 Lei Zhao1†
Lei Zhao1† Shifu Peng2†
Shifu Peng2† Muxi Ge3†Boming Xing1Xinyang Zhao1Tongquan Yang1Shenghui Yu4Cheng Zhang1Jinyang Liu1
Muxi Ge3†Boming Xing1Xinyang Zhao1Tongquan Yang1Shenghui Yu4Cheng Zhang1Jinyang Liu1 Ziwei Miao5*
Ziwei Miao5* Heyao Ma6*
Heyao Ma6*- 1Department of Hepatobiliary and Pancreatic Surgery, First Hospital of China Medical University, Shenyang, Liaoning, China
- 2Department of Environment and Health, Jiangsu Center for Disease Control and Prevention, Nanjing, China
- 3The First Clinical College of China Medical University, Shenyang, Liaoning, China
- 4Research Institute for Cancer Therapy, First Hospital of China Medical University, Shenyang, Liaoning, China
- 5Department of Developmental Cell Biology, Key Laboratory of Medical Cell Biology, Ministry of Education, China Medical University, Shenyang, China
- 6Department of Pharmacology, School of Pharmacy, China Medical University, Shenyang, China
Background: Despite advances and successes in precision oncology, pancreatic cancer (PC) remains a tumor with extremely low survival rates, and many of these cases experienced postoperative recurrence and metastasis. Alterations in the gut microbiota have been linked to the survival rates of PC patients. Nevertheless, the complexity of gut microbiota composition poses significant challenges in identifying definitive clinical biomarkers for PC.
Methods: Fecal samples were collected from PC patients, half of whom had metastasis, and their matched healthy controls (HCs). A metagenomic analysis was employed to further investigate the functional features of gut microbiota with both PC and metastatic PC. The clinical correlations, microbial metabolic pathways and antibiotic resistome were further assessed. In a follow-up validation, intraoperative tumor tissue and pancreatic fluid were sampled from PC patients and underwent comprehensive microbiological analysis, including bacterial culture, mass spectrometry-based identification, and third-generation whole-genome sequencing of Klebsiella pneumoniae isolates.
Results: We observed a significant alteration of the gut microbiota in PC patients, highlighted by an overall increase in microbial diversity compared to healthy controls (p < 0.05). Comparative abundance analysis identified 59 differentially abundant microbial species in non-metastatic pancreatic cancer (NMPC) (56 increased, 3 decreased) and 21 in metastatic pancreatic cancer (MPC) (19 increased, 2 decreased), alongside 18 significantly altered microbial metabolic pathways (FDR-adjusted p < 0.05). Notably, Klebsiella pneumoniae, Klebsiella oxytoca, and Akkermansia muciniphila were identified as prominent antibiotic resistance gene (ARG) carriers in the gut microbiota of PC patients, with 653 ARG subtypes detected across fecal samples, 38–47% of which were shared among groups. Strong co-occurrence patterns between ARGs (e.g., acrB, mdtC, cpxA, emr, pmrF) and the above species were observed predominantly in MPC samples (p < 0.05). Whole-genome sequencing of 14 isolates obtained from tumor tissue and pancreatic fluid revealed consistent ARG profiles and virulence genes, corroborating the metagenomic findings and supporting the hypothesis of gut-to-tumor translocation and potential intratumoral colonization.
Conclusion: This study provides a comprehensive microbiome-based insight into PC and its metastatic subtypes. By integrating microbiome analysis with microbial culture, this study provides direct evidence of gut-derived multidrug-resistant (MDR) K. pneumoniae colonization in PC tissues.
1 Introduction
Pancreatic cancer (PC) was considered to be one of the most lethal malignant tumors of the digestive tract, with a five-year overall survival rate of less than 12% (Siegel et al., 2023). This prognosis worsened with the onset of metastasis, dropping survival rate to a mere 3% (Siegel et al., 2022). Despite advancements in multimodal diagnostic and treatment strategies, the efficacy of surgical interventions, which were crucial for effective PC management was significantly hindered by the challenges in the early detection (Vuijk et al., 2020). Metastasis was recognized as another main cause of the death of PC patients and treatment failure (Zheng et al., 2020). Although immunotherapy was demonstrated to be effective in a wide variety of metastatic cancers, it failed in PC because of its inherently “non-immunogenic” nature. Therefore, it is still necessary to explore new diagnosis and treatment strategies for PC (Li et al., 2023; Ni et al., 2023). The leading gene therapy and precision medicine strategies for PC, including KRAS-targeted interventions, miRNA-based treatments, and immunotherapies such as CAR-T, hold great promise yet face substantial challenges due to significant inter-individual variability in treatment response (Dwivedi et al., 2025; Sharma et al., 2025). Emerging evidence now identifies the gut microbiome, particularly through gut-to-tumor translocation, as a clinically detectable and individualized factor in PC progression and treatment resistance (Sethi et al., 2018; Kirsoy et al., 2024; Dwivedi et al., 2025).
The gut microbiota, residing within a complex and dynamic ecosystem, was closely linked to human immunity, primarily through its influence on intestinal permeability (Pushalkar et al., 2018). Contrary to prior beliefs of a sterile pancreas, emerging evidence suggests that gut microbiota can migrate to the pancreas via lymph nodes and dendritic cells, thereby facilitating the colonization of the pancreas by various microorganisms (Fan et al., 2018). This migration was implicated in the neoplastic transformation, with significant increases in the abundance of specific microbial taxa like Proteobacteria, Synergistetes and Euryarchaeota observed in PC patients (Thomas and Jobin, 2020). Additionally, early PC stages have been associated with alterations in microbiota’s metabolic pathways, notably in polyamine and nucleotide biosynthesis, establishing a link between these microbial processes and PC development (Mendez et al., 2020). Moreover, carbohydrates produced by Malassezia have been identified to stimulate PC cell growth through interaction with mannose-binding lectin, thereby initiating inflammatory immune responses (Zhong et al., 2023). Recent therapeutic strategies aimed at microbiota modulation, including fecal transplants and probiotic supplementation, have shown promise in offering new therapeutic benefits for PC patients, with fecal transplant therapies currently undergoing phase one clinical trials. The potential therapeutic impact of beneficial bacteria, such as Aspergillus oryzae and Lactobacillus, in inducing PC cell death and reducing gemcitabine drug resistance, brought new hope for cancer treatments (Chen et al., 2020).
In recent years, antibiotics have gradually been utilized to sensitize anti-tumor drugs and assist in prolonging the survival of cancer patients. However, the presence of antibiotic-resistant bacteria posed a significant challenge, potentially weakening antibiotic effectiveness and, in some cases, promoting tumor progression. Studies have shown that the survival rates of PC patients can be positively influenced by the application of quinolones in cases with high levels of K. pneumoniae, whereas resistance to these antibiotics can adversely affect patient outcomes (Konishi et al., 2021). The adjunct use of antibiotics, such as ciprofloxacin, has been observed to mitigate drug resistance, particularly against gemcitabine in colon cancer, highlighting the role of antibiotics in enhancing the response to immunosuppressive treatment (Mohindroo et al., 2021). The recent surge in extensive drug resistance and pan-drug resistance strains posed a significant global health challenge (Weniger et al., 2021), emphasizing the importance of identifying resistance genes for informed antibiotic selection in PC treatment. Despite the critical need, research into the resistome of tumor-associated bacteria remained limited. Moreover, few studies have validated the presence and genomic features of such tumor-associated bacteria using culture-based approaches combined with high-resolution sequencing. This study aims to systematically investigate the gut microbial composition and resistome in both metastatic and non-metastatic PC patients, identify potential microbial biomarkers and functional pathways, and provide evidence for the translocation and colonization of antibiotic-resistant K. pneumoniae from the gut to the tumor microenvironment through culture-based and genomic validation. By integrating metagenomic analysis of the fecal microbiome with whole-genome sequencing of tumor-derived isolates, this study seeks to investigate the microbial signatures associated with metastasis, chemoresistance, and tumor microenvironment modulation in PC.
2 Materials and methods
2.1 Ethical considerations
This investigation was conducted with strict adherence to ethical guidelines, ensuring informed consent and voluntary participation from all involved. The study cohort comprised 24 participants, including 13 pancreatic cancer (PC) patients, 1 intraductal papillary mucinous neoplasm (IPMN) patient, and 10 cohabitating spouses or family members of the PC patients. The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Medical Science Research Ethics Committee of the First Hospital of China Medical University (protocol code [2022] 329 in July 2022). Informed consent was obtained from all subjects involved in the study.
2.2 Participant recruitment and sample collection
For metagenomic study, ten PC patients with a confirmed PC diagnosis with surgery and pathology were recruited between 2022 and 2023 from the First Hospital of China Medical University in Shenyang (Table 1). This included 5 patients with metastases (Group MPC: A1-A5) and 5 without (Group NMPC: B1-B5). Group MPC, A1, A4, and A5 were identified as those who experienced rapid recurrence within three months following surgery. 10 healthy controls (HCs), matched on age, gender, lifestyle, and dietary habits, were selected from the patient’s immediate circles (CA1-CA5 and CB1-CB3, CB5), with one exception where a patient’s daughter served as the control (CB4). To minimize inter-individual variability and control for lifestyle-related confounding factors, controls were selected from the patients’ immediate social networks (household members or close contacts). This matching strategy was chosen to ensure that the control group shared similar dietary habits, living environment, and healthcare access with the PC patients. Although this approach may reduce between-group differences, it maximizes the likelihood of identifying microbial signatures and antibiotic resistance gene (ARG) profiles specifically associated with PC rather than external exposures. Exclusion criteria encompassed recent antibiotic use within 3 months, probiotic or antifungal use within 1 month, significant physical impairments, and the presence of postoperative infections. The control group was screened according to the same fundamental criteria applied to PC patients, with the added stipulation of a minimum six-month antibiotic-free period. Fecal samples were collected 8 weeks after the discontinuation of antibiotics, with antibiotic use restricted to a maximum of 5 days, in postoperative patients. One patient (A3) in the MPC group did not undergo surgery; thus, samples were collected prior to the initiation of treatment. All samples were prepared in duplicate for analytical and backup purposes, and stored at −80 °C in sterile tubes.

Table 1. Clinical and demographic features of PCs and HCs.
For bacterial culture and following third-generation sequencing, pancreatic tumor tissue and pancreatic fluid were collected intraoperatively from four patients diagnosed with PC and IPMN under approved institutional ethical guidelines. Tissue homogenates and fluid samples were inoculated onto Columbia blood agar plates and incubated at 37°C for 18–24 hours under aerobic conditions. Control samples included operating room and non-tumorous surgical materials.
2.3 Metagenomic study for fecal samples
2.3.1 DNA extraction and metagenomic sequencing
Genomic DNA was extracted using the TIANamp Stool DNA Kit as per the manufacturer’s guidelines, with its concentration and purity verified through agarose gel electrophoresis and UV absorbance measurements (NanoDrop ND1000). Sequencing libraries were prepared using the Illumina TruSeq® DNA PCR-Free Sample Preparation Kit, assessed for quality, and sequenced on an Illumina platform to achieve 150 bp paired-end reads, which were filtered based on quality score (minimum Q30 for 90% of bases), removal of adapter sequences, and minimum read length (≥50 bp), generating approximately 10 GB of clean data per sample. The clean data were evaluated for sequencing quality using MultiQC. Data output statistics for both the raw and cleaned data were generated using ReSeqTools. Further, host-derived reads were removed by aligning sequences against the human genome using BMTagger, as recommended by NCBI.
2.3.2 Microbiome characterization
Kraken2 (Simpson et al., 2021) was used to process all the metagenomic sequencing data and Bracken (Lu et al., 2017) was for correction. A cladogram was produced by GraPhlAn (Wood et al., 2019). HUMAnN3 (Franzosa et al., 2018) (nucleotide-database: chocophlan; protein-database: uniref 90) software was performed to determine microbial pathways and abundances.
2.3.3 Metagenome assembly and identification of ARGs
Assigned based on specific barcode and primer sequences, paired-end reads underwent stringent quality control. High-quality reads were assembled using MEGAHIT, with open reading frames predicted by MetaGeneMark and subsequently clustered to minimize redundancy. The assembled genomes were evaluated for completeness and contamination, with taxonomic profiling of open reading frames conducted via DIAMOND against a comprehensive microbial database. ARG sequences were classified into more than 20 categories using data from the Comprehensive Antibiotic Resistance Database (CARD), followed by taxonomic assignment of ARG-carrying contigs using Kraken2. Host-ARG associations were further refined using stringent thresholds and strong correlation analyses, including sequence coverage (≥90%) and identity (≥95%). Additionally, the identified ARGs were cross-checked against two public databases: NT (Nucleotide Sequence Database) and RefSeq (NCBI Reference Sequence Database).
2.4 Isolation and characterization of bacteria from PC tissue and pancreatic fluid
2.4.1 Whole genome sequencing and assembly
To validate the presence of K. pneumoniae identified via metagenomics, we employed aerobic culture using blood agar medium, guided by the taxonomic and resistance profiles derived from fecal sequencing, a strategy focused on this specific facultative pathogen rather than on broad microbial diversity. Given that K. pneumoniae was the most prominent species enriched in PC patients and showed strong ARG associations, the culture conditions were specifically optimized for its isolation and downstream genomic confirmation. A total of 14 K. pneumoniae isolates from tumor tissue (PANC strains) and pancreatic fluid (PANF strains) were subjected to whole-genome sequencing using the Oxford Nanopore MinION platform. Genomic DNA was extracted using a QIAamp DNA Mini Kit (Qiagen) and quantified using a Qubit fluorometer. Libraries were prepared with the Rapid Barcoding Kit (Oxford Nanopore Technologies) and sequenced with R9.4.1 flow cells. Base calling was performed using Guppy, and reads were assembled de novo with Flye (v2.9).
2.4.2 Genome annotation and functional gene prediction
The assembled contigs were annotated using ABRicate (v1.0.1). ARGs were identified using the CARD (Comprehensive Antibiotic Resistance Database), while virulence factors were annotated against the VFDB (Virulence Factors Database). Genomic islands and gene clusters of interest were visualized on circular genome plots using CGView.
2.4.3 Phylogenetic analysis and comparative genomics
To investigate evolutionary relationships among isolates, core genome alignments were generated using Roary and multiple sequence alignments were performed with MAFFT. A maximum likelihood phylogenetic tree was constructed using FastTree and visualized with iTOL. Tree topologies were annotated with presence/absence heatmaps for virulence and resistance genes, as well as predicted antibiotic resistance phenotypes.
2.5 Statistical analysis
Alpha diversity was quantified using the Shannon index and analyzed with R software, employing the Wilcox.test for between-group comparisons. Metastats facilitated the identification of significant taxonomic differences, while network analysis, supported by Pearson’s rank correlations in Cytoscape, elucidated microbial and ARGs co-occurrence patterns. Significance was established at a p-value threshold of <0.05. To compare the species differences of group PC with group HC, categorical data were tested using the”edgeR”package (Pereira et al., 2018) (calcNormFactors: trimmed mean of M-values method). The statistical analysis for differentially expressed (DE) was done using edgeR (glmLRT test). Significant differences of in functional pathways among groups were determined by performing the”EasyAovWlxPlot.R” package. The microbial community heatmap was clustered and visualized by the “ComplexHeatmap” package. To account for potential false positives arising from multiple hypothesis testing, we have applied false discovery rate (FDR) corrections using the Benjamini-Hochberg procedure throughout the study. The Spearman rank correlation coefficient was used to evaluate the correlation between phenotypes and the correlation between microbiome features. Correlations with corresponding empirical p-values less than 0.05 were retained.
3 Results
3.1 Illumina sequencing read statistics
Illumina sequencing generated on average 11.6 GB of base-called data across 20 metagenomics libraries (Supplementary Table S1). The sequencing depth was verified to be adequate, as indicated by the stability of rarefaction curves using the Shannon index, observed species, and the Chao1 estimator (Supplementary Figure S1). Approximately 91.2% of bases across all samples achieved an average Phred score of Q30 or above, indicating high-quality of nucleobase that generated by DNA. A total 1,561,203 high-quality (length > 500bp) assembled contigs were generated from all 20 samples with a range of 23,678–154,881 sequences per sample (Supplementary Table S2). Regarding the contigs number and length of all samples, no significant differences were observed between cases and controls either between the MPC group NMPC group.
3.2 Alterations of microbiome communities
To investigate the differences in gut microbiota between PC and the paired HC group, a metagenomic analysis was conducted. The whole bacterial diversity of the gut microbiota in PC and HC groups was shown in Figure 1B. Dividing the PC group into NMPC and MPC subgroups, we observed distinct microbial communities. The top ten phylum and genus of bacteria (Figures 1C, D) particularly revealed differences at the genus levels between NMPC, MPC and HC groups. Dominant genera such as Bacteroides fragilis, Escherichia coli and Parabacteroides merdae in NMPC fecal samples, and Phoceaicola dorei, Bacteroides uniformis and Bacteroides thetaiotaomicron in MPC fecal samples were particularly contrasting with those in HC fecal samples (Figures 1E, F).

Figure 1. Compositional analysis of bacterial gut microbiota. (A) Metagenomic study design process diagram. (B) The taxonomic tree for different samples at genus taxonomic level. Different taxonomic type was represented by different color of nodes. (C, D) Different compositions of gut microbiota from HC, MPC and NMPC groups’ fecal sample at the phylum level (C), at the genus level (D). (E, F) The corresponding microbial composition analysis of each sample and relative frequency of the top 10 at the phylum level (E), at the genus level (F). Certain icons used in subfigure A were obtained from Flaticon (https://www.flaticon.com) under proper license.
3.3 Analysis of microbiome diversity
Extending our examination to the role of the microbiome in PC patients, principal coordinate analysis was utilized to assess beta diversity. Our results indicated distinct clustering within the NMPC group, in contrast to the MPC and HC groups (Figure 2A). Then alpha diversity at the phylum and class level were conducted for diversity comparison. The Shannon index value for the NMPC group was significantly higher than that for the HC group at phylum level, indicating increased bacterial diversity in fecal samples of the NMPC than in the HC group. No significant differences were observed between MPC and HC groups (Figure 2B). These data suggested enhanced microbial gut microbiota diversity within gut microbiota of PC patients, particularly in NMPC subgroup.
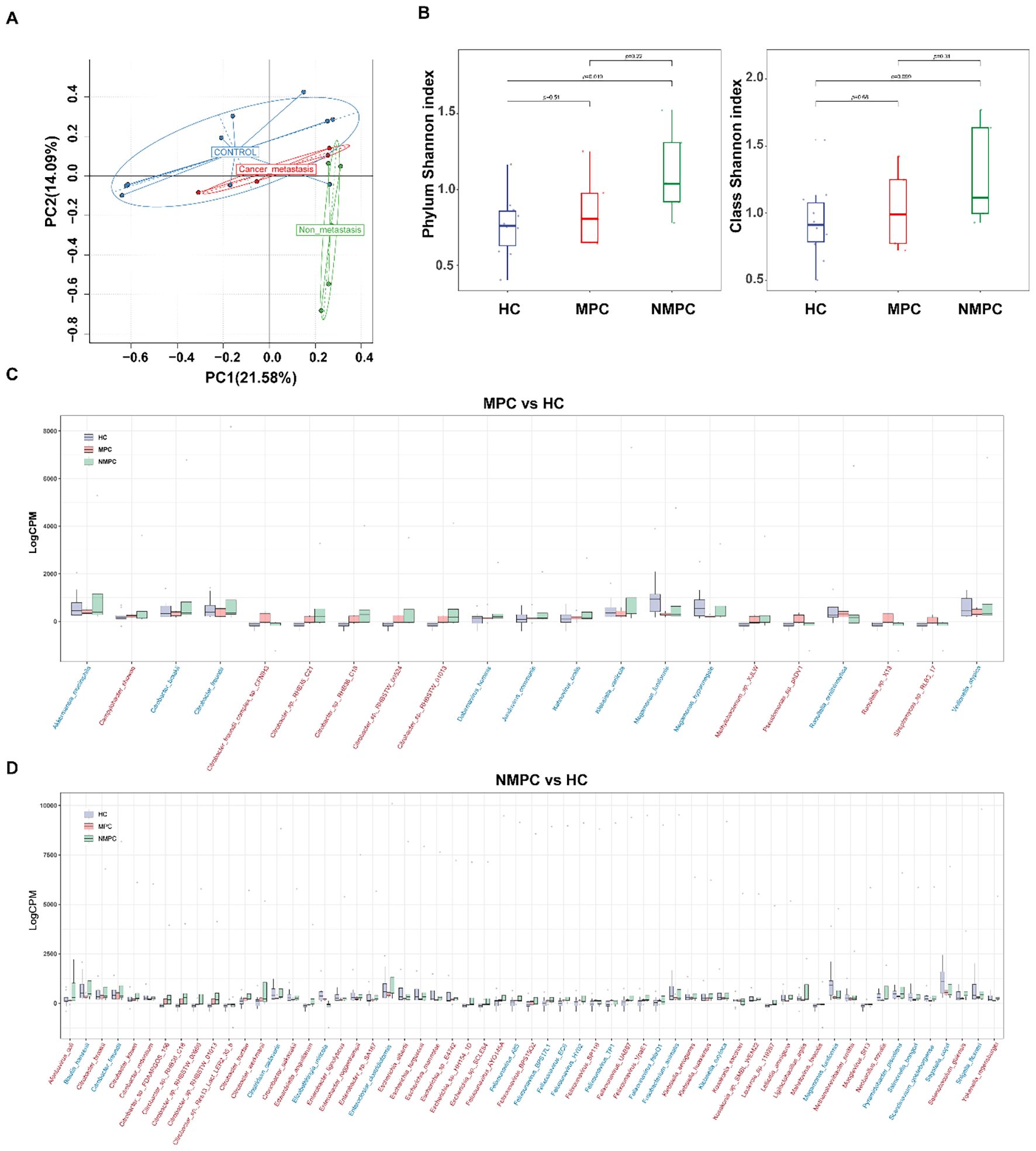
Figure 2. Changes of gut microbiota in PC patients. (A) Principal component analysis (PCA) of gut microbiota profiles (species level) from NMPC, MPC, and HC groups. The first two principal components (PC1 and PC2) explain ~36% of the total variance across samples and reflect the major axes of microbial community differentiation. (B) Alpha diversity assessed by the Shannon index across NMPC, MPC, and HC groups. Boxplots show medians and interquartile ranges; statistical significance determined by Wilcoxon test. (C, D) Differentially abundant bacterial species in fecal samples from MPC (C, n = 21 species) and NMPC (D, n = 59 species) compared to HC, identified using the edgeR package (glmLRT test, p < 0.05). Each dot represents a sample’s relative abundance. Horizontal axes list species names, with red labels indicating increased abundance and blue labels indicating decreased abundance in MPC or NMPC versus HC. Vertical axes show normalized relative abundance. Horizontal lines denote the median values across samples in each group.
3.4 Changes of species abundance
Upon examining changes of relative abundance in bacterial community, we noted distinct patterns between MPC (Figure 2C) and NMPC (Figure 2D) when compared to HC group. Significant differences in 21 species were observed between MPC and HCs, with 19 increased and 2 decreased species. Conversely, the NMPC and HC comparison revealed 59 species with significant abundance shifts, with 56 increased and 3 decreased species. Notably, both MPC and NMPC groups exhibited significant depletion of Megamonas funiformis, known for its probiotic properties. Moreover, the presence of Felixounavirus, Citrobacter, Klebsiella, Escherichia and Raoultella was most significantly linked to PC. The normalized abundance of dominant species in the gut microbiota across samples was shown as a heatmap (p < 0.05) in Figure 3A. Correlations between dominant species and clinical features of PC patients were further explored. The results included the strong and positive correlation of the genus Citrobacter (especially Citrobacter sp. RHB35_C21, Citrobacter sp. RHB36_C18 and Citrobacter sp. RHBSTW 01013) with both total and direct bilirubin levels, however, a relatively weak and positive correlation with CEA in blood. It’s also worthy to mention that the significant correlations were observed between PC biomarkers of CEA, CA12-5, CA15–3 and Citrobacter freundii or Citrobacter braakii species. A microbial community heatmap with cluster analysis, and the color intensity in each grid shows the percentage in a sample, referring to the color key at the right (Figure 3B). Microbial genus Citrobacter was significantly clustered in the PC group. These results suggested the potential role of the differential bacterial species within the gut microbiota in the diagnosis of PC patients.
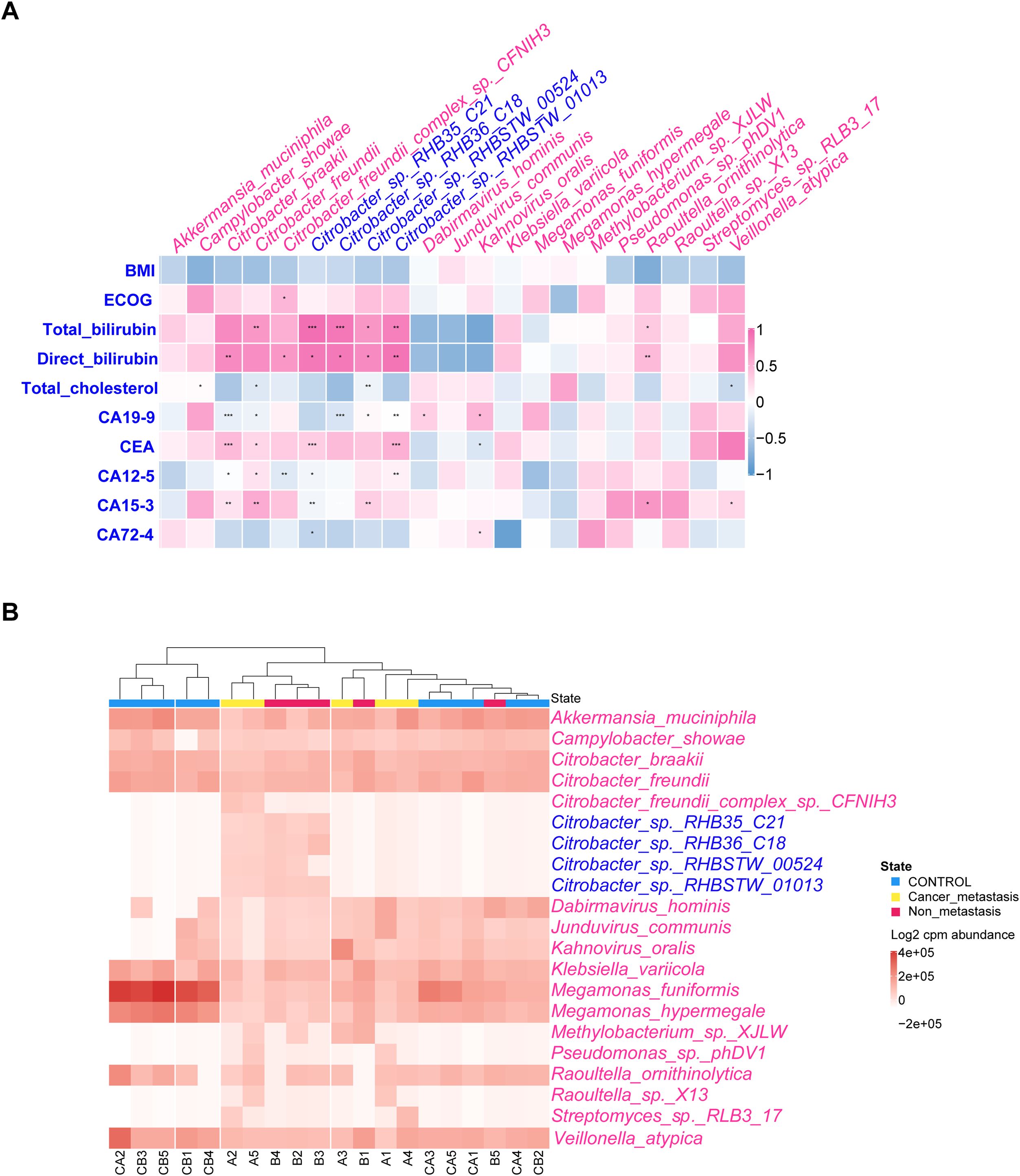
Figure 3. Correlation between dominant species and clinical features of PC patients. (A) Correlation of relative abundance of differentially expressed bacterial species and PC’s clinical features by Spearman’s rank correlation. The pink and blue heat map representing positive and negative correlation, respectively, *p < 0.05, **p < 0.01, ***p < 0.001. (B) Microbial community heatmap with cluster analysis, and the color intensity in each grid showed the percentage in a sample, referring to color key at the right.
3.5 Functional enrichment analysis of microbial metabolic pathways
We further explored the enrichment of functional pathways in microbial metabolism. Analysis of metagenomic data identified 18 significantly altered metabolic pathways (p < 0.05, Figure 4A), with the MPC group showing a significant increase in HSERMETANA-PWY compared to the HC group, and in PWY 7456 compared to the NMPC group. Among the pathways showing significant changes in the NMPC group, there were 9 increased pathways and 7 decreased pathways. The classification of functional pathways as amino acid (ARGININE SYN4 PWY, BRANCHED CHAIN AA SYN PWY, PWY 6292, COBALSYN PWY, HSERMETANA PWY), carbohydrate (PWY 6588, CENTFERM PWY, GLYCOLYSIS, PWY 5484, ANAEROFRUCAT PWY, PWY 6590), nucleotide metabolism (PWY 7220, 7221, 7222, 7228), and fatty acid (PWY66 429, PWY0 1477) were summarized in Table 2. Their corresponding stratified contributions analysis of HSERMETANA-PWY (Figure 4B), ARGININE SYN4 PWY (Figure 4C) and PWY 6588 pyruvate (Figure 4D) and other pathways (Supplementary Figure S2) also demonstrated the significant metabolic alterations among MPC and NMPC patients.
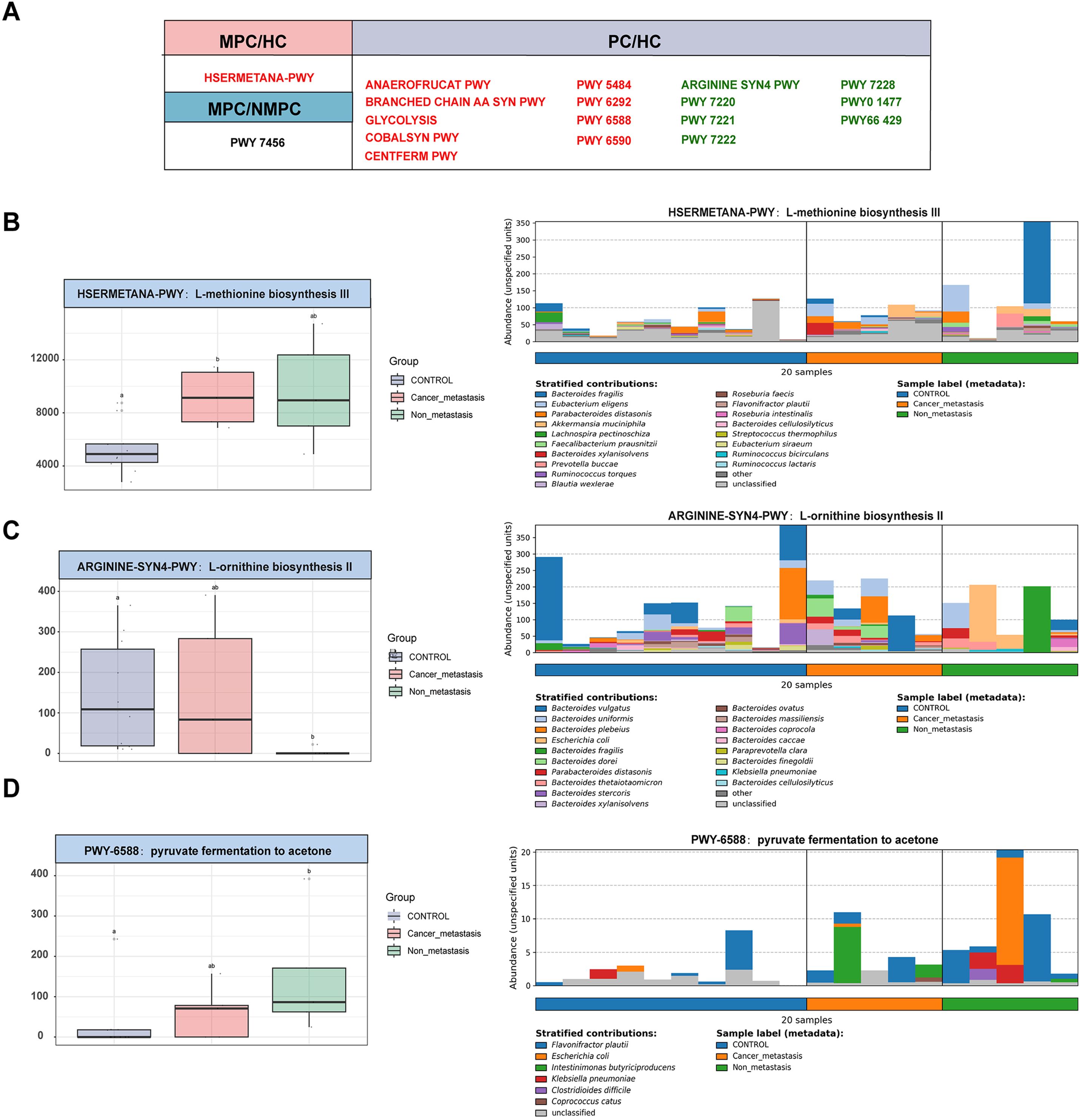
Figure 4. Enrichment of microbial metabolic pathways. (A) Significant alterations of 18 metabolic pathway of each sample among gut microbiota in MPC, NMPC group and paired HCs. Red color represented a significant increase of microbial abundance compared to HC, while green color represented a decrease. Stratified contributions of the microbial pathways of (B) HSERMETANA-PWY, (C) ARGININE_SYN4 PWY and (D) PWY 6588 pyruvate were showed as examples.
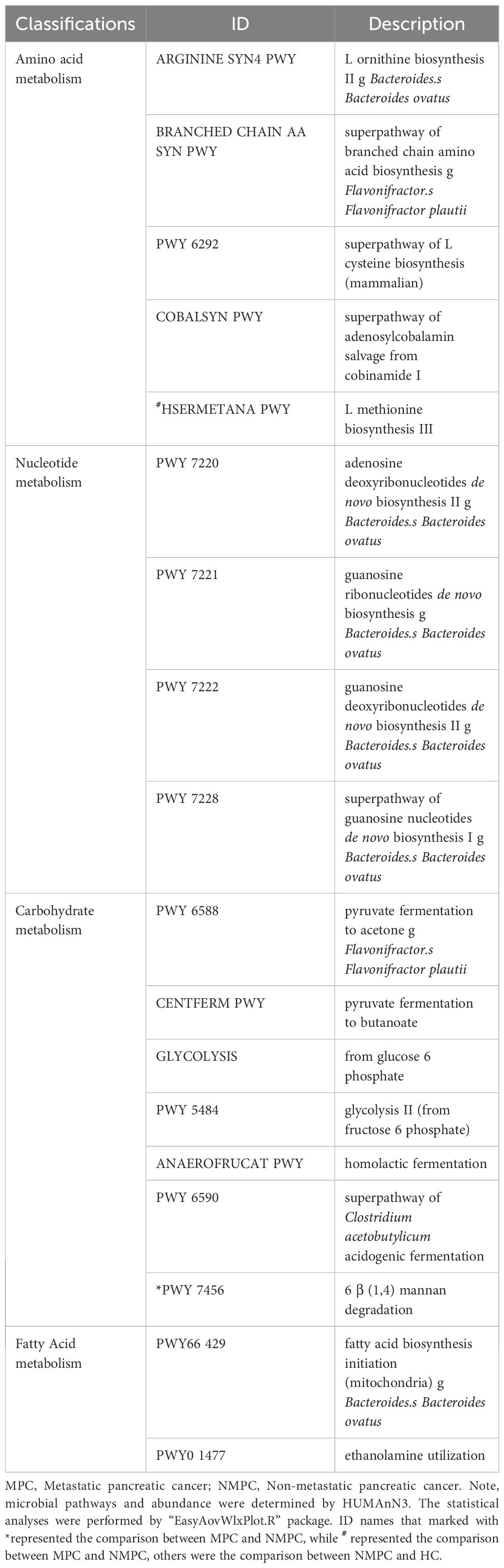
Table 2. Enriched 18 differently expressed metabolic pathways in HC, MPC and NMPC groups.
3.6 Occurrence of gut microbial ARG profiles
A total of 653 ARG subtypes conferring resistance to 37 different antibiotic classes were detected across all fecal samples, with 130 overlapping ARGs present in all samples, accounting for 38%-47% of ARGs detected in each group (Figure 5A). The gene tetQ, encoding resistance to tetracycline, was the most common across samples, except in group NMPC (Figure 5B). Resistance genes to tetracycline had the highest frequency in all sample types, especially in HC group samples. In addition to tetQ, adeF (the multidrug efflux transporter) and macrolide resistance genes (ErmB and ErmF) were also prevalent (Figure 5B). The detected ARG subtypes represent major resistance mechanisms, including cellular protection, antibiotic inactivation, efflux pumps, and antibiotic target alteration.
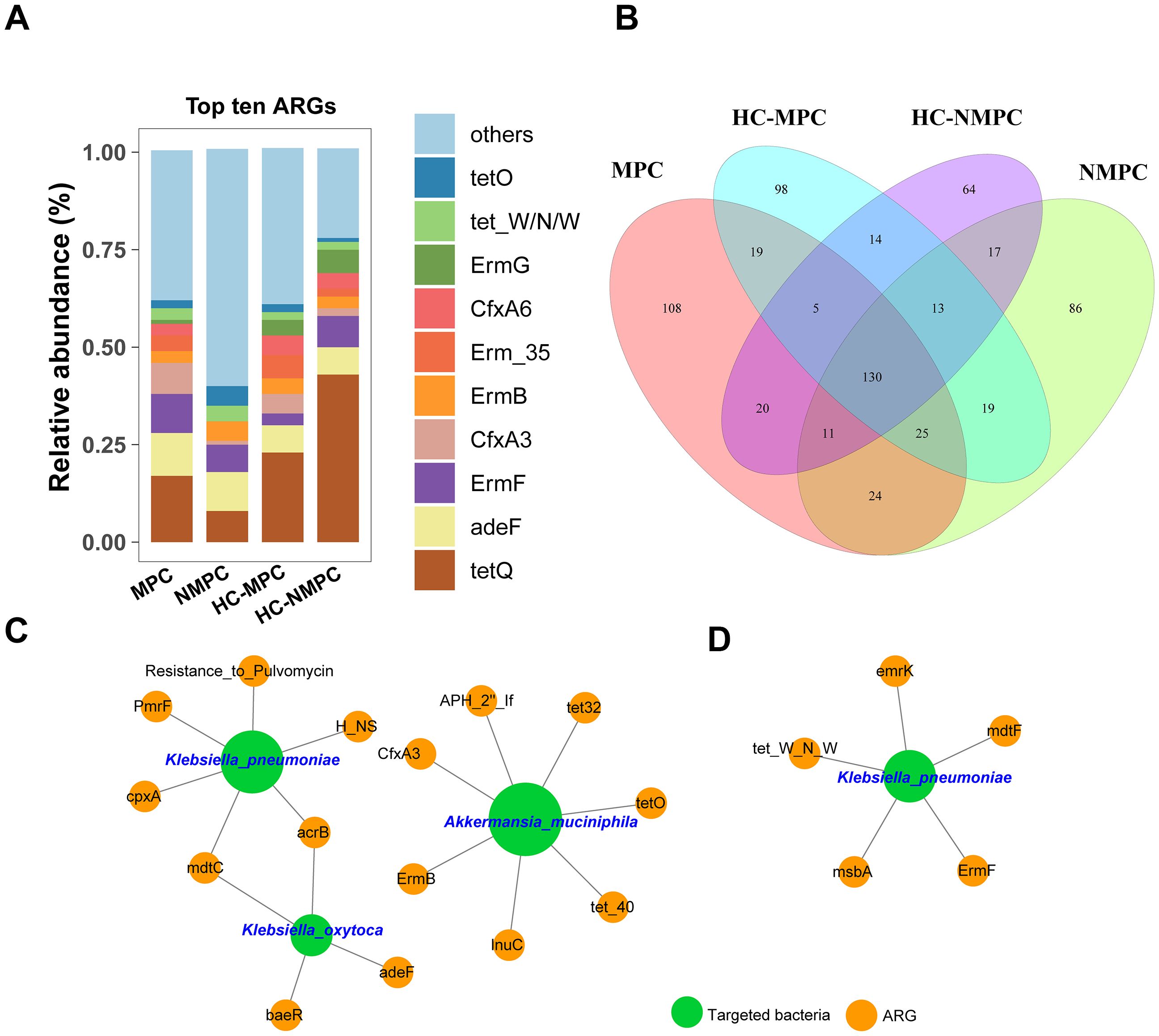
Figure 5. Profiling and network analysis of antibiotic resistance genes (ARGs) in fecal samples from different participant groups. (A) Stacked bar chart showing the relative abundance of the top 10 dominant ARG subtypes across four groups. (B) Venn diagram depicting the distribution of shared and distinct ARG subtypes within the samples of four groups. (C, D) representing the network analysis between the targeted bacterial species and ARGs conducted in fecal samples of group MPC and NMPC, respectively. The charts have specifically been drawn to highlight which ARGs and targeted bacteria are actually linked. A connection represents an extremely strong (Pearson’s r >0.9) and significant (p < 0.001) correlation. The nodes with green and orange colors represent targeted bacteria and ARGs, respectively. The size of each node is proportional to the number of connections between nodes.
3.7 Co-occurrence pattern between targeted bacteria and ARGs
Network analysis was employed to investigate co-occurrence patterns between microbial taxa and ARG subtypes. Three bacterial species were identified as probable ARG hosts based on co-occurrence analysis results (Figures 5C, D), specifically K. pneumoniae, Klebsiella oxytoca, and A.muciniphila, all of which have been implicated in the differentially expressed species of PC patients. Notably, these strong connections were observed exclusively in samples from the NMPC group, with stronger correlations in samples from PC with metastases (Figure 5C) and K. pneumoniae, K. oxytoca, and A. muciniphila, all of which have been implicated in the differentially expressed species of PC patients. Notably, these strong connections were observed exclusively in samples from the PC group, with stronger correlations in samples from PC with metastases (Figure 5C, Supplementary Table S3) compared to those without metastases (Figure 5D, Supplementary Table S4). Both K. pneumoniae and K. oxytoca, classified as Gammaproteobacteria, commonly carried the MdtC and ArcB genes, associated with multidrug resistance and the regulation of detoxification-related genes, respectively. K. pneumoniae was also a potential host for the polymyxin resistance gene (pmrF) and an efflux pump pump gene (cpxA), whereas A. muciniphila harbored up to seven ARGs, including those conferring resistance to tetracycline (tet32, tet40, tetO), MLS (ermB), β-lactam (cfxA3), and lincomycin (lnuC). Moreover, K. pneumoniae was associated with five ARG subtypes, including resistance genes for tetracycline (tet_W_N_W), MLS (ermF), and multidrugs (mdtF, msbA, and emrK). Finally, we filtered and validated the ARGs carried by the targeted host bacteria, including K. pneumoniae, Klebsiella oxytoca, and A.muciniphila. The results were presented in Supplementary Table S5, with associations between ARGs and host bacteria in samples from PC patients highlighted in bold for clarity.
3.8 Isolation and identification of K. pneumoniae from PC tissue and pancreatic fluid
In the initial stage of this study, fecal metagenomic sequencing of PC patients revealed a strong positive correlation between K. pneumoniae and a broad array of ARGs, including key efflux pump-related genes such as acrB, mdtC, cpxA, baeR, and H-NS. These genes are known to play critical roles in multidrug resistance mechanisms and suggested a potential for stable intestinal colonization and resistance development. To validate these findings and assess possible bacterial translocation beyond the gut, we collected tumor tissue and pancreatic fluid samples intraoperatively from 3 PC patients (Table 3). Additionally, samples obtained from a patient with IPMN were utilized as negative controls to ensure the specificity of our observations. Blood agar culturing revealed that in one patient (Case 1), K. pneumoniae was successfully isolated from both tumor tissue and pancreatic fluid (Figure 6A). Mass spectrometry-based species identification confirmed high-confidence hits of K. pneumoniae in both compartments (PANC1 and PANF1), while no bacterial growth was observed in the control group or other patients (Figure 6B). This result suggested that K. pneumoniae may colonize the tumor-associated microenvironment or migrate retrogradely through the pancreatic duct system.
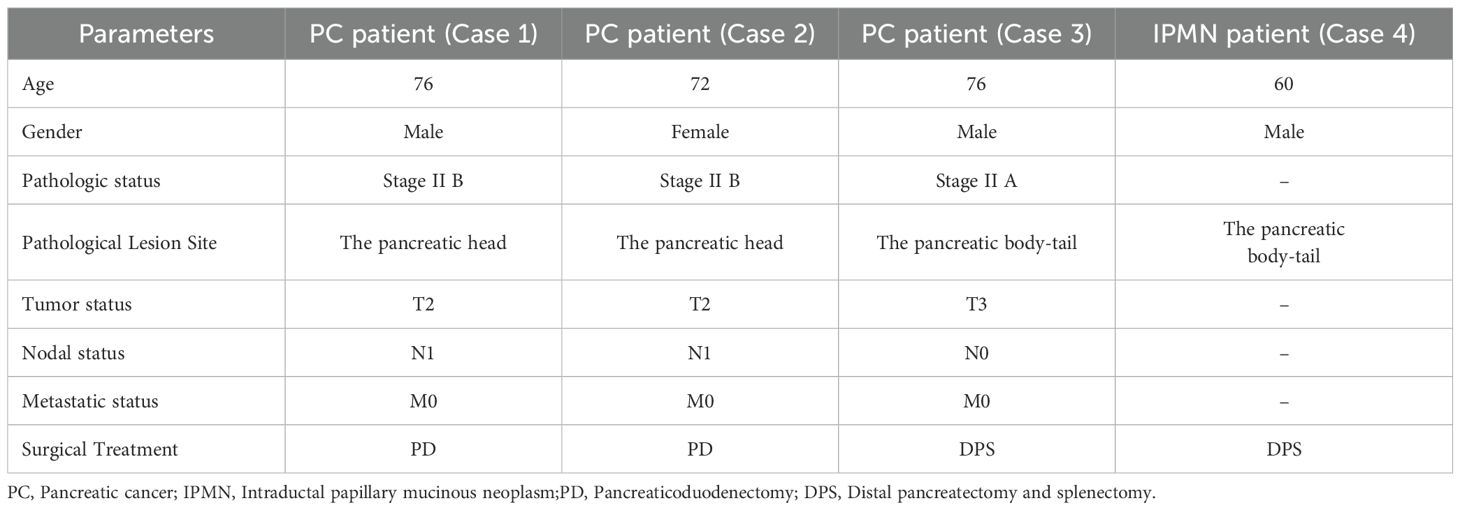
Table 3. Clinical and demographic features of PC and IPMN patients.
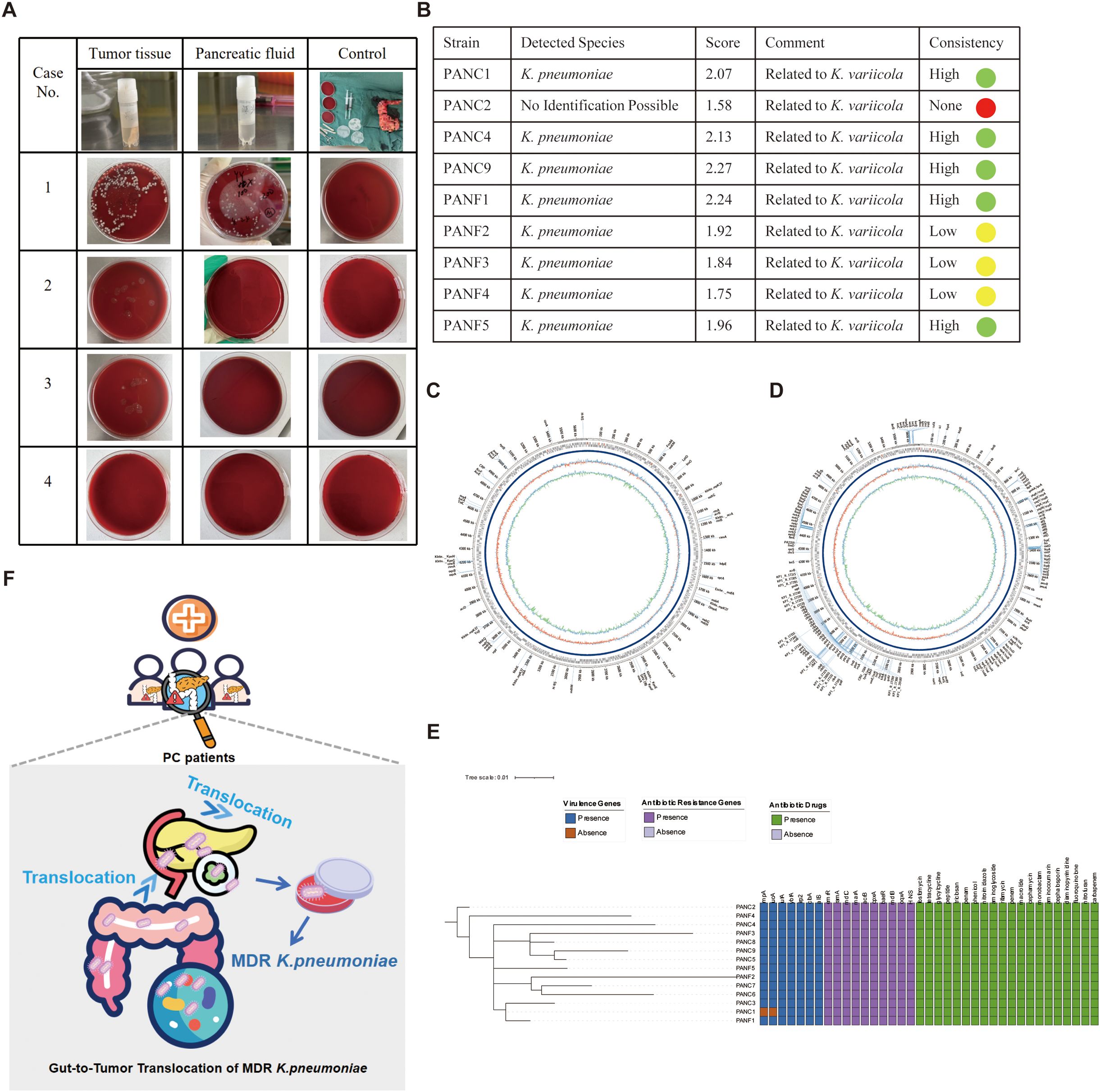
Figure 6. Isolation, identification, genomic and functional profiling of K. pneumoniae from pancreatic tumor tissue and pancreatic fluid. (A) Blood agar culture results of tumor tissue and pancreatic fluid from four patients with pancreatic cancer. Among them, only Case 1 yielded colonies from both tumor tissue and pancreatic fluid, while the other three cases showed no visible growth. Controls from operating room remained sterile. (B) MALDI-TOF MS (Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry) identification of bacterial isolates. Eight strains were confirmed as K. pneumoniae with high identification scores and high consistency, and most were closely related to K. variicola, indicating potential phylogenetic affiliation. (C–D) Circular genome maps of one K. pneumoniae strain (PANC1 from tumor tissue) generated using Oxford Nanopore long-read sequencing. The assembled genome was annotated using ABRicate with two different databases: CARD for (C) and VFDB (Virulence Factors Database) for virulence genes (D). The outermost rings display coding sequences (CDSs) on both DNA strands, with annotated resistance and virulence genes labeled. The inner rings represent GC content (green/red) and GC skew (purple/blue). Resistance genes and virulence genes are prominently marked, illustrating the multidrug-resistant and hypervirulent potential of the strain. (E) Phylogenetic tree and heatmap of 14 K. pneumoniae strains (PANC, from tumor tissue; PANF, from pancreatic fluid) based on whole genome sequencing. The left dendrogram shows genomic relationships, while heatmaps to the right illustrate the presence (colored) or absence (blank) of virulence genes (blue), antibiotic resistance genes (purple), and corresponding antibiotic resistance phenotypes (green). The PANC1 and PANF1 strains from the same patient clustered closely, suggesting a common origin. The figure highlights diverse resistance and virulence profiles among the isolates. (F) Schematic illustration of the translocation of MDR K. pneumoniae from the gut to pancreatic tumor tissue. Certain icons used in subfigure A were obtained from Flaticon (https://www.flaticon.com) under proper license.
3.9 Whole-genome sequencing confirms ARG and virulence profiles consistent with fecal metagenomics
To further characterize the genetic basis of these strains, we performed whole-genome sequencing (WGS) of 14 K. pneumoniae isolates using Oxford Nanopore long-read technology. The assembled circular genomes of PANC1 and PANF1 demonstrated conserved genomic architecture with rich annotations of both ARGs and virulence factors (Figures 6C, D). Notably, long-read WGS confirmed the presence of several resistance determinants previously identified in fecal metagenomes, including acrB, mdtC, cpxA, baeR, H-NS, and multiple emr-family efflux genes (emrK, emrD, etc.), validating the consistency of ARG profiles across intestinal and extraintestinal compartments. In addition, core genome phylogenetic analysis (Figure 6E, left) revealed high genomic similarity between tumor- and fluid-derived isolates, supporting a common clonal origin or intra-host migration. The heatmap summary (Figure 6E, right) showed that nearly all strains carried classic hypervirulence genes (rmpA, iucA, ybtA, wzi) and a wide spectrum of ARGs, including blaSHV-190, oqxA/B, and fosA, as well as resistance to aminoglycosides, cephalosporins, and carbapenems. Taken together, these results suggest that K. pneumoniae can translocate from the gut to tumor tissues and pancreatic fluid, maintaining its MDR and virulent nature. The convergence of metagenomic and isolate-based ARG detection underscores the potential clinical risk posed by intratumoral colonization of MDR K. pneumoniae, which may impair the efficacy of chemotherapeutic agents such as gemcitabine and increase the likelihood of treatment resistance.
4 Discussion
Pancreatic cancer (PC) remains one of the deadliest malignancies in the digestive tract, with limited survival improvements despite advances in precision oncology. Although surgery remains the most effective treatment for PC, postoperative recurrence and metastasis remain urgent clinical challenges. The absence of precise biomarkers and specific early symptoms contributes to delayed diagnosis and poor prognosis. While smoking, alcohol consumption, type 2 diabetes, chronic pancreatitis, and familial genetic predisposition are recognized as key risk factors for PC (Rawla et al., 2019), many patients develop the disease without presenting these feature. Recent evidence has highlighted the influence of host-associated microbial communities on PC development, suggesting their potential as early diagnostic markers for PC (Riquelme et al., 2019). In this study, we systematically analyzed gut microbiota signatures in metastatic and non-metastatic PC patients through metagenomic profiling. Given the impact of host environment on microbiota (Lao et al., 2024), we included matched HCs to control for inter-individual variation. The strict inclusion criteria and pair-matched design, while constraining the sample size, were instrumental in identifying a reliable microbial signature with minimal confounding. The agreement between computational and culture-based evidence not only confirms the gut-to-tumor translocation of MDR (Figure 7). Although aerobic culture but also provides compelling biological validation for this process. Therefore, this work should be regarded as a foundational study that provides a validated target and a methodological framework for subsequent large-scale validation. Our data revealed significantly increased microbial diversity in PC patients compared to HCs, with distinct community structures between NMPC and MPC.
Figure 7. Certain icons used in subfigure A were obtained from Flaticon (https://www.flaticon.com) under proper license.
Importantly, the MPC group included patients with confirmed metastasis and those who experienced rapid postoperative recurrence, suggesting that gut microbiota profiles may carry prognostic value. These results support previous studies linking microbiota to PC progression (Liu et al., 2024). Specifically, 59 and 21 species showed significant differences between PC/HC and MPC/HC, respectively. We confirmed the depletion of M. funiformis (Zhou et al., 2021) in PC patients and observed a reduction in Citrobacter freundii, a species previously linked to methionine γ-lyase with potential anti-tumor effects (Kharofa et al., 2023). Interestingly, Veillonella atypica, previously reported in tumor and oral microbiota (Raboni et al., 2018), was significantly reduced in the gut of MPC patients. Additionally, pathogens such as Edwardsiella anguillarum, Lelliottia amnigena, Klebsiella variicola, and Shigella flexneri showed altered abundance, which has not been reported in PC before (McKinley et al., 2023). Microbiota signatures also revealed clinical associations that may provide functional insights. The infection-associated genera Citrobacter (Koeninger et al., 2021) and Raoultella ornithinolytica (Seixas et al., 2021), both known gastrointestinal pathogens, were positively associated with elevated bilirubin levels in PC patients. These results further emphasized the interaction between gut-derived bacteria and systemic inflammation in PC. Our analysis also revealed significant alterations in 18 metabolic pathways in PC patients, with notable contributions from specific microbial species, particularly A. muciniphila and K. pneumoniae, to these metabolic changes. Of particular significance was the marked alteration of the L-methionine pathway in MPC patients. This observation was of substantial clinical relevance, as methionine has been well-documented to play crucial roles in both intestinal function and barrier integrity (Chen et al., 2014), as well as in tumor metastasis, including PC progression (Govaerts et al., 2021; He et al., 2022). These findings were further substantiated by emerging evidence demonstrating the pivotal role of gut microbiota in host methionine metabolism (Sanderson et al., 2019; Kaiser, 2020; Zhao and Lum, 2022).
Currently, the growing recognition of the role of pathogenic bacterial overgrowth within tumors in tumorigenesis and chemoresistance (Cruz et al., 2024).The intratumoral microbiota plays an emerging role in shaping chemotherapy response and tumor immune environment. Gammaproteobacteria in tumors have been shown to inactivate gemcitabine via cytidine deaminase isoforms (Sayin and Mitchell, 2023), reducing its efficacy. On the other hand, tryptophan-derived indole-3-acetic acid has been reported to amplify gemcitabine effects (Tintelnot et al., 2023). Another study of high ethanolamine (EA) levels not only proved to be associated with worse survival (Battini et al., 2017), but also proved to participate in Klebsiella’s adaptive drug resistance (Norsigian et al., 2019). Adjunctive antibiotics like quinolones have been proposed to overcome resistance (Weniger et al., 2021). However, the wide spread of ARGs among gut microbes may hinder these interventions. Our results further implicated K. pneumoniae as a central contributor to ARG burden and potential chemoresistance in PC. Both K. pneumoniae and K.oxytoca (Gammaproteobacteria) demonstrated significant enrichment in PC samples, particularly among patients in the MPC patients. Notably, these bacterial species exhibited strong associations with diverse antibiotic ARGs, with K. pneumoniae showing prominent correlation. Moreover, the intraoperative isolation of MDR K. pneumoniae from matched tumor and pancreatic fluid provides direct microbiological evidence for our central hypothesis of gut-to-tumor translocation, confirming its role as a clinically significant pathogen in PC. A previous study suggested bacteria class Gammaproteobacteria (e.g., K. pneumoniae) harbored in PC tumor was able to metabolize the gemcitabine to the inactive 2’, 2’-difluorodeoxyuridine through a long isoform of the enzyme cytidine deaminase (Ertz-Archambault et al., 2017; Geller et al., 2017; Sayin and Mitchell, 2023; Horvat et al., 2024). Moreover, bacteria within tumors can inactivate chemotherapeutic agents such as gemcitabine via bacterial enzymes like cytidine deaminase (CDD) (Pushalkar et al., 2018). Notably, ARGs such as acrB, mdtC, cpxA, baeR, H-NS, and emr genes were frequently co-localized with K.pneumoniae, suggesting potential roles in chemoresistance.
With an intact intestinal barrier, the pancreas and its alkaline secretions were historically considered a sterile environment. However, studies have identified the presence of intratumoral microbiota within PC tissues, which has been linked to PC outcomes (Riquelme et al., 2019; Abe et al., 2024). Moreover, the presence of gut microbiota in pancreatic tissues under normal conditions (Sammallahti et al., 2021) highlights the physiological relevance of the gut–pancreas axis (Ahuja et al., 2017). Although these results provide valuable insights, the role of ARGs within intratumoral microbiota in pancreatic tissues remains significantly understudied in current research literature. To directly validate gut–tumor translocation, we isolated K. pneumoniae strains from tumor and pancreatic fluid in PC patients (Figure 6F). Out of four patients sampled, one yielded positive cultures from both tumor tissue and pancreatic fluid; 14 isolates underwent Nanopore sequencing. Although aerobic culture on a single medium may limit the detection of obligate anaerobes or fastidious organisms, our approach was hypothesis-driven: based on metagenomic findings indicating K. pneumoniae as a key ARG carrier in PC patients, we designed culture conditions specifically to isolate and validate this species from tumor and pancreatic fluid samples. The successful recovery of 14 strains with concordant genomic features supports the targeted nature of our cultivation strategy.” Core genome-based phylogeny revealed close relatedness between PANC and PANF strains, indicating intra-patient spread. These isolates harbored the same resistance genes identified in metagenomic analysis, including efflux pump (acrB, mdtC) cells, regulators (H-NS, cpxA) (Nikaido, 2009; Paczosa and Mecsas, 2016), and membrane transport genes (emr family). These findings align with the concept of gut microbes seeding the tumor microenvironment via anatomical continuity or barrier disruption (Pushalkar et al., 2018; Riquelme et al., 2019). Tumor-associated hypervirulence loci (rmpA, iucA, ybtA) further suggest these bacteria may evade host defenses and persist in hostile environments (Lam et al., 2019). Consequently, our findings provided a focused validation of a key pathobiont but do not represent a comprehensive census of the viable tumor microbiota. Future studies employing multi-media culturomics and anaerobic techniques will be crucial to fully elucidate the ecological complexity and functional roles of the entire microbial community in PC. As the antibiotic treatment also required the involvement of adaptive immunity thereby improving the tumor microenvironment, suggesting that the action was not simply through a direct inhibitory effect on tumorigenesis (Horvat et al., 2024) Therefore, with consideration of ARGs, the presence of resistant K. pneumoniae in pancreatic fluid increases the risk of post-operative infections and may necessitate broader-spectrum prophylaxis.
While computational biology has successfully identified genetic biomarkers and immunomodulatory targets in PC (Kaviyaprabha et al., 2025; Tian et al., 2025), our findings have important implications for understanding cancer metastasis, chemoresistance management, and microbial biomarker development in PC. Through non-invasive fecal metagenomic screening, we identified PC patients carrying tumor-resident K. pneumoniae with coexisting ARGs and virulence factors, raising concerns about both postoperative infections and reduced chemotherapy efficacy. Our phylogenetic data support intra-patient spread and point to the need for gut microbiota surveillance. More importantly, by employing an innovative approach to track ARG transmission patterns, we provide compelling evidence that the translocation and intratumoral colonization of MDR K. pneumoniae implicates the gut-tumor axis as a clinically relevant pathway influencing both chemoresistance and immune microenvironment remodeling. This evidence supported a multidimensional therapeutic strategy integrating conventional treatments with microbiota-targeting interventions to overcome chemoresistance rooted in bacterial colonization. Future multi-center studies that also account for genetic influences will be essential to validate and generalize these findings. In conclusion, K. pneumoniae may act as a gut-derived pathobiont in PC, bridging microbial dysbiosis and therapeutic resistance.
5 Conclusion
By integrating metagenomic data with genomic evidence from isolates, this work provides direct evidence of the gut-to-tumor translocation of MDR K. pneumoniae in pancreatic cancer. The presence of these ARG-carrying strains links the gut microbiome to chemoresistance and postoperative infection risk, suggesting that fecal metagenomic monitoring could serve as a predictive tool for personalizing patient management.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA1032163.
Ethics statement
The studies involving humans were approved by The Medical Science Research Ethics Committee of the First Hospital of China Medical University, affiliated to the Ethics Committee of China Medical University. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
LZ: Funding acquisition, Writing – original draft, Writing – review & editing. SP: Data curation, Methodology, Writing – review & editing. MG: Methodology, Data curation, Writing – original draft. BX: Visualization, Investigation, Writing – original draft. XZ: Investigation, Writing – original draft. TY: Software, Writing – original draft. SY: Writing – original draft, Investigation. CZ: Writing – original draft, Methodology. JL: Writing – original draft. ZM: Writing – review & editing, Project administration. HM: Supervision, Funding acquisition, Writing – review & editing, Conceptualization.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This research was funded by (Natural Science Foundation of Liaoning Province, China) grant number (No. 2023JH2/20200047), (No. LJKMZ20221213) and (Jiangsu Commission of Health, China) grant number (No.M2022069). HM was the principal investigator of Project No. LJKMZ20221213, with primary responsibilities in Conceptualization, Supervision, and Writing – review & editing. LZ contributed to Project No. LJKMZ20221213 as the lead researcher in Methodology, Resources, Writing – original draft, and Writing – review & editing. SP served as the principal investigator of Project No. M2022069, overseeing Methodology, Data curation, and Writing – review & editing.
Acknowledgments
We also thank E-GENE (Shenzhen) for technical assistance of sequence data analyzing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2025.1694479/full#supplementary-material
Abbreviations
PC, pancreatic cancer; MPC, metastatic pancreatic cancer; NMPC, non-metastatic pancreatic cancer; ARG, antibiotic resistance gene; TME, tumor microenvironment; MDR, multidrug-resistant.
References
Abe, S., Masuda, A., Matsumoto, T., Inoue, J., Toyama, H., Sakai, A., et al. (2024). Impact of intratumoral microbiome on tumor immunity and prognosis in human pancreatic ductal adenocarcinoma. J. Gastroenterol. 59, 250–262. doi: 10.1007/s00535-023-02069-5
Ahuja, M., Schwartz, D. M., Tandon, M., Son, A., Zeng, M., Swaim, W., et al. (2017). Orai1-mediated antimicrobial secretion from pancreatic acini shapes the gut microbiome and regulates gut innate immunity. Cell Metab. 25, 635–646. doi: 10.1016/j.cmet.2017.02.007
Battini, S., Faitot, F., Imperiale, A., Cicek, A. E., Heimburger, C., Averous, G., et al. (2017). Metabolomics approaches in pancreatic adenocarcinoma: tumor metabolism profiling predicts clinical outcome of patients. BMC Med. 15, 56. doi: 10.1186/s12916-017-0810-z
Chen, S. M., Chieng, W. W., Huang, S. W., Hsu, L. J., and Jan, M. S. (2020). The synergistic tumor growth-inhibitory effect of probiotic Lactobacillus on transgenic mouse model of pancreatic cancer treated with gemcitabine. Sci. Rep. 10, 20319. doi: 10.1038/s41598-020-77322-5
Chen, Y., Li, D., Dai, Z., Piao, X., Wu, Z., Wang, B., et al. (2014). L-methionine supplementation maintains the integrity and barrier function of the small-intestinal mucosa in post-weaning piglets. Amino Acids 46, 1131–1142. doi: 10.1007/s00726-014-1675-5
Cruz, M. S., Tintelnot, J., and Gagliani, N. (2024). Roles of microbiota in pancreatic cancer development and treatment. Gut Microbes 16, 2320280. doi: 10.1080/19490976.2024.2320280
Dwivedi, M., Sanyal, S., Singh, S., Dwivedi, M., and Sanyal, S. (2025). Target and gene-based therapeutic strategies against pancreatic cancer: current and future prospects. Curr. Gene Ther. 25, 417–432. doi: 10.2174/0115665232320846240910055032
Ertz-Archambault, N., Keim, P., and Von Hoff, D. (2017). Microbiome and pancreatic cancer: A comprehensive topic review of literature. World J. Gastroenterol. 23, 1899–1908. doi: 10.3748/wjg.v23.i10.1899
Fan, X., Alekseyenko, A. V., Wu, J., Peters, B. A., Jacobs, E. J., Gapstur, S. M., et al. (2018). Human oral microbiome and prospective risk for pancreatic cancer: a population-based nested case-control study. Gut 67, 120–127. doi: 10.1136/gutjnl-2016-312580
Franzosa, E. A., McIver, L. J., Rahnavard, G., Thompson, L. R., Schirmer, M., Weingart, G., et al. (2018). Species-level functional profiling of metagenomes and metatranscriptomes. Nat. Methods 15, 962–968. doi: 10.1038/s41592-018-0176-y
Geller, L. T., Barzily-Rokni, M., Danino, T., Jonas, O. H., Shental, N., Nejman, D., et al. (2017). Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 357, 1156–1160. doi: 10.1126/science.aah5043
Govaerts, C. W., van Dijken, B. R., Stormezand, G. N., van der Weide, H. L., Wagemakers, M., Enting, R. H., et al. (2021). (11)C-methyl-L-methionine PET measuring parameters for the diagnosis of tumour progression against radiation-induced changes in brain metastases. Br J Radiol. 94, 20210275. doi: 10.1259/bjr.20210275
He, D., Feng, H., Sundberg, B., Yang, J., Powers, J., Christian, A. H., et al. (2022). Methionine oxidation activates pyruvate kinase M2 to promote pancreatic cancer metastasis. Mol. Cell. 82, 3045–3060. doi: 10.1016/j.molcel.2022.06.005
Horvat, N. K., Karpovsky, I., Phillips, M., Wyatt, M. M., Hall, M. A., Herting, C. J., et al. (2024). Clinically relevant orthotopic pancreatic cancer models for adoptive T cell transfer therapy. J. Immunother. Cancer 12, e008086. doi: 10.1136/jitc-2023-008086
Kaviyaprabha, R., Miji, T. V., Sreelakshmi, P. S., Muthusami, S., Arulselvan, P., and Bharathi, M. (2025). Unveiling the potential role of hesperetin and emodin as a combination therapy to inhibit the pancreatic cancer progression against the C-met gene. Protein Pept. Lett. 32, 280–298. doi: 10.2174/0109298665363165250225100109
Kharofa, J., Haslam, D., Wilkinson, R., Weiss, A., Patel, S., Wang, K., et al. (2023). Analysis of the fecal metagenome in long-term survivors of pancreas cancer. Cancer 129, 1986–1994. doi: 10.1002/cncr.34748
Kirsoy, F., Yalniz, M., Bahcecioglu, I. H., Artas, H., Turkoglu, S., Solmaz, O., et al. (2024). The gut-pancreas axis: investigating the relationship between microbiota metabolites and pancreatic steatosis. Intern. Emerg. Med. 19, 1887–1896. doi: 10.1007/s11739-024-03685-6
Koeninger, L., Osbelt, L., Berscheid, A., Wendler, J., Berger, J., Hipp, K., et al. (2021). Curbing gastrointestinal infections by defensin fragment modifications without harming commensal microbiota. Commun. Biol. 4, 47. doi: 10.1038/s42003-020-01582-0
Konishi, H., Isozaki, S., Kashima, S., Moriichi, K., Ichikawa, S., Yamamoto, K., et al. (2021). Probiotic Aspergillus oryzae produces anti-tumor mediator and exerts anti-tumor effects in pancreatic cancer through the p38 MAPK signaling pathway. Sci. Rep. 11, 11070. doi: 10.1038/s41598-021-90707-4
Lam, M. M. C., Wyres, K. L., Wick, R. R., Judd, L. M., Fostervold, A., Holt, K. E., et al. (2019). Convergence of virulence and MDR in a single plasmid vector in MDR Klebsiella pneumoniae ST15. J. Antimicrob. Chemother. 74, 1218–1222. doi: 10.1093/jac/dkz028
Lao, I. J., Berry, J., Li, J., Balogun, Z., Elgohari, B., Skinner, H., et al. (2024). Prognostic factors and outcomes associated with neck lymphedema in head and neck cancer survivors. Laryngoscope 134, 3656–3663. doi: 10.1002/lary.31396
Li, Y., Liang, X., Li, H., and Chen, X. (2023). Comparative efficacy and safety of immune checkpoint inhibitors for unresectable advanced melanoma: A systematic review and network meta-analysis. Int. Immunopharmacol 115, 109657. doi: 10.1016/j.intimp.2022.109657
Liu, X., Li, K., Yang, Y., Cao, D., Xu, X., He, Z., et al. (2024). Gut resistome profiling reveals high diversity and fluctuations in pancreatic cancer cohorts. Front. Cell Infect. Microbiol. 14. doi: 10.3389/fcimb.2024.1354234
Lu, J., Breitwieser, F. P., Thielen, P., and Salzberg, S. L. (2017). Bracken: estimating species abundance in metagenomics data. PeerJ Comput. Sci. 3, e104. doi: 10.7717/peerj-cs.104
McKinley, K. N. L., Herremans, K. M., Riner, A. N., Vudatha, V., Freudenberger, D. C., Hughes, S. J., et al. (2023). Translocation of oral microbiota into the pancreatic ductal adenocarcinoma tumor microenvironment. Microorganisms 11, 1466. doi: 10.3390/microorganisms11061466
Mendez, R., Kesh, K., Arora, N., Di Martino, L., McAllister, F., Merchant, N., et al. (2020). Microbial dysbiosis and polyamine metabolism as predictive markers for early detection of pancreatic cancer. Carcinogenesis 41, 561–570. doi: 10.1093/carcin/bgz116
Mohindroo, C., Hasanov, M., Rogers, J. E., Dong, W., Prakash, L. R., Baydogan, S., et al. (2021). Antibiotic use influences outcomes in advanced pancreatic adenocarcinoma patients. Cancer Med. 10, 5041–5050. doi: 10.1002/cam4.3870
Ni, Y., Lei, J., Huang, W., Wang, J., Guo, H., Lv, F., et al. (2023). Systematic review of the perioperative immunotherapy in patients with non-small cell lung cancer: evidence mapping and synthesis. Front. Oncol. 13. doi: 10.3389/fonc.2023.1092663
Nikaido, H. (2009). Multidrug resistance in bacteria. Annu. Rev. Biochem. 78, 119–146. doi: 10.1146/annurev.biochem.78.082907.145923
Norsigian, C. J., Attia, H., Szubin, R., Yassin, A. S., Palsson, B. O., Aziz, R. K., et al. (2019). Comparative genome-scale metabolic modeling of metallo-beta-lactamase-producing multidrug-resistant klebsiella pneumoniae clinical isolates. Front. Cell Infect. Microbiol. 9. doi: 10.3389/fcimb.2019.00161
Paczosa, M. K. and Mecsas, J. (2016). Klebsiella pneumoniae: going on the offense with a strong defense. Microbiol. Mol. Biol. Rev. 80, 629–661. doi: 10.1128/mmbr.00078-15
Pereira, M. B., Wallroth, M., Jonsson, V., and Kristiansson, E. (2018). Comparison of normalization methods for the analysis of metagenomic gene abundance data. BMC Genomics 19, 274. doi: 10.1186/s12864-018-4637-6
Pushalkar, S., Hundeyin, M., Daley, D., Zambirinis, C. P., Kurz, E., Mishra, A., et al. (2018). The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discov. 8, 403–416. doi: 10.1158/2159-8290.CD-17-1134
Raboni, S., Revtovich, S., Demitri, N., Giabbai, B., Storici, P., Cocconcelli, C., et al. (2018). Engineering methionine gamma-lyase from Citrobacter freundii for anticancer activity. Biochim. Biophys. Acta Proteins Proteom 1866, 1260–1270. doi: 10.1016/j.bbapap.2018.09.011
Rawla, P., Sunkara, T., and Gaduputi, V. (2019). Epidemiology of pancreatic cancer: global trends, etiology and risk factors. World J. Oncol. 10, 10–27. doi: 10.14740/wjon1166
Riquelme, E., Zhang, Y., Zhang, L., Montiel, M., Zoltan, M., Dong, W., et al. (2019). Tumor microbiome diversity and composition influence pancreatic cancer outcomes. Cell 178, 795–806 e712. doi: 10.1016/j.cell.2019.07.008
Sammallahti, H., Kokkola, A., Rezasoltani, S., Ghanbari, R., Asadzadeh Aghdaei, H., Knuutila, S., et al. (2021). Microbiota alterations and their association with oncogenomic changes in pancreatic cancer patients. Int. J. Mol. Sci. 22, 12978. doi: 10.3390/ijms222312978
Sanderson, S. M., Gao, X., Dai, Z., and Locasale, J. W. (2019). Methionine metabolism in health and cancer: a nexus of diet and precision medicine. Nat. Rev. Cancer 19, 625–637. doi: 10.1038/s41568-019-0187-8
Sayin, S. and Mitchell, A. (2023). Functional assay for measuring bacterial degradation of gemcitabine chemotherapy. Bio Protoc. 13, e4797. doi: 10.21769/BioProtoc.4797
Seixas, R., Alves, A., Selaru, A., Vanzeller, M., Shiang, T., and Conde, S. (2021). Raoultella ornithinolytica in MALT-type non-Hodgkin Lymphoma. Eur. J. Case Rep. Intern. Med. 8, 3023. doi: 10.12890/2021_003023
Sethi, V., Kurtom, S., Tarique, M., Lavania, S., Malchiodi, Z., Hellmund, L., et al. (2018). Gut microbiota promotes tumor growth in mice by modulating immune response. Gastroenterology 155, 33–37 e36. doi: 10.1053/j.gastro.2018.04.001
Sharma, R., Kumar, S., Ghosh, R., Komal, K., and Kumar, M. (2025). Gene therapy: transforming the battle against pancreatic cancer. Curr. Gene Ther. doi: 10.2174/0115665232364196250131102330
Siegel, R. L., Miller, K. D., Fuchs, H. E., and Jemal, A. (2022). Cancer statistics 2022. CA Cancer J. Clin. 72, 7–33. doi: 10.3322/caac.21708
Siegel, R. L., Miller, K. D., Wagle, N. S., and Jemal, A. (2023). Cancer statistics 2023. CA Cancer J. Clin. 73, 17–48. doi: 10.3322/caac.21763
Simpson, R. C., Shanahan, E., Scolyer, R. A., and Long, G. V. (2021). Targeting the microbiome to overcome resistance. Cancer Cell 39, 151–153. doi: 10.1016/j.ccell.2021.01.016
Thomas, R. M. and Jobin, C. (2020). Microbiota in pancreatic health and disease: the next frontier in microbiome research. Nat. Rev. Gastroenterol. Hepatol. 17, 53–64. doi: 10.1038/s41575-019-0242-7
Tian, G., Liu, C., Che, C., Ren, S., Zhang, K., Zhou, P., et al. (2025). Identifying ARRB2 as a Prognostic Biomarker and Key Player in the Tumor Microenvironment of Pancreatic Cancer through scPagwas Methodology. Curr. Gene Ther. doi: 10.2174/0115665232427614250904061700
Tintelnot, J., Xu, Y., Lesker, T. R., Schonlein, M., Konczalla, L., Giannou, A. D., et al. (2023). Microbiota-derived 3-IAA influences chemotherapy efficacy in pancreatic cancer. Nature 615, 168–174. doi: 10.1038/s41586-023-05728-y
Vuijk, F. A., de Muynck, L., Franken, L. C., Busch, O. R., Wilmink, J. W., Besselink, M. G., et al. (2020). Molecular targets for diagnostic and intraoperative imaging of pancreatic ductal adenocarcinoma after neoadjuvant FOLFIRINOX treatment. Sci. Rep. 10, 16211. doi: 10.1038/s41598-020-73242-6
Weniger, M., Hank, T., Qadan, M., Ciprani, D., Michelakos, T., Niess, H., et al. (2021). Influence of Klebsiella pneumoniae and quinolone treatment on prognosis in patients with pancreatic cancer. Br. J. Surg. 108, 709–716. doi: 10.1002/bjs.12003
Wood, D. E., Lu, J., and Langmead, B. (2019). Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257. doi: 10.1186/s13059-019-1891-0
Zhao, T. and Lum, J. J. (2022). Methionine cycle-dependent regulation of T cells in cancer immunity. Front. Oncol. 12. doi: 10.3389/fonc.2022.969563
Zheng, B., Qu, J., Ohuchida, K., Feng, H., Chong, S. J. F., Yan, Z., et al. (2020). LAMA4 upregulation is associated with high liver metastasis potential and poor survival outcome of Pancreatic Cancer. Theranostics 10, 10274–10289. doi: 10.7150/thno.47001
Zhong, H., Liu, S., Zhu, J., and Wu, L. (2023). Associations between genetically predicted levels of blood metabolites and pancreatic cancer risk. Int. J. Cancer 153, 103–110. doi: 10.1002/ijc.34466
Keywords: antibiotic resistance gene (ARG), gut microbiome, pancreatic cancer, whole-genome sequencing, metagenomic analysis, Klebsiella pneumoniae
Citation: Zhao L, Peng S, Ge M, Xing B, Zhao X, Yang T, Yu S, Zhang C, Liu J, Miao Z and Ma H (2025) Gut-to-tumor translocation of multidrug-resistant Klebsiella pneumoniae shapes the microbiome and chemoresistance in pancreatic cancer. Front. Cell. Infect. Microbiol. 15:1694479. doi: 10.3389/fcimb.2025.1694479
Received: 28 August 2025; Accepted: 04 November 2025; Revised: 03 November 2025;
Published: 02 December 2025.
Edited by:
Samantha Flores-Treviño, Autonomous University of Nuevo León, MexicoReviewed by:
Chen Xue, Gene Hospital of Henan Province, ChinaBaraa Saeed, Ibn Sina University for Medical and Pharmaceutical Sciences, Iraq
Copyright © 2025 Zhao, Peng, Ge, Xing, Zhao, Yang, Yu, Zhang, Liu, Miao and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Heyao Ma, MjAxNDIwNjlAY211LmVkdS5jbg==; Ziwei Miao, endtaWFvQGNtdS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship