 Zhi Li1
Zhi Li1 Yuan Han1Yong Fu1Qing Yuan1
Yuan Han1Yong Fu1Qing Yuan1 Shuqin Wang2Xingye Pan3Wanchao Xue3
Shuqin Wang2Xingye Pan3Wanchao Xue3 Hong Yin4
Hong Yin4 Shandian Gao4*
Shandian Gao4* Ru Meng2*
Ru Meng2*- 1Qinghai Provincial Key Laboratory of Pathogen Diagnosis for Animal Diseases and Green Technical Research for Prevention and Control, Academy of Animal Sciences and Veterinary Medicine, Qinghai University, Xining, China
- 2Xining Animal Disease Prevention and Control Center, Xining, China
- 3Animal Husbandry and Veterinary Station of Huangyuan County, Xining, China
- 4Gansu Province Research Center for Basic Disciplines of Pathogen Biology, State Key Laboratory for Animal Disease Control and Prevention, Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences, Lanzhou, China
Introduction: Bovine viral diarrhea virus (BVDV) is a major pathogen affecting livestock health in China. However, the current epidemiological status in yaks (Bos grunniens), particularly in Qinghai Province, remains insufficiently understood.
Methods: In the present study, a comprehensive serological and molecular investigation of BVDV in yaks was conducted across broad geographic areas of eight administrative regions including Yushu, Guoluo, Huangnan, Hainan, Haidong, Haixi, Haibei, and Xining in Qinghai Province.
Results: The results revealed widespread BVDV exposure in Qinghai yak, with an overall antibody prevalence of 84.52% (1158/1370) and substantial herd variation (12.00~98.07%). Active infections were confirmed through antigen detection, revealing prevalence ranging from 0.34% (Haixi) to 4.90% (Huangnan). Genetic characterization identified two circulating subgenotypes: BVDV-1a (n=3) and the predominant BVDV-1u (n=30), with the latter dominating across all regions.
Discussion: These results highlight the endemic circulation of BVDV in Qinghai yak populations and uncover unexpected genetic diversity, emphasizing the need for control measures to mitigate the adverse impacts of BVDV infection in yaks in high-altitude pastoral systems.
1 Introduction
Bovine Viral Diarrhea (BVD), caused by the Bovine Viral Diarrhea Virus (BVDV), is classified as a notifiable infectious disease by the World Organization for Animal Health (WOAH). Cattle infected with BVDV may exhibit subclinical infection or symptoms, such as transient fever, diarrhea, leukopenia, respiratory signs and hemorrhagic syndrome, while transplacental infections in pregnant cattle can lead to embryonic death, abortions or the birth of persistently infected (PI) calves that are prone to mucosal disease (MD) (Baker, 1995). BVDV is a member of the Flaviviridae family, specifically within the Pestivirus genus and the viruses can be classified into several distinct genotypes, namely BVDV-1 (Pestivirus A), BVDV-2 (Pestivirus B), and BVDV-3/HoBi-like pestivirus (Pestivirus H) (Liu et al., 2009; Bauermann and Ridpath, 2015; Postel et al., 2021). Among these, BVDV-1 is widely prevalent worldwide with multiple proposed subgenotypes (1a to 1x), whereas BVDV-2 and BVDV-3 has comparatively limited subgenotypes (2a to 2e for BVDV-2 and HobiPeV a-e for BVDV-3) (Yeşilbağ et al., 2017; Deng et al., 2020; de Oliveira et al., 2022; Kalaiyarasu et al., 2022; Mucellini et al., 2023).
In recent years, BVDV has become a growing concern in cattle industry in China, posing substantial economic challenges. Systematic reviews and meta-analysis have provided robust epidemiological insights and demonstrated pooled BVDV prevalence of 53.0% and 36.0% in dairy cattle and yaks (Bos gruniens) respectively (Ran et al., 2019; Diao et al., 2020). The genetic diversity of BVDV in China has been characterized, revealing the presence of multiple subgenotypes, including 1a, 1b, 1c, 1d, 1m, 1o, 1p, 1q, 1u, 1v, 1w, 2a and 2b, as well as BVDV-3 (Xue et al., 2010; Yeşilbağ et al., 2017; Deng et al., 2020; Chen et al., 2021; Yang et al., 2023).
Yaks mainly inhabit the high-altitude regions of the Qinghai-Tibet Plateau and its surrounding areas. In China, yaks are distributed across the provinces and autonomous regions of Qinghai, Tibet, Sichuan, Gansu, Xinjiang, and Yunnan, with an amount of approximately 16.36 million that accounts for over 94% of the global yak population. Qinghai Province maintains China’s largest yak population at 6.15 million (37.57% of national total) (Han and Liang, 2024). Previous studies have demonstrated a wide seroprevalence range of BVDV (44.86-100%) among yak populations across different regions of Qinghai (Gao et al., 2013), with infections involving the BVDV-1b, BVDV-1d, and BVDV-1q subgenotypes (Gong et al., 2014). However, the current epidemiological status in yaks across broad geographic areas of Qinghai remains poorly characterized, which prompt our comprehensive serological and molecular epidemiological investigation of BVDV in local yak populations, with the aim of providing critical data for effective prevention and control strategies.
2 Materials and methods
2.1 Collection of serum and blood samples
From April 2024 to May 2025, a total of 1,370 yaks (aged ≥1 year) exhibiting respiratory symptoms or diarrhea but with no history of BVD vaccination were selected for this study. The animals were sampled from 19 grazing herds (total population: 15726) across eight administrative regions including Yushu, Guoluo, Huangnan, Hainan, Haidong, Haixi, Haibei, and Xining in Qinghai Province; and the sampling size exceeded 5% of the population, with additional samples collected from selected herds based on owners’ voluntary participation in the surveillance program (Table 1; Supplementary Figure 1). Non-anticoagulated whole blood samples were collected by local veterinary practitioners. The serum were isolated via centrifugation at 4, 000 rpm for 10 min and transported to the laboratory on ice. The serum samples were preserved at -80 °C until detection.
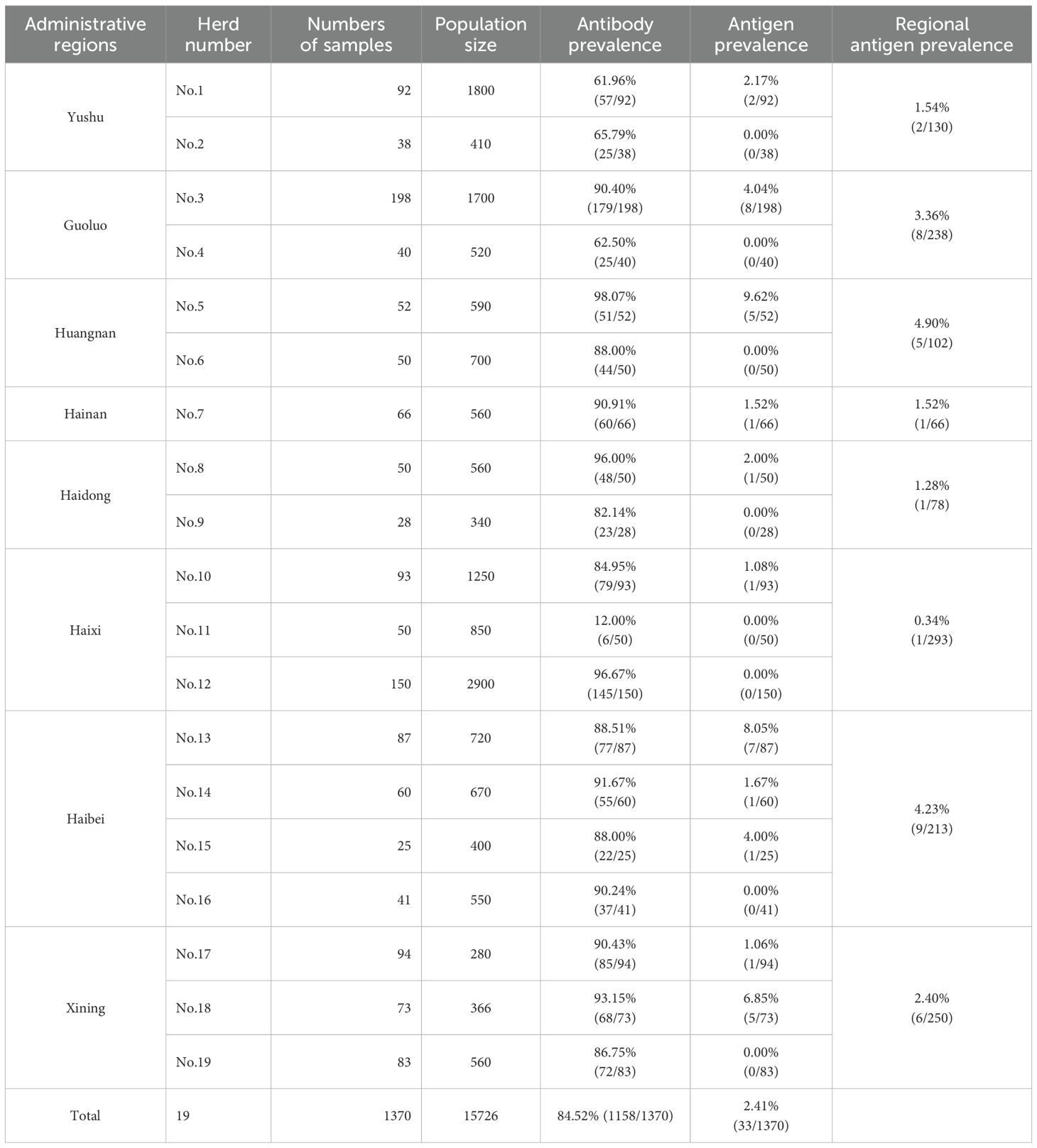
Table 1. Summary of sera sample and prevalence of BVDV in 19 yak herds in Qinghai province.
2.2 BVDV antibody ELISA
The serum samples were detected of antibodies against the BVDV NSP2–3 protein by using ID Screen® BVD p80 Antibody Competition Kit (IDvet Innovative Diagnostics, France) according to the user manual. Samples with competition percentage (ODsample/ODnegative%) less than 40% was classified as positive.
2.3 BVDV antigen ELISA
All blood samples were examined for the BVDV NSP2–3 protein via ID Screen® BVD p80 Antigen Capture Kit (IDvet Innovative Diagnostics, France) based on the manufacturer’s instructions. The ratio of the mean values of ((ODsample - ODnegative control)/(ODpositive control - ODnegative control)) higher than 50% were considered as positive.
2.4 One step RT-PCR assays
The RNA was extracted from 400 μL of antigen positive serum samples using Quick-RNA/DNA Viral Kit (Zymo Research, USA) according to the manufacturer’s instructions. The viral 5-untranslated region (5’-UTR) was detected by RT-PCR using primers F (5’-CTAGCCATGCCCTTAGTAGGACTA-3’) and R (5’-CAACTCCATGTGCCATGTACAGCA-3’) (Mahony et al., 2005). The RT-PCR reaction was performed using the PrimeScript One Step RT-PCR Kit Ver.2 (Dye Plus) (TaKaRa, Dalian, China), with 50°C for 30 min, followed by 35 cycles at 94°C for 30 s, 55°C for 30 s, 72°C for 30 s, and a final extension at 72°C for 10 min. The PCR products were checked by electrophoresis on a 1% agarose gel. In order to confirm the detection result and subsequent sequence analysis based on the 5’ UTR, additional RT-PCR reactions for the Npro coding region were performed with selected RNA samples, using the primers BD1 (5’-TCTCTGCTGTACATGGCACATG-3’) and BD2 (5’- TTGTTRTGGTACARRCCGTC -3’) (Vilcek et al., 1997). The reaction was carried out with identical RT-PCR reagents and a modified protocol (72°C extension for 1 min per cycle).
2.5 Phylogenetic analysis
The PCR products were recovered by using the Zymoclean Gel DNA Recovery Kit (Zymo Research, USA) and cloned into pMD19-T vector (TaKaRa, Dalian, China) for DNA sequencing. The obtained sequences were analyzed using NCBI BLAST program. To investigate the genetic diversity and evolutionary relationships of the identified BVDV strains, the assembled 5’-UTR and Npro coding sequences were aligned against the reference NADL strain sequences (245 bp for 5’-UTR, genome positions 130-347; 385 bp for Npro, genome positions 386-770) using MAFFT v7.505 (Katoh and Standley, 2013). Phylogenetic trees were conducted by the MEGA software (version 12) using the neighbor-joining (NJ) method and bootstrap analysis (n = 1,000).
3 Results
3.1 Seroprevalence of BVDV antibodies
The antibody ELISA results showed an overall BVDV antibody seroprevalence of 84.52% (1158/1370) in Qinghai yaks, with variations among herds and regions (Table 1). Two herds (No.1 and No.2) in Yushu exhibited seropositivity rates of 61.96% and 65.79% respectively. Yaks in Guoluo demonstrated antibody prevalence of 90.40% in one herd (Herd No.3) and 62.50% in another (Herd No.4). Except for low-prevalence in one herd (Herd No.11) in Haixi (12.00%), the BVDV antibody seroprevalence of other surveyed herds were 98.07% and 88.00% respectively in two herds (No.5 and No.6) in Huangnan, 90.91% in Hainan (Herd No.7), 96.00% and 82.14% in two herds (No.9 and No.10) in Haidong, 84.95% in one herd (No.10) and 96.67% in another (No.12) in Haixi, 88.51% (herd No.13), 91.67% (herd No.14), 88.00% (herd No.15), and 90.24% (herd No.16) in Haibei, as well as 90.43% (herd No.17), 93.15% (herd No.18) and 86.75% (herd No.19) in Xining (Table 1; Supplementary Figure 1). Since these animals were not immunized with BVD vaccine, the high antibody seroprevalence are more likely to indicates widespread transmission of BVDV among the Qinghai yak population.
3.2 Prevalence of BVDV antigen
The viral antigen detection results revealed a relatively high proportion of BVDV antigen-positive herds in Haibei and Xining, with herd-level prevalence of 75% (3/4) and 66.67% (2/3), respectively. This was followed by herd-level prevalences in Yushu (50%), Guoluo (50%), Huangnan (50%), Haidong (50%), and Haixi (33.33%) (Table 1). Additionally, one yak from the single selected herd in Hainan also tested positive for BVDV antigen, albeit at a low antigen prevalence of 1.52% for the herd (Table 1). It will be necessary to conduct more extensive herd testing to more accurately evaluate the antigen prevalence of BVDV in Hainan. The highest BVDV antigen prevalence (9.62%, 5/52) was observed in one herd (No.5) in Huangnan, followed by herd No. 13 in Haibei (8.05%, 7/87) and No. 18 in Xining (6.85%, 5/73). Other herds with relatively high BVDV antigen prevalence included one herd (No.3) in Guoluo (4.04%, 8/198) and one (No.15) in Haibei (4.00%, 1/25). Comparatively, the prevalence was lower in some herds of Yushu (No.1, 2.17%, 2/92), Haidong (No. 8, 2.00%, 1/50), Hainan (No.7, 1.52%, 1/66), Haixi (No.10, 1.08%, 1/93), and Xining (No.17, 1.06%, 1/94) (Supplementary Figure 1; Table 1).
As shown in supplementary Figure 1 and Table 1, the region level prevalence of BVDV antigen in yaks was observed in Huangnan (4.90%, 5/102) and Haibei (4.23%, 9/213), which likely resulted from higher breeding density or frequent animal movement. Intermediate viral antigen prevalence levels were exhibited in yaks form Guoluo (3.36%, 8/238) and Xining (2.40%, 6/250), but the inter-herd differences should not be neglected. Notably, the prevalence of viral antigen in Haixi was lowest the 0.34% (1/293), which may be attributed to better biosecurity measures or lower farm density.
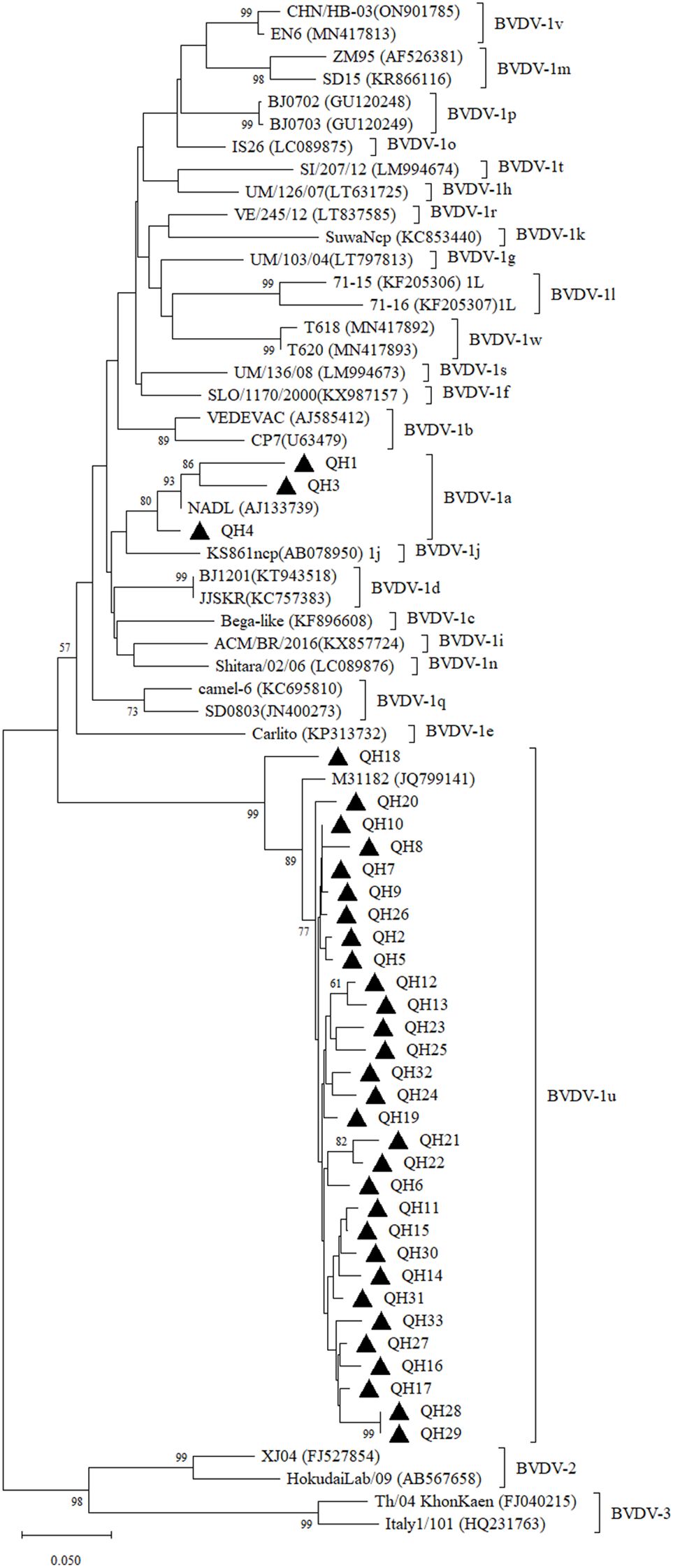
Figure 1. Neighbor-joining phylogeny of BVDV isolates based on the 5’-UTR sequences (245 bp). The phylogenetic tree was constructed using the neighbor-joining method implemented in MEGA version 12. Bootstrap values are percentage of 1000 replicates and are shown below the branches. Bar indicates substitutions per site. Accession numbers representative subgenotypes are listed in the brackets after the isolate names.
3.3 Sequencing and phylogenetic analysis
We further validated the virus in antigen-positive samples using RT-PCR. The results showed that the 5’-UTR fragment was successfully amplified in all 33 RNA samples prepared from antigen-positive sera. The obtained 5’-UTR sequences were submitted to GenBank under accession number PV806868-PV806900. The 5’-UTR sequences were aligned and a sequence identity matrix was generated, demonstrating that the 5’-UTR of the Qinghai strains shared nucleotide identity ranged from 74.90-100% (Supplementary Figure 2). Phylogenetic analysis revealed that these 33 samples belonged to the BVDV-1a (n=3) and BVDV-1u (n=30) subgenotypes (Figure 1). Among them, BVDV-1a was detected in yaks from two regions including Guoluo and Yushu, while BVDV-1u was distributed in yaks from all the eight administrative regions in Qinghai Province. The 5’-UTR sequence of BVDV-1a strains (QH1, QH3, QH4) shared 92.10-92.50% nucleotide identity, while BVDV-1u strains (QH2, QH5 to QH33) displayed much wider variability, sharing nucleotide identity ranging from 90.10% to 100%. These 5’-UTR sequences of Qinghai BVDV-1u strains shared 93.70-97.60% nucleotide identity with an BVDV isolate M31182 (GenBank accession number: JQ799141) originated from yaks in neighboring Sichuan Province, which was identified as BVDV-1u in a previous study (Deng et al., 2020). Notably, the 5’-UTR of QH18 exhibited significant divergence from other BVDV-1u strains, sharing only 90.10-93.70% nucleotide identity, whereas the 5’-UTR sequences of the remaining BVDV-1u strains showed much higher intra-group similarity (93.7-100%). The phylogenetic tree based on the 5’-UTR demonstrated that QH18 occupied a distinct phylogenetic position, clearly separated from other BVDV-1u strains (Figure 1).
To further validate the subgenotyping analysis based on the 5’-UTR, we conducted complementary sequencing and phylogenetic analysis of the Npro gene from representative regional samples. The obtained Npro gene coding sequences were submitted to GenBank under accession number and PV806901-PV806909. The QH1 strain had 95.20% and 93.0% nucleotide identity with the Npro gene sequences of the Chinese BVDV-1a strains BJ-2013 (GenBank No. MH490942) and HLJ-2018 (GenBank No. PV168122). Compared with the M31182 isolate, the Npro gene of Qinghai BVDV-1u strains shared 89.00~94.00% nucleotide identity, with QH18 displaying the lowest homology (89.00%). The phylogenetic analysis of the Npro gene yielded results consistent with those based on the 5’UTR region, with QH18 similarly positioned on a distant evolutionary branch (Figure 2). These findings suggest the potential existence of undetected genetically divergent BVDV-1u strains in yak populations in Qinghai, emphasizing the critical need for further molecular epidemiological studies to enable more detailed characterization of BVDV in Qinghai yaks.

Figure 2. Neighbor-joining phylogeny of BVDV isolates based on Npro coding region (385 bp). The phylogenetic tree was constructed using the neighbor-joining method implemented in MEGA version 12. Bootstrap values are percentage of 1000 replicates and are shown below the branches. Bar indicates substitutions per site. Accession numbers representative subgenotypes are listed in the brackets after the isolated names.
4 Discussion
Since the first report in 1946, BVD continues to be one of the primary causes of significant economic and production losses in the cattle industry (Newcomer, 2021). BVDV infects not only cattle but also a wide range of artiodactyls, including swine, domestic ruminants such as sheep and goats, camelids like alpacas and camels, and wildlife such as cervids, antelope, wild goats and sheep, and American bison (Bison bison), and infections in heterologous hosts may result in mild or similar clinical signs to BVDV infections in cattle and generate persistently infected carriers, posing risks as viral reservoirs (Passler and Walz, 2010; Van Campen and Rhyan, 2010; Nelson et al., 2016; Hause et al., 2021). In this study, we conducted a comprehensive investigation of (BVDV) in yak populations in the most important province for yak farming in China utilizing serological antibody detection, viral antigen profiling, and molecular genotyping. Prior to our investigation, BVDV infection in yaks had indeed attracted considerable attention. The initial detection of BVDV in China occurred during the 1980s, with near-simultaneous identifications in both cattle and yak (Li et al., 1983; Liu, 1984). However, the majority of studies of BVD in yak were published in Chinese journals, significantly limiting their accessibility to the international readers. A recently systematic review and meta-analysis illustrated that BVDV in yaks between 1987 and 2019 in China had a variable prevalence of 24.4%-67.5% and viral antigen-positive rate of 13.8% (Diao et al., 2020). In our study, the overall observed seroprevalence of BVD (84.52%) was comparable with the 72.14% prevalence reported in a previous study (Gao et al., 2013). Some discrepancies can be attributed to our targeted selection of yaks exhibiting certain clinical symptoms rather than random screening. Additionally, since classical swine fever (CSF)vaccines are sometimes used for prevention yaks from BVD in China (Ji et al., 1995; Liu et al., 2013), which may interfere with antibody detection. Therefore, we specifically selected yak herds that had not been vaccinated against either BVD or CSF vaccines for our seroprevalence study to accurately reflect the true infection risk within the population.
Notably, one herd in Haixi (No. 11) showed relatively low seroprevalence at the animal level, suggesting a reduced exposure to circulating virus. In cattle, BVDV transmission primarily relies on high viral shedding from persistently infected animals, with transiently infected animals playing a secondary role due to their lower and shorter-term viral excretion (Evans et al., 2019), and a similar dynamic occurs in non-bovine ruminant species (Passler and Walz, 2010; Van Campen and Rhyan, 2010; Nelson et al., 2016; Hause et al., 2021). The observation of seroprevalence in the herd in Haixi correlated with local BVDV antigen testing results, which indicated a markedly lower prevalence of BVDV antigen in this region compared with other study areas. Due to the free-range grazing practices of yaks, the epidemiological characteristics of diseases may differ from those in intensive indoor feeding systems. Previous studies showed yaks are prone to co-infections with bovine rotavirus, bovine enterovirus, bovine astrovirus, bovine coronavirus, bovine parainfluenza virus, Pasteurella multocida and Escherichia coli alongside BVDV (Yuan et al., 2015; Yang et al., 2019). Consequently, in clinically affected yak populations exhibiting low BVDV prevalence, potential co-infecting pathogens should be systematically evaluated in future studies.
The identification of BVDV antigen provided direct and definitive evidence for confirming the presence of progressive BVDV infection in eight yak herds. Compared with provincial survey data from a decade ago (Fu et al., 2013), the results obtained in our study were generally consistent while exhibiting certain discrepancies. The yaks in Huangnan Prefecture continued to exhibit the highest BVD prevalence (2.21% vs. 4.90%), while Haixi Prefecture maintains the lowest prevalence (0.43% vs. 0.34%), suggesting that stable factors such as climatic characteristics, livestock production practices may influence BVD prevalence in these two regions. Notably, increases in BVDV antigen prevalence in yaks in Haibei (0.43% to 4.23%) and Guoluo Prefecture (0.43% to 3.36%) were observed. Although we cannot rule out biases caused by different sampling methods and reagent sensitivities, the detection of antigens in the yak population indicates that the monitoring and prevention of BVDV in yaks must not be overlooked. Moreover, the detection of BVDV antigen in yaks from all regions suggests that all newly introduced yaks should be quarantined to confirm their BVDV-negative status before herd introduction.
The conserved 5’-UTR and the Npro coding region are widely used for genotyping phylogenetic analysis for BVDV, providing critical insights into the genetic relatedness, evolutionary divergence, and possible origins of the circulating BVDV strains. Consistent with the national BVDV epidemiological pattern (Xiao et al., 2025), our study confirmed BVDV-1 as the predominant genotype, while revealing unique regional characteristics. Notably, although BVDV-1m has been reported in Qinghai dairy cattle (Xiao et al., 2025), this subgenotype remains rarely documented in yaks. Alongside earlier reports of BVDV-1b, -1d, and -1q (Gong et al., 2014) and the recent identification of BVDV-1a in Haibei (Lin et al., 2025), our findings reveal that BVDV-1u has emerged as the dominant subgenotype across Qinghai yaks, while BVDV-1a remains at low prevalence. In comparison with previous studies, our findings revealed a shift in subgenotypes in yaks in Qinghai Province, suggesting potential ongoing viral evolution or host adaptation. Although relatively few Qinghai BVDV-1a strains were obtained in neighboring Yushu and Guoluo prefectures, the sequences among these strains were not identical. Therefore, the polymorphism of the Qinghai 1a subtype cannot be overlooked, and more sequences need to be acquired in the future for further verification. Furthermore, the genetic analysis of BVDV-1u sequences obtained in this study revealed significant polymorphism within this subgenotype, with QH18 exhibiting notable divergence from other strains, possibly resulting from unique selective pressures due to host adaptation or geographic isolation. The QH18 strain displays the lowest nucleotide similarity among BVDV-1u strains, with only 90.1% identity in the 5’UTR and 90.2–90.8% in the Npro gene compared to other Qinghai 1u isolates, followed by the Sichuan yak strain (M31182). Such divergence could have implications for viral pathogenicity and vaccine efficacy. Despite its current classification within subgenotype 1u, the considerable divergence observed in QH18 suggests that ongoing evolution may lead to the emergence of a distinct subgenotype, emphasizing the need for sustained surveillance. Given the vast territory of Qinghai and the free-range grazing practices of yaks across wide areas, we were unable to conduct systematic monitoring of persistent infection status BVDV in yaks. However, a recent study in Tibet revealed a persistent BVDV infection rate of 1.55% in yaks through antigen testing of paired sera samples collected at 3-week intervals (Luo et al., 2023). On the Qinghai-Tibet Plateau, the yak population substantially exceeds that of domesticated cattle, and persistent infection of BVDV in yak may suggest that the yak can sustain BVDV transmission cycles, but whether they can act as true reservoirs remains unclear. The limited BVDV data resulted in inconsistent subgenotype comparation between yak and cattle, highlighting the need to investigate potential epidemiological links between yaks under free-range grazing systems and intensively managed domestic cattle in Qinghai, which would be a crucial direction for future molecular epidemiological research on BVDV.
5 Conclusion
In conclusion, this study systematically elucidates the epidemiological characteristics and predominant subgenotypes of BVDV in yaks in Qinghai Province, highlighting the need for investigation of the transmission dynamics of BVDV under the unique ecological conditions and the development of effective intervention measures for preventing and controlling BVD for yak.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The animal study was approved by Institutional Animal Care and Use Committee of the Qinghai University. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
ZL: Data curation, Investigation, Writing – original draft. YH: Formal Analysis, Methodology, Writing – review & editing. YF: Investigation, Methodology, Visualization, Writing – review & editing. QY: Investigation, Methodology, Writing – review & editing. SW: Investigation, Validation, Writing – review & editing. XP: Formal Analysis, Investigation, Writing – review & editing. WX: Investigation, Visualization, Writing – review & editing. HY: Investigation, Methodology, Supervision, Writing – review & editing. SG: Conceptualization, Writing – review & editing. RM: Funding acquisition, Investigation, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study was supported by the Key R&D and Transformation Plan of Qinghai Province (Grant No. 2024-NK-103), the Qinghai Province “Kulun Talents High-end innovation and Entrepreneurial Talents” Talent Training Project, the Agricultural Science and Technology Innovation Program (CAAS-ASTIP), and NBCIS (CARS-37).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2025.1652023/full#supplementary-material
Supplementary Figure 1 | The geographic distribution of BVDV from the nineteen herds of yaks in Qinghai Province.
References
Baker, J. C. (1995). The clinical manifestations of bovine viral diarrhea infection. Vet. Clin. North Am. Food Anim. Pract. 11, 425–445. doi: 10.1016/s0749-0720(15)30460-6
Bauermann, F. V. and Ridpath, J. F. (2015). HoBi-like viruses–the typical ‘atypical bovine pestivirus’. Anim. Health Res. Rev. 16, 64–69. doi: 10.1017/S146625231500002X
Chen, M., Liu, M., Liu, S., and Shang, Y. (2021). HoBi-like pestivirus infection leads to bovine death and severe respiratory disease in China. Transbound Emerg. Dis. 68, 1069–1074. doi: 10.1111/tbed.13832
Deng, M., Chen, N., Guidarini, C., Xu, Z., Zhang, J., Cai, L., et al. (2020). Prevalence and genetic diversity of bovine viral diarrhea virus in dairy herds of China. Vet. Microbiol. 242, 108565. doi: 10.1016/j.vetmic.2019.108565
de Oliveira, P. S. B., Silva Júnior, J. V. J., Weiblen, R., and Flores, E. F. (2022). A new (old) bovine viral diarrhea virus 2 subtype: BVDV-2e. Arch. Virol. 167, 2545–2553. doi: 10.1007/s00705-022-05565-w
Diao, N. C., Gong, Q. L., Li, J. M., Zhao, D., Li, D., Zhao, B., et al. (2020). Prevalence of bovine viral diarrhea virus (BVDV) in yaks between 1987 and 2019 in mainland China: A systematic review and meta-analysis. Microb. Pathog. 144, 104185. doi: 10.1016/j.micpath.2020.104185
Evans, C. A., Pinior, B., Larska, M., Graham, D., Schweizer, M., Guidarini, C., et al. (2019). Global knowledge gaps in the prevention and control of bovine viral diarrhoea (BVD) virus. Transbound Emerg. Dis. 66, 640–652. doi: 10.1111/tbed.13068
Fu, Y. J., Wang, S. X., Li, X. Y., and Lin, Y. Q. (2013). Investigation on the prevalence of bovine viral diarrhea/mucosal disease in yaks in Qinghai Province. Anim. Husb. Vet. Med. 45, 621–623.
Gao, J., Liu, M., Meng, X., Han, Z., Zhang, D., and Hou, B. (2013). Seroprevalence of bovine viral diarrhea infection in Yaks (Bos grunniens) on the Qinghai-Tibetan Plateau of China. Trop. Anim. Health Prod. 45, 791–793. doi: 10.1007/s11250-012-0290-2
Gong, X., Liu, L., Zheng, F., Chen, Q., Li, Z., Cao, X., et al. (2014). Molecular investigation of bovine viral diarrhea virus infection in yaks (Bos gruniens) from Qinghai, China. Virol. J. 11, 29. doi: 10.1186/1743-422X-11-29
Han, X. and Liang, D. (2024). Economic analysis of yak industry in China. Food. Nutr. China 30, 17–20. doi: 10.19870/j.cnki.11-3716/ts.20231122.002
Hause, B. M., Pillatzki, A., Clement, T., Bragg, T., Ridpath, J., and Chase, C. C. L. (2021). Persistent infection of American bison (Bison bison) with bovine viral diarrhea virus and bosavirus. Vet. Microbiol. 252, 108949. doi: 10.1016/j.vetmic.2020.108949
Ji, J. C., Yan, G. F., Wang, B. L., Deng, C. H., Jinmei, D. B., Zha, X., et al. (1995). Intermediate test for prevention of viral diarrhea/mucosal disease in yaks. Qinghai J. Anim. Husb. Vet. Med. 2, 5–7.
Kalaiyarasu, S., Mishra, N., Jayalakshmi, K., Selvaraj, P., Sudhakar, S. B., Jhade, S. K., et al. (2022). Molecular characterization of recent HoBi-like pestivirus isolates from cattle showing mucosal disease-like signs in India reveals emergence of a novel genetic lineage. Transbound Emerg. Dis. 69, 308–326. doi: 10.1111/tbed.13981
Katoh, K. and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Li, Y. M., Liu, Z. R., and Wu, Y. L. (1983). Isolation and identification of bovine viral diarrhea-mucosal disease virus strain (Changchun 184). Chin. J. Vet. Sci. 3, 113–120. doi: 10.16303/j.cnki.1005-4545.1983.02.001
Lin, W. S., Ma, D. D., Lei, M. T., Wei, B., Li, G. C., Wang, G. H., et al. (2025). Epidemiological investigation and 5′-UTR phylogenetic analysis of bovine viral diarrhea virus in yaks from Haibei region, Qinghai Province. Chin. J. Anim. Sci. Vet. Med. 52, 1241–1249. doi: 10.16431/j.cnki.1671-7236.2025.03.026
Liu, S. J. (1984). Study on bovine viral diarrhea/mucosal disease in yaks. J. Southwest Univ. Nationalities 4, 1–5.
Liu, L., Xia, H., Wahlberg, N., Belák, S., and Baule, C. (2009). Phylogeny, classification and evolutionary insights into pestiviruses. Virology 385, 351–357. doi: 10.1016/j.virol.2008.12.004
Liu, M. Y., Zhang, K. R., Han, Z. Q., Li, K., Zhang, D., Danba, C. R., et al. (2013). Field immunization efficacy of bovine viral diarrhea vaccine and classical swine fever vaccine against yak viral diarrhea. Chin. Dairy Cattle, 17, 14–15.
Luo, R., Wu, D., Cai, C., Huang, J., Li, Y., Li, X., et al. (2023). Epidemiological investigation of bovine viral diarrhea in yaks and vaccine immunity duration study in the Qinghai-Tibet Plateau. Anim. Husb. Vet. Med. 55, 79–85.
Mahony, T. J., McCarthy, F. M., Gravel, J. L., Corney, B., Young, P. L., and Vilcek, S. (2005). Genetic analysis of bovine viral diarrhoea viruses from Australia. Vet. Microbiol. 106, 1–6. doi: 10.1016/j.vetmic.2004.10.024
Mucellini, C. I., Silva Júnior, J. V. J., de Oliveira, P. S. B., Weiblen, R., and Flores, E. F. (2023). Novel genomic targets for proper subtyping of bovine viral diarrhea virus 1 (BVDV-1) and BVDV-2. Virus Genes 59, 836–844. doi: 10.1007/s11262-023-02022-x
Nelson, D. D., Duprau, J. L., Wolff, P. L., and Evermann, J. F. (2016). Persistent bovine viral diarrhea virus infection in domestic and wild small ruminants and camelids including the mountain goat (Oreamnos americanus). Front. Microbiol. 6. doi: 10.3389/fmicb.2015.01415
Newcomer, B. W. (2021). 75 years of bovine viral diarrhea virus: Current status and future applications of the use of directed antivirals. Antiviral Res. 196, 105205. doi: 10.1016/j.antiviral.2021.105205
Passler, T. and Walz, P. H. (2010). Bovine viral diarrhea virus infections in heterologous species. Anim. Health Res. Rev. 11, 191–205. doi: 10.1017/S1466252309990065
Postel, A., Smith, D. B., and Becher, P. (2021). Proposed update to the taxonomy of pestiviruses: eight additional species within the genus pestivirus, family flaviviridae. Viruses 13, 1542. doi: 10.3390/v13081542
Ran, X., Chen, X., Ma, L., Wen, X., Zhai, J., Wang, M., et al. (2019). A systematic review and meta-analysis of the epidemiology of bovine viral diarrhea virus (BVDV) infection in dairy cattle in China. Acta Trop. 190, 296–303. doi: 10.1016/j.actatropica.2018.08.031
Van Campen, H. and Rhyan, J. (2010). The role of wildlife in diseases of cattle. Vet. Clin. North Am. Food Anim. Pract. 26, 147–161. doi: 10.1016/j.cvfa.2009.10.008
Vilcek, S., Nettleton, P. F., Paton, D. J., and Belák, S. (1997). Molecular characterization of ovine pestiviruses. J. Gen. Virol. 78, 725–735. doi: 10.1099/0022-1317-78-4-725
Xiao, Y., Liu, Y., Chi, T., Jiang, W., He, T., Xu, L., et al. (2025). Prevalence and genetic characterization of bovine viral diarrhea virus in dairy cattle in northern China. BMC Vet. Res. 21, 250. doi: 10.1186/s12917-025-04491-8
Xue, F., Zhu, Y. M., Li, J., Zhu, L. C., Ren, X. G., Feng, J. K., et al. (2010). Genotyping of bovine viral diarrhea viruses from cattle in China between 2005 and 2008. Vet. Microbiol. 143, 379–383. doi: 10.1016/j.vetmic.2009.11.010
Yang, X. L., Li, Z. Q., Luo, Y. Z., and Lu, G. H. (2019). Etiological investigation and analysis of bovine viral diarrhea in yaks in Xining, Qinghai Province. Chin. J. Vet. Med. 55, 8–12.
Yang, N., Xu, M., Ma, Z., Li, H., Song, S., Gu, X., et al. (2023). Detection of emerging HoBi-like Pestivirus (BVD-3) during an epidemiological investigation of bovine viral diarrhea virus in Xinjiang: a first-of-its-kind report. Front. Microbiol. 14. doi: 10.3389/fmicb.2023.1222292
Yeşilbağ, K., Alpay, G., and Becher, P. (2017). Variability and global distribution of subgenotypes of bovine viral diarrhea virus. Viruses 9, 128. doi: 10.3390/v9060128
Keywords: Bovine Viral Diarrhea Virus, yak (Bos grunniens), prevalence, phylogenetic analysis, subgenotypes
Citation: Li Z, Han Y, Fu Y, Yuan Q, Wang S, Pan X, Xue W, Yin H, Gao S and Meng R (2025) Comprehensive molecular epidemiology of BVDV in yaks (Bos gruniens) in Qinghai, China: high prevalence and dominance of BVDV-1u. Front. Cell. Infect. Microbiol. 15:1652023. doi: 10.3389/fcimb.2025.1652023
Received: 23 June 2025; Accepted: 29 July 2025;
Published: 14 August 2025.
Edited by:
Wenliang Li, Jiangsu Academy of Agricultural Sciences (JAAS), ChinaReviewed by:
Chris Chase, South Dakota State University, United StatesFaxing Wu, China Animal Health and Epidemiology Center, China
Copyright © 2025 Li, Han, Fu, Yuan, Wang, Pan, Xue, Yin, Gao and Meng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shandian Gao, Z2Fvc2hhbmRpYW5AY2Fhcy5jbg==; Ru Meng, bXItMDUyMkAxNjMuY29t