 Marcos E. Ramos Lopes
Marcos E. Ramos Lopes João Neves-da-Rocha
João Neves-da-Rocha Pablo R. Sanches
Pablo R. Sanches Vanderci M. Oliveira
Vanderci M. Oliveira Antonio Rossi
Antonio Rossi Nilce M. Martinez-Rossi*
Nilce M. Martinez-Rossi*- Department of Genetics, Ribeirão Preto Medical School, University of Sao Paulo, USP, Ribeirão Preto, SP, Brazil
Introduction: Alternative splicing (AS), a common process in pathogenic fungal species, is not fully understood. We hypothesized that AS is a critical regulatory mechanism that enables species to undergo continuous adaptations during interactions with challenging host environments.
Methods: We utilized the model species Trichophyton rubrum to contextualize the role of AS in fungal physiology and virulence. We performed transcriptome-wide splicing analysis to search for AS events in RNA-sequencing data of T. rubrum grown in keratin. This scenario mimicked infection in vitro and allowed us to map biologically relevant splicing events.
Results and discussion: Overall, the results showed that AS is recruited to regulate approximately 12.6% of the T. rubrum genome under an infection-like scenario. We extended this analysis to ex vivo infection models of T. rubrum grown on human nails and cocultured them with human HaCaT keratinocytes. We found that AS affects a wide range of cellular processes, including amino acid and carbohydrate metabolism, cell signaling, protein folding and transport, transcription, and translation. We showed that transcription factors such as PacC and Ap1 govern the major features of fungal virulence and metabolism and are controlled by the spliceosome machinery under different infection-like conditions.
Conclusions: Our data indicate that mRNA isoforms originating from AS contribute to the adaptation of T. rubrum, demonstrating that AS of transcription factor genes plays a central role in fungal pathogenesis. The transcription and splicing machinery tune fungal physiology to achieve an optimal metabolic balance in virulence traits during infection.
1 Introduction
Dermatophytes are opportunistic pathogens that specifically colonize keratinized tissues. These pathogens engage in complex coevolutionary dynamics with their hosts, shaping their ability to colonize and infect keratinized tissues, such as the skin and nails, while influencing host immune responses (Deng et al., 2023; Gupta et al., 2023; Peres et al., 2010). Anthropophilic dermatophytes are an increasingly relevant medical concern affecting approximately 25% of the world’s population and cause chronic infections that can progress to invasive diseases in patients who are immunocompromised (Martinez-Rossi et al., 2021). Trichophyton rubrum is the most prevalent and clinically significant species worldwide, accounting for over 60% of all dermatophytosis cases (Zheng et al., 2020; Wang et al., 2021). Moreover, the emergence of resistant strains poses a challenge to the effective management of fungal infections (Bristow and Joshi, 2023; Martinez-Rossi et al., 2018; Kruithoff et al., 2023). Continuous adaptation of these organisms and their ability to resist conventional antifungal treatments emphasizes the need for ongoing research to determine the mechanisms underlying their infection persistence and develop novel therapeutic approaches.
Carbohydrate substrates are scarce in host tissues; therefore, dermatophytes degrade more complex molecules during infection. The main sources of available nitrogen and carbon are proteins, notably keratin. T. rubrum metabolic plasticity occurs via nitrogen catabolite repression, which is coordinated by the GATA zinc-finger transcription factors (TFs) (Martins et al., 2020). This process allows the use of complex nitrogen sources when preferential nitrogen sources such as glutamine are unavailable. However, keratin has a rigid structure formed by disulfide bonds, which prevent the usual proteolytic digestion of this molecule. Dermatophytes can produce urea and sulfites secreted by the Ssu1 efflux pump, which reduces covalent bonding (Martins et al., 2020; Grumbt et al., 2013). Keratin degradation and the metabolism of specific amino acids such as glycine to generate acetyl-CoA promote ammonia release and consequent alkalinization of the medium, which can increase pH values to as high as 9.0 (Ferreira-Nozawa et al., 2003; Maranhão et al., 2007). As the infection progresses, repression/activation of pH-regulated pathways and enzymes occurs in response to cell demands and environmental changes.
In this scenario, effective adaptation during infection requires fungi to coordinate diverse metabolic responses. Fungal cells activate transcriptional programs according to the surrounding conditions, such as cellular stress and host infection sites (Martinez-Rossi et al., 2021; Park et al., 2013). Arguably, the most important components governing these response programs are TFs, which regulate gene expression through hierarchical interactions between signaling and response networks (Song et al., 2016). In dermatophytes, many attributes related to virulence and susceptibility to stressors, as well as tolerance to different antifungals, are regulated by TFs such as PacC, Dnr1, Ap1, HacA, SteA, and StuA (Kröber et al., 2017; Yamada et al., 2006; Ferreira-Nozawa et al., 2006; Peres et al., 2022; Bitencourt et al., 2020; Lang et al., 2020; Martins-Santana et al., 2025).
The post-transcriptional regulation of gene expression by alternative splicing (AS) is a common process in fungi (Grützmann et al., 2014). AS may influence the activity of many genes in response to the various conditions imposed on these organisms (Grützmann et al., 2014; Mendes et al., 2018; Neves-da-Rocha et al., 2019; Lopes et al., 2022). Intron retention (IR) events are the most common type of AS in fungi (Grützmann et al., 2014; Kempken, 2013; Jeon et al., 2022; Li et al., 2021). In addition, important correlations exist between AS and distinct fungal species, with multicellular complexity, pathogen lifestyle, and a younger evolutionary age being associated with higher AS rates (Grützmann et al., 2014). Nevertheless, the extent of AS and the biological relevance of this post-transcriptional mechanism in fungal pathogenesis remain unclear. To investigate and correlate AS with fungal biology, we assessed global AS patterns in the transcriptome of T. rubrum exposed to keratin, which enabled mapping of biologically relevant AS events in an infection-like manner. We further performed functional enrichment analysis to investigate the physiological consequences of AS regulation. IR events in major fungal TFs were validated using in vitro and ex-vivo infection models to demonstrate the practical relevance of our findings. Because dermatophytes are representative filamentous pathogenic fungi in which AS is prevalent, this study improved the understanding of the strategies employed by pathogens to adapt to and colonize host tissues. Furthermore, our data provide critical fundamental information for guiding future studies on AS and medical mycology.
2 Materials and methods
2.1 In vitro culture conditions
Trichophyton rubrum strain CBS118892 from the Westerdijk Fungal Biodiversity Institute, Netherlands, was pre-cultivated in malt extract agar medium (2% glucose, 2% malt extract, 0.1% peptone, and 2% agar, pH 5.7) at 28°C for 21 days. Subsequently, 1 × 106 conidia were germinated in 100 mL Sabouraud dextrose broth for 96 h and then transferred to Cove’s minimal medium at pH 5.0, containing either 50 mM glucose (Sigma–Aldrich, St. Louis, MO, USA) (control) or 0.5% (m/v) of powdered ox hull keratin as the carbon source and cultivated for 24 and 96 h (Peres et al., 2016). The cultures were filtered after each growth period, and mycelia were stored at −80°C until RNA extraction. All experiments were conducted in triplicate.
2.2 Ex vivo infection of human nail fragments
Human nail fragments obtained from healthy donors were autoclaved and placed in Eppendorf tubes. Conidia (1 × 104 in 20 µL of sterilized water) of T. rubrum were mixed with the nail fragments (approximately 1 mm2). After 1 h of incubation at room temperature, 200 μL of sterilized water was added, and the tubes were incubated at 28°C for 96 h (Peres et al., 2016). The infected nail fragments were vortexed to release the fungal mycelia and extract total RNA. The supernatant containing fungal cells was centrifuged at 12,000 ×g for 10 min, frozen in liquid nitrogen, and stored at −80°C until RNA extraction. Fungi grown in glucose (described in Section 2.1) were used as a reference. The assay was performed in triplicate.
2.3 Coculture of T. rubrum with human keratinocytes (HaCaT)
For coculture experiments, 1 × 107 conidia/mL and 2 × 105 HaCaT cells/mL were incubated for 24 h in RPMI medium (Sigma–Aldrich, St. Louis, MO, USA) supplemented with 5% fetal bovine serum at 37°C in 5% CO2, as previously reported (Komoto et al., 2015). Plates containing fungal cells without keratinocytes were used as controls. The assay was performed in triplicate.
2.4 Extraction, cDNA synthesis, and RT-qPCR analysis
Total RNA was extracted using an Illustra Spin RNA Isolation Kit (GE Healthcare, Chicago, IL, USA) according to the manufacturer’s instructions. RNA samples were treated with DNAse I (Sigma-Aldrich, St. Louis, MO, USA) and converted to cDNA using a high-capacity reverse transcription kit (Applied Biosystems, Foster City, CA, USA). To evaluate the quality of the cDNAs, PCR was conducted using oligonucleotides flanking intron-1 of the constitutive gene encoding β-tubulin, and the results were visualized on an agarose gel.
Transcripts were quantified using a QuantStudio v. 3 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). Primers were designed within intron regions to detect IR events. Exon regions were used as primer annealing sites to evaluate total gene expression. The construction and quality analysis of the primers used in RT-qPCR assays were performed using the PrimerQuest Tool (http://www.idtdna.com/primerquest/Home/, accessed on June 12, 2023) and OligoAnalyzer Tool (https://www.idtdna.com/calc/analyzer, accessed on June 14, 2023) from Integrated DNA Technologies (IDT), and the BLAST tool (http://blast.ncbi.nlm.nih.gov/Blast.cgi, accessed on June 14, 2023).
The primers used and their standardized concentrations are listed in Supplementary Table S1. Reactions were performed using Power SYBR Green PCR Master Mix (Life Technologies, Carlsbad, CA, USA). Relative quantification was performed using the 2−ΔΔCt method (Livak and Schmittgen, 2001), with gapdh and rpb2 as reference genes for expression normalization (Jacob et al., 2012). The results are expressed as mean relative expression values from three independent replicates, with standard deviations. Alternatively, spliced transcripts were evaluated as a percentage of RNA isoforms, calculated using the ratio of mean ΔCT values of IR to total expression.
2.5 Global AS analysis and functional enrichment
High-throughput RNA-sequencing data were analyzed to assess transcriptome-wide AS regulation in T. rubrum exposed to keratin (Gene Expression Omnibus database, accession number GSE134406). Reads were mapped to the reference genome using STAR aligner v2.7.10a (Dobin et al., 2013). Reads mapped to multiple locations were excluded using the STAR – out Filter Multimap N max 1 parameter, and gene-level read counts were quantified using the STAR – quant Mode Gene Counts parameter. Visual inspection was performed using Integrative Genomics Viewer (IGV) v2.16.2 (Robinson et al., 2011).
The biological replicates were inspected using principal component analysis. We processed and analyzed the reads in R v4.3.1 using the ASpli Bioconductor package v2.10.0 to identify AS events (Mancini et al., 2021) and the DESeq2 Bioconductor package v1.40.2 to perform differential expression analysis (Love et al., 2014). The ASpli output file is shown in Supplementary File S1. Events with a Benjamini–Hochberg adjusted p-value lower than 0.05 and a log2 fold change greater than | ± 1.0| were considered significantly modulated. Functional categorization was conducted according to Gene Ontology (GO) terms assigned using the OmicsBox tools v3.1 (Conesa et al., 2005). The FunRich tool v3.1.4 (Pathan et al., 2015) was used to perform an enrichment analysis on the identified dataset.
2.6 In silico analyses
Data from Ensembl fungi were used to predict the transcripts and proteins resulting from conventional and alternative isoforms (https://fungi.ensembl.org/index.html, accessed on June 25, 2023). Reading frames and domains were predicted using the Expasy Translate tool (https://web.expasy.org/translate/, accessed on June 25, 2023) and InterPro database (https://www.ebi.ac.uk/interpro/ accessed on June 25, 2023), respectively. Graphical representations of the isoforms and resulting proteins were generated using Illustrator of Biological Sequence (IBS) v1.0 software (Liu et al., 2015).
To detect putative phosphorylation sites on serine, threonine, and tyrosine residues in the TFsproteic isoforms, the generated amino acid sequences were used as inputs to the NetPhos tool v3.1 (https://services.healthtech.dtu.dk/services/NetPhos-3.1/, accessed on June 27, 2023) (Chun et al., 2021). Conventional and alternative isoforms of the evaluated genes were used for the predictions, and sites with scores higher than 0.5 were defined as candidate phosphorylation sites.
3 Results
3.1 Trichophyton rubrum coordinates global AS patterns in the presence of keratin
We applied high-confidence statistical thresholds to evaluate AS in an infection-like scenario, which enabled mapping of bona fide AS events that could be critical for dermatophyte pathogenesis. AS analysis was primarily designed to identify IR events, which are the most prevalent type of AS in fungi (Grützmann et al., 2014). IR events influence translation and commonly change the open reading frame structure.
To mimic infection progression and analyze AS over time, we compared fungal growth in keratin and glucose media at 24, 48, and 96 h. Overall, we detected AS events in 1085 genes, representing approximately 12.6% of the T. rubrum genome. The number of alternatively spliced genes (ASGs) was lower than that of differentially expressed genes (DEGs). Although most genes were exclusively regulated by AS or differential expression, 421 genes were simultaneously regulated by both processes (Figure 1A). The total number of ASGs represented a significant fraction of all gene expression changes in response to keratin, revealing that splicing plays a prominent role during T. rubrum infection (Figure 1A). Over time, AS events were prevalent at 24 h, and many ASGs were regulated at two or more time points (Figures 1B, C), reinforcing the importance of AS modulation in the infection process. Functional enrichment analysis of the genes that undergo modulation of AS expression in keratin revealed distinct molecular functions, such as splicing factor binding, helicase, kinase, phosphatase activities, and TF binding (Figure 1D). Remarkably, modulation of 132 ASGs at all incubation times indicated that these genes are involved in the virulence of T. rubrum and are mostly related to energy and amino acid metabolism (Figures 1B, 2A).
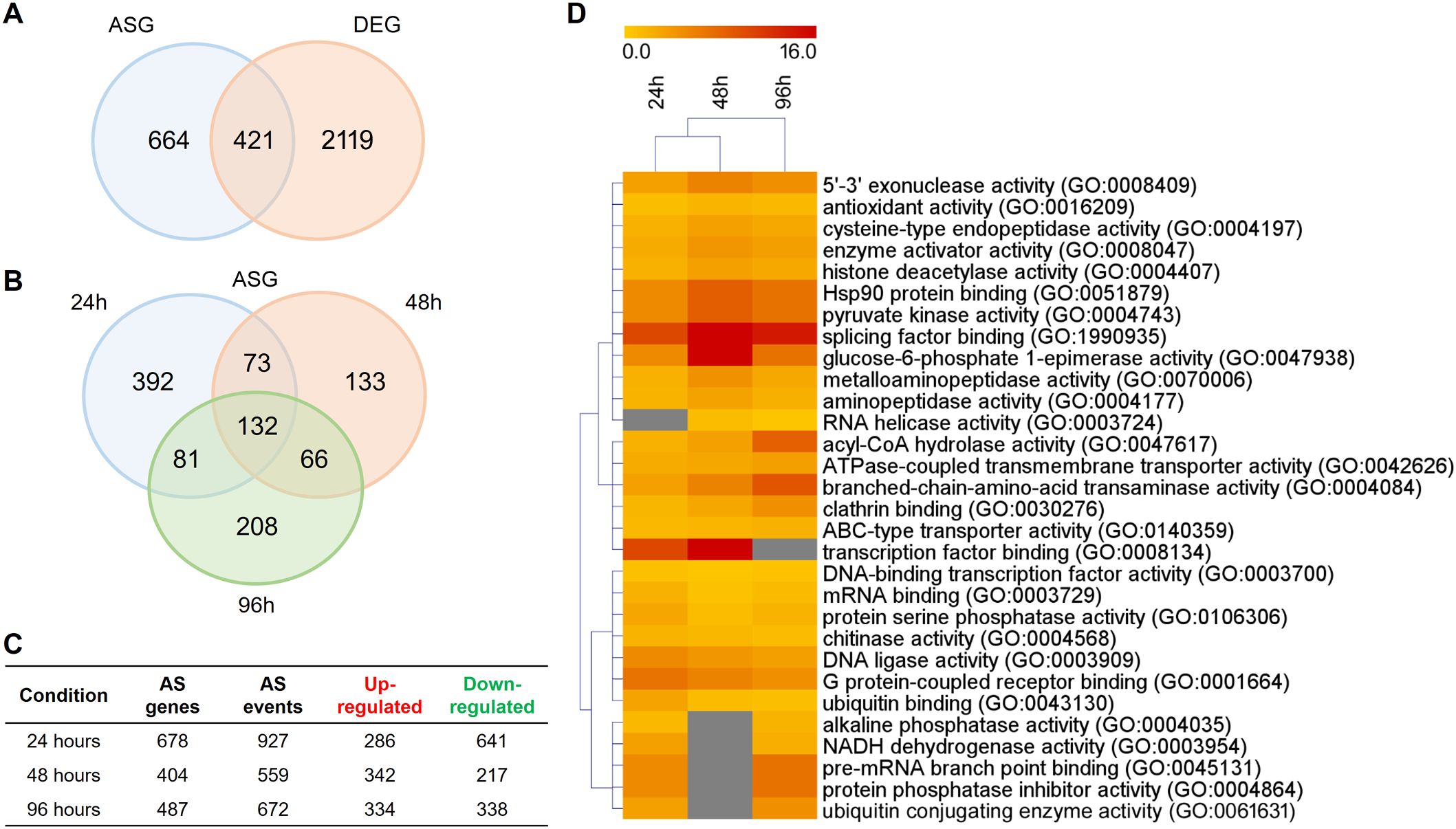
Figure 1. Distribution of genes modulated in response to keratin compared with the glucose (control). (A) Venn diagram of the number of alternative splicing genes (ASGs) and differentially expressed genes (DEGs) in response to keratin. (B) Venn diagram of alternative splicing (AS) events at 24, 48, and 96 h. (C) Total number of up- and down-regulated AS events at each time point. (D) Heatmap of functional enrichment analysis of ASGs based on Gene Ontology (GO) molecular function categories. The yellow-to-red color gradient indicates the fold-enrichment indices, which range from 0 to 16. Only the 30 most representative GO categories are displayed.
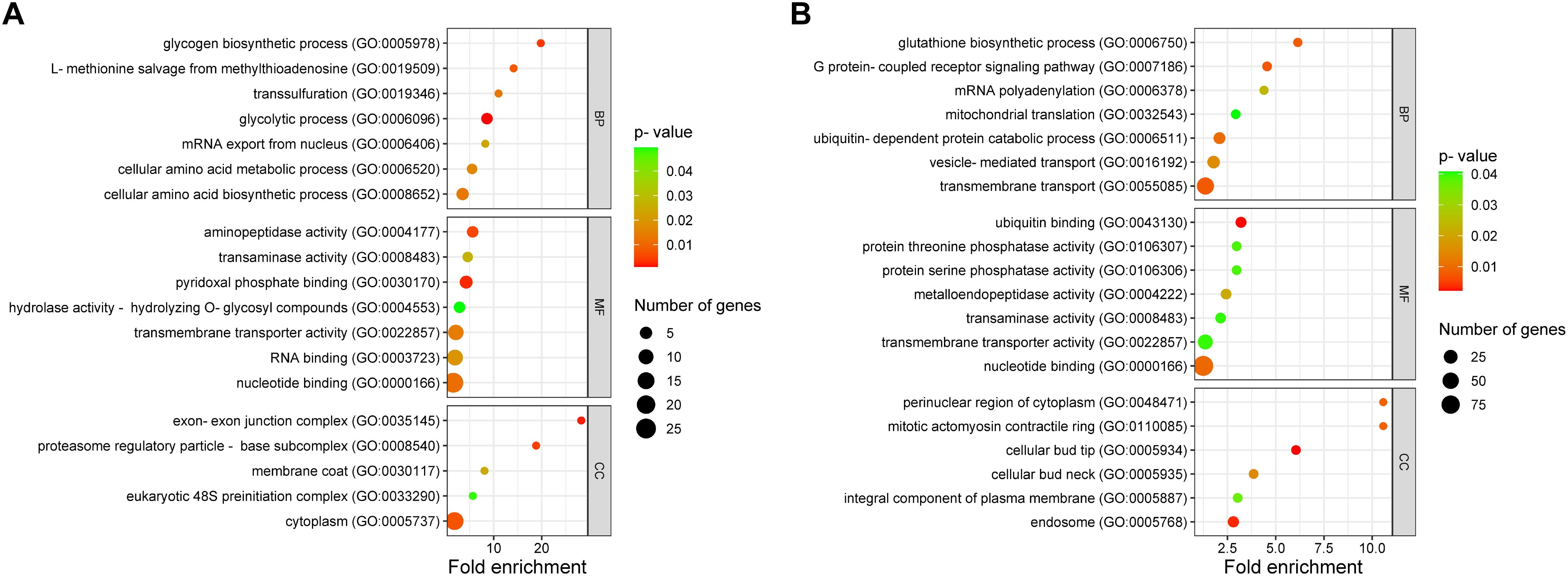
Figure 2. Functional characterization of genes with alternative splicing (AS) events in response to keratin. Functional annotation is categorized into biological processes (BP), molecular functions (MF), and cellular components (CC). (A) Functional characterization of 132 genes (from Figure 1B) that exhibited AS across all tested time points. (B) Functional categorization of genes with repressed AS in keratin. Sphere size represents the number of genes, whereas the color gradient, from red to green, indicates the p-value, ranging from smallest to largest.
The results also revealed that a large number of AS events were repressed in response to keratin (Figure 1C), indicating that AS governs many processes related to cell signaling, transmembrane transport, energy metabolism, autophagy, and other processes important for fungal physiology under control and unstressed conditions (Figure 2B). Based on the gene expression network, despite regulatory overlaps, AS and DE are distinct processes that coordinate different aspects of fungal metabolism (Figure 3).
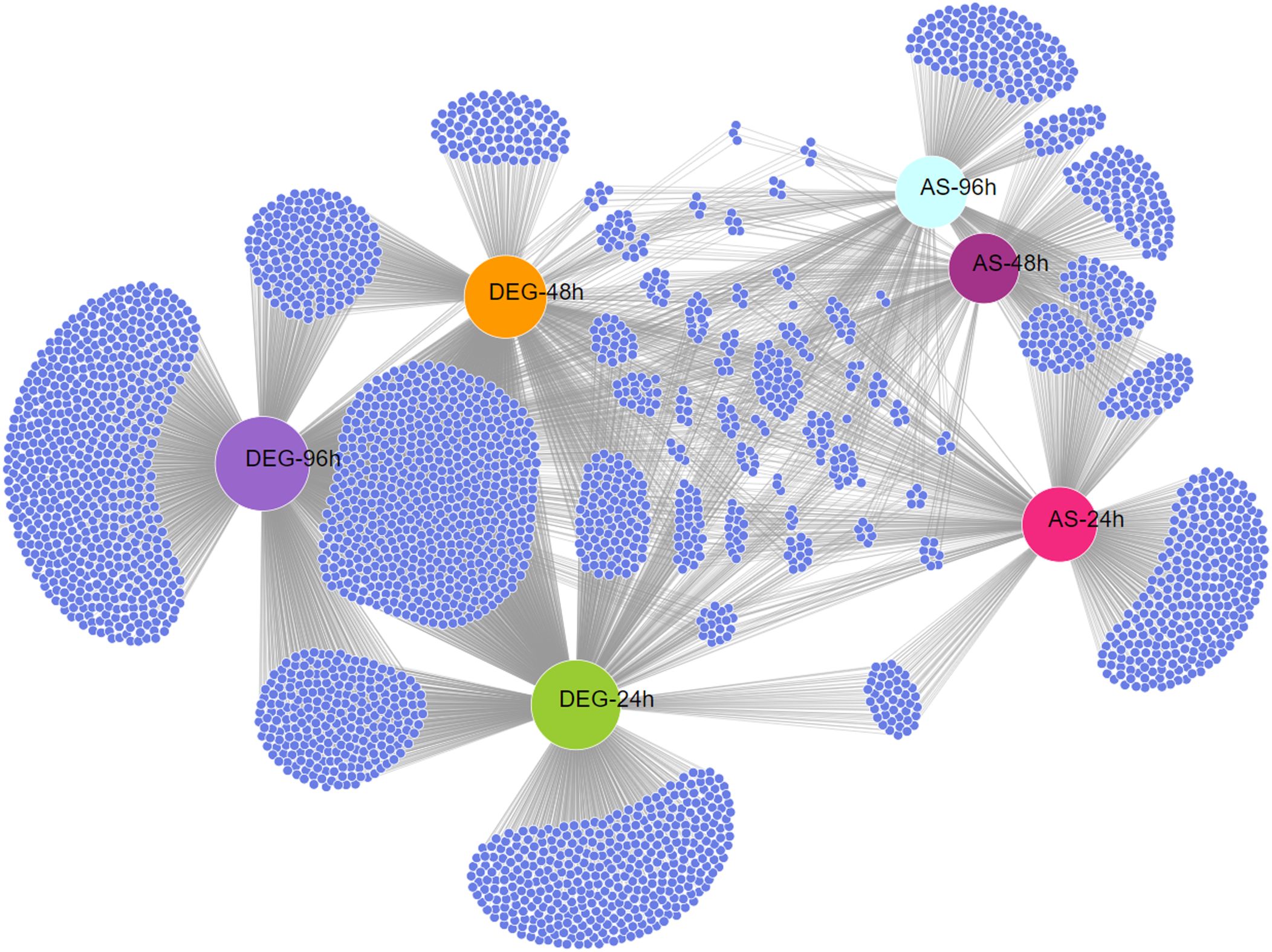
Figure 3. Expression network between differentially expressed genes (DEGs) and alternatively spliced genes (ASGs) in Trichophyton rubrum in response to keratin. Network representation of DEGs and ASGs across all experimental conditions. Small blue nodes represent individual genes, whereas gray lines indicate connections between regulatory mechanisms and conditions.
3.2 Keratin metabolism modulates expression and AS in TF genes
RNA-sequencing data from T. rubrum exposed to keratin revealed the differential expression of 57 TF-coding genes (Supplementary Figure S1). Because TFs are key regulators of several cellular processes, we evaluated whether AS mechanisms, particularly IR, are involved in this regulation. Our analyses revealed that 33 TF genes presented IR in response to keratin, 13 of which also presented DE, and 20 were exclusively controlled by AS (Figure 4). These results demonstrate the importance of this regulatory layer in the functionality of this gene family.

Figure 4. Venn diagrams showing the number of transcription factor (TF) genes differentially expressed (DEG) and alternatively spliced (ASG) in response to keratin. (A) Number of TF-coding genes showing alternative splicing (AS) events and were differentially expressed when Trichophyton rubrum was exposed to keratin. (B) Number of TF genes showing AS at different times of exposure to keratin.
We selected four TFs exhibiting IR and involved in virulence to determine the modulation of AS events under ex vivo conditions (keratinocytes and human nails) and compared them with those under keratin culture conditions. The four genes and their IR were pacC (TERG_00838, IR-2), con7 (TERG_03087, IR-3), ap1 (TERG_02940, IR-1), and c6 (TERG_06830, IR-2). We employed in silico approaches to investigate the effects of IR on pre-RNA of the selected genes. IR events generated premature stop codons in the sequences of retained introns, resulting in specific outcomes such as changes in phosphorylation profiles and protein shortening with partial or total domain loss (Figure 5; Supplementary Figure S2). The presence and modulation of IR events were validated for all genes tested (Figures 6A). Figure 6A also shows that pacC was highly expressed in medium containing keratin or in human nail fragments, con7 and ap1 were overexpressed in keratin and keratinocytes, and c6 was overexpressed only in keratin, indicating the specificity of each gene in the infection process.

Figure 5. Schematization of transcription factor isoforms after conventional or alternative splicing. The transcripts and putative proteins resulting from each isoform are shown. Blue boxes represent exons, and introns are depicted as lines. Domains are indicated in the protein structure. IR shows the number of retained introns.
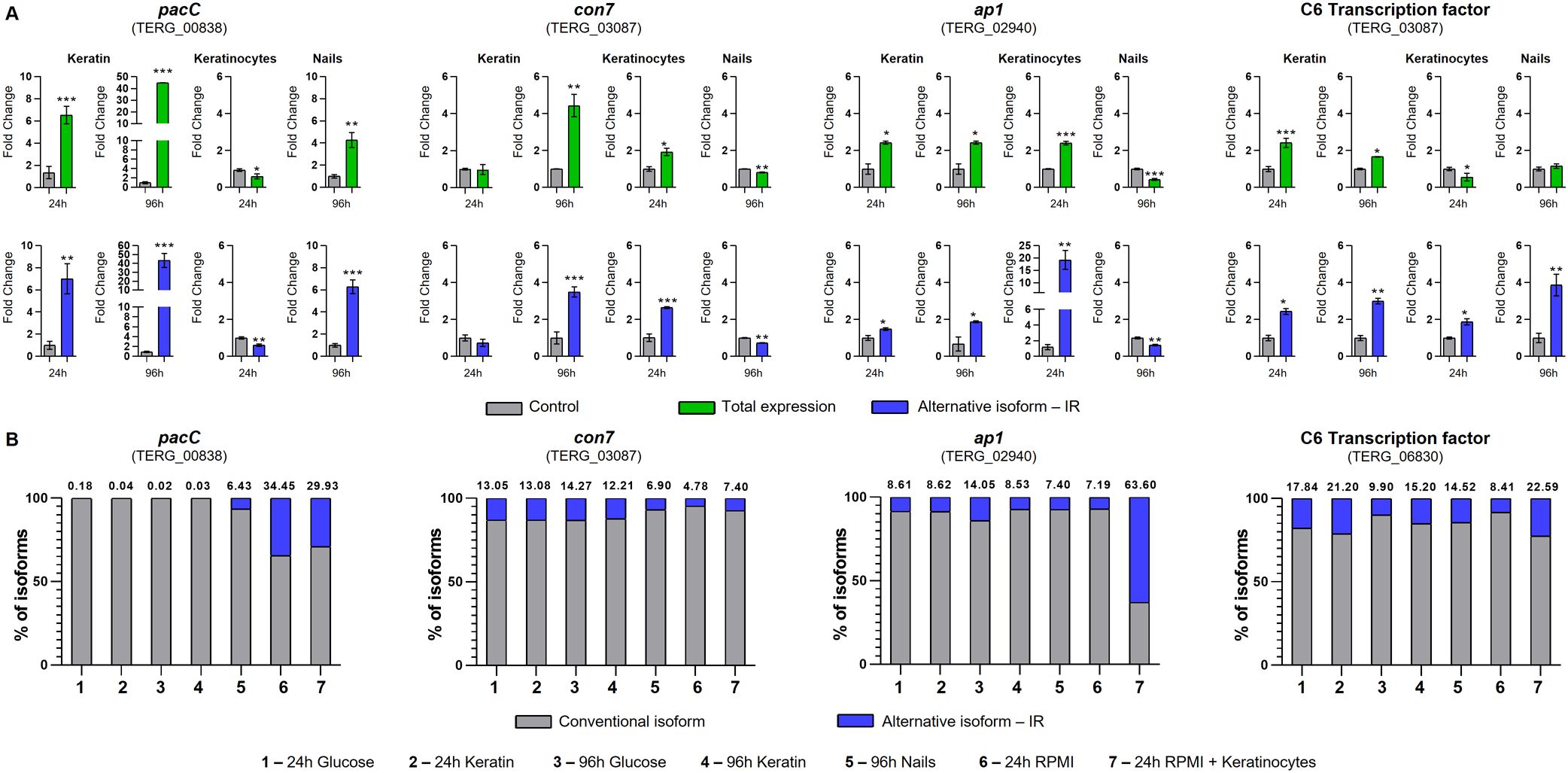
Figure 6. Expression of transcription factor isoforms of Trichophyton rubrum under in vitro and ex vivo conditions. (A) Expression analysis of pacC, con7, ap1, and c6 isoforms in T. rubrum upon exposure to several challenges. The control conditions were as follows: keratin (glucose 24 and 96 h), keratinocytes (RPMI 24 h), and nails (glucose 96 h). Paired controls, indicated by gray bars in each graph, were used as modulation references. Statistical analyses were performed using t-tests. *p < 0.05, **p < 0.01, ***p < 0.001. (B) Intron retention (IR) percentage of pacC, con7, ap1, and c6 transcripts of T. rubrum exposed to different conditions. The proportions of alternative isoform IR are represented in blue, and the conventional isoform is in gray. The numbers above each bar represent the percentage of alternate splicing isoforms.
To compare the levels of transcripts that exhibited IR with those that were fully processed (Figure 6A; Supplementary Figure S3), we calculated the percentage of IR under each experimental condition (Figure 6B). Under several assayed conditions (control or treatment), the pacC, con7, ap1, and c6 transcripts presented IR levels greater than 5%, indicating that these ASs are not random events.
3.3 Splicing factor genes are induced upon exposure to keratin
To verify whether keratin metabolism modulates the expression of splicing factor genes, we assayed four genes following T. rubrum cultivation in keratin, using glucose as a control. These genes were induced by exposure to keratin, mainly at 24 h (Figure 7).
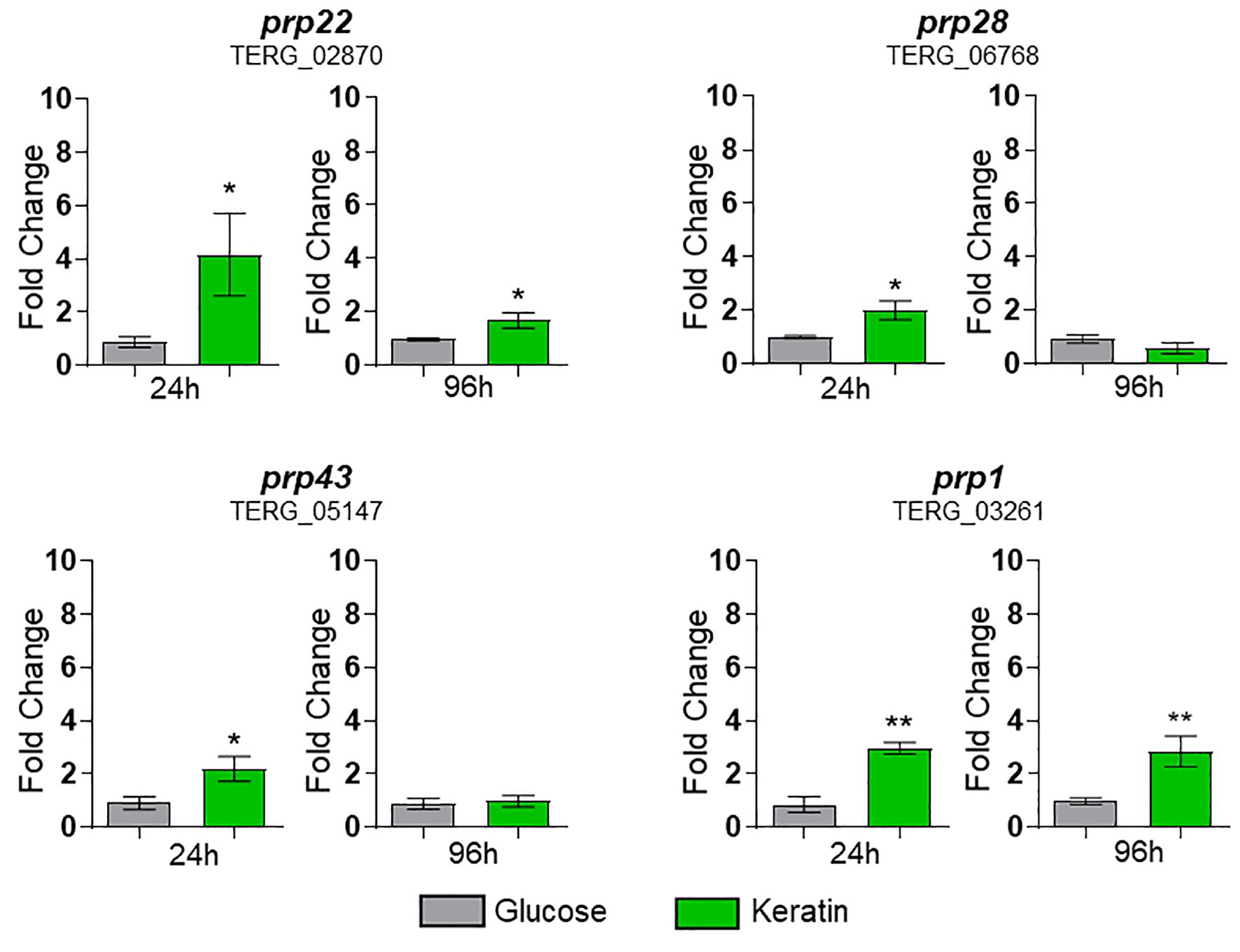
Figure 7. Expression of prps (splicing factors) of Trichophyton rubrum exposed to keratin. Statistical analysis was performed using t-test. *p < 0.05, **p < 0.01. Paired controls, indicated by the gray bar on each graph, were used as a modulation reference.
4 Discussion
We comprehensively evaluated the functional categories of genes that undergo AS in the human pathogen T. rubrum using a model that mimics infection. Our findings revealed that a large proportion of genes, approximately 12.6% of the genome, underwent AS, and approximately 39% of these genes are differentially expressed. This percentage of 12.6% is high considering the rate of AS observed in other pathogenic fungi, such as Sclerotinia sclerotiorum (5.6%) (Cheng et al., 2022), Aspergillus oryzae (8.55%) (Wang et al., 2010), and Ustilago maydis 3.6% (Ho et al., 2007), assayed under different conditions. Indeed, identification of TF genes that undergo many AS events in other pathogenic fungi suggests that AS regulates a broad spectrum of processes, highlighting the complex interplay of mechanisms regulating these genes in T. rubrum. Notably, at least 61% of ASGs did not exhibit differential expression, suggesting that AS plays a crucial role in regulating T. rubrum infection beyond the level of gene expression. These findings have significant implications for the understanding of pathogen–host interactions.
As a major step towards characterizing the role of the spliceosome machinery in dermatophytes, we assessed the global impacts of AS regulation on T. rubrum physiology. Functional enrichment analysis revealed that AS might contribute extensively to fungal metabolic adaptation during changes in nutrition sources, with direct impacts on the infection dynamics. During keratin degradation compared to glucose growth, AS influenced sensing and regulatory processes that are required for efficient physiological responses, such as kinase and phosphatase activities, transcription factors and splicing factors, transporters, enzyme activators, chaperones, and histone modifications (Figure 1D). Interestingly, considering the genes modulated in common in all time points in response to keratin, T. rubrum showed increased spliceosome influence over central carbon and nitrogen metabolism, affecting glycolysis, glycogen biosynthesis, amino acid metabolism, and proteasome function (Figure 2A). Those pathways indicate that dermatophyte physiology requires specific tuning of function to degrade complex substrates such as keratin, which is potentially influenced by nutrient limitation during keratin-based host colonization. Those findings are in accordance with previous data, which demonstrate keratin influence over dermatophyte central metabolism, including the control of nitrogen catabolite repression for regulating the TCA and urea cycles (Martins et al., 2020).
The number of ASGs in T. rubrum is particularly important during the initial phase of infection, when the pathogen must rapidly adapt to the new environment. Based on these findings, splicing factor genes such as prp1, prp22, prp28, and prp43, which are crucial in this process, were induced upon exposure to keratin, mainly at 24 h. This activation underscores the pivotal role of these four splicing factors in adaptation. Furthermore, AS events in TFs were present at all three tested time points, indicating that this phenomenon is an indispensable regulatory pathway for the adaptation, establishment, and maintenance of T. rubrum infection. The extensive range of categories that perform vital functions in cells under control of the splicing machinery in this infection-like context enhances the understanding of the crucial role of AS in fungal adaptation and pathogenicity. These findings may lead to the development of new strategies for combating fungal infections. The four selected TF genes revealed different expression profiles and IR in keratin medium, keratinocytes, and human nails. These results suggest that the fungus uses an intricate mechanism in its interaction with the host, depending on the tissue type and culture time.
The PacC signal transduction pathway, a crucial element in fungal infection, regulates the synthesis of a diverse range of enzymes and proteins with optimal activity at specific pH values dictated by fungal metabolism. This complex process enables the fungus to adhere to and penetrate host tissue, obtain nutrients, and subvert host defense mechanisms. Because human skin has an acidic pH, the first contact of dermatophytes during infection occurs in an acidic environment. We previously showed that the growth of pacC mutant strains of various fungi, including T. rubrum, is severely impaired under alkaline conditions (Ferreira-Nozawa et al., 2006; Trushina et al., 2013; Wiemann et al., 2009; Barda et al., 2020). The strong expression of pacC observed in keratin reinforces the involvement of this gene in keratin metabolism (Martins et al., 2020). The presence of IR-2 in pacC transcripts leads to the formation of protein isoforms that maintain three zinc-finger domains. In addition, in silico analyses indicated that the putative PacC protein generated by splicing (274 amino acids (aa)) resembled the activated form of the homolog protein of Aspergillus nidulans after proteolytic cleavage (254 aa) in terms of size, sequence, and domain number (three zinc finger domains) (Supplementary Figure S4). Functional activation of PacC in A. nidulans is a two-step proteolytic process, one of which is mediated by PalB, a calpain-family protease (Tilburn et al., 1995; Martinez-Rossi et al., 2012; Li et al., 2022), generating a minor protein containing three zinc fingers domains (Tilburn et al., 1995). Similarly, in silico data suggested that the putative version of PacC enzymatically activated in T. rubrum exhibited a conformation, size, and domain number similar to the putative isoform generated by IR-2 (Figure 8). Our RNA-seq data revealed high pacC expression, whereas differential expression of the T. rubrum palB homolog was not detected. This observation suggests the potential for alternative pathways to activate PacC protein at an acidic pH when culturing T. rubrum in glucose. Previous studies suggested that PacC protein in A. nidulans can be activated through an alternative pathway to PalB proteolysis due to the induction of an alternative palB isoform that is potentially nonfunctional, whereas PacC is functional (Trevisan et al., 2011). Our results suggest that IR-2 of the pacC pre-mRNA is an alternative mechanism to enzymatic activation of PacC at acidic pH in T. rubrum.
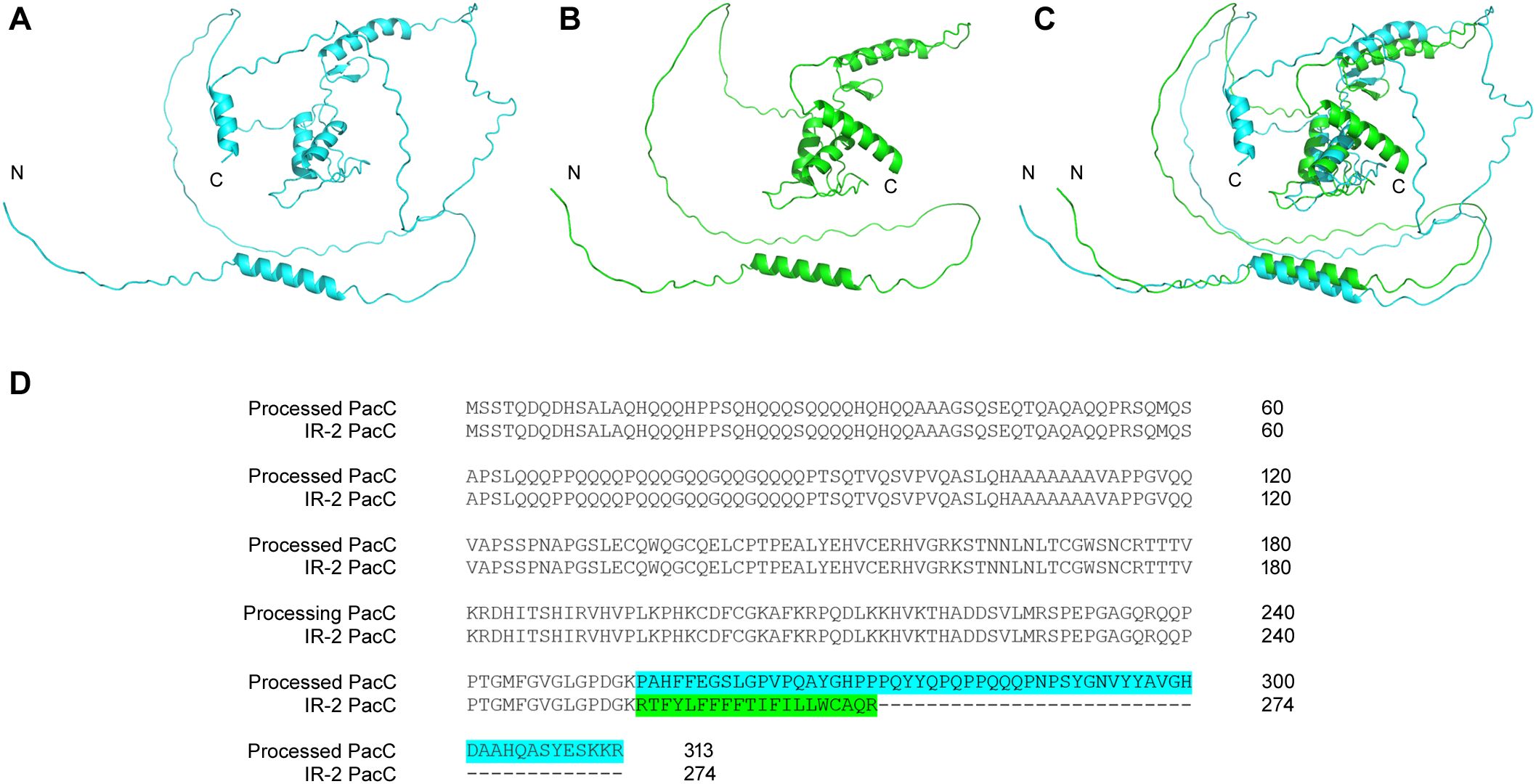
Figure 8. Comparison of putative PacC protein isoforms of Trichophyton rubrum. Three-dimensional structures of PacC protein enzymatically processed, in blue (A), from IR-2, in green (B), and their superposition (C). Additionally, alignment of the amino acid sequences of these two protein variants is shown (D).
Although IR-3 in the con7 transcripts resulted in a minor protein (197 aa) compared to the conventional isoform (263 aa), the zinc-finger domain was present in both isoforms. The prediction of phosphorylation sites in Con7 protein isoforms also revealed interesting differences between the two isoforms. These isoforms share 20 phosphorylation sites on the serine, threonine, and tyrosine residues. However, the alternative isoform had seven distinct phosphorylation sites resulting from the alternatively generated amino acid sequence, with five serine and two threonine residues (Supplementary Figure S2). These findings have important functional implications. Changes in the phosphorylation profile of TFs represent a mechanism employed by cells to mediate the activity of these proteins in response to environmental changes by regulating their transcriptional activity, cellular localization, stability, protein–protein interactions, DNA binding, and coregulators (Riedl and Egly, 2000; Whitmarsh and Davis, 2000; Zhou et al., 2020). Moreover, the IR-3 of con7 transcripts of T. rubrum was more prevalent when the fungus was cultured with keratinocytes than in the control (Figure 6B), suggesting a role of this isoform in tissue-specific interaction with the host. The role of this gene in fungal virulence and host interactions has been revealed in Fusarium oxysporum (Ruiz-Roldán et al., 2015). con7 in Magnaporthe grisea and F. oxysporum also have splicing variants, including isoforms generated through IR during vegetative growth and infection (Odenbach et al., 2007; Ruiz-Roldán et al., 2015).
IR-1 in ap1 transcripts generated a small protein of 153 amino acids lacking the essential leucine zipper domain, indicating loss of its function as a transcriptional regulator. However, notably, IR-1 isoform showed a high percentage among ap1 transcripts when cultured with keratinocytes (63%), suggesting that this isoform is essential for interactions with HaCaT cells (Figure 6B). This alternative isoform may lead to the formation a putative mini-protein without a typical Ap1 domain. However, we cannot rule out the possibility that the RNA produced participates in fungal interactions with keratinocytes. As previously reported, the Δap1 mutant of T. rubrum exhibited increased growth in cocultures with keratinocytes and nail fragments compared to the wild type, suggesting that ap1 participates in the negative control of virulence-related attributes and contributes to the chronicity of infection caused by this dermatophyte (Peres et al., 2022). The regulation of fungal virulence traits affects the chronicity of the disease and evasion of the host immune system (Martinez-Rossi et al., 2021; Peres et al., 2022). We hypothesized that the correct balance in the production of these ap1 isoforms contributes to the control of the chronic infection.
The presence of IR-2 in the c6 isoforms resulted in a minor protein, with a partial loss of the fungal-specific TF domain. However, the Zn(2) Cys(6) DNA-binding domain was maintained, suggesting that the function of this putative protein differed from that of the conventional isoform. In keratin, human nails, and keratinocytes, the IR-2 isoform of c6 was more prevalent than in its respective controls, suggesting that it functions under infectious conditions (Figure 6B). These data revealed the post-transcriptional regulation of the mechanisms involved in fungal pathogenicity. In Magnaporthe oryzae, TFs are a vital group of genes regulated by AS during fungal infections, with the C6 TF group being the most common (Jeon et al., 2022). The C6 TF of Aspergillus fumigatus (AFUB_043270) exhibits IR, which is a potential mechanism of self-protein activation control (Sieber et al., 2018). These data highlight the importance of C6-type TF AS events in several fungal infections.
Finally, the presence of alternative isoforms, even under control conditions (glucose or RPMI), indicates that infection conditions do not always dictate the presence of AS but instead influence the proportion of these events. These data underscore the importance of investigating the proportion of alternative isoforms compared to total isoforms, providing precise quantification of this phenomenon.
In conclusion, our data for these TF genes validate AS events and corroborate the idea that pathogenic fungi show different AS patterns in their infection environments (Jeon et al., 2022; Sieber et al., 2018; Cheng et al., 2022; Ibrahim et al., 2021). Our results also improve the understanding of the involvement of TF isoforms in this process, as pacC, con7, ap1, and c6 are crucial TFs for virulence and fungal adaptation to the host environment (van der Does and Rep, 2017; Wang et al., 2024; John et al., 2021). Exposure of the fungus to keratinolytic substrates induces the modulation of transcriptional and post-transcriptional responses, particularly of virulence factors and metabolic regulation. The regulatory amplitude exerted by AS under infection-like conditions suggests the impact of this mechanism on providing adequate responses to T. rubrum pathogenesis. The consequences observed in the TF genes from this regulation clarify how this mechanism contributes to the strategies employed by the fungus to adapt and colonize the host. This study provides a foundation for future fungal infection management by improving the understanding of the molecular mechanisms employed by pathogens to infect their hosts.
Data availability statement
The data presented in the study are deposited in Gene Expression Omnibus (GEO) database under accession number GSE134406.
Ethics statement
The studies involving humans were approved by The Ethics Committee of the Ribeirão Preto Medical School at the University of São Paulo approved this study (Protocol No. 5.868.345/2023). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
ML: Visualization, Formal analysis, Writing – review & editing, Writing – original draft, Investigation, Methodology, Conceptualization. JN-d-R: Visualization, Formal analysis, Writing – original draft, Investigation, Methodology. PS: Formal analysis, Methodology, Data curation, Software, Writing – original draft, Visualization. VO: Methodology, Investigation, Writing – original draft, Formal analysis. AR: Data curation, Formal analysis, Writing – review & editing, Conceptualization, Supervision, Funding acquisition. NM-R: Supervision, Conceptualization, Formal analysis, Investigation, Project administration, Writing – review & editing, Data curation, Funding acquisition.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The authors declare that financial support was received for the research from the Brazilian Funding Agencies, including the São Paulo Research Foundation -FAPESP (grant No. 2019/22596-9, and Fellowship No. 2021/04263-2 to JNR); the National Council for Scientific and Technological Development -CNPq (grant Nos. 307871/2021-5 and 307876/2021-7); CAPES (Finance Code 001), and the Fundação de Apoio ao Ensino, Pesquisa e Assistência -FAEPA.
Acknowledgments
We thank M. Mazzucato and M. D. Martins for technical support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor H.C. declared a shared parent affiliation with the authors at the time of review.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2025.1645525/full#supplementary-material
References
Barda, O., Maor, U., Sadhasivam, S., Bi, Y., Zakin, V., Prusky, D., et al. (2020). The pH-Responsive Transcription Factor PacC Governs Pathogenicity and Ochratoxin A Biosynthesis in. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.00210
Bitencourt, T. A., Lang, E. A. S., Sanches, P. R., Peres, N. T. A., Oliveira, V. M., Fachin, A. L., et al. (2020). HacA governs virulence traits and adaptive stress responses in Trichophyton rubrum. Front. Microbiol. 11, 193. doi: 10.3389/fmicb.2020.00193
Bristow, I. R. and Joshi, L. T. (2023). Dermatophyte resistance–on the rise. J. Foot Ankle Res. 16, 69. doi: 10.1186/s13047-023-00665-5
Cheng, X., Zhao, C., Gao, L., Zeng, L., Xu, Y., Liu, F., et al. (2022). Alternative splicing reprogramming in fungal pathogen Sclerotinia sclerotiorum at different infection stages on Brassica napus. Front. Plant Sci. 13. doi: 10.3389/fpls.2022.1008665
Chun, K.-H., Cho, S.-J., Lee, J.-W., Seo, J. H., Kim, K.-W., and Lee, S.-K. (2021). Protein kinase C-δ interacts with and phosphorylates ARD1. J. Cell. Physiol. 236, 379–391. doi: 10.1002/jcp.29866
Conesa, A., Götz, S., García-Gómez, J. M., Terol, J., Talón, M., and Robles., M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Deng, R., Wang, X., and Li., R. (2023). Dermatophyte infection: from fungal pathogenicity to host immune responses. Front. Immunol. 14. doi: 10.3389/fimmu.2023.1285887
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. doi: 10.1093/bioinformatics/bts635
Ferreira-Nozawa, M. S., Nozawa, S. R., Martinez-Rossi, N. M., and Rossi., A. (2003). The dermatophyte Trichophyton rubrum secretes an EDTA-sensitive alkaline phosphatase on high-phosphate medium. Braz. J. Microbiol. 34, 161–164. doi: 10.1590/S151783822003000200014
Ferreira-Nozawa, M. S., Silveira, H. C., Ono, C. J., Fachin, A. L., Rossi, A., and Martinez-Rossi., N. M. (2006). The pH signaling transcription factor PacC mediates the growth of Trichophyton rubrum on human nail in vitro. Med. Mycol. 44, 641–645. doi: 10.1080/13693780600876553
Grumbt, M., Monod, M., Yamada, T., Hertweck, C., Kunert, J., and Staib., P. (2013). Keratin degradation by dermatophytes relies on cysteine dioxygenase and a sulfite efflux pump. J. Invest. Dermatol. 133, 1550–1555. doi: 10.1038/jid.2013.41
Grützmann, K., Szafranski, K., Pohl, M., Voigt, K., Petzold, A., and Schuster., S. (2014). Fungal alternative splicing is associated with multicellular complexity and virulence: a genome-wide multi-species study. DNA Res. 21, 27–39. doi: 10.1093/dnares/dst038
Gupta, C., Das, S., Gaurav, V., Singh, P. K., Rai, G., Datt, S., et al. (2023). Review on host-pathogen interaction in dermatophyte infections. J. Mycol. Med. 33, 101331. doi: 10.1016/j.mycmed.2022.101331
Ho, E. C., Cahill, M. J., and Saville., B. J. (2007). Gene discovery and transcript analyses in the corn smut pathogen Ustilago maydis: expressed sequence tag and genome sequence comparison. BMC Genomics 8, 334. doi: 10.1186/1471-2164-8-334
Ibrahim, H. M. M., Kusch, S., Didelon, M., and Raffaele., S. (2021). Genome-wide alternative splicing profiling in the fungal plant pathogen Sclerotinia sclerotiorum during the colonization of diverse host families. Mol. Plant Pathol. 22, 31–47. doi: 10.1111/mpp.13006
Jacob, T. R., Peres, N. T. A., Persinoti, G. F., Silva, L. G., Mazucato, M., Rossi, A., et al. (2012). rpb2 is a reliable reference gene for quantitative gene expression analysis in the dermatophyte Trichophyton rubrum. Med. Mycol. 50, 368–377. doi: 10.3109/13693786.2011.616230
Jeon, J., Kim, K. T., Choi, J., Cheong, K., Ko, J., Choi, G., et al. (2022). Alternative splicing diversifies the transcriptome and proteome of the rice blast fungus during host infection. RNA Biol. 19, 373–385. doi: 10.1080/15476286.2022.2043040
John, E., Singh, K. B., Oliver, R. P., and Tan., K. C. (2021). Transcription factor control of virulence in phytopathogenic fungi. Mol. Plant Pathol. 22, 858–881. doi: 10.1111/mpp.13056
Kempken, F. (2013). Alternative splicing in ascomycetes. Appl. Microbiol. Biotechnol. 97, 4235–4241. doi: 10.1007/s00253-013-4841-x
Komoto, T. T., Bitencourt, T. A., Silva, G., Beleboni, R. O., Marins, M., and Fachin., A. L. (2015). Gene Expression Response of Trichophyton rubrum during Coculture on Keratinocytes Exposed to Antifungal Agents. Evid. Based Complement Alternat. Med. 2015, 180535. doi: 10.1155/2015/180535
Kröber, A., Etzrodt, S., Bach, M., Monod, M., Kniemeyer, O., Staib, P., et al. (2017). The transcriptional regulators SteA and StuA contribute to keratin degradation and sexual reproduction of the dermatophyte Arthroderma benhamiae. Curr. Genet. 63, 103–116. doi: 10.1007/s00294-016-0608-0
Kruithoff, C., Gamal, A., McCormick, T. S., and Ghannoum., M. A. (2023). Dermatophyte infections worldwide: increase in incidence and associated antifungal resistance. Life (Basel) 14, 1–12. doi: 10.3390/life14010001
Lang, E. A. S., Bitencourt, T. A., Peres, N. T. A., Lopes, L., Silva, L. G., Cazzaniga, R. A., et al. (2020). The stuA gene controls development, adaptation, stress tolerance, and virulence of the dermatophyte Trichophyton rubrum. Microbiol. Res. 241, 126592. doi: 10.1016/j.micres.2020.126592
Li, B., Chen, Y., and Tian., S. (2022). Function of pH-dependent transcription factor PacC in regulating development, pathogenicity, and mycotoxin biosynthesis of phytopathogenic fungi. FEBS J. 289, 1723–1730. doi: 10.1111/febs.15808
Li, Z., Yang, J., Peng, J., Cheng, Z., Liu, X., Zhang, Z., et al. (2021). Transcriptional landscapes of long non-coding RNAs and alternative splicing in Pyricularia oryzae revealed by RNA-seq. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.723636
Liu, W., Xie, Y., Ma, J., Luo, X., Nie, P., Zuo, Z., et al. (2015). IBS: an illustrator for the presentation and visualization of biological sequences. Bioinformatics 31, 3359–3361. doi: 10.1093/bioinformatics/btv362
Livak, K. J. and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lopes, M. E. R., Bitencourt, T. A., Sanches, P. R., Martins, M. P., Oliveira, V. M., Rossi, A., et al. (2022). Alternative splicing in Trichophyton rubrum occurs in efflux pump transcripts in response to antifungal drugs. J. Fungi (Basel) 8, 878–889. doi: 10.3390/jof8080878
Love, M. I., Huber, W., and Anders., S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Gen. Biol. 15, 550. doi: 10.1186/s13059-014-0550-8
Mancini, E., Rabinovich, A., Iserte, J., Yanovsky, M., and Chernomoretz., A. (2021). ASpli: integrative analysis of splicing landscapes through RNA-Seq assays. Bioinformatics 37, 2609–2616. doi: 10.1093/bioinformatics/btab141
Maranhão, F. C., Paião, F. G., and Martinez-Rossi., N. M. (2007). Isolation of transcripts over-expressed in human pathogen Trichophyton rubrum during growth in keratin. Microb. Pathog. 43, 166–172. doi: 10.1016/j.micpath.2007.05.006
Martinez-Rossi, N. M., Bitencourt, T. A., Peres, N. T. A., Lang, E. A. S., Gomes, E. V., Quaresemin, N. R., et al. (2018). Dermatophyte resistance to antifungal drugs: mechanisms and prospectus. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.01108
Martinez-Rossi, N. M., Peres, N. T. A., Bitencourt, T. A., Martins, M. P., and Rossi., A. (2021). State-of-the-art dermatophyte infections: epidemiology aspects, pathophysiology, and resistance mechanisms. J. Fungi (Basel) 7, 629–646. doi: 10.3390/jof7080629
Martinez-Rossi, N. M., Persinoti, G. F., Peres, N. T., and Rossi., A. (2012). Role of pH in the pathogenesis of dermatophytoses. Mycoses 55, 381–387. doi: 10.1111/j.1439-0507.2011.02162.x
Martins, M. P., Rossi, A., Sanches, P. R., Bortolossi, J. C., and Martinez-Rossi., N. M. (2020). Comprehensive analysis of the dermatophyte Trichophyton rubrum transcriptional profile reveals dynamic metabolic modulation. Biochem. J. 477, 873–885. doi: 10.1042/BCJ20190868
Martins-Santana, L., Petrucelli, M. F., Sanches, P. R., Martinez-Rossi, N. M., and Rossi., A. (2025). Unveiling the Role of StuA in Transcription Regulation in Trichophyton rubrum: From Global Regulation Dynamics to the pH−Responsive Program Control. Mycopathologia 190, 60. doi: 10.1007/s11046-025-00970-6
Mendes, N. S., Bitencourt, T. A., Sanches, P. R., Silva-Rocha, R., Martinez-Rossi, N. M., and Rossi., A. (2018). Transcriptome-wide survey of gene expression changes and alternative splicing in Trichophyton rubrum in response to undecanoic acid. Sci. Rep. 8, 2520. doi: 10.1038/s41598-018-20738-x
Neves-da-Rocha, J., Bitencourt, T. A., Oliveira, V. M., Sanches, P. R., Rossi, A., and Martinez-Rossi., N. M. (2019). Alternative splicing in heat shock protein transcripts as a mechanism of cell adaptation in Trichophyton rubrum. Cells 8, 1206–1219. doi: 10.3390/cells8101206
Odenbach, D., Breth, B., Thines, E., Weber, R. W., Anke, H., and Foster., A. J. (2007). The transcription factor Con7p is a central regulator of infection-related morphogenesis in the rice blast fungus Magnaporthe grisea. Mol. Microbiol. 64, 293–307. doi: 10.1111/j.1365-2958.2007.05643.x
Park, S. Y., Choi, J., Lim, S. E., Lee, G. W., Park, J., Kim, Y., et al. (2013). Global expression profiling of transcription factor genes provides new insights into pathogenicity and stress responses in the rice blast fungus. PloS Pathog. 9, e1003350. doi: 10.1371/journal.ppat.1003350
Pathan, M., Keerthikumar, S., Ang, C. S., Gangoda, L., Quek, C. Y. J., Williamson, N. A., et al. (2015). FunRich: an open access standalone functional enrichment and interaction network analysis tool. Proteomics 15, 2597–2601. doi: 10.1002/pmic.201400515
Peres, N. T., Lang, E. A. S., Bitencourt, T. A., Oliveira, V. M., Fachin, A. L., Rossi, A., et al. (2022). The bZIP Ap1 transcription factor is a negative regulator of virulence attributes of the anthropophilic dermatophyte Trichophyton rubrum. Curr. Res. Microbial. Sci. 3, 100132. doi: 10.1016/j.crmicr.2022.100132
Peres, N. T., Maranhão, F. C., Rossi, A., and Martinez-Rossi., N. M. (2010). Dermatophytes: host-pathogen interaction and antifungal resistance. Bras. Dermatol. 85, 657–667. doi: 10.1590/s0365-05962010000500009
Peres, N. T., Silva, L. G., Santos, R. S., Jacob, T. R., Persinoti, G. F., Rocha, L. B., et al. (2016). In vitro and ex vivo infection models help assess the molecular aspects of the interaction of Trichophyton rubrum with the host milieu. Med. Mycol. 54, 420–427. doi: 10.1093/mmy/myv113
Riedl, T. and Egly, J. M. (2000). Phosphorylation in transcription: the CTD and more. Gene Expr. 9, 3–13. doi: 10.3727/000000001783992704
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. doi: 10.1038/nbt.1754
Ruiz-Roldán, C., Pareja-Jaime, Y., González-Reyes, J. A., and Roncero., M. I. (2015). The transcription factor con7–1 is a master regulator of morphogenesis and virulence in. Fusarium Oxysporum Mol. Plant Microbe Interact. 28, 55–68. doi: 10.1094/MPMI-07-14-0205-R
Sieber, P., Voigt, K., Kämmer, P., Brunke, S., Schuster, S., and Linde., J. (2018). Comparative study on alternative splicing in human fungal pathogens suggests its involvement during host invasion. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.02313
Song, L., Huang, S. C., Wise, A., Castanon, R., Nery, J. R., Chen, H., et al. (2016). A transcription factor hierarchy defines an environmental stress response network. Science 354, 598–608. doi: 10.1126/science.aag1550
Tilburn, J., Sarkar, S., Widdick, D. A., Espeso, E. A., Orejas, M., Mungroo, J., et al. (1995). The Aspergillus PacC zinc finger transcription factor mediates regulation of both acid- and alkaline-expressed genes by ambient pH. EMBO J. 14, 779–790. doi: 10.1002/j.1460-2075.1995.tb07056.x
Trevisan, G. L., Oliveira, E. H., Peres, N. T., Cruz, A. H., Martinez-Rossi, N. M., and Rossi., A. (2011). Transcription of Aspergillus nidulans pacC is modulated by alternative RNA splicing of palB. FEBS Lett. 585, 3442–3445. doi: 10.1016/j.febslet.2011.09.037
Trushina, N., Levin, M., Mukherjee, P. K., and Horwitz., B. A. (2013). PacC and pH-dependent transcriptome of the mycotrophic fungus Trichoderma virens. BMC Genomics 14, 138. doi: 10.1186/1471-2164-14-138
van der Does, H. C. and Rep, M. (2017). Adaptation to the host environment by plant-pathogenic fungi. Annu. Rev. Phytopathol. 55, 427–450. doi: 10.1146/annurev-phyto-080516-035551
Wang, B., Guo, G., Wang, C., Lin, Y., Wang, X., Zhao, M., et al. (2010). Survey of the transcriptome of Aspergillus oryzae via massively parallel mRNA sequencing. Nucleic Acids Res. 38, 5075–5087. doi: 10.1093/nar/gkq256
Wang, Q., Huang, Z., Khan, I. A., Li, Y., Wang, J., Wang, J., et al. (2024). Key transcription factors required for outburst of rice blast disease in Magnaporthe oryzae. Phytopathol. Res. 6, 1–22. doi: 10.1186/s42483-024-00225-0
Wang, R., Huang, C., Zhang, Y., and Li., R. (2021). Invasive dermatophyte infection: A systematic review. Mycoses 64, 340–348. doi: 10.1111/myc.13212
Whitmarsh, A. J. and Davis, R. J. (2000). Regulation of transcription factor function by phosphorylation. Cell. Mol. Life Sci. CMLS 57, 1172–1183. doi: 10.1007/PL00000757
Wiemann, P., Willmann, A., Straeten, M., Kleigrewe, K., Beyer, M., Humpf, H. U., et al. (2009). Biosynthesis of the red pigment bikaverin in Fusarium fujikuroi: genes, their function and regulation. Mol. Microbiol. 72, 931–946. doi: 10.1111/j.1365-2958.2009.06695.x
Yamada, T., Makimura, K., and Abe., S. (2006). Isolation, characterization, and disruption of dnr1, the areA/nit-2-like nitrogen regulatory gene of the zoophilic dermatophyte, Microsporum canis. Med. Mycol. 44, 243–252. doi: 10.1080/13693780500410909
Zheng, H., Blechert, O., Mei, H., Ge, L., Liu, J., Tao, Y., et al. (2020). Assembly and analysis of the whole genome of Arthroderma uncinatum strain T10, compared with Microsporum canis and Trichophyton rubrum. Mycoses 63, 683–693. doi: 10.1111/myc.13079
Keywords: transcription factor, fungal pathogen, metabolism, intron retention, alternative splicing, PacC, Ap1, Con7
Citation: Lopes MER, Neves-da-Rocha J, Sanches PR, Oliveira VM, Rossi A and Martinez-Rossi NM (2025) Alternative splicing is a driving force that tunes metabolic adaptations to virulence traits in the dermatophyte Trichophyton rubrum. Front. Cell. Infect. Microbiol. 15:1645525. doi: 10.3389/fcimb.2025.1645525
Received: 11 June 2025; Accepted: 18 August 2025;
Published: 10 September 2025.
Edited by:
Hamilton Cabral, School of Pharmaceutical Sciences of Ribeirão Preto, University of São Paulo, BrazilReviewed by:
Prashant R. Desai, University of Wisconsin-Madison, United StatesChihiro Kadooka, Sojo University, Japan
Copyright © 2025 Lopes, Neves-da-Rocha, Sanches, Oliveira, Rossi and Martinez-Rossi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nilce M. Martinez-Rossi, bm1tcm9zc2lAdXNwLmJy