med. 353, 2433–2441. doi: 10.1056/NEJMoa051590
McGovern, B. H., Ford, C. B., Henn, M. R., Pardi, D. S., Khanna, S., Hohmann, E. L., et al. (2021). SER-109, an investigational microbiome drug to reduce recurrence after clostridioides difficile infection: lessons learned from a phase 2 trial. Clin. Infect. Dis. 72, 2132–2140. doi: 10.1093/cid/ciaa387
Mileto, S. J., Jarde, T., Childress, K. O., Jensen, J. L., Rogers, A. P., Kerr, G., et al. (2020). Clostridioides difficile infection damages colonic stem cells via TcdB, impairing epithelial repair and recovery from disease. Proc. Natl. Acad. Sci. U.S.A. 117, 8064–8073. doi: 10.1073/pnas.1915255117
Mondal, S. I., Draper, L. A., Ross, R. P., and Hill, C. (2020). Bacteriophage endolysins as a potential weapon to combat Clostridioides difficile infection. Gut Microbes 12, 1813533. doi: 10.1080/19490976.2020.1813533
Morteau, O., Castagliuolo, I., Mykoniatis, A., Zacks, J., Wlk, M., Lu, B., et al. (2002). Genetic deficiency in the chemokine receptor CCR1 protects against acute Clostridium difficile toxin A enteritis in mice. Gastroenterology 122, 725–733. doi: 10.1053/gast.2002.31873
Mullard, A. (2016). FDA approves antitoxin antibody. Nat. Rev. Drug Discov. 15, 811. doi: 10.1038/nrd.2016.257
Na, X., Kim, H., Moyer, M. P., Pothoulakis, C., and LaMont, J. T. (2008). gp96 is a human colonocyte plasma membrane binding protein for Clostridium difficile toxin A. Infect. Immun. 76, 2862–2871. doi: 10.1128/IAI.00326-08
Naz, F., Hagspiel, N., Xu, F., Thompson, B., Brett, M. G., Young, M., et al. (2025). Enhanced immunogenicity of a Clostridioides difficile TcdB vaccine adjuvanted with a synthetic dual-TLR ligand adjuvant. NPJ Vaccines 10, 33. doi: 10.1038/s41541-025-01075-3
Ng, J., Hirota, S. A., Gross, O., Li, Y., Ulke-Lemee, A., Potentier, M. S., et al. (2010). Clostridium difficile toxin-induced inflammation and intestinal injury are mediated by the inflammasome. Gastroenterology 139, 552.e1–552.e3. doi: 10.1053/j.gastro.2010.04.005
Nottrott, S., Schoentaube, J., Genth, H., Just, I., and Gerhard, R. (2007). Clostridium difficile toxin A-induced apoptosis is p53-independent but depends on glucosylation of Rho GTPases. Apoptosis 12, 1443–1453. doi: 10.1007/s10495-007-0074-8
Okhuysen, P. C., Ramesh, M. S., Louie, T., Kiknadze, N., Torre-Cisneros, J., de Oliveira, C. M., et al. (2024). A randomized, double-blind, phase 3 safety and efficacy study of ridinilazole versus vancomycin for treatment of clostridioides difficile infection: clinical outcomes with microbiome and metabolome correlates of response. Clin. Infect. Dis. 78, 1462–1472. doi: 10.1093/cid/ciad792
Olling, A., Huls, C., Goy, S., Muller, M., Krooss, S., Rudolf, I., et al. (2014). The combined repetitive oligopeptides of clostridium difficile toxin A counteract premature cleavage of the glucosyl-transferase domain by stabilizing protein conformation. Toxins (Basel) 6, 2162–2176. doi: 10.3390/toxins6072162
Orenstein, R., Dubberke, E. R., Khanna, S., Lee, C. H., Yoho, D., Johnson, S., et al. (2022). Durable reduction of Clostridioides difficile infection recurrence and microbiome restoration after treatment with RBX2660: results from an open-label phase 2 clinical trial. BMC Infect. Dis. 22, 245. doi: 10.1186/s12879-022-07256-y
Orrell, K. E. and Melnyk, R. A. (2021). Large clostridial toxins: mechanisms and roles in disease. Microbiol. Mol. Biol. Rev. 85, e0006421. doi: 10.1128/MMBR.00064-21
Orrell, K. E., Tellgren-Roth, A., Di Bernardo, M., Zhang, Z., Cuviello, F., Lundqvist, J., et al. (2018). Direct detection of membrane-inserting fragments defines the translocation pores of a family of pathogenic toxins. J. Mol. Biol. 430, 3190–3199. doi: 10.1016/j.jmb.2018.07.001
Orrell, K. E., Zhang, Z., Sugiman-Marangos, S. N., and Melnyk, R. A. (2017). Clostridium difficile toxins A and B: Receptors, pores, and translocation into cells. Crit. Rev. Biochem. Mol. Biol. 52, 461–473. doi: 10.1080/10409238.2017.1325831
Orth, P., Xiao, L., Hernandez, L. D., Reichert, P., Sheth, P. R., Beaumont, M., et al. (2014). Mechanism of action and epitopes of clostridium difficile toxin B-neutralizing antibody bezlotoxumab revealed by X-ray crystallography. J. Biol. Chem. 289, 18008–18021. doi: 10.1074/jbc.M114.560748
Pacifico, D. M., Costa, D., Lima, B. M., Reboucas, C., Simonato, S. G., Warren, C. A., et al. (2025). TRPV4 modulates inflammatory responses and apoptosis in enteric glial cells triggered by Clostridioides difficile toxins A and B. J. Inflammation (Lond) 22, 3. doi: 10.1186/s12950-024-00425-7
Pan, Z., Zhang, Y., Luo, J., Li, D., Zhou, Y., He, L., et al. (2021). Functional analyses of epidemic Clostridioides difficile toxin B variants reveal their divergence in utilizing receptors and inducing pathology. PloS Pathog. 17, e1009197. doi: 10.1371/journal.ppat.1009197
Papatheodorou, P., Song, S., Lopez-Urena, D., Witte, A., Marques, F., Ost, G. S., et al. (2019). Cytotoxicity of Clostridium difficile toxins A and B requires an active and functional SREBP-2 pathway. FASEB J. 33, 4883–4892. doi: 10.1096/fj.201801440R
Paredes-Sabja, D., Shen, A., and Sorg, J. A. (2014). Clostridium difficile spore biology: sporulation, germination, and spore structural proteins. Trends Microbiol. 22, 406–416. doi: 10.1016/j.tim.2014.04.003
Pepin, J. (2004). Clostridium difficile-associated diarrhea in a region of Quebec from 1991 to 2003: a changing pattern of disease severity. Can. Med. Assoc. J. 171, 466–472. doi: 10.1503/cmaj.1041104
Peritore-Galve, F. C., Shupe, J. A., Cave, R. J., Childress, K. O., Washington, M. K., Kuehne, S. A., et al. (2022). Glucosyltransferase-dependent and independent effects of Clostridioides difficile toxins during infection. PloS Pathog. 18, e1010323. doi: 10.1371/journal.ppat.1010323
Pfeifer, G., Schirmer, J., Leemhuis, J., Busch, C., Meyer, D. K., Aktories, K., et al. (2003). Cellular uptake of Clostridium difficile toxin B. Translocation of the N-terminal catalytic domain into the cytosol of eukaryotic cells. J. Biol. Chem. 278, 44535–44541. doi: 10.1074/jbc.M307540200
Phothichaisri, W., Chankhamhaengdecha, S., Janvilisri, T., Nuadthaisong, J., Phetruen, T., Fagan, R. P., et al. (2022). Potential role of the host-derived cell-wall binding domain of endolysin CD16/50L as a molecular anchor in preservation of uninfected clostridioides difficile for new rounds of phage infection. Microbiol. Spectr. 10, e0236121. doi: 10.1128/spectrum.02361-21
Pothoulakis, C., Gilbert, R. J., Cladaras, C., Castagliuolo, I., Semenza, G., Hitti, Y., et al. (1996). Rabbit sucrase-isomaltase contains a functional intestinal receptor for Clostridium difficile toxin A. J. Clin. Invest. 98, 641–649. doi: 10.1172/JCI118835
Pothoulakis, C., Karmeli, F., Kelly, C. P., Eliakim, R., Joshi, M. A., O’Keane, C. J., et al. (1993). Ketotifen inhibits Clostridium difficile toxin A-induced enteritis in rat ileum. Gastroenterology 105, 701–707. doi: 10.1016/0016-5085(93)90886-H
Pourliotopoulou, E., Karampatakis, T., and Kachrimanidou, M. (2024). Exploring the toxin-mediated mechanisms in clostridioides difficile infection. Microorganisms 12, 5. doi: 10.3390/microorganisms12051004
Pruitt, R. N., Chagot, B., Cover, M., Chazin, W. J., Spiller, B., and Lacy, D. B. (2009). Structure-function analysis of inositol hexakisphosphate-induced autoprocessing in Clostridium difficile toxin A, J Biol Chem, Vol. 284 No. 33 pp, 21934–21940. doi: 10.1074/jbc.M109.018929
Pruitt, R. N., Chumbler, N. M., Rutherford, S. A., Farrow, M. A., Friedman, D. B., Spiller, B., et al. (2012). Structural determinants of Clostridium difficile toxin A glucosyltransferase activity. J. Biol. Chem. 287, 8013–8020. doi: 10.1074/jbc.M111.298414
Qa’Dan, M., Ramsey, M., Daniel, J., Spyres, L. M., Safiejko-Mroczka, B., Ortiz-Leduc, W., et al. (2002). Clostridium difficile toxin B activates dual caspase-dependent and caspase-independent apoptosis in intoxicated cells. Cell Microbiol. 4, 425–434. doi: 10.1046/j.1462-5822.2002.00201.x
Qa’Dan, M., Spyres, L. M., and Ballard, J. D. (2000). pH-induced conformational changes in Clostridium difficile toxin B. Infect. Immun. 68, 2470–2474. doi: 10.1128/IAI.68.5.2470-2474.2000
Qiu, H., Cassan, R., Johnstone, D., Han, X., Joyee, A. G., McQuoid, M., et al. (2016). Novel Clostridium difficile Anti-Toxin (TcdA and TcdB) Humanized Monoclonal Antibodies Demonstrate In Vitro Neutralization across a Broad Spectrum of Clinical Strains and In Vivo Potency in a Hamster Spore Challenge Model. PloS One 11, e0157970. doi: 10.1371/journal.pone.0157970
Raeisi, H., Azimirad, M., Asadzadeh, A. H., Zarnani, A. H., Abdolalizadeh, J., Yadegar, A., et al. (2023). Development and characterization of phage display-derived anti-toxin antibodies neutralizing TcdA and TcdB of Clostridioides difficile. Microbiol. Spectr. 11, e0531022. doi: 10.1128/spectrum.05310-22
Razim, A., Gorska, S., and Gamian, A. (2023). Non-toxin-based clostridioides difficile vaccination approaches. Pathogens 12, 2. doi: 10.3390/pathogens12020235
Redelings, M. D., Sorvillo, F., and Mascola, L. (2007). Increase in Clostridium difficile-related mortality rates, United State-2004. Emerg. Infect. Dis. 13, 1417–1419. doi: 10.3201/eid1309.061116
Reineke, J., Tenzer, S., Rupnik, M., Koschinski, A., Hasselmayer, O., Schrattenholz, A., et al. (2007). Autocatalytic cleavage of Clostridium difficile toxin B. Nature 446, 415–419. doi: 10.1038/nature05622
Remich, S., Kitchin, N., Peterson, J., Li, P., Pride, M. W., Brock, L., et al. (2024). A phase 2 extension study evaluating the immunogenicity, safety, and tolerability of 3 or 4 doses of a clostridioides difficile vaccine in healthy US adults aged 65 to 85 years. J. Infect. Dis. 229, 367–375. doi: 10.1093/infdis/jiad307
Riegler, M., Sedivy, R., Pothoulakis, C., Hamilton, G., Zacherl, J., Bischof, G., et al. (1995). Clostridium difficile toxin B is more potent than toxin A in damaging human colonic epithelium in vitro. J. Clin. Invest. 95, 2004–2011. doi: 10.1172/JCI117885
Roberts, A. K., Harris, H. C., Smith, M., Giles, J., Polak, O., Buckley, A. M., et al. (2020). A novel, orally delivered antibody therapy and its potential to prevent clostridioides difficile infection in pre-clinical models. Front. Microbiol. 11, 578903. doi: 10.3389/fmicb.2020.578903
Rupnik, M. and Janezic, S. (2015). An update on clostridium difficile toxinotyping. J. Clin. Microbiol. 54, 13–18. doi: 10.1128/JCM.02083-15
Rupnik, M., Pabst, S., Rupnik, M., von Eichel-Streiber, C., Urlaub, H., and Soling, H. D. (2005). Characterization of the cleavage site and function of resulting cleavage fragments after limited proteolysis of Clostridium difficile toxin B (TcdB) by host cells. Microbiol. (Reading) 151, 199–208. doi: 10.1099/mic.0.27474-0
Rupnik, M., Wilcox, M. H., and Gerding, D. N. (2009). Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat. Rev. Microbiol. 7, 526–536. doi: 10.1038/nrmicro2164
Saavedra, P. H. V., Huang, L., Ghazavi, F., Kourula, S., Vanden Berghe, T., Takahashi, N., et al. (2018). Apoptosis of intestinal epithelial cells restricts Clostridium difficile infection in a model of pseudomembranous colitis. Nat. Commun. 9, 1. doi: 10.1038/s41467-018-07386-5
Sakisaka, T. and Takai, Y. (2004). Biology and pathology of nectins and nectin-like molecules. Curr. Opin. Cell Biol. 16, 513–521. doi: 10.1016/j.ceb.2004.07.007
Saleh, M. M., Frisbee, A. L., Leslie, J. L., Buonomo, E. L., Cowardin, C. A., Ma, J. Z., et al. (2019). Colitis-induced th17 cells increase the risk for severe subsequent clostridium difficile infection. Cell Host Microbe 25, 756–765.e5. doi: 10.1016/j.chom.2019.03.003
Savidge, T. C., Urvil, P., Oezguen, N., Ali, K., Choudhury, A., Acharya, V., et al. (2011). Host S-nitrosylation inhibits clostridial small molecule-activated glucosylating toxins. Nat. Med. 17, 1136–1141. doi: 10.1038/nm.2405
Schorch, B., Song, S., van Diemen, F. R., Bock, H. H., May, P., Herz, J., et al. (2014). LRP1 is a receptor for Clostridium perfringens TpeL toxin indicating a two-receptor model of clostridial glycosylating toxins. Proc. Natl. Acad. Sci. U.S.A. 111, 6431–6436. doi: 10.1073/pnas.1323790111
Schottelndreier, D., Langejurgen, A., Lindner, R., and Genth, H. (2020). Low density lipoprotein receptor-Related protein-1 (LRP1) is involved in the uptake of clostridioides difficile toxin A and serves as an internalizing receptor. Front. Cell Infect. Microbiol. 10, 565465. doi: 10.3389/fcimb.2020.565465
Schumacher, J., Nienhaus, A., Heber, S., Matylitsky, J., Chaves-Olarte, E., Rodriguez, C., et al. (2023). Exploring the inhibitory potential of the antiarrhythmic drug amiodarone against Clostridioides difficile toxins TcdA and TcdB. Gut Microbes 15, 2256695. doi: 10.1080/19490976.2023.2256695
Sehgal, K., Cifu, A. S., and Khanna, S. (2022). Treatment of clostridioides difficile infection. JAMA 328, 881–882. doi: 10.1001/jama.2022.12251
Sharma, D., Malik, A., Guy, C. S., Karki, R., Vogel, P., and Kanneganti, T. D. (2018). Pyrin inflammasome regulates tight junction integrity to restrict colitis and tumorigenesis. Gastroenterology 154, 948–964.e8. doi: 10.1053/j.gastro.2017.11.276
Sims, M. D., Khanna, S., Feuerstadt, P., Louie, T. J., Kelly, C. R., Huang, E. S., et al. (2023). Safety and tolerability of SER-109 as an investigational microbiome therapeutic in adults with recurrent clostridioides difficile infection: A phase 3, open-label, single-arm trial. JAMA Netw. Open 6, e2255758. doi: 10.1001/jamanetworkopen.2022.55758
Skinner, A. M., Phillips, S. T., Merrigan, M. M., O Leary, K. J., Sambol, S. P., Siddiqui, F., et al. (2021). The relative role of toxins A and B in the virulence of clotridioides difficile. J. Clin. Med. 10, 96. doi: 10.3390/jcm10010096
Smits, W. K., Lyras, D., Lacy, D. B., Wilcox, M. H., and Kuijper, E. J. (2016). Clostridium difficile infection. Nat. Rev. Dis. Primers 2, 16020. doi: 10.1038/nrdp.2016.20
Song, J., Shen, X., Huang, Z., Liu, Y., Cui, L., Cui, X., et al. (2021). Clostridium difficile toxin A and toxin B inhibit YAP in the colonic epithelial cells. J. Biochem. Mol. Toxicol. 35, e22652. doi: 10.1002/jbt.22652
Spiering, D. and Hodgson, L. (2011). Dynamics of the Rho-family small GTPases in actin regulation and motility. Cell Adh Migr 5, 170–180. doi: 10.4161/cam.5.2.14403
Steele, J., Mukherjee, J., Parry, N., and Tzipori, S. (2013). Antibody against TcdB, but not TcdA, prevents development of gastrointestinal and systemic Clostridium difficile disease. J. Infect. Dis. 207, 323–330. doi: 10.1093/infdis/jis669
Steinemann, M., Schlosser, A., Jank, T., and Aktories, K. (2018). The chaperonin TRiC/CCT is essential for the action of bacterial glycosylating protein toxins like Clostridium difficile toxins A and B. Proc. Natl. Acad. Sci. U.S.A. 115, 9580–9585. doi: 10.1073/pnas.1807658115
Stieglitz, F., Gerhard, R., Honig, R., Giehl, K., and Pich, A. (2022). TcdB of clostridioides difficile mediates RAS-dependent necrosis in epithelial cells. Int. J. Mol. Sci. 23, 8. doi: 10.3390/ijms23084258
Sun, X., He, X., Tzipori, S., Gerhard, R., and Feng, H. (2009). Essential role of the glucosyltransferase activity in Clostridium difficile toxin-induced secretion of TNF-alpha by macrophages. Microb. Pathog. 46, 298–305. doi: 10.1016/j.micpath.2009.03.002
Surawicz, C. M. (2009). Reining in recurrent Clostridium difficile infection–who’s at risk? Gastroenterology 136, 1152–1154. doi: 10.1053/j.gastro.2009.02.023
Tam, J., Hamza, T., Ma, B., Chen, K., Beilhartz, G. L., Ravel, J., et al. (2018). Host-targeted niclosamide inhibits C. difficile virulence and prevents disease in mice without disrupting the gut microbiota. Nat. Commun. 9, 5233. doi: 10.1038/s41467-018-07705-w
Tamburini, E., Dallatomasina, A., Quartararo, J., Cortelazzi, B., Mangieri, D., Lazzaretti, M., et al. (2019). Structural deciphering of the NG2/CSPG4 proteoglycan multifunctionality. FASEB J. 33, 3112–3128. doi: 10.1096/fj.201801670R
Tang, L., Que, H., Wei, Y., Yang, T., Tong, A., and Wei, X. (2025). Replicon RNA vaccines: design, delivery, and immunogenicity in infectious diseases and cancer. J. Hematol. Oncol. 18, 43. doi: 10.1186/s13045-025-01694-2
Tao, L., Tian, S., Zhang, J., Liu, Z., Robinson-McCarthy, L., Miyashita, S. I., et al. (2019). Sulfated glycosaminoglycans and low-density lipoprotein receptor contribute to Clostridium difficile toxin A entry into cells. Nat. Microbiol. 4, 1760–1769. doi: 10.1038/s41564-019-0464-z
Tao, L., Zhang, J., Meraner, P., Tovaglieri, A., Wu, X., Gerhard, R., et al. (2016). Frizzled proteins are colonic epithelial receptors for C. difficile toxin B. Nature 538, 350–355. doi: 10.1038/nature19799
Tian, S., Xiong, X., Zeng, J., Wang, S., Tremblay, B. J., Chen, P., et al. (2022). Identification of TFPI as a receptor reveals recombination-driven receptor switching in Clostridioides difficile toxin B variants. Nat. Commun. 13, 6786. doi: 10.1038/s41467-022-33964-9
Triadafilopoulos, G., Pothoulakis, C., O’Brien, M. J., and LaMont, J. T. (1987). Differential effects of Clostridium difficile toxins A and B on rabbit ileum. Gastroenterology 93, 273–279. doi: 10.1016/0016-5085(87)91014-6
Tucker, K. D. and Wilkins, T. D. (1991). Toxin A of Clostridium difficile binds to the human carbohydrate antigens I, X, and Y. Infect. Immun. 59, 73–78. doi: 10.1128/iai.59.1.73-78.1991
Van Gorp, H., Saavedra, P. H., de Vasconcelos, N. M., Van Opdenbosch, N., Vande, W. L., Matusiak, M., et al. (2016). Familial Mediterranean fever mutations lift the obligatory requirement for microtubules in Pyrin inflammasome activation. Proc. Natl. Acad. Sci. U.S.A. 113, 14384–14389. doi: 10.1073/pnas.1613156113
van Nood, E., Vrieze, A., Nieuwdorp, M., Fuentes, S., Zoetendal, E. G., de Vos, W. M., et al. (2013). Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl. J. Med. 368, 407–415. doi: 10.1056/NEJMoa1205037
van Prehn, J., Reigadas, E., Vogelzang, E. H., Bouza, E., Hristea, A., Guery, B., et al. (2021). European Society of Clinical Microbiology and Infectious Diseases: 2021 update on the treatment guidance document for Clostridioides difficile infection in adults. Clin. Microbiol. Infect. 27, Suppl 2S1–S21. doi: 10.1016/j.cmi.2021.09.038
von Eichel-Streiber, C., Laufenberg-Feldmann, R., Sartingen, S., Schulze, J., and Sauerborn, M. (1992). Comparative sequence analysis of the Clostridium difficile toxins A and B. Mol. Gen. Genet. MGG 233, 60–268. doi: 10.1007/BF00587587
Voth, D. E. and Ballard, J. D. (2005). Clostridium difficile toxins: mechanism of action and role in disease. Clin. Microbiol. Rev. 18, 247–263. doi: 10.1128/CMR.18.2.247-263.2005
Wang, Y., Chang, Y. J., Chen, J., Han, M., Hu, J., Hu, J., et al. (2024). Consensus on the monitoring, treatment, and prevention of leukaemia relapse after allogeneic haematopoietic stem cell transplantation in China: 2024 update. Cancer Lett. 605, 217264. doi: 10.1016/j.canlet.2024.217264
Wang, R., Hu, B., Pan, Z., Mo, C., Zhao, X., Liu, G., et al. (2025a). Antibody-Drug Conjugates (ADCs): current and future biopharmaceuticals. J. Hematol. Oncol. 18, 51. doi: 10.1186/s13045-025-01704-3
Wang, Q., Xu, H., Jin, J., Yang, Y., Jansch, L., and Li, S. (2025b). The battle between bacterial infection and autophagy in aquatic animals. Front. Immunol. 18 (1), 1614182. doi: 10.3389/fimmu.2025.1614182
Wang, L., Xu, T., Wu, S., Zhao, C., and Huang, H. (2025). The efficacy and underlying mechanisms of berberine in the treatment of recurrent Clostridioides difficile infection. Int. J. Antimicrob. Agents 65, 107468. doi: 10.1016/j.ijantimicag.2025.107468
Warny, M., Keates, A. C., Keates, S., Castagliuolo, I., Zacks, J. K., Aboudola, S., et al. (2000). p38 MAP kinase activation by Clostridium difficile toxin A mediates monocyte necrosis, IL-8 production, and enteritis. J. Clin. Invest. 105, 1147–1156. doi: 10.1172/JCI7545
Wen, X., Shen, C., Xia, J., Zhong, L., Wu, Z., Ahmed, M. A. E. E., et al. (2022). Whole-genome sequencing reveals the high nosocomial transmission and antimicrobial resistance of clostridioides difficile in a single center in China, a four-year retrospective study. Microbiol. Spectr. 10, e0132221. doi: 10.1128/spectrum.01322-21
Wilcox, M. H., Gerding, D. N., Poxton, I. R., Kelly, C., Nathan, R., Birch, T., et al. (2017). Bezlotoxumab for prevention of recurrent clostridium difficile infection. N Engl. J. Med. 376, 305–317. doi: 10.1056/NEJMoa1602615
Wilcox, M. H., McGovern, B. H., and Hecht, G. A. (2020). The efficacy and safety of fecal microbiota transplant for recurrent clostridium difficile infection: current understanding and gap analysis. Open Forum Infect. Dis. 7, ofaa114. doi: 10.1093/ofid/ofaa114
Wingen-Heimann, S. M., Davies, K., Viprey, V. F., Davis, G., Wilcox, M. H., Vehreschild, M., et al. (2023). Clostridioides difficile infection (CDI): A pan-European multi-center cost and resource utilization study, results from the Combatting Bacterial Resistance in Europe CDI (COMBACTE-CDI). Clin. Microbiol. Infect. 29, .e1–651.e8. doi: 10.1016/j.cmi.2022.12.019
Wohlan, K., Goy, S., Olling, A., Srivaratharajan, S., Tatge, H., Genth, H., et al. (2014). Pyknotic cell death induced by Clostridium difficile TcdB: chromatin condensation and nuclear blister are induced independently of the glucosyltransferase activity. Cell Microbiol. 16, 1678–1692. doi: 10.1111/cmi.12317
Xie, Y., Irwin, S., Nelson, B., van Daelen, M., Fontenot, L., Jacobs, J. P., et al. (2025). Citrulline inhibits clostridioides difficile infection with anti-inflammatory effects. Cell Mol. Gastroenterol. Hepatol. 19, 101474. doi: 10.1016/j.jcmgh.2025.101474
Xu, H., Yang, J., Gao, W., Li, L., Li, P., Zhang, L., et al. (2014). Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 513, 237–241. doi: 10.1038/nature13449
Yang, Z., Ramsey, J., Hamza, T., Zhang, Y., Li, S., Yfantis, H. G., et al. (2015a). Mechanisms of protection against Clostridium difficile infection by the monoclonal antitoxin antibodies actoxumab and bezlotoxumab. Infect. Immun. 83, 822–831. doi: 10.1128/IAI.02897-14
Yang, Z., Schmidt, D., Liu, W., Li, S., Shi, L., Sheng, J., et al. (2014). A novel multivalent, single-domain antibody targeting TcdA and TcdB prevents fulminant Clostridium difficile infection in mice. J. Infect. Dis. 210, 964–972. doi: 10.1093/infdis/jiu196
Yang, H., Wu, X., Li, X., Zang, W., Zhou, Z., Zhou, Y., et al. (2024). A commensal protozoan attenuates Clostridioides difficile pathogenesis in mice via arginine-ornithine metabolism and host intestinal immune response. Nat. Commun. 15, 2842. doi: 10.1038/s41467-024-47075-0
Yang, Z., Zhang, Y., Huang, T., and Feng, H. (2015b). Glucosyltransferase activity of Clostridium difficile Toxin B is essential for disease pathogenesis. Gut Microbes 6, 221–224. doi: 10.1080/19490976.2015.1062965
Yuan, P., Zhang, H., Cai, C., Zhu, S., Zhou, Y., Yang, X., et al. (2015). Chondroitin sulfate proteoglycan 4 functions as the cellular receptor for Clostridium difficile toxin B. Cell Res. 25, 157–168. doi: 10.1038/cr.2014.169
Zackular, J. P., Kirk, L., Trindade, B. C., Skaar, E. P., and Aronoff, D. M. (2019). Misoprostol protects mice against severe Clostridium difficile infection and promotes recovery of the gut microbiota after antibiotic perturbation. Anaerobe 58, 89–94. doi: 10.1016/j.anaerobe.2019.06.006
Zeiser, J., Gerhard, R., Just, I., and Pich, A. (2013). Substrate specificity of clostridial glucosylating toxins and their function on colonocytes analyzed by proteomics techniques. J. Proteome Res. 12, 1604–1618. doi: 10.1021/pr300973q
Zhang, Y., Li, S., Yang, Z., Shi, L., Yu, H., Salerno-Goncalves, R., et al. (2018). Cysteine protease-mediated autocleavage of clostridium difficile toxins regulates their proinflammatory activity. Cell Mol. Gastroenterol. Hepatol. 5, 611–625. doi: 10.1016/j.jcmgh.2018.01.022
Zhang, F., Wang, S., Yang, J., Fraser, K., Gibson, J. M., Wang, C., et al. (2025). Characterization of heparin interactions with Clostridioides difficile toxins and its potential as anti-CDI therapeutics. Carbohydr Polym 351, 123143. doi: 10.1016/j.carbpol.2024.123143
Keywords: Clostridioides difficile infection, glucosyltransferase toxins, TcdA and TcdB, pathogenicity, therapeutic strategies
Citation: Wen X, Liu X, Wan K, Liu H, Zhang C, Zhang X and Wen Q (2025) Mechanisms of Clostridioides difficile glucosyltransferase toxins and their roles in pathology: insights and emerging therapeutic strategies. Front. Cell. Infect. Microbiol. 15:1641564. doi: 10.3389/fcimb.2025.1641564
Received: 05 June 2025; Accepted: 23 September 2025;
Published: 13 October 2025.
Edited by:
Deiziane Viana da Silva Costa, University of Virginia, United StatesReviewed by:
Farha Naz, University of Virginia, United StatesMaria Luana Morais, University of Virginia, United States
Copyright © 2025 Wen, Liu, Wan, Liu, Zhang, Zhang and Wen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xi Zhang, emhhbmd4eGlAc2luYS5jb20=; Qin Wen, cWlxaTEwNUBzaW5hLmNvbQ==