 Wentao Zhu
Wentao Zhu Xi Chen
Xi Chen- Department of Infectious Diseases and Clinical Microbiology, Beijing Institute of Respiratory Medicine and Beijing Chao-Yang Hospital, Capital Medical University, Beijing, China
Background: Serratia spp. is an important nosocomial pathogen, with increasing threat of antimicrobial resistance. We aimed to describe the population structure, antimicrobial resistance and dissemination of Serratia isolates in ICUs of China.
Methods: Serratia spp. were isolated from patients admitted to ICUs of a large hospital between January 2014 and December 2024. Whole-genome and clinical data were collected to identify their epidemiological and evolutionary characteristics.
Results: 106 Serratia isolates was divided into five species based on phylogenomic and ANI analyses, namely S. sarumanii, S. ureilytica, S. marcescens, S. bockelmannii, and S. nevei. The predominant ST was ST595 (12.3%), followed by ST525 (10.4%) and ST428 (4.7%), all of which belonged to S. sarumanii. Based on a 16 SNPs threshold, 15 distinct clusters and 44 singleton strains were identified, with the largest cluster circulating in five different ICUs over the past 11 years. Notably, most grouped isolates within each cluster were isolated from different ICUs, indicative of potential inter-ICU transmission. The unique genes significantly enriched within each species contributed to their niche adaptation and plasticity. Various beta-lactamase genes, such as blaCTX-M and blaOXA, were detected, along with carbapenemase genes including blaKPC-2 and blaNDM-5 in nine isolates.
Conclusion: These results contribute to understanding the population structure and dissemination of Serratia spp. in ICUs, highlighting their ongoing evolution towards increasing resistance and outbreak potential.
Introduction
Serratia is a genus of Gram-negative, rod-shaped bacteria belonging to the family Yersiniaceae in the order Enterobacteriales (Adeolu et al., 2016). Serratia is considered to be ubiquitous in diverse environments, including water, soil, plants, and insects (Ono et al., 2022). However, it is not a common component of the human fecal microbiome (Bush and Vazquez-Pertejo, 2024). Serratia isolates can cause a range of nosocomial infections, including pneumonia, urinary tract infections, bacteremia, meningitis and endocarditis, particularly in immunocompromised patients (Mahlen, 2011). These infections account for about 1% to 2% of hospital-acquired infections (HAIs) and can lead to outbreaks with high morbidity and mortality rates, especially in pediatric or intensive care unit (ICU) wards (Mahlen, 2011; Aracil-Gisbert et al., 2024).
Taxonomically, the genus Serratia comprises twenty-four species (https://lpsn.dsmz.de/genus/serratia). S. marcescens, the most prevalent opportunistic human pathogen in this genus, frequently causes outbreaks of hospital-associated infections (Khanna et al., 2013; Van Goethem et al., 2024). Other members, such as S. rubidaea and S. liquefaciens, have also been reported to cause hospital-acquired infections, although less frequently (Dubouix et al., 2005; Karkey et al., 2018). Recently, a new species, S. sarumanii, was isolated from a wound swab (Klages et al., 2024). The transmission of some human-associated Serratia species via contaminated medical devices, such as nebulizers and catheters, is well known (Mahlen, 2011). However, most studies consist of individual case reports (Williams et al., 2022).
Clinical Serratia isolates are typically identified using MALDI-TOF MS (Aracil-Gisbert et al., 2024; Pérez-Viso et al., 2024; Van Goethem et al., 2024; Zhang et al., 2024). However, MALDI-TOF MS struggles to distinguish the species within Serratia marcescens complex (SMC), including S. marcescens, S. ureilytica, S. bockelmannii and S. nematodiphila (Harch et al., 2025). Furthermore, newly proposed species, such as S. montpellierensis (Blackburn et al., 2024) and S. sarumanii (Klages et al., 2024), may be sufficiently distinct at the molecular level, yet are not represented in current MALDI-TOF MS databases. Therefore, MALDI-TOF MS cannot reliably differentiate species within the SMC and should be complemented by whole genome sequencing (WGS) for species identification (Harch et al., 2025).
In this work, we aimed to provide a comprehensive understanding of Serratia marcescens complex in the ICUs of our hospital over an 11-year period (2014–2024). Based on WGS, we conducted a large-scale precision epidemiology analysis to decipher the evolution of clinical Serratia marcescens complex. Additionally, we compared the genomic features, in-hospital transmission patterns, antimicrobial resistance, and clinical characteristics of Serratia marcescens complex across different species.
Materials and methods
Study design and bacterial isolates
This retrospective study investigated inpatients infected with Serratia marcescens complex at Beijing Chao-Yang Hospital of Capital Medical University, a 2,500-bed public teaching hospital located in the capital of China that receives approximately four million patients per year. The study enrolled Serratia marcescens complex isolated from inpatients admitted to the ICUs from January 2014 to December 2024. Firstly, samples from patients suspected of having a bacterial infection were collected and send to the clinical microbiology laboratory for pathogen isolation and identification. The clinical samples included sputum, bronchoalveolar lavage fluid (BLAF), urine, drainage, body fluid, and wound. Suspected isolates that were cultured on China blue agar plates (Product code: HBPM6233, Thermo, USA) were initially identified using MALDI-TOF MS. Secondly, initial inclusion criteria varied across sample types. For non-sterile samples, such as sputum and BLAF, inclusion was only considered when the number of Serratia marcescens complex in the sample accounted for the vast majority. For sterile samples, inclusion was only considered when the number of Serratia marcescens complex in the sample was higher than 105 CFU/mL. Thirdly, the final inclusion criteria required that the patients received the appropriate treatment for the Serratia marcescens complex infection, as determined based on their electronic medical records. In total, non-duplicate isolates were mainly recovered from sputum (n = 89), followed by BLAF (n = 6), urine (n = 5), ascites (n = 3), drainage (n = 2), and wound (n = 1).
Antimicrobial susceptibility testing
Antimicrobial susceptibility testing of the bacterial isolates was investigated using the VITEK 2 system (bioMérieux, France) with GN09 cards following the guidelines provided by the Clinical and Laboratory Standards Institute (CLSI, 2025). In addition, the susceptibility of several antimicrobial agents, such as meropenem and imipenem, were determined or confirmed using the Kirby-Bauer method. Escherichia coli ATCC 8739 was used as the reference strain for quality control in all procedures. A total of 17 antimicrobial agents were tested, including amikacin (AK), aztreonam (ATM), ceftazidime (CAZ), Ciprofloxacin (CIP), gentamicin (CN), ceftriaxone (CRO), cefepime (FEP), imipenem (IPM), levofloxacin (LEV), meropenem (MEM), minocycline (MH), piperacillin (PRL), cefperazone-sulbactam (SCF), sulfamethoxazole-trimethoprim (SXT), tigecycline (TGC), tobramycin (TOB), and piperacillin-tazobactam (TZP).
Whole-genome sequencing and annotation
The genomic DNA of all bacterial isolates was extracted using Wizard Genomic DNA extraction kits (Promega, Wisconsin, USA) and stored at −80 °C until shipment to the Shanghai Majorbio Bio-pharm Technology Co., Ltd for whole genome sequencing on an Illumina HiSeq 2500 platform, resulting in a 2×150 bp paired-end library for each isolate. The quality control of raw sequenced reads was conducted as previously reported (Liu et al., 2025), which was used to eliminate sequences with low-quality (Q ≤ 20) and adapters. The clean data obtained was de novo assembled to generate draft contigs using SPAdes v4.1.0 (Prjibelski et al., 2020). The gaps of draft contigs were further filled using gapclose v1.12, resulting in draft genomes (Xu et al., 2020). Whole genome annotation, such as potential open reading frames and their functions, was investigated using Prokka pipeline v1.14.5 (Seemann, 2014). Taxonomic classification was determined using the GTDB Toolkit, which was based on the Genome Database Taxonomy (GTDB) (https://gtdb.ecogenomic.org/). The reference genomes for S. marcescens, S. ureilytica, S. nevei, S. bockelmannii, and S. sarumanii in the GTDB were GCF_017299535.1, GCF_013375155.1, GCF_008364245.1, GCF_008011855.1, and GCF_001537005.1, respectively. The species of sequenced genome was also identified using the rMLST scheme as previously proposed (Aracil-Gisbert et al., 2024; Harch et al., 2025), which is available at https://pubmlst.org/species-id.
Comparative genomic analysis
The sequence type (ST) of genomes was determined using PubMLST 7-loci schema (https://pubmlst.org/organisms/serratia-spp). Unassigned allele sequences were extracted and submitted to BIGSdb (Jolley and Maiden, 2010), and obtained 47 new STs. The whole-genome average nucleotide identity (ANI) was calculated as previously described (Bonnin et al., 2025), with intra-species strains displaying > 95% ANI. Antimicrobial resistance genes were identified using abricate v1.0.0 based on the NCBI AMRFinderPlus database (Feldgarden et al., 2019). The intrinsic genes were determined according to a previous report (Sandner-Miranda et al., 2018). The identified antimicrobial resistance genes were further classified into drug class based on the Comprehensive Antibiotic Resistance Database (CARD) (Alcock et al., 2020). Virulence genes were screened using abricate v1.0.0 based on virulence factor database (VFDB) with 80% coverage and 80% identity (Chen et al., 2016; Liu and Zhu, 2025; Zhu et al., 2025). The GFF3 format of each genome was obtained from Prokka annoation result and used to conduct core-pan genome analysis using the Roary pipeline v3.12.0 (Page et al., 2015). The Serratia core genome was defined by sequences present in ≥ 95% of these genomes as previously reported (Näpflin et al., 2025; Zhu et al., 2025).
Phylogenetic analysis
Single nucleotide polymorphism (SNP) analysis was conducted using Snippy v4.6.0 with default parameters (https://github.com/tseemann/snippy), utilizing the first identified genome (CY27690) from this study as the reference. The core genome alignment was then refined to eliminate elevated densities of base substitutions and recombination events using Gubbins v3.4 (Croucher et al., 2015). The core genome SNP (cgSNP) distance matrix of all paired genomes was calculated using snp-dists (https://github.com/tseemann/snp-dists). Paired genomes differing by no more than 16 SNPs were categorized as possible outbreaks (cluster groups) in the hospital, according to the previously recommended threshold (Chng et al., 2020), and were visualized using Standard Neighbour Joining algorithm of GrapeTree of (https://github.com/achtman-lab/GrapeTree). The maximum likelihood tree, based on core genome alignment, was built using IQtree v2.0.6 (https://github.com/Cibiv/IQ-TREE) and visualized using the Interactive Tree of Life (iTOL) web server (https://itol.embl.de/).
Statistical analysis
The clinical symptoms and demographic data were retrospectively extracted from the electronic medical record system. Categorical variables, such as age groups, were compared using the median and interquartile range (IQR). Violin plots were obtained using ggplot2 package. P values of < 0.05 indicated statistical significance. All statistics analyses were performed using R software package v4.3.3 (https://www.r-project.org/).
Results
Spatiotemporal distribution of Serratia marcescens complex in the ICUs
From January 2014 to December 2024, a total of 874 Serratia strains were isolated from all departments. The prevalence of S. marcescens complex in all bacterial strains isolated from the whole hospital ranged from 0.40% to 3.54% per year, with the highest prevalence occurring in 2014 (Supplementary Figure S1A). Among all the Serratia strains, the prevalence of these S. marcescens complex isolated from the ICUs was 4.6% to 30.4% per year, with the highest prevalence seen in 2024 (Supplementary Figure S1B). This suggested that the prevalence trend of S. marcescens complex in all bacterial strains was the opposite of that of S. marcescens complex from the ICU in Serratia strains per year (Supplementary Figure S1). In total, 106 non-repetitive Serratia spp. were included for further analysis (Supplementary Data Sheet 0), with isolation coming from five different ICUs: cardiac ICU (CICU, n = 8), emergency ICU (EICU, n = 18), neurology ICU (NICU, n = 30), respiratory ICU (RICU, n = 23), surgical ICU (SICU, n = 27).
Population structure of Serratia marcescens complex in the ICU
All the 106 Serratia spp. were initially identified as S. marcescens by the standard method in clinical labs (MALDI-TOF MS). To further confirm their species classification and genetic diversity, the whole genomes of these isolates were sequenced. The core genome among these isolates revealed a total of 469,717 core SNPs that were shared. Phylogenomic analysis based on core genome alignment indicated that the 106 Serratia isolates were clustered into five groups (Figure 1). ANI analysis of these 106 Serratia isolates also showed a diverse population structure, forming five groups (Supplementary Figure S2). The ANI values between different groups were all below the generally accepted 95-96% ANI threshold, suggesting that the five groups belonged to five different species, including S. sarumanii (n = 62), S. ureilytica (n = 17), S. marcescens (n = 13), S. bockelmannii (n = 11), and S. nevei (n = 3).
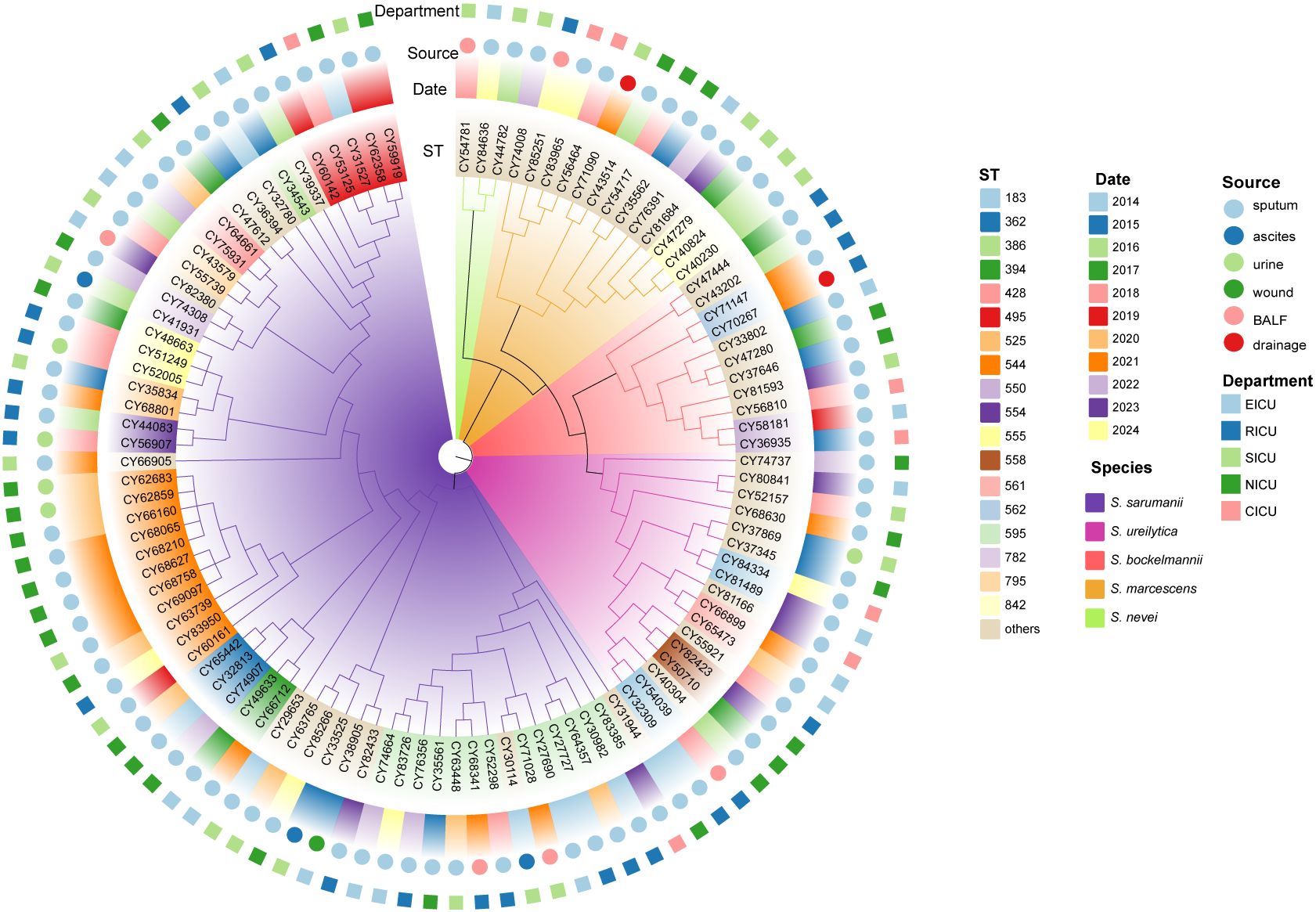
Figure 1. Phylogenetic analysis of Serratia clinical isolates based on core genome. The maximum-likelihood phylogenetic tree was constructed using 3151 core genes that were presented in ≥ 99% of genomes. The branch colors are labeled with corresponding species. The colored rings, from the inside out, represent the ST, collection date, source, and department of the strains isolated, respectively. BALF, bronchoalveolar lavage fluid; CICU, cardiac ICU; EICU, emergency ICU; NICU, neurology ICU; RICU, respiratory ICU; SICU, surgical ICU.
The in silico MLST analysis was performed utilizing the established Serratia spp. scheme. In total, these Serratia strains were categorized into 62 STs, with 43 STs represented by only one isolate each. The most prevalent ST was ST595 (12.3%, 13/106), followed by ST525 (10.4%, 11/106), ST428 (4.7%, 5/106), ST362 (2.8%, 3/106), ST554 (2.8%, 3/106), and ST842 (2.8%, 3/106). The ST diversity of Serratia strains from inpatients admitted to the SICU was more pronounced (Supplementary Figure S3). Notably, over a quarter of the isolates from patients in the NICU and RICU belonged to ST525 (26.7%) and ST595 (26.1%), respectively. Furthermore, all ST525 and ST595 isolates belonged to S. sarumanii (Figure 1). Interestingly, the STs of all the isolates from the CICU were different.
Genomic diversity of Serratia marcescens complex in the ICUs
The pairwise SNP distance matrix was estimated based on the core genome mentioned above, and used to construct a minimum spanning tree (MST). The isolates belonged to separate lineages that were identical to the species classification (Figure 2). The number of pairwise SNPs among the analyzed Serratia isolates ranged from 0 to 17723, with all isolates sharing a median of 3300 (IQR 3,197-14,535) SNPs with the first isolated strain (CY27690). Based on the threshold of 16 SNPs, these 106 isolates were assigned to 15 distinct genetic clusters (GCs) and 44 singleton strains. The majority of GCs were comprised of only two isolates (IQR 2-3). The largest GC was cluster 4, which included thirteen ST595 isolates and one ST839 (new ST identified in this study) isolate (CY83385) (Figure 3). These strains from cluster 4 circulated in five different ICUs over the past 11 years (Figure 3). Cluster 11, the second largest transmission chain, contained ten isolates from ST525, which were mainly prevalent in NICU between 2020 and 2021 before spreading to SICU in 2021 and RICU in 2024. Cluster 5 contained six isolates from three different STs (ST428, ST490, and ST525), spreading among four ICUs between 2014 and 2020. Cluster 6 had only five isolates from two ICUs, which belonged to two STs, including two from ST368 and three from ST362. Of particular interest was that isolates in each cluster were collected over a span of at least two different years (Supplementary Figure S4), suggesting the duration for which the strains had been circulating. Overall, most clustered isolates in each cluster were isolated from different ICUs, indicating of potential outbreaks and transmission.
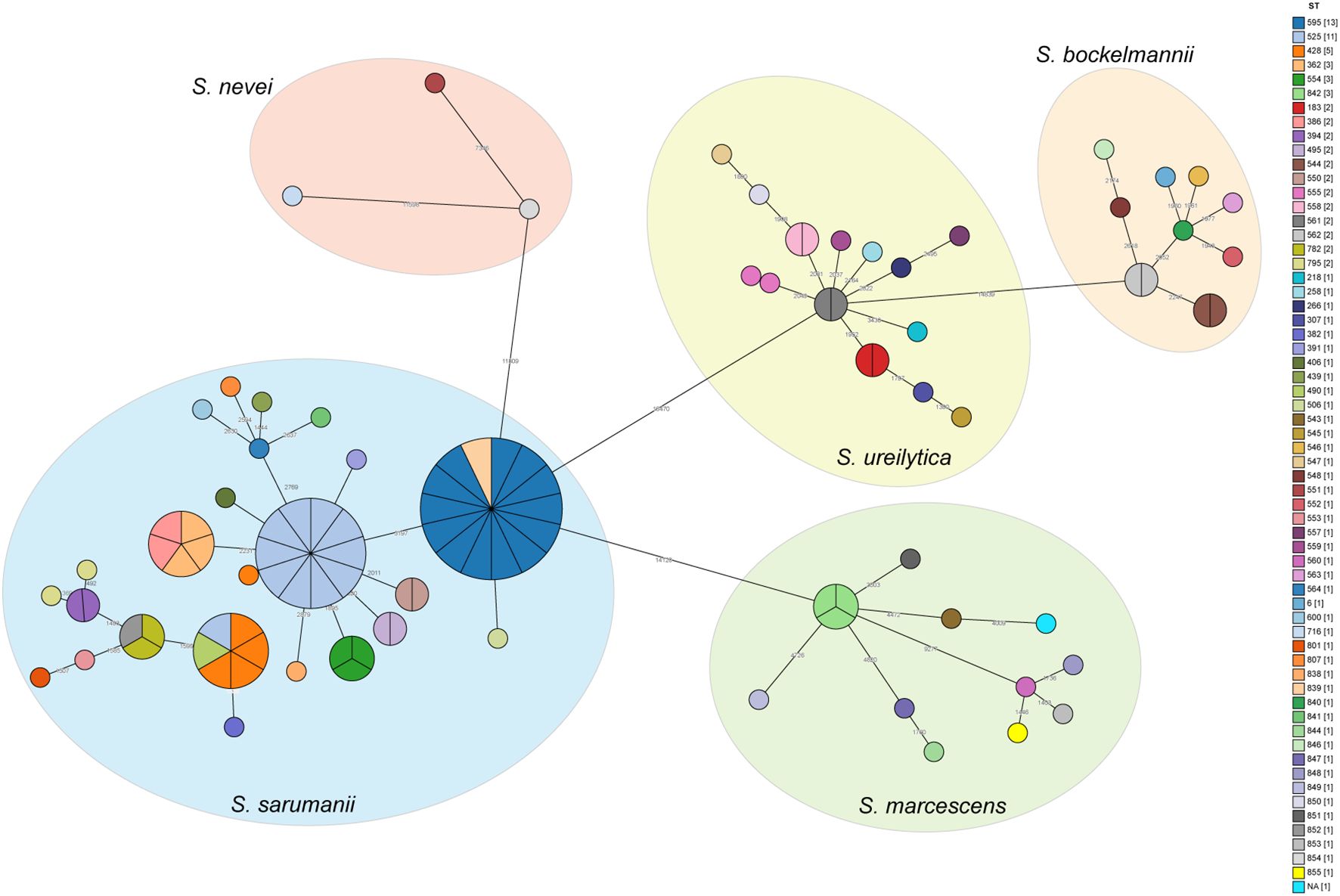
Figure 2. Minimum spanning trees showing the SNP difference among Serratia clinical isolates. Strains are grouped together based on having less than 16 SNPs. Each circle or section of the pie chart represents an individual strain, with the color indicating its ST. The number of SNPs between strains are showed on the connecting lines. The numbers in brackets in the legend indicate the number of strains in each ST.
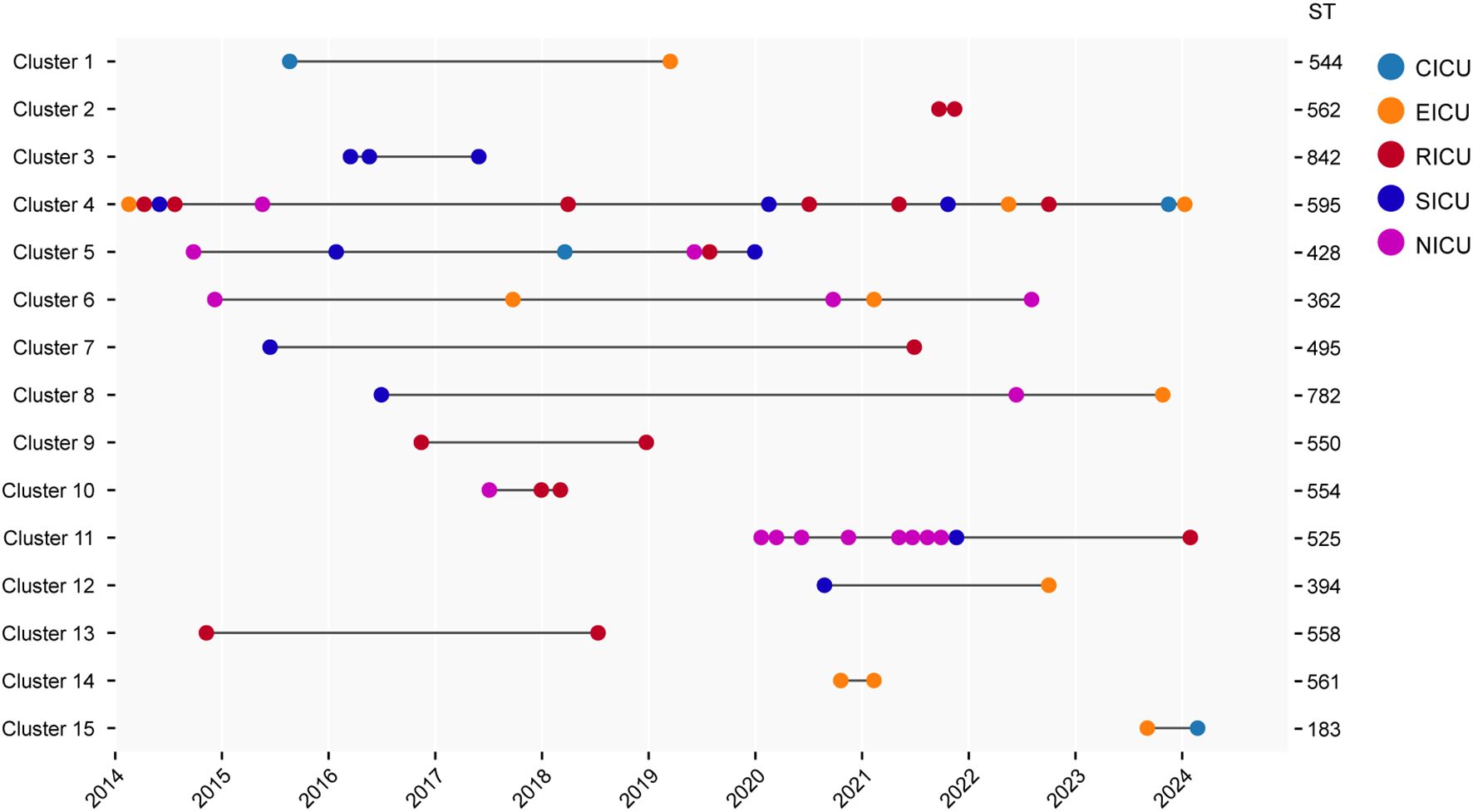
Figure 3. Timeline of detected cases in each cluster. The left Y axis is labeled with the name of each cluster, and the right Y axis is labeled with the ST of corresponding cluster. The X axis is labeled with collection year. CICU, cardiac ICU; EICU, emergency ICU; NICU, neurology ICU; RICU, respiratory ICU; SICU, surgical ICU.
Analysis of accessory genome
Accessory genes were analyzed to determine the presence of adaptive traits across different species. As the number of genomes examined increased, the number of core genes remained relatively stable, while the number of accessory genes showed an increasing trend (Supplementary Figure S5). Among 14242 (80.6%) accessory genes identified, less than 95% were shared across all Serratia spp. The average number of accessory genes per isolate within each species was determined (Figure 4A; Supplementary Data Sheets 2–5), with the highest number found in isolates of S. sarumanii (P < 0.05). Unique genes were notably enriched in isolates of S. sarumanii (n = 1075), the most represented species in the study, followed by S. ureilytica (n = 1063) (Figure 4B). Of particular interest within all S. marcescens isolates were the enrichment of adh3, coding for alcohol dehydrogenase, and fosB2, which imparts resistance to the antibiotic Fosfomycin. Moreover, isolates of S. sarumanii were enriched in various proteins, including hemolysin transporter protein ShlB, macrolide export ATP-binding/permease protein MacB, toxic protein HokC that required for toxin and drug export, as well as proteins associated with type II secretion system like type II secretion system protein E J, and L.
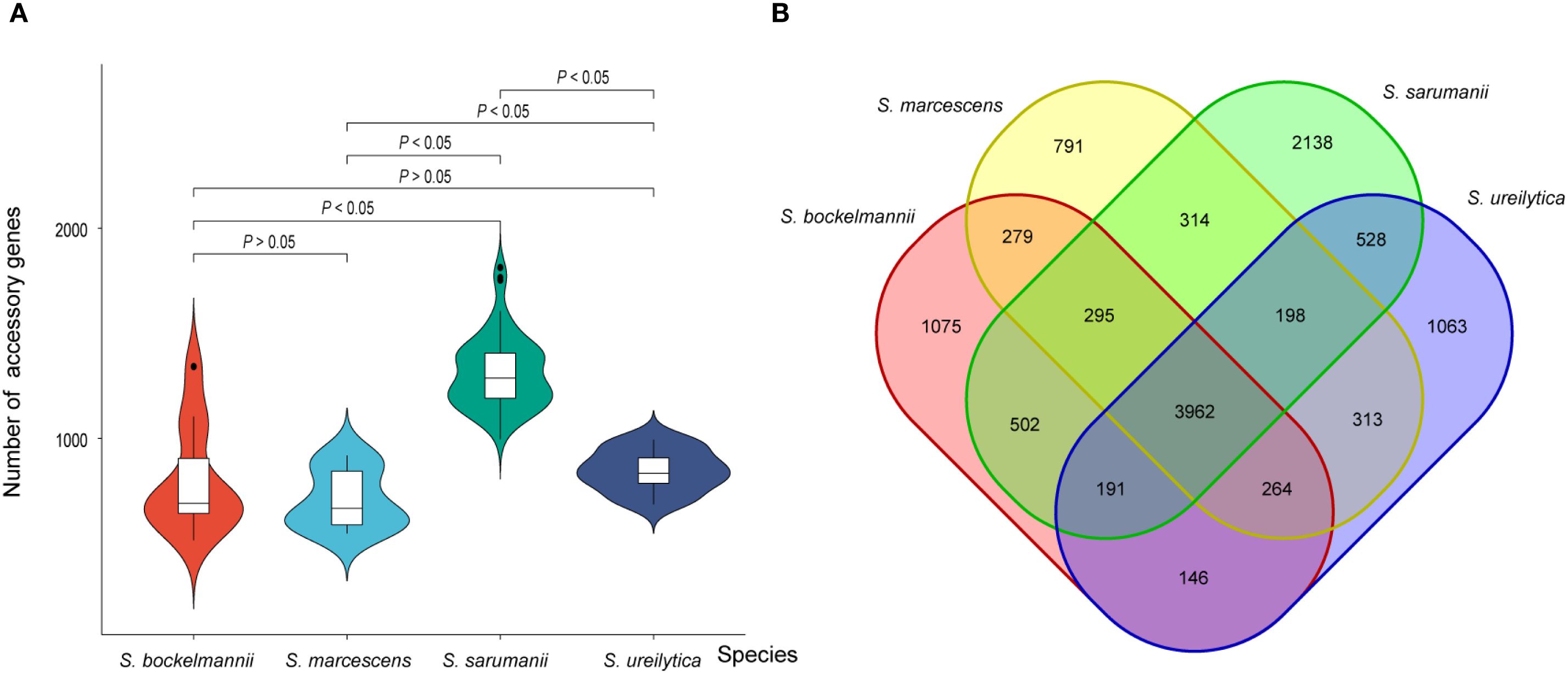
Figure 4. (A) Violin plot displaying the number of accessory genes for each species (A). The horizontal line within each box plot represents the median number of accessory genes for each species. Significance values are labeled on the top of plot. (B) Venn plot illustrating the shared accessory genes among each species.
Antimicrobial susceptibility and resistance phenotypes
The antimicrobial susceptible testing revealed that a total of 76 (71.7%) isolates were susceptibility to all 17 antimicrobial agents tested (Supplementary Data Sheet 1). The resistance of the remaining 30 strains ranged from 1 and 13 antimicrobial agents per isolate, with a median of 5 (IQR 3-9) (Figure 5). Among these Serratia spp., the most common resistance was observed against CRO (24.5%, 26/106), followed by PRL (23.6%, 25/106), ATM (21.7%, 23/106), CIP (14.1%, 15/106), LEV (11.3%, 12/106), CN (10.4%, 11/106), and FEP (9.4%, 10/106). Notably, nine isolates were resistant to IPM (8.5%, 9/106), indicating the emergence of carbapenem-resistant S. marcescens complex. The prevalence of other resistance phenotypes was all less than 10% (< 9 isolates). The most prevalent resistant profile included resistance to CRO, PRL, and ATM simultaneously. The resistant profile of most antimicrobial resistant isolates was unique among Serratia spp. in this study. Furthermore, isolates demonstrating antimicrobial resistance to the tested agents were primarily presented in S. sarumanii (83.3%, 25/30) and S. bockelmannii (10.0%, 3/30).
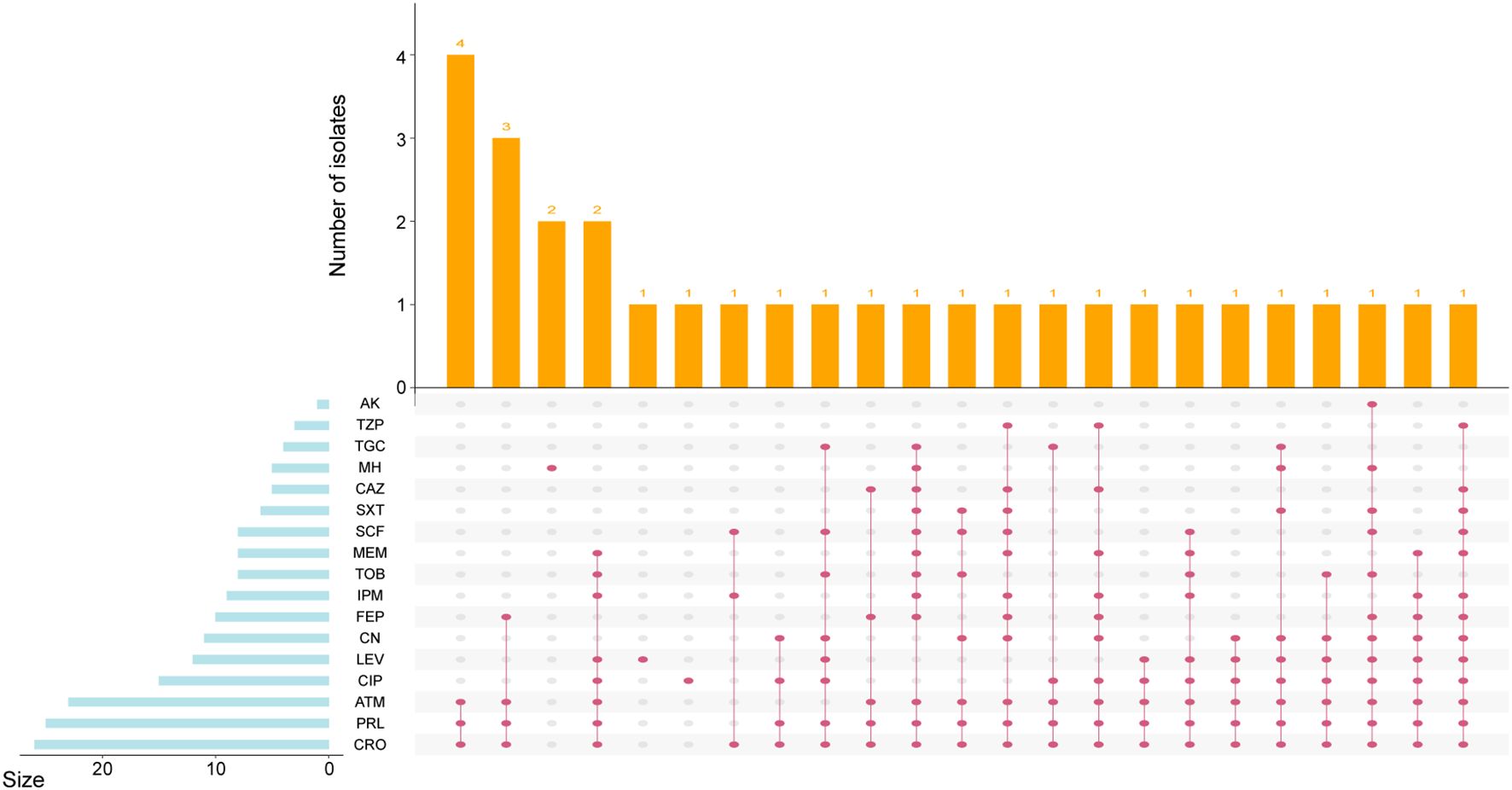
Figure 5. UpSetR plot showing the resistance phenotype of Serratia clinical isolates. The left histogram indicates the number of isolates with each antimicrobial resistance. The top histogram reveals the distribution of isolates by type. The phenotypic results indicate resistance as defined by the Clinical and Laboratory Standards Institute guidelines.
Patterns of antimicrobial resistance and virulence genes
To investigate their resistome, genomes were screened for the presence of antimicrobial resistance (AMR) genes (Figure 6). A total of 52 AMR genes were identified from the 106 Serratia isolates, which were classified into 14 drug classes. The number of AMR genes varied across isolates, ranging from 3 to 21, with a median of 4 (IQR 3-4). The most prevalent AMR gene was intrinsic aac(6’)_Serra gene (97/106), which was prevalent in all five species. However, the second most prevalent AMR gene, intrinsic oqxB9 gene (70/106), was absent in isolates belonging to S. bockelmannii and S. ureilytica, and present in several isolates of S. marcescens. The strains carrying AMR genes associated with cephalosporin, aminoglycoside, and antibiotic efflux were distributed in all isolates. Among 12 beta-lactamase genes conferring resistance to cephalosporin, intrinsic blaSRT gene was the most prevalent (56/106). Additionally, three acquired blaCTX-M genes, including blaCTX-M-14 (n = 12), blaCTX-M-3 (n = 12), and blaCTX-M-15 (n = 2), were prevalent in 26 isolates, which was consistent with the results of the CRO resistance genotypes. In addition, acquired carbapenemase genes were detected in nine Serratia isolates (8.5%, 9/106), including five blaKPC-2 (4.7%, 5/106) and four blaNDM-5 (3.8%, 4/106), which aligned with the results of the IPM resistance phenotypes.
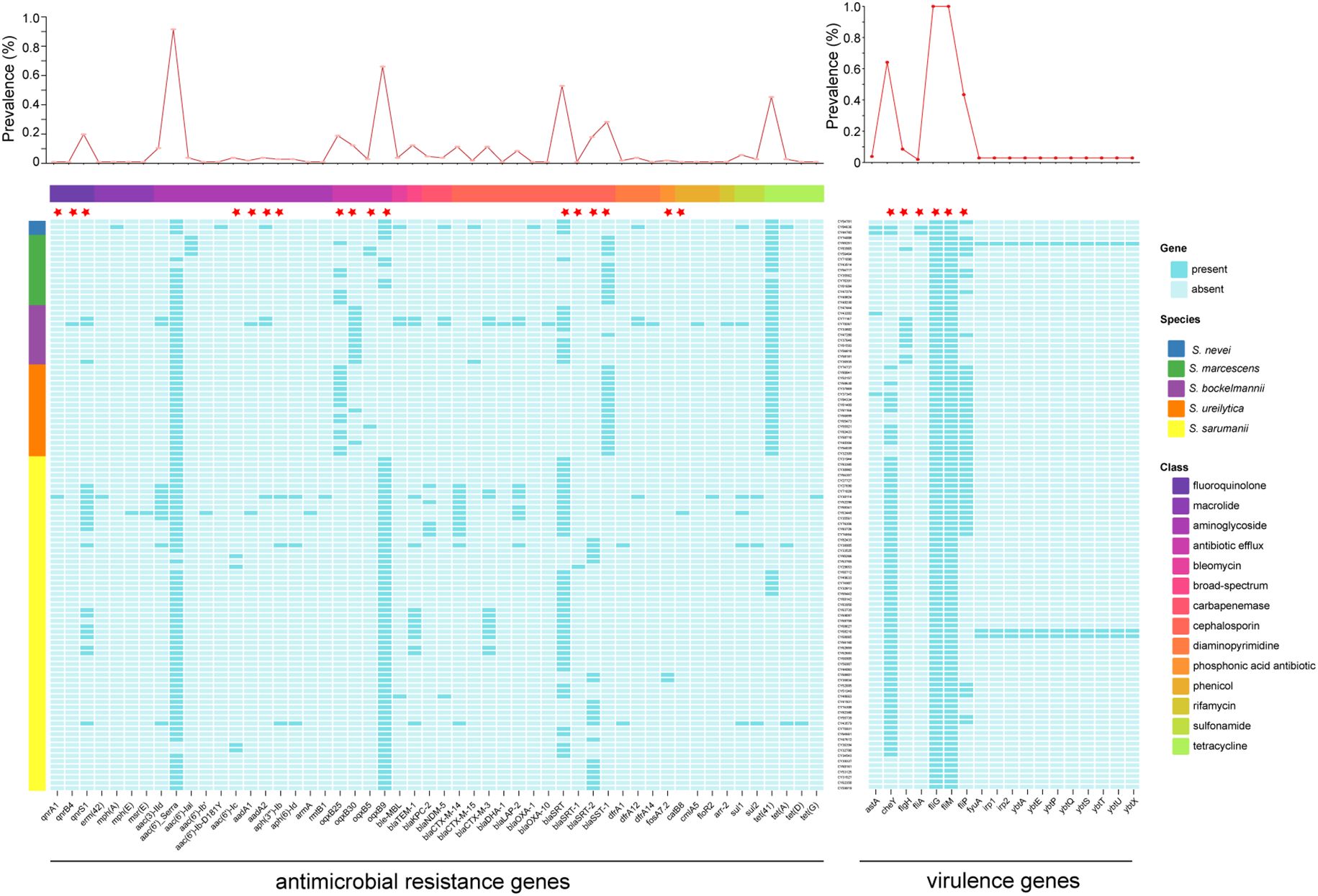
Figure 6. Distributions of antimicrobial resistance genes and virulence genes. The line chart at the top showed the incidence of each gene. The color of left rectangle represents the corresponding species, while the color of top rectangle is labeled with the corresponding drug class of antimicrobial resistance genes. The intrinsic antimicrobial resistance and virulence genes were labeled with red five-pointed stars.
The potential pathogenicity of Serratia spp. in the ICUs were determined based on the distribution of virulence genes (Figure 6). The most common virulence-related genes were intrinsic fliG and fliM genes, which were prevalent in all isolates. And, the intrinsic cheY gene, involved in the transmission of sensory signals to the flagellar motors, was the second common virulence-related gene (64.2%, 68/106), followed by intrinsic fliP gene (43.4%, 46/106), which were sporadically distributed across species. It is noteworthy that three isolates contained a high-pathogenicity island comprised of fyuA, irp1, irp2, ybtA, ybtE, ybtP, ybtQ, ybtS, ybtT, ybtU, ybtX. Furthermore, there was a significant difference in the number of virulence genes between several species (Supplementary Figure S6), as well as between isolates of several clusters (Supplementary Figure S7).
Clinical manifestations of Serratia marcescens complex in the ICU
To evaluate the differences in clinical manifestations, we retrieved the electronic medical records of inpatients infected with Serratia spp. The median age of all cases was 69 years (IQR 60-79), with 68.9% of them being male (73/106). We proceeded to compare the clinical manifestations caused by the four main species (Supplementary Table S1). Although the proportion of male patients infected with S. ureilytica seems to be similar to that of female patients, a larger proportion of males were infected with the remaining three species, with a significant difference noted in S. sarumanii (P = 0.0001). The median age of patients diagnosed with Serratia infection showed a significant difference among four species (P < 0.05), with the median age of patients diagnosed with S. bockelmannii being the youngest. Individuals infected with S. sarumanii, S. ureilytica and S. marcescens tended to be older. The clinical symptoms including fever (median 38°C, IQR 37.6-38.8°C) and cough had a higher prevalence. The incidence of severe pneumonia ranged from 16.7-28.6%. Dyspnea and chill were only observed in patients infected with S. sarumanii and S. ureilytica. The abnormality rate of clinical examination varied, with the highest WBC count observed in cases of S. marcescens and S. bockelmannii, neutrophilic granulocyte percentage in S. marcescens, and hemoglobin in S. sarumanii.
Discussion
Although a wide variety of species have been proposed in the genus Serratia, most of the understanding of Serratia has focused on the type species, S. marcescens (Williams et al., 2022). This study retrospectively investigated the Serratia species isolated from multiple sources, revealing the wide genomic diversity of human-associated Serratia within five ICU wards over the past 11 years. These Serratia spp. were genomically classified into five species, with the majority of them being S. sarumanii, which was identified as a novel species in the genus Serratia in 2024 (Klages et al., 2024). The findings of this study indicated that there were multiple species of Serratia can infected humans, and similar species may not be accurately distinguished by MALDI-TOF MS, but could be differentiated by conserved genomic regions.
Analysis of closely clustered isolates using cgSNP distance matrix with a threshold of 16 SNPs (David et al., 2019) revealed a high incidence of clonal dissemination of Serratia spp. Our study also showed that the largest cluster had spread over 11 years, including inter-ICU transmission, which exceeded previous reports (Zhang et al., 2024). Multiple simultaneous transmission chains were identified within the same ICU, raising concerns about ongoing dissemination. However, the full extent of Serratia spp. dissemination may be underestimated as not all isolates collected from 2014 to 2025 were included, nor those obtained prior to 2014. The exact routes of transmission remain unclear, but potential sources could include contact with caregivers, cleaning workers, shared bathrooms, and changes in hospital departments during a patient’s stay. In addition, more than 11,500 SNPs were identified between one isolate and the other two isolates within S. nevei, indicating the potential existence of a sixth group.
Both intrinsic and acquired AMR genes contributed to antibiotic resistance across Serratia species. The chromosomal AmpC-type β-lactamase genes, including blaSRT, blaSRT-1,and blaSRT-2, were present in all isolates, with each isolate having a resistance gene, consistent with previous research (Matteoli et al., 2021). The acquired β-lactamase genes in this study, such as blaCTX-M and blaOXA, were found primarily in S. sarumanii species. Notably, carbapenemase-producing Serratia spp. were detected, with blaKPC-2 found exclusively in S. sarumanii. Additionally, the carbapenemase gene, blaNDM-5, was prevalent in three Serratia species. Although previous reports have described the presence of blaKPC-2, blaVIM-1, and blaOXA-48 in Serratia spp (Matteoli et al., 2021; Pérez-Viso et al., 2024; Tang et al., 2024; Zhang et al., 2024), NDM-5-producing Serratia isolates have been rarely reported in China or elsewhere (Matteoli et al., 2021; Nakanishi et al., 2022; Pérez-Viso et al., 2024). Fortunately, none of these carbapenemase-producing S. marcescens complex caused clonal outbreaks in the hospital.
Analysis of the virulome of S. marcescens complex revealed a narrower range of features, with virulence genes related to flagella being the most common (Pérez-Viso et al., 2024). The intrinsic cheY gene, encoding proteins of chemotaxis, was present in most isolates, potentially delivering secondary signals to flagellar motors (Paul et al., 2010; Lazzaro, 2019). Furthermore, the Yersinia high-pathogenicity island, comprising genes related to the synthesis of the siderophore yersiniabactin (Carniel, 2001), was identified in S. sarumanii and S. marcescens in this study, and had previously been found in S. liquefaciens (Olsson et al., 2003). The transmission and exact virulence level of all these genes in Serratia spp. remain poorly understanded and require further investigation. Additionally, individual species contained unique genes compared with other three species, such as type II secretion system only presented in S. sarumanii.
In conclusion, we provided important insights into the population structure of S. marcescens complex were provided based on 106 isolates collected from ICUs in a single center over the past 11 years, representing five different species. Investigation into the potential transmissions of these Serratia isolates revealed that the majority of them were clonal related. Antimicrobial resistance analysis showed that while the overall incidence of resistance was not severe, carbapenemase-producing S. marcescens complex had emerged as early as 2014. To prevent the dissemination of these emergent and future clones, further research is needed to understand the transmission mechanisms and routes associated with these clinically significant Serratia clones.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: BioProject accession number PRJNA1191627, with genome accession numbers JBMIOK000000000, JBMIOL000000000, and JBJNLA000000000 to JBJNOZ000000000.
Ethics statement
This study was approved by the Ethics Committee of Beijing Chaoyang Hospital, Capital Medical University (2024-ke-381).
Author contributions
WZ: Conceptualization, Funding acquisition, Project administration, Writing – original draft, Writing – review & editing. XC: Formal analysis, Resources, Writing – original draft. HS: Formal analysis, Resources, Writing – original draft. MW: Resources, Writing – original draft. CY: Resources, Writing – review & editing. LG: Project administration, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the National Natural Science Foundation of China (82502766), and the Beijing Chao-Yang Hospital Golden Seeds Fundation (CYJZ202220). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgments
We would like to thank all of staff from the Department of Infectious Diseases and Clinical Microbiology at Beijing Chao-Yang Hospital for their contribution to this work.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2025.1672468/full#supplementary-material
References
Adeolu, M., Alnajar, S., Naushad, S., and Gupta, R. S. (2016). Genome-based phylogeny and taxonomy of the ‘enterobacteriales’: Proposal for enterobacterales ord. Nov. Divided into the families enterobacteriaceae, erwiniaceae fam. Nov., pectobacteriaceae fam. Nov., yersiniaceae fam. Nov., hafniaceae fam. Nov., morganellaceae fam. Nov., and budviciaceae fam. Nov. Int. J. Syst. Evol. Microbiol. 66, 5575–5599. doi: 10.1099/ijsem.0.001485
Alcock, B. P., Raphenya, A. R., Lau, T. T. Y., Tsang, K. K., Bouchard, M., Edalatmand, A., et al. (2020). Card 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 48, D517–d525. doi: 10.1093/nar/gkz935
Aracil-Gisbert, S., Fernández-De-Bobadilla, M. D., Guerra-Pinto, N., Serrano-Calleja, S., Pérez-Cobas, A. E., Soriano, C., et al. (2024). The ICU environment contributes to the endemicity of the “serratia marcescens complex” in the hospital setting. mBio 15, e0305423. doi: 10.1128/mbio.03054-23
Blackburn, M. B., Tannières, M., Sparks, M. E., Gundersen-Rindal, D. E., and Bon, M. C. (2024). Serratia montpellierensis sp. Nov., isolated from laboratory-reared parasitic wasps psyttalia lounsburyii silvestri and psyttalia ponerophaga silvestri (hymenoptera: Braconidae). Curr. Microbiol. 81, 146. doi: 10.1007/s00284-024-03666-0
Bonnin, R. A., Naas, T., and Dortet, L. (2025). Diversity of β-lactamase resistance and genome of morganella spp clinical isolates in China-authors’ reply. Lancet Microbe 6, 101004. doi: 10.1016/j.lanmic.2024.101004
Bush, L. and Vazquez-Pertejo, M. (2024). Klebsiella, enterobacter, and serratia infections (Merck Manual). Available online at: https://www.merckmanuals.com/professional/infectious-diseases/gram-negative-bacilli/klebsiella-enterobacter-and-serratia-infections.
Carniel, E. (2001). The yersinia high-pathogenicity island: An iron-uptake island. Microbes Infect. 3, 561–569. doi: 10.1016/S1286-4579(01)01412-5
Chen, L., Zheng, D., Liu, B., Yang, J., and Jin, Q. (2016). Vfdb 2016: Hierarchical and refined dataset for big data analysis–10 years on. Nucleic Acids Res. 44, D694–D697. doi: 10.1093/nar/gkv1239
Chng, K. R., Li, C., Bertrand, D., Ng, A. H. Q., Kwah, J. S., Low, H. M., et al. (2020). Cartography of opportunistic pathogens and antibiotic resistance genes in a tertiary hospital environment. Nat. Med. 26, 941–951. doi: 10.1038/s41591-020-0894-4
CLSI (2025). Performance standards for antimicrobial susceptibility testing. 35th ed Clsi supplement m100 (Wayne, PA: Clinical and Laboratory Standards Institute).
Croucher, N. J., Page, A. J., Connor, T. R., Delaney, A. J., Keane, J. A., Bentley, S. D., et al. (2015). Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using gubbins. Nucleic Acids Res. 43, e15. doi: 10.1093/nar/gku1196
David, S., Reuter, S., Harris, S. R., Glasner, C., Feltwell, T., Argimon, S., et al. (2019). Epidemic of carbapenem-resistant klebsiella pneumoniae in europe is driven by nosocomial spread. Nat. Microbiol. 4, 1919–1929. doi: 10.1038/s41564-019-0492-8
Dubouix, A., Roques, C., Segonds, C., Jeannot, M. J., Malavaud, S., Daude, S., et al. (2005). Epidemiological investigation of a serratia liquefaciens outbreak in a neurosurgery department. J. Hosp Infect. 60, 8–13. doi: 10.1016/j.jhin.2004.09.029
Feldgarden, M., Brover, V., Haft, D. H., Prasad, A. B., Slotta, D. J., Tolstoy, I., et al. (2019). Validating the amrfinder tool and resistance gene database by using antimicrobial resistance genotype-phenotype correlations in a collection of isolates. Antimicrob. Agents Chemother. 63 (11), e00483-19. doi: 10.1128/AAC.00483-19
Harch, S. A. J., Jenkins, F., Farhat, R., and van Hal, S. J. (2025). Complexities in species identification for serratia marcescens complex for the modern microbiology laboratory. Microbiol. Spectr. 13, e0136124. doi: 10.1128/spectrum.01361-24
Jolley, K. A. and Maiden, M. C. (2010). BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinf. 11, 595. doi: 10.1186/1471-2105-11-595
Karkey, A., Joshi, N., Chalise, S., Joshi, S., Shrestha, S., Thi Nguyen, T. N., et al. (2018). Outbreaks of serratia marcescens and serratia rubidaea bacteremia in a central kathmandu hospital following the 2015 earthquakes. Trans. R Soc. Trop. Med. Hyg 112, 467–472. doi: 10.1093/trstmh/try077
Khanna, A., Khanna, M., and Aggarwal, A. (2013). Serratia marcescens- a rare opportunistic nosocomial pathogen and measures to limit its spread in hospitalized patients. J. Clin. Diagn. Res. 7, 243–246. doi: 10.7860/JCDR/2013/5010.2737
Klages, L. J., Kaup, O., Busche, T., Kalinowski, J., and Rückert-Reed, C. (2024). Classification of a novel serratia species, isolated from a wound swab in north rhine-westphalia: Proposal of serratia sarumanii sp. nov. Syst. Appl. Microbiol. 47, 126527. doi: 10.1016/j.syapm.2024.126527
Liu, Q., Shen, H., Wei, M., Chen, X., Gu, L., and Zhu, W. (2025). Global phylogeography and antibiotic resistance characteristics of morganella: An epidemiological, spatial, comparative genomic study. Drug Resist. Update 78, 101180. doi: 10.1016/j.drup.2024.101180
Liu, Q. and Zhu, W. (2025). Diversity of β-lactamase resistance and genome of morganella clinical isolates in China. Lancet Microbe 6, 101003. doi: 10.1016/j.lanmic.2024.101003
Mahlen, S. D. (2011). Serratia infections: From military experiments to current practice. Clin. Microbiol. Rev. 24, 755–791. doi: 10.1128/CMR.00017-11
Matteoli, F. P., Pedrosa-Silva, F., Dutra-Silva, L., and Giachini, A. J. (2021). The global population structure and beta-lactamase repertoire of the opportunistic pathogen serratia marcescens. Genomics 113, 3523–3532. doi: 10.1016/j.ygeno.2021.08.009
Nakanishi, N., Komatsu, S., Iwamoto, T., and Nomoto, R. (2022). Characterization of a novel plasmid in serratia marcescens harbouring bla(ges-5) isolated from a nosocomial outbreak in Japan. J. Hosp Infect. 121, 128–131. doi: 10.1016/j.jhin.2021.11.022
Näpflin, N., Schubert, C., Malfertheiner, L., Hardt, W. D., and von Mering, C. (2025). Gene-level analysis of core carbohydrate metabolism across the enterobacteriaceae pan-genome. Commun. Biol. 8, 1241. doi: 10.1038/s42003-025-08640-5
Olsson, C., Olofsson, T., Ahrné, S., and Molin, G. (2003). The yersinia hpi is present in serratia liquefaciens isolated from meat. Lett. Appl. Microbiol. 37, 275–280. doi: 10.1046/j.1472-765X.2003.01387.x
Ono, T., Taniguchi, I., Nakamura, K., Nagano, D. S., Nishida, R., Gotoh, Y., et al. (2022). Global population structure of the serratia marcescens complex and identification of hospital-adapted lineages in the complex. Microb. Genom 8 (3), 000793. doi: 10.1099/mgen.0.000793
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Paul, K., Nieto, V., Carlquist, W. C., Blair, D. F., and Harshey, R. M. (2010). The c-di-GMP binding protein YcgR controls flagellar motor direction and speed to affect chemotaxis by a “backstop brake” mechanism. Mol. Cell 38, 128–139. doi: 10.1016/j.molcel.2010.03.001
Pérez-Viso, B., Hernández-García, M., Rodríguez, C. M., Fernández-de-Bobadilla, M. D., Serrano-Tomás, M. I., Sánchez-Díaz, A. M., et al. (2024). A long-term survey of serratia spp. Bloodstream infections revealed an increase of antimicrobial resistance involving adult population. Microbiol. Spectr. 12, e0276223. doi: 10.1128/spectrum.02762-23
Prjibelski, A., Antipov, D., Meleshko, D., Lapidus, A., and Korobeynikov, A. (2020). Using spades de novo assembler. Curr. Protoc. Bioinf. 70, e102. doi: 10.1002/cpbi.102
Sandner-Miranda, L., Vinuesa, P., Cravioto, A., and Morales-Espinosa, R. (2018). The genomic basis of intrinsic and acquired antibiotic resistance in the genus serratia. Front. Microbiol. 9, 828. doi: 10.3389/fmicb.2018.00828
Seemann, T. (2014). Prokka: Rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Tang, B., Zhao, H., Li, J., Liu, N., Huang, Y., Wang, J., et al. (2024). Detection of clinical serratia marcescens isolates carrying bla(kpc-2) in a hospital in China. Heliyon 10, e29702. doi: 10.1016/j.heliyon.2024.e29702
Van Goethem, S., Xavier, B. B., Glupczynski, Y., Berkell, M., Willems, P., Van Herendael, B., et al. (2024). Genomic epidemiological analysis of a single-centre polyclonal outbreak of serratia marcescens, Belgium 2022 to 2023. Euro Surveill 29 (48), 2400144. doi: 10.2807/1560-7917.ES.2024.29.48.2400144
Williams, D. J., Grimont, P. A. D., Cazares, A., Grimont, F., Ageron, E., Pettigrew, K. A., et al. (2022). The genus serratia revisited by genomics. Nat. Commun. 13, 5195. doi: 10.1038/s41467-022-32929-2
Xu, M., Guo, L., Gu, S., Wang, O., Zhang, R., Peters, B. A., et al. (2020). TGS-gapcloser: A fast and accurate gap closer for large genomes with low coverage of error-prone long reads. Gigascience 9 (9), giaa094. doi: 10.1093/gigascience/giaa094
Zhang, F., Li, Z., Liu, X., Li, Z., Lei, Z., Zhao, J., et al. (2024). In-host intra- and inter-species transfer of bla(KPC-2) and bla(NDM-1) in serratia marcescens and its local and global epidemiology. Int. J. Antimicrob. Agents 64, 107327. doi: 10.1016/j.ijantimicag.2024.107327
Zhu, W., Chen, X., Shen, H., Wei, M., Gu, L., and Liu, Q. (2025). Genomic evolution, antimicrobial resistance, and dissemination of global serratia spp. Unveil increasing species diversity and carbapenemae−resistance: A retrospective and genomic epidemiology study. Curr. Res. Microbial Sci. 9, 100456. doi: 10.1016/j.crmicr.2025.100456
Keywords: Serratia, genomic surveillance, dissemination, antimicrobial resistance, ICU - intensive care unit
Citation: Zhu W, Chen X, Shen H, Wei M, Yang C and Gu L (2025) Genomic diversity, antimicrobial resistance and dissemination of Serratia marcescens complex in patients admitted to ICUs. Front. Cell. Infect. Microbiol. 15:1672468. doi: 10.3389/fcimb.2025.1672468
Received: 24 July 2025; Accepted: 11 September 2025;
Published: 16 October 2025.
Edited by:
Valerio Baldelli, University of Milan, ItalyReviewed by:
Verónica Mixão, National Health Institute Doutor Ricardo Jorge (INSA), PortugalSonia Aracil-Gisbert, Instituto Ramón y Cajal de Investigación Sanitaria (IRYCIS), Spain
Copyright © 2025 Zhu, Chen, Shen, Wei, Yang and Gu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wentao Zhu, d2VudGFvemh1QDEyNi5jb20=; Li Gu, ZGlkY20yMDA2QG1haWwuY2NtdS5lZHUuY24=