 Luan Rocha Lima1
Luan Rocha Lima1 Yasmin Mucunã Mustafá1
Yasmin Mucunã Mustafá1 Paula Luize Camargos Fonseca2
Paula Luize Camargos Fonseca2 Sharton Vinícius Antunes Coelho1
Sharton Vinícius Antunes Coelho1 Pierina Lorencini Parisi3
Pierina Lorencini Parisi3 Camila Lopes Simeoni3
Camila Lopes Simeoni3 Lana Monteiro Meuren1
Lana Monteiro Meuren1 Bruno Braz Bezerra1
Bruno Braz Bezerra1 Nathane Cunha Mebus-Antunes4
Nathane Cunha Mebus-Antunes4 Flavio Matassoli1,5
Flavio Matassoli1,5 Jose Luiz Proença-Modena3
Jose Luiz Proença-Modena3 Renato Santana Aguiar2,6
Renato Santana Aguiar2,6 Luciana Barros de Arruda1*
Luciana Barros de Arruda1*- 1Departamento de Virologia, Instituto de Microbiologia Paulo de Góes, Universidade Federal do Rio de Janeiro (UFRJ), Rio de Janeiro, RJ, Brazil
- 2Departamento de Genética, Ecologia e Evolução, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Belo Horizonte, Brazil
- 3Departamento de Genética, Microbiologia e Imunologia, Instituto de Biologia, Universidade Estadual de Campinas (UNICAMP), Campinas, SP, Brazil
- 4Instituto de Bioquímica Médica Leopoldo De Meis (IBqM), Universidade Federal do Rio de Janeiro (UFRJ), Rio de Janeiro, RJ, Brazil
- 5Laboratory of Immunoregulation, National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health, Bethesda, MD, United States
- 6Instituto D’OR de Pesquisa e Ensino, Rio de Janeiro, Rio de Janeiro, Brazil
Introduction: Zika virus (ZIKV) infection has been associated with neurological syndromes, particularly during outbreaks caused by Asian lineage strains. However, experimental models suggest that African strains may exhibit an equal or more virulent profile. Neuroinvasion by systemic viruses often requires crossing the blood–brain barrier (BBB), which disruption amplifies viral dissemination and neuropathology. Type I interferons (IFNs) are key to restricting ZIKV replication, but their specific role in preserving BBB integrity remains poorly defined.
Methods: Here, we used human brain microvascular endothelial cells (HBMECs) as a simplified BBB model to compare transcriptional responses and IFN modulation following infection with either the African prototype strain ZIKVMR766 or the Asian epidemic strain ZIKVPE243. The role of endothelial cell–mediated IFN responses was further assessed in vivo by intravascular inoculation of mice with endothelial-specific IFNAR depletion using ZIKVMR766.
Results and discussion: Infection of HBMEC with ZIKVMR766 triggered a greater number and broader range of differentially expressed genes, especially ones associated with interferon signaling and translational pathways, whereas ZIKVPE243-infected samples clustered closer to non-infected ones. ZIKVMR766 infection also resulted in higher viral titers and faster dissemination across endothelial monolayers. Both strains induced IFN-β expression but suppressed downstream IFN signaling by reducing STAT1 phosphorylation and promoting STAT2 degradation, with these effects being more pronounced for ZIKVMR766. Despite these evasion mechanisms, neutralization assays revealed that endothelial cells-derived IFNs production and response partially restricted viral replication, preserved HBMEC viability, and protected against barrier disruption, with ZIKVPE243 showing greater sensitivity to IFN-β. Importantly, in vivo infection of mice lacking endothelial IFNAR signaling resulted in elevated CNS viral load and increased lethality following ZIKVMR766 infection, underscoring the pivotal role of endothelial IFN responses in viral control, maintenance of BBB integrity, and protection against neuroinvasion.
1 Introduction
Zika virus (ZIKV), recently renamed Orthoflavivirus zikaense by ICTV, is a single-stranded RNA virus of the Flaviviridae family. First isolated in Uganda in 1947 (Dick et al., 1952; Simpson, 1964), it circulated silently in Africa and Southeast Asia for decades (Saba Villarroel et al., 2024; WHO, 2016), until outbreaks in the Pacific, in 2007 (Duffy et al., 2009; Cao-Lormeau et al., 2014; rev in Weaver et al., 2016), and the Americas, in 2015. Different forms of transmission and association with neuropathologies were evidenced during this period (Schuler-Faccini et al., 2016; de Oliveira et al., 2017; Musso et al., 2019). Two distinct lineages, African and Asian, have been described, with the latter linked to recent epidemics (Haddow et al., 2012; Liu et al., 2019). Although incidence has declined, ZIKV remains endemic in tropical regions (Saba Villarroel et al., 2024; Lackritz et al., 2025), underscoring the importance of continuous surveillance and comparative studies between viral lineages to better understand pathogenesis.
ZIKV infection is usually associated to a mild febrile disease, however, a percentage of infected individuals develop neurologic manifestations, particularly, after vertical transmission (Brasil et al., 2016; Melo et al., 2016; Miranda-Filho et al., 2016). Congenital Zika syndrome (CZS) encompasses a broader spectrum of abnormalities, including cerebral atrophy, intracranial calcifications, ocular anomalies, and arthrogryposis (Brasil et al., 2016; Melo et al., 2016). In experimental models, impaired brain development by ZIKV was associated with abnormal differentiation and death of neural progenitor cells, massive neuronal death, and altered vascular density and diameter, resulting in permeable blood brain barrier (BBB) (Garcez et al., 2016; Li et al., 2016; Shao et al., 2016). Although less frequent, ZIKV can also cause meningitis and meningoencephalitis in adults, which are characterized by cortical and subcortical lesions, temporal lobes hyperintensities, leukocytosis and increased protein levels in the cerebrospinal fluid (CSF) (Carteaux et al., 2016; da Silva et al., 2017). Importantly, the neurotropic and neuroinvasive nature of ZIKV is evidenced by detection of viral RNA or particles in the brains and CSF of fetuses, stillborns, and adults (Mlakar et al., 2016; Carteaux et al., 2016).
Neuroinvasion by systemic viruses often involves virus crossing through the BBB, a complex structure composed of microvascular endothelial cells associated with astrocytes, microglia, and pericytes, which regulates the traffic of cells, solutes, and pathogens into the central nervous system (CNS) (rev in Abbott et al., 2010; Kadry et al., 2020). BBB dysfunction has been implicated as a major determinant of neurological outcomes of multiple viral infections, including herpesviruses, HIV, SARS-CoV-2, and several arboviruses (rev in Mustafa et al., 2019; Boardman et al., 2025). Nonetheless, the molecular pathways through which virus replication and endothelial cell activation impacts BBB integrity during ZIKV remain poorly characterized. So far, only Asian genotype isolates have been linked to congenital and neurological disorders (Musso et al., 2019; Liu et al., 2019). However, experimental models have demonstrated that both lineages can be neuroinvasive, with isolates from African lineages often showing greater pathogenicity (Anfasa et al., 2017; Papa et al., 2017; Bowen et al., 2017; Shao et al., 2017; Tripathi et al., 2017; Lucas et al., 2018; Smith et al., 2017). Therefore, investigating the interaction between ZIKV and the BBB is critical to understanding the mechanisms of ZIKV neuropathogenesis.
Type I interferons play a key role in restricting ZIKV replication in various experimental models (Lazear et al., 2016; Grant et al., 2016; Bayer et al., 2016). While wild-type immunocompetent adult mice are resistant to ZIKV, disruption of type I IFN signaling, through deletion of type I IFN receptor (IFNAR), signal transducer and activator of transcription (STAT1), or interferon regulatory factors (IRF), renders them highly susceptible to viral replication and disease (Dowall et al., 2016; Lazear et al., 2016; Kamiyama et al., 2017). In contrast, ZIKV efficiently replicates in human fibroblasts, monocytes, dendritic cells, and endothelial cells, despite IFN production following PRR activation (Hamel et al., 2015; Papa et al., 2017; Chazal et al., 2018; Hertzog et al., 2018; Luo et al., 2018). Immune evasion strategies such as STAT2 degradation by ZIKV-NS5, reduction of STAT1/2 phosphorylation, and inhibition of IFN by other viral proteins were reported (Grant et al., 2016; Wu et al., 2017; Hertzog et al., 2018; Xia et al., 2018; Lin et al., 2019; Li et al., 2020; Shu et al., 2021; Airo et al., 2022). Also, translational control by lineage-specific upstream open reading frames (uORFs) in the 5′UTR (Lefevre et al., 2024) may further sustain viral protein synthesis under IFN-induced stress. However, it is unclear whether differential modulation of IFN signaling contributes to strain-specific neuroinvasiveness. Indeed, the role of type I IFN signaling in brain endothelial cells, particularly at the BBB, remains poorly understood, not only during ZIKV infection, but also across other virus-induced neuropathological disease.
Human brain microvascular endothelial cell lines (HBMECs) are a simplified model of the BBB that is widely employed in studies of viral infection (Rust et al., 2012; da Conceição et al., 2013; Motta et al., 2023). We and others have demonstrated that HBMECs are permissive to ZIKV infection (Papa et al., 2017; Mladinich et al., 2017), which leads to the induction of type I and III IFNs and proinflammatory mediators (Papa et al., 2017; Mladinich et al., 2017). Notably, Asian lineage isolates (e.g., PE243, PRVABC59) did not induce marked cytopathic effect (CPE) or permeability changes, whereas the African prototype MR766 caused pronounced CPE. Additionally, systemic infection of adult IFNAR-deficient mice with ZIKVMR766, but not ZIKVPE243, resulted in a lethal outcome, with disrupted BBB (Lucas et al., 2018).
Considering the central role of endothelial cells as the structural and functional core of the BBB, and the limited understanding of how IFN responses and viral replication affect these cells relative to other systems, here, we compared the infection dynamics of ZIKVMR766 and ZIKVPE243 in HBMECs, aiming to identify molecular differences that could explain the distinct virulence profiles previously observed. To achieve this, we combined transcriptomic and functional approaches, ranging from global expression profiling in HBMECs to targeted in vitro and in vivo validation assays. Transcriptomic alterations induced by ZIKVMR766 infection in HBMECs were markedly more extensive than those triggered by ZIKVPE243, particularly in pathways related to interferon-mediated antiviral responses and host translational regulation. Functional validation demonstrated that, although ZIKVMR766 induces higher IFN-β levels, it remains more resistant to the antiviral response. Nevertheless, endogenous type I IFN partially limited viral replication and preserved HBMEC viability and barrier integrity. In vivo, mice with endothelial-restricted IFNAR-depletion succumbed to ZIKVMR766 infection, highlighting the critical role of type I IFN signaling in endothelial cells for controlling viral replication and preserving BBB integrity.
2 Materials and methods
2.1 Cells
Culture medium and supplements were acquired from Thermo Fisher Scientific Inc. (Pittsburgh, PA, USA). Vero cells (ATCC-CCL81) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% fetal bovine serum (FBS). BHK-21 cells (ATCC® CCL-10) were cultured in alpha minimum essential medium (α-MEM) supplemented with 10% FBS. Aedes albopictus clone C6/36 cells (ATCC-CLR1660) were cultured in Leibovitz (L15) medium supplemented with 5% FBS, 0.3% tryptose phosphate broth, 0.2% sodium bicarbonate, 0.2 mM non-essential amino acids, and 0.75% L-glutamine. Human brain microvascular endothelial cells (HBMEC) (Nikolskaia et al., 2006) were kindly provided by Dr. Dennis J. Grab (Uniformed Services University of the Health Sciences, Bethesda, MD). The cells were cultured in DMEM, supplemented with 10% FBS. HBMECpISREluc reporter cells, kindly provided by Dr. Laura Gil (IpqAM, FIOCRUZ, Recife, PE, Brazil), were generated from stable transfection of HBMEC with the pISRE-Luc-Hygro reporter vector containing a pISRE-Luc in the NdeI-Bst1107 site (Stratagene, La Jolla, CA) cloned into a pCEP4 vector (Thermo Fisher Scientific). Cells were grown in DMEM, supplemented with 20% FBS, 1% L-glutamine (GlutaMAX), and hygromycin B (50 mg/mL). All cells were maintained at 37°C with a 5% CO2 atmosphere, except for C6/36 cells, which were maintained at 28°C in the BOD incubator.
2.2 Virus
ZIKA strains MR766 (ZIKVMR766; ATCC VR1838); and PE243 (ZIKVPE243; gene bank ref. number KX197192) were kindly provided by Dr. Amilcar Tanuri (Universidade Federal do Rio de Janeiro) and Dr. Ernesto T.A. Marques Jr. (Center for Vaccine Research, University of Pittsburgh, PA), respectively. Stock samples were propagated in C6/36 cells, and the viral titers were determined by plaque assay in Vero cells, as previously described (Coelho et al., 2017). The viruses used in all the assays were from passages three or four. Viral inactivation was performed by 1 h UV exposure and confirmed by RT-qPCR in Vero cells. DENV serotype 2 (DENV-2), Asiatic strain 16681, was propagated in C6/36 cells, and viral titers were determined by plaque assay in BHK-21 cells, as previously described (Meuren et al., 2022). The supernatants of all infected cells were harvested, filtered, and stored at -80°C. The supernatants obtained from noninfected cell lines were used as mock control.
Genomic sequence analyses were performed using sequences deposited in the NCBI with accession codes NC_012532.1 and KX197192.1 for strains MR766 and Brazil/PE243/2015, respectively. Alignments of the 5′UTR nucleotide sequence and the NS5 and capsid protein amino acid sequences were performed to analyze variations between the ZIKV strains MR766 and PE243, using BioEdit (v7.2.5) with the ClustalW algorithm (default parameters), and validated by pairwise comparisons in Jalview (v2.11.5.0). Prediction of secondary structures of the 5’UTR was performed with the RNAfold WebServer, and compared with flaviviruses previously predicted sequences (Lodeiro et al., 2009). Access codes YP_009227196.1, YP_009227205.1, and ANC90426.1, were used for protein sequence analysis. PDB access codes 5U0B and 6C44 were used in this study, and protein structures were represented using PyMOL (v. 2.5).
2.3 In vitro infection
HBMECs were mock treated or inoculated with ZIKV at different MOIs, depending on the assay, and incubated for 2 h at 4°C for virus adsorption (0 hours post-infection - h.p.i.). The cells were then washed with phosphate-buffered saline (PBS) and maintained at 370C/5%CO2 afterwards. In one set of experiments, the cells were treated or not with IFN-β (1000 U/mL – PeproTech, Cranbury, BJ) or poly I:C (50 µg/mL; Merck, Burlington, MA) or with a combination of neutralizing anti-IFN-β and anti-IFN-α (0.2μg/mL; Thermo Fisher Scientific Inc; Cat# 16-9978-81; #MA1-35514) just after virus adsorption. In another experimental setting, the cells were pre-treated with IFN-β for 24 h, washed, and then infected with the indicated viruses. The cells were cultured for different time periods, according to the experiment to be performed. Cell lysates and supernatants were harvested, and viral RNA, cytokines, and ISGs were analyzed by RT-qPCR, whereas the titer of released infectious particles was evaluated by plaque assay (Coelho et al., 2017).
2.4 RNA sequencing and differential expression analysis
HBMECs were mock-treated or infected, as described, using a MOI of 10. At 24 h.p.i., cells were harvested, and total RNA was extracted at 24 hpi using the ReliaPrep™ RNA Miniprep System (Promega), following the manufacturer’s instructions. RNA concentration and purity were determined with a Qubit 4 fluorometer (Thermo Fisher Scientific) and RNA integrity was assessed on a 4150 TapeStation System (Agilent Technologies). Only samples with RNA Integrity Number (RIN) ≥ 8.7 were included.
For library preparation, 100 ng of DNase-treated RNA from three replicates of two independent experiments per group (non-infected, ZIKVPE243, or ZIKVMR766) were used. Sequencing libraries were generated with the Illumina Ribo-Zero Plus rRNA Depletion Kit, Illumina cDNA Synthesis followed by Illumina RNA Prep Ligation. Cleanup steps employed Agencourt RNAClean XP and AMPure XP beads (Beckman Coulter). Libraries were indexed with IDT for Illumina DNA/RNA UD Indexes Set A, quantified using the QIAseq Library Quant Assay Kit (Qiagen), and their average fragment size (~350 bp) was confirmed by High Sensitivity D1000 ScreenTape (Agilent). Sequencing was performed as paired-end (200 cycles) on a NextSeq 500 platform (Illumina).
The raw data obtained from the sequencing passed through an initial quality analysis step. Low-quality sequences (Phred below 30) and Illumina adapters were trimmed with the BBDuk program (Bushnell, 2014). With the trimmed libraries, we used the Bowtie2 program (Langmead and Salzberg, 2012) to map the reads to ZIKV genome (MR766: NC_012532.1; PE243: KX197192.1). The mapped reads were used as input for the SPADES program (Bushmanova et al., 2019) to assemble the viral genome in each condition and to confirm the viral infection.
After assembling the viral genome, we also mapped the reads from each library to the Homo Sapiens genome (GRCh38 - GCA_000001405.29) using the STAR program and quantified them using the Salmon software (Patro et al., 2017). The output generated is a counting matrix that was used as input for the R package DESeq2 (Love et al., 2014) to identify differentially expressed genes (DEGs). Genes with a log2FC ≥1 and adjusted padj<0.05 were used for further analysis. The volcano plot was generated using the R package EnhancedVolcano (Blighe et al., 2024). The annotation (Ensembl) of H. sapiens was obtained using the R package BiomaRt (Durinck et al., 2009), and the packages ClusterProfiler (Xu et al., 2024) and enrichplot (Yu, 2025) were used to generate the Gene-Set Enrichment plots. The libraries sequenced in our study are available at SRA (www.ncbi.nlm.nih.gov/sra) under BioProject accession number PRJNA1337481 and accession numbers SAMN52321194–SAMN52321202.
2.5 Evaluation of ZIKV RNA, IFN and interferon-stimulated genes by quantitative RT-PCR
HBMECs were infected as described, and, after different time points, cells and supernatants were harvested, total RNA was isolated using the TRIzol reagent, and cDNA syntheses were performed with 1 μg RNA and random primers using High-Capacity cDNA Archive Kit mix, following the manufacturer’s protocol (Thermo Fisher Scientific Inc.). The cDNA samples were submitted to RT-qPCR using the TaqMan Universal Master Mix kit or PowerUp SYBR Green Master Mix kit (Thermo Fisher Scientific Inc.) to quantify ZIKV RNA or intracellular expression of IFN-β, OAS1, MX1, IFIT1, ISG15, and GAPDH, respectively. All the primers and probes are described in Table 1, and the reactions were carried out in the AriaMX Real-time PCR System (Agilent Technologies Inc., Santa Clara, CA). The absolute quantification of the ZIKV mRNA copy number occurred from a standard curve established through serial dilutions of a synthetic RNA transcript copy of the ZIKV 2007 sequence (Lanciotti et al., 2008) corresponding to the target fragment of amplification by the set of primers and probes used.
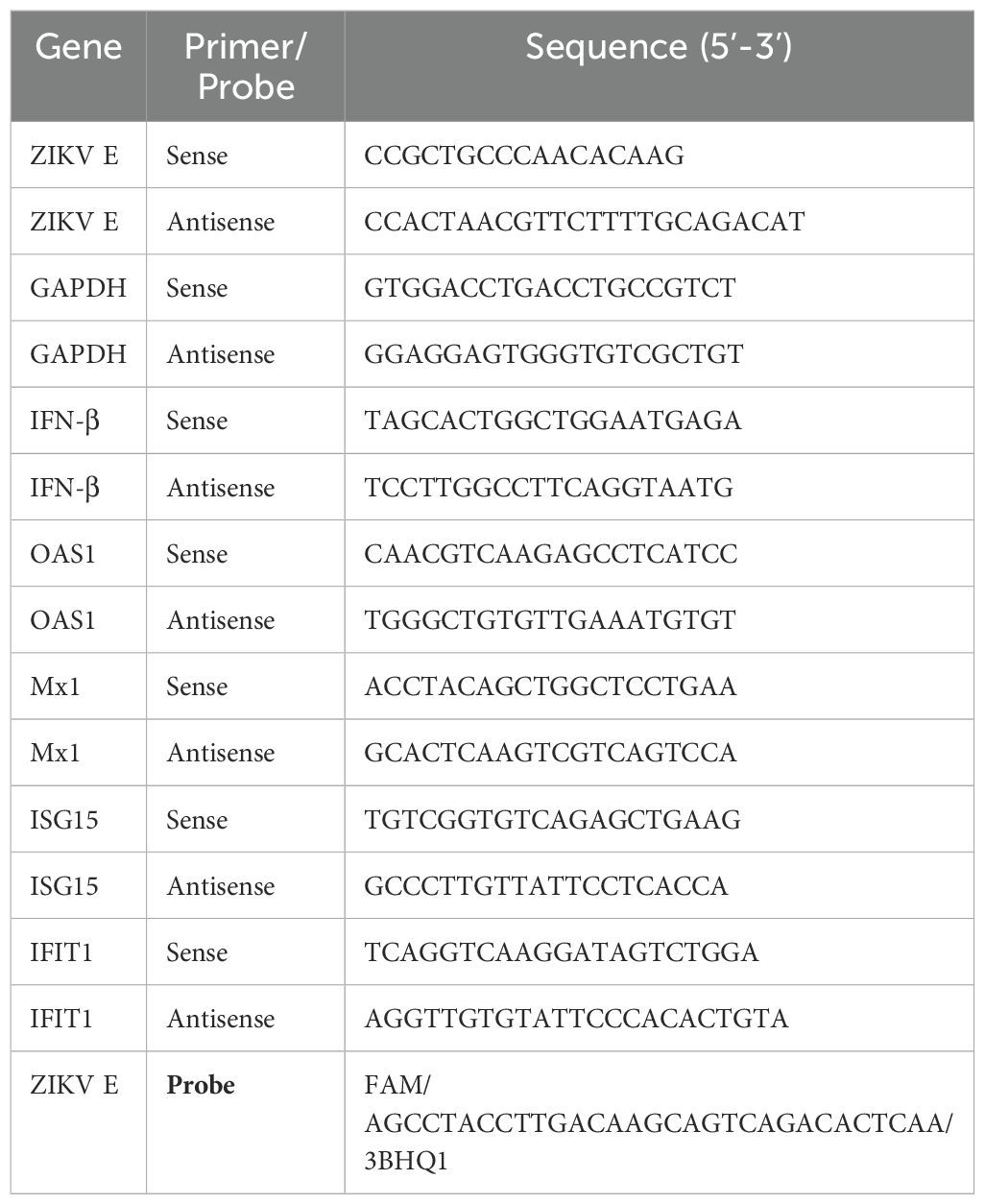
Table 1. Primers and probe sequences used for RT-qPCR assay.
Cytokines and ISGs expression quantification were performed by the comparative CT method (ΔΔCt), using GAPDH Ct for normalization (Livak and Schmittgen, 2001).
2.6 Immunofluorescence analysis
HBMECs were infected with ZIKVPE243 or ZIKMR766 at the indicated MOIs, in the presence or not of anti-IFN-α/IFN-β. At 24 and 48 h.p.i., the cells were fixed with 4% formaldehyde, blocked with 3% BSA for 30 min, and permeabilized with 0.1% saponin diluted in a blocking solution for 15 min. Then, the samples were incubated with 4G2 antibody (supernatant from hybridoma clone D1-4G2-4-15; Ref.: ATCC HB-112) for 2 h, followed by AlexaFluor594-conjugated anti-mouse IgG (1 μg/mL, Ref.: A32744, Thermo Fisher Scientific Inc). DAPI was used to stain cells’ nuclei. The images were obtained by fluorescence microscopy, using OLYMPUS IX81 equipment and the frequency of ZIKV-infected cells (4G2+) was measured using ImageJ software (Version 1.54k).
2.7 Cell viability assay
HBMECs were infected with ZIKVPE243 or ZIKMR766 at the indicated MOI, in the presence or not of anti-IFN-α/IFN-β. After different time points, from 24 h to 96 h, cell viability was assessed using Cell-Titer Aqueous One Solution (Promega, Madison, USA), according to the manufacturer’s protocols. Absorbance readings were taken using a spectrophotometer (GloMax®-Promega); 1% Triton X-100 solution was used as a positive control.
2.8 Depletion of RIG-I and TLR3 by siRNA
HBMECs were transiently transfected with small interfering RNA (siRNA) targeting TLR-3, RIG-I, or nontargeting scrambled siRNA (Santa Cruz Biotechnology, Dallas, TX), using lipofectamine 2000 (Thermo Fisher Scientific Inc.), as previously described (Conceição et al., 2013). Receptors’ depletion was confirmed by western blotting analysis. At 48 h post-transfection, the cells were mock-treated or infected with ZIKVMR766 or ZIKVPE243 at a MOI of 1; poly I:C was used as a positive control. At 24 h.p.i., the cells were harvested, and IFN-β production was measured by RT-qPCR.
2.9 Western blotting
The expression of RIG-I, TLR-3, and phosphorylated and nonphosphorylated STAT-1 and STAT-2 were evaluated by western blotting. RIG-I and TLR3 were analyzed after 48 hours post-siRNA transfection. To analyze the expression and phosphorylation of STAT-1 and STAT-2, HBMECs were infected with ZIKVMR766, ZIKVPE243, or DENV-2 (MOI=1) for 24 h and, then cultured with IFN-β (1000 U/ml), or poly I:C (50 mg/ml) for additional 45 minutes. For all the western blotting assays, the cells were harvested in RIPA lysis buffer (10 mM Tris-HCl (pH 7.5) with 150 mM NaCl, 1% sodium deoxycholate, 0.1% SDS, Triton X-100 1%), supplemented a cocktail of protease inhibitor (Roche Applied Science, Germany). Protein concentration was measured using the Bradford quantification method (Bio-Rad Laboratórios Brasil Ltda, SP, Brazil), and 20 μg of each sample was reduced and denatured in Laemmli sample buffer containing 2-mercapto-ethanol heated for 5 min at 95°C. The samples were subjected to a 10% polyacrylamide gel electrophoresis (SDS-PAGE), followed by transfer to a nitrocellulose membrane (Merck). The membranes were blocked with 10% TBS containing 5% bovine serum albumin (BSA; Merck) for 1 h, and incubated with the antibodies anti-p-STAT1, anti-STAT1, anti-p-STAT2, anti-STAT2, anti-RIG-I, anti-TLR-3, and anti-β-actin (Cell Signaling Technology, Danvers, MA), overnight at 40C. The membranes were washed and incubated with the respective HRP-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories Inc., West Grove, PA) for 1 h and developed using the ECL™ Prime Western Blotting System kit (GE Healthcare, Boston, MA). The ratio between phosphorylated and nonphosphorylated proteins, as well as the ratio of total protein expression (sum of phosphorylated and nonphosphorylated) and β-actin, were calculated using ImageJ software.
2.10 Evaluation of ISRE activation by luciferase assay
HBMECpISREluc reporter cells were infected with ZIKV strains in the presence or absence of IFN-β or poly I:C. At 24 and 48 h.p.i., cells were washed with PBS, lysed with Luciferase Cell Culture Lysis Reagent (CCLR; Promega, Madison, WI), vortexed, and centrifuged at 12,000 × g for 2 min at 4°C. Supernatants were collected, and luciferase activity was measured using the Luciferase Assay Reagent substrate in a GloMax® luminometer (Promega), following the manufacturer’s instructions. Results are expressed as relative luminescence units (RLU) normalized to the number of viable cells.
2.11 Analysis of type I IFN receptor by flow cytometry
HBMECs were infected with ZIKVMR766 or ZIKVPE243, at a MOI of 1 and, at 24 h.p.i., the cells were harvested, and the expression of IFNAR was evaluated by flow cytometry. Cells were fixed in 4% formaldehyde, permeabilized in 0.1% saponin with 2% FBS, and blocked in PBS with 2% FBS. Then, cells were incubated with rabbit anti-IFNAR1 antibody (0.25 mg/ml; Abcam, Cambridge, UK) for 1h, followed by incubation with PECy5-conjugated rabbit anti-IgG antibody (ThermoFisher Scientific Inc.) for 30 min. Cells stained in the absence of primary antibodies were used as controls. The samples were acquired in a FACSCanto™ cytometer and analyzed using FlowJo software (v10.8.1; Becton Dickson Immunocytometry System).
2.12 Analysis of BBB permeability and virus extravasation
HBMECs were seeded onto transwell inserts (Corning Costar, ME, USA; 0.4 µm membrane) and infected with ZIKVPE243 or ZIKVMR766 (MOI=0.1). The combination of neutralizing anti-IFN-β and anti-IFN-α was added to some wells just after adsorption. After 0, 24, 48 and 72 h.p.i. the transendothelial electric resistance (TEER) across cell monolayers were measured daily using a Voltohmmeter. Culture medium from the upper (luminal) and lower (abluminal) chambers were collected, and infectious virus particles were measured by plaque assay.
2.13 Mouse experiments and ethical statements
Male and female IFNAR-/- (B6(Cg)-Ifnar1tm1.2Ees/J), CDH5cre (B6.FVB-Tg (Cdh5-cre)7Mlia/J) (Alva et al., 2006) and Ifnarflox/flox (B6(Cg)-Ifnar1tm1.1Ees/J) (named IFNARfl/fl) (Prigge et al., 2015) mice were purchased from The Jackson Laboratories and housed/bred at the Animal Facility of the Institute of Biology, University of Campinas. Mice with IFNAR1 deficiency only in endothelial cells (CDH5cre+ IFNARfl/fl) were generated by mating CDH5cre and IFNARfl/fl. Mice CDH5cre(negative) IFNARfl/fl from the same litter were used as controls in all experiments. All animals were genotyped using tail samples digested by proteinase K as previously described (Alva et al., 2006). Animals were housed in groups of five per cage with free access to food and water, under a 12 h light/dark cycle, with controlled temperature and humidity. All procedures followed the “Principles of Laboratory Animal Care” (US National Institutes of Health) and institutional policies for animal care and usage and were approved by The Ethics Committee of Animal Care and Use from UNICAMP (protocol 4858-1/2018).
2.14 In vivo infection, tissue collection and analysis of viral load
Four-week-old mice with body weight between 11-13g, CDH5cre(negative) IFNARfl/fl (12 mice) and CDH5cre+ IFNARfl/fl mice (27 mice) were inoculated with 30μl (2x105 PFU) suspension of ZIKVMR766, as previously described (Lucas et al., 2018). Six mice per group were mock inoculated, as negative controls. After injection, the animals were monitored daily to assess body weight, neurological signs, and survival. Mice were euthanized at the indicated time points. In addition, mice presenting one or more physical or behavioral characteristics determined as humane endpoint (weight loss above 20%, intense lethargy or no mobility, or excessive agitation) were considered as end-stage illness or moribund, and euthanized, following the policies for animal care and usage. Dead and moribund mice were included as dead in the survival curves, and the ones found at end-stage illness were used to assess viral load in the tissues. The brains and spleens were harvested, weighed, and macerated in DMEM medium supplemented with 1% gentamicin, following the ratio of 0.2 mg of tissue to 1 μL of medium. After homogenization, centrifugation was performed at 4500 g for 5 min to remove tissue residue. Viral load in the tissues was determined by plaque assay.
2.15 Statistical analysis
Statistical analyses were performed using GraphPad Prism v.8 software. Differences between groups were analyzed by One-Way ANOVA or Two-Way ANOVA, followed by multiple comparisons posttests indicated in the legends; comparisons between two groups were performed by Student’s t-test for unpaired samples. IFN-β IC50 and IC90 were also calculated using GraphPad Prism after data normalization and nonlinear curve progression analyses. Values for all measurements are expressed as mean or mean ± standard deviation, and every statistical method is indicated in the corresponding legend. Analyses with p-value <0.05 were considered significant. (*) p ≤ .05; (**) p ≤ .01; (***) p ≤ .001; (****) p ≤ .0001; not significant (ns).
3 Results
3.1 Global transcript abundance and differential gene expression profiles induced by ZIKVMR766 and ZIKVPE243 replication in HBMECs
Aiming to investigate whether ZIKVMR766 and ZIKVPE243 replication could differentially impact BBB integrity, we performed comparative global gene expression and functional analyses of HBMEC infected with each strain, as a simplified model of BBB. The transcriptional landscape of ZIKV infection was assessed by RNAseq of control samples and HBMECs infected with each ZIKV strain. A MOI of 10 was chosen to synchronize infection for transcriptomic analysis, as this condition resulted in nearly all HBMECs were 4G2-positive at 24 h.p.i., with no significant cell death detected for either strain (Supplementary Figure S1A, B). Transcript abundance quantification in tpm (transcripts per million) revealed that ZIKVMR766 generated a higher number of reads compared to ZIKVPE243 (Supplementary Figure S1C). Indeed, differential expression analysis identified 121 differentially expressed genes (DEGs) in ZIKVPE243-infected samples compared to control, with 44 up-regulated and 77 down-regulated genes. In contrast, ZIKVMR766-infected samples displayed a markedly stronger transcriptional response, with a total of 285 DEGs, including 110 up-regulated and 175 down-regulated genes (Supplementary Figure S1D).
Principal component analysis (PCA) revealed three distinct clusters corresponding to the control, ZIKVMR766, and ZIKVPE243-infected samples (Figure 1A). Control and ZIKVPE243 samples clustered closely, whereas ZIKVMR766 showed a more divergent transcriptional profile. Volcano plots highlighted these differences, with ZIKVMR766 inducing substantially more DEGs (n=868) than ZIKVPE243 (n=430) (Figures 1B, C). In the ZIKVMR766-infected samples, numerous transcripts were significantly upregulated, including genes such as EGR1, EEF1A1, AMP1, and KIF4, which are commonly associated with cellular stress responses and viral replication. Several genes such as PBLD, EBP, and LINC00857 were markedly downregulated, suggesting the suppression of host pathways that may hinder viral propagation. In contrast, the ZIKVPE243 infection resulted in a lower number of significantly dysregulated genes, though a few exhibited large fold changes, including MD1, MT1P1, and FOSB. Genes such as FOS and RNU1–1 were significantly altered in both conditions, pointing to shared host response elements triggered by both viral lineages. To explore the overlap and specificity, we compared the sets of DEGs (Supplementary Figure S1E). Among upregulated genes, 94 were unique to ZIKVMR766, 26 to ZIKVPE243, and 18 shared (Supplementary Table S1). For downregulated genes, 162 were unique to ZIKVMR766, 63 to ZIKVPE243, and 14 were common to both (Supplementary Table S2).
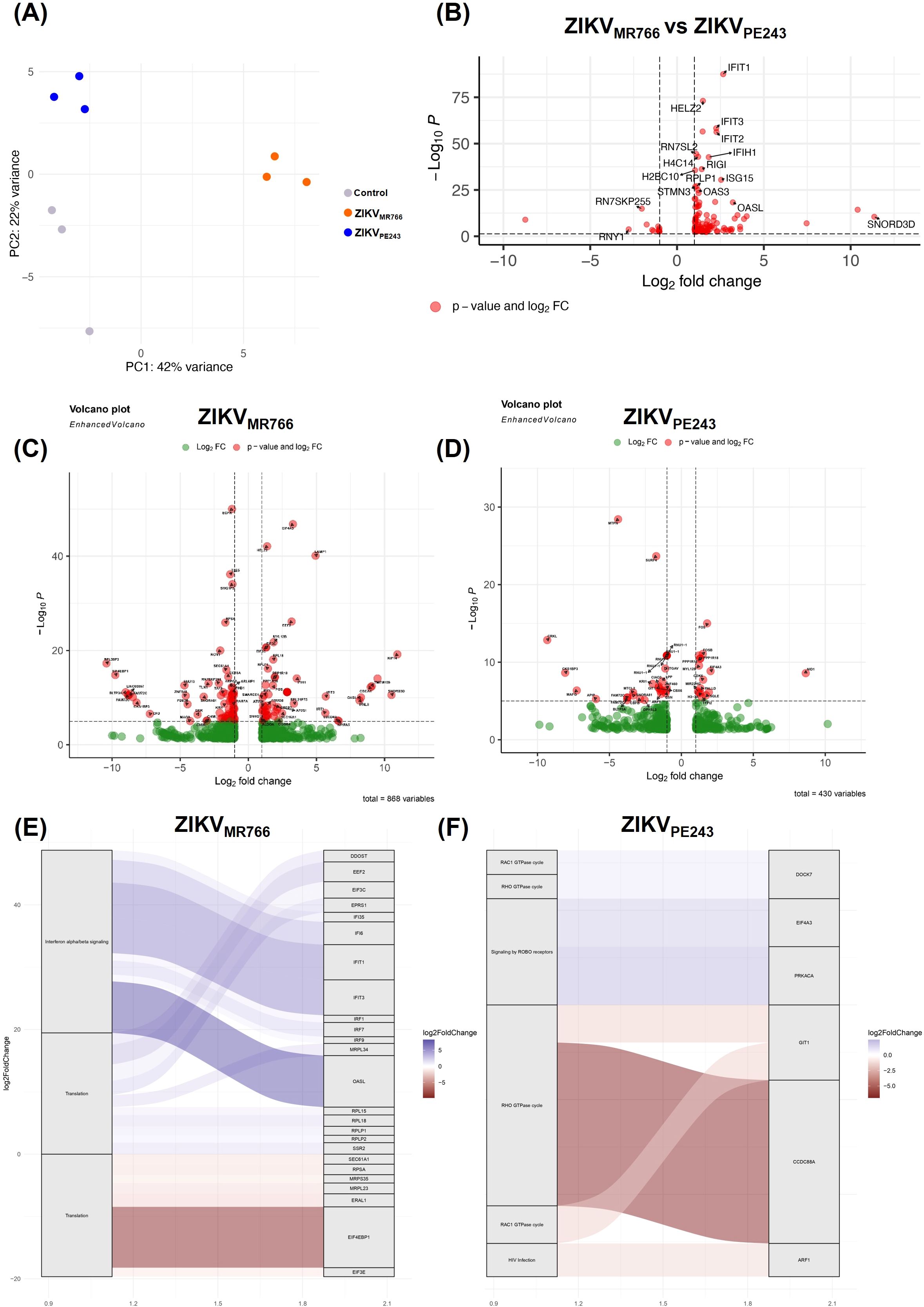
Figure 1. Global gene expression analysis in HBMECs infected with different ZIKV lineages. (A) Principal component analysis (PCA) showing control samples and cells infected with ZIKVMR766 and ZIKVPE243 strains. (B) Differentially expressed genes (DEGs) between ZIKVMR766 infection condition compared to ZIKVPE243 condition. (C, D) Volcano plots depicting DEGs in HBMECs infected with ZIKVMR766 (C) and ZIKVPE243 (D), compared to control. Three biological replicates were analyzed for each condition and significantly regulated genes according to p-values and log2 fold changes are labeled in each volcano plot. (E, F) Differentially expressed genes associated with biological pathways during ZIKV infection compared with control condition. The plot illustrates the relation between genes and enriched pathways in infections with the ZIKVMR766 (E) and ZIKVPE243 (F).
Direct comparison of lineages (Figure 1D) confirmed a distinct MR766 signature, with 117 genes significantly upregulated. Among the most prominently upregulated transcripts were classical interferon-stimulated genes (ISGs), including IFIT1, IFIT2, IFIT3, IFIH1, ISG15, OAS1, and OASL, as well as RIG-I (DDX58), reflecting the activation of innate antiviral responses. Other notable transcripts included HELZ2 and RPLP1, the latter implicated in viral translation and replication processes. SNORD3D also exhibited strong upregulation, suggesting potential modulation of small RNA processing during infection.
Pathway enrichment analysis (Figures 1E, F) also supported that MR766 predominantly activated interferon α/β signaling and translation, consistent with the upregulation of IRFs and ISGs (IFI6, IFIT1, IFIT3, IRF1, IRF7, ISG15) and genes involved in the translational machinery (EIF3C, RPL15, RPL2, MRPL23). In contrast, ZIKVPE243 elicited enrichment in distinct pathways, with prominent activation of RHO and RAC1 GTPase cycles, signaling by ROBO receptors, and HIV infection-related pathways. Key genes such as CCDC88A, GIT1, and ARF1 were implicated in cytoskeletal remodeling and intracellular trafficking, which may support viral entry or replication. The genes PRKACA and EIF4A3, involved in signaling and RNA processing, respectively, were also enriched. Together, these results indicate that ZIKVMR766 elicits a more robust and extensive transcriptional response than ZIKVPE243, predominantly activating immune-related and translational processes, whereas ZIKVPE243 modulates pathways linked to cytoskeletal dynamics and signal transduction. These distinct patterns potentially reflect distinct viral–host interaction dynamics between the African and Asian lineages.
To further characterize the model, we performed a series of time-course experiments using various MOIs to monitor key parameters of viral replication. No significant difference was observed in the levels of viral RNA released between the two ZIKV isolates when using the same MOI (Figure 2A). However, the concentration of infectious viral particles was significantly higher in cultures infected with ZIKVMR766 compared to those infected with ZIKVPE243 (Figure 2B). Consistently, the PFU-to-RNA ratio was significantly different between the two strains at all time points analyzed (Figure 2C), suggesting more efficient viral maturation by ZIKVMR766. Immunofluorescence analysis of 4G2-positive cells over time revealed a faster spread of ZIKVMR766 infection relative to ZIKVPE243 (Figure 2D), likely due to the earlier release of infectious particles. While ZIKVPE243-infected HBMECs remained viable throughout the infection course, ZIKVMR766 infection induced a modest but significant reduction in cell viability from 72 h.p.i. (Figure 2E), which correlated with viral replication (Figure 2F).
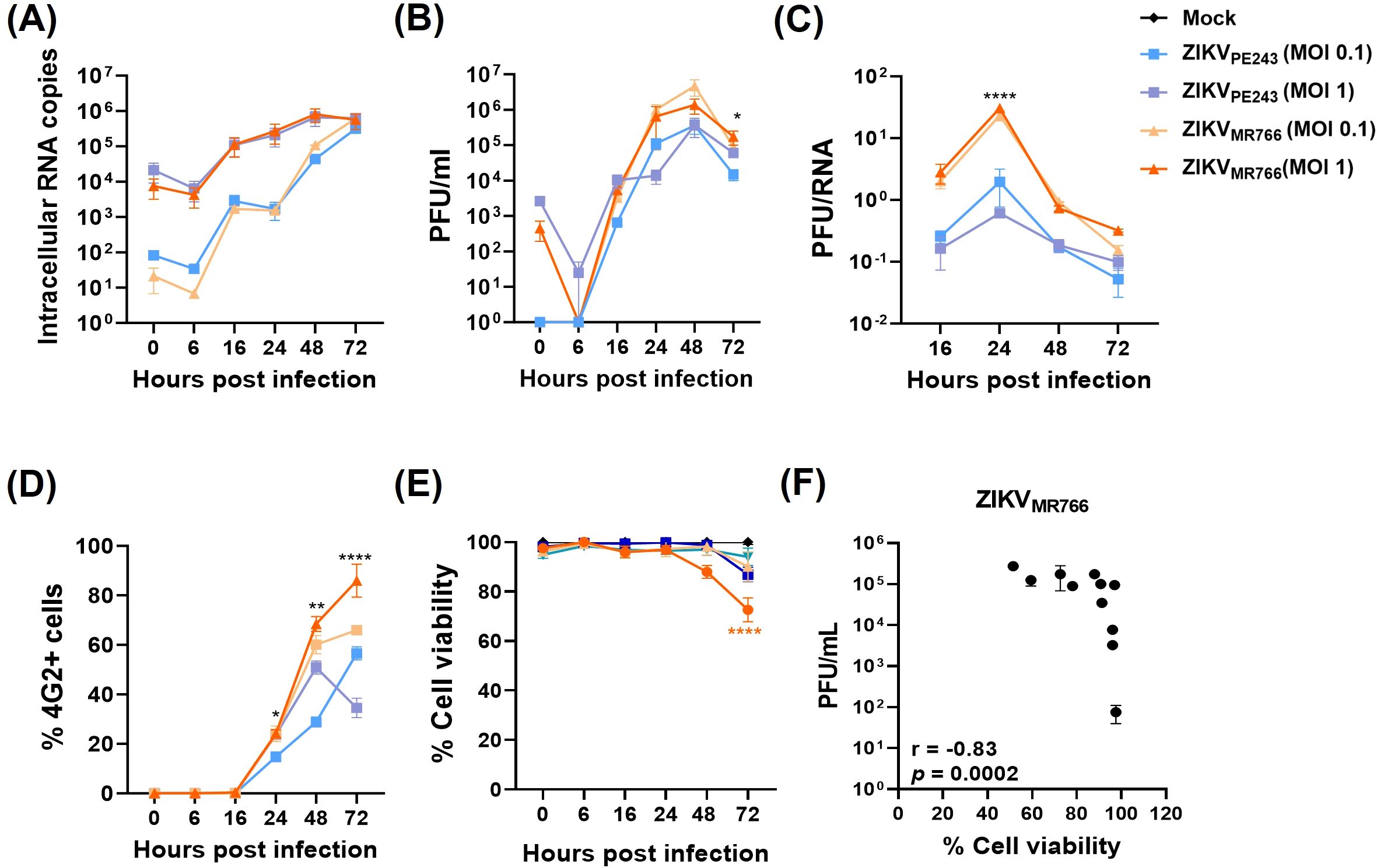
Figure 2. Replication, dissemination, and cytopathic effects of ZIKV strains in HBMECs. HBMECs were infected with ZIKVPE243 or ZIKVMR766, at a MOI of 1 or 0.1, and cell lysates and culture medium were collected at the indicated time points. (A) ZIKV mRNA copies were quantified in cell lysates by RT–qPCR. (B) Infectious viral titers were measured in the culture medium by plaque assay. (C) Ratio between PFU and viral RNA levels comparing ZIKVPE243 and ZIKVMR766. (D) Infected cells were stained with the 4G2 antibody, and the frequency of infected cells (%4G2+) was determined by immunofluorescence. (E) Cell viability was assessed using the CytoTox 96® Non-Radioactive Cytotoxicity Assay. (F) Correlation between viral titers (PFU) and cell viability in HBMECs infected with ZIKVPE243 and ZIKVMR766. Data represent mean ± SD of two independent experiments. *p<0.05; **p<0.01; ***p<0.001.
3.2 ZIKVMR766 evades IFN response more efficiently, through reduction of STAT1 phosphorylation and degradation of STAT2
Since many of the transcriptional changes observed in infected HBMECs were associated with IFN production and signaling, we next sought to investigate this pathway in greater detail. Analysis of IFN-β expression by RT-qPCR confirmed that both ZIKV strains induced IFN-β expression in HBMECs, with ZIKVMR766 eliciting a stronger and earlier response (Figure 3A). To identify the viral sensors responsible for IFN-β induction, we focused on key pattern-recognition receptors involved in RNA virus detection. While TLR3 recognizes viral dsRNA within endosomal vesicles, RIG-I and MDA5 act as cytoplasmic sensors that frequently function in a redundant manner, as reported in ZIKV infection models (Lin et al., 2019). Therefore, we selected TLR3 and RIG-I for depletion as representatives of endosomal and cytoplasmic sensing pathways, respectively. Poly I:C was added to the cultures, without transfection, as a positive control for TLR3, but not RIG-I activation. As expected, TLR3 depletion, but not RIG-I, significantly reduced poly I:C-induced IFN-β mRNA expression. On the other hand, while TLR3 silencing did not affect ZIKV-induced IFN-β transcription (Figures 3B, C), depletion of RIG-I almost abolished IFN-β expression in cells infected with either ZIKVPE243 (88% inhibition; p=0.008) or ZIKVMR766 (94% inhibition; p=0.0004) (Figures 3D, E), indicating RIG-I as a major RNA sensor related to this pathway.
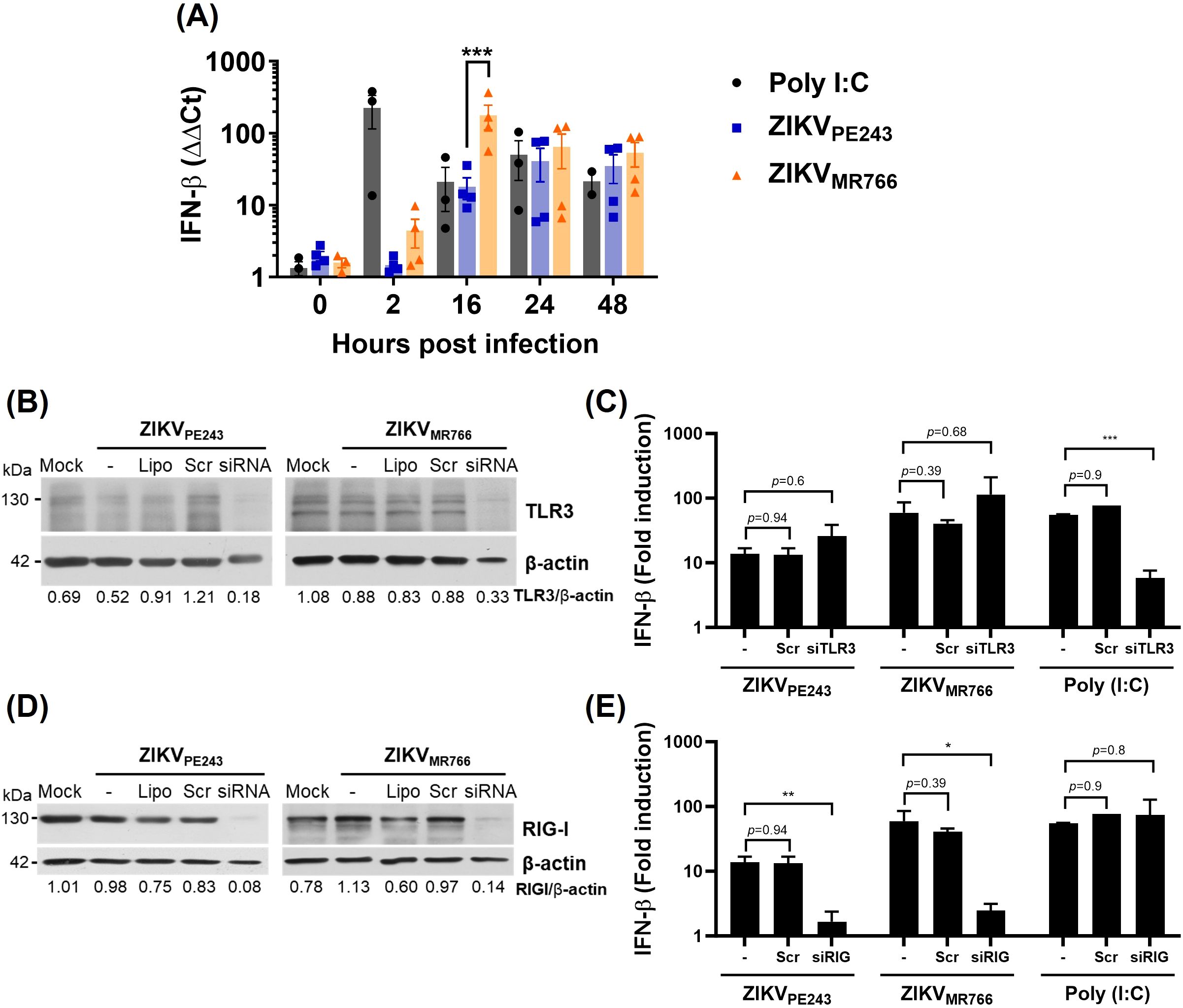
Figure 3. ZIKV induces RIG-I-dependent IFN-β production by HBMECs. (A) HBMECs were mock-treated or infected with ZIKVPE243 or ZIKVMR766 (MOI=1); poly (I:C) was used as positive control. The cells were harvested at the indicated time points and IFN-β mRNA was measured by RT-qPCR. Bars represent the mean and standard deviation of seven independent experiments; statistical analyses were performed by two-way anova and Bonferroni’s multiple comparisons test. (B-E) Depletion of TLR3 (B, C) and RIG-I (D, E) in HBMECs were performed by specific siRNA transfection; lipofectamine only (Lipo), and scramble siRNA (Scr) were used as controls. Receptor depletion was confirmed by western blotting using anti-TLR3 (B) or anti-RIG-I (D) specific antibodies, and anti-βactin as a loading control. After 48 h, the cells were mock-treated or infected with ZIKVPE243 or ZIKVMR766 and, at 24 hpi, IFN-β expression was evaluated by RT-qPCR (C, E). Statistical analysis were performed by unpaired t test; *p<0.05, **p<0.01, ***p<0.001.
Given the efficient ZIKV replication in HBMECs, we assessed whether the viruses could evade IFN-mediated antiviral response. HBMECpISREluc reporter cells were infected with ZIKVPE243 or ZIKVMR766 at MOI 1, and, after viral adsorption, cells were treated with IFN-β or poly I:C. DENV infection was used as a control for inhibition of IFN-response (Morrison et al., 2013). Infection of HBMECs with both virus strains significantly reduced the activation of ISRE induced by either IFN-β itself or by poly I:C stimulation (Figure 4A). Accordingly, IFN-β-induced expression of the interferon-stimulated genes (ISG) OAS1 (2’-5’-oligoadenylate synthetase 1), Mx1, ISG15, and IFIT1 (Interferon-induced protein with tetratricopeptide repeats 1) were inhibited when the cells were previously infected with ZIKVMR766 or ZIKVPE243 (Figure 4B). Addition of IFN-β after infection resulted in only ~20% inhibition of viral replication, further supporting that ZIKV can efficiently counteract late IFN signaling once infection is already established (Figure 4C).
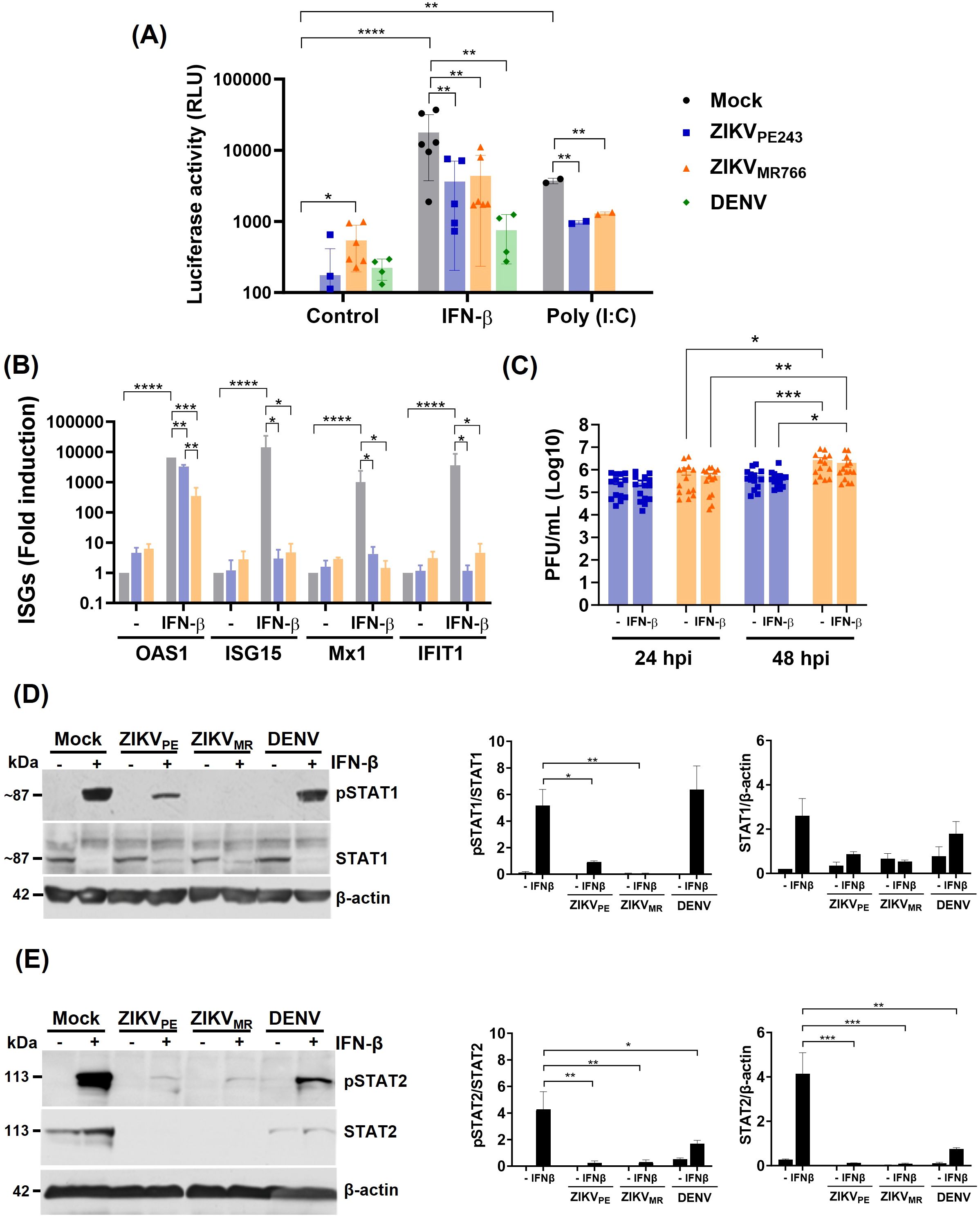
Figure 4. ZIKV infection inhibits IFN-β response in HBMEC. (A) pISRE-HBMEC-luc reporter cells were mock-treated or infected with ZIKVPE243, ZIKVMR766 or DENV-2 at a MOI of 1. Cells were treated with IFN-β or poly I:C after virus adsorption and cellular luciferase activity was measured at 24 hpi. (B, C) HBMECs were cultured as in (A). At 24 hpi, OAS1, ISG15, MX1, and IFIT1 mRNA was assessed by RT-qPCR (B) and released viral titers was measured by plaque assay (C). The data represent the mean and standard deviation of five independent experiments, and the statistical analyses were performed by two-way ANOVA and Dunnett’s multiple comparisons test. (D, E) The expression of phosphorylated or nonphosphorylated STAT1 (pSTAT1, STAT1) (D) and of pSTAT2 and STAT2 (E) were evaluated by western blotting; β-actin expression was measured as loading control. Representative membranes are demonstrated in the left panels; the ratio of pSTAT1 in relation to STAT1, as well as pSTAT2 in relation to STAT2 are indicated in the middle panels; and total STAT1 or STAT2 expression in relation to β-actin are demonstrated in right panels. Data represent mean and standard deviation of six independent experiments, and the statistical analysis were performed by one-way anova and Tukey’s multiple comparisons test; *p<0,05, **p<0,01, ***p<0,001, ****p<0,0001.
Viral replication did not alter IFNAR expression (Supplementary Figure S2) but led to reduced phosphorylation of STAT1 (Figure 4D), as well as both degradation and reduced phosphorylation of STAT2, similarly to DENV-infection (Figure 4E). Notably, inhibition of STAT activation and decreased expression of most ISGs were more pronounced in ZIKVMR766-infected cells (Figures 4B, D), prompting us to investigate potential viral determinants underlying these differences.
Comparison of the ZIKVPE243 and ZIKVMR766 genomes revealed several nucleotide and amino acid differences in regions potentially linked to IFN modulation. A few nucleotide substitutions were identified in the 5′ UTR of both ZIKV strains, most of which have not been associated with IFN modulation (Chazal et al., 2018). However, we identified a specific nucleotide insertion at position 81, consistent with the lineage-specific upstream open reading frames (uORFs) previously described (Lefevre et al., 2024). These uORFs have been implicated in modulating viral translation and conferring resistance to translational arrest under stress conditions, which could enhance IFN resistance. Sequence alignment confirmed the presence of this insertion in ZIKVPE243 but not in ZIKVMR766 (Supplementary Figure S3). We did not find any differences in the NS5 positions Y25-R327, D734, and H855, which were previously determined to bind to STAT2 (Wang et al., 2020), as well as in the K252 SUMOylation position (Conde et al., 2020). However, a few other substitutions were detected at methyltransferase and RdRp domains, although their role in NS5-STAT2 interactions had not been investigated (Figures 5C, D). Amino acid substitutions were also seen when comparing the capsid sequence of ZIKVPE243 and ZIKVMR766 (Figures 5E, F), which could affect RIG-I ubiquitination and IFN production (Airo et al., 2022). Those include K101R substitution, which was recently associated to enhanced neurovirulence of ZIKV African strains (Song et al., 2023).
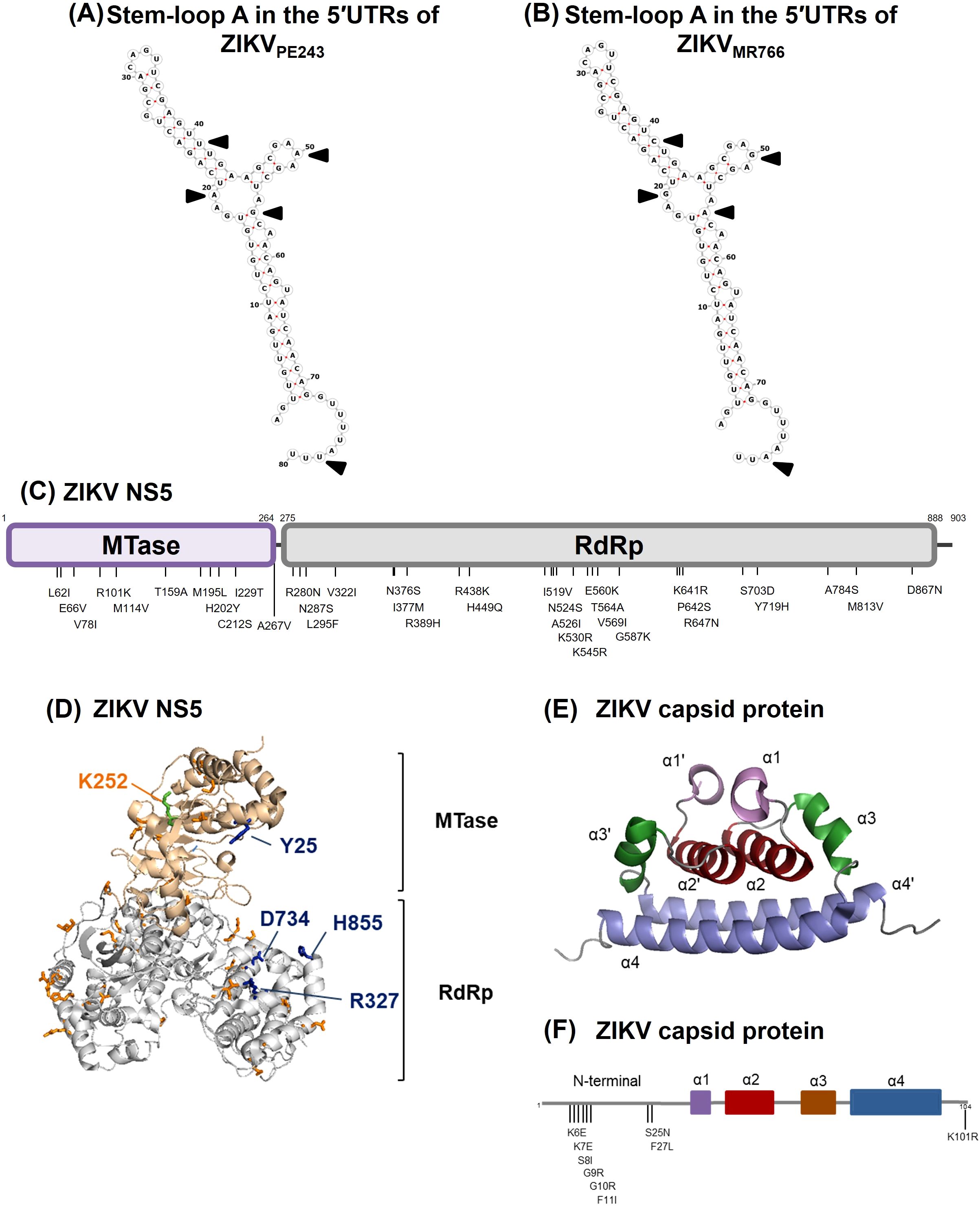
Figure 5. Comparison of genomic and amino acid sequences by the 5’UTR, NS5 and capsid protein from ZIKVMR766 (NC_012532.1) and ZIKVPE243 (GenBank KX197192.1). (A, B) Sequence and predicted secondary structure of stem-loop A in the 5′UTRs from ZIKVPE243 (A) and ZIKVMR766 (B). The black arrows indicate sequence variations in the 5′-UTR. (C) Schematic representation of ZIKV NS5 from ZIKVMR766 indicating the amino acid substitutions in the NS5 from ZIKVPE243. MTase and RdRp domains are colored purple and gray, respectively. (D) Ribbon representation showing the MTase (light pink) and RdRp (gray) domains of ZIKVMR766 NS5. The locations of residues that differed in NS5 from ZIKVPE243 in the context of ZIKVMR766 NS5 structure (PDB, 5U0B) are shown in orange sticks. The residues Y25, R327, D734 and H855 are highlighted in blue sticks, and the K252 residue (green stick) is labeled in orange. (E) Ribbon representation showing the structure of the ZIKV capsid protein (PDB, 6C44), with the α-helix pairs indicated: α1/α1’ (purple), α2/α2’ (red), α3/α3’ (brown), and α4/α4’ (blue). (F) Schematic representation of the capsid protein from ZIKVPE243 indicating the amino acid substitutions from ZIKVMR766. The α-helices are colored according to protein structure in (E).
3.3 IFN-β partially protects HBMEC from ZIKV replication
Despite the viral mechanisms of IFN evasion, we next assessed whether IFN-β could still confer protection to noninfected bystander endothelial cells when present prior to infection. To evaluate this, HBMECs were pretreated with the cytokine for 24 h before infection with ZIKVPE243 or ZIKVMR766. At this point, ZIKV infection did not affect IFN-β-induced ISG expression (Figure 6A), and viral replication was significantly reduced (Figure 6B). This indicates that IFN-β signaling before infection established an antiviral state in bystander cells, despite the virus’s ability to evade these responses once infection is established. To compare the sensitivity of each virus strain to IFN-β, the cells were cultured with different concentrations of the cytokine before infection, and the IC50 and IC90 were calculated. ZIKVMR766 showed higher resistance to IFN-β than ZIKVPE243, with IFN-β IC50/90 of 2.53/22.74 and 0.1/0.9 U/ml, respectively (Figures 6C, D).
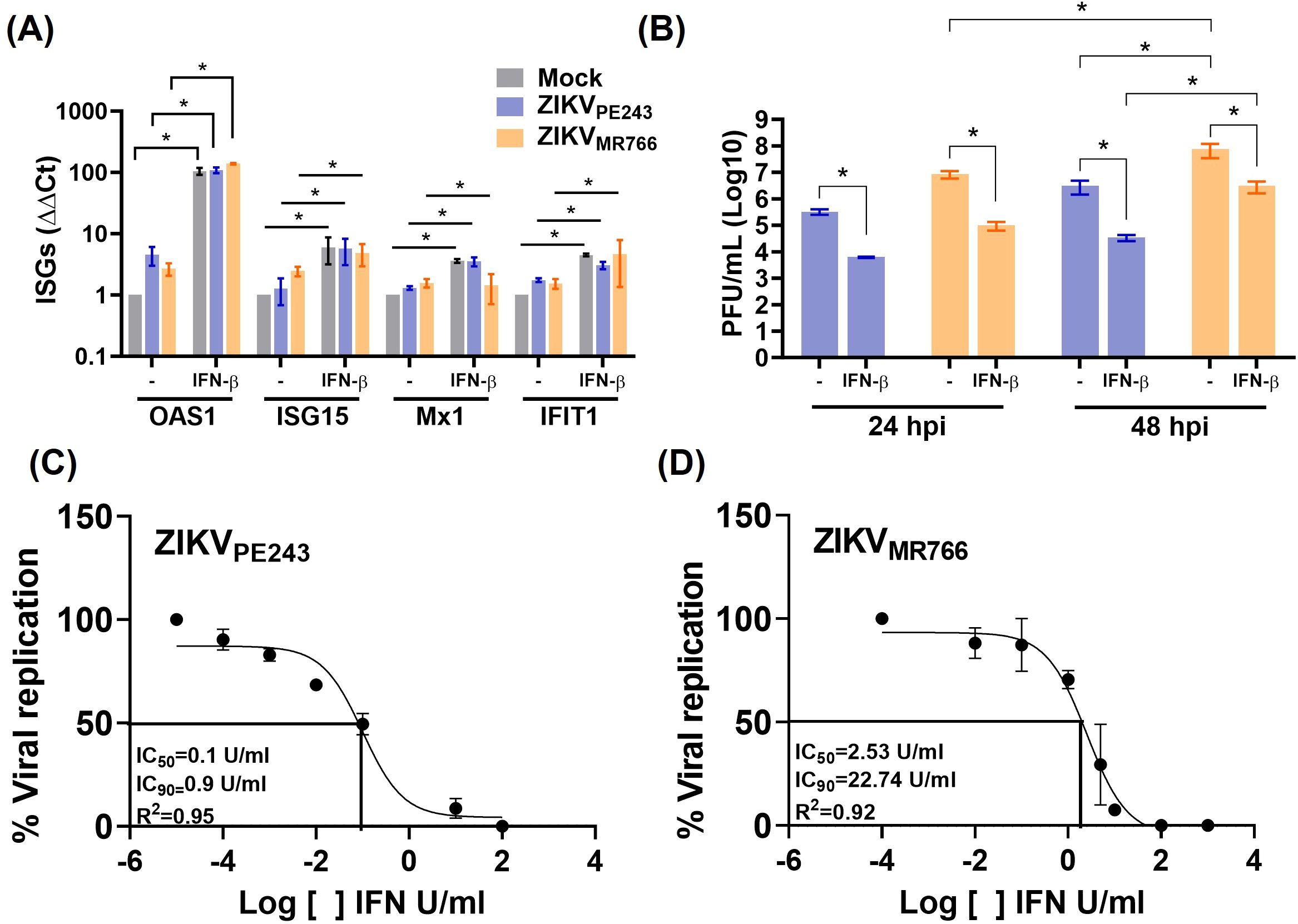
Figure 6. IFN-β treatment before ZIKV infection reduces viral replication. (A, B) HBMECs were treated with IFN-β (1000U/mL) and, after 24 h, cells were mock-treated or infected with ZIKVPE243 or ZIKVMR766 (MOI=1). Cells were harvested and mRNA expression of OAS1, ISG15, MX1, and IFIT1 were measured by RT-qPCR (A). Viral titers were measured by plaque assay (B). (C, D) HBMECs were treated with different concentration (10-3–104 U/mL) of IFN-β for 24 hours and were then infected with ZIKVPE243 (C) or ZIKVMR766 (D). At 24 hpi, the supernatants were harvested, virus titration was performed by plaque assay, and the IFN concentration inhibiting 50% and 90% of virus titer (IC50 and IC90) were calculated with GraphPad prism. The data represent mean and SD of three independent experiments. Bars indicate average and SD from three independent assays and statistical analysis were performed by two-way anova and Tukey’s multiple comparison test; *p<0.05; **p<0.01 ***p<0.001.
Using a different experimental setting, a combination of anti-IFN-α and anti-IFN-β neutralizing antibodies was added at the beginning of ZIKV infection to block the effect of endogenous virus-induced IFN. As controls, cell viability was evaluated in antibody-treated cultures, and no significant toxicity was detected from 24 h to 96 h (Supplementary Figure S3A). Also, neutralization activity at non-toxic concentration was confirmed by measuring IFN response in HBMECISREluc reporter cells treated with recombinant IFN-β or infected with DENV, in the presence of the neutralizing antibodies (Supplementary Figure S3B). Analysis of ZIKV-infected cultures revealed that type I IFN neutralization significantly increased virus replication and cell death (Figures 7A, B), supporting that endogenous IFN-signaling protects HBMEC from ZIKV-induced cytopathic effect.
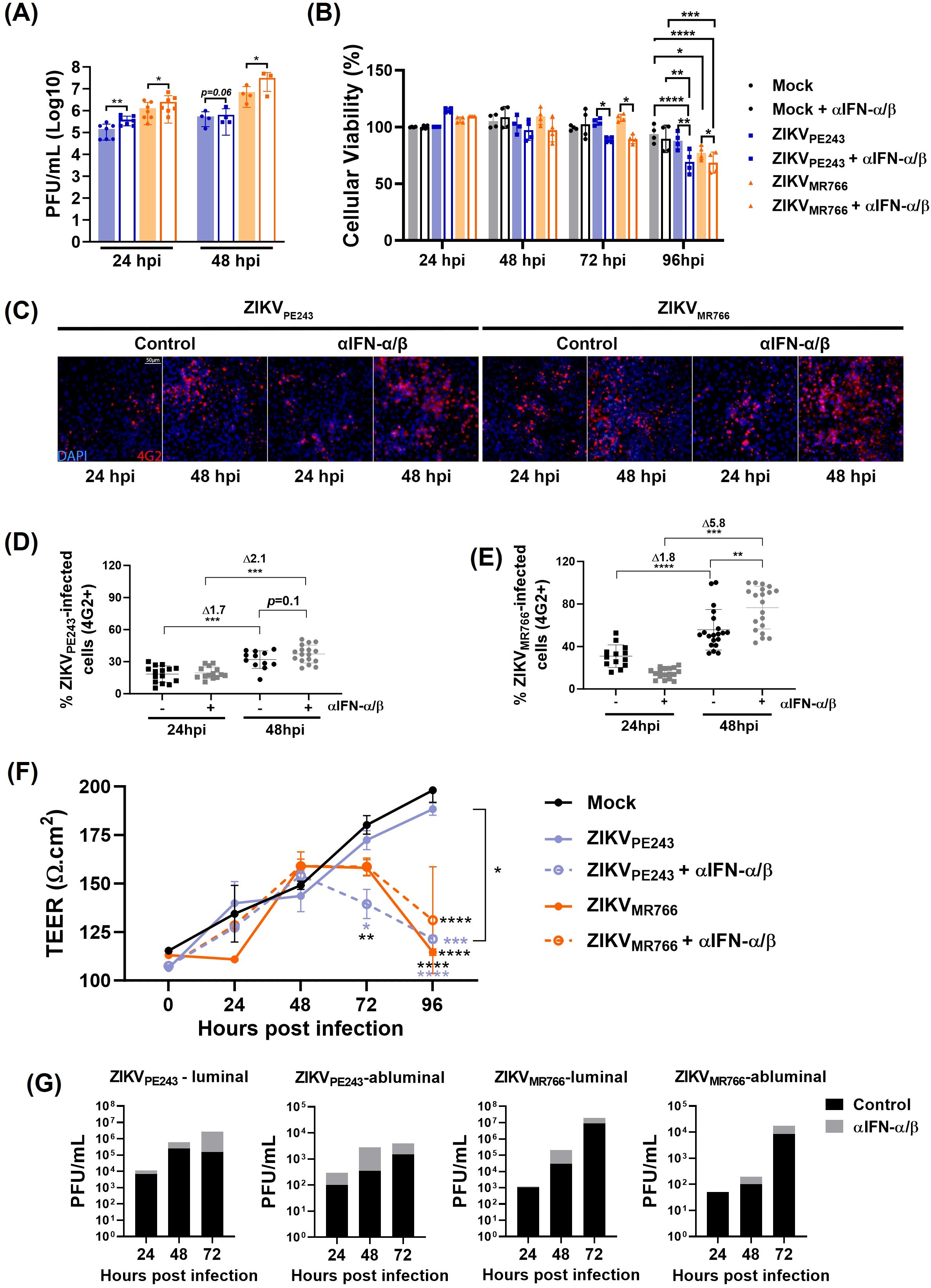
Figure 7. Type I IFN limits ZIKV replication, dissemination and preserves HBMECs barrier integrity. (A) HBMECs were infected with ZIKV strains (MOI=0.1) in the presence or absence of a combination of neutralizing anti-IFN-α and anti-IFN-β antibodies (αIFN-α/β). At 24 and 48 hpi, virus replication was evaluated by plaque assay; graph bars represent the mean and SD of four (24 h) and two (48 h) independent assays, and statistical analyses were performed by paired t-test. (B) Cells were cultured as in A, and viability was evaluated at the indicated time points using a Cell Titer assay. Data indicates the mean and SD of four independent experiments, and statistical analyses were performed by two-way ANOVA and Tukey’s multiple comparison test. (C–E) Cells were cultured as in (A) and, at 24 and 48hpi, the cells were stained with 4G2 antibody. Representative figures are shown in (C) and the average and SD of the frequency of ZIKV-infected cells (% 4G2+) is depicted in (D) and (E). Data was calculated from two experiments, including 20 images. Statistical analyses were performed by one-way ANOVA. (F, G) HBMECs were cultured onto transwell plates and the cells were mock-treated or infected with ZIKVPE243 or ZIKVMR766 and then treated or not with αIFN-α/β. Transendothelial electrical resistance (TEER) were measured in indicated time points (F) and viral titer was measured in the luminal and abluminal chambers of the transwell plates by plaque assay (G); statistical analyses were performed by two-way ANOVA, followed by Tukey’s multiple comparison test; *p<0.05; **p<0.005; ***p<0.0005; ****p<0.0001; blue and black stars (*) indicate comparison with ZIKVPE243 or mock, respectively.
The frequency of infected cells in these cultures was monitored through immunofluorescence analyses using the 4G2 antibody. The percentage of HBMECs infected with ZIKVPE243 and ZIKVMR766 increased by 1.7-fold and 1.8-fold, respectively, between 24 and 48 hours. In contrast, when the cells were infected in the presence of anti-IFN, the fold increases rose to 2 and 5.8, respectively (Figures 7C-E), suggesting that IFN neutralization enhanced virus dissemination.
Earlier data from our group showed that ZIKV strains can cross the HBMEC monolayer without causing its disruption (Papa et al., 2017). To investigate whether interferon produced by infected cells acts as an endogenous protective factor, we sought to evaluate its ability to preserve endothelial barrier integrity and to limit, at least partially, viral extravasation across the monolayer. To this end, HBMECs were seeded in a transwell system and transendothelial electric resistance (TEER) was measured for at various time points post infection, in the presence or absence of anti-IFN neutralizing antibodies. As expected, ZIKVPE243 infection did not impact HBMEC integrity, however, a significant TEER reduction was detected from 72h.p.i. in the wells treated with anti-IFN (Figure 7F). In contrast, ZIKVMR766-infected HBMECs showed a reduction in TEER after 72 and 96 h.p.i., which was not affected significantly by IFN neutralization.
Virus titers were assessed in the upper (luminal) and lower (abluminal) chambers. Increased concentration of infectious particles was detected in both compartments of ZIKVPE243 and ZIKVMR766 cultures, supporting that IFN neutralization facilitated ZIKV dissemination across and through the HBMEC monolayer (Figure 7G). These findings highlight the role of type I IFNs in preserving BBB endothelial cells viability, function and protection of against viral dissemination.
3.4 IFNAR expression in endothelial cells partially restricts ZIKV infection and disease in vivo
We had previously demonstrated that 4-week-old IFNAR-deficient SvA129 mice systemically infected (i.v.) with ZIKVMR766, but not with ZIKVPE243, resulted in increased virus dissemination to the CNS, followed by BBB disruption, development of neurological syndrome, and death (Papa et al., 2017; Lucas et al., 2018). To specifically address the role of endothelium-mediated IFN response, we performed the same infection protocol using a mouse model generated from the mating between IFNARflox/flox (IFNARfl/fl) and CDH5cre, which express Cre-recombinase under the regulatory control of the VE-Cadherin (Alva et al., 2006), thus restricting IFNAR deficiency to vascular endothelial cells. CDH5cre+ IFNARfl/fl were mock-inoculated or i.v. infected with ZIKVMR766, and mice were followed to assess survival, body weight, and viral load in different tissues. Fully deficient IFNAR-/- and IFNARfl/fl mice were used as controls. As expected, all mock-infected mice survived infection, whereas 100% of infected IFNAR-/- succumbed at 6 dpi. Interestingly, about 55% of CDH5cre+ IFNARfl/fl mice succumbed to infection or presented an end-stage illness and were euthanized from 6 to 9 dpi (Figures 8A, B). Indeed, we found striking differences between CDH5cre+ IFNARfl/fl and control IFNARfl/fl mice regarding survival (~75% vs ~45% survival, p=0.0142) and weight (% starting weight 117% vs 66% at each endpoint; p<0.0001) (Figures 7A, B). This data demonstrates that IFNAR response in endothelial cells partially protects mice from lethal ZIKV disease.
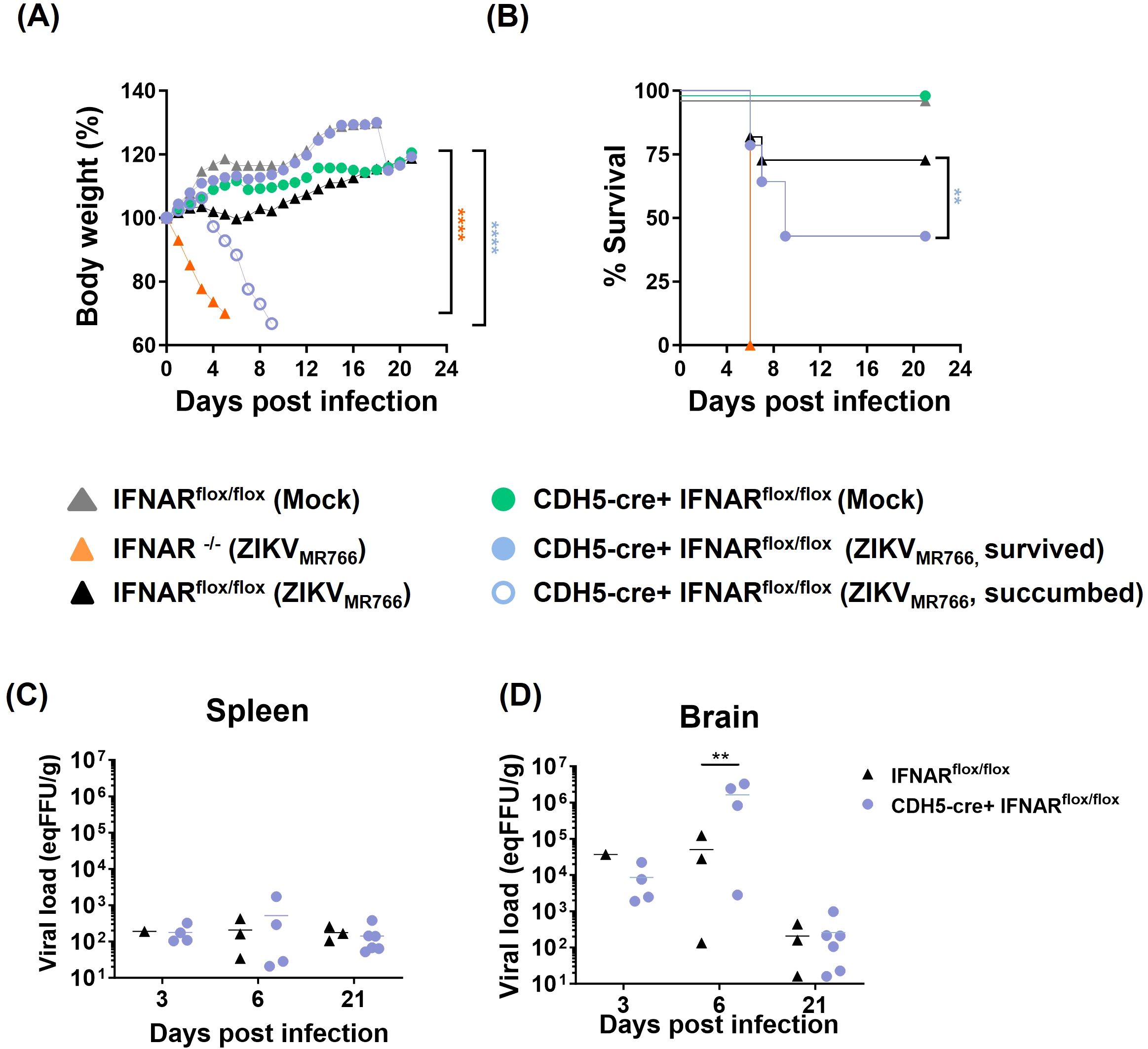
Figure 8. IFNAR deficiency conditioned to endothelial cells partially protects mice from ZIKV-mediated disease. Fully IFNAR-deficient mice (IFNAR-/-) and mice deficient in IFNAR only in endothelial cells (CDH5cre IFNARflox/flox), as well as IFNARflox/flox (IFNARflox/flox) control mice were intravenously infected with 2x105`PFU of ZIKVMR766 or inoculated with mock control. Mice were followed for 21 days. (A) Survival curve showed statistical significance (**p<0.01) as evaluated by log-rank (Mantel-Cox) tests. (B) Mice body weight was followed until the sacrifice and the results were statistically analyzed using two-way Anova and Tukey’s posttest; ****p<0.0001 in relation to IFNARflox/flox (mock and ZIKVMR766), CDH5-cre+IFNARflox/flox (mock and ZIKVMR766 survived). (C, D) Viral titer in the spleens (C) and the brains (D) were evaluated by plaque assay; statistical analyses were performed at 6 dpi by one-way anova and Tukey’s posttest. *p<0.05; **p<0.01 ***p<0.001; ****p<0.0001.
Finally, viral load in the brains and spleen were then accessed at different time points after infection. No difference was detected at 3 dpi, when all mice groups were clinically stable, with no weight loss nor clinical signs. At 6 dpi, four mice from the CDH5cre+ IFNARfl/fl group were euthanized, being two of them moribund, presenting weight loss above 20% and intense lethargy. Three mice from the control (IFNARf/f) group were also euthanized, for comparison. Increased viral load were detected in three out of four (75%) mice from the CDH5cre+ IFNARfl/fl group, with titers 30–120 times greater than the mouse with the highest viral load from the control group (p=0.0072) (Figures 8C, D). When moribund and survivors’ mice were independently analyzed, p values of 0.02 (CDH5cre+ IFNARfl/fl moribund vs IFNARf/f) and of 0.03 (CDH5cre+ IFNARfl/fl moribund vs CDH5cre+ IFNARfl/fl survivors) were determined. Other mice from CDH5cre+ IFNARfl/fl group were found critically ill at different days post infection and were also analyzed. One mouse was euthanized at 7 dpi and three others at 9 dpi. All those mice presented a higher viral load compared to control mice at 6 dpi (above 5x104 eq PFU/g tissue). At 21 dpi, viral titers decreased in both groups to 101–102 eqPFU/g, and no differences were further detected.
4 Discussion
This study demonstrates that two ZIKV strains, ZIKVPE243 (Asian/American lineage) and ZIKVMR766 (African prototype), induce distinct molecular signatures in HBMEC, an established model of the BBB. Among the pathways modulated, type I IFNs signaling stood out as one of the most marked differences between the strains. This prompted us to further investigate how strain-specific modulation of IFN responses in endothelial cells could influence viral replication, cytotoxicity, and BBB integrity, using an integrated approach that combined transcriptomic, functional, and in vivo analyses.
So far, only isolates belonging to the Asian genotype have been associated with congenital and neonatal problems and neurological disorders (Musso et al., 2019; Liu et al., 2019). However, experimental in vitro and in vivo models have consistently shown that African genotypes isolates may show a more virulent profile than the Asian strains (Anfasa et al., 2017; Bowen et al., 2017; Shao et al., 2017; Tripathi et al., 2017; Smith et al., 2017). These findings align with our previous work showing that ZIKVMR766 displays higher replication rates, cytotoxicity, and BBB disruption in vitro (Papa et al., 2017), correlating with increased neurological outcomes and mortality in vivo (Lucas et al., 2018). Similarly, Aubry et al. (2021) reported that recent African isolates caused higher viremia, morbidity, and fetal death in IFNAR/IFNAGR-deficient mice, compared to pandemic and nonpandemic Asian strains, suggesting that the neuropathogenic potential of first ones might have been underestimated or misdiagnosed.
Our transcriptome analysis showed broader and more pronounced transcriptional changes in ZIKVMR766–infected HBMECs, whereas ZIKVPE243 induced a profile closer to mock controls, consistent with milder cytopathic effects. Previous studies in ZIKV-infected patients and diverse in vitro models have reported heterogeneous activation of IFN-related pathways. Infection of primary macrophages with the Asian strain COL345Si induced upregulation of genes related to RNA modification and post-transcriptional regulation (Fernandez et al., 2022), together with enhanced expression of several ISGs, yet with only a slight induction of type I IFN transcripts. Infection of primary mDCs with another Asian strain (PRVABC59), however, had minimal impact on global transcriptional profile and did not enhance the expression of genes involved in pathogen sensing or interferon signaling (Sun et al., 2017), which closely resemble the profile of HBMECs infected with ZIKVPE243. Conversely, mDCs isolated from acutely infected patients showed major alterations in gene expression signatures; while IFN-related genes were downregulated (Sun et al., 2017). Consistent with our findings, these studies suggest that limited activation of sensing and innate immune pathways may represent an evasion strategy of epidemic ZIKV strains, thereby enabling more silent dissemination within individuals and across populations.
The distinct transcriptional modulation IRFs and ISGs led us to identify the upstream sensors responsible for this activation. Selective depletion of RIG-I or TLR3 revealed RIG-I as the primary sensor mediating IFN-β induction upon ZIKV infection, consistent with studies conducted with various human cell types (Hamel et al., 2015; Singh et al., 2017; Hertzog et al., 2018; Schilling et al., 2020; Plociennikowska et al., 2021). Indeed, a conserved stem-loop structure in the 5’ UTR region in the flaviviruses genome has been proposed as a RIG-I agonist (Chazal et al., 2018), supporting the role of RIG-I as a common viral RNA sensor during infection with both ZIKV strains. MDA5 expression was also upregulated in ZIKVMR766-infected cells, suggesting a possible contribution to IFN-β induction. However, since RIG-I and MDA5 share overlapping functions and are both IFN-inducible, this increase could reflect secondary signaling. Furthermore, RIG-I depletion nearly abolished IFN-β production, underscoring its predominant role in initiating antiviral signaling in our system.
ZIKVMR766 induced a more rapid and robust IFN-β response compared with ZIKVPE243, but it could not be attribute to higher viral RNA levels at early time points. Inhibition of IFN production by ZIKV nonstructural (NS) proteins was previously described (Bowen et al., 2017; Hertzog et al., 2018; Fanunza et al., 2021a, b), and may be triggered by several pathways, including interaction with RIG-I CARD domain, TBK1 or IRF3, hampering the activation of TBK1/IRF3 signaling (Wu et al., 2017; Xia et al., 2018; Lin et al., 2019; Li et al., 2020). Also, interaction of the viral structural capsid protein with TRIM25 has been shown to regulate RIG-I ubiquitination and downstream IFN production (Airo et al., 2022). We identified few substitutions in the capsid sequence, including the K101R, which was associated with enhanced neurovirulence of ZIKV African strains (Song et al., 2023), suggesting that strain-specific differences in capsid composition or structure may contribute to RIG-I activation and distinct IFN-β expression kinetics.
Despite efficient IFN-β production, both ZIKV strains productively replicated in HBMECs, indicating virus evasion from IFN-mediated antiviral signaling. Flaviviruses can counteract type I IFN response through multiple pathways. A major described mechanism involves STAT2 ubiquitination and proteasomal degradation by the NS5 protein, previously observed in DENV and ZIKV infections (Morrison et al., 2013; Grant et al., 2016). In ZIKV, STAT2 degradation depends on its interaction with NS5 RNA-dependent RNA polymerase (RdRP) domain (Grant et al., 2016; Wang et al., 2020; Parisien et al., 2022), and may be accelerated by downmodulation of de novo host proteins synthesis (Shu et al., 2021). Another important mechanism relies on inhibition of STAT1 or STAT2 phosphorylation by NS proteins, reported in DENV, WNV, Japanese encephalitis virus (JEV), and Tick-born encephalitis virus (TBEV) (Muñoz-Jordan et al., 2003, Lin et al., 2006; Laurent-Rolle et al., 2010; Yang et al., 2020). However, IFN evasion is not equally detected in all cell types (Yang et al., 2021).
In HBMECs, we observed a remarkable reduction of STAT1 phosphorylation and almost suppression of STAT2 expression, indicating these as major mechanisms hampering IFN-β signal transduction. Those events were even more pronounced in the cells infected with ZIKVMR766 in relation to ZIKVPE243. A variable magnitude of IFN signaling inhibition has also been reported following NS5 transfection or infection with distinct ZIKV strains (Grant et al., 2016; Xia et al., 2018). Although we have identified amino acid substitutions in the NS5 sequences of ZIKVMR766 and ZIKVPE243, none correspond to residues previously associated with RNA synthesis efficiency or with STAT2 interaction and inhibition (Zhao et al., 2017; Wang et al., 2020; Conde et al., 2020), and remains to be further explored.
ZIKV also produces 3’UTR derived sfRNA (Akiyama et al., 2016; Donald et al., 2016), which has been associated with cytopathic effect and modulation of antiviral responses in multiple flavivirus infection models (Pijlman et al., 2008; Schuessler et al., 2012). Transfection of A549 cells with a plasmid containing the sfRNA of ZIKVPE243 inhibited poly I:C-induced IFN response, mostly due to antagonizing RIG-I signaling (Donald et al., 2016). A comparison of different ZIKV isolates, including the ones investigated here, showed that despite nucleotide differences in the 3′ UTRs, these are unlikely to affect the predicted secondary structures responsible for sfRNA, suggesting conserved functions in antagonizing type I interferon responses.
Importantly, although HBMECs infected with ZIKV resist the effects mediated by type I IFN, IFN neutralization in low MOI infected cultures led to increased viral load, cytotoxicity and virus dissemination. In agreement with our previous findings (Papa et al., 2017) and others (Wang et al., 2023), ZIKVPE243 infection alone did not affect endothelial permeability throughout the experiment. However, IFN neutralization rendered ZIKVPE243-infected monolayers permeable, indicating that the preserved integrity observed under normal conditions depends on type I IFN signaling. Differently from earlier studies (Papa et al., 2017; Wang et al., 2023), where TEER values remained stable across the entire culture period, here we observed an initial increase in TEER during the early phase of the assay. This is likely due to the monolayers starting slightly below full confluence, as a lower MOI was applied in these conditions. Despite this initial rise, TEER values were not influenced by infection up to 48 h. Thereafter, however, neutralization of IFN led to a gradual decline in TEER, more clearly evident in ZIKVPE243-infected cultures, where barrier integrity is otherwise preserved.
Also, addition of IFN-β 24 h before infection inhibited virus replication. IFN-β IC50 and IC90 were significantly higher in the cultures infected with ZIKVMR766 in relation to the ones infected with ZIKVPE423, indicating greater resistance to IFN-mediated antiviral activity, consistent with higher virus titers later upon infection. Accordingly, pretreating fibroblasts with type I IFN also reduced ZIKV replication (Hamel et al., 2015), and increased resistance of ZIKVMR766 in comparison with the Asian strain PRBAVC59, had been previously reported in A549 cells (Gobillot et al., 2020).
Recent findings by Lefevre et al. (2024) identified lineage-specific upstream open reading frames (uORFs) in the 5′ untranslated region (UTR) of ZIKV that may modulate viral replication and tropism. In African isolates, a single uORF is present, whereas in Asian/American strain, including ZIKVPE243, a nucleotide insertion at position 81 divides this uORF into two distinct uORFs - uORF1 and uORF2. Using human brain organoids and glioblastoma U251 cells, the authors showed that viruses bearing the African-like uORF or lacking uORF1 reached higher titers than the American wild-type strain. While direct evidence was not demonstrated, the data suggest that the African-like uORF arrangement may confer resistance to eIF2α-mediated translational arrest, potentially favoring replication under IFN-induced stress. Our genomic analysis confirmed the presence of nt81 insertion in ZIKVPE243, but not in ZIKVMR766, possibly implicating lineage-specific uORF as an additional mechanism of IFN evasion. In fact, our comparative genomic analysis identified amino acid changes in the 5′ UTR, capsid, and NS5 regions of ZIKVPE243 and ZIKVMR766, including ones previously associated with differences in viral biology, as well as novel substitutions whose functional relevance remains unknown. While reverse-genetic or gene-editing approaches will be required to define their precise roles, it is plausible that sequence variations across these genomic regions act synergistically to modulate IFN signaling, viral replication, and disease outcome, thereby shaping strain-specific virulence.
Evasion of type I IFN signaling has been linked to epidemiological fitness and disease severity during flavivirus infections, including ZIKV (Manokaran et al., 2015; Castro-Jimenez et al., 2022). To specifically evaluate the contribution of endothelial cells in these processes, we employed an in vivo model in which IFNAR deficiency is restricted to the endothelium. Our previous studies showed that intravenous infection of IFNAR-deficient mice with ZIKVMR766, but not ZIKVPE243, led to BBB disruption and lethality (Lucas et al., 2018). Using identical infection parameters (4-week-old mice; 2×105 PFU) for direct comparison, we now demonstrated, for the first time, that endothelial-specific IFNAR depletion also rendered mice susceptible to ZIKVMR766-induced lethality, accompanied by greater neuroinvasiveness.
In an in vivo context, however, other components of the neurovascular unit might play an additional role in regulating BBB integrity and antiviral response. Previous studies have reported that pericytes, astrocytes and microglial cells are susceptible to be ZIKV, potentially contributing to IFN-β-mediated protection (Chen et al., 2018; Kim et al., 2020; Stefanik et al., 2018; Ojha et al., 2019). Astrocytes have been recently identified as innate immune sentinel cells capable of sensing ZIKV and producing IFN-β in human fetal brain explants and iPSC-derived neural cultures (Stokes et al., 2025), and infection of astrocytic endfeet has been associated with BBB disruption in IFNAR-deficient mice (Jurado et al., 2018). However, in contrast to our observations in HBMECs, infection of human astrocytes with Asian ZIKV strains resulted in higher infectivity and cell death than with African isolates (Ojha et al., 2019). Also, transcriptome analysis of primary astrocytes infected with ZIKVPE243 showed downregulation of IFN-signaling pathways (Geddes et al., 2021), in accordance with the observed profile in HBMECs. Microglia are also recognized targets of ZIKV infection, and interferon responses influence viral replication dynamics (Hanrath et al., 2022). Lineage-specific differences have been reported in this cell type, although the patterns diverge between human and mouse microglial models. Asian strains consistently elicit weaker inflammatory activation than African isolates in both species; however, they replicate more efficiently in human microglia (Bird et al., 2024), but not in mouse microglia (Oliveira et al., 2023). In pericyte cultures, neutralization of type I IFNs increased viral replication at later time points, suggesting a protective antiviral effect (Kim et al., 2020).
Still, in our model, other BBB-associated cells were not included in the in vitro setting; therefore, IFN neutralization primarily blocked autocrine signaling in endothelial cells. Furthermore, although IFNAR deficiency in vivo was not restricted to endothelial cells from the BBB, these findings collectively reinforce the importance of endothelial IFN signaling in maintaining barrier integrity and controlling ZIKV neuroinvasiveness and lethality, even though direct evidence linking viral replication in these cells remains to be established.
Other in vitro models have also supported a protective role for endothelial IFN responses in restricting viral replication and neuroinvasion. Using iPSC-derived human BMECs infected with a panel of alphaviruses and flaviviruses, Cheng et al. (2022) showed an inverse correlation between IFITM1 expression and both viral replication and the ability to cross the iBMEC monolayer. Similarly, BMEC derived from IFNAR-deficient mice infected with WNV exhibit reduced IFN and increased inflammatory cytokines, leading to barrier dysfunction (Daniels et al., 2014). Co-culture experiments with astrocytes further demonstrated that endothelial IFN signaling played a predominant role in preventing BBB breakdown. Therefore, studies using IFNAR-deficient HBMECs will contribute of dissect the impact of endothelial IFN signaling to ZIKV replication, metabolic modulation, and BBB integrity.
In summary, our findings demonstrate that type I IFN signaling in brain endothelial cells is a key determinant of BBB integrity and resistance to ZIKV infection. By inducing classical antiviral response, endothelial IFN limit viral replication, cytotoxicity, and barrier permeability, thereby contributing to disease resistance. However, this protection is only partial, since ZIKV proteins interfere with IFN signaling, rendering infected cells less responsive to its antiviral effects. The two viral strains analyzed exhibited distinct biological profiles: ZIKVMR766 induced stronger IFN and inflammatory responses yet showed greater resistance to IFN-mediated restriction, correlating with enhanced BBB disruption and disease severity in vivo.
Although BBB dysfunction is a hallmark of several viral infections associated with neurological disease (Mustafa et al., 2019; Greene et al., 2024; Coelho et al., 2025; Boardman et al., 2025), the specific contribution of endothelial cells and their interferon-mediated responses remains poorly understood. As the structural and immunological core of the BBB, endothelial cells actively coordinate antiviral defense, inflammatory regulation, and barrier preservation. Our study thus reinforces their role as central players in CNS protection. Beyond ZIKV, these findings provide a conceptual framework for investigating IFN-dependent mechanisms of BBB protection and neuroinvasion across other neurotropic viral infections, guiding future development of therapeutic strategies aiming to maintain endothelial integrity and mitigate virus-induced neurological disorders.
Data availability statement
The libraries sequenced in our study are available at SRA (www.ncbi.nlm.nih.gov/sra) under BioProject accession number PRJNA1337481 and accession numbers SAMN52321194–SAMN52321202.
Ethics statement
Ethical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used. The animal study was approved by Ethics Committee of Animal Care and Use from UNICAMP (protocol 4858-1/2018). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
LL: Data curation, Formal analysis, Investigation, Methodology, Validation, Writing – original draft, Writing – review & editing. YM: Formal analysis, Investigation, Methodology, Writing – original draft. PF: Data curation, Formal analysis, Investigation, Methodology, Software, Writing – review & editing. SC: Investigation, Methodology, Supervision, Writing – review & editing. PP: Data curation, Formal analysis, Investigation, Methodology, Writing – review & editing. CS: Investigation, Methodology, Writing – review & editing. LM: Investigation, Methodology, Writing – review & editing. BB: Formal analysis, Investigation, Methodology, Writing – review & editing. NC: Formal analysis, Investigation, Methodology, Software, Writing – review & editing. FM: Investigation, Methodology, Writing – review & editing. JP: Resources, Writing – review & editing, Formal analysis, Funding acquisition, Investigation, Supervision. RA: Funding acquisition, Resources, Writing – review & editing, Data curation, Formal analysis, Software, Supervision. LA: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work received financial support from Rede Corona-ômica BR MCTI/FINEP affiliated to Re-deVírus/MCTI: (01.20.0029.000462/20 404096/2020-4; 01.22.0074.00 (1227/21)); INCT program award (grant 408527/2024-2); Coordination for the Improvement of Higher Education Personnel: CAPES; Brazilian National Council for Scientific and Technological Development: CNPq (LA 404852/2024-6; 306368/2022-6); Carlos Chagas Filho Research Support Foundation: FAPERJ (LA (E-26/201.206/2021, E-26/204.307/2024; LL (E-26/201.918/2024); SC (E-26/20.746/2022); LM (E-26/200.441/2025); BB (E-26/202.346/2024)); Division of Intramural Research, NIAID, NIH; Instituto Todos pela Saúde - ITpS (C1294; C2024-0779, A113); CNPq (315592/2021-4, INCT-One Health CNPq 405786/2022-0); FAPEMIG (APQ-02826-24; APD-00520-25); FAPEMIG (BPD-00820-22).
Acknowledgments
The authors wish to thank Dr. Dennis J. Grab (Uniformed Services University of the Health Sciences, Bethesda (USUHS)) for kindly providing us with HBMEC, to Dr. Ernesto Marques for kindly providing ZIKVPE243 strain, and to Dr. Laura Gil for the construction of HBMECISREluc reporter cell. The authors also wish to thank Ronaldo Rocha (UFRJ) for technical support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. Generative AI was used only to improve grammar and language clarity and the authors are fully responsible for the content and interpretation of the work.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2025.1726007/full#supplementary-material
References
Abbott, N. J., Patabendige, A. A., Dolman, D. E., Yusof, S. R., and Begley, D. J. (2010). Structure and function of the blood-brain barrier. Neurobiol. Dis. 37, 13–25. doi: 10.1016/j.nbd.2009.07.030
Airo, A. M., Felix-Lopez, A., Mancinelli, V., Evseev, D., Lopez-Orozco, J., Shire, K., et al. (2022). Flavivirus capsid proteins inhibit the interferon response. Viruses 14, 968. doi: 10.3390/v14050968
Akiyama, B. M., Laurence, H. M., Massey, A. R., Costantino, D. A., Xie, X., Yang, Y., et al.(2016).Zika virus produces non-coding RNAs using a multi-pseudoknot structure that confounds a cellular exonuclease. Science 354, 1148–1152. doi: 10.1126/science.aah3963
Alva, J. A., Zovein, A. C., Monvoisin, A., Murphy, T., Salazar, A., Harvey, N. L., et al. (2006). VE-Cadherin-Cre-recombinase transgenic mouse: a tool for lineage analysis and gene deletion in endothelial cells. Dev. Dyn. 235, 759–767. doi: 10.1002/dvdy.20643
Anfasa, F., Siegers, J. Y., van der Kroeg, M., Mumtaz, N., Raj, V. S., de Vrij, F. M. S., et al.(2017).Phenotypic differences between Asian and African lineage Zika viruses in human neural progenitor cells. mSphere 2, e00292–17. doi: 10.1128/mSphere.00292-17
Aubry, F., Jacobs, S., Darmuzey, M., Lequime, S., Delang, L., Fontaine, A., et al. (2021). Recent African strains of Zika virus display higher transmissibility and fetal pathogenicity than Asian strains. Nat. Commun. 12, 916. doi: 10.1038/s41467-021-21199-z
Bayer, A., Lennemann, N. J., Ouyang, Y., Bramley, J. C., Morosky, S., Marques, E. T. D. A., et al. (2016). Type III interferons produced by human placental trophoblasts confer protection against zika virus infection. Cell Host Microbe 19, 705–712. doi: 10.1016/j.chom.2016.03.008
Bird, I. M., Cavener, V., Surendran Nair, M., Nissly, R. H., Chothe, S. K., Jacob, J., et al. (2024). Distinct replication kinetics, cytopathogenicity, and immune gene regulation in human microglia cells infected with Asian and African lineages of zika virus. Microorganisms 12, 1840. doi: 10.3390/microorganisms12091840
Blighe, K., Rana, S., and Lewis, M. (2024). EnhancedVolcano: Publication-ready volcano plots with enhanced colouring and labeling (R package version 1.24.0). Available online at: https://github.com/kevinblighe/EnhancedVolcano (Accessed July 14, 2025).
Boardman, S. A., Hetherington, C., Hughes, T., Cook, C., Galea, I., Hilton, O., et al. (2025). Viral infection and the blood-brain barrier: molecular research insights and therapies. J. Infect. Dis., jiaf455. doi: 10.1093/infdis/jiaf455
Bowen, J. R., Quicke, K. M., Maddur, M. S., O’Neal, J. T., McDonald, C. E., Fedorova, N. B., et al. (2017). Zika virus antagonizes type I interferon responses during infection of human dendritic cells. PloS Pathog. 13, e1006164. doi: 10.1371/journal.ppat.1006164
Brasil, P., Pereira, J. P., Jr, Moreira, M. E., Ribeiro Nogueira, R. M., Damasceno, L., Wakimoto, M., et al. (2016). Zika virus infection in pregnant women in Rio de Janeiro. N Engl. J. Med. 375, 2321–2334. doi: 10.1056/NEJMoa1602412
Bushmanova, E., Antipov, D., Lapidus, A., and Prjibelski, A. D. (2019). rnaSPAdes: a de novo transcriptome assembler and its application to RNA-Seq data. Gigascience 8, giz100. doi: 10.1093/gigascience/giz100
Bushnell, B. (2014). BBMap: A Fast, Accurate, Splice-Aware Aligner (Berkeley, CA: Ernest Orlando Lawrence Berkeley National Laboratory).
Cao-Lormeau, V. M., Roche, C., Teissier, A., Robin, E., Berry, A. L., Mallet, H. P., et al. (2014). Zika virus, French polynesia, South pacifi. Emerg. Infect. Dis. 20, 1085–1086. doi: 10.3201/eid2006.140138
Carteaux, G., Maquart, M., Bedet, A., Contou, D., Brugières, P., Fourati, S., et al. (2016). Zika virus associated with meningoencephalitis. N Engl. J. Med. 374, 1595–1596. doi: 10.1056/NEJMc1602964
Castro-Jiménez, T. K., Gómez-Legorreta, L. C., López-Campa, L. A., Martínez-Torres, V., Alvarado-Silva, M., Posadas-Mondragón, A., et al. (2022). Variability in susceptibility to type I interferon response and subgenomic RNA accumulation between clinical isolates of dengue and zika virus from oaxaca Mexico correlate with replication efficiency in human cells and disease severity. Front. Cell Infect. Microbiol. 12. doi: 10.3389/fcimb.2022.890750
Chazal, M., Beauclair, G., Gracias, S., Najburg, V., Simon-Lorière, E., Tangy, F., et al. (2018). RIG-I recognizes the 5’ Region of dengue and zika virus genomes. Cell Rep. 24, 320–328. doi: 10.1016/j.celrep.2018.06.047
Chen, J., Yang, Y. F., Yang, Y., Zou, P., Chen, J., He, Y., et al. (2018). AXL promotes Zika virus infection in astrocytes by antagonizing type I interferon signalling. Nat. Microbiol. 3, 302–309. doi: 10.1038/s41564-017-0092-4
Cheng, Y., Medina, A., Yao, Z., Basu, M., Natekar, J. P., Lang, J., et al. (2022). Intrinsic antiviral immunity of barrier cells revealed by an iPSC-derived blood-brain barrier cellular model. Cell Rep. 39, 110885. doi: 10.1016/j.celrep.2022.110885
Coelho, S. V. A., Neris, R. L. S., Papa, M. P., Schnellrath, L. C., Meuren, L. M., Tschoeke, D. A., et al. (2017). Development of standard methods for Zika virus propagation, titration, and purification. J. Virol. Methods 246, 65–74. doi: 10.1016/j.jviromet.2017.04.011
Coelho, S. V. A., Souza, G. L. E., Bezerra, B. B., Lima, L. R., Correa, I. A., de Almeida, D. V., et al.(2025).SARS-Cov-2 Replication in a Blood-Brain Barrier Model Established with Human Brain Microvascular Endothelial Cells Induces Permeability and Disables ACE2-Dependent Regulation of Bradykinin B1 Receptor. Int. J. Mol. Sci. 26, 5540. doi: 10.3390/ijms26125540
Conceição, T. M., Rust, N. M., Berbel, A. C., Martins, N. B., do Nascimento Santos, C. A., Da Poian, A. T., et al. (2013). Essential role of RIG-I in the activation of endothelial cells by dengue virus. Virology 435, 281–292. doi: 10.1016/j.virol.2012.09.038
Conde, J. N., Schutt, W. R., Mladinich, M., Sohn, S. Y., Hearing, P., and Mackow, E. R. (2020). NS5 sumoylation directs nuclear responses that permit zika virus to persistently infect human brain microvascular endothelial cells. J. Virol. 94, e01086–e01020. doi: 10.1128/JVI.01086-20
Daniels, B. P., Holman, D. W., Cruz-Orengo, L., Jujjavarapu, H., Durrant, D. M., and Klein, R. S. (2014). Viral pathogen-associated molecular patterns regulate blood-brain barrier integrity via competing innate cytokine signals. mBio 5, e01476–e01414. doi: 10.1128/mBio.01476-14
da Silva, I. R. F., Frontera, J. A., Bispo de Filippis, A. M., Nascimento, O. J. M. D., and RIO-GBS-ZIKV Research Group (2017). Neurologic complications associated with the zika virus in Brazilian adults. JAMA Neurol. 74, 1190–1198. doi: 10.1001/jamaneurol.2017.1703
de Oliveira, W. K., Carmo, E. H., Henriques, C. M., Coelho, G., Vazquez, E., Cortez-Escalante, J., et al.(2017).Zika virus infection and associated neurologic disorders in Brazil. N. Engl. J. Med.376, 1591–1593. doi: 10.1056/NEJMc1608612
Dick, G. W., Kitchen, S. F., Haddow, A. J., and Isolations, I. (1952). and serological specificity. Trans. R Soc. Trop. Med. Hyg. 46, 509–520. doi: 10.1016/0035-9203(52)90042-4
Donald, C. L., Brennan, B., Cumberworth, S. L., Rezelj, V. V., Clark, J. J., Cordeiro, M. T., et al. (2016). Full genome sequence and sfRNA interferon antagonist activity of zika virus from Recife, Brazil. PloS Negl. Trop. Dis. 10, e0005048. doi: 10.1371/journal.pntd.0005048
Dowall, S. D., Graham, V. A., Rayner, E., Hunter, L., Atkinson, B., Hall, G., et al. (2016). A susceptible mouse model for Zika virus infection. PloS Negl. Trop. Dis. 10, e0004658. doi: 10.1371/journal.pntd.0004658
Duffy, M. R., Chen, T. H., Hancock, W. T., Powers, A. M., Kool, J. L., Lanciotti, R. S., et al. (2009). Zika virus outbreak on Yap Island, Federated States of Micronesia. N Engl. J. Med. 360, 2536–2543. doi: 10.1056/NEJMoa0805715
Durinck, S., Spellman, P., Birney, E., and Huber, W. (2009). Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 4, 1184–1191. doi: 10.1038/nprot.2009.97
Fanunza, E., Carletti, F., Quartu, M., Grandi, N., Ermellino, L., Milia, J., et al. (2021a). Zika virus NS2A inhibits interferon signaling by degradation of STAT1 and STAT2. Virulence 12, 1580–1596. doi: 10.1080/21505594.2021.1935613
Fanunza, E., Grandi, N., Quartu, M., Carletti, F., Ermellino, L., Milia, J., et al. (2021b). INMI1 zika virus NS4B antagonizes the interferon signaling by suppressing STAT1 phosphorylation. Viruses 13, 2448. doi: 10.3390/v13122448
Fernandez, G. J., Ramírez-Mejía, J. M., and Urcuqui-Inchima, S. (2022). Transcriptional and post-transcriptional mechanisms that regulate the genetic program in Zika virus-infected macrophages. Int. J. Biochem. Cell Biol. 153, 106312. doi: 10.1016/j.biocel.2022.106312
Garcez, P. P., Loiola, E. C., da Costa, R. M., Higa, L. M., Trindade, P., Delvecchio, R., et al. (2016). Zika virus impairs growth in human neurospheres and brain organoids. Science 352, 816–818. doi: 10.1126/science.aaf6116
Geddes, V. E. V., Brustolini, O. J. B., Cavalcante, L. T. F., Moreira, F. R. R., de Castro, F. L., Guimarães, A. P. C., et al. (2021). Common dysregulation of innate immunity pathways in human primary astrocytes infected with chikungunya, mayaro, oropouche, and zika viruses. Front. Cell Infect. Microbiol. 11. doi: 10.3389/fcimb.2021.641261
Gobillot, T. A., Humes, D., Sharma, A., Kikawa, C., and Overbaugh, J. (2020). The robust restriction of zika virus by type-I interferon in A549 cells varies by viral lineage and is not determined by IFITM3. Viruses 12, 503. doi: 10.3390/v12050503
Grant, A., Ponia, S. S., Tripathi, S., Balasubramaniam, V., Miorin, L., Sourisseau, M., et al. (2016). Zika virus targets human STAT2 to inhibit type I interferon signaling. Cell Host Microbe 19, 882–890. doi: 10.1016/j.chom.2016.05.009
Greene, C., Connolly, R., Brennan, D., Laffan, A., O’Keeffe, E., Zaporojan, L., et al. (2024). Blood–brain barrier disruption and sustained systemic inflammation in individuals with long COVID-associated cognitive impairment. Nat. Neurosci. 27, 421–432. doi: 10.1038/s41593-024-01576-9
Haddow, A. D., Schuh, A. J., Yasuda, C. Y., Kasper, M. R., Heang, V., Huy, R., et al. (2012). Genetic characterization of Zika virus strains: geographic expansion of the Asian lineage. PloS Negl. Trop. Dis. 6, e1477. doi: 10.1371/journal.pntd.0001477
Hamel, R., Dejarnac, O., Wichit, S., Ekchariyawat, P., Neyret, A., and Luplertlop, N. (2015). Biology of Zika virus infection in human skin cells. J. Virol. 89, 8880–8896. doi: 10.1128/JVI.00354-15
Hanrath, A. T., Hatton, C. F., Gothe, F., Browne, C., Vowles, J., Leary, P., et al. (2022). Type I interferon receptor (IFNAR2) deficiency reveals Zika virus cytopathicity in human macrophages and microglia. Front. Immunol. 13. doi: 10.3389/fimmu.2022.1035532
Hertzog, J., Dias Junior, A. G., Rigby, R. E., Donald, C. L., Mayer, A., Sezgin, E., et al. (2018). Infection with a Brazilian isolate of Zika virus generates RIG-I stimulatory RNA and the viral NS5 protein blocks type I IFN induction and signaling. Eur. J. Immunol. 48, 1120–1136. doi: 10.1002/eji.201847483
Jurado, K. A., Yockey, L. J., Wong, P. W., Lee, S., Huttner, A. J., and Iwasaki, A. (2018). Antiviral CD8 T cells induce Zika-virus-associated paralysis in mice. Nat. Microbiol. 3, 141–147. doi: 10.1038/s41564-017-0060-z
Kadry, H., Noorani, B., and Cucullo, L. (2020). A blood-brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 17, 69. doi: 10.1186/s12987-020-00230-3
Kamiyama, N., Soma, R., Hidano, S., Watanabe, K., Umekita, H., Fukuda, C., et al. (2017). Ribavirin inhibits Zika virus (ZIKV) replication in vitro and suppresses viremia in ZIKV-infected STAT1-deficient mice. Antiviral Res. 146, 1–11. doi: 10.1016/j.antiviral.2017.08.007
Kim, J., Alejandro, B., Hetman, M., Hattab, E. M., Joiner, J., Schroten, H., et al. (2020). Zika virus infects pericytes in the choroid plexus and enters the central nervous system through the blood-cerebrospinal fluid barrier. PloS Pathog. 16, e1008204. doi: 10.1371/journal.ppat.1008204
Lackritz, E. M., Ng, L.-C., Marques, E. T. A., Rabe, I. B., Bourne, N., Staples, J. E., et al. (2025). Zika virus: advancing a priority research agenda for preparedness and response. Lancet Infect. Dis. 25, e390–e401. doi: 10.1016/S1473-3099(24)00794-1
Lanciotti, R. S., Kosoy, O. L., Laven, J. J., Velez, J. O., Lambert, A. J., Johnson, A. J., et al. (2008). Genetic and serologic properties of Zika virus associated with an epidemic, Yap State, Micronesia 2007. Emerg. Infect. Dis. 14, 1232–1239. doi: 10.3201/eid1408.080287
Langmead, B. and Salzberg, S. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Laurent-Rolle, M., Boer, E. F., Lubick, K. J., Wolfinbarger, J. B., Carmody, A. B., Rockx, B., et al. (2010). The NS5 protein of the virulent West Nile virus NY99 strain is a potent antagonist of type I interferon-mediated JAK-STAT signaling. J. Virol. 84, 3503–3515. doi: 10.1128/JVI.01161-09
Lazear, H. M., Govero, J., Smith, A. M., Platt, D. J., Fernandez, E., Miner, J. J., et al. (2016). A mouse model of zika virus pathogenesis. Cell Host Microbe 19, 720–730. doi: 10.1016/j.chom.2016.03.010
Lefèvre, C., Cook, G. M., Dinan, A. M., Torii, S., Stewart, H., Gibbons, G., et al. (2024). Zika viruses encode 5’ upstream open reading frames affecting infection of human brain cells. Nat. Commun. 15, 8822. doi: 10.1038/s41467-024-53085-9
Li, A., Wang, W., Wang, Y., Chen, K., Xiao, F., Hu, D., et al. (2020). NS5 conservative site is required for zika virus to restrict the RIG-I signaling. Front. Immunol. 11. doi: 10.3389/fimmu.2020.00051
Li, C., Xu, D., Ye, Q., Hong, S., Jiang, Y., Liu, X., et al.(2016).Zika virus disrupts neural progenitor development and leads to microcephaly in mice. Cell Stem Cell19, 120–126. doi: 10.1016/j.stem.2016.04.017
Lin, J. Y., Kuo, R. L., and Huang, H. I. (2019). Activation of type I interferon antiviral response in human neural stem cells. Stem Cell Res. Ther. 10, 387. doi: 10.1186/s13287-019-1521-5
Lin, R. J., Liao, C. L., Lin, E., and Lin, Y. L.(2006). Blocking of interferon-induced Jak-Stat signaling by Japanese encephalitis virus NS5 through a protein tyrosine phosphatase-mediated mechanism.J. Virol. 80, 5908–5918. doi: 10.1128/JVI.02714-05
Liu, Z. Y., Shi, W. F., and Qin, C. F. (2019). The evolution of Zika virus from Asia to the Americas. Nat. Rev. Microbiol. 17, 131–139. doi: 10.1038/s41579-018-0134-9
Livak, K. J. and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lodeiro, M. F., Filomatori, C. V., and Gamarnik, A. V. (2009). Structural and functional studies of the promoter element for dengue virus RNA replication. J. Virol. 83, 993–1008. doi: 10.1128/JVI.01647-08
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. doi: 10.1186/s13059-014-0550-8
Lucas, C. G. O., Kitoko, J. Z., Ferreira, F. M., Suzart, V. G., Papa, M. P., Coelho, S. V. A., et al. (2018). Critical role of CD4+ T cells and IFNγ signaling in antibody-mediated resistance to Zika virus infection. Nat. Commun. 9, 3136. doi: 10.1038/s41467-018-05519-4
Luo, H., Winkelmann, E. R., Fernandez-Salas, I., Li, L., Mayer, S. V., Danis-Lozano, R., et al. (2018). Zika, dengue and yellow fever viruses induce differential anti-viral immune responses in human monocytic and first trimester trophoblast cells. Antiviral Res. 151, 55–62. doi: 10.1016/j.antiviral.2018.01.003
Manokaran, G., Finol, E., Wang, C., Gunaratne, J., Bahl, J., Ong, E. Z., et al. (2015). Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science 350, 217–221. doi: 10.1126/science.aab3369
Melo, A. S., Aguiar, R. S., Amorim, M. M., Arruda, M. B., Melo, F. O., Ribeiro, S. T., et al. (2016). Congenital zika virus infection: beyond neonatal microcephaly. JAMA Neurol. 73, 1407–1416. doi: 10.1001/jamaneurol.2016.3720
Meuren, L. M., Prestes, E. B., Papa, M. P., de Carvalho, L. R. P., Mustafá, Y. M., da Costa, L. S., et al. (2022). Infection of endothelial cells by dengue virus induces ROS production by different sources affecting virus replication, cellular activation, death and vascular permeability. Front. Immunol. 13. doi: 10.3389/fimmu.2022.810376
Miranda-Filho, D., de, B., Martelli, C. M., Ximenes, R. A., Araújo, T. V., Rocha, M. A., et al. (2016). Initial description of the presumed congenital zika syndrome. Am. J. Public Health 106, 598–600. doi: 10.2105/AJPH.2016.303115
Mladinich, M. C., Schwedes, J., and Mackow, E. R. (2017). Zika virus persistently infects and is basolaterally released from primary human brain microvascular endothelial cells. mBio 8, e00952–e00917. doi: 10.1128/mBio.00952-17
Mlakar, J., Korva, M., Tul, N., Popović, M., Poljšak-Prijatelj, M., Mraz, J., et al. (2016). Zika virus associated with microcephaly. N Engl. J. Med. 374, 951–958. doi: 10.1056/NEJMoa1600651
Morrison, J., Laurent-Rolle, M., Maestre, A. M., Rajsbaum, R., Pisanelli, G., Simon, V., et al. (2013). Dengue virus co-opts UBR4 to degrade STAT2 and antagonize type I interferon signaling. PloS Pathog. 9, e1003265. doi: 10.1371/journal.ppat.1003265
Motta, C. S., Torices, S., da Rosa, B. G., Marcos, A. C., Alvarez-Rosa, L., Siqueira, M., et al. (2023). Human brain microvascular endothelial cells exposure to SARS-coV-2 leads to inflammatory activation through NF-κB non-canonical pathway and mitochondrial remodeling. Viruses 15, 745. doi: 10.3390/v15030745
Muñoz-Jordan, J. L., Sánchez-Burgos, G. G., Laurent-Rolle, M., and García-Sastre, A. (2003). Inhibition of interferon signaling by dengue virus. Proc. Natl. Acad. Sci. U S A 100, 14333–14338. doi: 10.1073/pnas.2335168100
Musso, D., Ko, A. I., and Baud, D. (2019). Zika virus infection - after the pandemic. N Engl. J. Med. 381, 1444–1457. doi: 10.1056/NEJMra1808246
Nikolskaia, O. V., Kim, Y. V., Kovbasnjuk, O., Kim, K. J., and Grab, D. J.(2006).Entry of Trypanosoma brucei gambiense into microvascular endothelial cells of the human blood-brain barrier. Int. J. Parasitol. 36, 513–519. doi: 10.1016/j.ijpara.2006.01.011
Mustafá, Y. M., Meuren, L. M., Coelho, S. V. A., and de Arruda, L. B.(2019).Pathways exploited by flaviviruses to counteract the blood-brain barrier and invade the central nervous system. Front. Microbiol. 10, 525. doi: 10.3389/fmicb.2019.00525
Ojha, C. R., Rodriguez, M., Karuppan, M. K. M., Lapierre, J., Kashanchi, F., and El-Hage, N. (2019). Toll-like receptor 3 regulates Zika virus infection and associated host inflammatory response in primary human astrocytes. PloS One 14, e0208543. doi: 10.1371/journal.pone.0208543
Oliveira, F. B. C., Sá Freire, V. P. A. S., Coelho, S. V. A., Meuren, L. M., Palmeira, J. F., Cardoso, A. L., et al. (2023). ZIKV Strains Elicit Different Inflammatory and anti-viral responses in microglia cells. Viruses 15, 1250. doi: 10.3390/v15061250
Papa, M. P., Meuren, L. M., Coelho, S. V. A., de Oliveira Lucas, C. G., Mustafá, Y. M., Matassoli, F. L., et al(2017).Zika virus infects, activates, and crosses brain microvascular endothelial cells, without barrier disruption.Front. Microbiol. 8, 2557. doi: 10.3389/fmicb.2017.02557
Parisien, J. P., Lenoir, J. J., Alvarado, G., and Horvath, C. M. (2022). The human STAT2 coiled-coil domain contains a degron for zika virus interferon evasion. J. Virol. 96, e0130121. doi: 10.1128/JVI.01301-21
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A., and Kingsford, C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. doi: 10.1038/nmeth.4197
Pijlman, G. P., Funk, A., Kondratieva, N., Leung, J., Torres, S., van der Aa, L., et al. (2008). A highly structured, nuclease-resistant, noncoding RNA produced by flaviviruses is required for pathogenicity. Cell Host Microbe 4, 579–591. doi: 10.1016/j.chom.2008.10.007
Plociennikowska, A., Frankish, J., Moraes, T., Del Prete, D., Kahnt, F., Acuna, C., et al. (2021). TLR3 activation by Zika virus stimulates inflammatory cytokine production which dampens the antiviral response induced by RIG-I-like receptors. J. Virol. 95, e01050–e01020. doi: 10.1128/JVI.01050-20
Prigge, J. R., Hoyt, T. R., Dobrinen, E., Capecchi, M. R., Schmidt, E. E., and Meissner, N. (2015). Type I IFNs Act upon Hematopoietic Progenitors To Protect and Maintain Hematopoiesis during Pneumocystis Lung Infection in Mice. J. Immunol. 195, 5347–5357. doi: 10.4049/jimmunol.1501553
Rust, N. M., Papa, M. P., Scovino, A. M., da Silva, M. M., Calzavara-Silva, C. E., Marques, E. T., Jr, et al. (2012). Bradykinin enhances Sindbis virus infection in human brain microvascular endothelial cells. Virology 422, 81–91. doi: 10.1016/j.virol.2011.10.003
Saba Villarroel, P. M., Hamel, R., Gumpangseth, N., Yainoy, S., Koomhin, P., Missé, D., et al. (2024). Global seroprevalence of Zika virus in asymptomatic individuals: A systematic review. PloS Negl. Trop. Dis. 18, e0011842. doi: 10.1371/journal.pntd.0011842
Schilling, M., Bridgeman, A., Gray, N., Hertzog, J., Hublitz, P., Kohl, A., et al. (2020). RIG-I plays a dominant role in the induction of transcriptional changes in zika virus-infected cells, which protect from virus-induced cell death. Cells 9, 1476. doi: 10.3390/cells9061476
Schuessler, A., Funk, A., Lazear, H. M., Cooper, D. A., Torres, S., Daffis, S., et al. (2012). West Nile virus noncoding subgenomic RNA contributes to viral evasion of the type I interferon-mediated antiviral response. J. Virol. 86, 5708–5718. doi: 10.1128/JVI.00207-12
Schuler-Faccini, L., Ribeiro, E. M., Feitosa, I. M., Horovitz, D. D., Cavalcanti, D. P., Pessoa, A., et al. (2016). Possible association between zika virus infection and microcephaly - Brazil 2015. MMWR Morb. Mortal Wkly Rep. 65, 59–62. doi: 10.15585/mmwr.mm6503e2
Shao, Q., Herrlinger, S., Yang, S.-L., Lai, F., Moore, J. M., Brindley, M. A., et al.(2016).Zika virus infection disrupts neurovascular development and results in postnatal microcephaly with brain damage.Development 143, 4127–4136. doi: 10.1242/dev.143768
Shao, Q., Herrlinger, S., Zhu, Y. N., Yang, M., Goodfellow, F., Stice, S. L., et al.(2017).The African Zika virus MR-766 is more virulent and causes more severe brain damage than current Asian lineage and dengue virus. Development 144, 4114–4124. doi: 10.1242/dev.156752
Shu, J., Ma, X., Zhang, Y., Zou, J., Yuan, Z., and Yi, Z. (2021). NS5-independent Ablation of STAT2 by Zika virus to antagonize interferon signalling. Emerg. Microbes Infect. 10, 1609–1625. doi: 10.1080/22221751.2021.1964384
Simpson, D. I. (1964). ZIKA VIRUS INFECTION IN MAN. Trans. R Soc. Trop. Med. Hyg. 58, 335–338. doi: 10.1016/0035-9203(64)90201-9
Singh, P. K., Guest, J. M., Kanwar, M., Boss, J., Gao, N., Juzych, M. S., et al.(2017).Zika virus infects cells lining the blood-retinal barrier and causes chorioretinal atrophy in mouse eyes. JCI Insight 2, e92340. doi: 10.1172/jci.insight.92340
Smith, D. R., Hollidge, B., Daye, S., Zeng, X., Blancett, C., Kuszpit, K., et al.(2017).Neuropathogenesis of Zika virus in a highly susceptible immunocompetent mouse model after antibody blockade of type I interferon. PLoS Negl. Trop. Dis.11, e0005296. doi: 10.1371/journal.pntd.0005296
Song, G. Y., Huang, X. Y., He, M. J., Zhou, H. Y., Li, R. T., Tian, Y., et al. (2023). A single amino acid substitution in the capsid protein of Zika virus contributes to a neurovirulent phenotype. Nat. Commun. 14, 6832. doi: 10.1038/s41467-023-42676-7
Stefanik, M., Formanova, P., Bily, T., Vancova, M., Eyer, L., Palus, M., et al. (2018). Characterisation of Zika virus infection in primary human astrocytes. BMC Neurosci. 19, 5. doi: 10.1186/s12868-018-0407-2
Stokes, C., Whitmore, L. S., Moreno, D., Malhotra, K., Tisoncik-Go, J., Tran, E., et al. (2025). The human neural cell atlas of Zika virus infection in developing brain tissue. Cell Rep. Med. 6, 102189. doi: 10.1016/j.xcrm.2025.102189
Sun, X., Hua, S., Chen, H. R., Ouyang, Z., Einkauf, K., Tse, S., et al. (2017). Transcriptional changes during naturally acquired zika virus infection render dendritic cells highly conducive to viral replication. Cell Rep. 21, 3471–3482. doi: 10.1016/j.celrep.2017.11.087
Tripathi, S., Balasubramaniam, V. R., Brown, J. A., Mena, I., Grant, A., Bardina, S. V., et al. (2017). A novel Zika virus mouse model reveals strain specific differences in virus pathogenesis and host inflammatory immune responses. PloS Pathog. 13, e1006258. doi: 10.1371/journal.ppat.1006258
Wang, B., Thurmond, S., Zhou, K., Sánchez-Aparicio, M. T., Fang, J., Lu, J., et al. (2020). Structural basis for STAT2 suppression by flavivirus NS5. Nat. Struct. Mol. Biol. 27, 875–885. doi: 10.1038/s41594-020-0472-y
Wang, K., Zou, S., Chen, H., Higazy, D., Gao, X., Zhang, Y., et al. (2023). Zika virus replication on endothelial cells and invasion into the central nervous system by inhibiting interferon β translation. Virology 582, 23–34. doi: 10.1016/j.virol.2023.03.006
Weaver, S. C., Costa, F., Garcia-Blanco, M. A., Ko, A. I., Ribeiro, G. S., Saade, G., et al. (2016). Zika virus: History, emergence, biology, and prospects for control. Antiviral Res. 130, 69–80. doi: 10.1016/j.antiviral.2016.03.010
World Health Organization(2016).Zika virus technical report.https://www.who.int/docs/librariesprovider2/default-document-library/zika-virus-echnical-report.pdf (Accessed November 2025).
Wu, Y., Liu, Q., Zhou, J., Xie, W., Chen, C., Wang, Z., et al. (2017). Zika virus evades interferon-mediated antiviral response through the co-operation of multiple nonstructural proteins in vitro. Cell Discov. 3, 17006. doi: 10.1038/celldisc.2017.6
Xia, H., Luo, H., Shan, C., Muruato, A. E., Nunes, B. T. D., and Medeiros, D. B. A. (2018). An evolutionary NS1 mutation enhances Zika virus evasion of host interferon induction. Nat. Commun. 9, 414. doi: 10.1038/s41467-017-02816-2
Xu, S., Hu, E., Cai, Y., Xie, Z., Luo, X., Zhan, L., et al. (2024). Using clusterProfiler to characterize multiomics data. Nat. Protoc. 19, 3292–3320. doi: 10.1038/s41596-024-01020-z
Yang, D., Chu, H., Lu, G., Shuai, H., Wang, Y., Hou, Y., et al. (2021).STAT2-dependent restriction of Zika virus by human macrophages but not dendritic cells. Emerg. Microbes. Infect. 10, 1024–1037. doi: 10.1080/22221751.2021.1929503
Yang, Q., You, J., Zhou, Y., Wang, Y., Pei, R., Chen, X., et al. (2020). Tick-borne encephalitis virus NS4A ubiquitination antagonizes type I interferon-stimulated STAT1/2 signalling pathway. Emerg. Microbes Infect. 9, 714–726. doi: 10.1080/22221751.2020.1745094
Yu, G. (2025). enrichplot: visualization of functional enrichment result (R package version 1.26.6). Available online at: https://yulab-smu.top/biomedical-knowledge-mining-book/ (Accessed July 17, 2025).
Keywords: Zika virus, interferon, endothelial cells, blood brain barrier, neuroinvasion
Citation: Lima LR, Mustafá YM, Fonseca PLC, Coelho SVA, Parisi PL, Simeoni CL, Meuren LM, Bezerra BB, Mebus-Antunes NC, Matassoli F, Proença-Modena JL, Aguiar RS and de Arruda LB (2025) Distinct ZIKV strain signatures and type I IFN modulation reveal a protective role of brain endothelial interferon signaling in vitro and in vivo. Front. Cell. Infect. Microbiol. 15:1726007. doi: 10.3389/fcimb.2025.1726007
Received: 15 October 2025; Accepted: 17 November 2025; Revised: 13 November 2025;
Published: 03 December 2025.
Edited by:
Milan Surjit, Translational Health Science and Technology Institute (THSTI), IndiaReviewed by:
Ke Wang, Huazhong Agricultural University, ChinaZhang Yage, Huazhong Agricultural University, China
Copyright © 2025 Lima, Mustafá, Fonseca, Coelho, Parisi, Simeoni, Meuren, Bezerra, Mebus-Antunes, Matassoli, Proença-Modena, Aguiar and de Arruda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luciana Barros de Arruda, YXJydWRhQG1pY3JvLnVmcmouYnI=