 Anna E. Harris1
Anna E. Harris1 Veronika M. Metzler1Jennifer Lothion-Roy1Dhruvika Varun1Corinne L. Woodcock1Daisy B. Haigh1Chantelle Endeley1Maria Haque1Michael S. Toss1Mansour Alsaleem1,2Jenny L. Persson3,4
Veronika M. Metzler1Jennifer Lothion-Roy1Dhruvika Varun1Corinne L. Woodcock1Daisy B. Haigh1Chantelle Endeley1Maria Haque1Michael S. Toss1Mansour Alsaleem1,2Jenny L. Persson3,4 Lorraine J. Gudas5Emad Rakha1
Lorraine J. Gudas5Emad Rakha1 Brian D. Robinson6
Brian D. Robinson6 Francesca Khani6Laura M. Martin7
Francesca Khani6Laura M. Martin7 Jenna E. Moyer7Juliette Brownlie1Srinivasan Madhusudan1
Jenna E. Moyer7Juliette Brownlie1Srinivasan Madhusudan1 Cinzia Allegrucci1
Cinzia Allegrucci1 Victoria H. James1
Victoria H. James1 Catrin S. Rutland1
Catrin S. Rutland1 Rupert G. Fray8
Rupert G. Fray8 Atara Ntekim9*
Atara Ntekim9* Simone de Brot10
Simone de Brot10 Nigel P. Mongan1,5*
Nigel P. Mongan1,5* Jennie N. Jeyapalan1*
Jennie N. Jeyapalan1*- 1University of Nottingham Biodiscovery Institute, University of Nottingham, University Park, Nottingham, United Kingdom
- 2Department of Applied Medical Science, Applied College, Qassim University, Qassim, Saudi Arabia
- 3Department of Molecular Biology, Umeå University, Umeå, Sweden
- 4Department of Biomedical Sciences, Malmö Universitet, Malmö, Sweden
- 5Department of Pharmacology, Weill Cornell Medicine, New York, NY, United States
- 6Department of Urology, Weill Cornell Medicine, New York, NY, United States
- 7Englander Institute for Precision Medicine, Weill Cornell Medicine, New York, NY, United States
- 8School of Biosciences, University of Nottingham, Nottingham, United Kingdom
- 9Department of Oncology, University Hospital Ibadan, Ibadan, Nigeria
- 10Comparative Pathology Platform (COMPATH), Institute of Animal Pathology, University of Bern, Bern, Switzerland
Androgen deprivation therapies (ADTs) are important treatments which inhibit androgen-induced prostate cancer (PCa) progression by either preventing androgen biosynthesis (e.g. abiraterone) or by antagonizing androgen receptor (AR) function (e.g. bicalutamide, enzalutamide, darolutamide). A major limitation of current ADTs is they often remain effective for limited durations after which patients commonly progress to a lethal and incurable form of PCa, called castration-resistant prostate cancer (CRPC) where the AR continues to orchestrate pro-oncogenic signalling. Indeed, the increasing numbers of ADT-related treatment-emergent neuroendocrine-like prostate cancers (NePC), which lack AR and are thus insensitive to ADT, represents a major therapeutic challenge. There is therefore an urgent need to better understand the mechanisms of AR action in hormone dependent disease and the progression to CRPC, to enable the development of new approaches to prevent, reverse or delay ADT-resistance. Interestingly the AR regulates distinct transcriptional networks in hormone dependent and CRPC, and this appears to be related to the aberrant function of key AR-epigenetic coregulator enzymes including the lysine demethylase 1 (LSD1/KDM1A). In this review we summarize the current best status of anti-androgen clinical trials, the potential for novel combination therapies and we explore recent advances in the development of novel epigenetic targeted therapies that may be relevant to prevent or reverse disease progression in patients with advanced CRPC.
Introduction
Prostate cancer (PCa) is the most commonly diagnosed cancer in individuals with a prostate, with an estimated 268,490 new cases and 34,500 deaths predicted to occur in the USA alone over 2022 (1). Acinar adenocarcinoma is the most common type of all prostate cancers and accounts for around 95% of cases, whereas ductal adenocarcinoma accounts for only 0.4-0.8% (2). The epithelium of the ducts and acini is mainly composed of luminal and basal cells, but also intermediate, neuroendocrine and stem cells (Figure 1) (3). Primary PCa predominantly has a luminal cell-like phenotype with atypical glands, enhanced androgen signalling and absence of a continuous basal cell layer (4, 5). Luminal cells are terminally differentiated secretory cells of the prostate gland that are responsible for the exocrine activity of the prostate. They express high levels of Androgen receptor (AR) and secrete proteins including the prostate specific antigen (PSA) and prostate acid phosphatase (PacP) into the lumen of the gland (3). Basal cells, which express very little or no AR, are in direct contact with luminal cells via gap junctions and form a barrier between the luminal cells and the stroma and, like luminal cells, basal cells can give rise to cancers (4, 6). Prostate stem cells make up ~0.1-3% of the epithelial cell population and are believed to reside within “niches” near the basement membrane. The third most common cell type in the prostate are neuroendocrine cells which have neuronal characteristics (7–9). However, only about ~0.5-2% of prostate cancer patients have neuroendocrine prostate cancer (NePC) at initial diagnosis and NePC emergence often occurs after patients have received androgen deprivation therapies (ADTs), consequently NePC accounts for ~25% of all metastatic prostate cancers (10–12). NePC accounts for ~25% of all metastatic prostate cancers, is highly proliferative, aggressive and can lack AR expression, thus represents a major therapeutic challenge (12, 13). This review will discuss the androgen receptor signalling pathway, current treatments targeting androgen production and AR antagonists, moving forwards towards emerging treatment options for advanced castration resistant PCa (CRPC) and will consider how novel approaches to suppress androgen signalling may reduce or delay development of treatment-emergent NE-like PCa.
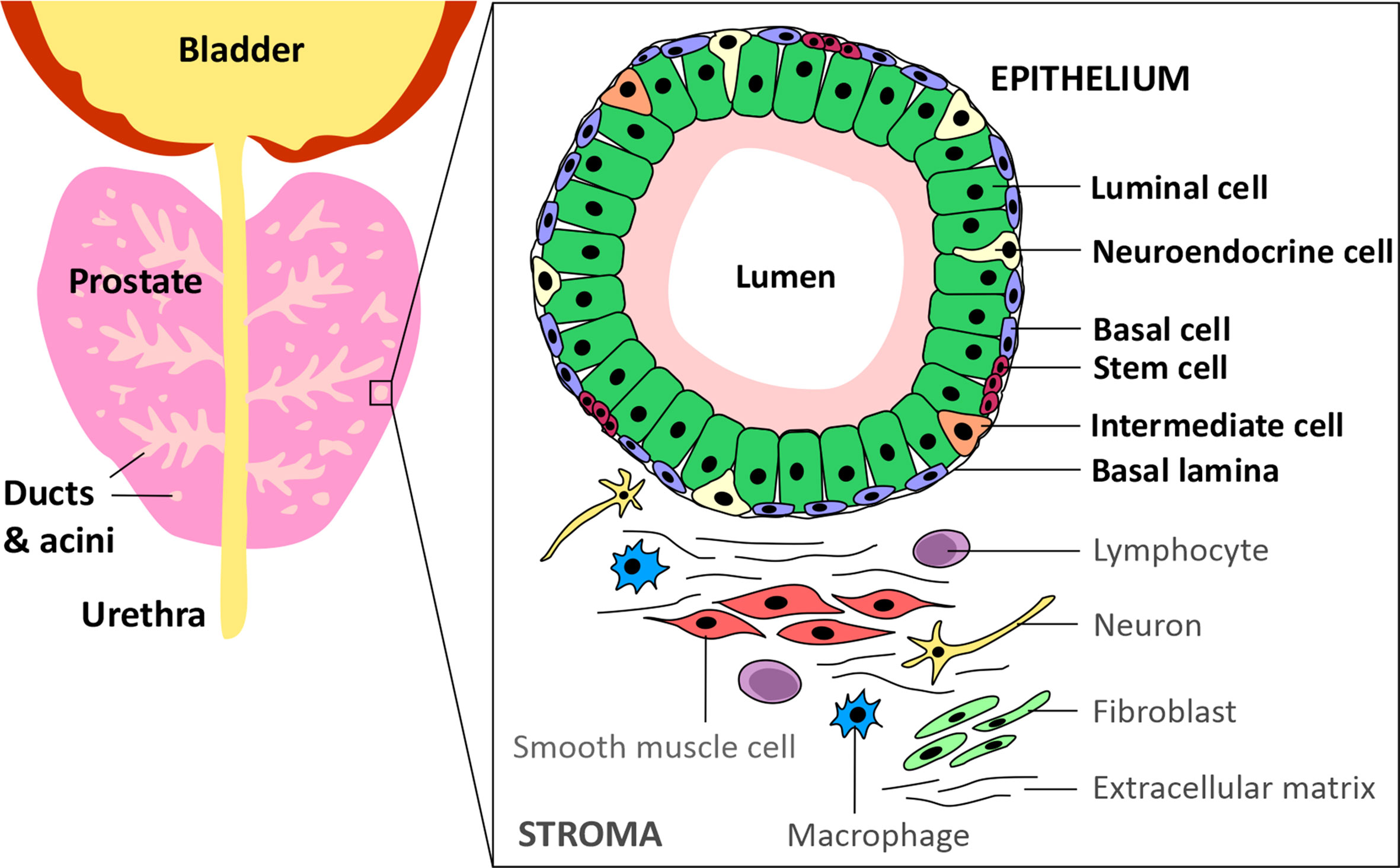
Figure 1 Schematic of the prostate epithelium and stroma. The prostate glandular structure is made up of ducts and acini. The epithelium that surrounds the lumen contains luminal, intermediate, basal, neuroendocrine and stem cells. The stroma contains extracellular matrix, immune cells, fibroblasts, neurons and smooth muscle cells.
Mechanisms of androgen receptor signaling
The androgen receptor (AR) is the primary mediator of androgen action. The main physiological androgens, testosterone and it more potent derivative, dihydrotestosterone (5-α-DHT), have long been recognized to play an important role in the initiation and progression of PCa (14). The transcriptional effects of androgens are mediated by the androgen receptor (AR), a member of the ligand dependent superfamily of transcription factors (15). The AR gene is located on chromosome X and spans ~186,587 base pairs (bp). The protein coding region is ~2757 nucleotides long and the protein is comprised of ~920 amino acids and possess polymorphic glutamine and glycine tracts (NM_000044.3, Figure 2). The 110 kDa AR protein is encoded by eight exons and contains four domains (i) the N-terminal domain (NTD), (ii) the DNA-binding domain (DBD), (iii) the hinge region and (iv) the ligand binding domain (LBD) (16). Within the COOH-terminus of the DBD and hinge region there is a so-called nuclear localization signal (NLS) which is responsible for the transport of the AR into the nucleus (17) (Figure 2). Agonist binding to the AR induces homodimerization, conformational changes and nuclear translocation (Figure 3). The AR homodimer usually is a “head-to-head” formation (18) and preferentially binds to the androgen response element (ARE) on the DNA (19, 20). The AR harbors two activation functions, the constitutively active activation function-1 (AF-1) is located in the NTD, whereas the ligand-dependent activation function-2 (AF-2) is situated in the LBD (16, 17). AR coregulator proteins bind to the hydrophobic pocket of AF-2 through their LxxLL (L = leucine, x = any amino acid) motif. This motif is highly conserved between nuclear receptor interacting proteins such as the steroid receptor coactivator-1 (SRC-1) and was shown to be necessary and sufficient to mediate coregulator binding to the LBD of the nuclear receptor (21). He et al. found a second motif, namely FXXLF, which is also present in the NTD of the AR (FQNLF) (22). This motif is therefore important for both interactions within AR itself and AR interaction with coregulator proteins. Coregulator proteins often share the same binding motif, however, the way the binding pocket of AR can bind coregulator proteins specifically is by electrostatic interactions with positively or negatively charged amino acids flanking the motif (23). The transcriptional activity of AR requires the recruitment of pioneer factors such as FoxA1 (24) and epigenetic co-regulator proteins (Figure 3), (25–30).
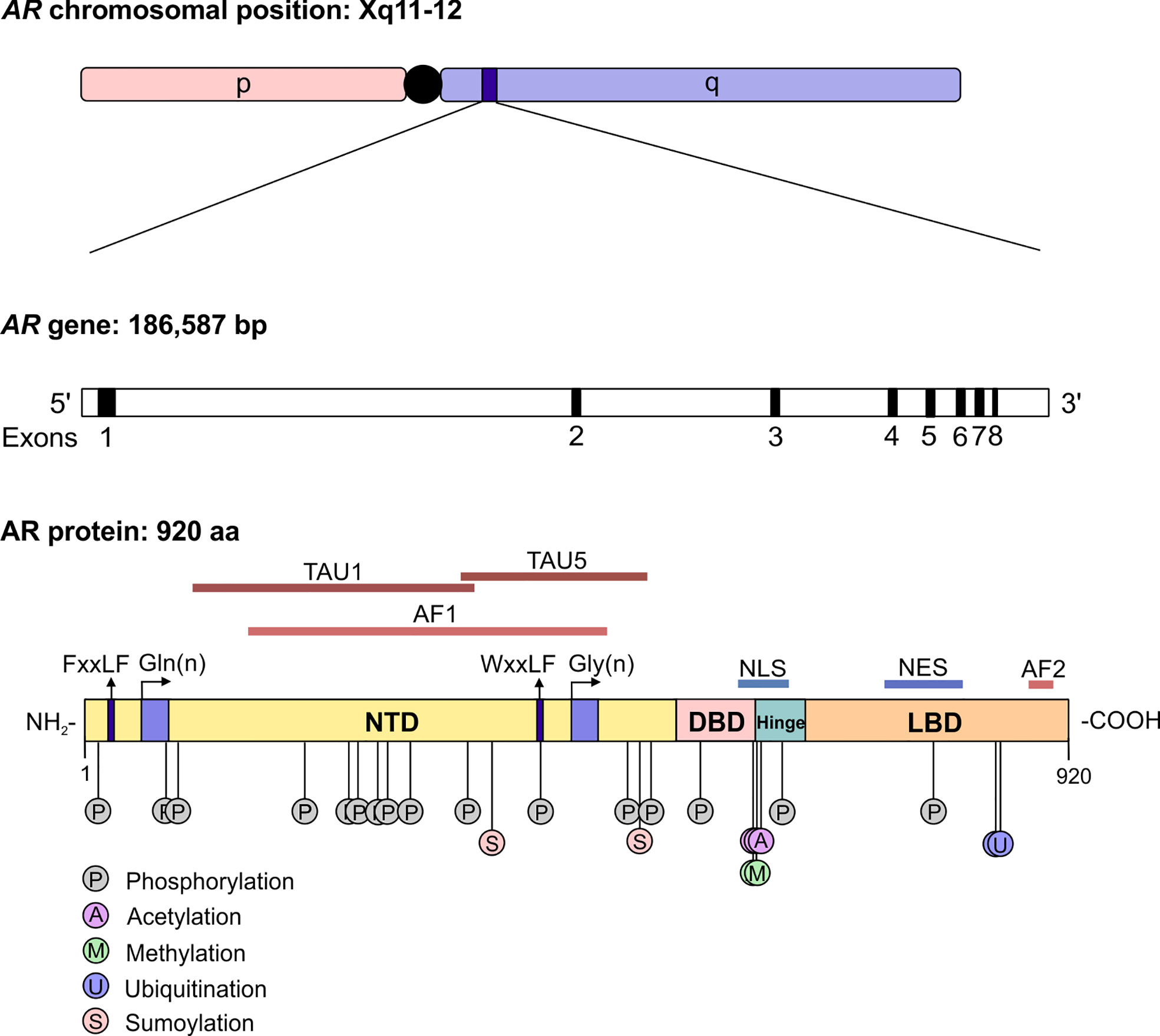
Figure 2 The structure of the androgen receptor gene and protein. The AR gene is situated on position q11-12 of chromosome X and contains 8 exons. The protein reference sequence NM_000044.3 is comprised of 920 amino acids and is composed of different domains which are depicted. In addition, posttranslational modifications known to influence AR function are shown. AR, androgen receptor; bp, base pair; NTD, N-terminal domain; DBD, DNA binding domain; LBD, ligand binding domain; AF, activation function; TAU, transcription activation unit; NLS, nuclear localisation signal; NES, nuclear export signal.
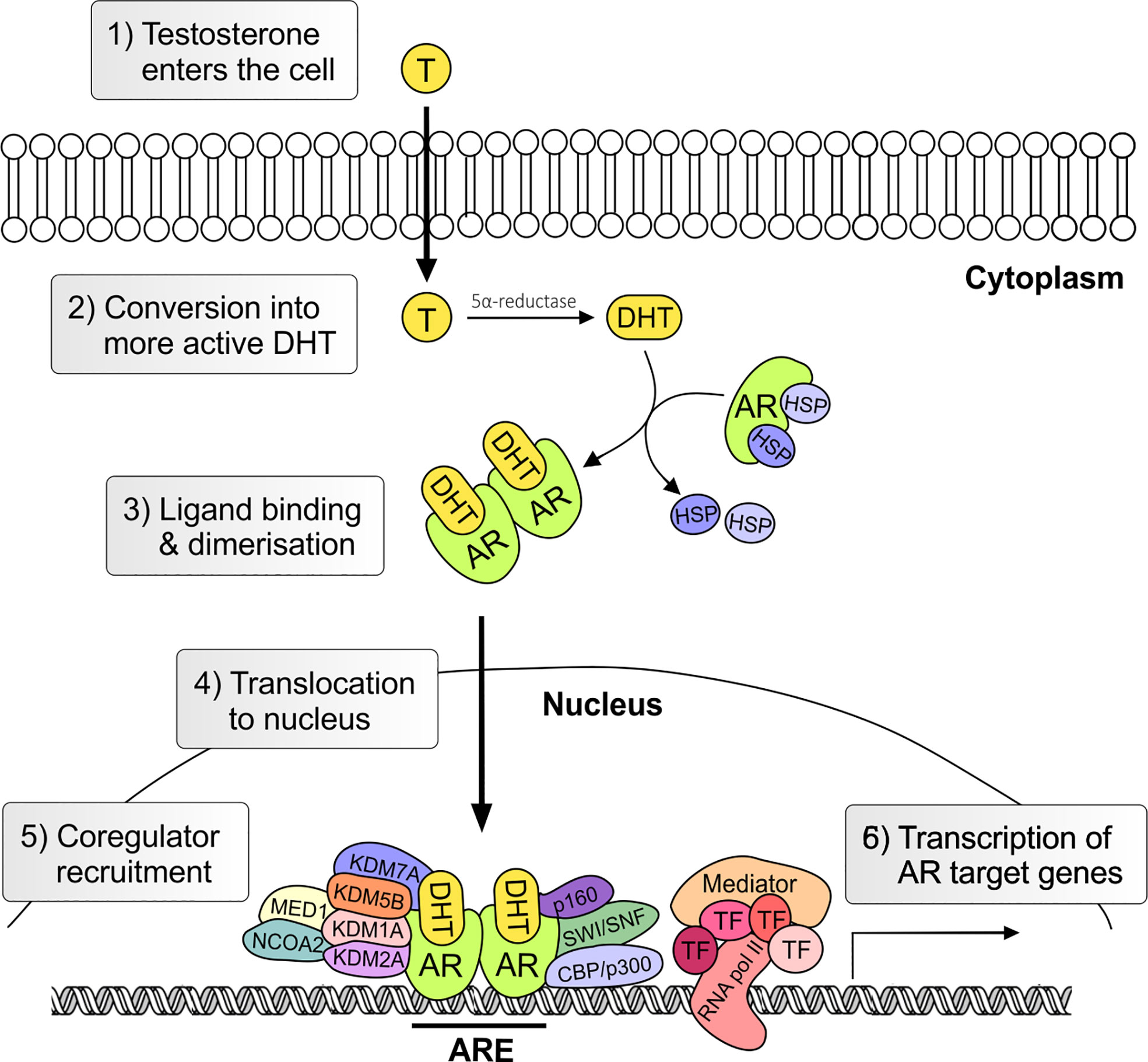
Figure 3 Genomic androgen receptor signaling pathway. Androgens, such as testosterone and dihydrotestosterone, enter the cell and are converted in the more active metabolite (5α-DHT) by the steroid-5α-reductase. Upon ligand binding heat stress protein (HSP) chaperones are released and AR undergoes conformational change and dimerization. In the nucleus the AR together with co-regulators activates the transcription of androgen regulated genes. T, testosterone; AR, androgen receptor; DHT, 5α-dihydrotestosterone; HSP, heat shock; TF, transcription factor; ARE, Androgen Response Element.
Androgen deprivation therapies
It has long been recognized that PCa initiation and progression is androgen-dependent and for this reason depriving PCa cells of androgen and androgen receptor function through inhibition of androgen biosynthesis or AR function will act to suppress tumorigenesis. Luteinizing hormone releasing hormone (LHRH) (also referred to as Gonadotropin Hormone-Releasing Hormone (GHRH)) agonists are androgen synthesis blockers which suppress androgen production by acting on the hypothalamus–pituitary–gonadal axis, whereby sustained stimulation of the LHRH receptor results in the downregulation of testosterone after an initial surge (31). LHRH antagonists bind directly to the GnRH receptor so act faster and without causing a testosterone surge and may be protective against neurodegenerative and cardiovascular disease compared to agonists (32–34). However, in LHRH agonists (e.g. Goserelin) and antagonist (e.g. Degarelix) treatments, androgen precursors released from the adrenal glands remain unaffected and can be metabolized into 5α-DHT, an AR agonist (35). Abiraterone irreversibly inhibits the Cytochrome P450 17 α-hydroxysteroid dehydrogenase (CYP17A) enzyme which is responsible for the conversion of pregnenolone to 17-OH Pregnenolone and a lyase activity to further convert 17-OH Pregnenolone into dehydroepiandrosterone (36). These are precursor molecules required for androgen biosynthesis in both testes and adrenal glands (36). Androgen synthesis blockers are often combined with androgen receptor antagonists, which compete with physiological androgens for AR binding sites. Flutamide was one of the first anti-androgens and has been shown to have positive effects on therapy response and PCa patient survival when combined with surgical or chemical castration (37). Bicalutamide is thought to have a more favorable tolerability profile relative to Flutamide (38). Later studies with advanced prostate cancer patients have shown that Bicalutamide combined with an LHRH agonist resulted in improved PSA levels and overall survival compared to LHRH agonist treatment alone (39, 40). The newer generation AR antagonists, Enzalutamide, Apalutamide, and Darolutamide, have a greater affinity for AR than Flutamide and Bicalutamide, and additionally block nuclear translocation of the AR, coactivator recruitment and DNA binding (41–43) (Figure 4). These newer generation of AR antagonists are discussed later in the review.
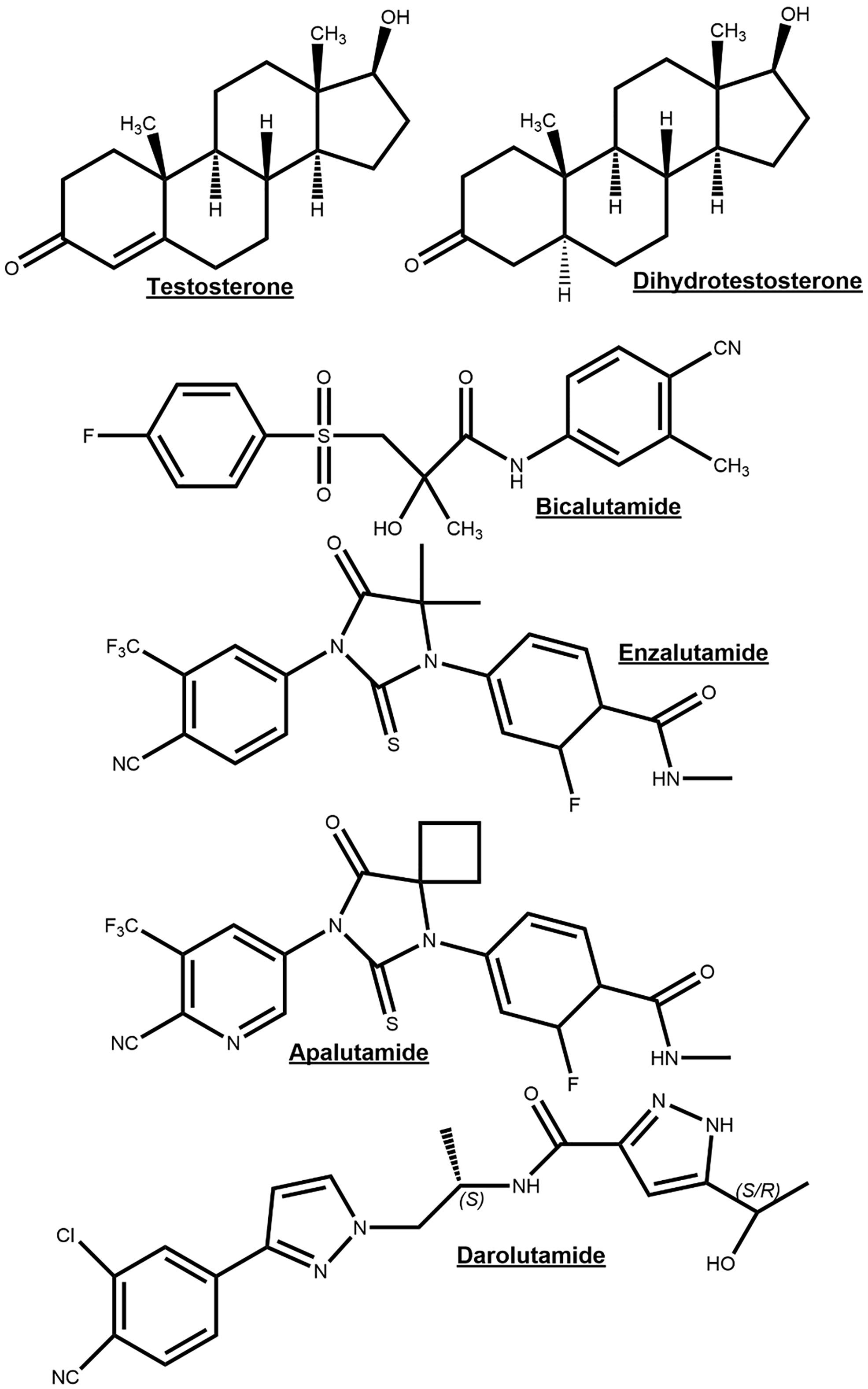
Figure 4 Chemical structures of the endogenous androgen receptor (AR) agonists testosterone and dihydrotestosterone for comparison to AR-antagonists bicalutamide, enzalutamide, apalutamide and darolutamide.
Current and emerging therapies for CRPC
Despite recent success in developing more specific and effective ADTs, current ADTs are still only effective for ~24 months after which patients commonly progress to a lethal and incurable form of PCa, termed castration-resistant prostate cancer (CRPC) (44). Previously CRPC has also been referred to as hormone-refractory or androgen-independent PCa, but it has become clear that most CRPC cases remain influenced by androgen receptor signaling (45, 46). The mechanisms behind persistent AR signaling with concomitant ADT-resistance are the topic of many recent reviews and include AR overexpression, hypersensitivy to low androgen, increased androgen levels, AR gene mutations, drug antagonist-to-agonist switching, ligand promiscuity, bypassing the AR signaling pathway, AR splice variants, AR genomic structural rearrangements (AR-GSRs), and alterations in transcription factors and AR co-regulators (44, 47, 48). These mechanisms are thought to be the result of selective pressure of ADTs and provide the tumor with a selective advantage in the absence of androgen (49).
Several phase I and II studies demonstrated reductions in PSA (with varying success across cohorts) with abiraterone acetate treatment, often in conjunction with prednisone and subsequently is a standard of care treatment for metastatic castration resistant prostate cancer (mCRPC) (NCT00473512, NCT02217566, NCT01309672). These results also confirm that CRPC commonly remains hormone driven (50, 51). Additionally, clinical trials have concluded that the addition of abiraterone acetate and prednisone to conventional ADT significantly increased overall survival and radiographic progression-free survival in men with locally advanced and metastatic hormone naïve prostate cancer (HNPC) [NCT01715285, NCT00268476 (51, 52)]. Many trials remain ongoing including a Phase I trial to determine the feasibility of using PSA response and testosterone level to guide the treatment with ADT and/or abiraterone plus prednisone (NCT03511196), which if successful could present a more personalized approach and a minimalization of side effects. Abiraterone is currently used as a standard treatment for patients with metastatic CRPC (53). A screen for higher affinity, more affective non-steroidal antiandrogens identified Enzalutamide as the most promising candidate molecule (43). It is currently approved for the treatment of advanced CRPC after having demonstrated in clinical trials its effectiveness after resistance to traditional ADT has occurred. Several trials have shown a significant reduction in risk of progression or death compared to first generation antiandrogens (NCT02446405, NCT01664923) (54, 55). It is currently in phase II/III trials for both localized and metastatic hormone sensitive PCa as a component of first line ADT (NCT02446405, NCT02677896, NCT02446444, NCT03860987, NCT03809000) (54, 56). The combination of abiraterone with prednisone and enzalutamide is also in phase 2 trials for preoperative neoadjuvant treatment (NCT03860987). Another recent phase II trial supported the use of abiraterone with prednisone as a first-line therapy followed by enzalutamide as a second-line drug for better clinical outcomes for mCRPC as abiraterone was found to retain no second-line activity following enzalutamide [NCT02125357, (57)]. This may represent a method of prolonging sensitivity to hormonal therapy. Even though there are promising outcomes, Enzalutamide has been shown to promote NePC trans-differentiation (58).
Following Enzalutamide, another second-generation anti-androgen, Apalutamide, was developed. The addition of Apalutamide to conventional ADT was found to significantly improve patient outcome in non-metastatic CRPC (nmCRPC; NCT01946204) (59). In addition, initial results from the TITAN trial (NCT02489318) showed the addition of apalutamide to ADT significantly improved the primary endpoints of overall survival and radiographic progression-free survival in patients with metastatic castration-sensitive PCa (60, 61). Apalutamide is US FDA approved for use in both nmCRPC and metastatic castration sensitive PCa. Ongoing trials featuring apalutamide include for neoadjuvant ADT prior to radical prostatectomy for patients diagnosed with localized high-risk prostate cancer (NCT03767244) and as part of a combination therapy for patients with rising PSA after radiation therapy and ADT (NCT03777982) or radical prostatectomy (NCT03141671).
Another promising drug is darolutamide (ODM-201) which is a structurally distinct androgen receptor antagonist, which holds a significant advantage over other anti-androgens, as it shows negligible brain penetrance, does not increase serum testosterone levels in mice, reduces the risk of seizures and was found to inhibit AR variants (62). A recent study in men with nmCRPC (ARAMIS trial, phase III, NCT02200614) showed a significant improvement of metastasis-free survival in patients who received darolutamide (n = 955) compared to patients who received placebo (n = 554) (63). Darolutamide was approved in July 2019 by the US FDA for treatment of nmCRPC in combination with a GnRH analog or in patients who have had a bilateral orchiectomy. Currently it is in phase 3 trials for treatment in combination with an LHRH agonist/antagonist or orchiectomy with radiation therapy for localised prostate cancer at very high risk of recurrence (NCT04136353) and with docetaxel in patients with metastatic hormone sensitive prostate cancer (NCT02799602) (64). It is also now in a phase 2 trial as a monotherapy for hormone naïve PCa with limited metastases (NCT02972060).
Therapies such as Abiraterone and androgen receptor antagonists (Enzalutamide, Apalutamide, and Darolutamide) have been used to treat CRPC but are not curative (65). Efforts to target the AR NTD have generally been unsuccessful. For this reason there is an urgent need for new therapies targeting AR variants in CRPC, including potentially blocking AR variant expression (66). Another potential avenue for future therapeutics is targeted degradation of AR. PROTAC (Proteolysis targeting chimera) is a novel small molecule technology that targets a specific protein of interest for ubiquitination by E3 ubiquitin ligases and subsequently degradation. Several potent PROTAC degraders of AR have been developed including ARV-110 which is currently in phase 1/2 clinical trial for mCRPC (NCT03888612).
Current combination therapies
Despite the challenges of resistance, there are benefits of hormonal therapies and there are many on-going clinical trials to identify optimal combinations of hormonal therapies with chemotherapy, radiotherapy, and immunotherapy.
Chemotherapy, commonly docetaxel or cabazitaxel, is often given only in late stages of prostate cancer disease to prolong the life of patients. Recently it has been shown Cabazitaxel plus prednisone use as a third line treatment after disease progression with docetaxel and abiraterone or enzalutamide, is significantly more effective than the hormonal therapy (NCT02485691) (67).
Radiotherapy is both given after neoadjuvant treatment to treat localized prostate cancers and at late-stage prostate cancer to target metastatic sites. The Phase III RADICALS trial (NCT00541047) found that precautionary radiotherapy after surgery, resulted in no significant difference in recurrence after 5 years compared with observation (68). This provides a strong case that radiotherapy should only be given if the cancer is found to have progressed. Stereotactic body radiation therapy (SBRT), also known as stereotactic ablative radiotherapy (SABR), is a method of delivering very high doses of radiation precisely to the tumor. It is emerging from phase 2 trials (NCT02680587 ORIOLE) as a treatment to target oligometastases that has low-toxicity and significantly increases progression free survival compared to observational arm (69). It has the potential to be an effective treatment that could delay ADT and thus maintain patient quality of life. Other trials remain ongoing (NCT04115007).
Another potential new therapeutic approach, recently reviewed by Loizzo et al., is targeting autophagy to sensitize prostate cancer to other drugs (70). Autophagy has a pro-tumorigenic role by promoting survival through enabling prostate cancer cells to meet metabolic demand and avoid apoptosis. Inhibitors of autophagy have been investigated in clinical trials, including hydroxychloroquine, which was shown in a phase 2 trial to have some reducing effect on PSA progression in localized PCa with PSA progression after local treatment (NCT00726596) (71).
Theranostics (or Theragnostics) is a novel therapeutic strategy in the move towards precision medicine to co-deliver therapeutic and imaging functions within a single agent. This includes delivering cytotoxic levels of radiation which are targeted at a cellular level to specific biomarkers. Prostate-specific membrane antigen (PSMA) presents a potential target for this approach, [reviewed by (72)]. It has shown promising results over Cabazitaxel in mCRPC in phase II trials (73). However, as not all PCa express PSMA, this method is not a universal treatment (74).
Immunotherapies and DNA repair targeted therapies: Precision medicine in practice?
The identification of clinically relevant biomarkers and/or genotypes is critical for effective personalized treatment approaches to enable and inform prioritizing patients for immuno-and targeted therapies. While there is great interest in the development of immunotherapies for cancer, PCa is considered to have an immunosuppressive microenvironment, with few infiltrative lymphocytes. Recently the use of high dose-rate brachytherapy was able to alter this with increase in immune infiltrates (75). There are currently two FDA approved immunotherapies. Sipuleucel-T is a cell-based immunotherapy approach in which a patient’s own antigen presenting cells (APCs) are harvested and activated against prostatic acid phosphatase (PAP), a highly expressed antigen in most prostate cancer cells, then infused back into the patient. It was approved for the treatment of mCRPC following a series of successful phase III trials (76). Secondly, Pembrolizumab is a monoclonal antibody that targets the anti-programmed cell death protein 1 (PD-1) receptor has shown efficacy in several cancer types (NCT02578680, NCT02256436, NCT02362594) (77). A recent study identified a subset of super responders’ patients, of approximately 1 in 20 men with mCRPC that had an increase in life expectancy of two years (NCT02787005) (78). Several phase 2 and 3 trials featuring Pembrolizumab are ongoing including a Phase 2 combining ADT, SBRT and pembrolizumab with or without intra-tumoral SD-101, a synthetic CpG oligonucleotide that enhances immune response, in patients with newly diagnosed hormone-naive oligometastatic prostate cancer (NCT03007732). Immunotherapies are generally well tolerated and there are many ongoing trials for the treatment of mPCa and localised PCa alongside radiotherapy. Identification of biomarkers which identify individuals likely to benefit from immunotherapies will enable more personalized use of such treatments.
Recent advances in biomarker discovery have enabled the more precise use of targeted therapies in PCa. A new class of anticancer drugs called PARP inhibitors (PARPi) have been developed and trialled successfully on several types of cancers including PCa. Poly (ADP-ribose) polymerases 1 and 2 (PARP1 and 2) play a crucial role in DNA damage repair (DDR) by sensing DNA damage, binding to the damaged DNA and recruiting DNA repair proteins to the damage site, through a process called PARylation (79). PARP inhibitors bind to the catalytic site of PARP1 and trap the enzyme at the location of DNA damage. This blocks the replication fork and eventually causes double strand breaks (DSB) in the DNA. Since the catalytic site is also responsible for PARylation, blocking of this site inhibits the recruitment of DNA repair factors, inhibiting PARP-mediated DDR (80).
A clinical trial in 2015 (NCT01682772) tested the efficacy of olaparib, a PARP inhibitor, in treating mCRPC and found that treatment with olaparib prolonged radiographic progression–free survival (rPFS) and reduced circulating tumor-cell counts. Importantly, a better response was observed in patients with tumors that had aberrations in DNA repair genes (81). An alternative PARP inhibitor, veliparib, was tested for its efficacy with abiraterone acetate and prednisone therapy (AAP) in a phase 2 clinical trial (NCT01576172) (82). Although no significant benefits were observed with veliparib in addition to AAP in mCRPC, the study reported better treatment outcomes with AAP in tumors where DNA repair gene defects were found on metastatic tumor tissue sequencing as compared to tumours without these defects (82).
Indeed, the success of PARP inhibition as a treatment strategy has been linked to the genetic context of the tumor and more specifically to the functionality of certain genes involved in DDR. PARP inhibition in breast and ovarian cancers with BRCA1/2 mutation represents the first clinical application of synthetic lethality; a term used to describe cell death following the genetic or pharmacological inhibition of a cellular pathway in the context of a distinct loss of function mutation in a separate pathway (where neither the primary mutation nor the treatment alone would be lethal to the cell) (83). BRCA 1 and 2 are involved in the resolution of DSBs via the homologous recombination repair (HRR) pathway. Where homologous repair of DSBs cannot be carried out, cells are forced to use more error-prone non-homologous end joining (NHEJ) which results in chromosomal instability and increased sensitivity to PARPi (83). BRCA germline mutations are present in a subset of patients with PCa and identifying these patients would allow the use of PARP inhibitors (84). Additionally, BRCA2 mutation has been identified in 2.5-5.3% of aggressive prostate cancers (85, 86) and the standardized incidence ratio of prostate cancer in males known to carry a pathogenic BRCA2 mutation was found to be 4.45 (87). Overall, mutations in DDR have been identified in 11-30% of prostate cancers and represent possible targets for synthetically lethal treatment approaches (86, 88, 89).
The profound trial (NCT02987543) aimed to compare olaparib against enzalutamide or abiraterone for the treatment of mCRPC with HRR gene alterations (79). It found that olaparib improved rPFS and objective response rate in comparison to treatment with enzalutamide or abiraterone, although concluded that further studies are required to better define the genetic mutations that will sensitize a cancer to olaparib. For patients with PCa and at least one alteration in BRCA1, BRCA2 or ATM (a serine threonine kinase involved in the processing of DSBs and in HRR) (90, 91), overall survival was significantly longer for patients that received olaparib (79). However, analysis from the TRITON2 study of PARP inhibition with rucaparib found that of the 49 patients with mCRPC identified to have ATM mutation, only 4.1% showed a significant decline in PSA following treatment (92). Neeb et al. identified ATM loss in 11% of a cohort of 631 patients with advanced prostate cancer and found that ATM knockout in a human prostate carcinoma cell line was associated with genetic instability. HRR was found to be reduced but not absent in these cells and response to rucaparib was inconstant between clones of the same cell line. Chemical inhibition of ATR (a kinase involved in DNA repair via cell cycle checkpoint control) was found to inhibit HRR in the ATM knockout cells and although cytotoxic effect was modest in the context of ATM knockout alone, addition of PARP inhibition significantly increased cell death. The efficacy of inhibiting both PARP and ATR was then confirmed in a xenographic model of a patient derived mCRPC cell line with loss of ATM (93). This study identifies the possible need for the HRR pathway to be completely inhibited in order for olaparib treatment to be most successful. ATM inhibition represents another potential application of precision medicine and a phase 2 clinical trial of combined treatment with olaparib alongside ATR inhibition in mCRPC is currently ongoing (NCT03787680). Other possible DDR genes of interest in PCa have been identified and include PALB2, FANCA, BRIP1 and RAD51 (92).
So far, olaparib has been approved by the FDA for use in mCRPC with identified mutation in HRR and rucaparib has been approved for mCRPC with BRCA mutation. Ongoing trials of PARP inhibition in prostate cancer and completed trials awaiting results are included in Table 1. Current evidence from both pre-clinical and clinical trials indicates that tumours with DNA repair gene deficiency often respond better to PARP inhibitors and other DDR-targeted therapies, due to possible synthetically lethal interactions. This paves a new direction for the use of precision medicine in treating PCa.
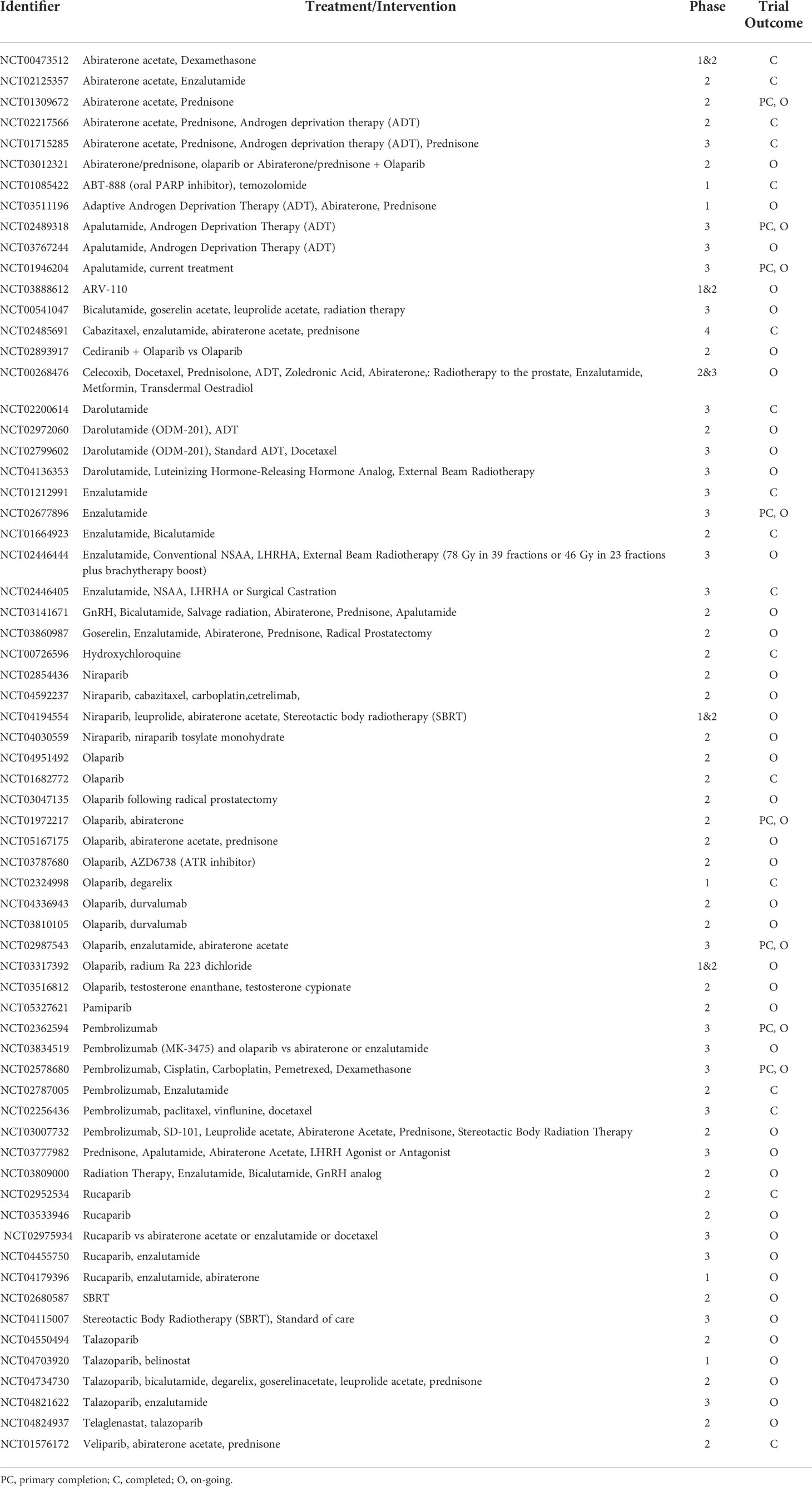
Table 1 Ongoing androgen deprivation and targeted therapies and clinical trials in prostate cancer– ordered by drug name.
Emerging epigenetic therapies for advanced prostate cancer
In recent years the focus on understanding cancer progression has led to several multicenter studies being able to stratify prostate cancer. This stratification has also highlighted the crucial role of epigenetic factors in cancer progression (94–100). Here we introduce the epigenetic factors that have been well- studied in prostate cancer and their potential therapeutic role as future treatment options.
As noted earlier, the AR depends on multiple enzymatic distinct epigenetic coregulators, many of which are independently implicated in PCa. For this reason the focus is now shifting to the chromatin modifying coregulator proteins such as lysine demethylases (KDMs) that are key cofactors for cancer drivers such as AR (26, 27, 101–104). For these reasons, the recent entry of KDM1A pharmaco-inhibitors into cancer clinical trials herald a new step in the translation of innovative epigenetic therapies to the clinic (105), even though some of the trials were terminated due to no beneficial effects in the cancer types treated. Some KDM1A inhibitors being investigated in clinical trials to treat cancers are: ORY-1001, ORY-2001, GSK-2879552, IMG7289, INCB059872 and CC-90011 (chemical structure shown in Figure 5) (106). Other histone demethylases like KDM3A, KDM4B/KDM4C, KDM6A/KDM6B have also been associated with regulating the transcriptional activity of AR (107–110). Moreover, inhibitors simultaneously targeting KDM4 and KDM1 family enzymes have been developed and shown to be effective in inducing apoptosis of PCa and colon cancer cell lines (111). Thus, these KDMs form interesting epigenetic therapeutic targets in PCa.
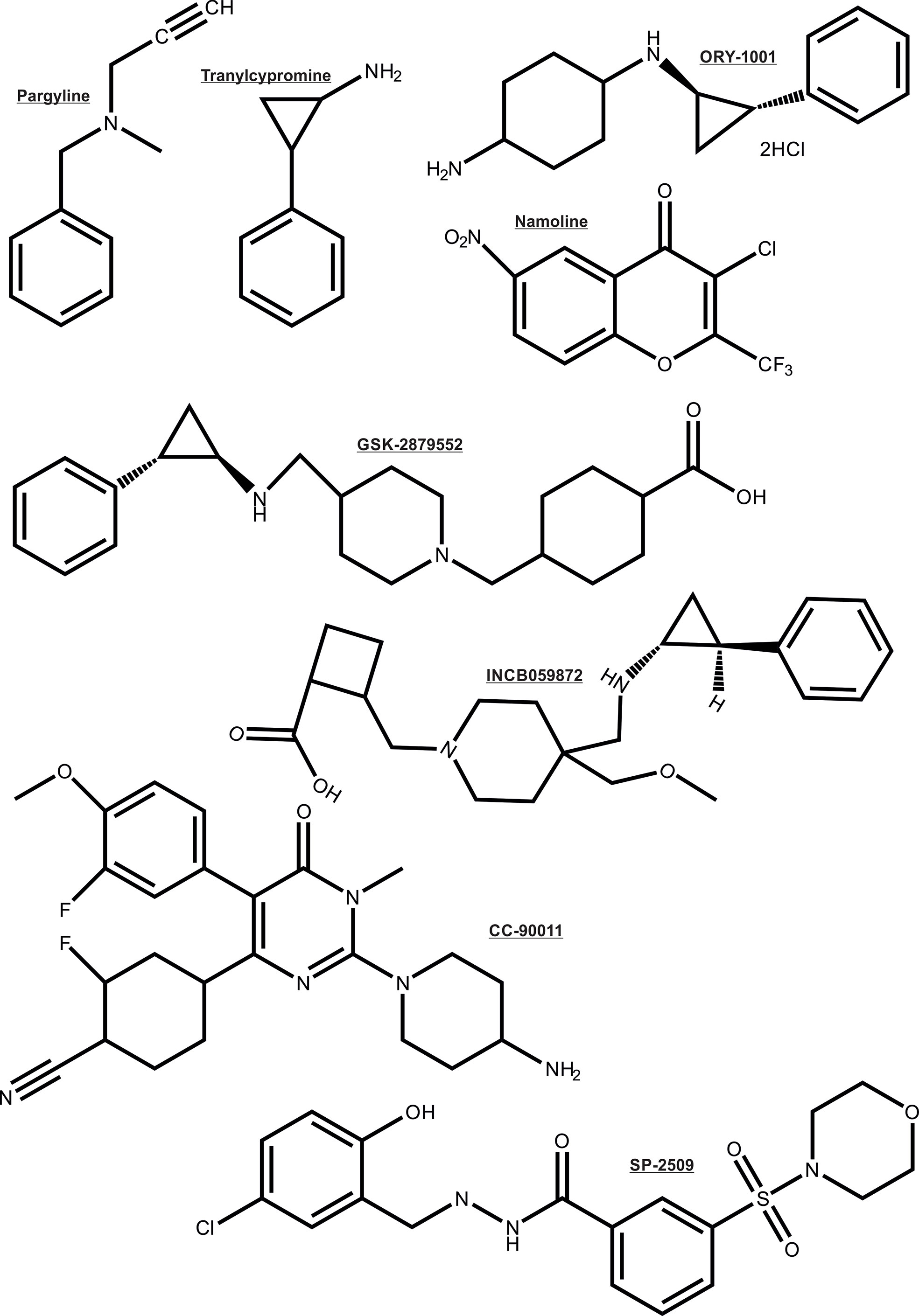
Figure 5 Chemical structure of the KDM1A inhibitors.
Another important epigenetic factor in PCa is BRD4, a member of BET (bromodomain and extra terminal domain) subfamily. It binds acetylated histone lysine residues and has been shown to be important for recruiting RNA polymerase II to facilitate transcription. In CRPC, BRD4 interacts with AR and inhibition of BRD4 prevents AR from binding to its target genes, disrupting AR transcriptional activity [nicely reviewed by Pawar et al.; (112)]. Thus, BRD4 is being investigated as a potential therapeutic target in PCa. JQ1 and AZD5153 are BET inhibitors that are effective at selectively inhibiting BRD4 and have been shown to reduce proliferation and survival of PCa cells (113). It has been shown that mutations in SPOP gene in PCa often impart resistance to BET inhibitors (114). However, a molecule called PROTAC-BETd (ZBC260) has been shown to re-sensitize the resistant cells to BET inhibitors. Hence, it has been suggested that ZBC260 and BET inhibitor combination therapy may yield better treatment outcomes in cancers (115). BRD4 works together with E2F1 transcription factor, the activation of which induces lineage plasticity in treatment-emergent neuroendocrine PCa (t-NEPC). BET inhibitors have been reported to block this E2F1/BRD4 dependent mechanism and reduce tumor growth in t-NEPC (116). BRD4 also plays a role in transcriptional regulation of KDM5C, which is often overexpressed in CRPC. Knockdown of KDM5C tends to sensitize CRPC cells to BET inhibitors, suggesting an interplay between these proteins and uncovering a new pathway that could be targeted to develop treatments for CRPC (117). Recently, JQ1 and a KDM1A inhibitor, SP-2509, were tested together in CRPC and showed a synergistic effect at inhibiting growth of AR positive CRPC cells. Although, the sensitivity to these inhibitors was lost on AR knockdown (118).
EZH2 (enhancer of zeste homolog 2) is a methyltransferase that typically functions as part of the polycomb repressor complex 2 (PRC2). However, recently it was shown that it also exhibits a separate activation function when it binds to the promoter region of AR and plays a role in AR signaling. It also acts as a co-activator of AR in CRPC [reviewed by Jones et al. (110)]. Recently, a study found that inhibition of EZH2 enhanced enzalutamide activity and could potential overcome enzalutamide resistance in CRPC. This was thought to be due to inhibition of PSA transcription by EZH2 directly binding to PSA promoter region (119). Furthermore, EZH2 and EED (embryonic ectoderm development) can modulate AR expression and AR signaling pathways. A drug called astemizole disturbs EZH2-EED interaction and inhibits both, EZH2 and AR expression. Astemizole has been reported to promote autophagy in PCa cells and reduce tumor growth in castration-resistant mouse xenograft model with implanted VCaP cells (120). It has also been recognised that EZH2 is a major player in the evolution of NEPC-CRPC (121). A study recently showed how understanding the ECM components within the tumor environment can be utilized in organoid models and identified ECM-dependent EZH2 inhibition was able to sensitize NEPC to treatment (122). This has also brought to the forefront the regulation of EZH2 in PCa. The methylation and subsequent degradation of EZH2 by the methyltransferase SETD2, was recently shown to block metastasis in PCa, further suggesting that EZH2 inhibition in PCa could be of therapeutic advantage (123).
These studies provide hope for the development of combined epigenetic therapies in the future that effectively target two or more epigenetic factors significant in PCa.
Conclusion
In conclusion, therapeutic strategies targeting the AR function remain the cornerstone for treating men with advanced PCa. However, the emergence of castrate resistant PCa and indeed treatment emergent neuroendocrine-like PCa, highlights the need for the development of newer more targeted pharmacological approaches to suppress pro-proliferative androgen signaling. With the emerging role of DNA repair and epigenetic factors in mCRPC and NEPC, identifying and repurposing drugs for these new factors in PCa could lead to better precision treatment options for individuals with advanced PCa.
The development of epigenetic targeting therapies represents a novel approach to suppress androgen signaling and could be an additional line of therapy after resistance has occurred to other approaches. Indeed, a precision medicine approach could also be applied to identify and treat individuals that would respond to immunotherapies, including methods of enhancing immune response and immune infiltration for effective immunotherapy. The numerous ongoing clinical trials will continue to identify the most effective combinations of hormonal therapies with chemotherapy, radiotherapy, immunotherapy and other novel therapies to improve both survival and quality of life of prostate cancer patients.
Author contributions
AH, AN, and NM contributed to conception and design of the study. AH, VM, JL-R, DV, NM, and JJ review preparation. AH, VM, DV, NM, CR, AN, SB, and JJ wrote first draft. AH, DV, CW, CE, MH, MT, MA, JB, SM, AN, SB, and JJ wrote sections of the review. JP, LG, ER, BR, FK, LM, JM, JB, SM, CA, VJ, CR, RF, AN, SB and NM critical revision of review. All authors contributed to the article and approved the submitted version.
Funding
We gratefully acknowledge the financial support of the University of Nottingham, the BBSRC; Doctoral training program (BB/M008770/1: AEH, VMM, DBH, CLW, JL-R, CSR, NPM) and (BB/T008369/1: DV, CE, MH) and Prostate Cancer UK RIA15-ST2-005 (NPM) and Prostate Cancer Foundation & John Black Charitable Foundation (20CHAL06: NPM).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistic. CA Cancer J Clin (2022) 72:7–33. doi: 10.3322/caac.21708
2. Orihuela E, Green JM. Ductal prostate cancer: Contemporary management and outcomes. Urologic Oncol: Semin And Orig Investigations (2008) 26(4):368–71. doi: 10.1016/j.urolonc.2007.05.028
3. Shen MM, Abate-Shen C. Molecular genetics of prostate cancer: New prospects for old challenges. Genes Dev (2010) 24:1967–2000. doi: 10.1101/gad.1965810
4. Wang ZA, Toivanen R, Bergren SK, Chambon P, Shen MM. Luminal cells are favored as the cell of origin for prostate cancer. Cell Rep (2014) 8:1339–46. doi: 10.1016/j.celrep.2014.08.002
5. Zhang D, Zhao S, Li X, Kirk JS, Tang DG. Prostate luminal progenitor cells in development and cancer. Trends In Canc (2018) 4(11):769–83. doi: 10.1016/j.trecan.2018.09.003
6. El-Alfy M, Pelletier G, Hermo LS, Labrie F. Unique features of the basal cells of human prostate epithelium. Microscopy Res And Technique (2000) 51:436–46. doi: 10.1002/1097-0029(20001201)51:5<436::AID-JEMT6>3.0.CO;2-T
7. Abrahamsson PA. Neuroendocrine differentiation in prostatic carcinoma. Prostate (1999) 39:135–48. doi: 10.1002/(SICI)1097-0045(19990501)39:2<135::AID-PROS9>3.0.CO;2-S
8. Aprikian AG, Cardon-Cardo C, Fair WR, Reuter VE. Characterization of neuroendocrine differentiation in human benign prostate and prostatic adenocarcinoma. Cancer (1993) 71:3952–65. doi: 10.1002/1097-0142(19930615)71:12<3952::AID-CNCR2820711226>3.0.CO;2-X
9. Bonkhoff H, Remberger K. Differentiation pathways and histogenetic aspects of normal and abnormal prostatic growth: A stem cell model. Prostate (1996) 28:98–106. doi: 10.1002/(SICI)1097-0045(199602)28:2<98::AID-PROS4>3.0.CO;2-J
10. Humphrey PA. Histological variants of prostatic carcinoma and their significance. Histopathology (2012) 60:59–74. doi: 10.1111/j.1365-2559.2011.04039.x
11. Mucci NR, Akdas G, Manely S, Rubin MA. Neuroendocrine expression in metastaticprostate cancer: Evaluation of high throughput tissue microarrays to detect heterogeneous protein expression. Hum Pathol (2000) 31:406–14. doi: 10.1053/hp.2000.7295
12. Aggarwal R, Huang J, Alumkal JJ, Zhang L, Feng FY, Thomas GV, et al. Clinical and genomic characterization of treatment-emergent small-cell neuroendocrine prostate cancer: A multi-institutional prospective study. J Clin Oncol (2018) 36:2492–503. doi: 10.1200/JCO.2017.77.6880
13. Beltran H, Tomlins S, Aparicio A, Arora V, Rickman D, Ayala G, et al. Aggressive variants of castration-resistant prostate cancer. Clin Cancer Res (2014) 20:2846–50. doi: 10.1158/1078-0432.CCR-13-3309
14. Huggins C, Hodges CV. Studies on prostatic cancer - I the effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res (1941) 1:293–7. doi: 10.3322/canjclin.22.4.232
15. Whirledge S, Cidlowski JA. Steroid hormone action. In: Yen and jaffe's reproductive endocrinology. (Philadelphia, PA: Elsevier) (2019).
16. Simental J, Sar M, Lane M, French F, Wilson E. Transcriptional activation and nuclear targeting signals of the human androgen receptor. J Of Biol Chem (1991) 266:510–8. doi: 10.1016/S0021-9258(18)52466-2
17. Zhou Z, Sar M, Simental JA, Lane MV, Wilson EM. A ligand-dependent bipartite nuclear targeting signal in the human androgen receptor. Requirement for the dna-binding domain and modulation by Nh2-terminal and carboxyl-terminal sequences. J Of Biol Chem (1994) 269:13115–23. doi: 10.1016/S0021-9258(17)36806-0
18. Nadal M, Prekovic S, Gallastegui N, Helsen C, Abella M, Zielinska K, et al. Structure of the homodimeric androgen receptor ligand-binding domain. Nat Commun (2017) 8:14388. doi: 10.1038/ncomms14388
19. Bennett NC, Gardiner RA, Hooper JD, Johnson DW, Gobe GC. Molecular cell biology of androgen receptor signalling. Int J Of Biochem Cell Biol (2010) 42:813–27. doi: 10.1016/j.biocel.2009.11.013
20. Nelson PS, Clegg N, Arnold H, Ferguson C, Bonham M, White J, et al. The program of androgen-responsive genes in neoplastic prostate epithelium. Proc Of Natl Acad Of Sci (2002) 99:11890–5. doi: 10.1073/pnas.182376299
21. Heery DM, Kalkhoven E, Hoare S, Parker MG. A signature motif in transcriptional Co-activators mediates binding to nuclear receptors. Nature (1997) 387:733–6. doi: 10.1038/42750
22. He B, Minges JT, Lee LW, Wilson EM. The fxxlf motif mediates androgen receptor-specific interactions with coregulators. J Biol Chem (2002) 277:10226–35. doi: 10.1074/jbc.M111975200
23. Darimont BD, Wagner RL, Apriletti JW, Stallcup MR, Kushner PJ, Baxter JD, et al. Structure and specificity of nuclear receptor–coactivator interactions. Genes Dev (1998) 12:3343–56. doi: 10.1101/gad.12.21.3343
24. Jin HJ, Zhao JC, Wu L, Kim J, Yu J. Cooperativity and equilibrium with Foxa1 define the androgen receptor transcriptional program. Nat Commun (2014) 5:3972. doi: 10.1038/ncomms4972
25. Bevan CL, Hoare S, Claessens F, Heery DM, Parker MG. The Af1 and Af2 domains of the androgen receptor interact with distinct regions of Src1. Mol Cell Biol (1999) 19:8383–92. doi: 10.1128/MCB.19.12.8383
26. Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH, et al. Lsd1 demethylates repressive histone marks to promote androgen-Receptor-Dependent transcription. Nature (2005) 437:436–9. doi: 10.1038/nature04020
27. Wissmann M, Yin N, Muller JM, Greschik H, Fodor BD, Jenuwein T, et al. Cooperative demethylation by Jmjd2c and Lsd1 promotes androgen receptor-dependent gene expression. Nat Cell Biol (2007) 9:347–53. doi: 10.1038/ncb1546
28. Clark EL, Coulson A, Dalgliesh C, Rajan P, Nicol SM, Fleming S, et al. The rna helicase P68 is a novel androgen receptor coactivator involved in splicing and is overexpressed in prostate cancer. Cancer Res (2008) 68:7938–46. doi: 10.1158/0008-5472.CAN-08-0932
29. Zhou XE, Suino-Powell KM, Li J, He Y, Mackeigan JP, Melcher K, et al. Identification of Src3/Aib1 as a preferred coactivator for hormone-activated androgen receptor. J Biol Chem (2010) 285:9161–71. doi: 10.1074/jbc.M109.085779
30. Jin F, Claessens F, Fondell JD. Regulation of androgen receptor-dependent transcription by coactivator Med1 is mediated through a newly discovered noncanonical binding motif. J Biol Chem (2012) 287:858–70. doi: 10.1074/jbc.M111.304519
31. Crawford ED, Hou AH. The role of lhrh antagonists in the treatment of prostate cancer. Prostate (2009) 23(7):626–30.
32. Cook T, Sheridan WP. Development of gnrh antagonists for prostate cancer: New approaches to treatment. Oncol (2000) 5:162–8. doi: 10.1634/theoncologist.5-2-162
33. Sciarra A, Busetto GM, Salciccia S, Del Giudice F, Maggi M, Crocetto F, et al. Does exist a differential impact of degarelix versus lhrh agonists on cardiovascular safety? evidences from randomized and real-world studies. Front Endocrinol (Lausanne) (2021) 12:695170. doi: 10.3389/fendo.2021.695170
34. Branigan GL, Torrandell-Haro G, Soto M, Gelmann EP, Vitali F, Rodgers KE, et al. Androgen-targeting therapeutics mitigate the adverse effect of gnrh agonist on the risk of neurodegenerative disease in men treated for prostate cancer. Cancer Med (2022) 11:2687–98. doi: 10.1002/cam4.4650
35. Labrie F, Belanger A, Dupont A, Simard J, Labrie C. Science behind total androgen blockade: From gene to combination therapy. Clin And Invest Med Med Clinique Et Experimentale (1993) 16:475–92.
36. Rowlands MG, Barrie SE, Chan F, Houghton J, Jarman M, Mccague R, et al. Esters of 3-pyridylacetic acid that combine potent inhibition of 17. alpha.-Hydroxylase/C17, 20-lyase (Cytochrome P45017. alpha.) with resistance to esterase hydrolysis. J Of Medi Chem (1995) 38:4191–7. doi: 10.1021/jm00021a008
37. Labrie F, Dupont A, Belanger A, Giguere M, Lacoursiere Y, Emond J, et al. Combination therapy with flutamide and castration (Lhrh agonist or orchiectomy) in advanced prostate cancer: A marked improvement in response and survival. J Of Steroid Biochem (1985) 23:833–41. doi: 10.1016/S0022-4731(85)80024-8
38. Gillatt D. Antiandrogen treatments in locally advanced prostate cancer: Are they all the same? J Of Cancer Res And Clin Oncol (2006) 132:17–26. doi: 10.1007/s00432-006-0133-5
39. Akaza H, Yamaguchi A, Matsuda T, Igawa M, Kumon H, Soeda A, et al. Superior anti-tumor efficacy of bicalutamide 80 mg in combination with a luteinizing hormone-releasing hormone (Lhrh) agonist versus lhrh agonist monotherapy as first-line treatment for advanced prostate cancer: Interim results of a randomized study in Japanese patients. Japanese J Of Clin Oncol (2004) 34:20–8. doi: 10.1093/jjco/hyh001
40. Akaza H, Hinotsu S, Usami M, Arai Y, Kanetake H, Naito S, et al. Combined androgen blockade with bicalutamide for advanced prostate cancer: Long-term follow-up of a phase 3, double-blind, randomized study for survival. Canc: Interdiscip Int J Of Am Cancer Soc (2009) 115:3437–45. doi: 10.1002/cncr.24395
41. Rathkopf D, Scher HI. Androgen receptor antagonists in castration-resistant prostate cancer. Cancer J (Sudbury Mass.) (2013) 19:43. doi: 10.1097/PPO.0b013e318282635a
42. Semenas J, Dizeyi N, Persson JL. Enzalutamide as a second generation antiandrogen for treatment of advanced prostate cancer. Drug Design Dev And Ther (2013) 7:875. doi: 10.2147/dddt.s45703
43. Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science (2009) 324:787–90. doi: 10.1126/science.1168175
44. Chandrasekar T, Yang JC, Gao AC, Evans CP. Mechanisms of resistance in castration-resistant prostate cancer (Crpc). Trans Andrology And Urol (2015) 4:365. doi: 10.3978/j.issn.2223-4683.2015.05.02
45. Coutinho I, Day TK, Tilley WD, Selth LA. Androgen receptor signaling in castration-resistant prostate cancer: A lesson in persistence. Endocrine-Related Cancer (2016) 23:T179–97. doi: 10.1530/ERC-16-0422
46. Sharma NL, Massie CE, Ramos-Montoya A, Zecchini V, Scott HE, Lamb AD, et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell (2013) 23:35–47. doi: 10.1016/j.ccr.2012.11.010
47. Seruga B, Ocana A, Tannock IF. Drug resistance in metastatic castration-resistant prostate cancer. Nat Rev Clin Oncol (2011) 8:12. doi: 10.1038/nrclinonc.2010.136
48. Ferraldeschi R, Welti J, Luo J, Attard G, De Bono J. Targeting the androgen receptor pathway in castration-resistant prostate cancer: Progresses and prospects. Oncogene (2015) 34(14):1745–57. doi: 10.1038/onc.2014.115
49. Kumari S, Senapati D, Heemers HV. Rationale for the development of alternative forms of androgen deprivation therapy. Endocrine-Related Cancer (2017) 24:R275–95. doi: 10.1530/ERC-17-0121
50. Ryan CJ, Smith MR, De Bono JS, Molina A, Logothetis CJ, De Souza P, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. New Engl J Of Med (2013) 368:138–48. doi: 10.1056/NEJMoa1209096
51. Fizazi K, Tran N, Fein L, Matsubara N, Rodriguez-Antolin A, Alekseev BY, et al. Abiraterone plus prednisone in metastatic, castration-sensitive prostate cancer. New Engl J Of Med (2017) 377:352–60. doi: 10.1056/NEJMoa1704174
52. James ND, De Bono JS, Spears MR, Clarke NW, Mason MD, Dearnaley DP, et al. Abiraterone for prostate cancer not previously treated with hormone therapy. New Engl J Of Med (2017) 377:338–51. doi: 10.1056/NEJMoa1702900
53. Ramaekers BL, Riemsma R, Tomini F, Van Asselt T, Deshpande S, Duffy S, et al. Abiraterone acetate for the treatment of chemotherapy-naïve metastatic castration-resistant prostate cancer: An evidence review group perspective of an nice single technology appraisal. Pharmacoeconomics (2017) 35:191–202. doi: 10.1007/s40273-016-0445-5
54. Davis ID, Martin AJ, Stockler MR, Begbie S, Chi KN, Chowdhury S, et al. Enzalutamide with standard first-line therapy in metastatic prostate cancer. N Engl J Med (2019) 381:121–31. doi: 10.1056/NEJMoa1903835
55. Penson DF, Armstrong AJ, Concepcion RS, Agarwal N, Olsson CA, Karsh LI, et al. Enzalutamide versus bicalutamide in patients with nonmetastatic castration-resistant prostate cancer: A prespecified subgroup analysis of the strive trial. Prostate Cancer Prostatic Dis (2022) 25:363–5. doi: 10.1038/s41391-021-00465-7
56. Armstrong AJ, Szmulewitz RZ, Petrylak DP, Holzbeierlein J, Villers A, Azad A, et al. Arches: A randomized, phase iii study of androgen deprivation therapy with enzalutamide or placebo in men with metastatic hormone-sensitive prostate cancer. J Clin Oncol (2019) 37:2974–86. doi: 10.1200/JCO.19.00799
57. Khalaf DJ, Annala M, Taavitsainen S, Finch DL, Oja C, Vergidis J, et al. Optimal sequencing of enzalutamide and abiraterone acetate plus prednisone in metastatic castration-resistant prostate cancer: A multicentre, randomised, open-label, phase 2, crossover trial. Lancet Oncol (2019) 20:1730–9. doi: 10.1016/S1470-2045(19)30688-6
58. Dang Q, Li L, Xie H, He D, Chen J, Song W, et al. Anti-androgen enzalutamide enhances prostate cancer neuroendocrine (Ne) differentiation via altering the infiltrated mast cells→ androgen receptor (Ar)→ Mirna32 signals. Mol Oncol (2015) 9:1241–51. doi: 10.1016/j.molonc.2015.02.010
59. Smith MR, Saad F, Chowdhury S, Oudard S, Hadaschik BA, Graff JN, et al. Apalutamide treatment and metastasis-free survival in prostate cancer. N Engl J Med (2018) 378:1408–18. doi: 10.1056/NEJMoa1715546
60. Chi KN, Chowdhury S, Bjartell A, Chung BH, Pereira De Santana Gomes AJ, Given R, et al. Apalutamide in patients with metastatic castration-sensitive prostate cancer: Final survival analysis of the randomized, double-blind, phase iii titan study. J Clin Oncol (2021) 39:2294–303. doi: 10.1200/JCO.20.03488
61. Chi KN, Agarwal N, Bjartell A, Chung BH, Pereira De Santana Gomes AJ, Given R, et al. Apalutamide for metastatic, castration-sensitive prostate cancer. New Engl J Of Med (2019) 381:13–24. doi: 10.1056/NEJMoa1903307
62. Moilanen A-M, Riikonen R, Oksala R, Ravanti L, Aho E, Wohlfahrt G, et al. Discovery of odm-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Sci Rep (2015) 5:12007. doi: 10.1038/srep12007
63. Fizazi K, Shore N, Tammela TL, Ulys A, Vjaters E, Polyakov S, et al. Darolutamide in nonmetastatic, castration-resistant prostate cancer. New Engl J Of Med (2019) 380:1235–46. doi: 10.1056/NEJMoa1815671
64. Smith MR, Hussain M, Saad F, Fizazi K, Sternberg CN, Crawford ED, et al. Darolutamide and survival in metastatic, hormone-sensitive prostate cancer. N Engl J Med (2022) 386:1132–42. doi: 10.1056/NEJMoa2119115
65. Nelson WG, Yegnasubramanian S. Resistance emerges to second-generation antiandrogens in prostate cancer. Cancer Discovery (2013) 3:971–4. doi: 10.1158/2159-8290.CD-13-0405
66. Paschalis A, Sharp A, Welti JC, Neeb A, Raj GV, Luo J, et al. Alternative splicing in prostate cancer. Nat Rev Clin Oncol (2018) 15:663–75. doi: 10.1038/s41571-018-0085-0
67. De Wit R, De Bono J, Sternberg CN, Fizazi K, Tombal B, Wulfing C, et al. Cabazitaxel versus abiraterone or enzalutamide in metastatic prostate cancer. N Engl J Med (2019) 381:2506–18. doi: 10.1056/NEJMoa1911206
68. Parker CC, Clarke NW, Cook AD, Kynaston HG, Petersen PM, Catton C, et al. Timing of radiotherapy after radical prostatectomy (Radicals-rt): A randomised, controlled phase 3 trial. Lancet (2020) 396:1413–21. doi: 10.1016/S0140-6736(20)31553-1
69. Phillips R, Shi WY, Deek M, Radwan N, Lim SJ, Antonarakis ES, et al. Outcomes of observation vs stereotactic ablative radiation for oligometastatic prostate cancer: The oriole phase 2 randomized clinical trial. JAMA Oncol (2020) 6:650–9. doi: 10.1001/jamaoncol.2020.0147
70. Loizzo D, Pandolfo SD, Rogers D, Cerrato C, Di Meo NA, Autorino R, et al. Novel insights into autophagy and prostate cancer: A comprehensive review. Int J Mol Sci (2022) 23(7):3826. doi: 10.3390/ijms23073826
71. George MA, Mayer TM, Moore D, Chen C, White E, Dipaola RS, et al. Autophagic cell death with hydroxychloroquine in patients with hormone-dependent prostate-specific antigen progression after local therapy for prostate cancer. J Of Clin Oncol (2017) 35:102–2. doi: 10.1200/JCO.2017.35.6_suppl.102
72. Iravani A, Violet J, Azad A, Hofman MS. Lutetium-177 prostate-specific membrane antigen (Psma) theranostics: Practical nuances and intricacies. Prostate Cancer And Prostatic Dis (2019) 23(1):38–52. doi: 10.1038/s41391-019-0174-x
73. Hofman MS, Emmett L, Sandhu S, Iravani A, Joshua AM, Goh JC, et al. [(177)Lu]Lu-Psma-617 versus cabazitaxel in patients with metastatic castration-resistant prostate cancer (Therap): A randomised, open-label, phase 2 trial. Lancet (2021) 397:797–804. doi: 10.1016/S0140-6736(21)00237-3
74. Wright GL Jr., Haley C, Beckett ML, Schellhammer PF. Expression of prostate-specific membrane antigen in normal, benign, and malignant prostate tissues. Urol Oncol (1995) 1:18–28. doi: 10.1016/1078-1439(95)00002-Y
75. Keam SP, Halse H, Nguyen T, Wang M, Van Kooten Losio N, Mitchell C, et al. High dose-rate brachytherapy of localized prostate cancer converts tumors from cold to hot. J Immunother Cancer (2020) 8. doi: 10.1101/2020.03.02.20030346
76. Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med (2010) 363:411–22. doi: 10.1056/NEJMoa1001294
77. Patnaik A, Kang SP, Rasco D, Papadopoulos KP, Elassaiss-Schaap J, Beeram M, et al. Phase I study of pembrolizumab (Mk-3475; anti-Pd-1 monoclonal antibody) in patients with advanced solid tumors. Clin Cancer Res (2015) 21:4286–93. doi: 10.1158/1078-0432.CCR-14-2607
78. Antonarakis ES, Piulats JM, Gross-Goupil M, Goh J, Ojamaa K, Hoimes CJ, et al. Pembrolizumab for treatment-refractory metastatic castration-resistant prostate cancer: Multicohort, open-label phase ii keynote-199 study. J Clin Oncol (2020) 38:395–405. doi: 10.1200/JCO.19.01638
79. De Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med (2020) 382:2091–102. doi: 10.1056/NEJMoa1911440
80. Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of Parp1 and Parp2 by clinical parp inhibitors. Cancer Res (2012) 72:5588–99. doi: 10.1158/0008-5472.CAN-12-2753
81. Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. Dna-repair defects and olaparib in metastatic prostate cancer. N Engl J Med (2015) 373:1697–708. doi: 10.1056/NEJMoa1506859
82. Hussain M, Daignault-Newton S, Twardowski PW, Albany C, Stein MN, Kunju LP, et al. Targeting androgen receptor and dna repair in metastatic castration-resistant prostate cancer: Results from nci 9012. J Clin Oncol (2018) 36:991–9. doi: 10.1200/JCO.2017.75.7310
83. Lord CJ, Ashworth A. Parp inhibitors: Synthetic lethality in the clinic. Science (2017) 355:1152–8. doi: 10.1126/science.aam7344
84. Crocetto F, Barone B, Caputo VF, Fontana M, De Cobelli O, Ferro M. Brca germline mutations in prostate cancer: The future is tailored. Diagnostics (Basel) (2021) 11(5):908. doi: 10.3390/diagnostics11050908
85. Pritchard CC, Mateo J, Walsh MF, De Sarkar N, Abida W, Beltran H, et al. Inherited dna-repair gene mutations in men with metastatic prostate cancer. N Engl J Med (2016) 375:443–53. doi: 10.1056/NEJMoa1603144
86. Darst BF, Dadaev T, Saunders E, Sheng X, Wan P, Pooler L, et al. Germline sequencing dna repair genes in 5545 men with aggressive and nonaggressive prostate cancer. J Natl Cancer Inst (2021) 113:616–25. doi: 10.1093/jnci/djaa132
87. Nyberg T, Frost D, Barrowdale D, Evans DG, Bancroft E, Adlard J, et al. Prostate cancer risks for Male Brca1 and Brca2 mutation carriers: A prospective cohort study. Eur Urol (2020) 77:24–35. doi: 10.1016/j.eururo.2019.08.025
88. Castro E, Romero-Laorden N, Del Pozo A, Lozano R, Medina A, Puente J, et al. Prorepair-b: A prospective cohort study of the impact of germline dna repair mutations on the outcomes of patients with metastatic castration-resistant prostate cancer. J Clin Oncol (2019) 37:490–503. doi: 10.1200/JCO.18.00358
89. Kim IE Jr., Kim S, Srivastava A, Saraiya B, Mayer T, Kim WJ, et al. Similar incidence of dna damage response pathway alterations between clinically localized and metastatic prostate cancer. BMC Urol (2019) 19:33. doi: 10.1186/s12894-019-0453-9
90. Klinakis A, Karagiannis D, Rampias T. Targeting dna repair in cancer: Current state and novel approaches. Cell Mol Life Sci (2020) 77:677–703. doi: 10.1007/s00018-019-03299-8
91. Johnson DP, Chandrasekharan MB, Dutreix M, Bhaskara S. Targeting dna repair and chromatin crosstalk in cancer therapy. Cancers (Basel) (2021) 13(3):381. doi: 10.3390/cancers13030381
92. Abida W, Campbell D, Patnaik A, Shapiro JD, Sautois B, Vogelzang NJ, et al. Non-brca dna damage repair gene alterations and response to the parp inhibitor rucaparib in metastatic castration-resistant prostate cancer: Analysis from the phase ii Triton2 study. Clin Cancer Res (2020) 26:2487–96. doi: 10.1158/1078-0432.CCR-20-0394
93. Neeb A, Herranz N, Arce-Gallego S, Miranda S, Buroni L, Yuan W, et al. Advanced prostate cancer with atm loss: Parp and atr inhibitors. Eur Urol (2021) 79:200–11. doi: 10.1016/j.eururo.2020.10.029
94. Cancer Genome Atlas Research, N The molecular taxonomy of primary prostate cancer. Cell (2015) 163:1011–25. doi: 10.1016/j.cell.2015.10.025
95. Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med (2016) 22:298–305. doi: 10.1038/nm.4045
96. Armenia J, Wankowicz SAM, Liu D, Gao J, Kundra R, Reznik E, et al. The long tail of oncogenic drivers in prostate cancer. Nat Genet (2018) 50:645–51. doi: 10.1038/s41588-018-0078-z
97. Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci U.S.A. (2019) 116:11428–36. doi: 10.1073/pnas.1902651116
98. Zhao SG, Chen WS, Li H, Foye A, Zhang M, Sjostrom M, et al. The dna methylation landscape of advanced prostate cancer. Nat Genet (2020) 52:778–89. doi: 10.1038/s41588-020-0648-8
99. Tang F, Xu D, Wang S, Wong CK, Martinez-Fundichely A, Lee CJ, et al. Chromatin profiles classify castration-resistant prostate cancers suggesting therapeutic targets. Science (2022) 376:Eabe1505. doi: 10.1126/science.abe1505
100. Grbesa I, Augello MA, Liu D, Mcnally DR, Gaffney CD, Huang D, et al. Reshaping of the androgen-driven chromatin landscape in normal prostate cells by early cancer drivers and effect on therapeutic sensitivity. Cell Rep (2021) 36:109625. doi: 10.1016/j.celrep.2021.109625
101. Nilsson EM, Laursen KB, Whitchurch J, Mcwilliam A, Odum N, Persson JL, et al. Mir137 is an androgen regulated repressor of an extended network of transcriptional coregulators. Oncotarget (2015) 6:35710–25. doi: 10.18632/oncotarget.5958
102. Kashyap V, Ahmad S, Nilsson EM, Helczynski L, Kenna S, Persson JL, et al. The lysine specific demethylase-1 (Lsd1/Kdm1a) regulates vegf-a expression in prostate cancer. Mol Oncol (2013) 7:555–66. doi: 10.1016/j.molonc.2013.01.003
103. Cai C, He HH, Gao S, Chen S, Yu Z, Gao Y, et al. Lysine-specific demethylase 1 has dual functions as a major regulator of androgen receptor transcriptional activity. Cell Rep (2014) 9:1618–27. doi: 10.1016/j.celrep.2014.11.008
104. Kahl P, Gullotti L, Heukamp LC, Wolf S, Friedrichs N, Vorreuther R, et al. Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half lim domain protein 2 predict risk of prostate cancer recurrence. Cancer Res (2006) 66:11341–7. doi: 10.1158/0008-5472.CAN-06-1570
105. Mohammad HP, Smitheman KN, Kamat CD, Soong D, Federowicz KE, Van Aller GS, et al. A dna hypomethylation signature predicts antitumor activity of Lsd1 inhibitors in sclc. Cancer Cell (2015) 28:57–69. doi: 10.1016/j.ccell.2015.06.002
106. Fang Y, Liao G, Yu B. Lsd1/Kdm1a inhibitors in clinical trials: Advances and prospects. J Hematol Oncol (2019) 12:129. doi: 10.1186/s13045-019-0811-9
107. Chu CH, Wang LY, Hsu KC, Chen CC, Cheng HH, Wang SM, et al. Kdm4b as a target for prostate cancer: Structural analysis and selective inhibition by a novel inhibitor. J Med Chem (2014) 57:5975–85. doi: 10.1021/jm500249n
108. Wilson S, Fan L, Sahgal N, Qi J, Filipp FV. The histone demethylase Kdm3a regulates the transcriptional program of the androgen receptor in prostate cancer cells. Oncotarget (2017) 8:30328–43. doi: 10.18632/oncotarget.15681
109. Lin CY, Wang BJ, Chen BC, Tseng JC, Jiang SS, Tsai KK, et al. Histone demethylase Kdm4c stimulates the proliferation of prostate cancer cells via activation of akt and c-myc. Cancers (Basel) (2019) 11(11):1785. doi: 10.3390/cancers11111785
110. Jones K, Zhang Y, Kong Y, Farah E, Wang R, Li C, et al. Epigenetics in prostate cancer treatment. J Transl Genet Genom (2021) 5:341–56. doi: 10.20517/jtgg.2021.19
111. Rotili D, Tomassi S, Conte M, Benedetti R, Tortorici M, Ciossani G, et al. Pan-histone demethylase inhibitors simultaneously targeting jumonji c and lysine-specific demethylases display high anticancer activities. J Med Chem (2014) 57:42–55. doi: 10.1021/jm4012802
112. Pawar A, Gollavilli PN, Wang S, Asangani IA. Resistance to bet inhibitor leads to alternative therapeutic vulnerabilities in castration-resistant prostate cancer. Cell Rep (2018) 22:2236–45. doi: 10.1016/j.celrep.2018.02.011
113. Shen G, Chen J, Zhou Y, Wang Z, Ma Z, Xu C, et al. Azd5153 inhibits prostate cancer cell growth In vitro and In vivo. Cell Physiol Biochem (2018) 50:798–809. doi: 10.1159/000494244
114. Dai X, Gan W, Li X, Wang S, Zhang W, Huang L, et al. Prostate cancer-associated spop mutations confer resistance to bet inhibitors through stabilization of Brd4. Nat Med (2017) 23:1063–71. doi: 10.1038/nm.4378
115. Zhou B, Hu J, Xu F, Chen Z, Bai L, Fernandez-Salas E, et al. Discovery of a small-molecule degrader of bromodomain and extra-terminal (Bet) proteins with picomolar cellular potencies and capable of achieving tumor regression. J Med Chem (2018) 61:462–81. doi: 10.1021/acs.jmedchem.6b01816
116. Kim DH, Sun D, Storck WK, Welker Leng K, Jenkins C, Coleman DJ, et al. Bet bromodomain inhibition blocks an ar-repressed, E2f1-activated treatment-emergent neuroendocrine prostate cancer lineage plasticity program. Clin Cancer Res (2021) 27:4923–36. doi: 10.1158/1078-0432.CCR-20-4968
117. Hong Z, Wu G, Xiang ZD, Xu CD, Huang SS, Li C, et al. Kdm5c is transcriptionally regulated by Brd4 and promotes castration-resistance prostate cancer cell proliferation by repressing pten. BioMed Pharmacother (2019) 114:108793. doi: 10.1016/j.biopha.2019.108793
118. Wang J, Yu Q, Qiu Z, Dai T, Wang S, Yang X, et al. The combined effect of epigenetic inhibitors for Lsd1 and Brd4 alters prostate cancer growth and invasion. Aging (Albany Ny) (2020) 12:397–415. doi: 10.18632/aging.102630
119. Bai Y, Zhang Z, Cheng L, Wang R, Chen X, Kong Y, et al. Inhibition of enhancer of zeste homolog 2 (Ezh2) overcomes enzalutamide resistance in castration-resistant prostate cancer. J Biol Chem (2019) 294:9911–23. doi: 10.1074/jbc.RA119.008152
120. Liu Q, Wang G, Li Q, Jiang W, Kim JS, Wang R, et al. Polycomb group proteins Ezh2 and eed directly regulate androgen receptor in advanced prostate cancer. Int J Cancer (2019) 145:415–26. doi: 10.1002/ijc.32118
121. Davies AH, Beltran H, Zoubeidi A. Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat Rev Urol (2018) 15:271–86. doi: 10.1038/nrurol.2018.22
122. Mosquera MJ, Kim S, Bareja R, Fang Z, Cai S, Pan H, et al. Extracellular matrix in synthetic hydrogel-based prostate cancer organoids regulate therapeutic response to Ezh2 and Drd2 inhibitors. Adv Mater (2022) 34:E2100096. doi: 10.1002/adma.202100096
Keywords: Therapy, anti-androgen, castration resistant prostate cancer, PARP inhibitors, epigenetic targeted treatment
Citation: Harris AE, Metzler VM, Lothion-Roy J, Varun D, Woodcock CL, Haigh DB, Endeley C, Haque M, Toss MS, Alsaleem M, Persson JL, Gudas LJ, Rakha E, Robinson BD, Khani F, Martin LM, Moyer JE, Brownlie J, Madhusudan S, Allegrucci C, James VH, Rutland CS, Fray RG, Ntekim A, de Brot S, Mongan NP and Jeyapalan JN (2022) Exploring anti-androgen therapies in hormone dependent prostate cancer and new therapeutic routes for castration resistant prostate cancer. Front. Endocrinol. 13:1006101. doi: 10.3389/fendo.2022.1006101
Received: 09 August 2022; Accepted: 16 September 2022;
Published: 03 October 2022.
Edited by:
Erika Di Zazzo, University of Molise, ItalyReviewed by:
Biagio Barone, University of Naples Federico II, ItalySavio Domenico Pandolfo, Federico II University Hospital, Italy
Copyright © 2022 Harris, Metzler, Lothion-Roy, Varun, Woodcock, Haigh, Endeley, Haque, Toss, Alsaleem, Persson, Gudas, Rakha, Robinson, Khani, Martin, Moyer, Brownlie, Madhusudan, Allegrucci, James, Rutland, Fray, Ntekim, de Brot, Mongan and Jeyapalan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jennie N. Jeyapalan, amVubmllLmpleWFwYWxhbkBub3R0aW5naGFtLmFjLnVr; Nigel P. Mongan, bmlnZWwubW9uZ2FuQG5vdHRpbmdoYW0uYWMudWs=; bnBtMjAwMUBtZWQuY29ybmVsbC5lZHU=; Atara Ntekim, a2ltYXRhMjAwMEBob3RtYWlsLmNvbQ==