 Saad Khan
Saad Khan Yi Tao Chan
Yi Tao Chan Xavier S. Revelo
Xavier S. Revelo Daniel A. Winer1,2,5,6,7*‡
Daniel A. Winer1,2,5,6,7*‡- 1Department of Immunology, University of Toronto, Toronto, ON, Canada
- 2Division of Cellular & Molecular Biology, Diabetes Research Group, Toronto General Hospital Research Institute (TGHRI), University Health Network, Toronto, ON, Canada
- 3Center for Immunology, University of Minnesota, Minneapolis, MN, United States
- 4Department of Integrative Biology and Physiology, University of Minnesota, Minneapolis, MN, United States
- 5Department of Pathology, University Health Network, Toronto, ON, Canada
- 6Buck Institute for Research on Aging, Novato, CA, United States
- 7Department of Laboratory Medicine and Pathobiology, University of Toronto, Toronto, ON, Canada
Obesity and aging represent major health burdens to the global adult population. Both conditions promote the development of associated metabolic diseases such as insulin resistance. The visceral adipose tissue (VAT) is a site that becomes dysfunctional during obesity and aging, and plays a significant role during their pathophysiology. The changes in obese and aging VAT are now recognized to be partly driven by a chronic local inflammatory state, characterized by immune cells that typically adopt an inflammatory phenotype during metabolic disease. Here, we summarize the current knowledge on the immune cell landscape of the VAT during lean, obese, and aged conditions, highlighting their similarities and differences. We also briefly discuss possible linked mechanisms that fuel obesity- and age-associated VAT dysfunction.
Introduction
Obesity and aging represent two of the largest global health issues of our time. Obesity currently affects over a third of the world's population. Alarmingly, 57.8% of the global adult population are estimated to be overweight or obese by 2030 (1–3). Obesity is part of a condition known as Metabolic Syndrome (MetS), which is defined as a cluster of metabolic risk factors such as abdominal obesity, dyslipidemia, hyperglycemia and insulin resistance (IR) (4). MetS is characterized by a state of low-grade inflammation that is implicated in the development of chronic diseases such as type 2 diabetes (T2D), non-alcoholic fatty liver disease (NAFLD) and cardiovascular disease (CVD) (5–7).
The biological processes of aging and senescence are accelerated during MetS and the risk of developing chronic diseases increases with age (8). In the years to come, aging is expected to represent an enormous challenge for social and health systems. Without adjusting for the rise in longevity, the median age of the world's population is expected to increase from 26.6 to 37.3 years between 2000 and 2050 and to 45.6 years by 2100 (9). This increase in aging will be likely associated with an elevated risk of developing chronic diseases that will aggravate the functional abilities and quality of life of the elderly (10). The changes in the immune system associated with aging are characterized by an imbalance between inflammatory and anti-inflammatory pathways, leading to low-grade inflammation and a greater susceptibility to chronic disease (11).
Obesity and aging contribute to the development of chronic low-grade inflammation in multiple tissues. This low-grade inflammation is recognized as an important factor that promotes downstream consequences of obesity and aging, including the control of whole-body metabolism. During obesity and aging, cells of the innate and adaptive systems accumulate inside the VAT where they alter the local inflammatory environment and impact insulin sensitivity (12). Indeed, the transition from lean to obese or aged states is accompanied by distinct and associated immune cell-driven inflammatory processes. This review will focus on the immune changes that occur in adipose tissue and the means by which immune cells shape the progression of VAT-specific obesity-associated inflammation (OAI) and age-associated inflammation (AAI).
Vat Plasticity and Remodeling
The adipose tissue is considered an endocrine organ that becomes remodeled during metabolic diseases and is involved in the progression of obesity- and aging-associated IR. There are two distinct types of adipose tissue: white adipose tissue (WAT) and brown adipose tissue (BAT). WAT acts an energy store by accumulating free fatty acids whereas BAT has the capacity of undergoing thermogenesis to dissipate energy (13, 14). WAT can be further divided into two major depots: subcutaneous and visceral. Subcutaneous WAT forms a layer under the skin in the hypodermis while visceral WAT surrounds the inner organs in the abdominal cavity and mediastinum (14, 15). VAT is deposited in certain locations such as the mesenteric fat between the intestines, and the retroperitoneal fat surrounding the kidneys; each VAT store consists of adipocytes and the stromal vascular fraction (SVF) (14). The mesenteric fat pads in mice are the most analogous to human VAT but are not well-studied due to limitations in surgical access (16). Perigonadal fat pads in mice are the most accessible and are used in the majority of mouse VAT studies; however, humans do not have such identical fat depots as mice and as such, these differences should be considered when comparing humans and mice (16). While adipocytes are tightly packed unilocular cells that are supported by a dense network of capillaries, the SVF consists of extra-cellular matrix (ECM) that holds together various cells such as pre-adipocytes, stem cells, fibroblasts, vascular endothelial cells and immune cells (13, 17–19).
During obesity, adipose tissue expansion is characterized by adipocyte hyperplasia (increase in adipocyte numbers) and hypertrophy (increase in adipocyte size), which results in increased adipocyte hypoxia, dysregulation of fatty acid fluxes, increased chemokine secretion, adipocyte cell death, and the recruitment of pro-inflammatory cells (20–23). In turn, pro-inflammatory cytokine release mediated by immune cells induces serine phosphorylation of insulin receptor substrate-1 (IRS-1) leading to local and systemic insulin resistance, which is thought to contribute toward whole body glucose and fatty acid metabolic dysregulation (24, 25) Furthermore, the obese adipose tissue can exhibit a pro-fibrotic phenotype characterized by an increased expression of ECM proteins such as collagen, which can impact adiposity, glucose homeostasis, and susceptibility to metabolic disease (26–29). On the other hand, aging results in an increase in body fat percentage, expansion of the VAT due to a shift in lipid storage from the SAT to the VAT, increased accumulation of senescent cells, and an altered pre-adipocyte cell phenotype (30). During aging, the accumulation of senescent preadipocytes in the adipose tissue leads to increased production of pro-inflammatory cytokines in a process dependent on the JAK pathway (31). It has been hypothesized that an elevated presence of senescent cells and reprogrammed pre-adipocytes in the aged VAT results in the generation of chemokines, pro-inflammatory cytokines, and ECM modifiers which contribute to VAT inflammaging (30).
The innate and adaptive immune systems are now widely accepted as forces that respond to and participate in the remodeling processes taking place in the VAT during metabolic diseases. While inflammatory changes in fat are likely required for proper adipose tissue remodeling and expansion, it is the chronic nature of the inflammation which ultimately drives metabolic disease during obesity and aging (32). In this review, we will discuss the role of immune cells in these two conditions with a focus on their function in the VAT.
Innate Immune Cells
Macrophages
Homeostasis
Macrophages are a fundamental component of the innate immune system, with the ability to phagocytose harmful pathogens and apoptotic/necrotic cells. Historically, macrophages have been broadly categorized into two categories: the pro-inflammatory “classically activated” M1-like and the anti-inflammatory “alternatively activated” M2-like macrophages. However, macrophages are now thought to exist on a spectrum of functionalities based on their resident and recruited status (33). Macrophages were the first immune cell to be characterized inside VAT during obesity and, until recently, have been the primary focus of most studies. Under homeostatic conditions, the VAT is home to a group of macrophages known as Adipose Tissue Macrophages (ATMs), which represent roughly 5–10% of the stromal vascular fraction, display an M2-like phenotype, depend on the expression of peroxisome proliferator activated receptor γ (PPARγ), and secrete anti-inflammatory IL-10 (34–36). Fate mapping techniques have revealed that not all tissue resident macrophages terminally differentiate from monocyte precursors or emerge from adult hematopoiesis; indeed, some ATMs may develop from bone marrow (BM)-independent progenitors in the embryonic yolk sac and possess the ability to self-renew (37, 38).
OAI
During obesity, the population of ATMs within the VAT increases up to 40–50% of the stromal vascular fraction, become metabolically activated, secrete pro-inflammatory cytokines, and engage in “inflammatory cross-talk” with other immune cells, notably CD4+ T cells (34, 35, 39–43). Obesity-associated ATMs accumulate primarily at crown like structures (CLS), characterized by ATMs surrounding dead or dying adipocytes (44). This accumulation of ATMs is the result of increased infiltration due to chemo-attractive gradients and higher proliferation, both partly driven by the monocyte chemoattractant protein 1 (MCP1) (45–47). It is possible that these mechanisms of ATM accumulation are reflective of the presence of distinct self-renewing yolk sac-derived and BM monocyte-derived ATMs. Efforts are in progress to reconcile these observations. Recent work has implicated secreted molecules in skewing ATM population phenotypes. In addition to pro-inflammatory cytokines such as TNFα and IFNγ, the transmembrane activator and calcium modulator and cyclophiliin ligand interactor (TACI) has been implicated in the skewing of macrophages toward an M1-like phenotype. Indeed, ATMs from TACI−/− mice were biased toward an M2-like phenotype and their adoptive transfer into obese mice rescue their dysregulated metabolic parameters (48, 49). Hormones and/or growth factors, such as insulin growth factor 1 (IGF1), can also impact the balance between M1- and M2-like phenotypes in ATMs (50).
However, as mentioned earlier, segregating macrophages into M1-like and M2-like subsets is not reflective of macrophage heterogeneity and efforts are being made to avoid this categorization (51). The markers and phenotypes of VAT monocytes, macrophages and DCs in the context of obesity have been described (52, 53). Further, macrophage classification is moving toward a genomics approach to identification in order to delineate the many heterogeneous cell subsets (54). Conditions during obesity may produce a “metabolically-activated' phenotype in macrophages that is mechanistically distinct from classical activation, which may explain the complexity in macrophage phenotypes seen in mice and humans (55). In light of this consensus, it was recently reported that the obese VAT contains a heterogenous group of macrophages, with BM-derived CD9+ and Ly6C+ ATMs representing the predominant populations (56). CD9+ ATMs exhibit pro-inflammatory gene signatures, localize in the CLS, are lipid-laden, secrete pathogenic exosomes, and induce a pro-inflammatory response in lean adipose tissue upon adoptive transfer (56). In contrast, Ly6C+ ATMs reside outside the CLS, express factors that support vascular development and organization, contain less lipids, and activate gene programs typical of normal adipocyte physiology upon adoptive transfer into lean mice (56). An expansion of the monocyte-derived CD9+ ATM population was recently confirmed via unbiased single-cell RNA sequencing. This population also displays a transcriptional signature associated with lipid metabolism and phagocytosis, consequently being termed lipid-associated macrophages (LAMs) (54). LAMs express Trem2, depletion of which resulted in their loss, decreasing the formation of CLSs observed during diet-induced obesity (54). However, in the absence of Trem2, mice fed a HFD displayed worsened metabolic parameters, implicating a role of these CD9+ Trem2+ LAMs in “buffering” against CLS-associated lipid plaques (54). While an increase in CLS surrounding lipid metabolizing CD9+ ATMs was reported in both of these studies, there is a need to reach a consensus with regards to their inflammatory profiles, their role in VAT inflammation, and whether lipid metabolism can directly regulate the inflammatory output of LAMs. Recent reports also propose the accumulation of sympathetic neuron/nerve-associated macrophages (NAMs) during diet-induced obesity which mediate the clearance of norepinephrine, a catecholamine that has been implicated in lipolysis and fat mass reduction (57).
AAI
In aged mice, the proportion of M2-like ATMs reportedly decreases, M1-like macrophages remains unchanged, and CD11c CD206 double negative (DN) ATMs trended to increase, suggesting that aging also skews ATM phenotypes toward a pro-inflammatory phenotype (58). These changes contrast to obesity where a large increase in numbers of VAT macrophages are seen, though aged macrophages still show an overall inflammatory shift. Similar to obesity, aged ATMs display an elevated expression of the chemokine receptor CCR2, possessed enhance secretion of pro-inflammatory cytokines IL-6, MCP-1, and TNFα, and a decrease in expression of PPARγ, which mechanistically accounts for the loss in M2 ATMs. (58). Recent studies propose that although there is a decrease in the proportions of ATMs in aged mice, aged ATMs lack M1- or M2-like polarization and showed a diversity of activation states (59). Furthermore, NAMs are also detected within the aged VAT, which were activated in an NLRP3-dependent manner to regulate lipolysis and fatty acid release through altering catecholamine bioavailability, thereby affecting VAT lipolysis (59). NLRP3 activity was further linked to the production of cytokines IL-1β and IL-18, which contributes to B cell accumulation in the VAT, as discussed below (60). Notably, NLRP3-deficient mice had restored proportions of ATMs which was postulated to reflect an exhausted senescent-like profile driven by chronic activation of the NLRP3 inflammasome (59). It remains to be determined whether levels of CD9+ LAMs and Ly6C+ATMs are altered in the aged VAT; future research in aging should focus on the heterogeneity of the ATM population within the adipose tissue, similar to what has been done for studies in obesity.
Innate Lymphoid Cells
Homeostasis
Innate lymphoid cells (ILCs) play a crucial role in providing defense against a wide array of pathogens such as parasites, microbes, and viruses as well as demonstrating cytotoxic activity toward tumors (61). ILCs are widely categorized into three groups based on their functional differences: group 1 ILCs (ILC-1), group 2 ILCs (ILC-2), and group 3 ILCs (ILC-3). In mice, ILC2 and ILC3 are the most abundant ILC subsets, and often residing in WAT and the intestine, respectively. In humans, however, ILC1 and ILC3 subsets are the predominant ILC subsets which are found in higher frequencies in mucosal and lymphoid sites such as the colon and ileum (62).
ILC-1s are defined by their ability to produce the pro-inflammatory cytokine IFNγ and have been widely studied in natural killer (NK) cells and mixed ILC-1 cells, which develop from different progenitors and possess unique tissue distribution (63). Under homeostatic conditions, ILC-1s are present in lean VAT, where they are resident cells and rely less on infiltration from the periphery, unlike splenic ILC-1s that constantly recirculate (64, 65). Interestingly, compared to their peripheral counterparts, murine NK1.1+ NKp46+ VAT ILC-1s express low levels of Ly49 and consist primarily of three major populations: immature NK (iNK cells), mature NK (mNK) cells, and mixed ILC-1s (64, 65). Parabiosis studies have revealed that while mNK cells recirculate, mixed ILC-1s and iNK cells are mostly resident within the VAT (64, 65). Under homeostatic conditions, VAT ILC-1s were observed to surround ATMs and regulate their numbers by killing both M1-like and M2-like ATMs (65).
ILC-2s are known for producing T helper 2 (Th2) cytokines such as IL-5 and IL-13, and a large body of work conducted on ILC-2s has focused on their role in promoting “beiging” within the adipose tissue (61). Beiging is a process whereby fat-accumulating white adipose tissue is converted into energy-dissipating brown-like adipose tissue (66, 67). Under homeostatic conditions, the presence of ILC-2s in the VAT is maintained by IL-33. ILC-2s have been implicated in promoting the beiging of adipocytes and the accumulation of eosinophils and M2-like macrophages in an IL-4/IL-13-dependent manner (68–70). Additionally, ILC-2s promote beiging of white adipose tissue through the production and release of methionine-enkephalin peptides that upregulate UCP1 expression in adipocytes (71).
ILC-3s contain various populations of RORγt-expressing ILCs that can produce T helper 17 (Th17) cytokines such as IL-17 and IL-22 (61). It remains to be determined whether ILC-3s are found within the adipose tissue, whether they are tissue resident or recruited from the periphery, and finally, what their role is in the context of diet-induced obesity, aging, and chronic inflammation in the VAT.
OAI
In mice, during the early stage of diet-induced obesity, there is a transient increase in VAT ILC-1s, although their proportion decreases within the VAT during chronic obesity (64, 65). ILC-1s from chronically obese VAT show a reduced ability to regulate ATMs which may contribute to their uncontrolled expansion (65). Additionally, IFNγ secretion by VAT ILC-1s is also elevated during short-term HFD feeding in an IL-12/STAT-4-dependent manner, which further contributes to the polarization of macrophages to an M1-like phenotype (65). Furthermore, VAT ILC-1s, and potentially their IFNγ, have been shown to promote adipose fibrogenesis by activating pro-inflammatory CD11c+ macrophages and the TGF-β1 pathway in adipocytes. These changes may serve to impair adipose function and glycemic tolerance in both mice and humans (72).
The early stage expansion of ILC-1s is primarily a consequence of the recruitment of mNK cells from the periphery followed by a smaller contribution of local proliferation (64, 65). A pathogenic role for the accumulation of mNK cells is likely, as their depletion improved metabolic parameters and decreased macrophage infiltration in obese mice (73–75). During obesity, adipocytes increase their expression of NK cell activating receptor (NCR1) which may be responsible for the expansion of local VAT NK cells and their IFNγ production (74, 76, 77). While increased IFNγ secretion by NK cells drives metabolic dysfunction during early-stage obesity, TNFα secretion by NK cells may play a larger role in driving inflammation and metabolic dysfunction during chronic obesity (78). During chronic obesity, NK cells have been shown to have an impaired capacity to degranulate or produce pro-inflammatory cytokines, which is linked to their uptake of lipids and subsequent metabolic reprogramming (79).
Chronic obesity also promotes the expansion of a distinct IL-6 receptor (IL-6R)- and colony-stimulating factor 1 receptor (csf1r)-expressing myeloid-signature NK cell (myNK) subpopulation in the perigonadal adipose tissue (PGAT) and blood circulation (80). Specific depletion of myNK cells led to reduced inflammation, obesity, and systemic insulin resistance, which could also be recapitulated through abrogation of IL-6 and STAT-3 signaling that is required for the formation of these unique cells (80). Future research should focus on determining the factors responsible for the change in ILC-1 functionality in the transition between early and late chronic obesity. Identifying the factors involved in NK cell recruitment early in obesity is needed as inhibiting NK cell accumulation within the VAT may serve as a therapeutic avenue to prevent elevated IFNγ levels within the adipose tissue; as well, understanding mechanisms to spare NK cells from becoming metabolically dysfunctional due to the lipid-laden adipose environment during chronic obesity.
ILC-2s have been shown to decrease in frequency and numbers in the obese WAT of humans and mice, which may impair the thermogenic ability of fat (68). As such, ILC-2s, along with eosinophils, discussed below, represent potential targets that can be manipulated to promote beiging and thermogenesis of white adipose tissue.
AAI
The role of VAT ILCs in aging is largely unknown. As myNK cells are among the cell types that potently respond to IL-6, a cytokine that is elevated in the VAT and circulation during aging, it is possible that aging affects the number and function of these cells (81). However, a large study in humans recently cataloged the different ILC subsets in humans with obesity and age (62). While age showed only marginal mixed up and down correlations of ILC-1/2/3s in abdominal and mesenteric fat, there was a significant correlative decrease in the presence of intestinal ILC-3s with age, highlighting tissue specific changes of these cells during aging (62). More work is needed to tease out how aging impacts VAT ILC accumulation and their inflammatory potential in mice and humans.
Neutrophils
Homeostasis
Neutrophils are responsible for providing protection against pathogens through the release of secretory granules containing a diverse array of antimicrobial proteins and pro-inflammatory mediators (82). Neutrophils can release reactive oxygen species (ROS) and cytokines to kill extracellular bacteria and recruit additional leukocytes to the region of inflammation (82, 83). Neutrophils can also kill extracellular bacteria through the generation of a web of extracellular fibers known as neutrophil extracellular traps (NETs), which are composed of DNA, histones, and antimicrobial proteins (82–84). Much of the work that has been conducted on VAT neutrophils has focused on their role in obesity. Their frequency inside VAT is very low (<1% of non-adipocyte cells) in the lean state and their role in homeostatic functions in fat is unclear (85).
OAI
Neutrophils have been shown to enter the adipose tissue during early high fat diet (HFD) feeding in mice. The frequency of VAT neutrophils increase from <1% of non-adipocyte cells in lean mice to ~2% within 1 week after initiating HFD feeding (85). Their crosstalk with adipocytes is sustained through neutrophil CD11b and adipocyte ICAM-1 interactions (85, 86). VAT neutrophils show increased production of the serine protease, elastase, which promotes inflammation in a toll like receptor (TLR)-dependent manner, and deletion of elastase in vivo in mice results in improved metabolic parameters (85). It has also been shown that elastase can decrease the amounts of proteins involved in the insulin signaling pathway such as IRS-1 (87, 88). In humans, clinical evidence points toward increased numbers and activation of neutrophils in obese patients. Neutrophils from obese patients possessed enhanced chemotactic activity and produced elevated amounts of superoxide molecules (87, 89, 90).
HFD-fed mice showed increased release and decreased clearance of NETs and increased autoantibodies against nuclear antigens (86). The excess nucleic acids and related protein antigens worsened metabolic parameters through the activation of VAT macrophages and plasmacytoid dendritic cells in the liver through a TLR-dependent manner while treatment of HFD-fed mice with inhibitors against TLR7/9 or NET formation improved metabolic parameters (86). Future work should aim to understand mechanisms and subsequently design therapies that can be used to reduce the accumulation of these cells within the adipose tissue or inhibit their ability to secrete NETs or elastase during obesity and metabolic disease.
AAI
There are no data regarding the role for neutrophils in the adipose tissue during the aging process, though few studies have explored the effect of aging in neutrophils. Neutrophils show age-related impairments in phagocytosis, degranulation, ROS generation, migration, and neutrophil microbicidal activity, which can contribute to the poor resolution of infections in the elderly (91–97). Future research should aim to address what factors contribute to the dysregulation of neutrophils in aged individuals, and whether these changes manifest inside fat.
Dendritic Cells
Homeostasis
Dendritic cells (DCs) are considered the bridge between the innate and adaptive immune system due to their antigen presentation role to prime T cells (98). There are two main subsets of DCs that have been well-studied: antigen presenting classical or conventional DCs (cDCs) and plasmacytoid DCs (pDCs) (98). pDCs are significantly less efficient at presenting antigen and stimulating T cells as compared to cDCs but can secrete copious amounts of type 1 interferon (IFN-1) when activated (98). Recently, it was suggested that pDCs emerge from lymphoid progenitors that are distinct from the myeloid lineage and hence share a different ontogeny from cDCs (99).
Two main populations of cDCs are found under homeostatic conditions in murine VAT, namely CD103+ cDC-1s and CD11b+ cDC-2s, both of which promote a tolerogenic, anti-inflammatory environment in the VAT (100). cDC-1s primarily activate the Wnt/β-catenin pathway whereas VAT cDC-2s upregulate the PPARγ pathway. Depletion of β-catenin and PPARγ in VAT cDCs stimulates a pro-inflammatory response in a mouse model of obesity, suggesting a role of these pathways in cDCs in delaying the onset of metabolic disease (100).
OAI
Chronic obesity and expansion of the VAT interfere with β-catenin and PPARγ pathways and abrogate the anti-inflammatory function of cDCs, furthering meta-inflammation (100). Earlier studies in humans and mice demonstrated that obesity is associated with an expansion of VAT DCs, mainly cDCs that accumulate in the VAT in a CCR7-dependent and CCR2-independent manner (101, 102). Another study showed that VAT cDCs have the ability to promote pro-inflammatory Th17 responses (53). pDCs have also been implicated in the pathogenesis of VAT meta-inflammation as they are recruited to the tissue due to elevated levels of the adipokine chemerin, and subsequently activated to promote IFN-1 signals in VAT, resulting in the polarization of ATMs to an M1-like state (103). Furthermore, depletion of IFN signaling by genetic deletion of IFNAR or genetic ablation of pDCs resulted in improved metabolic parameters in HFD-fed mice, strongly indicating the role for this subset in contributing to meta-inflammation (104, 105).
AAI
Current research on peripheral DCs suggests that aging alters DC function in humans, including defective phagocytosis of antigen, migratory capacity, and enhanced secretion of pro-inflammatory cytokines upon stimulation with TLR agonists (106). While this change in function may contribute to DC mediated inflammatory change inside VAT with age, the roles of cDCs and pDCs in aged VAT remains to be determined.
Eosinophils
Homeostasis
Eosinophils are major producers of IL-4 and IL-13 and play a significant role in host defense, notably against helminth infections (107). Under homeostatic conditions, eosinophils are abundant in the adipose tissue and participate in the beiging process through their production of IL-4 and IL-13 and subsequent activation and accumulation of M2-like ATMs. The activation of M2-like ATMs may be one factor that regulates the expression of tyrosine hydroxylase and production of catecholamines inside VAT, enhancing thermogenesis. However, the details surrounding the cells involved, including NAMs, and the mechanisms of action, such as altering catecholamine bioavailability, require further study (59, 108, 109). Furthermore, IL-5 within the VAT is believed to be responsible for recruiting eosinophils via IL-5 producing cells, which are predominantly ILC-2s (70, 108).
OAI
As a consequence of obesity, there is a decline in eosinophils in the VAT; eosinophil-deficient mice are more susceptible to weight gain and show impaired glucose tolerance, greater insulin resistance, and fewer anti-inflammatory M2-like ATMs (108). Treating obese mice with recombinant IL-5 for 8 weeks successfully restored eosinophil numbers within the VAT but did not reduce weight gain, glucose intolerance, insulin resistance, or alter energy expenditure and beiging capacity, implying that rescuing this immune population is not sufficient in the context of VAT dysfunction; thus, the mechanism of eosinophils in regulating VAT health and function therefore appears to be more complex than initially understood and must be further studied (110).
AAI
Knowledge on the role of VAT eosinophils during aging is very limited and requires additional work. In mice, aging has been associated with only a modest change in VAT eosinophils, in contrast with obesity where eosinophils decrease in numbers (111). It has also been indicated that aged human eosinophils possess altered degranulation abilities though with regular adhesive and chemotactic properties (112). Aging also has been recently described to prevent formation of cold-induced beige adipocytes in mice and humans and it remains to be determined whether eosinophils are implicated in this defect, along with other beiging-inducing immune cells (112, 113).
Mast Cells
Homeostasis
Mast cells play important roles during allergy and inflammation, and are components of the myeloid cell population in mouse VAT under homeostatic conditions. Mast cells have been shown to facilitate the preadipocyte to adipocyte transition in VAT (114, 115).
OAI
The VAT mast cell population increases during obesity, although they initially decrease slightly in the intermediate stages of HFD feeding in WT mice (12 weeks) but rebound in the later stages (114, 116). In humans, mast cell proportions positively correlated with the typical features of expanded obese VAT such as inflammation of endothelium, ATM build-up, and formation of fibrous tissue (117).
Mast cells can contribute to VAT dysfunction through degranulation and release of proteases such as tryptase, chymase, cathepsins, and matrix metalloprotease-9. These enzymes regulate adiponectin action and catabolize ECM collagens and fibronectin to promote adipogenesis and infiltration of pro-inflammatory cells into the tissue (117–120). Furthermore, mast cell-derived IL-6 and IFNγ contribute to adipose tissue cysteine cathepsin expression, which promotes VAT angiogenesis and growth in obese mice (118, 121). Mast cell-derived cathepsins can also degrade adipocyte insulin receptor and the glucose transporter Glut-4, leading to impaired glucose physiology in adipocytes (122).
Earlier reports indicated that mice deficient in mast cells fed a HFD displayed improved metabolic parameters and reduced pro-inflammatory cytokines in the VAT (118). It was further shown that mast cells regulate metabolism through IL-6 and IFNγ (118). Additionally, KitW−sh/W−sh mice, which are deficient in mast cells, reconstituted with mast cells from Il6 or Ifng knockout mice were protected from metabolic dysregulation (118). However, other models of mast cell deficiency reported no differences in weight gain, glucose physiology, or VAT inflammation (123, 124). The differences observed between studies are likely due to the use of different models of mast cells deficiency such as the KitW−sh/W−sh mast cell-deficient mice (which are not specific to mast cells), Kit-independent Cpa3Cre/+ and Mcpt5-Cre R-DTA mice, and mast cell stabilizer disodium cromoglycate treated mice (118, 123, 124).
AAI
To our knowledge, no studies have explored the presence of mast cells in the aged VAT and whether they contribute to tissue specific inflammaging. In mice, aging has been shown to promote the degranulation of mast cells upon exposure to prostaglandin E (PGE), which is not observed in mast cells from young animals (125). Furthermore, aged adipocytes secrete more PGE than young adipocytes and thus this warrants the examination of mast cell degranulation within the adipose tissue and the potential role of various proteases released, in the context of inflammaging and age associated IR (126).
Myeloid Derived Suppressor Cells
Homeostasis
Myeloid derived suppressor cells (MDSCs) are a heterogenous population of immature myeloid cells with anti-inflammatory functions including suppression of adaptive immunity, modulation of macrophage cytokine production, and elevated expression of immunosuppressive factors such as arginase 1 (Arg1) and inducible nitric oxide synthase (iNOS) (127). MDSCs tend to typically develop during a variety of inflammatory conditions such as sepsis, cancer, and instances of autoimmunity but little is known about their presence and/or role in the lean adipose tissue.
OAI
MDSCs have been shown to increase in the adipose tissue of leptin-deficient obese mice, while transferring MDSCs into obese mice improves parameters associated with metabolic disease (128). Accordingly, tumor-bearing mice fed a HFD display an enhanced accumulation of MDSCs. While MDSCs protected against metabolic disease and VAT inflammation, they have a detrimental effect on tumor progression and overall reduced animal survival time (129). Several factors have been implicated in the increased accumulation of MDSCs, including the adipokine leptin, polyunsaturated fats, and exogenous lipids, which could potentially be harnessed to boost MDSC activity and deter inflammation (129–131).
AAI
In mice, MDSCs have been shown to accumulate in the spleen, lymph nodes, and bone marrow of aged mice and also possess greater suppressive activity in T cell proliferation, which is associated with defective PI3K-Akt signaling pathway (132). Furthermore, MDSCs have been linked to limiting B lymphopoiesis during aging, which is driven via their production of IL-1 (133). Whether MDSCs accumulate in the aged VAT to suppress inflammatory cells or to impact insulin resistance remains to be evaluated.
Adaptive Immune Cells
B Cells
Homeostasis
B cells are a crucial component of the adaptive immune response that achieve their functions via cytokine secretion, antibody secretion, and modulation of the function of other cells (134). The majority of B cells can be categorized into B1 and B2 cells. B1 cells are enriched in the pleural and peritoneal cavities, while B2 cells are found abundantly in secondary lymphoid organs such as the spleen (134). Regulatory B cells (Bregs) are a collection of heterogenous IL-10 producing B cells that can ameliorate inflammation. Inside VAT, a subset of these Bregs are maintained by CXCL12 and free fatty acids and Breg deletion of IL-10 results in aggravated VAT inflammation, insulin resistance, and loss of metabolic homeostasis (135). However, the majority of B cell-derived IL-10 in VAT at steady state is derived from B1 cells, the B cell population that is also predominantly abundant within fat-associated lymphoid clusters in the VAT (136, 137).
OAI
Obesity induces an accumulation of total B cells in the VAT, including the proportion and absolute number of class switched mature IgM– IgD– IgG+ B2 cells (138). IgG isolated from obese mice is capable of driving metabolic disease due to their hyposialylated profile which activates the endothelial IgG receptor FCγRIIB (138, 139). In humans, insulin resistance is associated with a distinct profile of IgG autoantibodies, arguing for (self) antigen-specific targets contributing to B cell-mediated insulin resistance (138). A recent paper extended these findings by characterizing self-antigens targeted by IgG inside human adipose tissue (140). Moreover, B cells isolated from obese mice are responsible for modulating T cells within the VAT in an MHC-I/II-dependent manner by priming both CD4+ and CD8+ T cells. Ablation of the antigen-presenting complex from B cells is sufficient to improve metabolic parameters, indicating a role of B-T cognate interactions in the VAT in modulating metabolic disease (138). Obese B cells also secrete elevated levels of pro-inflammatory cytokines and in line with these various observations, both B cell-deficient μMT mice and anti-CD20-treated mice display improved metabolic and inflammatory parameters (138, 141). Notably, the LTB4/LTB4R1 chemokine/receptor axis has been shown to promote the activation and recruitment of VAT B2 cells, highlighting its potential as a target for insulin sensitizing therapies (142). Unlike B2 cells, tolerogenic B1a cells are reduced in frequency during obesity and produce less IL-10 and transferring B1a cells from lean mice into HFD-fed B null mice can improve metabolic parameters through IL-10- and polyclonal IgM-dependent mechanisms (136). Obesity also induces compromised functionality in natural Bregs (135).
Given the potential pathogenicity of B2 cells and beneficial effects of Bregs and B1 cells, targeting B2-driven VAT inflammation while enhancing the anti-inflammatory function of B1/Breg cells would help alleviate obesity-related adipose pathophysiology. For example, one possible B cell targeting therapy involves the use of B-cell Activating Factor (BAFF) antagonists as BAFF deficiency or inhibition ameliorates obesity-induced inflammation in mice (136, 143). Despite the growing interest in VAT B cells, the role of antibody secreting cells (ASCs) in the VAT is unknown. ASCs are terminally differentiated B cells that secrete antibodies and cytokines and can be further categorized into short-lived plasmablasts and long-lived plasma cells (144, 145). While VAT IgG increase during obesity, it is unclear whether this increase is due to a recruitment of long-lived plasma cells from the bone marrow or from other tissue such as intestines (146).
AAI
Like obesity, aging is characterized by a similar increase in mature B2 cells in the VAT, as well as plasma IgG as early as 12 months in mice (147). Furthermore, aging is associated with an increase in the expression of the B cell-specific nuclear co-factor Oct coactivator from B cells (OcaB) in the VAT. Depletion of OcaB in mice improved metabolic parameters, prevented the accumulation of B2 cells within the VAT, and decreased circulating levels of IgG2c and pro-inflammatory cytokines (147). An increase in the number of B1a and B1b cells has also been observed in the aged VAT. However, this accumulation was much subtler and the role of these innate B cells in aging requires further examination (147). Interestingly, some B1 cells develop pathological features during aging. Indeed, monocytes can convert omental B1a cells into 4-1BBL-expressing B1a cells which promotes immune activation and insulin resistance (148).
Interestingly, aging drives the formation of a unique circulating B cell subset known as age-associated B cells (ABCs), which are defined by their overexpression of the transcription factor T-bet (149–151). ABCs are further characterized by their ability to respond to nucleic acid antigen, and produce pro-inflammatory TNFα and IgG2c, making them prime candidates for mediating VAT inflammaging and age-associated insulin resistance (152, 153). Some evidence suggests that ABCs may represent either a memory B cell population or an ASC population as they express elevated levels of transcription factors involved in ASC formation, namely Prdm1, Irf4, and Xbp1, as well as expressing the ASC surface marker CD138 (151, 153, 154). Moreover, members of the SWEF family of Rho guanine exchange factor (GEF) proteins have been implicated in regulating the expansion of ABCs through the activity of IRF5 (155). Such ABCs have also been implicated in autoimmune disorders such as lupus in young mice, suggesting that aging does not serve as a prerequisite for their induction. This notion warrants the need to determine whether they are present within the VAT during diet-induced obesity independently of aging (154). Interestingly, ABC expansion is observed more consistently in female mice and it remains to be examined whether sex-specific hormone or other factors are at the root of these differences (149). Recently, another unique population of B cell, aged adipose B cells (AABs), have been shown to accumulate in aged VAT, which are memory-like B cells that expand within the fat-associated lymphoid clusters (FALCs) in aged VAT in an NLRP3 dependent manner; depletion of these cells can improve insulin resistance in mice and reverse age-induced lipolytic dysfunction, and future work will need to assess triggers and targets regulating these cells function during aging (60, 156).
T Cells
Homeostasis
T cells are a major component of the adaptive immune system and can be categorized into various subsets based on their expression of surface markers, the composition of T cell antigen receptors (TCR), and the secretion of different cytokines. T cells with the αβ TCR rearrangement can be categorized according to their expression of CD4 or CD8, with the former being further sub-divided into various subsets based on their effector cytokine profiles including IFNγ-producing T helper 1 (Th1) cells, IL-4-producing Th2 cells, IL-17-producing Th17 cells and IL-10-producing Foxp3+ T regulatory cells (Tregs).
Tregs are highly enriched in the lean adipose tissue, uniquely regulated by PPARγ, and promote skewing of macrophages to an M2-like state (157–160). Furthermore, the accumulation of Tregs depends on antigens presented in an MHC-II manner and the release of soluble mediators, notably IL-33, the majority of which is secreted by mesenchymal stromal cells (161–163). There is a growing appreciation of an evolutionarily conserved requirement for IL-33 in VAT Treg function, as demonstrated by their dependency upon the IL-33 receptor ST2, and downstream transcription factors BATF and IRF4 (164, 165). Interestingly, recent work has shown that sexual dimorphism has also been reported to play a role in VAT Treg function in this axis (166). VAT inflammation is increased with testosterone and limited with estrogen in males. Increased VAT inflammation and male-specific IL-33-producing stromal cells mediate the recruitment and local expansion of Tregs in a BLIMP-1-dependent manner, constituting a male-specific feedback circuit that potentially limits VAT inflammation (166). Research on Th2 cells in the adipose tissue also points toward a protective role, and human VAT is also thought to be enriched in Th2 cells expressing IL-13 under insulin sensitive conditions (157, 167).
OAI
Generally, T cells are known to accumulate within the VAT in obese mice and humans. Depleting T cells with an anti-CD3 antibody improves insulin sensitivity and limits VAT meta-inflammation in obese mice (157, 168). CD4+ cells within the obese VAT produce higher amounts of IFNγ than those from lean VAT, indicating an overall Th1 polarization of CD4+ T cells (157, 169, 170). Indeed, IL-12p35null mice, which are deficient in Th1 cells, show improved insulin sensitivity as the main Th1 effector cytokine, IFNγ, has been shown to be directly responsible for affecting insulin signaling, lipid storage, and differentiation of adipocytes via sustained JAK-STAT1 pathway activation (157, 171).
Obesity results in increased accumulation of VAT Th17 cells in an IL-6-dependent manner, although these cells are present at a lower frequency compared with Th1 cells (157, 170). Mechanistically, extracellular ATP acts through the P2X7 receptor pathway promotes a Th17-polarizing microenvironment (172). This was also accompanied by a greater frequency in Th17 cells and higher expression of Th17 markers such as RORC (RORγt in mice), IL-17, and IL-23R in VAT explants from metabolically unhealthy obese donors compared to metabolically healthy obese and lean donors (172, 173). Despite this research, the role of IL-17 in obesity-induced metabolic disease is unclear. Despite reports of increased IL-17 in obese individuals, mouse models have shown that IL-17 deficiency worsens the effects of diet-induced obesity, accelerates adiposity in mice fed a low-fat diet, and elevates circulating leptin levels (172, 174, 175). However, it is important to note that γδ T cells and MAIT cells are also sources of IL-17 and all studies regarding the impact of IL-17 on metabolic disease cannot be attributed to Th17 cells alone (175). More work is required to distinguish and elucidate the cell specific role of IL-17 in the adipose tissue during metabolic disease.
During obesity, adipose tissue Tregs decrease in number and are outweighed by the function of pro-inflammatory T cells (157, 176). As such, rescuing the Treg population may represent a potential therapeutic strategy in improving metabolic parameters during obesity; treatments with PPARγ agonists such as thiazolidinedione, 5-aminosalicylic acid (5ASA) or IL-33 administration in vivo were shown to regulate VAT Treg numbers and improve metabolic parameters in obese mouse models (160, 164, 165, 177).
CD8+ T cells, or cytotoxic T lymphocytes (CTLs), increase in the VAT during obesity and have an enhanced capacity to secrete IFNγ (167, 178). Despite no differences in body weight, obese mice deficient in CD8+ T cells display improved glucose tolerance and insulin sensitivity, suggesting a pathogenic role for CD8+ T cells in impairing metabolic health (105, 178). The cytotoxic activity of CD8+ T cells has been demonstrated to be regulated by perforin, as perforin-deficient CD8+ T cells show an elevated proliferative and inflammatory capacity. Transfer of these perforin-deficient CD8+ T cells into CD8-deficient mice significantly worsened metabolic parameters compared to those transferred with perforin-sufficient CD8+ T cells (75).
Several outstanding questions that remain to be explored include the mechanisms by which co-stimulatory molecules induce T cell pathogenicity during obesity, as well as how T cell metabolism is altered during adipose tissue inflammation. The 4-1BB-4-1BBL checkpoint pathway has been reported to be important in Th1-skewing of CD4+ cells and enhancing CD8+ T cell proliferation (179, 180). In obese mice, there is an increase in the expression of both the receptor and ligand; mice deficient in 4-1BB are reportedly protected from HFD-induced metabolic dysregulation and adipose macrophage and T cell infiltration (181, 182). Thus, it would be possible to target pro-inflammatory T cell function via blockade of this pathway. Recently, our group has reported that insulin receptor signaling modulates T cell inflammatory function and subset skewing via the control of intrinsic cellular metabolism, though it remains to be determined whether insulin signaling in VAT T cells is impacted during chronic obesity (183). Finally, it will be important to identify which antigens are targeted by T cells within the VAT. Indeed, a large majority of T cells in the VAT are effector memory T cells that exhibit a markedly restricted TCR diversity (157, 184). This, along with the presence of pathogenic IgG autoantibodies produced by B cells, suggests that there may potentially be specific target antigens within the VAT that drives an antigen-specific T cell response (140, 185). Whether these antigens are bystanders or true drivers of disease remain to be seen.
AAI
Within the aged VAT, similar to obesity, there is an expansion of various T cell populations including CD8+ T cells and CD4+ T cells. However, unlike obesity, Treg numbers increase in VAT with age (58, 111). Age-dependent accumulation of CD8+ T cells appears to be dependent on biological sex as aged CD8+ T cells from the VAT of female mice display an elevated activation status and secrete more pro-inflammatory cytokines than their male counterparts (186). Future work needs to explore the distribution of CD4+ T helper subsets within the VAT micro-environment and what signals are responsible for driving the accumulation of T cells within the aged VAT. Interestingly, Tregs within the VAT are enriched in aged male mice and selectively depleting them improves glucose uptake and age-associated insulin resistance (111), though the mechanisms are unclear. It should be noted that aged females have lower levels of VAT Tregs compared to male counterparts and the mechanism driving this difference is poorly understood (186), though recent work in describing sex differences in VAT Tregs may provide insight to these differences (166). Finally, further work should explore the role of the IL-33-Treg axis in the context of healthy aging, as well as determine whether dysfunctional stromal cell activity contributes toward the expansion of VAT Tregs during aging.
Innate Like T Cells
Homeostasis
There is an emerging appreciation for the role of innate-like T (ILT) cells that possess innate mechanisms to respond rapidly to stress and pathogens while concurrently expressing antigen receptors reminiscent of adaptive immunity (187). There are three main subsets of ILT cells that we will be discussing: natural killer T (NKT) cells, γδ T cells, and mucosal-associated invariant T (MAIT) cells.
NKT cells are a group of ILT cells that are activated upon recognizing glycolipid antigens presented on the non-classical MHC-I-like protein, CD1d, and can be further categorized into two subtypes based on their TCR diversity: type 1 or invariant NKT (iNKT) cells, which express an invariant TCRα chain and limited numbers of TCRβ chains, and type 2 or diverse NKT (dNKT) cells, which show more diverse usage of TCRα and β chains (188, 189). The VAT is enriched with iNKT cells which help maintain the adipose tissue microenvironment under homeostatic conditions by promoting IL-4-dependent M2-like macrophage polarization and sustaining Treg activity in an IL-10/IL-2-dependent manner; indeed, depletion of CD1d in adipocytes worsened inflammation and insulin resistance (190–192). Surprisingly, VAT iNKT cells in mice constitute a specialized tissue resident subset that express the transcription factor E4BP4 and lack PLZF expression, unlike circulating iNKT, and produce IL-2 and IL-10 to regulate immune homeostasis within the VAT (193, 194).
γδ T cells also possess innate-like features that allow for their activation upon recognition of conserved stress-induced ligands and subsequent rapid secretion of cytokines such as IFNγ, TNFα, and IL-17A (195). In lean wild type mice, adipose tissue resident PLZF+ γδ T cells were shown to produce TNFα and IL-17A which together promoted IL-33 production by adipose stromal cells (196). IL-33 production induced by γδ T cells was shown to promote thermogenesis as mice lacking γδ T cells or IL-17A showed impaired adipocyte UCP1 expression and thermoregulation (196).
Mucosal associated invariant T (MAIT) cells are found primarily in peripheral blood, the intestinal mucosa, and the liver and are evolutionary restricted by the MHC-related molecule 1 (MR1) (197). Upon stimulation, human peripheral MAIT cells produce IFNγ, TNFα, IL-17, IL-2, and granzyme B, whereas mouse spleen MAIT produce higher levels of IL-17 and lower levels of IL-10, IFNγ, and TNFα (198–202). In humans, adipose tissue MAIT cells but not peripheral blood MAIT cells produce more IL-10 than IL-17, highlighting site-specific differences in MAIT function (202). Although the role of MAIT cells inside VAT is poorly understood, they are potent secretors of IFNγ and IL-17 and warrant further investigation under homeostatic conditions (197).
OAI
During obesity, there is reportedly a decrease in the numbers of iNKT cells; furthermore, CD1d−/− mice deficient in iNKT cells display an insulin resistant phenotype even in the absence of a high fat diet and adipose inflammation, providing evidence for their protective function in the VAT during obesity (190, 203, 204). Conversely, in a Vα14 transgenic mouse model, which has elevated levels of iNKT cells, having increased iNKT cells on a Ldlr−/− background resulted in worsened metabolic parameters when mice were placed on an obesogenic diet. These findings suggest that all iNKT cells might not be outright protective, but rather the reduction in iNKT cells might be a compensatory mechanism in response to obesity (205). This observation could also be explained by the differential roles between peripheral and local iNKT subsets in maintaining tissue homeostasis. Further, most of these studies did not assess the role of type 2 dNKT cells as CD1d−/− mice lack both NKT subsets. dNKT cells have been shown to play a protective role during obesity as adoptive transfer of dNKT cells into obese mice improved weight loss and glucose homeostasis (206). Future studies should aim to use newly developed mouse strains to better address the specific roles of each NKT subsets in obesity (207). The observation that VAT PLZF+ γδ T cells promote the IL-33-Treg axis and thermogenesis under homeostatic conditions warrants the need for studies to explore the functions of PLZF+ γδ T cells in obese adipose tissue, whether they are lost during obesity, and whether IL-17 secretion by γδ T cells is impaired during obesity (196).
Patients with T2D and/or obesity show decreased frequency of peripheral MAIT cells but increased frequency in adipose tissue MAIT cells (202, 208, 209). Further, MAIT cells in both the adipose tissue and periphery from obese patients produced higher levels of IL-17 and reduced levels of IL-10, different from the lean state, suggesting a potential role for MAIT cells in obesity-associated inflammation (210). The mechanisms underlying the alterations in MAIT cell numbers, activation, and IL-17 production need to be further elucidated, with a need to determine the ontogeny of adipose tissue MAIT cells and their role in the adipose environment during obesity.
AAI
Only a few studies have explored the role of ILT cells within the aged VAT. In mice, NKT cell numbers were increased 2- to 3-fold in the secondary lymphoid organs of aged mice compared to young mice, suggesting that a similar increase or accumulation of peripheral PLZF+ NKT cells might be seen within the VAT (211). However, it remains to be determined how the population of adipose resident E4BP4+ iNKT cells are affected and whether the distribution of peripheral vs. resident NKT cells is shifted (193). Similar to obesity, it has also been suggested that peripheral MAIT cells are reduced in aged individuals and future studies need to determine whether they are being recruited to metabolic tissues such as the liver and/or VAT (212).
Concluding Remarks
Aging and diet-induced obesity present themselves as metabolic diseases that are characterized by an alteration of the VAT immune landscape and display an activation of chronic inflammatory pathways that contribute to insulin resistance and diabetes (Figure 1, Tables 1, 2). There are several overlapping mechanisms of VAT inflammation in obesity and aging that could help direct future research. One common driver of VAT inflammation in both conditions is the altered gut and the resident host microbiota. Obesity is associated with microbial dysbiosis and an overall reduction in bacterial diversity, which imparts features of the metabolic syndrome as the obese phenotype can be transferred from obese humans to mice through transplant of the gut microbiota (213–216). A consequence of microbial dysbiosis is increased intestinal permeability, characterized by leakage of bacterial antigen or their products such as metabolites or pathogen associated molecular patterns including lipopolysaccharide (LPS) across the intestinal epithelial barrier. These products can access metabolic tissues such as the VAT by entering systemic circulation and also being taken up by chylomicrons, which further drives meta-inflammation by activating pro-inflammatory cascades in immune cells via pattern recognition receptor signaling (177, 217–219). Aging is also potentially associated with intestinal microbial dysbiosis that contributes to worsened intestinal permeability, serum endotoxemia and inflammation, which can be recapitulated upon transfer of gut microbiota from aged mice into young germ-free mice, as seen during obesity (219–221). Low grade chronic intestinal inflammation is an early manifestation of obesity which precedes systemic metabolic disease and contributes to worsened barrier function and insulin resistance via secretion of pro-inflammatory cytokines (213). Moreover, an increased flux of bacterial product entering the VAT may be one common mechanism linking aging and obesity related insulin resistance to distinct downstream inflammasome receptors such as the NLRP3 inflammasome, which ultimately represent immune activation in response to gut bacterial products in obesity and aging (60, 156).
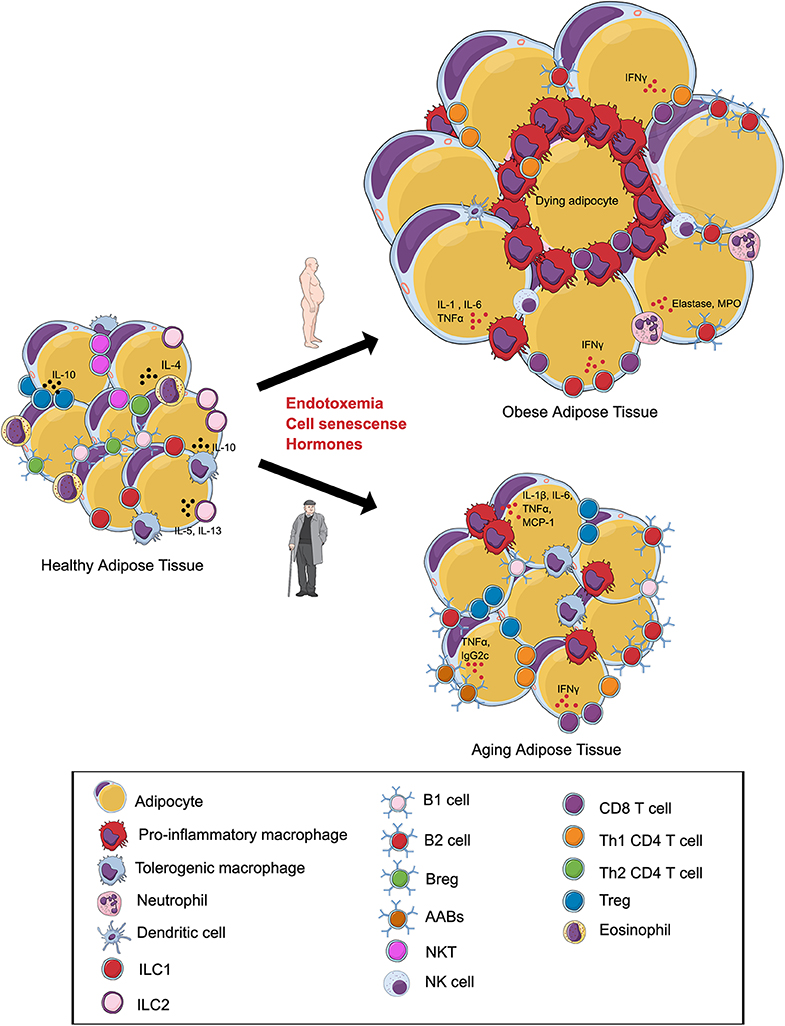
Figure 1. Alterations in the visceral adipose tissue (VAT) immune cells during obesity and aging. In lean VAT, homeostasis is maintained via the secretion of anti-inflammatory cytokines by regulatory T cells (Tregs), T helper 2 (Th2) cells, tolerogenic macrophages, group 1 innate lymphoid cells (ILC1s), ILC2s, regulatory B cells (Bregs), B1 cells, and eosinophils. (Top) Obesity induces an expansion in adipocyte size and promotes a shift in the phenotype of local immune cells toward a pro-inflammatory state with increases in pro-inflammatory macrophages, NK Cells, B2 cells, Th1 CD4 cells, CD8+ T cells, and neutrophils. Inflammation of adipose tissue leads to tissue damage, cell death, and metabolic disturbances. (Bottom) During aging, the VAT is characterized by alterations in the immune cell environment. Emerging evidence indicates that these changes are associated with a shift in the phenotype of macrophages, expansion of B2 cells, age-associated B cells (AABs), CD8+ T cells and, paradoxically, regulatory T cells (Tregs). Changes in the composition of adipose tissue immune cells during aging may contribute to insulin resistance and ectopic lipid storage. Illustration created in the Mind the Graph platform: www.mindthegraph.com.

Table 1. Comparison between innate immune cells in OAI and AAI.

Table 2. Comparison between adaptive immune cells in OAI and AAI.
Another shared factor that could potentially link obesity and age driven VAT inflammation is cellular senescence. Senescence is regarded as a hallmark of aging, and metabolically active senescent cells secrete a variety of cytokines and chemokines, in an NFKB dependent manner, which further drives local inflammation (222, 223). Senescent cells have been demonstrated to also accumulate within the VAT during obesity, and their clearance is associated with improved metabolic parameters and decreased macrophage homing (224). Interestingly, within the VAT, CD4+ T cells adopt a senescent phenotype which drives VAT inflammation and insulin resistance in an osteopontin dependent manner (225). A combination of cell senescence and inflammatory cell death within the fat might represent a source of shared antigenic targets between obesity and aging, which likely underpins a mechanism fueling local immune cells.
Finally, both obesity and aging are associated with changes in the levels of hormones that could impact immune cell function in the VAT. For instance, leptin and insulin typically rise with obesity and aging. Leptin metabolically activates T cells and skews them toward a Th1 cytokine secretion profile, promotes pro-inflammatory cytokine secretion by circulating monocytes, and enhances expression of perforin in NK cells (226–229). Insulin has been shown to facilitate T cell glycolytic programming and IFNγ mediated effector functions (183). The interactions of these hormones and adipokines in an environment of increasing cell senescence and bacterial products likely represents additional common drivers between aging and obesity related VAT inflammation.
Given these potential underlying common drivers of obesity- and aging-related IR, it will be important to understand the mechanistic differences between both conditions. For instance, why do VAT Tregs function differently with age compared to obesity? What are the underlying reasons behind the differences in VAT macrophage numbers? It will also be important to tease out the differences in inflammatory responses between mouse models and humans. In particular, do aged humans show similar changes to VAT immune cells as seen in aged mice? Understanding the differences and similarities between VAT immune populations with age and obesity will help identify unifying pathophysiological root causes of the associated metabolic disease.
Over the past 20 years, our knowledge about the immune landscape of the VAT has grown from the simple identification of the role of cytokines and macrophages, to include adaptive immunity, and lately most known immune cell populations. The level of cellular characterization will continue to improve to further refine these populations using single-cell genomics and advanced cytometry methods. Such analyses will ultimately yield new insights into disease pathogenesis and may lead to new therapies to combat obesity and aging related metabolic disease.
Author Contributions
SK, YC, XR, and DW contributed to the design and writing of the manuscript and the generation of the figures.
Funding
SK was a recipient of the Queen Elizabeth II Graduate Scholarship in Science and Technology (QEII-GSST)/Aventis Pasteur, and the Banting & Best Centre (BBDC)-Novo Nordisk Studentship. XR was funded by a National Institute of Health (NIH) Research Project Grant 1R01DK122056 (XR). DW was funded by a Canadian Institutes of Health Research (CIHR) New Investigator Foundation Grant FDN-148385 (DW) and a Canadian Liver Foundation operating grant (2017). DW holds an Ontario Ministry of Innovation Early Researcher Award.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Stevens GA, Singh GM, Lu Y, Danaei G, Lin JK, Finucane MM, et al. National, regional, and global trends in adult overweight and obesity prevalences. Popul Health Metr. (2012) 10:22. doi: 10.1186/1478-7954-10-22
2. Kelly T, Yang W, Chen CS, Reynolds K, He J. Global burden of obesity in 2005 and projections to 2030. Int J Obes. (2008) 32:1431–7. doi: 10.1038/ijo.2008.102
3. Hruby A, Hu FB. The epidemiology of obesity: a big picture. Pharmacoeconomics. (2015) 33:673–89. doi: 10.1007/s40273-014-0243-x
4. Cornier MA, Dabelea D, Hernandez TL, Lindstrom RC, Steig AJ, Stob NR, et al. The metabolic syndrome. Endocr Rev. (2008) 29:777–822. doi: 10.1210/er.2008-0024
5. Esser N, Legrand-Poels S, Piette J, Scheen AJ, Paquot N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res Clin Pract. (2014) 105:141–50. doi: 10.1016/j.diabres.2014.04.006
6. Birkenfeld AL, Shulman GI. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology. (2014) 59:713–23. doi: 10.1002/hep.26672
7. Wilson PW, D'Agostino RB, Parise H, Sullivan L, Meigs JB. Metabolic syndrome as a precursor of cardiovascular disease and type 2 diabetes mellitus. Circulation. (2005) 112:3066–72. doi: 10.1161/CIRCULATIONAHA.105.539528
8. Bonomini F, Rodella LF, Rezzani R. Metabolic syndrome, aging and involvement of oxidative stress. Aging Dis. (2015) 6:109–20. doi: 10.14336/AD.2014.0305
9. Lutz W, Sanderson W, Scherbov S. The coming acceleration of global population ageing. Nature. (2008) 451:716–9. doi: 10.1038/nature06516
10. Ferrucci L, Giallauria F, Guralnik JM. Epidemiology of aging. Radiol Clin North Am. (2008) 46:643–52. doi: 10.1016/j.rcl.2008.07.005
11. Goronzy JJ, Weyand CM. Understanding immunosenescence to improve responses to vaccines. Nat Immunol. (2013) 14:428–36. doi: 10.1038/ni.2588
12. Frasca D, Blomberg BB, Paganelli R. Aging, obesity, and inflammatory age-related diseases. Front Immunol. (2017) 8:1745. doi: 10.3389/fimmu.2017.01745
13. Cinti S. The adipose organ at a glance. Dis Model Mech. (2012) 5:588–94. doi: 10.1242/dmm.009662
14. Wronska A, Kmiec Z. Structural and biochemical characteristics of various white adipose tissue depots. Acta Physiol. (2012) 205:194–208. doi: 10.1111/j.1748-1716.2012.02409.x
15. Ibrahim MM. Subcutaneous and visceral adipose tissue: structural and functional differences. Obes Rev. (2010) 11:11–8. doi: 10.1111/j.1467-789X.2009.00623.x
16. Chusyd DE, Wang D, Huffman DM, Nagy TR. Relationships between rodent white adipose fat pads and human white adipose fat depots. Front Nutr. (2016) 3:10. doi: 10.3389/fnut.2016.00010
17. Divoux A, Clément K. Architecture and the extracellular matrix: the still unappreciated components of the adipose tissue. Obes Rev. (2011) 12:e494–503. doi: 10.1111/j.1467-789X.2010.00811.x
18. Bora P, Majumdar AS. Adipose tissue-derived stromal vascular fraction in regenerative medicine: a brief review on biology and translation. Stem Cell Res Ther. (2017) 8:145. doi: 10.1186/s13287-017-0598-y
19. Tholpady SS, Llull R, Ogle RC, Rubin JP, Futrell JW, Katz AJ. Adipose tissue: stem cells and beyond. Clin Plast Surg. (2006) 33:55–62. doi: 10.1016/j.cps.2005.08.004
20. Sun K, Kusminski CM, Scherer PE. Adipose tissue remodeling and obesity. J Clin Invest. (2011) 121:2094–101. doi: 10.1172/JCI45887
21. Halberg N, Khan T, Trujillo ME, Wernstedt-Asterholm I, Attie AD, Sherwani S, et al. Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol Cell Biol. (2009) 29:4467–83. doi: 10.1128/MCB.00192-09
22. Berger JJ, Barnard RJ. Effect of diet on fat cell size and hormone-sensitive lipase activity. J Appl Physiol. (1999) 87:227–32. doi: 10.1152/jappl.1999.87.1.227
23. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. (1993) 259:87–91. doi: 10.1126/science.7678183
24. Copps KD, White MF. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia. (2012) 55:2565–82. doi: 10.1007/s00125-012-2644-8
25. Tanti JF, Ceppo F, Jager J, Berthou F. Implication of inflammatory signaling pathways in obesity-induced insulin resistance. Front Endocrinol. (2012) 3:181. doi: 10.3389/fendo.2012.00181
26. Khan T, Muise ES, Iyengar P, Wang ZV, Chandalia M, Abate N, et al. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol Cell Biol. (2009) 29:1575–91. doi: 10.1128/MCB.01300-08
27. Nakajima I, Muroya S, Tanabe R, Chikuni K. Positive effect of collagen V and VI on triglyceride accumulation during differentiation in cultures of bovine intramuscular adipocytes. Differentiation. (2002) 70:84–91. doi: 10.1046/j.1432-0436.2002.700203.x
28. Hasegawa Y, Ikeda K, Chen Y, Alba DL, Stifler D, Shinoda K, et al. Repression of adipose tissue fibrosis through a PRDM16-GTF2IRD1 complex improves systemic glucose homeostasis. Cell Metab. (2018) 27:180–94.e6. doi: 10.1016/j.cmet.2017.12.005
29. O'Hara A, Lim FL, Mazzatti DJ, Trayhurn P. Microarray analysis identifies matrix metalloproteinases (MMPs) as key genes whose expression is up-regulated in human adipocytes by macrophage-conditioned medium. Pflugers Arch. (2009) 458:1103–14. doi: 10.1007/s00424-009-0693-8
30. Tchkonia T, Morbeck DE, Von Zglinicki T, Van Deursen J, Lustgarten J, Scrable H, et al. Fat tissue, aging, and cellular senescence. Aging Cell. (2010) 9:667–84. doi: 10.1111/j.1474-9726.2010.00608.x
31. Xu M, Tchkonia T, Ding H, Ogrodnik M, Lubbers ER, Pirtskhalava T, et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc Natl Acad Sci USA. (2015) 112:E6301–10. doi: 10.1073/pnas.1515386112
32. Wernstedt Asterholm I, Tao C, Morley TS, Wang QA, Delgado-Lopez F, Wang ZV, et al. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab. (2014) 20:103–18. doi: 10.1016/j.cmet.2014.05.005
33. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. (2014) 40:274–88. doi: 10.1016/j.immuni.2014.01.006
34. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. (2003) 112:1796–808. doi: 10.1172/JCI200319246
35. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. (2007) 117:175–84. doi: 10.1172/JCI29881
36. Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. (2007) 447:1116–20. doi: 10.1038/nature05894
37. Davies LC, Taylor PR. Tissue-resident macrophages: then and now. Immunology. (2015) 144:541–8. doi: 10.1111/imm.12451
38. Hassnain Waqas SF, Noble A, Hoang AC, Ampem G, Popp M, Strauß S, et al. Adipose tissue macrophages develop from bone marrow-independent progenitors in. J Leukoc Biol. (2017) 102:845–55. doi: 10.1189/jlb.1A0317-082RR
39. Lumeng CN, Deyoung SM, Bodzin JL, Saltiel AR. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes. (2007) 56:16–23. doi: 10.2337/db06-1076
40. Jang JE, Ko MS, Yun JY, Kim MO, Kim JH, Park HS, et al. Nitric oxide produced by macrophages inhibits adipocyte differentiation and promotes profibrogenic responses in preadipocytes to induce adipose tissue fibrosis. Diabetes. (2016) 65:2516–28. doi: 10.2337/db15-1624
41. Boutens L, Hooiveld GJ, Dhingra S, Cramer RA, Netea MG, Stienstra R. Unique metabolic activation of adipose tissue macrophages in obesity promotes inflammatory responses. Diabetologia. (2018) 61:942–53. doi: 10.1007/s00125-017-4526-6
42. Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. (2015) 42:419–30. doi: 10.1016/j.immuni.2015.02.005
43. Morris DL, Cho KW, Delproposto JL, Oatmen KE, Geletka LM, Martinez-Santibanez G, et al. Adipose tissue macrophages function as antigen-presenting cells and regulate adipose tissue CD4+ T cells in mice. Diabetes. (2013) 62:2762–72. doi: 10.2337/db12-1404
44. Cinti S, Mitchell G, Barbatelli G, Murano I, Ceresi E, Faloia E, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res. (2005) 46:2347–55. doi: 10.1194/jlr.M500294-JLR200
45. Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. (2006) 116:1494–505. doi: 10.1172/JCI26498
46. Amano SU, Cohen JL, Vangala P, Tencerova M, Nicoloro SM, Yawe JC, et al. Local proliferation of macrophages contributes to obesity-associated adipose tissue inflammation. Cell Metab. (2014) 19:162–71. doi: 10.1016/j.cmet.2013.11.017
47. Nomiyama T, Perez-Tilve D, Ogawa D, Gizard F, Zhao Y, Heywood EB, et al. Osteopontin mediates obesity-induced adipose tissue macrophage infiltration and insulin resistance in mice. J Clin Invest. (2007) 117:2877–88. doi: 10.1172/JCI31986
48. Allman WR, Dey R, Liu L, Siddiqui S, Coleman AS, Bhattacharya P, et al. TACI deficiency leads to alternatively activated macrophage phenotype and susceptibility to Leishmania infection. Proc Natl Acad Sci USA. (2015) 112:E4094–103. doi: 10.1073/pnas.1421580112
49. Liu L, Inouye KE, Allman WR, Coleman AS, Siddiqui S, Hotamisligil GS, et al. TACI-Deficient macrophages protect mice against metaflammation and obesity-induced dysregulation of glucose homeostasis. Diabetes. (2018) 67:1589–603. doi: 10.2337/db17-1089
50. Spadaro O, Camell CD, Bosurgi L, Nguyen KY, Youm YH, Rothlin CV, et al. IGF1 shapes macrophage activation in response to immunometabolic challenge. Cell Rep. (2017) 19:225–34. doi: 10.1016/j.celrep.2017.03.046
51. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. (2014) 6:13. doi: 10.12703/P6-13
52. Russo L, Lumeng CN. Properties and functions of adipose tissue macrophages in obesity. Immunology. (2018) 155:407–17. doi: 10.1111/imm.13002
53. Bertola A, Ciucci T, Rousseau D, Bourlier V, Duffaut C, Bonnafous S, et al. Identification of adipose tissue dendritic cells correlated with obesity-associated insulin-resistance and inducing Th17 responses in mice and patients. Diabetes. (2012) 61:2238–47. doi: 10.2337/db11-1274
54. Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, et al. Lipid-associated macrophages control metabolic homeostasis in a trem2-dependent manner. Cell. (2019) 178:686–98.e14. doi: 10.1016/j.cell.2019.05.054
55. Kratz M, Coats BR, Hisert KB, Hagman D, Mutskov V, Peris E, et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. (2014) 20:614–25. doi: 10.1016/j.cmet.2014.08.010
56. Hill DA, Lim HW, Kim YH, Ho WY, Foong YH, Nelson VL, et al. Distinct macrophage populations direct inflammatory versus physiological changes in adipose tissue. Proc Natl Acad Sci USA. (2018) 115:E5096–105. doi: 10.1073/pnas.1802611115
57. Pirzgalska RM, Seixas E, Seidman JS, Link VM, Sánchez NM, Mahú I, et al. Sympathetic neuron-associated macrophages contribute to obesity by importing and metabolizing norepinephrine. Nat Med. (2017) 23:1309–18. doi: 10.1038/nm.4422
58. Lumeng CN, Liu J, Geletka L, Delaney C, Delproposto J, Desai A, et al. Aging is associated with an increase in T cells and inflammatory macrophages in visceral adipose tissue. J Immunol. (2011) 187:6208–16. doi: 10.4049/jimmunol.1102188
59. Camell CD, Sander J, Spadaro O, Lee A, Nguyen KY, Wing A, et al. Inflammasome-driven catecholamine catabolism in macrophages blunts lipolysis during ageing. Nature. (2017) 550:119–23. doi: 10.1038/nature24022
60. Camell CD, Günther P, Lee A, Goldberg EL, Spadaro O, Youm YH, et al. Aging induces an Nlrp3 inflammasome-dependent expansion of adipose B cells that impairs metabolic homeostasis. Cell Metab. (2019) 30:1024–39.e6. doi: 10.1016/j.cmet.2019.10.006
61. Diefenbach A, Colonna M, Koyasu S. Development, differentiation, and diversity of innate lymphoid cells. Immunity. (2014) 41:354–65. doi: 10.1016/j.immuni.2014.09.005
62. Yudanin NA, Schmitz F, Flamar AL, Thome JJC, Tait Wojno E, Moeller JB, et al. Spatial and temporal mapping of human innate lymphoid cells reveals elements of tissue specificity. Immunity. (2019) 50:505–19.e4. doi: 10.1016/j.immuni.2019.01.012
63. Fuchs A. ILC1s in tissue inflammation and infection. Front Immunol. (2016) 7:104. doi: 10.3389/fimmu.2016.00104
64. O'Sullivan TE, Rapp M, Fan X, Weizman OE, Bhardwaj P, Adams NM, et al. Adipose-resident group 1 innate lymphoid cells promote obesity-associated insulin resistance. Immunity. (2016) 45:428–41. doi: 10.1016/j.immuni.2016.06.016
65. Boulenouar S, Michelet X, Duquette D, Alvarez D, Hogan AE, Dold CJ. Adipose type one innate lymphoid cells regulate macrophage homeostasis through targeted cytotoxicity. Immunity. (2017) 46:273–86. doi: 10.1016/j.immuni.2017.01.008
66. Kim BS, Artis D. Group 2 innate lymphoid cells in health and disease. Cold Spring Harb Perspect Biol. (2015) 7:a016337. doi: 10.1101/cshperspect.a016337
67. Montanari T, Pošćić N, Colitti M. Factors involved in white-to-brown adipose tissue conversion and in thermogenesis: a review. Obes Rev. (2017) 18:495–513. doi: 10.1111/obr.12520
68. Brestoff JR, Kim BS, Saenz SA, Stine RR, Monticelli LA, Sonnenberg GF, et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature. (2015) 519:242–6. doi: 10.1038/nature14115
69. Price AE, Liang HE, Sullivan BM, Reinhardt RL, Eisley CJ, Erle DJ, et al. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad Sci USA. (2010) 107:11489–94. doi: 10.1073/pnas.1003988107
70. Molofsky AB, Nussbaum JC, Liang HE, Van Dyken SJ, Cheng LE, Mohapatra A, et al. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J Exp Med. (2013) 210:535–49. doi: 10.1084/jem.20121964
71. Lee MW, Odegaard JI, Mukundan L, Qiu Y, Molofsky AB, Nussbaum JC, et al. Activated type 2 innate lymphoid cells regulate beige fat biogenesis. Cell. (2015) 160:74–87. doi: 10.1016/j.cell.2014.12.011
72. Wang H, Shen L, Sun X, Liu F, Feng W, Jiang C, et al. Adipose group 1 innate lymphoid cells promote adipose tissue fibrosis and diabetes in obesity. Nat Commun. (2019) 10:3254. doi: 10.1038/s41467-019-11270-1
73. O'Rourke RW, Meyer KA, Neeley CK, Gaston GD, Sekhri P, Szumowski M, et al. Systemic NK cell ablation attenuates intra-abdominal adipose tissue macrophage infiltration in murine obesity. Obesity. (2014) 22:2109–14. doi: 10.1002/oby.20823
74. Wensveen FM, Jelencic V, Valentic S, Sestan M, Wensveen TT, Theurich S, et al. NK cells link obesity-induced adipose stress to inflammation and insulin resistance. Nat Immunol. (2015) 16:376–85. doi: 10.1038/ni.3120
75. Revelo XS, Tsai S, Lei H, Luck H, Ghazarian M, Tsui H, et al. Perforin is a novel immune regulator of obesity-related insulin resistance. Diabetes. (2015) 64:90–103. doi: 10.2337/db13-1524
76. O'Rourke RW, Metcalf MD, White AE, Madala A, Winters BR, Maizlin II, et al. Depot-specific differences in inflammatory mediators and a role for NK cells and IFN-gamma in inflammation in human adipose tissue. Int J Obes. (2009) 33:978–90. doi: 10.1038/ijo.2009.133
77. O'Rourke RW, Gaston GD, Meyer KA, White AE, Marks DL. Adipose tissue NK cells manifest an activated phenotype in human obesity. Metabolism. (2013) 62:1557–61. doi: 10.1016/j.metabol.2013.07.011
78. Lee BC, Kim MS, Pae M, Yamamoto Y, Eberlé D, Shimada T, et al. Adipose natural killer cells regulate adipose tissue macrophages to promote insulin resistance in obesity. Cell Metab. (2016) 23:685–98. doi: 10.1016/j.cmet.2016.03.002
79. Michelet X, Dyck L, Hogan A, Loftus RM, Duquette D, Wei K, et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat Immunol. (2018) 19:1330–40. doi: 10.1038/s41590-018-0251-7
80. Theurich S, Tsaousidou E, Hanssen R, Lempradl AM, Mauer J, Timper K, et al. IL-6/Stat3-dependent induction of a distinct, obesity-associated NK cell subpopulation deteriorates energy and glucose homeostasis. Cell Metab. (2017) 26:171–84.e6. doi: 10.1016/j.cmet.2017.05.018
81. Starr ME, Saito M, Evers BM, Saito H. Age-associated increase in cytokine production during systemic inflammation-II: the role of IL-1β in age-dependent IL-6 upregulation in adipose tissue. J Gerontol A Biol Sci Med Sci. (2015) 70:1508–15. doi: 10.1093/gerona/glu197
82. Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil function: from mechanisms to disease. Annu Rev Immunol. (2012) 30:459–89. doi: 10.1146/annurev-immunol-020711-074942
83. Lacy P. Mechanisms of degranulation in neutrophils. Allergy Asthma Clin Immunol. (2006) 2:98–108. doi: 10.1186/1710-1492-2-3-98
84. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. (2018) 18:134–47. doi: 10.1038/nri.2017.105
85. Elgazar-Carmon V, Rudich A, Hadad N, Levy R. Neutrophils transiently infiltrate intra-abdominal fat early in the course of high-fat feeding. J Lipid Res. (2008) 49:1894–903. doi: 10.1194/jlr.M800132-JLR200
86. Revelo XS, Ghazarian M, Chng MH, Luck H, Kim JH, Zeng K, et al. Nucleic acid-targeting pathways promote inflammation in obesity-related insulin resistance. Cell Rep. (2016) 16:717–30. doi: 10.1016/j.celrep.2016.06.024
87. Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med. (2012) 18:1407–12. doi: 10.1038/nm.2885
88. Houghton AM, Rzymkiewicz DM, Ji H, Gregory AD, Egea EE, Metz HE, et al. Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat Med. (2010) 16:219–23. doi: 10.1038/nm.2084
89. Nijhuis J, Rensen SS, Slaats Y, van Dielen FM, Buurman WA, Greve JW. Neutrophil activation in morbid obesity, chronic activation of acute inflammation. Obesity. (2009) 17:2014–8. doi: 10.1038/oby.2009.113
90. Brotfain E, Hadad N, Shapira Y, Avinoah E, Zlotnik A, Raichel L, et al. Neutrophil functions in morbidly obese subjects. Clin Exp Immunol. (2015) 181:156–63. doi: 10.1111/cei.12631
91. Esparza B, Sanchez H, Ruiz M, Barranquero M, Sabino E, Merino F. Neutrophil function in elderly persons assessed by flow cytometry. Immunol Invest. (1996) 25:185–90. doi: 10.3109/08820139609059301
92. Wenisch C, Patruta S, Daxböck F, Krause R, Hörl W. Effect of age on human neutrophil function. J Leukoc Biol. (2000) 67:40–5. doi: 10.1002/jlb.67.1.40
93. Butcher SK, Chahal H, Nayak L, Sinclair A, Henriquez NV, Sapey E, et al. Senescence in innate immune responses: reduced neutrophil phagocytic capacity and CD16 expression in elderly humans. J Leukoc Biol. (2001) 70:881–6. doi: 10.1189/jlb.70.6.881
94. McLaughlin B, O'Malley K, Cotter TG. Age-related differences in granulocyte chemotaxis and degranulation. Clin Sci. (1986) 70:59–62. doi: 10.1042/cs0700059
95. Fulop T, Larbi A, Douziech N, Fortin C, Guérard KP, Lesur O, et al. Signal transduction and functional changes in neutrophils with aging. Aging Cell. (2004) 3:217–26. doi: 10.1111/j.1474-9728.2004.00110.x
96. Simell B, Vuorela A, Ekström N, Palmu A, Reunanen A, Meri S, et al. Aging reduces the functionality of anti-pneumococcal antibodies and the killing of Streptococcus pneumoniae by neutrophil phagocytosis. Vaccine. (2011) 29:1929–34. doi: 10.1016/j.vaccine.2010.12.121
97. Brubaker AL, Rendon JL, Ramirez L, Choudhry MA, Kovacs EJ. Reduced neutrophil chemotaxis and infiltration contributes to delayed resolution of cutaneous wound infection with advanced age. J Immunol. (2013) 190:1746–57. doi: 10.4049/jimmunol.1201213
98. Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, et al. Immunobiology of dendritic cells. Annu Rev Immunol. (2000) 18:767–811. doi: 10.1146/annurev.immunol.18.1.767
99. Dress RJ, Dutertre CA, Giladi A, Schlitzer A, Low I, Shadan NB, et al. Plasmacytoid dendritic cells develop from Ly6D+ lymphoid progenitors disctinct from the myeloid lineage. Nat Immunol. (2019) 20:852–64. doi: 10.1038/s41590-019-0420-3
100. Macdougall CE, Wood EG, Loschko J, Scagliotti V, Cassidy FC, Robinson ME, et al. Visceral adipose tissue immune homeostasis is regulated by the crosstalk between adipocytes and dendritic cell subsets. Cell Metab. (2018) 27:588–601.e4. doi: 10.1016/j.cmet.2018.02.007
101. Cho KW, Zamarron BF, Muir LA, Singer K, Porsche CE, DelProposto JB, et al. Adipose tissue dendritic cells are independent contributors to obesity-induced inflammation and insulin resistance. J Immunol. (2016) 197:3650–61. doi: 10.4049/jimmunol.1600820
102. Hellmann J, Sansbury BE, Holden CR, Tang Y, Wong B, Wysoczynski M, et al. CCR7 maintains nonresolving lymph node and adipose inflammation in obesity. Diabetes. (2016) 65:2268–81. doi: 10.2337/db15-1689
103. Ghosh AR, Bhattacharya R, Bhattacharya S, Nargis T, Rahaman O, Duttagupta P, et al. Adipose recruitment and activation of plasmacytoid dendritic cells fuel metaflammation. Diabetes. (2016) 65:3440–52. doi: 10.2337/db16-0331
104. Hannibal TD, Schmidt-Christensen A, Nilsson J, Fransen-Pettersson N, Hansen L, Holmberg D. Deficiency in plasmacytoid dendritic cells and type I interferon signalling prevents diet-induced obesity and insulin resistance in mice. Diabetologia. (2017) 60:2033–41. doi: 10.1007/s00125-017-4341-0
105. Ghazarian M, Revelo XS, Nøhr MK, Luck H, Zeng K, Lei H, et al. Type I interferon responses drive intrahepatic t cells to promote metabolic syndrome. Sci Immunol. (2017) 2:eaai7616. doi: 10.1126/sciimmunol.aai7616
106. Agrawal A, Agrawal S, Cao JN, Su H, Osann K, Gupta S. Altered innate immune functioning of dendritic cells in elderly humans: a role of phosphoinositide 3-kinase-signaling pathway. J Immunol. (2007) 178:6912–22. doi: 10.4049/jimmunol.178.11.6912
107. Klion A. Recent advances in understanding eosinophil biology. F1000Res. (2017) 6:1084. doi: 10.12688/f1000research.11133.1
108. Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science. (2011) 332:243–7. doi: 10.1126/science.1201475
109. Fischer K, Ruiz HH, Jhun K, Finan B, Oberlin DJ, van der Heide V, et al. Alternatively activated macrophages do not synthesize catecholamines or contribute to adipose tissue adaptive thermogenesis. Nat Med. (2017) 23:623–30. doi: 10.1038/nm.4316
110. Bolus WR, Peterson KR, Hubler MJ, Kennedy AJ, Gruen ML, Hasty AH. Elevating adipose eosinophils in obese mice to physiologically normal levels does not rescue metabolic impairments. Mol Metab. (2018) 8:86–95. doi: 10.1016/j.molmet.2017.12.004
111. Bapat SP, Myoung Suh J, Fang S, Liu S, Zhang Y, Cheng A, et al. Depletion of fat-resident Treg cells prevents age-associated insulin resistance. Nature. (2015) 528:137–41. doi: 10.1038/nature16151
112. Mathur SK, Schwantes EA, Jarjour NN, Busse WW. Age-related changes in eosinophil function in human subjects. Chest. (2008) 133:412–9. doi: 10.1378/chest.07-2114
113. Berry DC, Jiang Y, Arpke RW, Close EL, Uchida A, Reading DV. Cellular aging contributes to failure of cold-induced beige adipocyte formation in old mice and humans. Cell Metab. (2017) 25:481. doi: 10.1016/j.cmet.2017.01.011
114. Kumar D, Pandya SK, Varshney S, Shankar K, Rajan S, Srivastava A, et al. Temporal immmunometabolic profiling of adipose tissue in HFD-induced obesity: manifestations of mast cells in fibrosis and senescence. Int J Obes. (2019) 43:1281–94. doi: 10.1038/s41366-018-0228-5
115. Ishijima Y, Ohmori S, Ohneda K. Mast cell deficiency results in the accumulation of preadipocytes in adipose tissue in both obese and non-obese mice. FEBS Open Bio. (2013) 4:18–24. doi: 10.1016/j.fob.2013.11.004
116. Altintas MM, Azad A, Nayer B, Contreras G, Zaias J, Faul C, et al. Mast cells, macrophages, and crown-like structures distinguish subcutaneous from visceral fat in mice. J Lipid Res. (2011) 52:480–8. doi: 10.1194/jlr.M011338
117. Divoux A, Moutel S, Poitou C, Lacasa D, Veyrie N, Aissat A, et al. Mast cells in human adipose tissue: link with morbid obesity, inflammatory status, and diabetes. J Clin Endocrinol Metab. (2012) 97:E1677–85. doi: 10.1210/jc.2012-1532
118. Liu J, Divoux A, Sun J, Zhang J, Clement K, Glickman JN, et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat Med. (2009) 15:940–5. doi: 10.1038/nm.1994
119. Moreno M, Puig J, Serrano M, Moreno-Navarrete JM, Ortega F, Ricart W, et al. Circulating tryptase as a marker for subclinical atherosclerosis in obese subjects. PLoS ONE. (2014) 9:e97014. doi: 10.1371/journal.pone.0097014
120. Pejler G, Rönnberg E, Waern I, Wernersson S. Mast cell proteases: multifaceted regulators of inflammatory disease. Blood. (2010) 115:4981–90. doi: 10.1182/blood-2010-01-257287
121. Sun J, Sukhova GK, Wolters PJ, Yang M, Kitamoto S, Libby P, et al. Mast cells promote atherosclerosis by releasing proinflammatory cytokines. Nat Med. (2007) 13:719–24. doi: 10.1038/nm1601
122. Yang M, Sun J, Zhang T, Liu J, Zhang J, Shi MA, et al. Deficiency and inhibition of cathepsin K reduce body weight gain and increase glucose metabolism in mice. Arterioscler Thromb Vasc Biol. (2008) 28:2202–8. doi: 10.1161/ATVBAHA.108.172320
123. Gutierrez DA, Muralidhar S, Feyerabend TB, Herzig S, Rodewald HR. Hematopoietic kit deficiency, rather than lack of mast cells, protects mice from obesity and insulin resistance. Cell Metab. (2015) 21:678–91. doi: 10.1016/j.cmet.2015.04.013
124. Chmelar J, Chatzigeorgiou A, Chung KJ, Prucnal M, Voehringer D, Roers A, et al. No role for mast cells in obesity-related metabolic dysregulation. Front Immunol. (2016) 7:524. doi: 10.3389/fimmu.2016.00524
125. Nguyen M, Pace AJ, Koller BH. Age-induced reprogramming of mast cell degranulation. J Immunol. (2005) 175:5701–7. doi: 10.4049/jimmunol.175.9.5701
126. Wu D, Ren Z, Pae M, Guo W, Cui X, Merrill AH, et al. Aging up-regulates expression of inflammatory mediators in mouse adipose tissue. J Immunol. (2007) 179:4829–39. doi: 10.4049/jimmunol.179.7.4829
127. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. (2009) 9:162–74. doi: 10.1038/nri2506
128. Xia S, Sha H, Yang L, Ji Y, Ostrand-Rosenberg S, Qi L. Gr-1+ CD11b+ myeloid-derived suppressor cells suppress inflammation and promote insulin sensitivity in obesity. J Biol Chem. (2011) 286:23591–9. doi: 10.1074/jbc.M111.237123
129. Clements VK, Long T, Long R, Figley C, Smith DMC, Ostrand-Rosenberg S. Frontline science: high fat diet and leptin promote tumor progression by inducing myeloid-derived suppressor cells. J Leukoc Biol. (2018) 103:395–407. doi: 10.1002/JLB.4HI0517-210R
130. Yan D, Yang Q, Shi M, Zhong L, Wu C, Meng T, et al. Polyunsaturated fatty acids promote the expansion of myeloid-derived suppressor cells by activating the JAK/STAT3 pathway. Eur J Immunol. (2013) 43:2943–55. doi: 10.1002/eji.201343472
131. Al-Khami AA, Zheng L, Del Valle L, Hossain F, Wyczechowska D, Zabaleta J, et al. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology. (2017) 6:e1344804. doi: 10.1080/2162402X.2017.1344804
132. Enioutina EY, Bareyan D, Daynes RA. A role for immature myeloid cells in immune senescence. J Immunol. (2011) 186:697–707. doi: 10.4049/jimmunol.1002987
133. Kennedy DE, Knight KL. Inhibition of B lymphopoiesis by adipocytes and IL-1-producing myeloid-derived suppressor cells. J Immunol. (2015) 195:2666–74. doi: 10.4049/jimmunol.1500957
134. Winer DA, Winer S, Chng MH, Shen L, Engleman EG. B lymphocytes in obesity-related adipose tissue inflammation and insulin resistance. Cell Mol Life Sci. (2014) 71:1033–43. doi: 10.1007/s00018-013-1486-y
135. Nishimura S, Manabe I, Takaki S, Nagasaki M, Otsu M, Yamashita H, et al. Adipose natural regulatory B cells negatively control adipose tissue inflammation. Cell Metab. (2013) 18:759–66. doi: 10.1016/j.cmet.2013.09.017
136. Shen L, Chng MH, Alonso MN, Yuan R, Winer DA, Engleman EG. B-1a lymphocytes attenuate insulin resistance. Diabetes. (2015) 64:593–603. doi: 10.2337/db14-0554
137. Jackson-Jones LH, Duncan SM, Magalhaes MS, Campbell SM, Maizels RM, McSorley HJ, et al. Fat-associated lymphoid clusters control local IgM secretion during pleural infection and lung inflammation. Nat Commun. (2016) 7:12651. doi: 10.1038/ncomms12651
138. Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med. (2011) 17:610–7. doi: 10.1038/nm.2353
139. Tanigaki K, Sacharidou A, Peng J, Chambliss KL, Yuhanna IS, Ghosh D, et al. Hyposialylated IgG activates endothelial IgG receptor FcγRIIB to promote obesity-induced insulin resistance. J Clin Invest. (2018) 128:309–22. doi: 10.1172/JCI89333
140. Frasca D, Diaz A, Romero M, Garcia D, Jayram D, Thaller S, et al. Identification and characterization of adipose tissue-derived human antibodies with “anti-self” specificity. Front Immunol. (2020) 11:392. doi: 10.3389/fimmu.2020.00392
141. DeFuria J, Belkina AC, Jagannathan-Bogdan M, Snyder-Cappione J, Carr JD, et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc Natl Acad Sci USA. (2013) 110:5133–8. doi: 10.1073/pnas.1215840110
142. Ying W, Wollam J, Ofrecio JM, Bandyopadhyay G, El Ouarrat D, Lee YS, et al. Adipose tissue B2 cells promote insulin resistance through leukotriene LTB4/LTB4R1 signaling. J Clin Invest. (2017) 127:1019–30. doi: 10.1172/JCI90350
143. Kim DH, Do MS. BAFF knockout improves systemic inflammation via regulating adipose tissue distribution in high-fat diet-induced obesity. Exp Mol Med. (2015) 47:e129. doi: 10.1038/emm.2014.98
144. Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The generation of antibody-secreting plasma cells. Nat Rev Immunol. (2015) 15:160–71. doi: 10.1038/nri3795
145. Gommerman JL, Rojas OL, Fritz JH. Re-thinking the functions of IgA(+) plasma cells. Gut Microbes. (2014) 5:652–62. doi: 10.4161/19490976.2014.969977
146. Luck H, Khan S, Kim JH, Copeland JK, Revelo XS, Tsai S, et al. Gut-associated IgA+ immune cells regulate obesity-related insulin resistance. Nat Commun. (2019) 10:3650. doi: 10.1038/s41467-019-11370-y
147. Carter S, Miard S, Caron A, Salle-Lefort S, St-Pierre P, Anhe FF, et al. Loss of OcaB prevents age-induced fat accretion and insulin resistance by altering B-lymphocyte transition and promoting energy expenditure. Diabetes. (2018) 67:1285–96. doi: 10.2337/db17-0558
148. Bodogai M, O'Connell J, Kim K, Kim Y, Moritoh K, Chen C, et al. Commensal bacteria contribute to insulin resistance in aging by activating innate B1a cells. Sci Transl Med. (2018) 10:eaat4271. doi: 10.1126/scitranslmed.aat4271
149. Rubtsova K, Rubtsov AV, Cancro MP, Marrack P. Age-associated B cells: a T-bet-dependent effector with roles in protective and pathogenic immunity. J Immunol. (2015) 195:1933–7. doi: 10.4049/jimmunol.1501209
150. Hao Y, O'Neill P, Naradikian MS, Scholz JL, Cancro MP. A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood. (2011) 118:1294–304. doi: 10.1182/blood-2011-01-330530
151. Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, et al. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c? B-cell population is important for the development of autoimmunity. Blood. (2011) 118:1305–15. doi: 10.1182/blood-2011-01-331462
152. Ratliff M, Alter S, Frasca D, Blomberg BB, Riley RL. In senescence, age-associated B cells secrete TNFα and inhibit survival of B-cell precursors. Aging Cell. (2013) 12:303–11. doi: 10.1111/acel.12055
153. Frasca D, Diaz A, Romero M, Vazquez T, Blomberg BB. Obesity induces pro-inflammatory B cells and impairs B cell function in old mice. Mech Ageing Dev. (2017) 162:91–9. doi: 10.1016/j.mad.2017.01.004
154. Rubtsova K, Rubtsov AV, Thurman JM, Mennona JM, Kappler JW, Marrack P. B cells expressing the transcription factor T-bet drive lupus-like autoimmunity. J Clin Invest. (2017) 127:1392–404. doi: 10.1172/JCI91250
155. Manni M, Gupta S, Ricker E, Chinenov Y, Park SH, Shi M, et al. Regulation of age-associated B cells by IRF5 in systemic autoimmunity. Nat Immunol. (2018) 19:407–19. doi: 10.1038/s41590-018-0056-8
156. Khan S, Tsai S, Winer DA. Adipose tissue B cells come of age: the AABs of fat inflammation. Cell Metab. (2019) 30:997–99. doi: 10.1016/j.cmet.2019.11.007
157. Winer S, Chan Y, Paltser G, Truong D, Tsui H, Bahrami J, et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med. (2009) 15:921–9. doi: 10.1038/nm.2001
158. Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. (2009) 15:930–9. doi: 10.1038/nm.2002
159. Tiemessen MM, Jagger AL, Evans HG, van Herwijnen MJ, John S, Taams LS. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc Natl Acad Sci USA. (2007) 104:19446–51. doi: 10.1073/pnas.0706832104
160. Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, et al. PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature. (2012) 486:549–53. doi: 10.1038/nature11132
161. Kolodin D, van Panhuys N, Li C, Magnuson AM, Cipolletta D, Miller CM, et al. Antigen- and cytokine-driven accumulation of regulatory T cells in visceral adipose tissue of lean mice. Cell Metab. (2015) 21:543–57. doi: 10.1016/j.cmet.2015.03.005
162. Mahlakõiv T, Flamar AL, Johnston LK, Moriyama S, Putzel GG, Bryce PJ, et al. Stromal cells maintain immune cell homeostasis in adipose tissue via production of interleukin-33. Sci Immunol. (2019) 4:eaax0416. doi: 10.1126/sciimmunol.aax0416
163. Spallanzani RG, Zemmour D, Xiao T, Jayewickreme T, Li C, Bryce PJ, et al. Distinct immunocyte-promoting and adipocyte-generating stromal components coordinate adipose tissue immune and metabolic tenors. Sci Immunol. (2019) 4:eaaw3658. doi: 10.1126/sciimmunol.aaw3658
164. Vasanthakumar A, Moro K, Xin A, Liao Y, Gloury R, Kawamoto S, et al. The transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of adipose tissue-resident regulatory T cells. Nat Immunol. (2015) 16:276–85. doi: 10.1038/ni.3085
165. Han JM, Wu D, Denroche HC, Yao Y, Verchere CB, Levings MK. IL-33 reverses an obesity-induced deficit in visceral adipose tissue ST2+ T regulatory cells and ameliorates adipose tissue inflammation and insulin resistance. J Immunol. (2015) 194:4777–83. doi: 10.4049/jimmunol.1500020
166. Vasanthakumar A, Chisanga D, Blume J, Gloury R, Britt K, Henstridge DC, et al. Sex-specific adipose tissue imprinting of regulatory T cells. Nature. (2020) 579:581–5. doi: 10.1038/s41586-020-2040-3
167. McLaughlin T, Liu LF, Lamendola C, Shen L, Morton J, Rivas H, et al. T-cell profile in adipose tissue is associated with insulin resistance and systemic inflammation in humans. Arterioscler Thromb Vasc Biol. (2014) 34:2637–43. doi: 10.1161/ATVBAHA.114.304636
168. Wu H, Ghosh S, Perrard XD, Feng L, Garcia GE, Perrard JL, et al. T-cell accumulation and regulated on activation, normal T cell expressed and secreted upregulation in adipose tissue in obesity. Circulation. (2007) 115:1029–38. doi: 10.1161/CIRCULATIONAHA.106.638379
169. Strissel KJ, DeFuria J, Shaul ME, Bennett G, Greenberg AS, Obin MS. T-cell recruitment and Th1 polarization in adipose tissue during diet-induced obesity in C57BL/6 mice. Obesity. (2010) 18:1918–25. doi: 10.1038/oby.2010.1
170. Cho KW, Morris DL, DelProposto JL, Geletka L, Zamarron B, Martinez-Santibanez G, et al. An MHC II-dependent activation loop between adipose tissue macrophages and CD4+ T cells controls obesity-induced inflammation. Cell Rep. (2014) 9:605–17. doi: 10.1016/j.celrep.2014.09.004
171. McGillicuddy FC, Chiquoine EH, Hinkle CC, Kim RJ, Shah R, Roche HM, et al. Interferon gamma attenuates insulin signaling, lipid storage, and differentiation in human adipocytes via activation of the JAK/STAT pathway. J Biol Chem. (2009) 284:31936–44. doi: 10.1074/jbc.M109.061655
172. Pandolfi JB, Ferraro AA, Sananez I, Gancedo MC, Baz P, Billordo LA, et al. ATP-induced inflammation drives tissue-resident Th17 cells in metabolically unhealthy obesity. J Immunol. (2016) 196:3287–96. doi: 10.4049/jimmunol.1502506
173. Jagannathan-Bogdan M, McDonnell ME, Shin H, Rehman Q, Hasturk H, Apovian CM, et al. Elevated proinflammatory cytokine production by a skewed T cell compartment requires monocytes and promotes inflammation in type 2 diabetes. J Immunol. (2011) 186:1162–72. doi: 10.4049/jimmunol.1002615
174. Sumarac-Dumanovic M, Stevanovic D, Ljubic A, Jorga J, Simic M, Stamenkovic-Pejkovic D, et al. Increased activity of interleukin-23/interleukin-17 proinflammatory axis in obese women. Int J Obes. (2009) 33:151–6. doi: 10.1038/ijo.2008.216
175. Ahmed M, Gaffen SL. IL-17 in obesity and adipogenesis. Cytokine Growth Factor Rev. (2010) 21:449–53. doi: 10.1016/j.cytogfr.2010.10.005
176. Cipolletta D, Kolodin D, Benoist C, Mathis D. Tissular T(regs): a unique population of adipose-tissue-resident Foxp3+CD4+ T cells that impacts organismal metabolism. Semin Immunol. (2011) 23:431–7. doi: 10.1016/j.smim.2011.06.002
177. Luck H, Tsai S, Chung J, Clemente-Casares X, Ghazarian M, Revelo XS, et al. Regulation of obesity-related insulin resistance with gut anti-inflammatory agents. Cell Metab. (2015) 21:527–42. doi: 10.1016/j.cmet.2015.03.001
178. Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. (2009) 15:914–20. doi: 10.1038/nm.1964
179. Shuford WW, Klussman K, Tritchler DD, Loo DT, Chalupny J, Siadak AW, et al. 4-1BB costimulatory signals preferentially induce CD8+ T cell proliferation and lead to the amplification in vivo of cytotoxic T cell responses. J Exp Med. (1997) 186:47–55. doi: 10.1084/jem.186.1.47
180. Kim YJ, Kim SH, Mantel P, Kwon BS. Human 4-1BB regulates CD28 co-stimulation to promote Th1 cell responses. Eur J Immunol. (1998) 28:881–90.
181. Tu TH, Kim CS, Kang JH, Nam-Goong IS, Nam CW, Kim ES, et al. Levels of 4-1BB transcripts and soluble 4-1BB protein are elevated in the adipose tissue of human obese subjects and are associated with inflammatory and metabolic parameters. Int J Obes. (2014) 38:1075–82. doi: 10.1038/ijo.2013.222
182. Kim CS, Kim JG, Lee BJ, Choi MS, Choi HS, Kawada T, et al. Deficiency for costimulatory receptor 4-1BB protects against obesity-induced inflammation and metabolic disorders. Diabetes. (2011) 60:3159–68. doi: 10.2337/db10-1805
183. Tsai S, Clemente-Casares X, Zhou AC, Lei H, Ahn JJ, Chan YT, et al. Insulin receptor-mediated stimulation boosts t cell immunity during inflammation and infection. Cell Metab. (2018) 28:922–34.e4. doi: 10.1016/j.cmet.2018.08.003
184. Yang H, Youm YH, Vandanmagsar B, Ravussin A, Gimble JM, Greenway F, et al. Obesity increases the production of proinflammatory mediators from adipose tissue T cells and compromises TCR repertoire diversity: implications for systemic inflammation and insulin resistance. J Immunol. (2010) 185:1836–45. doi: 10.4049/jimmunol.1000021
185. Tsai S, Clemente-Casares X, Revelo XS, Winer S, Winer DA. Are obesity-related insulin resistance and type 2 diabetes autoimmune diseases? Diabetes. (2015) 64:1886–97. doi: 10.2337/db14-1488
186. Ahnstedt H, Roy-O'Reilly M, Spychala MS, Mobley AS, Bravo-Alegria J, Chauhan A, et al. Sex differences in adipose tissue CD8+ T cells and regulatory T cells in middle-aged mice. Front Immunol. (2018) 9:659. doi: 10.3389/fimmu.2018.00659
187. Wencker M, Turchinovich G, Di Marco Barros R, Deban L, Jandke A, Cope A, et al. Innate-like T cells straddle innate and adaptive immunity by altering antigen-receptor responsiveness. Nat Immunol. (2014) 15:80–7. doi: 10.1038/ni.2773
188. Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol. (2007) 25:297–336. doi: 10.1146/annurev.immunol.25.022106.141711
189. Dhodapkar MV, Kumar V. Type II NKT cells and their emerging role in health and disease. J Immunol. (2017) 198:1015–21. doi: 10.4049/jimmunol.1601399
190. Lynch L, Nowak M, Varghese B, Clark J, Hogan AE, Toxavidis V. Adipose tissue invariant NKT cells protect against diet-induced obesity and metabolic disorder through regulatory cytokine production. Immunity. (2012) 37:574–87. doi: 10.1016/j.immuni.2012.06.016
191. Huh JY, Park J, Kim JI, Park YJ, Lee YK, Kim JB. Deletion of CD1d in adipocytes aggravates adipose tissue inflammation and insulin resistance in obesity. Diabetes. (2017) 66:835–47. doi: 10.2337/db16-1122
192. Ji Y, Sun S, Xu A, Bhargava P, Yang L, Lam KSL. Activation of natural killer T cells promotes M2 Macrophage polarization in adipose tissue and improves systemic glucose tolerance via interleukin-4 (IL-4)/STAT6 protein signaling axis in obesity. J Biol Chem. (2012) 287:13561–71. doi: 10.1074/jbc.M112.350066
193. Lynch L, Michelet X, Zhang S, Brennan PJ, Moseman A, Lester C, et al. Regulatory iNKT cells lack expression of the transcription factor PLZF and control the homeostasis of T(reg) cells and macrophages in adipose tissue. Nat Immunol. (2015) 16:85–95. doi: 10.1038/ni.3047
194. Satoh M, Iwabuchi K. Role of natural killer T cells in the development of obesity and insulin resistance: insights from recent progress. Front Immunol. (2018) 9:1314. doi: 10.3389/fimmu.2018.01314
195. Bonneville M, O'Brien RL, Born WK. Gammadelta T cell effector functions: a blend of innate programming and acquired plasticity. Nat Rev Immunol. (2010) 10:467–78. doi: 10.1038/nri2781
196. Kohlgruber AC, Gal-Oz ST, LaMarche NM, Shimazaki M, Duquette D, Koay HF. γδ T cells producing interleukin-17A regulate adipose regulatory T cell homeostasis and thermogenesis. Nat Immunol. (2018) 19:464–74. doi: 10.1038/s41590-018-0094-2
197. Le Bourhis L, Guerri L, Dusseaux M, Martin E, Soudais C, Lantz O. Mucosal-associated invariant T cells: unconventional development and function. Trends Immunol. (2011) 32:212–8. doi: 10.1016/j.it.2011.02.005
198. Chiba A, Murayama G, Miyake S. Mucosal-associated invariant T cells in autoimmune diseases. Front Immunol. (2018) 9:1333. doi: 10.3389/fimmu.2018.01333
199. Dusseaux M, Martin E, Serriari N, Péguillet I, Premel V, Louis D, et al. Human MAIT cells are xenobiotic-resistant, tissue-targeted, CD161hi IL-17-secreting T cells. Blood. (2011) 117:1250–9. doi: 10.1182/blood-2010-08-303339
200. Miyazaki Y, Miyake S, Chiba A, Lantz O, Yamamura T. Mucosal-associated invariant T cells regulate Th1 response in multiple sclerosis. Int Immunol. (2011) 23:529–35. doi: 10.1093/intimm/dxr047
201. Le Bourhis L, Martin E, Péguillet I, Guihot A, Froux N, Coré M, et al. Antimicrobial activity of mucosal-associated invariant T cells. Nat Immunol. (2010) 11:701–8. doi: 10.1038/ni.1890
202. Rahimpour A, Koay HF, Enders A, Clanchy R, Eckle SB, Meehan B, et al. Identification of phenotypically and functionally heterogeneous mouse mucosal-associated invariant T cells using MR1 tetramers. J Exp Med. (2015) 212:1095–108. doi: 10.1084/jem.20142110
203. Schipper HS, Rakhshandehroo M, van de Graaf SF, Venken K, Koppen A, Stienstra R, et al. Natural killer T cells in adipose tissue prevent insulin resistance. J Clin Invest. (2012) 122:3343–54. doi: 10.1172/JCI62739
204. Lynch L, O'Shea D, Winter DC, Geoghegan J, Doherty DG, O'Farrelly C. Invariant NKT cells and CD1d(+) cells amass in human omentum and are depleted in patients with cancer and obesity. Eur J Immunol. (2009) 39:1893–901. doi: 10.1002/eji.200939349
205. Subramanian S, Turner MS, Ding Y, Goodspeed L, Wang S, Buckner JH, et al. Increased levels of invariant natural killer T lymphocytes worsen metabolic abnormalities and atherosclerosis in obese mice. J Lipid Res. (2013) 54:2831–41. doi: 10.1194/jlr.M041020
206. Hams E, Locksley RM, McKenzie AN, Fallon PG. Cutting edge: IL-25 elicits innate lymphoid type 2 and type II NKT cells that regulate obesity in mice. J Immunol. (2013) 191:5349–53. doi: 10.4049/jimmunol.1301176
207. Chandra S, Zhao M, Budelsky A, de Mingo Pulido A, Day J, Fu Z, et al. A new mouse strain for the analysis of invariant NKT cell function. Nat Immunol. (2015) 16:799–800. doi: 10.1038/ni.3203
208. Magalhaes I, Pingris K, Poitou C, Bessoles S, Venteclef N, Kiaf B, et al. Mucosal-associated invariant T cell alterations in obese and type 2 diabetic patients. J Clin Invest. (2015) 125:1752–62. doi: 10.1172/JCI78941
209. Magalhaes I, Kiaf B, Lehuen A. iNKT and MAIT cell alterations in diabetes. Front Immunol. (2015) 6:341. doi: 10.3389/fimmu.2015.00341
210. Carolan E, Tobin LM, Mangan BA, Corrigan M, Gaoatswe G, Byrne G, et al. Altered distribution and increased IL-17 production by mucosal-associated invariant T cells in adult and childhood obesity. J Immunol. (2015) 194:5775–80. doi: 10.4049/jimmunol.1402945
211. Faunce DE, Palmer JL, Paskowicz KK, Witte PL, Kovacs EJ. CD1d-restricted NKT cells contribute to the age-associated decline of T cell immunity. J Immunol. (2005) 175:3102–9. doi: 10.4049/jimmunol.175.5.3102
212. Novak J, Dobrovolny J, Novakova L, Kozak T. The decrease in number and change in phenotype of mucosal-associated invariant T cells in the elderly and differences in men and women of reproductive age. Scand J Immunol. (2014) 80:271–5. doi: 10.1111/sji.12193
213. Winer DA, Luck H, Tsai S, Winer S. The intestinal immune system in obesity and insulin resistance. Cell Metab. (2016) 23:413–26. doi: 10.1016/j.cmet.2016.01.003
214. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. (2006) 444:1027–31. doi: 10.1038/nature05414
215. Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, et al. consortium, richness of human gut microbiome correlates with metabolic markers. Nature. (2013) 500:541–6. doi: 10.1038/nature12506
216. Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. (2013) 341:1241214. doi: 10.1126/science.1241214
217. Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. (2007) 56:1761–72. doi: 10.2337/db06-1491
218. Ghoshal S, Witta J, Zhong J, de Villiers W, Eckhardt E. Chylomicrons promote intestinal absorption of lipopolysaccharides. J Lipid Res. (2009) 50:90–7. doi: 10.1194/jlr.M800156-JLR200
219. Thevaranjan N, Puchta A, Schulz C, Naidoo A, Szamosi JC, Verschoor CP, et al. Age-associated microbial dysbiosis promotes intestinal permeability, systemic inflammation, and macrophage dysfunction. Cell Host Microbe. (2017) 21:455–66.e4. doi: 10.1016/j.chom.2017.03.002
220. Fransen F, van Beek AA, Borghuis T, Aidy SE, Hugenholtz F, van der Gaast-de Jongh C, et al. Aged gut microbiota contributes to systemical inflammaging after transfer to germ-free mice. Front Immunol. (2017) 8:1385. doi: 10.3389/fimmu.2017.01385
221. Li H, Qi Y, Jasper H. Preventing age-related decline of gut compartmentalization limits microbiota dysbiosis and extends lifespan. Cell Host Microbe. (2016) 19:240–53. doi: 10.1016/j.chom.2016.01.008
222. Lasry A, Ben-Neriah Y. Senescence-associated inflammatory responses: aging and cancer perspectives. Trends Immunol. (2015) 36:217–28. doi: 10.1016/j.it.2015.02.009
223. Rodier F, Coppé JP, Patil CK, Hoeijmakers WA, Muñoz DP, Raza SR, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. (2009) 11:973–9. doi: 10.1038/ncb1909
224. Palmer AK, Xu M, Zhu Y, Pirtskhalava T, Weivoda MM, Hachfeld CM, et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell. (2019) 18:e12950. doi: 10.1111/acel.12950
225. Shirakawa K, Yan X, Shinmura K, Endo J, Kataoka M, Katsumata Y, et al. Obesity accelerates T cell senescence in murine visceral adipose tissue. J Clin Invest. (2016) 126:4626–39. doi: 10.1172/JCI88606
226. Saucillo DC, Gerriets VA, Sheng J, Rathmell JC, Maciver NJ. Leptin metabolically licenses T cells for activation to link nutrition and immunity. J Immunol. (2014) 192:136–44. doi: 10.4049/jimmunol.1301158
227. Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature. (1998) 394:897–901. doi: 10.1038/29795
228. Santos-Alvarez J, Goberna R, Sánchez-Margalet V. Human leptin stimulates proliferation and activation of human circulating monocytes. Cell Immunol. (1999) 194:6–11. doi: 10.1006/cimm.1999.1490
Keywords: metabolism, obesity, aging, diabetes, insulin resistance, immunology, immunometabolism, visceral adipose tissue
Citation: Khan S, Chan YT, Revelo XS and Winer DA (2020) The Immune Landscape of Visceral Adipose Tissue During Obesity and Aging. Front. Endocrinol. 11:267. doi: 10.3389/fendo.2020.00267
Received: 16 January 2020; Accepted: 14 April 2020;
Published: 15 May 2020.
Edited by:
Bruno Guigas, Leiden University, NetherlandsReviewed by:
Michèle Guerre-Millo, INSERM UMRS1269 Nutrition et obésités (nutriomique), FranceAnne Bouloumie, INSERM U1048 Institut des Maladies Métaboliques et Cardiovasculaires, France
Arion Kennedy, North Carolina State University, United States
Copyright © 2020 Khan, Chan, Revelo and Winer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xavier S. Revelo, eHJldmVsb0B1bW4uZWR1; Daniel A. Winer, ZHdpbmVyQGJ1Y2tpbnN0aXR1dGUub3Jn
†These authors share first authorship
‡These authors share senior authorship