 Jan Slunečko1
Jan Slunečko1 Rok Kogoj1
Rok Kogoj1 Samo Zakotnik1
Samo Zakotnik1 Alen Suljič1Nataša Knap1Martin Bosilj1
Alen Suljič1Nataša Knap1Martin Bosilj1 Franc Strle2
Franc Strle2 Tatjana Avšič-Županc1
Tatjana Avšič-Županc1 Petra Bogovič2*
Petra Bogovič2* Miša Korva1*
Miša Korva1*- 1Institute of Microbiology and Immunology, Faculty of Medicine, University of Ljubljana, Ljubljana, Slovenia
- 2Department of Infectious Diseases, University Medical Center Ljubljana, Ljubljana, Slovenia
Introduction: Blood culture is the cornerstone of microbiological diagnostics for patients with acute undifferentiated fever and no obvious localization of infection; however, up to 50% of cases remain undiagnosed. Infections caused by arboviruses, fastidious or even uncultivable bacteria, or parasites often go undiagnosed without the use of target-specific molecular methods. These are typically performed in a stepwise manner, increasing cost and delaying results. Metagenomic next-generation sequencing (mNGS) has recently gained recognition as a potential universal pathogen detection tool for such cases. Our study aimed to develop a streamlined mNGS workflow for simultaneous detection of intracellular and cell-free pathogens within a single sequencing library.
Methods: Total nucleic acid was isolated separately from 200 EDTA blood samples. The plasma isolate was processed with DNase, followed by the depletion of host ribosomal and messenger RNA, reverse transcription, and sequence-independent single primer amplification (SISPA). The whole blood isolate was only reverse transcribed, with no other pre-processing manipulation. Finally, the two fractions were combined prior to library preparation and sequencing using either Oxford Nanopore Technologies or Illumina. Following established bioinformatics analysis, we developed a mathematical ranking approach (ClinSeq score) that enabled quick identification of relevant pathogens in approximately one hour.
Results: The mNGS workflow reached 79.5% (159/200) overall sensitivity. For bacteria the sensitivity was 88.6% (70/79), DNA viruses, 66.7% (10/15) and for RNA viruses 73.8% (76/103). Pathogen detections by individual sequencing methods showed overall sensitivity of Illumina and ONT to be 80.0% (76/95) and 79.1% (83/105) respectively. The ClinSeq score correctly highlighted the pathogen in 126/200 (63.0%) samples effectively with a Cohen’s kappa (κ) agreement of 0.61 with manual analysis.
Conclusion: Developed comprehensive mNGS workflow detects a wide range of pathogens in patients with acute undifferentiated fever. The unified workflow improves sensitivity for intracellular bacteria and RNA viruses, reduces time, cost and complexity by eliminating the need for separate library preparations, enabling faster turnaround suitable for clinical settings. The ClinSeq score effectively differentiates true pathogen signals from background noise, reducing false positives and manual interpretation time. Overall, the workflow demonstrates flexible, and efficient pathogen detection, supporting its potential for clinical diagnostics and improved patient management.
1 Introduction
Clinical metagenomic next-generation sequencing (mNGS) is emerging as a powerful diagnostic tool for the diagnostics of undifferentiated fever, which can have numerous infectious or non-infectious causes (Fu et al., 2022). Contemporary microbiological laboratory diagnostic methods are able to identify a causative agent in up to 50% of patients with acute undifferentiated fever with no obvious localization of infection (Bleeker-Rovers et al., 2007). While up to 15% of these cases can be resolved by blood cultures (Foong et al., 2022), they are often limited by collection procedure issues i.e. one set instead of the recommended two or three, under- or overfilling, contamination (Doern et al., 2019; Khare et al., 2020), antibiotic treatment prior to blood drawing (Fabre et al., 2020; Lin et al., 2023; Zeng et al., 2025), and its inherent inability to detect bacteria with special atmosphere (microaerophilic) growth requirements, slow growing bacteria, intracellular bacteria, or biochemically inactive bacteria (Fenollar and Raoult, 2007; Lamy et al., 2016; Zeng et al., 2025). Parallelly, PCR tests are limited by target specificity, pathogen nucleic acid integrity and a priori suspicion of the causative agent (Batool and Galloway-Peña, 2023). In such cases, several recent studies have demonstrated the added value of mNGS (Hong et al., 2020; Hu et al., 2021; Lu et al., 2022; Qi et al., 2023; Zhou et al., 2023). Metagenomic NGS has been used effectively to uncover viral, bacterial and parasitic etiologies in patients with febrile illness (Jerome et al., 2019; Kandathil et al., 2024; Ashraf et al., 2025; Lai et al., 2025). However, the variety of protocols in terms of sample preparation, nucleic acid extraction strategies, sequencing depth, and data interpretation pipelines makes direct comparison difficult and the general application of mNGS to clinical diagnostics challenging (Korzeniewski et al., 2015; Marra et al., 2024). Most of the protocols developed so far focus exclusively on targeting viral pathogens, particularly in cases of febrile illness in returning travelers, where arboviruses are commonly suspected to be the cause of fever (Jerome et al., 2019; Kandathil et al., 2024). Conversely, studies that rely solely on DNA sequencing are inherently limited in their ability to detect RNA viruses. While some studies have attempted to address this issue and demonstrated the feasibility of sequencing both DNA and RNA pathogens simultaneously (Carbo et al., 2020; Lopez-Labrador et al., 2024), other studies have shown that separating the sample into two sub-samples, which are then processed separately, enhances pathogen detection (Lewandowska et al., 2017; Miller et al., 2019; Arroyo Mühr et al., 2021; Cebriá-Mendoza et al., 2021; Atkinson et al., 2023; Buddle et al., 2024; Lopez-Labrador et al., 2024). Although some studies have investigated a wide variety of clinical sample types, plasma is generally favored due to its lower human background (Lewandowska et al., 2017; Cheng et al., 2019; Niles et al., 2023; Lopez-Labrador et al., 2024). However, this approach reduces the sensitivity in detecting intracellular pathogens, such as Babesia sp., Ehrlichia sp. and Plasmodium sp (Cunha et al., 2008; Wass et al., 2018; Erdem et al., 2024). As preparing DNA/RNA in parallel and duplicating NGS libraries increases the costs of mNGS, sample pooling was investigated (Teufel and Sobetzko, 2022). However, this is only possible when large batches of clinical samples are processed at once, which rarely occur in diagnostics. Conversely, the bioinformatic processing of sequencing data is equally challenging. This involves setting the correct thresholds for the number of reads needed to distinguish between background noise, contamination and pathogens. The outcome of taxonomic classification is highly sensitive to variability across bioinformatic pipelines (Bosilj et al., 2024). The choice of reference databases, classification algorithms, quality filtering, and downstream processing steps can all influence the number of reads assigned to a given taxon. These factors are compounded by upstream variables, including the biological background of the sample, laboratory procedures and sequencing technology. Consequently, standardized pipelines are required, and the interpretation of processed data must be as unbiased as possible to establish reporting criteria capable of distinguishing true positives from false ones (Bosilj et al., 2024).
The aim of the study was to develop a streamlined mNGS workflow comprising of a wet-lab protocol, and dry-lab analysis for the detection of fastidious and uncultivable pathogens, enabling the simultaneous detection of cell-free and intracellular pathogens, within a single sequencing library. To facilitate laborious result analysis and interpretation, a data driven mathematical ranking approach (ClinSeq score) has been developed.
2 Materials and methods
2.1 Sample selection
A total of 200 EDTA blood samples collected from patients with acute undifferentiated fever (body temperature > 38.3 °C; > 3 days) and no obvious localization of infection were included in the validation of the developed mNGS workflow. As an additional inclusion criterion, the etiology of the disease had to be confirmed with validated molecular tests during routine diagnostics. Information regarding the molecular tests employed in this study can be found in Supplementary Material (Supplementary Table S1). The list of pathogens in individual samples was: fastidious bacteria (Anaplasma phagocytophilum, Bartonella quintana, Coxiella burnettii, Francisella tularensis, Leptospira sp., Neoehrlichia mikurensis, Capnocytophaga canimorsus), viruses with RNA genome (chikungunya virus (CHIKV), dengue virus (DENV), Dobrava virus (DOBV), Puumala virus (PUUV), tick-borne encephalitis virus (TBEV), yellow fever virus (YFV) and Zika virus (ZIKV)), viruses with DNA genome (cytomegalovirus (CMV), Epstein–Barr virus (EBV) and parvovirus B19 (PB19)) and parasites (Babesia sp. and Plasmodium falciparum). Detailed sample data can be found in the Supplementary Material (Supplementary Table S2).
2.2 Molecular detection assays
All molecular detections assays, except for EBV and CMV, are qualitative assays and the results are reported as positive or negative (Supplementary Table S1). Only for the purpose of this study, to compare relative abundance of pathogens, we retrieved cycle threshold (Ct) values from each assay. For EBV and CMV Ct values and quantitative values, expressed in international units per ml (IU/ml), were retrieved.
2.3 Sample processing
A total of 600 μl of blood sample per patient was retrieved from storage at −80 °C. Total nucleic acid (NA) was isolated separately from 300 μl of EDTA whole blood and from 300 μl of plasma using the TANBead OptiPure Viral Auto Plate kit (TANbead inc., Taoyuan City, Taiwan) on the Maelstrom 9600 instrument (TANbead inc., Taoyuan City, Taiwan). In both cases, the elution volume was 60 μl. Following NA isolation, only the plasma isolates underwent treatment with the TURBO DNA-free™ Kit (Thermo Fisher Scientific, Waltham, MA, USA). DNA content was measured using the Qubit dsDNA High Sensitivity Assay (Thermo Fisher Scientific, Waltham, MA, USA) on a Qubit 3.0 fluorometer (Thermo Fisher Scientific, Waltham, MA, USA). Depletion of host ribosomal RNA (rRNA) and globin messenger RNA (mRNA) was achieved using the QIAseq FastSelect -rRNA/Globin kit (QIAGEN, Hilden, Germany) at a 1:10 dilution of each QIAseq reagent (Ramachandran et al., 2022).
2.4 Random primer amplification
For complementary DNA (cDNA) generation and subsequent random primer amplification, we employed the sequence-independent, single-primer amplification (SISPA) protocol. Total NA isolated from whole blood was processed directly with the SISPA-A protocol without any pretreatment. DNase- and QIAseq-treated NA isolated from plasma was processed with both SISPA-A and SISPA-B (Reyes and Kim, 1991; Chrzastek et al., 2017; Moore et al., 2020). Briefly, double-stranded complementary DNA (ds-cDNA) was generated using a SISPA-A primer (5′-GTTTCCCAGTCACGATC-N9-3′). Amplification of the resulting ds-cDNA was performed by utilizing the barcoded SISPA-A primer and its complementary SISPA-B primer (5′-GTTTCCCAGTCACGATC-3′). The necessary purification steps were carried out using AMPure XP magnetic beads (Beckman Coulter, Brea, CA, USA) at a 1:1 ratio, according to the manufacturer’s instructions. Purified SISPA-A from whole blood and purified SISPA-B from plasma were then mixed at a 1:1 ratio prior to NGS library generation. The detailed protocol for both (plasma and whole blood) is available in the Supplementary Material (Supplementary Data S3).
2.5 NGS library preparation
Libraries were prepared using either the NexteraXT DNA library preparation kit (Illumina, San Diego, CA, USA) or Native Barcoding Kit v14 (Oxford Nanopore Technologies, Oxford, UK), according to the manufacturer’s instructions. The necessary purification steps were carried out using AMPure XP magnetic beads (Beckman Coulter, Brea, CA, USA). Final pool concentration was measured using the Qubit dsDNA High Sensitivity Assay (Thermo Fisher Scientific, Waltham, MA, USA) on a Qubit 3.0 instrument (Thermo Fisher Scientific, Waltham, MA, USA), and the fragment size was analyzed using the Agilent High Sensitivity DNA Kit on the Bioanalyzer 2100 (both Agilent Technologies, Santa Clara, CA, USA).
2.6 Sequencing
Samples prepared with the NexteraXT DNA library preparation kit (Illumina, San Diego, CA, USA) were sequenced on a NextSeq 500/550 HighOutput Kit v2.5 (300 cycles) cartridge (Illumina, San Diego, CA, USA) with a target of 5 million reads per sample. Samples prepared with Native Barcoding Kit v14 (Oxford Nanopore Technologies, Oxford, UK) were analyzed in batches of 20 samples on PromethION (R.10.4.1) flow cells (Oxford Nanopore Technologies, Oxford, UK) for a run time of 72 hours, which achieved a comparable yield. On both instruments, for each sequencing run, positive controls (a mixture of Equid alphaherpesvirus 1 and Equine arteritis virus) and negative controls (NA isolated from Nuclease-Free Water (QIAGEN, Hilden, Germany)) were added.
2.7 Bioinformatics
All bioinformatic analyses were performed with a pipeline developed in-house, as previously published (Bosilj et al., 2024). First, adapter sequences were removed using BBDuk (v.39.01) (Bushnell, 2014) and host depletion was performed using bowtie2 (v2.50) (Langmead and Salzberg, 2012) by mapping trimmed reads to the human genome (GRCh38). Read classification was performed with the KrakenUniq (v1.0.2) (Breitwieser et al., 2018) tool. KronaTools (v.2.8.1) (Ondov et al., 2011) and Pavian (v1.0) (Breitwieser and Salzberg, 2020) were used to visualize KrakenUniq results. Because the pathogens in question were previously confirmed with molecular tests, during manual analysis of results we considered one read as detected and mNGS-positive (mREV(+)). If no reads were detected for the target pathogen, the results were considered as mNGS-negative (mREV(-)).
2.8 Automated score-based result ranking: ClinSeq score
From the KrakenUniq tool output file, the number of classified reads per taxon and kmer count, duplicity, and coverage were incorporated into a scoring system (ClinSeq score; CS), which allows ranking the results (Equation 1) and is calculated as follows:
represents the normalized read counts per 10 million total reads, where w is the weight factor, which can be adjusted (in this study w = 2 was used), K is the kmer count, C is the kmer coverage, and D is the duplicity of kmers. Furthermore, the presence of individual pathogens was assessed on an intra-run, cross samples basis. Microorganisms detected in more than 90% of the samples in the same run were flagged as background noise (likely contaminants) but not excluded from analysis. In addition, further data polishing was performed based on the reads present in the negative control. The top five ClinSeq score ranks were classified as likely or unlikely true positive. The ClinSeq score uses an adaptive threshold of 0.6× standard deviation above the mean. The adaptive threshold was determined experimentally by iterative testing by balancing CS(+)/CS(-) with manual analysis mREV(+)/mREV(-) detections.
2.9 Statistical analysis
Differences between groups were evaluated using the Wilcoxon rank sum (Mann–Whitney) test. The differences were considered significant at p< 0.05. Cohen’s kappa was employed to calculate the agreement between manually analyzed mNGS results and the ClinSeq score.
3 Results
3.1 Clinical sensitivity of developed mNGS workflow
We developed a comprehensive mNGS workflow to detect a wide range of pathogens in patients with acute undifferentiated fever and no obvious localization of infection. First, to minimize the human background and increase the detection of RNA viruses, an EDTA blood sample was centrifuged to separate the plasma from the cells. After this, the plasma isolate was subjected to a series of treatment steps and amplified using the SISPA-B protocol. The whole blood isolate was left untreated and processed with the SISPA-A step. Finally, both fractions were mixed in equal volumes prior to NGS library construction. This strategy enhances the detection of DNA pathogens, particularly intracellular bacteria, and the treated plasma fraction guarantees amplification of RNA pathogens and minimization of the host background (Figure 1).
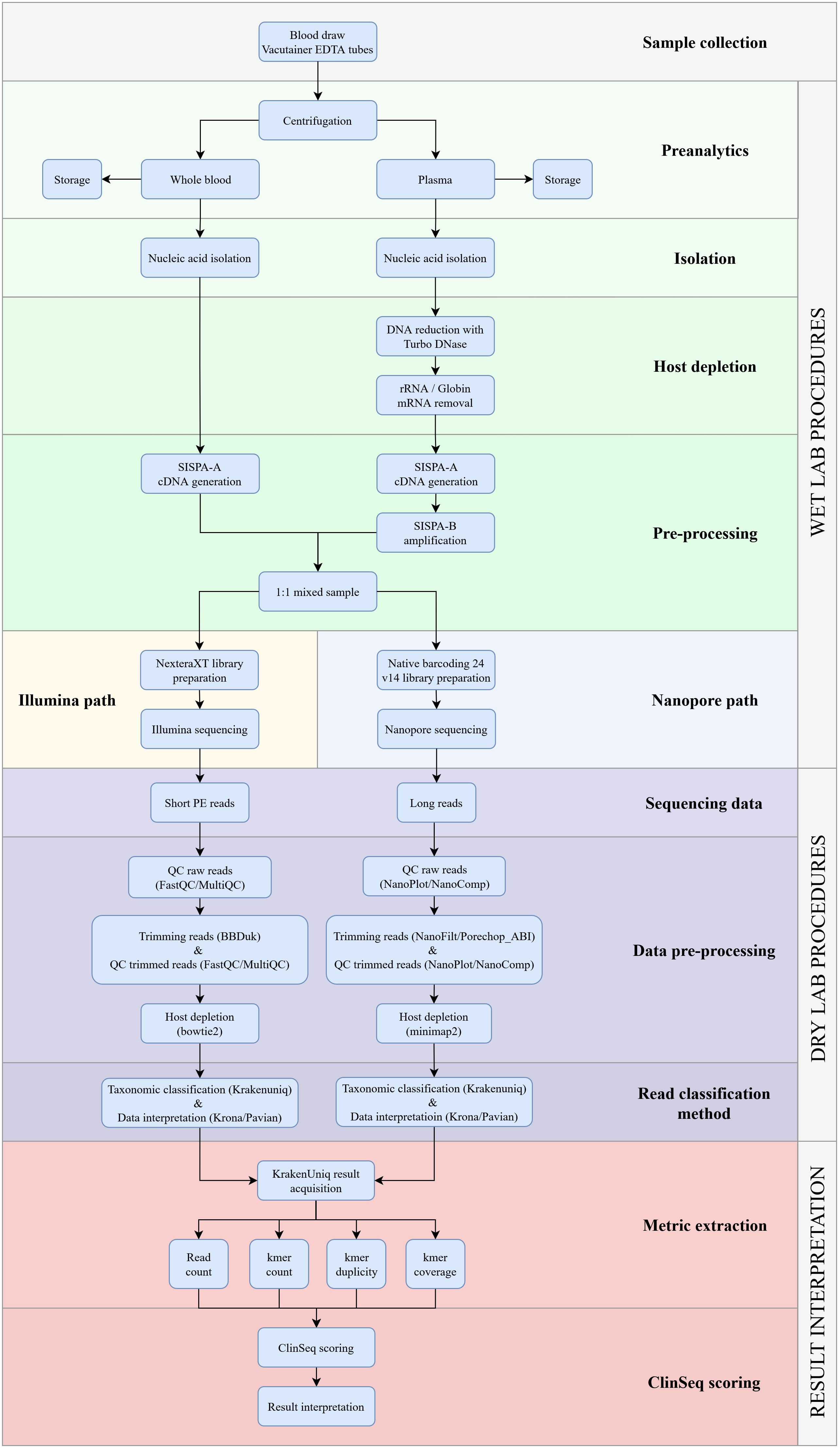
Figure 1. Detailed overview of the clinical metagenomics NGS workflow for identifying pathogens in EDTA blood from patients with undifferentiated fever. The workflow is divided into three sections: the laboratory protocol, the bioinformatics pipeline, and mNGS results analysis. The bioinformatics pipeline has been described in detail previously (Bosilj et al., 2024).
We sequenced 200 samples from patients with acute undifferentiated fever with known etiology either with the Illumina or ONT platforms. In total, the mNGS workflow identified pathogens in 159 of the PCR positive samples, corresponding to a sensitivity of 79.5% (159/200). The mean Ct value of mNGS positive samples was 24.5 (range: 6.2 to 33.5). On the other hand, the mean Ct value of samples for which a pathogen was not detected by mNGS results was 30.6 (range: 25.0–37.3; Figure 2). Unlike other pathogens where Ct values were used as semi-quantitative indicators, EBV and CMV were quantified using IU/mL. For CMV, detected samples had viral loads ranging from 2.39 × 10³ to 2.16 × 106 IU/mL (mean 5.59 × 105 IU/mL), whereas samples that were not detected by mNGS had uniformly lower loads of 1.97 × 10³ IU/mL. For EBV, detected samples had viral loads ranging from 2.94 × 106 to 1.81 × 107 IU/mL (mean 1.05 × 107 IU/mL), while mNGS negative samples ranged from 6.36 × 104 to 3.45 × 105 IU/mL (mean 1.62 × 105 IU/mL) (Table 1).
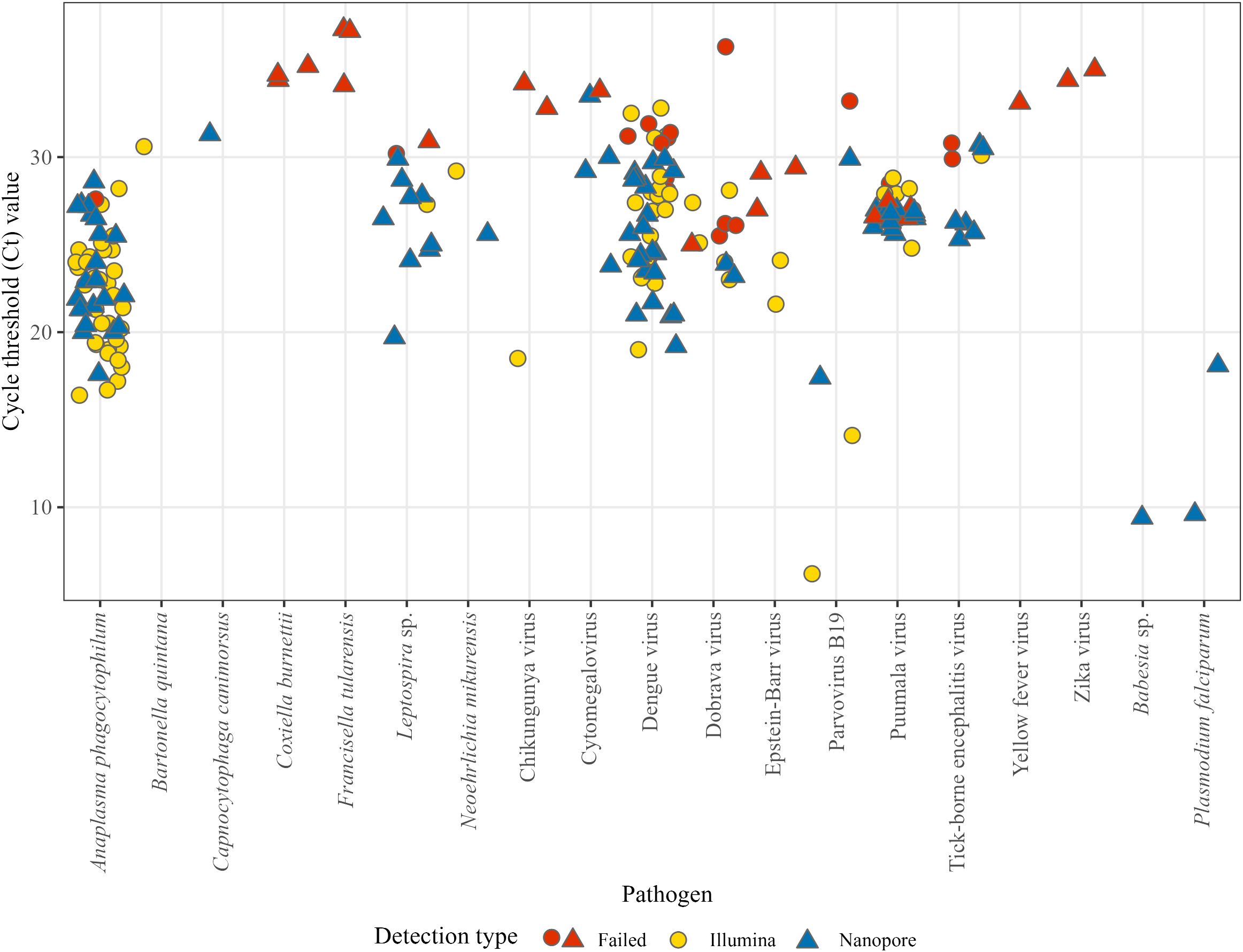
Figure 2. The performance of the developed mNGS workflow depending on the relative abundance of the pathogen, represented by Ct value from standardized in vitro diagnostic methods. Blue (n = 83) are mNGS positives with ONT, yellow (n = 76) are mNGS positives with Illumina and red (n = 41) represents failed mNGS detections.
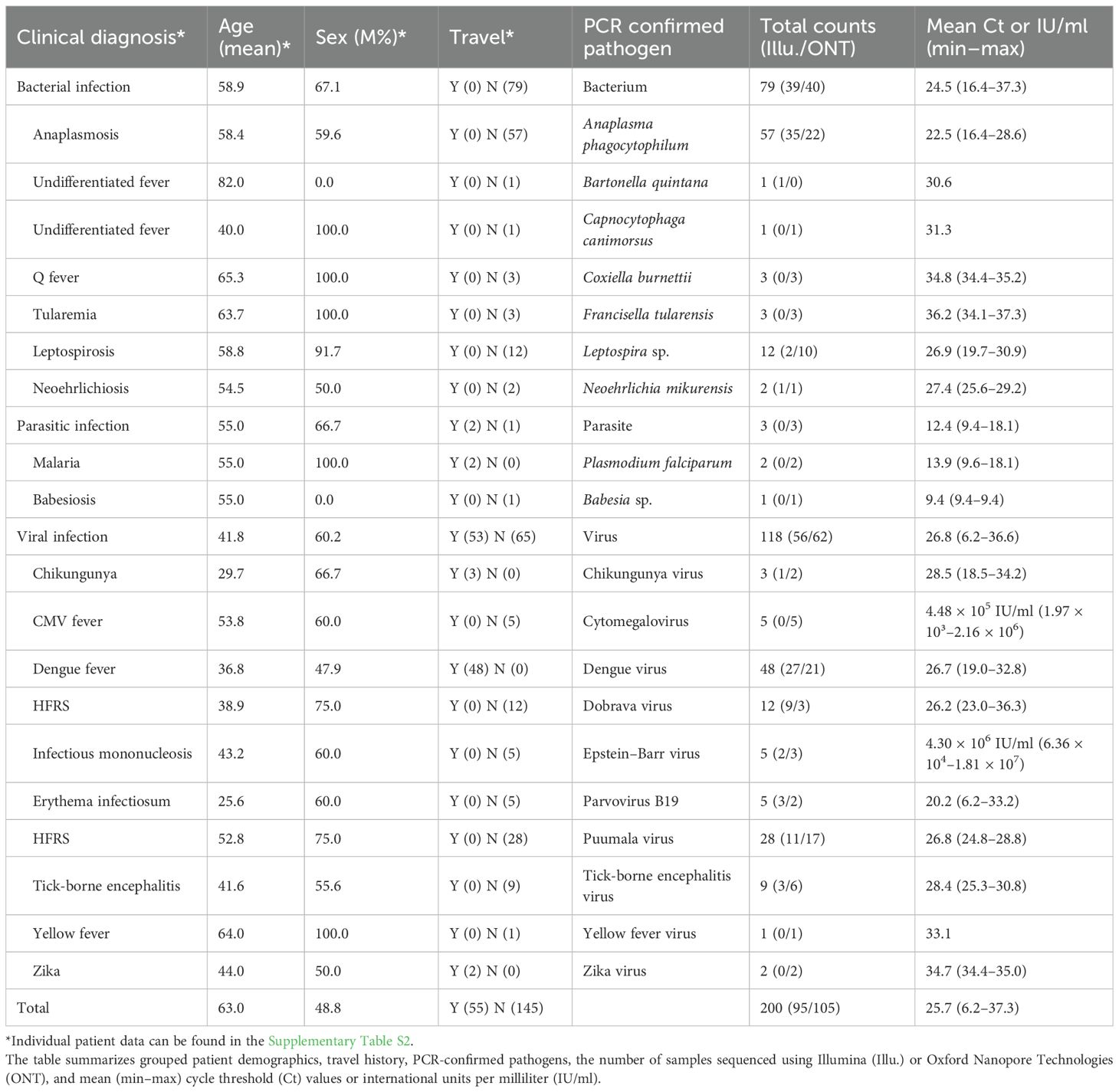
Table 1. List of included EDTA blood samples collected from patients with undifferentiated fever and molecularly confirmed etiology of the disease.
For bacteria, the sensitivity was 88.6% (70/79). Successful pathogen detections by mNGS were as follows: A. phagocytophilum (56/57), B. quintana, C. canimorsus, Leptospira sp. (10/12), and N. mikurensis. Besides individual failed detections, we couldn’t detect cases with C. burnettii and F. tularensis infections. Both samples with P. falciparum and the one with Babesia sp. were successfully detected with mNGS. For DNA viruses, we observed sensitivity of 66.7% (10/15), while for RNA viruses, the sensitivity was 73.8% (76/103). Successful viral detections by mNGS were CMV (4/5), EBV (2/5), PB19 (4/5), CKIHV (1/3), DENV (40/48), DOBV (7/12), PUUV (21/28), and TBEV (7/9). Besides individual failed detections, unsuccessful detections were also in one YFV case and both ZIKV cases. Analyzing pathogen detections by individual sequencing methods, we observed the overall sensitivity of Illumina and ONT to be 80.0% (76/95) and 79.1% (83/105) respectively.
3.2 Automated ClinSeq score
With ClinSeq score we detected the pathogen in 126/200 samples, resulting in 63.0% sensitivity. The comparative evaluation of ClinSeq score and manual analysis showed an overall percent agreement of 83.5% (95% CI; 77.6%–88.4%), a positive percent agreement of 79.2% (95% CI; 72.1%–85.3%), a negative percent agreement of 100% (95% CI; 91.4%–100%), and a κ value of 0.61 (95% CI; 0.49–0.73), which reflects a substantial agreement between the two methods (Table 2).
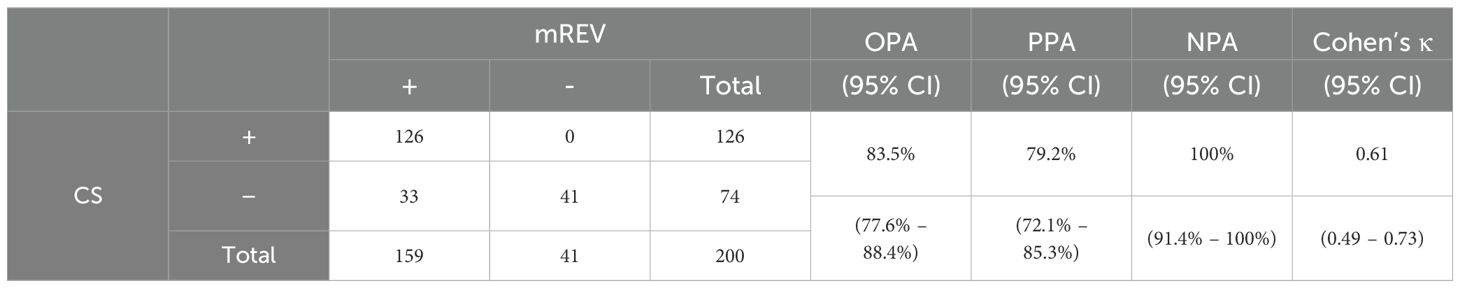
Table 2. Comparison of mNGS manual analysis (mREV) and ClinSeq (CS) score, with calculated overall percent agreement (OPA), positive percent agreement (PPA), negative percent agreement (NPA), and Cohen’s kappa (κ).
We investigated the reason for missed detections in the ClinSeq score (CS(-) | mREV(+)). When we compared the mean Ct between CS(-) and CS(+) samples, we observed a ΔCt of 2.8. This translated also to pathogen specific read counts, where we observed a statistically significant difference between CS(-) and CS(+) in normalized read counts, kmer count, duplicity and coverage (p< 0.001; Figure 3).
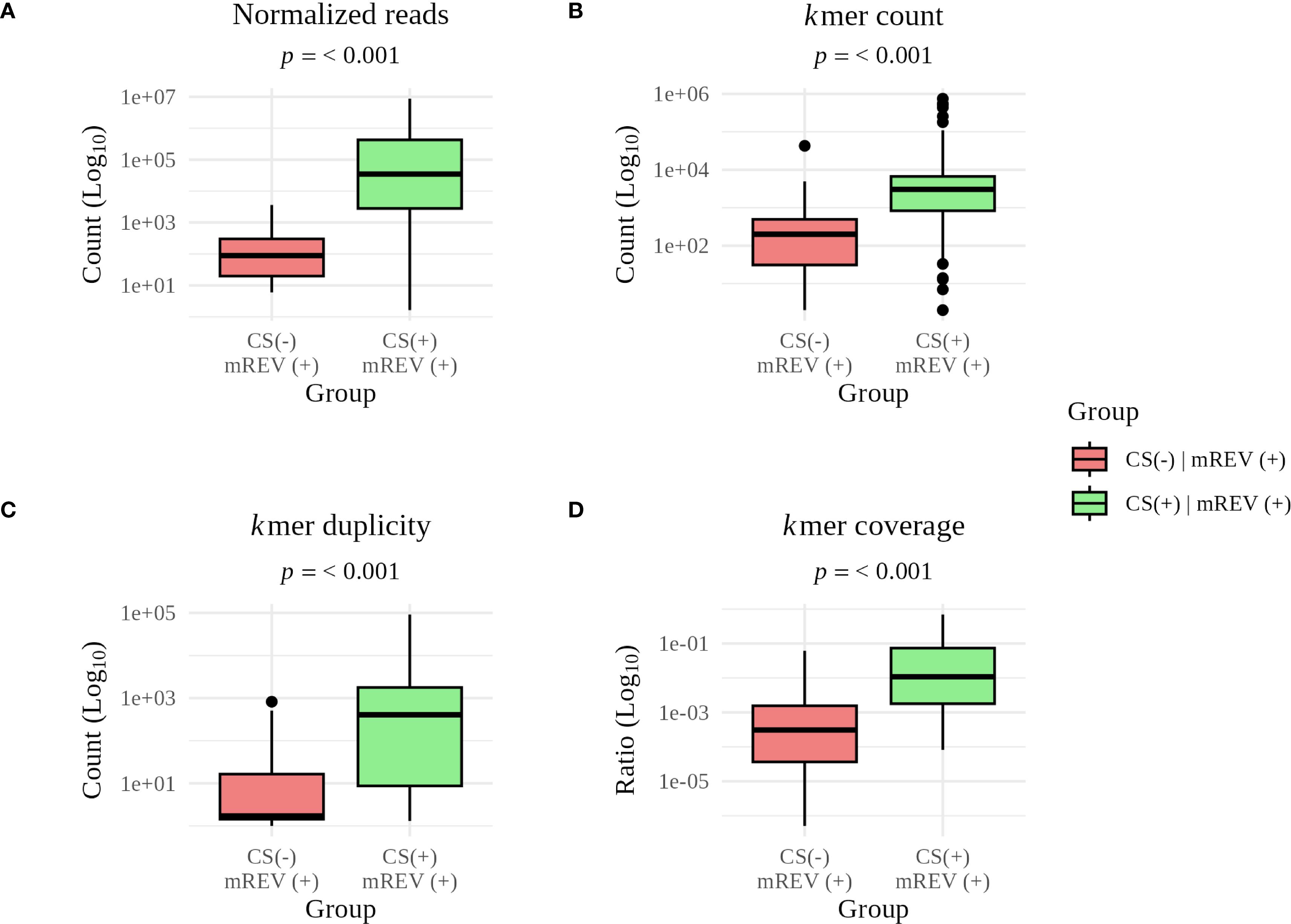
Figure 3. Comparison of individual metrics for concordant and discordant results between the ClinSeq score (CS) and manual analysis (mREV). Statistically significant differences are pathogen specific (A) read count (p = 1.9 × 10-14), (B) kmer count (p = 4.1 × 10-9), (C) kmer duplicity (p = 9.4 × 10-10), and (D) kmer coverage (p = 1.5 × 10-11). The long whiskers reflect the natural variability in read counts, kmer content, duplicity, and coverage within individual groups.
4 Discussion
The primary goal of this study was to develop a comprehensive mNGS workflow that maximizes pathogen detection while minimizing background noise, reducing costs, and ensuring a rapid turnaround time suitable for clinical applications. We developed an innovative approach to pre-processing EDTA blood samples, beginning with the separation of the sample into whole blood and plasma. These fractions undergo tailored pretreatment processes to optimize the enrichment of pathogen nucleic acids. Whole blood enables the detection of intracellular pathogens, including DNA viruses, intracellular bacteria and parasites. Conversely, the plasma fraction is processed separately to amplify pathogens with RNA genome. Because the fractions are mixed in equal volumes prior to library preparation, a reduction in hands-on time, turnaround time, cost, and complexity is achieved.
Overall, the developed mNGS workflow demonstrated a sensitivity of 79.5% (159/200) in comparison to conventional molecular diagnostic approach. This sensitivity is consistent with studies comparing blood pathogen detection with mNGS to droplet digital PCR (Hu et al., 2021) and to qPCR (Lu et al., 2022). The majority of standardized molecular methods for in vitro diagnostics are qualitative, so absolute quantification of the pathogen genome is not possible. However, the reported Ct values provide a relative estimation of the abundance of the pathogen (Kogoj et al., 2022). Bearing these limitations in mind, we compared the Ct values of mNGS-positive and -negative samples, observing that mNGS detection is less consistent when Ct > 30, a finding comparable to other studies (Gauthier et al., 2021, 2024; Koh et al., 2023; Pichler et al., 2023; Kandathil et al., 2024). Nevertheless, eleven samples with Ct > 30 in our dataset still yielded a reliable pathogen detection with mNGS. This indicates that other parameters also influence the success of detection. These include sample composition, nucleic acid integrity, pathogen type, amplification efficiency and biases introduced during procedures, such as the SISPA protocol (Bustin and Mueller, 2005; Rao et al., 2020). While the detection of potential pathogens can be improved with higher sequencing depth (Resman Rus et al., 2025), this however, comes at a higher cost, as a lower number of samples can be included per run.
Besides the challenge of sequencing the samples with a low pathogen load, they are equally fastidious for bioinformatics analysis, as their pathogen-to-background reads ratio is low. In the absence of standardized detection thresholds in such cases, interpretation becomes subjective and prone to bias. Although defining consistent thresholds is essential for objective data interpretation, the optimal cut-off point often varies depending on the pathogen and between sequencing runs, which presents a major analytical challenge. For this reason, we have developed an automated, score-based result ranking system called the ClinSeq score. This is a mathematical, data-driven algorithm that adapts to each sequencing run. Unlike fixed thresholds, the ClinSeq score sets an adaptive threshold defined as the mean read count plus 0.6× standard deviation for each classified taxon. Although manual mNGS analysis was ultimately superior to the ClinSeq score (79.5% versus 63.0%), the clinical microbiology interpretation was achieved in just under an hour after completing the bioinformatic workflow for 126 samples. Manual analysis, on the other hand, took between 30 minutes and 1 hour per sample, depending on the sample complexity and intra-run variability. Furthermore, as the ClinSeq score produced no false positive results, the manual analysis could focus solely on CS(-) samples, reducing labor and time. A similar mathematical algorithm was also developed by Guellil et al. and further modified by Borry (Guellil et al., 2022; Borry, 2022). However, when applied to our dataset both yielded lower sensitivity of 22.0% and 12.0%, respectively. While the PPA (79.2%) of our mNGS pipeline might not yet be considered as an excellent result and therefore suitable for replacing current microbiological methods, it is comparable to available data as summarized by Liu et al (Liu et al., 2025). Nevertheless, the benefits of the current mNGS pipeline are most prominent in the ability to detect pathogens which cannot be identified in blood cultures or need initial suspicion from the clinician.
5 Conclusion
In conclusion, the developed mNGS workflow demonstrates reliable and comprehensive detection of a wide range of pathogens, including uncultivable bacteria, viruses, and parasites, with promising sensitivity and efficiency. The innovative approach of combining plasma and whole blood samples prior to library preparation enhances detection capabilities while streamlining processing, reducing costs, and enabling flexible sequencing options. The implementation of a mathematical ranking method further improves interpretative efficiency. These findings provide a strong foundation for integrating mNGS into routine diagnostic settings. Future studies will focus on validating the workflow across additional sample types, as well as refining the analytic methods to optimize clinical utility.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/bioproject, BioProject ID: PRJNA1275620.
Ethics statement
The study was performed retrospectively, and no additional sample was taken for the purpose of the study. The use of archived samples, from the Institute of Microbiology and Immunology, Faculty of Medicine, was approved by the National Medical Ethics Committee of the Republic of Slovenia, which waived the need for additional written informed consent due to anonymized use of data, no use of demographic or any other patient data and retrospective analysis with no impact on patient care (ethical approval no. 0120-253/2023/3). The study was conducted according to the Declaration of Helsinki and the Patient Rights Law of the Republic of Slovenia.
Author contributions
JS: Investigation, Methodology, Visualization, Conceptualization, Validation, Writing – original draft, Writing – review & editing, Formal Analysis. RK: Conceptualization, Writing – review & editing, Writing – original draft, Investigation, Formal Analysis, Validation, Methodology. SZ: Validation, Conceptualization, Methodology, Writing – review & editing, Formal Analysis, Writing – original draft, Software, Investigation. AS: Formal Analysis, Visualization, Data curation, Writing – review & editing, Software, Methodology. NK: Conceptualization, Methodology, Investigation, Writing – review & editing. MB: Writing – review & editing, Methodology, Formal Analysis, Software. FS: Funding acquisition, Writing – review & editing, Supervision, Resources, Conceptualization. TA-Ž: Supervision, Funding acquisition, Resources, Writing – review & editing, Conceptualization. PB: Writing – review & editing, Supervision, Conceptualization, Project administration, Resources. MK: Validation, Methodology, Project administration, Formal Analysis, Supervision, Conceptualization, Investigation, Funding acquisition, Writing – review & editing, Resources.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the Slovenian Research and Innovation Agency (ARIS; grants P3-0083, P3-0296, J3-50101), a tertiary project of the University Medical Center Ljubljana (grant number 20230154), and the Network of Infrastructure Centers of the University of Ljubljana (MRIC-UL-IC-BSL3+, grant number IP-022). The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Acknowledgments
We thank Mateja Jelovšek, Martin Sagadin, Doroteja Vlaj, and Patricija Pozvek for their technical support. The authors would like to thank Tina Triglav, MD, PhD, specialist in clinical microbiology, for her extensive help with interpreting and discussing the bacterial mNGS results.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2025.1667422/full#supplementary-material
References
Arroyo Mühr, L. S., Dillner, J., Ure, A. E., Sundström, K., and Hultin, E. (2021). Comparison of DNA and RNA sequencing of total nucleic acids from human cervix for metagenomics. Sci. Rep. 11, 18852. doi: 10.1038/s41598-021-98452-4
Ashraf, S., Jerome, H., Bugembe, D. L., Ssemwanga, D., Byaruhanga, T., Kayiwa, J. T., et al. (2025). Uncovering the viral aetiology of undiagnosed acute febrile illness in Uganda using metagenomic sequencing. Nat. Commun. 16, 1–12. doi: 10.1038/s41467-025-57696-8
Atkinson, L., Lee, J. C., Lennon, A., Shah, D., Storey, N., Morfopoulou, S., et al. (2023). Untargeted metagenomics protocol for the diagnosis of infection from CSF and tissue from sterile sites. Heliyon 9, e19854. doi: 10.1016/J.HELIYON.2023.E19854
Batool, M. and Galloway-Peña, J. (2023). Clinical metagenomics-challenges and future prospects. Front. Microbiol. 14. doi: 10.3389/FMICB.2023.1186424
Bleeker-Rovers, C. P., Vos, F. J., De Kleijn, E. M. H. A., Mudde, A. H., Dofferhoff, T. S. M., Richter, C., et al. (2007). A prospective multicenter study on fever of unknown origin: The yield of a structured diagnostic protocol. Medicine 8, 26–38. doi: 10.1097/MD.0B013E31802FE858
Borry, M. (2022). A new E-score for KrakenUniq. Available online at: https://maximeborry.com/post/kraken-uniq/ (Accessed May 21, 2025).
Bosilj, M., Suljič, A., Zakotnik, S., Slunečko, J., Kogoj, R., and Korva, M. (2024). MetaAll: integrative bioinformatics workflow for analysing clinical metagenomic data. Brief Bioinform. 25, bbae597. doi: 10.1093/BIB/BBAE597
Breitwieser, F. P., Baker, D. N., and Salzberg, S. L. (2018). KrakenUniq: Confident and fast metagenomics classification using unique k-mer counts. Genome Biol. 19, 198. doi: 10.1186/S13059-018-1568-0
Breitwieser, F. P. and Salzberg, S. L. (2020). Pavian: interactive analysis of metagenomics data for microbiome studies and pathogen identification. Bioinformatics 36, 1303–1304. doi: 10.1093/BIOINFORMATICS/BTZ715
Buddle, S., Forrest, L., Akinsuyi, N., Martin Bernal, L. M., Brooks, T., Venturini, C., et al. (2024). Evaluating metagenomics and targeted approaches for diagnosis and surveillance of viruses. Genome Med. 16, 111. doi: 10.1186/S13073-024-01380-X
Bushnell, B. (2014). BBMap: A fast, accurate, splice-aware aligner. Available online at: https://www.osti.gov/biblio/1241166 (Accessed May 21, 2025).
Bustin, S. A. and Mueller, R. (2005). Real-time reverse transcription PCR (qRT-PCR) and its potential use in clinical diagnosis. Clin. Sci. 109, 365–379. doi: 10.1042/CS20050086
Carbo, E. C., Buddingh, E. P., Karelioti, E., Sidorov, I. A., Feltkamp, M. C. W., von dem Borne, P. A., et al. (2020). Improved diagnosis of viral encephalitis in adult and pediatric hematological patients using viral metagenomics. J. Clin. Virol. 130, 104566. doi: 10.1016/j.jcv.2020.104566
Cebriá-Mendoza, M., Arbona, C., Larrea, L., Díaz, W., Arnau, V., Peña, C., et al. (2021). Deep viral blood metagenomics reveals extensive anellovirus diversity in healthy humans. Sci. Rep. 11, 6921. doi: 10.1038/S41598-021-86427-4
Cheng, A. P., Burnham, P., Lee, J. R., Cheng, M. P., Suthanthiran, M., Dadhania, D., et al. (2019). A cell-free DNA metagenomic sequencing assay that integrates the host injury response to infection. Proc. Natl. Acad. Sci. U.S.A. 116, 18738–18744. doi: 10.1073/pnas.1906320116
Chrzastek, K., Lee, D., Smith, D., Sharma, P., Suarez, D. L., Pantin-Jackwood, M., et al. (2017). Use of Sequence-Independent, Single-Primer-Amplification (SISPA) for rapid detection, identification, and characterization of avian RNA viruses. Virology 509, 159–166. doi: 10.1016/J.VIROL.2017.06.019
Cunha, B. A., Cohen, Y. Z., and McDermott, B. (2008). Fever of unknown origin (FUO) due to babesiosis in a immunocompetent host. Heart Lung 37, 481–484. doi: 10.1016/j.hrtlng.2008.01.003
Doern, G. V., Carroll, K. C., Diekema, D. J., Garey, K. W., Rupp, M. E., Weinstein, M. P., et al. (2019). Practical guidance for clinical microbiology laboratories: A comprehensive update on the problem of blood culture contamination and a discussion of methods for addressing the problem. Clin. Microbiol. Rev. 33, e00009–e00019. doi: 10.1128/CMR.00009-19
Erdem, H., Al-Tawfiq, J. A., Abid, M., Yahia, W.B., Akafity, G., Ramadan, M. E., et al. (2024). Infectious causes of fever of unknown origin in developing countries: An international ID-IRI study. J. Intensive Med. 4, 94–100. doi: 10.1016/j.jointm.2023.07.004
Fabre, V., Sharara, S. L., Salinas, A. B., Carroll, K. C., Desai, S., and Cosgrove, S. E. (2020). Does this patient need blood cultures? A scoping review of indications for blood cultures in adult nonneutropenic inpatients. Clin. Infect. Dis. 71, 1339–1347. doi: 10.1093/CID/CIAA039
Fenollar, F. and Raoult, D. (2007). Molecular diagnosis of bloodstream infections caused by non-cultivable bacteria. Int. J. Antimicrob. Agents 30, 7–15. doi: 10.1016/J.IJANTIMICAG.2007.06.024
Foong, K. S., Munigala, S., Kern-Allely, S., and Warren, D. K. (2022). Blood culture utilization practices among febrile and/or hypothermic inpatients. BMC Infect. Dis. 22, 779. doi: 10.1186/S12879-022-07748-X
Fu, Z. F., Zhang, H. C., Zhang, Y., Cui, P., Zhou, Y., Wang, H. Y., et al. (2022). Evaluations of clinical utilization of metagenomic next-generation sequencing in adults with fever of unknown origin. Front. Cell Infect. Microbiol. 11. doi: 10.3389/FCIMB.2021.745156
Gauthier, N. P. G., Chan, W., Locher, K., Smailus, D., Coope, R., Charles, M., et al. (2024). Validation of an automated, end-to-end metagenomic sequencing assay for agnostic detection of respiratory viruses. J. Infect. Dis. 230, e1245–e1253. doi: 10.1093/infdis/jiae226
Gauthier, N. P. G., Nelson, C., Bonsall, M. B., Locher, K., Charles, M., MacDonald, C., et al. (2021). Nanopore metagenomic sequencing for detection and characterization of SARS-CoV-2 in clinical samples. PloS One 16, e0259712. doi: 10.1371/JOURNAL.PONE.0259712
Guellil, M., Keller, M., Dittmar, J. M., Inskip, S. A., Cessford, C., Solnik, A., et al. (2022). An invasive Haemophilus influenzae serotype b infection in an Anglo-Saxon plague victim. Genome Biol. 23, 22. doi: 10.1186/S13059-021-02580-Z
Hong, N. T. T., Anh, N. T., Mai, N. T. H., Nghia, H. D. T., Nhu, L. N. T., Thanh, T. T., et al. (2020). Performance of metagenomic next-generation sequencing for the diagnosis of viral meningoencephalitis in a resource-limited setting. Open Forum Infect. Dis. 7, ofaa046. doi: 10.1093/OFID/OFAA046
Hu, B., Tao, Y., Shao, Z., Zheng, Y., Zhang, R., Yang, X., et al. (2021). A comparison of blood pathogen detection among droplet digital PCR, metagenomic next-generation sequencing, and blood culture in critically ill patients with suspected bloodstream infections. Front. Microbiol. 12. doi: 10.3389/FMICB.2021.641202
Jerome, H., Taylor, C., Sreenu, V. B., Klymenko, T., Filipe, A. D. S., Jackson, C., et al. (2019). Metagenomic next-generation sequencing aids the diagnosis of viral infections in febrile returning travellers. J. Infection 79, 383–388. doi: 10.1016/j.jinf.2019.08.003
Kandathil, A. J., Blair, P. W., Lu, J., Anantharam, R., Kobba, K., Robinson, M. L., et al. (2024). Metagenomic next generation sequencing of plasma RNA for diagnosis of unexplained, acute febrile illness in Uganda. PloS Negl. Trop. Dis. 18, e0012451. doi: 10.1371/JOURNAL.PNTD.0012451
Khare, R., Kothari, T., Castagnaro, J., Hemmings, B., Tso, M., and Juretschko, S. (2020). Active monitoring and feedback to improve blood culture fill volumes and positivity across a large integrated health system. Clin. Infect. Dis. 70, 262–268. doi: 10.1093/CID/CIZ198
Kogoj, R., Korva, M., Knap, N., Rus, K. R., Pozvek, P., Avšič-županc, T., et al. (2022). Comparative evaluation of six SARS-coV-2 real-time RT-PCR diagnostic approaches shows substantial genomic variant-dependent intra- and inter-test variability, poor interchangeability of cycle threshold and complementary turn-around times. Pathogens 11, 462. doi: 10.3390/PATHOGENS11040462
Koh, W. L. C., Poh, S. E., Lee, C. K., Chan, T. H. M., Yan, G., Kong, K. W., et al. (2023). Towards a rapid-turnaround low-depth unbiased metagenomics sequencing workflow on the illumina platforms. Bioengineering 10, 520. doi: 10.3390/BIOENGINEERING10050520/S1
Korzeniewski, K., Gaweł, B., Krankowska, D., and Wasilczuk, K. (2015). Fever of unknown origin in returning travellers. Int. Marit. Health 66, 77–83. doi: 10.5603/IMH.2015.0019
Lai, L. M., Chen, Q. G., Liu, Y., Zhao, R., Cao, M. L., and Yuan, L. (2025). The value of metagenomic next-generation sequencing in the diagnosis of fever of unknown origin. Sci. Rep. 15, 1–10. doi: 10.1038/s41598-025-86295-2
Lamy, B., Dargère, S., Arendrup, M. C., Parienti, J. J., and Tattevin, P. (2016). How to optimize the use of blood cultures for the diagnosis of bloodstream infections? A state-of-the art. Front. Microbiol. 7. doi: 10.3389/FMICB.2016.00697
Langmead, B. and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lewandowska, D. W., Zagordi, O., Geissberger, F. D., Kufner, V., Schmutz, S., Böni, J., et al. (2017). Optimization and validation of sample preparation for metagenomic sequencing of viruses in clinical samples. Microbiome 5, 94. doi: 10.1186/S40168-017-0317-Z
Lin, K. P., Yeh, T. K., Chuang, Y. C., Wang, L. A., Fu, Y. C., and Liu, P. Y. (2023). Blood culture negative endocarditis: A review of laboratory diagnostic approaches. Int. J. Gen. Med. 16, 317. doi: 10.2147/IJGM.S393329
Liu, C., Song, X., Liu, J., Zong, L., Xu, T., Han, X., et al. (2025). Consistency between metagenomic next-generation sequencing versus traditional microbiological tests for infective disease: systemic review and meta-analysis. Crit. Care 29, 55. doi: 10.1186/S13054-025-05288-9
Lopez-Labrador, F. X., Huber, M., Sidorov, I. A., Brown, J. R., Cuypers, L., Laenen, L., et al. (2024). Multicenter benchmarking of short and long read wet lab protocols for clinical viral metagenomics. J. Clin. Virol. 173, 105695. doi: 10.1016/j.jcv.2024.105695
Lu, H., Ma, L., Zhang, H., Feng, L., Yu, Y., Zhao, Y., et al. (2022). The comparison of metagenomic next-generation sequencing with conventional microbiological tests for identification of pathogens and antibiotic resistance genes in infectious diseases. Infect. Drug Resist. 15, 6115–6128. doi: 10.2147/IDR.S370964
Marra, A. R., Lopes, G. O. V., Pardo, I., Hsieh, M. K., Kobayashi, T., Marra, P. S., et al. (2024). Metagenomic next-generation sequencing in patients with fever of unknown origin: A comprehensive systematic literature review and meta-analysis. Diagn. Microbiol. Infect. Dis. 110, 116465. doi: 10.1016/j.diagmicrobio.2024.116465
Miller, S., Naccache, S. N., Samayoa, E., Messacar, K., Arevalo, S., Federman, S., et al. (2019). Laboratory validation of a clinical metagenomic sequencing assay for pathogen detection in cerebrospinal fluid. Genome res. 29, 831–842. doi: 10.1101/gr.238170.118
Moore, S. C., Penrice-Randal, R., Alruwaili, M., Randle, N., Armstrong, S., Hartley, C., et al. (2020). Amplicon-based detection and sequencing of SARS-coV-2 in nasopharyngeal swabs from patients with COVID-19 and identification of deletions in the viral genome that encode proteins involved in interferon antagonism. Viruses 12, 1164. doi: 10.3390/V12101164
Niles, D. T., Lee, R. A., Lamb, G. S., Al Dhaheri, F., and Boguniewicz, J. (2023). Plasma cell-free metagenomic next generation sequencing in the clinical setting for the diagnosis of infectious diseases: a systematic review and meta-analysis. Diagn. Microbiol. Infect. Dis. 105, 115838. doi: 10.1016/j.diagmicrobio.2022.115838
Ondov, B. D., Bergman, N. H., and Phillippy, A. M. (2011). Interactive metagenomic visualization in a Web browser. BMC Bioinf. 12, 385. doi: 10.1186/1471-2105-12-385
Pichler, I., Schmutz, S., Ziltener, G., Zaheri, M., Kufner, V., Trkola, A., et al. (2023). Rapid and sensitive single-sample viral metagenomics using Nanopore Flongle sequencing. J. Virol. Methods 320, 114784. doi: 10.1016/j.jviromet.2023.114784
Qi, Y., Lin, W. Q., Liao, B., Chen, J. W., and Chen, Z. S. (2023). Blood plasma metagenomic next-generation sequencing for identifying pathogens of febrile neutropenia in acute leukemia patients. Sci. Rep. 13, 1–8. doi: 10.1038/s41598-023-47685-6
Ramachandran, P. S., Ramesh, A., Creswell, F. V., Wapniarski, A., Narendra, R., Quinn, C. M., et al. (2022). Integrating central nervous system metagenomics and host response for diagnosis of tuberculosis meningitis and its mimics. Nat. Commun. 13, 1675. doi: 10.1038/s41467-022-29353-x
Rao, S. N., Manissero, D., Steele, V. R., and Pareja, J. (2020). A narrative systematic review of the clinical utility of cycle threshold values in the context of COVID-19. Infect. Dis. Ther. 9, 573–586. doi: 10.1007/s40121-020-00324-3
Resman Rus, K., Bosilj, M., Triglav, T., Jereb, M., Zalaznik, M., Klešnik, M., et al. (2025). Metagenomic sequencing for diagnosing listeria-induced rhombencephalitis in patient and contaminated cheese samples: A case report. Int. J. Mol. Sci. 26, 655. doi: 10.3390/IJMS26020655
Reyes, G. R. and Kim, J. P. (1991). Sequence-independent, single-primer amplification (SISPA) of complex DNA populations. Mol. Cell Probes 5, 473–481. doi: 10.1016/S0890-8508(05)80020-9
Teufel, M. and Sobetzko, P. (2022). Reducing costs for DNA and RNA sequencing by sample pooling using a metagenomic approach. BMC Genomics 23, 613. doi: 10.1186/S12864-022-08831-Y
Wass, L., Grankvist, A., Mattsson, M., Gustafsson, H., Krogfelt, K., Olsen, B., et al. (2018). Serological reactivity to Anaplasma phagocytophilum in neoehrlichiosis patients. Eur. J. Clin. Microbiol. Infect. Dis. 37, 1673–1678. doi: 10.1007/s10096-018-3298-3
Zeng, X., Lloyd, K. M., Hamdy, R. F., Shapiro, C. A., Fisher, M. A., Lin, J., et al. (2025). Identification and characterization of an invasive, hyper-aerotolerant Campylobacter jejuni strain causing bacteremia in a pediatric leukemia patient. ASM Case Rep. 1, e00060–e00024. doi: 10.1128/ASMCR.00060-24
Keywords: mNGS, clinical metagenomics, molecular diagnostics, universal pathogen detection, enhanced RNA virus detection
Citation: Slunečko J, Kogoj R, Zakotnik S, Suljič A, Knap N, Bosilj M, Strle F, Avšič-Županc T, Bogovič P and Korva M (2025) Development and performance evaluation of a clinical metagenomics approach for identifying pathogens in the whole blood from patients with undifferentiated fever. Front. Cell. Infect. Microbiol. 15:1667422. doi: 10.3389/fcimb.2025.1667422
Received: 16 July 2025; Accepted: 27 August 2025;
Published: 15 September 2025.
Edited by:
Stefano Marletta, University of Verona, ItalyReviewed by:
Benjamin M. Liu, George Washington University, United StatesVladimir Gostev, Children’s Scientific Clinical Centre for Infectious Diseases, Russia
Copyright © 2025 Slunečko, Kogoj, Zakotnik, Suljič, Knap, Bosilj, Strle, Avšič-Županc, Bogovič and Korva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miša Korva, bWlzYS5rb3J2YUBtZi51bmktbGouc2k=; Petra Bogovič, cGV0cmEuYm9nb3ZpY0BrY2xqLnNp