 Indrani Ray
Indrani Ray Sushil K. Mahata
Sushil K. Mahata Rajat K. De
Rajat K. De- 1Machine Intelligence Unit, Indian Statistical Institute, Kolkata, India
- 2Metabolic Physiology & Ultrastructural Biology Laboratory, VA San Diego Healthcare System, La Jolla, CA, USA
- 3Metabolic Physiology & Ultrastructural Biology Laboratory, University of California San Diego, La Jolla, CA, USA
Obesity, characterized by chronic activation of inflammatory pathways, is a critical factor contributing to insulin resistance (IR) and type 2 diabetes (T2D). Free fatty acids (FFAs) are increased in obesity and are implicated as proximate causes of IR and induction of inflammatory signaling in adipose, liver, muscle, and pancreas. Cells of the innate immune system produce cytokines, and other factors that affect insulin signaling and result in the development of IR. In the lean state, adipose tissue is populated by adipose tissue macrophage of the anti-inflammatory M2 type (ATM2) and natural killer (NK) cells; this maintains the insulin-sensitive phenotype because ATM2 cells secrete IL10. In contrast, obesity induces lipolysis and release of pro-inflammatory FFAs and factors, such as chemokine (C–C motif) ligand 2 (CCL2) and tumor necrosis factor alpha (TNF-α), which recruit blood monocytes in adipose tissue, where they are converted to macrophages of the highly pro-inflammatory M1-type (ATM1). Activated ATM1 produce large amounts of pro-inflammatory mediators such as TNF-α, interleukin-1β, IL-6, leukotriene B4, nitric oxide (NO), and resistin that work in a paracrine fashion and cause IR in adipose tissue. In the liver, both pro-inflammatory Kupffer cells (M1-KCs) and recruited hepatic macrophages (Ly6Chigh) contribute to decreased hepatic insulin sensitivity. The present mini-review will update the bidirectional interaction between the immune system and obesity-induced changes in metabolism in adipose tissue and liver and the metabolic consequences thereof.
Introduction
Multicellular organisms rely on two highly conserved mechanisms for their survival: the ability to store energy to prevent starvation (metabolic pathways) and the ability to fight infection (immune pathways). When nutrients are in excess, adipose tissue stores lipids and the liver stores glycogen for use during starvation or to combat stressful situations. In addition, both adipose tissue and liver are populated with innate and adaptive immune cells. Thus, immune cells modulate whole-body metabolism [in metabolic syndromes such as type 2 diabetes (T2D) and obesity] via effects on adipocytes and hepatocytes, and reciprocally, host nutrition and commensal microbiota-derived metabolites modulate immunological homeostasis. This bidirectional interaction between the immune system and whole-body metabolism has created the field of immunometabolism, which has witnessed a renaissance in the past 15 years. The landmark discovery by Hotamisligil et al. in 1993 suggested that tumor necrosis factor (TNF) levels are elevated in the adipose tissue of obese and diabetic rodents and that its neutralization improves insulin-stimulated glucose uptake, which formed the cornerstone for immunometabolism (1). The second groundbreaking discovery in the field of immunometabolism came from Ferrante and Chen’s group, who reported simultaneously that adipose tissue of obese mice is infiltrated with macrophages that contribute to adipose tissue inflammation and IR (2, 3). Since these initial discoveries in immunometabolism, it has been shown that a large number of immune cells and pathways regulate metabolic homeostasis in obese animals (4–11).
Obesity, an epidemic of the twenty-first century, continues to rise throughout the world, even in the countries where poverty and malnutrition are major problems. The World Health Organization estimates that globally there are more than 1.9 billion overweight adults [body mass index (BMI) > 27 kg/m2]. Of them, 600 million people are obese with BMI more than 30 kg/m2 (WHO obesity and overweight fact sheet, updated in June 2016: http://www.who.int/mediacentre/factsheets/fs311/en/). Obesity provides bacterial and metabolic danger signals that activate a plethora of inflammatory cascades that drives M1 macrophage phenotype. In addition, immune and metabolic pathways are tightly balanced in that the immune response is highly energy demanding and shifts energy away from non-essential functions (12). In contrast, infection and sepsis often result in metabolic disruptions including IR (13). Obesity- and T2D-induced alterations in components of the immune system are most apparent in adipose tissue, the liver, and the pancreatic islets. Therefore, this review will focus on obesity-induced changes in immune system and metabolism in adipose tissue and liver and the consequent development of disease states such as IR, T2D, non-alcoholic fatty liver disease (NAFLD), and non-alcoholic steatohepatitis (NASH).
Obesity: Innate and Adaptive Immune Responses and Their Signaling
The mammalian immune system consists of two types of immune responses: innate and adaptive. Innate immune cells include neutrophils, dendritic cells, macrophages, mast cells, and eosinophils, which respond to general danger signals associated with invading pathogens. Neutrophils are the first responders to invading pathogens and are generally among the first immune cells to arrive at the site of inflammation. Macrophages are long lived and highly dynamic. They readily switch from anti-inflammatory M2 type to pro-inflammatory M1-type in resident tissues. Besides bacterial danger signals mediated by lipopolysaccharide (LPS), the toll-like receptor 4 (TLR4) ligand, obesity-associated metabolic danger signals also play an important role in macrophage polarization. To provide local immune responses, macrophages get assistance from other immune cells, such as TLR-proficient mast cells (14). Eosinophils are anti-inflammatory in nature and maintain the M2 macrophage population. Adaptive immune cells include B-2 and T lymphocytes, which exert specific and decisive adaptive immune functions and provide immunological memory (15). B-2 and T lymphocytes are also involved in sterile inflammation and autoimmune disorders (16, 17). TNF-α released by M1 macrophage initiates inflammatory signaling through its receptor TNFR1 with consequent regulation of gene expression. In the cytoplasm, NF-κB is sequestered by the inhibitor of κB (IκB) to prevent nuclear translocation. The activation of the IκB kinase leads to phosphorylation of IκB and release of NF-κB, which then translocate to the nucleus and bind to the promoters of pro-inflammatory genes and initiates transcription (9, 18) (Figure 1). Alternatively, the inflammatory signaling can be initiated by the microbial-derived LPS, which acts through the TLRs. TLRs can sense lipids and saturated fatty acids and are able to induce activation of TLR2 and TLR4 through myeloid differentiation primary response protein 88-dependent pathways, whereas unsaturated fatty acids block TLR-mediated signaling pathways and gene expression (Figure 1). Receptors of advanced glycation end product bind to lipids and nucleic acids resulting in oxidative stress, activate NF-κB, and promote transcription of pro-inflammatory factors (19, 20) (Figure 1). The inflammasome, an oligomeric protein complex, comprises scaffold, adaptor, and caspase proteins that mediate the maturation and secretion of inflammatory cytokines interleukin-1β (IL-1β) and IL-18 (21). The NLR family pyrin domain containing 3 inflammasome recruits and activates pro-caspase 1 to produce caspase-1, which then cleaves pro-IL-1β and pro-IL-18 to mature IL-1β and IL-18, respectively (22).
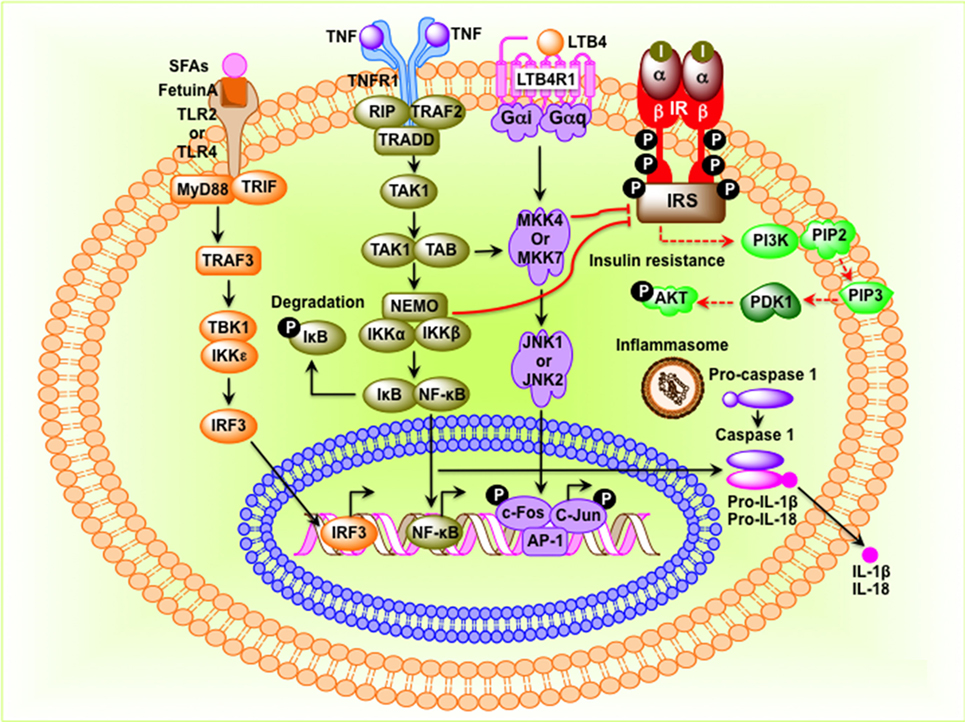
Figure 1. Molecular events that connect inflammation to insulin resistance in obesity. Saturated fatty acids (SFAs) bind to Fetuin-A, an endogenous ligand of toll-like receptor 4 (TLR4) and TLR2, and initiate transcription of interferon regulatory factor 3 (IRF3) in a myeloid differentiation primary response protein 88 (MyD88)–TIR-domain-containing adapter-inducing interferon-β-dependent pathway. Activated IRF3 then translocates to the nucleus and binds to target DNA sequences. Tumor necrosis factor (TNF) protein binds to its receptor and initiates inhibitor of κB (IκB)–NF-κB signaling pathway leading to translocation of NF-κB to the nucleus where it binds to AP-1 DNA sequences. Stimulation leukotriene B4 receptor 1 (LTB41) activates the c-Jun N-terminal kinase pathway, leading to phosphorylation and binding of the c-Jun–c-Fos heterodimer to target genes. NF-κB, c-Jun–c-Fos, and IRF3 induce expression of inflammatory factors such as cytokines, chemokines, and components of the inflammasome. When inflammasome is assembled, pro-caspase-1 is converted to caspase-1, which then converts pro-interleukin-1β (IL-1β) and pro-IL-18 to IL-1β and IL-18, respectively. I, insulin; insulin receptor; IRS, insulin receptor substrate.
Immune Cells and Their Polarization in Adipose Tissue
The adipose tissue comprises adipocytes, immune cells (macrophages and lymphocytes), pre-adipocytes, and endothelial cells. Under lean conditions, Th2 T cells, Treg cells, eosinophils, and ATM2-like resident macrophages predominate in the adipose tissue (Figure 2). ATM2 macrophages express CD11b, F4/80, CD301, and CD206 and promote local insulin sensitivity through production of anti-inflammatory cytokines, such as IL-10 (18). Treg cells not only secrete IL-10 but also stimulate ATM2 macrophage to secrete IL-10. Eosinophils, on the other hand, secrete IL-4 and IL-13. In the lean state, IL-4, IL-10, and IL-13 maintain the anti-inflammatory and insulin-sensitive phenotype. In contrast, obesity induces lipolysis and release of pro-inflammatory free fatty acids (FFAs) and factors such as C–C motif ligand 2 (CCL2) and TNF-α that recruit blood monocytes in adipose tissue, where they become polarized to the highly pro-inflammatory M1-like state (Figure 2). FFAs serve as ligands for the TLR4 complex (23), activate classical inflammatory response, and drive accumulation of ATM (24, 25). Activated ATM1 express CD11c in addition to CD11b and F4/80 and produce large amounts of pro-inflammatory mediators such as TNF-α, IL-1β, IL-6, leukotriene B4, NO, and resistin that work in a paracrine fashion and causes IR in adipose tissue (26). The anti-inflammatory eosinophil population declines in obese adipose tissue. In addition, obesity decreases Treg content and an increase in CD4+ Th1 and CD8+ effector T cells, which also secrete pro-inflammatory cytokines. Obesity increases B cell numbers and activates T cells, which potentiate M1-like macrophage polarization, inflammation, and IR. Cytokines and chemokines are also released from the adipose tissue and promote inflammation and consequent IR in liver, muscle, and pancreas.
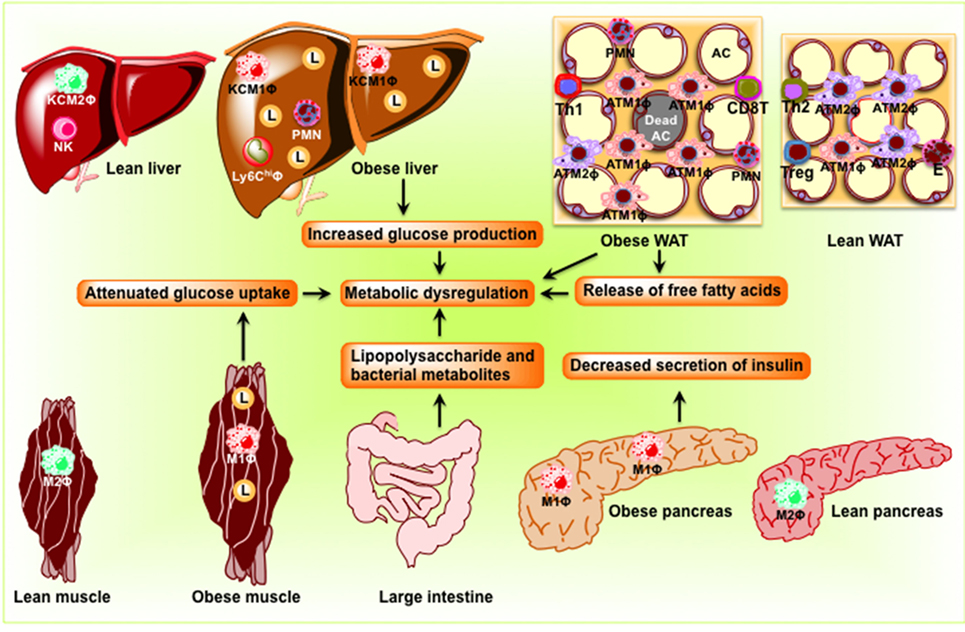
Figure 2. Schematic diagram showing obesity-induced inflammation in peripheral organs including adipose tissue, the liver, skeletal muscle, and the pancreas to cause dysbiosis in the intestine. In adipose tissue, pro-inflammatory signaling induces lipolysis and release of free fatty acids eventuating in the development of insulin resistance. In the liver, obesity induces pro-inflammatory cytokine production and M1 macrophage recruitment, resulting in insulin resistance and steatosis. In skeletal muscle of obese rodents, accumulations of lipid and pro-inflammatory macrophage inhibit insulin signaling, which result in the development of insulin resistance. In the pancreas, obesity induces macrophage infiltration, interleukin-1β secretion, and decreases insulin secretion. Because of the change in the composition of the microbial population, dysbiosis occurs in the intestine. AC, adipocyte; KC, Kupffer cell; L, lipid droplets; M1Φ, classically activated macrophages/pro-inflammatory macrophages; M2Φ, alternatively activated macrophages/anti-inflammatory macrophages; NK, natural killer cell; PMN, polymorphonuclear neutrophil; WAT, white adipose tissue.
Immune Cells and Their Polarization in Liver
In the lean liver, hepatocytes are the major parenchymal cells, while the non-parenchymal cells integrate five cell populations including resident macrophages of M2-type or Kupffer cells (27), recruited hepatic macrophages, resident innate lymphocytes or natural killer cells (NKs) (28, 29), fat storing cells termed Ito or stellate cells (HSCs) (30), and liver sinusoidal endothelial cells (LSECs) (31). Under lean conditions, Kupffer cells (KCs) in collaboration with other hepatic immune cell populations clear microbial material while maintaining the inflammatory tone of the liver at a level sufficient for essential functions such as pathogen killing, tissue remodeling, and sinusoidal permeability, but below that they would result in overt inflammation and tissue damage (32–34). NKs eliminate virus-infected or transformed cells and regulate adaptive immune responses via contact-dependent signals and the secretion of cytokines (35–38).
Hepatic lipid accumulation and peroxidation lead to chronic hepatocyte endoplasmic reticulum stress, the production of reactive oxygen species, and TLR activation, which converts KCs into an M1 phenotype defined by production of pro-inflammatory cytokines, oncostatin, and prostaglandins (PGE2) (39–41). Circulating cytokines, adipokines, and FFAs released from inflamed adipose tissue in the obese state or immunogenic material derived from an altered intestinal microbiota can also contribute to KC polarization. M1-KCs secrete chemokine CCL2 (also known as MCP1), pro-inflammatory cytokines (TNF-α, IL-1β, and IL-6), macrophage inflammatory protein (MIP)-1a, MIP1b, RANTES, oncostatin, and PGE2, which contribute to the alteration of the liver homeostasis and worsen the hepatic inflammatory response (42). PGE2 regulates cytokine production (IL-1β, IL-6, TNF-α, and TGF-β) (43, 44), acts synergistically with IL-6 to induce IR (45), and induces production of oncostatin M (OSM) in KCs (46). Increased OSM contributes to hepatic IR and the development of NASH (46). High levels of TNF-α released by M1-KCs stimulates hepatic expression of CCL2, a powerful monocyte chemoattractant, which recruits CCR2+Ly6Chigh monocytes from the vasculature into the liver (47), where they differentiate into Ly6Chigh macrophages. The Ly6Chigh macrophages amplify the severity of obesity-induced inflammation and hepatic IR through secretion of TNF-α and IL-6 (48).
Adipose Tissue Fibrosis and Metabolic Dysfunction
Adipocytes and their progenitor cells (pre-adipocytes) are embedded in a network of extracellular matrix (ECM), which tightly regulates the function of adipose tissue (49). Fibrosis, the excessive accumulation of ECM components, is a highly conserved and coordinated protective response to tissue injury and is a common pathological consequence of inflammatory diseases (50). Fibrosis develops from an imbalance between excess synthesis of ECM components including collagens (I, III, and VI), elastins, and proteoglycans (51, 52), and an impairment in degradation of these proteins. Fibrosis limits the expandability of adipose tissue and contributes to ectopic fat accumulation and the development of IR (53). It has been recently shown that treatment with the antidiabetic drug metformin inhibits excessive ECM deposition in white adipose tissue (WAT) of leptin-deficient ob/ob mice and mice with diet-induced obesity (54). Fibrotic disorders cause 45% deaths in the United States (52). In adipose tissues, ECM undergoes constant remodeling to allow adipocytes to rapidly expand and shrink in parallel with weight gain and loss and function in adaptation to nutritional clues (55). Adipocytes undergo dramatic expansion during the development of obesity. Macrophages are believed to be the master “regulators” of fibrosis as they produce soluble mediators including TGF-β1 and platelet-derived growth factor (PDGF), which directly activate fibroblasts and control ECM dynamics by regulating the balance of various matrix metalloproteinases (MMPs) and tissue inhibitors of MMP (TIMP) (56). Myofibroblasts, macrophages, and endothelial cells also produce MMP and TIMP for ECM regulation (57). While MMPs are responsible for the degradation of virtually all ECM proteins (58), TIMP inhibits MMPs and is responsible for degrading excess ECM (59). Macrophages also regulate fibrogenesis by releasing chemokines and attract fibroblasts and other inflammatory cells. Thus, IL-13 produced by Th2 CD4+ T cells (52, 60, 61) and TGF-β1 activate fibroblasts to differentiate into α-smooth muscle actin (α-SMA) expressing myofibroblasts to produce ECM (62–64).
Liver Fibrosis and Metabolic Dysfunction
Liver fibrosis results from the would-healing response of the liver to repeated injury such as hepatitis C virus (HCV) infection, alcohol abuse, and NASH (65, 66). Fibrosis is increasingly appreciated as a major contributor to metabolic dysregulation in obese humans and T2D patients (67). Advanced liver fibrosis leads to cirrhosis and death (68). Increased gut permeability and hepatic TLR4 signaling promotes fibrogenesis. Both KCs and recruited Ly6Chigh macrophages contribute to the development of hepatic fibrosis (69). HSCs are the main collagen-producing cells in liver (70, 71). KCs activate HSCs through increased production of profibrotic cytokine TGF-β and mitogenic PDGF (72) leading to fibrosis. TGF-β leads to transdifferentiation of HSCs into myofibroblasts. PDGF stimulates myofibroblast proliferation. Inhibition of PDGF by anti-sense strategy attenuates liver fibrogenesis (73). HSC-derived myofibroblasts express α-SMA and collagen I. During fibrogenesis, LY6Chigh monocytes are recruited to the inflamed liver via the CCL2/CCR2 (C–C chemokine receptor type 2) axis, forming a profibrotic Ly6Chigh macrophage, which has been shown to be the predominant pro-fibrogenic population in the liver (74, 75). These cells express TNF-α and IL-1β, which perpetuate hepatocellular injury and enhance the survival of hepatic myofibroblasts. In addition, Ly6Chigh macrophages express high levels of TGF-β-activating thrombospondin 1 (76). Macrophages also express the potent mitogen PDGF and the Th2 cell cytokines IL-4 and IL-13, which directly stimulate collagen synthesis in myofibroblasts. Chemokine expression such as CCL8 (also known as MCP2) and CCL7 (also known as MCP3) by these macrophages promotes the recruitment of monocytes, other inflammatory cells, and HSCs (77). Ly6Chigh macrophages also interact with HSCs to promote fibrosis through increased production of TGF-β, connective tissue growth factor (CTGF), and PDGF (78). Inhibition of the main monocyte chemoattractant CCL2 in rats or genetic deletion of its receptor CCR2 in mice decreased macrophage infiltration in response to injury and markedly inhibited liver fibrosis, implicating monocyte recruitment as an essential component in liver fibrogenesis (78–82). In addition, pharmacological inhibition of CCL2 by the RNA-aptamer mNOX-E36 attenuates liver fibrosis, thereby strengthening a profibrotic function of Ly6Chigh macrophages (83, 84). Hepatic myofibroblasts express TIMP1, which inhibits MMP activity and augments the accumulation of ECM in the scar tissue.
Obesity, Tissue Inflammation, and Insulin Resistance
Components of the immune system are affected in obesity and T2D and inflammation participates in the pathogenesis of T2D. Thus, obesity affects the immune system and promotes inflammation with consequent development of IR (85–87). Obesity-induced increased levels of glucose and FFAs create stress in pancreatic islets, adipose tissue, liver, and muscle, resulting in increased local production and release of cytokines and chemokines such as IL-1β, TNFα, CCL2, CCL3, and CXC-chemokine ligand 8 (CXCL8, also known as IL-8). These changes promote recruitment of immune cells in insulin-sensitive tissues and contribute to tissue inflammation and further production and release of cytokines and chemokines. The augmented release of cytokines and chemokines promotes inflammation in liver, muscle, and pancreatic islets. Obesity affects insulin signaling and causes IR by the following mechanisms: (i) inflammatory stimuli phosphorylate IκB resulting its dissociation from IκB/NF-κB complex followed by degradation in the cytoplasm. This allows translocation of free NF-κB to the nucleus, where it binds to cognate DNA response elements and transactivates the transcription of inflammatory genes. (ii) Phosphorylation and activation of c-Jun N-terminal kinase (JNK) leading to phosphorylation of the N-terminus of c-Jun. This initiates a switch of c-Jun dimers for c-Jun–c-Fos heterodimers with consequent stimulation of transcription of inflammatory target genes. (iii) Production of “second messengers,” such as FFAs, that promote IR. (iv) Augmented transcription of genes involved in lipid processing, including the enzymes that synthesize ceramide, which inhibits the activation of AKT (88, 89).
Recent studies in both rodents and humans implicate gut microbiota as a contributor to metabolic disorders (90). The gut microbiota plays a part in the host’s genomic profile and metabolic efficiency (91). Obesity in humans and rodents is associated with changes in the composition of the intestinal microbiota (92, 93). Dysbiotic microbiota in obesity enhances the digestion of complex carbohydrates and macronutrient absorption, leading to the development of obesity (94). In addition, gut microbiota has the capacity to harvest energy from nutrients and stores energy in the form of fat (95). The gut microbiota is also capable of inducing “metabolic endotoxemia” by increasing exposure to bacterial LPS coming from gut (96). LPS in the bloodstream contributes to IR by promoting tissue inflammation (97, 98).
Obesity and NAFLDs
Non-alcoholic fatty liver disease, the liver manifestation of the metabolic syndrome, has become the most common disorder in the United States and other developed countries, affecting over a third of the population (99). NAFLD begins with a simple steatosis that may evolve into NASH, a medley of inflammation, hepatocellular injury, and fibrosis, often resulting in cirrhosis and even hepatocellular cancer (100–102). KCs differ in their population density, morphological characteristics, and physiological functions depending on their position within the liver sinusoids (103, 104). Severity of human NAFLD is associated with higher population of KCs (105). However, NASH is associated with aggregates of enlarged KCs (106). Selective depletion of large KCs by administration of gadolinium chloride markedly attenuates liver injury induced by thioacetamide (107), carbon tetrachloride (108), alcohol (109), and ischemia/reperfusion (110), indicating the critical roles played by larger KCs in liver damage in these condition. In experimental NAFLD induced by methionine/choline deficient diet, liposome-encapsulated dichloromethylene bisphosphonate (clodronate) effective blunts all histological evidence of NASH (111). These findings indicate that the activation of KCs positioned at the “frontline” is an essential element in the pathogenesis of NAFLD similar to other types of liver injury.
Therapeutic Perspectives on Immunomodulation
Although it is yet to be definitely established whether tissue inflammation causes IR in humans, several anti-inflammatory approaches have been tested in clinical studies of obese individuals with IR. Thus, salsalate, an analog of salicylate, has been shown to improve insulin clearance and insulin sensitivity (112–115). Anti-TNF antibodies were found to decrease blood glucose in obese individuals (116). Anti-IL-1β monoclonal antibody therapy improved glycemic condition and β-cell insulin secretion (117–119). The antidiabetic thiazolidinediones (e.g., rosiglitazone and pioglitazone) decreased adipose tissue macrophage content (120, 121) and increase circulating levels of adiponectin and FGF21, thereby mediating redistribution of adipose tissue lipid stores (122, 123). Orexin-1 receptor antagonist has been shown to exert anti-obesity effects in obese leptin-deficient ob/ob mice (124, 125). While obese mice fed a high-fat diet supplemented with ω-3 fatty acids caused a decrease in inflammation, improved insulin sensitivity, and normalized glucose tolerance (126), fish-oil supplementation yielded mixed results on metabolic end points in human studies (127, 128).
Conclusion and Future Perspectives
Although the last 15 years has witnessed a renaissance in the field of immunology and metabolism, immunometabolism is still a young field with many questions to be answered. (i) To what extent are obesity and inflammation triggered in parallel or in sequence? (ii) What is the ontogeny and fate of stromal cells that populate WAT and liver? (iii) Do macrophage localization and origin regulate immunometabolic phenotype? (iv) By what pathway(s) does inflammation provoke T2D? (v) Can genetic and environmental factors reinforce or dissociate the link between metabolic and immunological abnormalities? (vi) Do anti-inflammatory strategies target the underlying mechanisms of the disease, and if so, would starting these therapies early prevent progression or even the overt manifestation of the disease? Answers to the above questions and a more detailed understanding of immunometabolism will permit more focused immune therapies to target metabolic diseases.
Author Contributions
IR and RD researched data and wrote the first draft of the article. SM researched data and extensively revised the draft, and made both the figures.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Sumana Mahata for editing the review article. Indrani Ray gratefully acknowledges CSIR, India, for providing her a Senior Research Fellowship (09/093(0156)/2014-EMR-I).
Funding
There is no financial support from funding organization. The research was supported by Mahata’s personal funding.
References
1. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science (1993) 259(5091):87–91. doi: 10.1126/science.7678183
2. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest (2003) 112(12):1796–808. doi:10.1172/JCI200319246
3. Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest (2003) 112(12):1821–30. doi:10.1172/JCI200319451
4. Hotamisligil GS. Inflammation and metabolic disorders. Nature (2006) 444(7121):860–7. doi:10.1038/nature05485
5. Mathis D. Immunological goings-on in visceral adipose tissue. Cell Metab (2013) 17(6):851–9. doi:10.1016/j.cmet.2013.05.008
6. Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med (2012) 18(3):363–74. doi:10.1038/nm.2627
7. Odegaard JI, Chawla A. Pleiotropic actions of insulin resistance and inflammation in metabolic homeostasis. Science (2013) 339(6116):172–7. doi:10.1126/science.1230721
8. McPhee JB, Schertzer JD. Immunometabolism of obesity and diabetes: microbiota link compartmentalized immunity in the gut to metabolic tissue inflammation. Clin Sci (Lond) (2015) 129(12):1083–96. doi:10.1042/CS20150431
9. Lackey DE, Olefsky JM. Regulation of metabolism by the innate immune system. Nat Rev Endocrinol (2016) 12(1):15–28. doi:10.1038/nrendo.2015.189
10. Newton R, Priyadharshini B, Turka LA. Immunometabolism of regulatory T cells. Nat Immunol (2016) 17(6):618–25. doi:10.1038/ni.3466
11. Cousin B, Casteilla L, Laharrague P, Luche E, Lorsignol A, Cuminetti V, et al. Immuno-metabolism and adipose tissue: the key role of hematopoietic stem cells. Biochimie (2016) 124:21–6. doi:10.1016/j.biochi.2015.06.012
12. Wolowczuk I, Verwaerde C, Viltart O, Delanoye A, Delacre M, Pot B, et al. Feeding our immune system: impact on metabolism. Clin Dev Immunol (2008) 2008:639803. doi:10.1155/2008/639803
13. Schaffler A, Scholmerich J. Innate immunity and adipose tissue biology. Trends Immunol (2010) 31(6):228–35. doi:10.1016/j.it.2010.03.001
14. Abraham SN, St John AL. Mast cell-orchestrated immunity to pathogens. Nat Rev Immunol (2010) 10(6):440–52. doi:10.1038/nri2782
15. Boehm T. Design principles of adaptive immune systems. Nat Rev Immunol (2011) 11(5):307–17. doi:10.1038/nri2944
16. Mbitikon-Kobo FM, Vocanson M, Michallet MC, Tomkowiak M, Cottalorda A, Angelov GS, et al. Characterization of a CD44/CD122int memory CD8 T cell subset generated under sterile inflammatory conditions. J Immunol (2009) 182(6):3846–54. doi:10.4049/jimmunol.0802438
17. Mills KH. TLR-dependent T cell activation in autoimmunity. Nat Rev Immunol (2011) 11(12):807–22. doi:10.1038/nri3095
18. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol (2010) 72:219–46. doi:10.1146/annurev-physiol-021909-135846
19. Hudson BI, Carter AM, Harja E, Kalea AZ, Arriero M, Yang H, et al. Identification, classification, and expression of RAGE gene splice variants. FASEB J (2008) 22(5):1572–80. doi:10.1096/fj.07-9909com
20. Curtiss LK, Tobias PS. Emerging role of toll-like receptors in atherosclerosis. J Lipid Res (2009) 50(Suppl):S340–5. doi:10.1194/jlr.R800056-JLR200
21. Westwell-Roper C, Nackiewicz D, Dan M, Ehses JA. Toll-like receptors and NLRP3 as central regulators of pancreatic islet inflammation in type 2 diabetes. Immunol Cell Biol (2014) 92(4):314–23. doi:10.1038/icb.2014.4
22. Haneklaus M, O’Neill LA. NLRP3 at the interface of metabolism and inflammation. Immunol Rev (2015) 265(1):53–62. doi:10.1111/imr.12285
23. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest (2006) 116(11):3015–25. doi:10.1172/JCI28898
24. Suganami T, Nishida J, Ogawa Y. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: role of free fatty acids and tumor necrosis factor alpha. Arterioscler Thromb Vasc Biol (2005) 25(10):2062–8. doi:10.1161/01.ATV.0000183883.72263.13
25. Nguyen MT, Satoh H, Favelyukis S, Babendure JL, Imamura T, Sbodio JI, et al. JNK and tumor necrosis factor-alpha mediate free fatty acid-induced insulin resistance in 3T3-L1 adipocytes. J Biol Chem (2005) 280(42):35361–71. doi:10.1074/jbc.M504611200
26. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest (2007) 117(1):175–84. doi:10.1172/JCI29881
27. Widmann JJ, Cotran RS, Fahimi HD. Mononuclear phagocytes (Kupffer cells) and endothelial cells. Identification of two functional cell types in rat liver sinusoids by endogenous peroxidase activity. J Cell Biol (1972) 52(1):159–70.
28. Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or adaptive immunity? The example of natural killer cells. Science (2011) 331(6013):44–9. doi:10.1126/science.1198687
29. Yokoyama WM, Sojka DK, Peng H, Tian Z. Tissue-resident natural killer cells. Cold Spring Harb Symp Quant Biol (2013) 78:149–56. doi:10.1101/sqb.2013.78.020354
30. Hautekeete ML, Geerts A. The hepatic stellate (Ito) cell: its role in human liver disease. Virchows Arch (1997) 430(3):195–207. doi:10.1007/BF01324802
31. DeLeve LD. Liver sinusoidal endothelial cells and liver regeneration. J Clin Invest (2013) 123(5):1861–6. doi:10.1172/JCI66025
32. McNelis JC, Olefsky JM. Macrophages, immunity, and metabolic disease. Immunity (2014) 41(1):36–48. doi:10.1016/j.immuni.2014.05.010
33. Robinson MW, Harmon C, O’Farrelly C. Liver immunology and its role in inflammation and homeostasis. Cell Mol Immunol (2016) 13(3):267–76. doi:10.1038/cmi.2016.3
34. Devisscher L, Verhelst X, Colle I, Van Vlierberghe H, Geerts A. The role of macrophages in obesity-driven chronic liver disease. J Leukoc Biol (2016) 99(5):693–8. doi:10.1189/jlb.5RU0116-016R
35. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol (2008) 9(5):503–10. doi:10.1038/ni1582
36. Peng H, Jiang X, Chen Y, Sojka DK, Wei H, Gao X, et al. Liver-resident NK cells confer adaptive immunity in skin-contact inflammation. J Clin Invest (2013) 123(4):1444–56. doi:10.1172/JCI66381
37. Fasbender F, Widera A, Hengstler JG, Watzl C. Natural killer cells and liver fibrosis. Front Immunol (2016) 7:19. doi:10.3389/fimmu.2016.00019
38. Peng H, Wisse E, Tian Z. Liver natural killer cells: subsets and roles in liver immunity. Cell Mol Immunol (2016) 13(3):328–36. doi:10.1038/cmi.2015.96
39. Dixon LJ, Barnes M, Tang H, Pritchard MT, Nagy LE. Kupffer cells in the liver. Compr Physiol (2013) 3(2):785–97. doi:10.1002/cphy.c120026
40. Jager J, Aparicio-Vergara M, Aouadi M. Liver innate immune cells and insulin resistance: the multiple facets of Kupffer cells. J Intern Med (2016) 280(2):209–20. doi:10.1111/joim.12483
41. Kolios G, Valatas V, Kouroumalis E. Role of Kupffer cells in the pathogenesis of liver disease. World J Gastroenterol (2006) 12(46):7413–20. doi:10.3748/wjg.v12.i46.7413
42. Dey A, Allen J, Hankey-Giblin PA. Ontogeny and polarization of macrophages in inflammation: blood monocytes versus tissue macrophages. Front Immunol (2014) 5:683. doi:10.3389/fimmu.2014.00683
43. Goss JA, Mangino MJ, Callery MP, Flye MW. Prostaglandin E2 downregulates Kupffer cell production of IL-1 and IL-6 during hepatic regeneration. Am J Physiol (1993) 264(4 Pt 1):G601–8.
44. Roland CR, Goss JA, Mangino MJ, Hafenrichter D, Flye MW. Autoregulation by eicosanoids of human Kupffer cell secretory products. A study of interleukin-1, interleukin-6, tumor necrosis factor-alpha, transforming growth factor-beta, and nitric oxide. Ann Surg (1994) 219(4):389–99.
45. Henkel J, Neuschafer-Rube F, Pathe-Neuschafer-Rube A, Puschel GP. Aggravation by prostaglandin E2 of interleukin-6-dependent insulin resistance in hepatocytes. Hepatology (2009) 50(3):781–90. doi:10.1002/hep.23064
46. Henkel J, Gartner D, Dorn C, Hellerbrand C, Schanze N, Elz SR, et al. Oncostatin M produced in Kupffer cells in response to PGE2: possible contributor to hepatic insulin resistance and steatosis. Lab Invest (2011) 91(7):1107–17. doi:10.1038/labinvest.2011.47
47. Klueh U, Czajkowski C, Ludzinska I, Qiao Y, Frailey J, Kreutzer DL. Impact of CCL2 and CCR2 chemokine/receptor deficiencies on macrophage recruitment and continuous glucose monitoring in vivo. Biosens Bioelectron (2016) 86:262–9. doi:10.1016/j.bios.2016.06.026
48. Morinaga H, Mayoral R, Heinrichsdorff J, Osborn O, Franck N, Hah N, et al. Characterization of distinct subpopulations of hepatic macrophages in HFD/obese mice. Diabetes (2015) 64(4):1120–30. doi:10.2337/db14-1238
49. Chun TH, Hotary KB, Sabeh F, Saltiel AR, Allen ED, Weiss SJ. A pericellular collagenase directs the 3-dimensional development of white adipose tissue. Cell (2006) 125(3):577–91. doi:10.1016/j.cell.2006.02.050
50. Ricard-Blum S. The collagen family. Cold Spring Harb Perspect Biol (2011) 3(1):a004978. doi:10.1101/cshperspect.a004978
51. Meneghin A, Hogaboam CM. Infectious disease, the innate immune response, and fibrosis. J Clin Invest (2007) 117(3):530–8. doi:10.1172/JCI30595
52. Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol (2004) 4(8):583–94. doi:10.1038/nri1412
53. Buechler C, Krautbauer S, Eisinger K. Adipose tissue fibrosis. World J Diabetes (2015) 6(4):548–53. doi:10.4239/wjd.v6.i4.548
54. Luo T, Nocon A, Fry J, Sherban A, Rui X, Jiang B, et al. AMPK activation by metformin suppresses abnormal extracellular matrix remodeling in adipose tissue and ameliorates insulin resistance in obesity. Diabetes (2016) 65(8):2295–310. doi:10.2337/db15-1122
55. Mariman EC, Wang P. Adipocyte extracellular matrix composition, dynamics and role in obesity. Cell Mol Life Sci (2010) 67(8):1277–92. doi:10.1007/s00018-010-0263-4
56. Song E, Ouyang N, Horbelt M, Antus B, Wang M, Exton MS. Influence of alternatively and classically activated macrophages on fibrogenic activities of human fibroblasts. Cell Immunol (2000) 204(1):19–28. doi:10.1006/cimm.2000.1687
57. Pessin JE, Kwon H. How does high-fat diet induce adipose tissue fibrosis? J Investig Med (2012) 60(8):1147–50. doi:10.2310/JIM.0b013e318271fdb9
58. Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol (2014) 15(12):786–801. doi:10.1038/nrm3904
59. Brew K, Dinakarpandian D, Nagase H. Tissue inhibitors of metalloproteinases: evolution, structure and function. Biochim Biophys Acta (2000) 1477(1–2):267–83. doi:10.1016/S0167-4838(99)00279-4
60. Chiaramonte MG, Donaldson DD, Cheever AW, Wynn TA. An IL-13 inhibitor blocks the development of hepatic fibrosis during a T-helper type 2-dominated inflammatory response. J Clin Invest (1999) 104(6):777–85. doi:10.1172/JCI7325
61. Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, et al. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest (1999) 103(6):779–88. doi:10.1172/JCI5909
62. Lee CG, Homer RJ, Zhu Z, Lanone S, Wang X, Koteliansky V, et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1). J Exp Med (2001) 194(6):809–21. doi:10.1084/jem.194.6.809
63. Fichtner-Feigl S, Strober W, Kawakami K, Puri RK, Kitani A. IL-13 signaling through the IL-13alpha2 receptor is involved in induction of TGF-beta1 production and fibrosis. Nat Med (2006) 12(1):99–106. doi:10.1038/nm1332
64. Aliprantis AO, Wang J, Fathman JW, Lemaire R, Dorfman DM, Lafyatis R, et al. Transcription factor T-bet regulates skin sclerosis through its function in innate immunity and via IL-13. Proc Natl Acad Sci U S A (2007) 104(8):2827–30. doi:10.1073/pnas.0700021104
65. Friedman SL. Liver fibrosis – from bench to bedside. J Hepatol (2003) 38(Suppl 1):S38–53. doi:10.1016/S0168-8278(02)00429-4
66. Brunt EM. Nonalcoholic steatohepatitis. Semin Liver Dis (2004) 24(1):3–20. doi:10.1055/s-2004-823098
67. Sun K, Tordjman J, Clement K, Scherer PE. Fibrosis and adipose tissue dysfunction. Cell Metab (2013) 18(4):470–7. doi:10.1016/j.cmet.2013.06.016
68. Bataller R, Brenner DA. Liver fibrosis. J Clin Invest (2005) 115(2):209–18. doi:10.1172/JCI24282
69. Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol (2014) 14(3):181–94. doi:10.1038/nri3623
70. Friedman SL, Roll FJ, Boyles J, Bissell DM. Hepatic lipocytes: the principal collagen-producing cells of normal rat liver. Proc Natl Acad Sci U S A (1985) 82(24):8681–5. doi:10.1073/pnas.82.24.8681
71. Gabele E, Brenner DA, Rippe RA. Liver fibrosis: signals leading to the amplification of the fibrogenic hepatic stellate cell. Front Biosci (2003) 8:d69–77. doi:10.2741/887
72. Pradere JP, Kluwe J, De Minicis S, Jiao JJ, Gwak GY, Dapito DH, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology (2013) 58(4):1461–73. doi:10.1002/hep.26429
73. Borkham-Kamphorst E, Stoll D, Gressner AM, Weiskirchen R. Antisense strategy against PDGF B-chain proves effective in preventing experimental liver fibrogenesis. Biochem Biophys Res Commun (2004) 321(2):413–23. doi:10.1016/j.bbrc.2004.06.153
74. Lin SL, Castano AP, Nowlin BT, Lupher ML Jr, Duffield JS. Bone marrow Ly6Chigh monocytes are selectively recruited to injured kidney and differentiate into functionally distinct populations. J Immunol (2009) 183(10):6733–43. doi:10.4049/jimmunol.0901473
75. Gibbons MA, MacKinnon AC, Ramachandran P, Dhaliwal K, Duffin R, Phythian-Adams AT, et al. Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am J Respir Crit Care Med (2011) 184(5):569–81. doi:10.1164/rccm.201010-1719OC
76. Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci U S A (2012) 109(46):E3186–95. doi:10.1073/pnas.1119964109
77. Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis (2010) 30(3):245–57. doi:10.1055/s-0030-1255354
78. Karlmark KR, Weiskirchen R, Zimmermann HW, Gassler N, Ginhoux F, Weber C, et al. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology (2009) 50(1):261–74. doi:10.1002/hep.22950
79. Imamura M, Ogawa T, Sasaguri Y, Chayama K, Ueno H. Suppression of macrophage infiltration inhibits activation of hepatic stellate cells and liver fibrogenesis in rats. Gastroenterology (2005) 128(1):138–46. doi:10.1053/j.gastro.2004.10.005
80. Mitchell C, Couton D, Couty JP, Anson M, Crain AM, Bizet V, et al. Dual role of CCR2 in the constitution and the resolution of liver fibrosis in mice. Am J Pathol (2009) 174(5):1766–75. doi:10.2353/ajpath.2009.080632
81. Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut (2012) 61(3):416–26. doi:10.1136/gutjnl-2011-300304
82. Ehling J, Bartneck M, Wei X, Gremse F, Fech V, Mockel D, et al. CCL2-dependent infiltrating macrophages promote angiogenesis in progressive liver fibrosis. Gut (2014) 63(12):1960–71. doi:10.1136/gutjnl-2013-306294
83. Baeck C, Wei X, Bartneck M, Fech V, Heymann F, Gassler N, et al. Pharmacological inhibition of the chemokine C-C motif chemokine ligand 2 (monocyte chemoattractant protein 1) accelerates liver fibrosis regression by suppressing Ly-6C(+) macrophage infiltration in mice. Hepatology (2014) 59(3):1060–72. doi:10.1002/hep.26783
84. Zimmermann HW, Tacke F. In search of the magic bullet: can liver inflammation and fibrosis be reversed with medications? Expert Rev Gastroenterol Hepatol (2015) 9(9):1139–41. doi:10.1586/17474124.2015.1063417
85. Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Invest (2000) 106(4):473–81. doi:10.1172/JCI10842
86. Wellen KE, Hotamisligil GS. Obesity-induced inflammatory changes in adipose tissue. J Clin Invest (2003) 112(12):1785–8. doi:10.1172/JCI20514
87. Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest (2005) 115(5):1111–9. doi:10.1172/JCI25102
88. Schubert KM, Scheid MP, Duronio V. Ceramide inhibits protein kinase B/Akt by promoting dephosphorylation of serine 473. J Biol Chem (2000) 275(18):13330–5. doi:10.1074/jbc.275.18.13330
89. Holland WL, Bikman BT, Wang LP, Yuguang G, Sargent KM, Bulchand S, et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J Clin Invest (2011) 121(5):1858–70. doi:10.1172/JCI43378
90. Khan MT, Nieuwdorp M, Backhed F. Microbial modulation of insulin sensitivity. Cell Metab (2014) 20(5):753–60. doi:10.1016/j.cmet.2014.07.006
91. Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science (2005) 307(5717):1915–20. doi:10.1126/science.1104816
92. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature (2006) 444(7122):1022–3. doi:10.1038/4441022a
93. Tremaroli V, Backhed F. Functional interactions between the gut microbiota and host metabolism. Nature (2012) 489(7415):242–9. doi:10.1038/nature11552
94. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature (2006) 444(7122):1027–31. doi:10.1038/nature05414
95. Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A (2004) 101(44):15718–23. doi:10.1073/pnas.0407076101
96. Caesar R, Reigstad CS, Backhed HK, Reinhardt C, Ketonen M, Lunden GO, et al. Gut-derived lipopolysaccharide augments adipose macrophage accumulation but is not essential for impaired glucose or insulin tolerance in mice. Gut (2012) 61(12):1701–7. doi:10.1136/gutjnl-2011-301689
97. Amar J, Chabo C, Waget A, Klopp P, Vachoux C, Bermudez-Humaran LG, et al. Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: molecular mechanisms and probiotic treatment. EMBO Mol Med (2011) 3(9):559–72. doi:10.1002/emmm.201100159
98. Teixeira TF, Souza NC, Chiarello PG, Franceschini SC, Bressan J, Ferreira CL, et al. Intestinal permeability parameters in obese patients are correlated with metabolic syndrome risk factors. Clin Nutr (2012) 31(5):735–40. doi:10.1016/j.clnu.2012.02.009
99. McCullough AJ. Pathophysiology of nonalcoholic steatohepatitis. J Clin Gastroenterol (2006) 40(Suppl 1):S17–29. doi:10.1097/01.mcg.0000168645.86658.22
100. Marrero JA, Fontana RJ, Su GL, Conjeevaram HS, Emick DM, Lok AS. NAFLD may be a common underlying liver disease in patients with hepatocellular carcinoma in the United States. Hepatology (2002) 36(6):1349–54. doi:10.1002/hep.1840360609
101. Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology (2010) 52(5):1836–46. doi:10.1002/hep.24001
102. Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell (2010) 140(2):197–208. doi:10.1016/j.cell.2009.12.052
103. Sleyster EC, Knook DL. Relation between localization and function of rat liver Kupffer cells. Lab Invest (1982) 47(5):484–90.
104. Laskin DL, Weinberger B, Laskin JD. Functional heterogeneity in liver and lung macrophages. J Leukoc Biol (2001) 70(2):163–70.
105. Park JW, Jeong G, Kim SJ, Kim MK, Park SM. Predictors reflecting the pathological severity of non-alcoholic fatty liver disease: comprehensive study of clinical and immunohistochemical findings in younger Asian patients. J Gastroenterol Hepatol (2007) 22(4):491–7. doi:10.1111/j.1440-1746.2006.04758.x
106. Lefkowitch JH, Haythe JH, Regent N. Kupffer cell aggregation and perivenular distribution in steatohepatitis. Mod Pathol (2002) 15(7):699–704. doi:10.1097/01.MP.0000019579.30842.96
107. Andres D, Sanchez-Reus I, Bautista M, Cascales M. Depletion of Kupffer cell function by gadolinium chloride attenuates thioacetamide-induced hepatotoxicity. Expression of metallothionein and HSP70. Biochem Pharmacol (2003) 66(6):917–26. doi:10.1016/S0006-2952(03)00443-X
108. Muriel P, Escobar Y. Kupffer cells are responsible for liver cirrhosis induced by carbon tetrachloride. J Appl Toxicol (2003) 23(2):103–8. doi:10.1002/jat.892
109. Zhong Z, Connor HD, Mason RP, Qu W, Gao W, Lemasters JJ, et al. Role of Kupffer cells in reperfusion injury in fat-loaded livers from ethanol-treated rats. J Pharmacol Exp Ther (1995) 275(3):1512–7.
110. Tsung A, Hoffman RA, Izuishi K, Critchlow ND, Nakao A, Chan MH, et al. Hepatic ischemia/reperfusion injury involves functional TLR4 signaling in nonparenchymal cells. J Immunol (2005) 175(11):7661–8. doi:10.4049/jimmunol.175.11.7661
111. Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol (2007) 47(4):571–9. doi:10.1016/j.jhep.2007.04.019
112. Goldfine AB, Silver R, Aldhahi W, Cai D, Tatro E, Lee J, et al. Use of salsalate to target inflammation in the treatment of insulin resistance and type 2 diabetes. Clin Transl Sci (2008) 1(1):36–43. doi:10.1111/j.1752-8062.2008.00026.x
113. Goldfine AB, Fonseca V, Jablonski KA, Pyle L, Staten MA, Shoelson SE, et al. The effects of salsalate on glycemic control in patients with type 2 diabetes: a randomized trial. Ann Intern Med (2010) 152(6):346–57. doi:10.7326/0003-4819-152-6-201003160-00004
114. Goldfine AB, Conlin PR, Halperin F, Koska J, Permana P, Schwenke D, et al. A randomised trial of salsalate for insulin resistance and cardiovascular risk factors in persons with abnormal glucose tolerance. Diabetologia (2013) 56(4):714–23. doi:10.1007/s00125-012-2819-3
115. Barzilay JI, Jablonski KA, Fonseca V, Shoelson SE, Goldfine AB, Strauch C, et al. The impact of salsalate treatment on serum levels of advanced glycation end products in type 2 diabetes. Diabetes Care (2014) 37(4):1083–91. doi:10.2337/dc13-1527
116. Stanley TL, Zanni MV, Johnsen S, Rasheed S, Makimura H, Lee H, et al. TNF-alpha antagonism with etanercept decreases glucose and increases the proportion of high molecular weight adiponectin in obese subjects with features of the metabolic syndrome. J Clin Endocrinol Metab (2011) 96(1):E146–50. doi:10.1210/jc.2010-1170
117. Hensen J, Howard CP, Walter V, Thuren T. Impact of interleukin-1beta antibody (canakinumab) on glycaemic indicators in patients with type 2 diabetes mellitus: results of secondary endpoints from a randomized, placebo-controlled trial. Diabetes Metab (2013) 39(6):524–31. doi:10.1016/j.diabet.2013.07.003
118. Cavelti-Weder C, Babians-Brunner A, Keller C, Stahel MA, Kurz-Levin M, Zayed H, et al. Effects of gevokizumab on glycemia and inflammatory markers in type 2 diabetes. Diabetes Care (2012) 35(8):1654–62. doi:10.2337/dc11-2219
119. Sloan-Lancaster J, Abu-Raddad E, Polzer J, Miller JW, Scherer JC, De Gaetano A, et al. Double-blind, randomized study evaluating the glycemic and anti-inflammatory effects of subcutaneous LY2189102, a neutralizing IL-1beta antibody, in patients with type 2 diabetes. Diabetes Care (2013) 36(8):2239–46. doi:10.2337/dc12-1835
120. Esterson YB, Zhang K, Koppaka S, Kehlenbrink S, Kishore P, Raghavan P, et al. Insulin sensitizing and anti-inflammatory effects of thiazolidinediones are heightened in obese patients. J Investig Med (2013) 61(8):1152–60. doi:10.2310/JIM.0000000000000017
121. Koppaka S, Kehlenbrink S, Carey M, Li W, Sanchez E, Lee DE, et al. Reduced adipose tissue macrophage content is associated with improved insulin sensitivity in thiazolidinedione-treated diabetic humans. Diabetes (2013) 62(6):1843–54. doi:10.2337/db12-0868
122. Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, et al. PPARgamma signaling and metabolism: the good, the bad and the future. Nat Med (2013) 19(5):557–66. doi:10.1038/nm.3159
123. Dutchak PA, Katafuchi T, Bookout AL, Choi JH, Yu RT, Mangelsdorf DJ, et al. Fibroblast growth factor-21 regulates PPARgamma activity and the antidiabetic actions of thiazolidinediones. Cell (2012) 148(3):556–67. doi:10.1016/j.cell.2011.11.062
124. Haynes AC, Chapman H, Taylor C, Moore GB, Cawthorne MA, Tadayyon M, et al. Anorectic, thermogenic and anti-obesity activity of a selective orexin-1 receptor antagonist in ob/ob mice. Regul Pept (2002) 104(1–3):153–9. doi:10.1016/S0167-0115(01)00358-5
125. Thompson MD, Xhaard H, Sakurai T, Rainero I, Kukkonen JP. OX1 and OX2 orexin/hypocretin receptor pharmacogenetics. Front Neurosci (2014) 8:57. doi:10.3389/fnins.2014.00057
126. Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, et al. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell (2010) 142(5):687–98. doi:10.1016/j.cell.2010.07.041
127. Spencer M, Finlin BS, Unal R, Zhu B, Morris AJ, Shipp LR, et al. Omega-3 fatty acids reduce adipose tissue macrophages in human subjects with insulin resistance. Diabetes (2013) 62(5):1709–17. doi:10.2337/db12-1042
128. Tousoulis D, Plastiras A, Siasos G, Oikonomou E, Verveniotis A, Kokkou E, et al. Omega-3 PUFAs improved endothelial function and arterial stiffness with a parallel antiinflammatory effect in adults with metabolic syndrome. Atherosclerosis (2014) 232(1):10–6. doi:10.1016/j.atherosclerosis.2013.10.014
Keywords: obesity, insulin resistance, macrophages, ER stress, reactive oxygen species, type 2 diabetes, non-alcoholic fatty liver diseases
Citation: Ray I, Mahata SK and De RK (2016) Obesity: An Immunometabolic Perspective. Front. Endocrinol. 7:157. doi: 10.3389/fendo.2016.00157
Received: 29 September 2016; Accepted: 29 November 2016;
Published: 12 December 2016
Edited by:
Jae B. Kim, Seoul National University, South KoreaReviewed by:
Miles Douglas Thompson, University of Toronto, CanadaJiyoung Park, Ulsan National Institute of Science and Technology, South Korea
Copyright: © 2016 Ray, Mahata and De. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sushil K. Mahata, c21haGF0YUB1Y3NkLmVkdQ==;
Rajat K. De, cmFqYXRAaXNpY2FsLmFjLmlu