 Jan-Inge Bjune1,2†‡
Jan-Inge Bjune1,2†‡ Pouda Panahandeh Strømland1†‡
Pouda Panahandeh Strømland1†‡ Regine Åsen Jersin1,2‡
Regine Åsen Jersin1,2‡ Gunnar Mellgren1,2
Gunnar Mellgren1,2 Simon Nitter Dankel1,2*
Simon Nitter Dankel1,2*- 1Hormone Laboratory, Department of Biochemistry and Pharmacology, Haukeland University Hospital, Bergen, Norway
- 2Mohn Nutrition Research Laboratory, Department of Clinical Science, University of Bergen, Bergen, Norway
Sex hormones contribute to differences between males and females in body fat distribution and associated disease risk. Higher concentrations of estrogens are associated with a more gynoid body shape and with more fat storage on hips and thighs rather than in visceral depots. Estrogen-mediated protection against visceral adiposity is shown in post-menopausal women with lower levels of estrogens and the reduction in central body fat observed after treatment with hormone-replacement therapy. Estrogen exerts its physiological effects via the estrogen receptors (ERα, ERβ and GPR30) in target cells, including adipocytes. Studies in mice indicate that estrogen protects against adipose inflammation and fibrosis also before the onset of obesity. The mechanisms involved in estrogen-dependent body fat distribution are incompletely understood, but involve, e.g., increased mTOR signaling and suppression of autophagy and adipogenesis/lipid storage. Estrogen plays a key role in epigenetic regulation of adipogenic genes by interacting with enzymes that remodel DNA methylation and histone tail post-translational modifications. However, more studies are needed to map the differential epigenetic effects of ER in different adipocyte subtypes, including those in subcutaneous and visceral adipose tissues. We here review recent discoveries of ER-mediated transcriptional and epigenetic regulation in adipocytes, which may explain sexual dimorphisms in body fat distribution and obesity-related disease risk.
Introduction
Sexual dimorphism in obesity and related cardiometabolic risk involves differences in fat distribution (1, 2), described by Vague already in 1947 (3). Most body fat is stored in two main white adipose tissue (WAT) depots; subcutaneous adipose tissue (SAT) and visceral adipose tissue (VAT). Increased visceral adiposity is particularly associated with increased mortality and risk of a range of metabolic conditions including insulin resistance, type 2 diabetes, and cardiovascular disease (4–11). In contrast, preferential fat accumulation on the hips, thighs and other subcutaneous sites in females compared to males may help explain the lower risk of metabolic diseases generally seen in females (11). VAT (omental and mesenteric fat) normally constitutes about 10-20% of total body fat in males and 5-10% in females (12), although these percentages vary greatly for different individuals (4–6, 8, 13). There is a relative increase in adipose tissue (AT) mass and decrease in muscle mass with age (14, 15), which is associated with altered concentrations and activity of sex hormones (16), including testosterone and estrogens, which are potent regulators of adipogenesis and energy metabolism (17, 18). Importantly, with loss of estrogens after menopause, females often begin storing more VAT and have higher risk of metabolic diseases, more like males (1, 19, 20). This shift in AT function and distribution can in turn alter the metabolic functions of other tissues, in part via changes in adipokine secretion, release of lipids for energy expenditure or storage in tissues such as liver, muscle and heart, and other mechanisms (2, 21).
Among all natural or synthetic estrogens (22), endogenous estrogens in humans consist of estrone (E1), estriol (E3) and 17β-estradiol (E2), the latter being the most biologically active (22, 23). In premenopausal women, E2 is the dominating estrogen, while E1 produced by adipose tissue is more important after menopause (24). Androgens are converted to estrogens by the enzyme aromatase, thus linking the sex hormones in both males and females (25). Estrogens bind to two ‘classical’ estrogen receptor (ER) subtypes, alpha (ERα) and beta (ERβ), which have multiple isoforms and exhibit distinct tissue expression patterns (26). E2 has similar affinity to both receptors (26, 27). Estrogen-mediated activation of ER-dependent transcriptional activity alters epigenetic programming and global gene expression patterns, contributing critically to the cellular effects of estrogens, such as in breast cancer (28) and hippocampal memory formation (29). Thus, in breast cancer cells, estrogen deprivation has been found to cause DNA hypermethylation and histone deacetylation and consequent downregulation of global gene expression, which was largely reversed by E2 re-stimulation (30). It is possible that such epigenetic mechanisms are central in ER subtype-specific effects, given tissue differences in ER subtype expression levels (26).
Studies on mechanisms of sexual dimorphism in body fat distribution have pointed to the role of sex hormones as well as the microenvironment and cell-specific properties within fat depots (31). Due to the importance of epigenetic/transcriptional programming for the unique functional properties of different adipocyte subtypes (32), it may be critical to determine how and to what extent estrogens contribute to these distinct properties, and consequently to sex differences in body fat distribution and associated risk of metabolic diseases. In this review, we discuss the role of estrogens in adipose tissue distribution and function, and emphasize emerging knowledge of estrogen-dependent epigenetic mechanisms that may govern sexual dimorphism in obesity and adipogenesis.
Role of Estrogen in Adipose Tissue
ERα expression is inversely associated with obesity in both females (33), males (34) and over 100 different strains of inbred mice (34). In human (20) as well as rodent (35) females, the decline in circulating E2 after menopause corresponds to increased fat mass and lower glucose tolerance. Conversely, estrogen replacement therapy reverses these effects (35–38). Moreover, E2 treatment in nutritionally challenged female mice reduced VAT mass and adipocyte size, and altered gene expression of lipogenic markers, adipokines, specific nuclear receptors, and thermogenic markers (39). However, effects of estrogen-ERα signaling often differ greatly, both between the two sexes, and between SAT and VAT (as described in detail in the sections below and summarized in Figure 1).
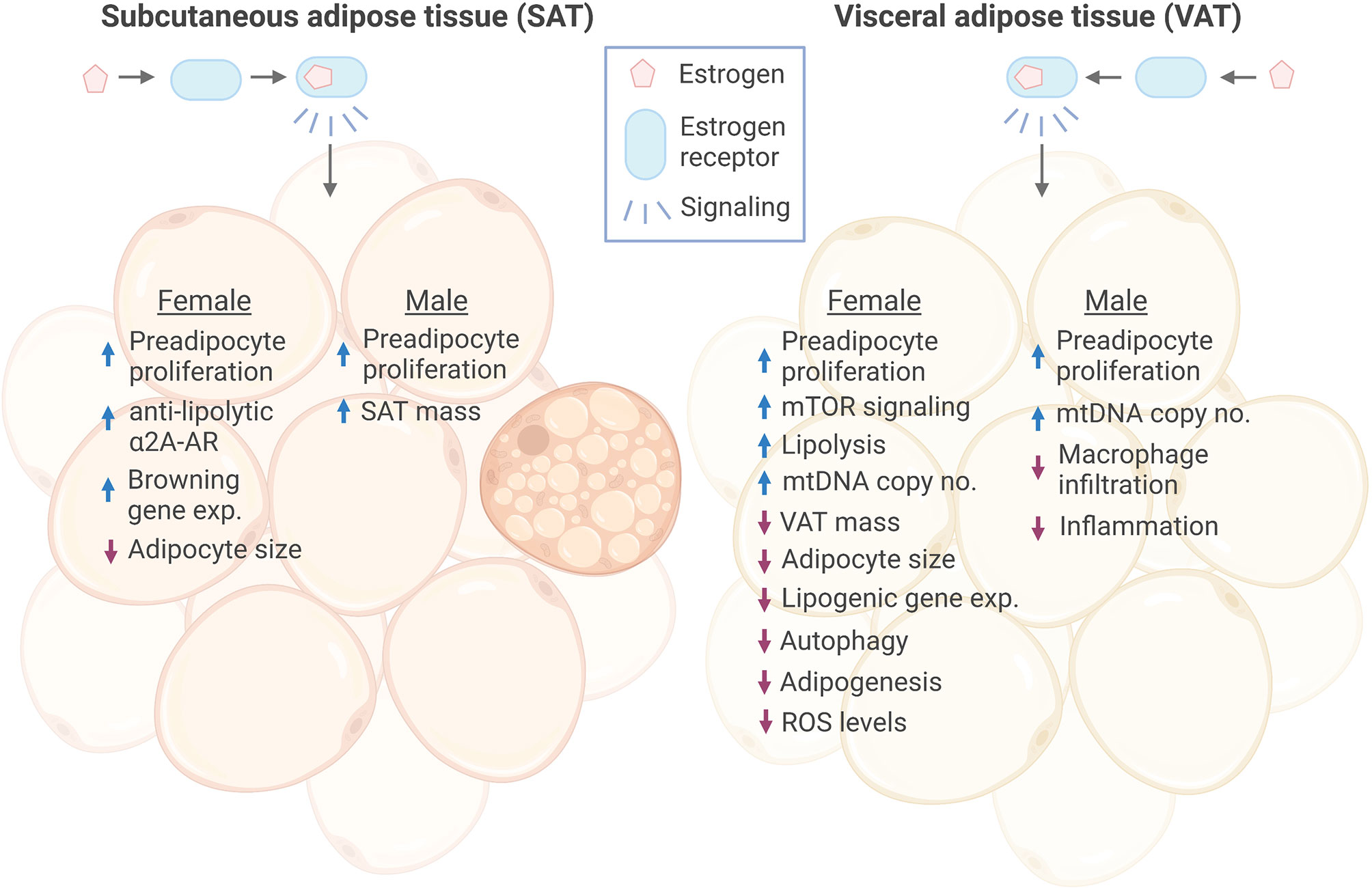
Figure 1 Effects of estrogen signaling in female and male SAT and VAT. Estrogen signaling in subcutaneous adipose tissue (SAT) and visceral adipose tissue (VAT) of both sexes has been found to promote increased proliferation of preadipocytes. Estrogen has been shown to promote anti-lipolytic effects through increasing the expression of a2A-AR in female SAT, which may, at least in part, explain the concomitant increase in SAT mass and overall anti-obesogenic effect of estrogen. In addition, Estrogen induced expression of several browning genes in female SAT. In response to estrogen, female VAT showed increased lipolysis, while lipogenic gene expression was decreased, together resulting in reduced VAT mass. On the contrary, estrogen increased male SAT volume. Adipocyte size was reduced in both female SAT and VAT by estrogen, while there were no reports of this in males. However, estrogen decreased macrophage infiltration and inflammation in male VAT. Female VAT has been shown to have reduced autophagy, adipogenesis and ROS levels in response to estrogen treatment. ER, estrogen receptor; a2A-AR, alpha2A-adrenergic receptors; exp., expression. Figure created in BioRender.com.
Estrogen exerts anti-obesity effects through multiple mechanisms, such as central regulation of energy intake and expenditure (reviewed in (40)). However, estrogen also has direct effects in WAT, and it has been shown that loss of estrogens has a much stronger effect on gene expression in WAT compared to for example the hypothalamus (41). In WAT, E2 is shown to decrease expression of genes involved in triglyceride synthesis (lipogenesis) and promote catecholamine-induced lipolysis (42, 43). While estrogens can affect adiposity, adiposity can also increase the production of estrogens locally in several tissues. Although AT is not steroidogenic, it is the most important site for steroid production outside the gonads due to the presence of the aromatase cytochrome P450 enzyme, which converts androgens taken up from the circulation into estrogens (44, 45). Due to the presence of aromatase in AT, the locally produced estrogen can affect metabolism independent of plasma E2 levels (44). In rats, it has been shown that local E2 levels are about tenfold higher in AT compared to the circulation (44). Conversely, another study observed no statistically significant differences between the sexes in neither plasma nor overall adipose E2 levels between male and female rats, while a significant depot-dependent effect was found in both sexes, where E2 levels showed 1.5-2-fold higher levels in SAT compared to different VAT depots (44).
Female ovariectomized mice display increased VAT and reduced leptin sensitivity compared to controls, which upon E2 administration can be restored to levels seen in intact cycling females (46). Interestingly, male mice given E2 show decreased insulin sensitivity, increased SAT volume, higher sensitivity to leptin, and overall increased body fat (46), at least in part explained by reduced physical activity and energy expenditure (46). No stimulatory effects on food intake were seen, and estrogen may rather have leptin mimetic/anorectic functions that suppress food uptake (46, 47), pointing to other tissue-specific obesogenic effects of E2 in males.
In male humans, an increase in AT mass is associated with increased levels of aromatase (48, 49), and hence increased ability to synthesize estrogens (50). Conversely, administration of aromatase inhibitors increases the testosterone-estrogen ratio and reverses hypogonadal obesity, resulting in the stimulation of muscle protein synthesis and increased muscle mass (51, 52). The aromatization process progressively reduces testosterone levels and elevates estrogen levels in males (52). Decreased testosterone concentrations in males are associated with elevated concentrations of leptin, which is produced by fat cells as a reflection of fat stores. Further expansion of visceral AT and production of aromatase through this hypogonadal-obesity cycle may result in a vicious cycle of continued visceral AT expansion and insulin resistance (52). On the other hand, higher levels of AT aromatase activity in male mice leads to a decreased adipose tissue inflammation and improved insulin sensitivity (53). Given the generally protective effect of estrogen against visceral adiposity, an important question is whether estrogen has different effects in males compared to females. To answer this question, we need more detailed insight into how estrogen exerts its biological effects, and whether there are differences in intracellular signaling mechanisms in relevant metabolic cells between the sexes.
Mechanisms of Action and Metabolic Regulation by Estrogen in Adipose Tissue
In the early 1990s, Mizutani et al. and Pedersen et al. reported the presence of ER along with other steroid receptors such as glucocorticoid and androgen receptors, but not progesterone receptors, in human mature adipocytes (54, 55). The effect of estrogen on AT distribution is mainly controlled by the adipocyte ERα (56), and the estrogen-ERα signaling has anti-obesity effects (57). In a rat study by Rodriguez-Cuenca et al., VAT from both males and females exhibited lower levels of E2, but higher expression levels of ERα and ERβ compared to SAT (44). These data suggest that VAT is more sensitive to E2 than SAT (44), which supports the observation that estrogen-stimulated lipolysis occurs mainly in visceral compartments (58). Moreover, it may explain why ERα-KO mice of both sexes gain weight only in visceral compartments (56).
Estrogen has been shown to reduce adipogenesis through activation of mTOR signaling, promoting inhibition of PPARγ (40, 59–62) or reduction of autophagy in female VAT (63). Importantly, the pro-lipolytic effect of E2 has been found to be blunted specifically in female SAT (64), via estrogen-mediated increase in anti-lipolytic α2A-adrenergic receptors (59, 64). Interestingly, this was not observed in VAT (64) which may help to explain why only SAT and not VAT in females is affected by changes in serum levels of estrogen and how estrogen overall has anti-obesity effects but at the same time promotes fat storage subcutaneously (59, 64). These effects of estrogen may explain some of the findings in genome-wide association studies with more than 224,000 individuals (65), showing that metabolic changes are likely involved in the sexual dimorphism of obesity and fat distribution, implicating mechanisms via differential control of adipogenesis and insulin resistance between sexes (1, 65, 66).
Studies have previously shown that estrogen and its receptors are involved in regulating preadipocyte and adipocyte growth and function, and some differences between the sexes are reported (67–69). Interestingly, E2 stimulates the proliferation of preadipocytes from both sexes (67). However, subcutaneous and visceral preadipocytes from females were more responsive to E2 and proliferated faster compared to preadipocytes from males (67). Both male and female mice harboring a knockout (KO) of ERα showed increased levels of body fat compared to their wild-type (WT) littermates, despite similar body weights (68). The same study reported that these male and female ERα KO mice had larger adipocytes, and higher expression of markers of macrophage infiltration and markers of fibrosis than WT mice (68). Another report found that female whole body ERα KO mice also showed reduced adiponectin expression, and increased fibrosis and inflammation (69).
Furthermore, similar phenotypes were observed in both male and female adipocyte-specific ERα knockout (AdipoERα) mice compared to whole-body KOs, with some exceptions (69). Despite no increase in weight gain, the male AdipoERα mice showed reduced glucose clearance as measured by an oral glucose tolerance test, suggesting adipocyte dysfunction in the absence of estrogen-ERα signaling in males. Surprisingly, glucose clearance in female AdipoERα mice, showing increased weight gain compared to WTs, was not affected (69). While both male and female AdipoERα mice showed increased adipocyte size compared to their WT counterparts, only adipocytes of male mice had increased expression of markers of macrophage infiltration, inflammation and fibrosis, indicating sex-dependent regulation of adipocyte function (68). Interestingly, adipocyte-specific loss of ERα in ERβ deficient mice leads to lower glucose tolerance also in female mice (as seen for male AdipoERα mice with expression of ERβ), as well as increased markers of inflammation and fibrosis. These findings suggest that ERβ may regulate glucose homeostasis, fibrosis and inflammation in female AdipoERα mice but not in males (68).
Moreover, E2, via ERβ signaling, increased the expression of thermogenic uncoupling protein-1 (UCP-1) in mouse brown adipose tissue (BAT), leading to increased energy expenditure and thus reduced fat mass (60). In 2018, it was shown that activation of ERs in white adipocytes in both humans and mice increased markers of beiging (70). However, whether there are sex differences in this regulation remains to be determined. Of note, both the anorectic function of E2 as well as its role in increasing the energy expenditure can also be mediated through both ERα and β in the hypothalamic area of the brain (71).
Estrogen signaling also is best known to affect gene expression in target tissues, but can also affect processes outside the nucleus, involving ion channels and protein kinases, which is so-called non-genomic or non-nuclear signaling. In contrast to the relatively slow activation of gene transcription, these non-genomic pathways occur rapidly (within seconds or minutes) via membrane-associated forms of the ERs (72). It has been shown that E2 treatment of ovariectomized mice rapidly increased fat oxidation through activation of AMPK (42). Moreover, E2 can inhibit glucose oxidation in adipocytes through non-genomic mechanisms (73). Estrogen may also bind other non-classical receptors, including GPR30, which is a G protein-coupled estrogen receptor (GPER) in the endoplasmic reticulum that has a high affinity for E2 (74, 75). These pathways have been mostly studied in neurons or pancreatic β cells, and have been suggested to be the most important mediators of estrogen signaling in these tissues (40, 76). However, recent in vitro and in vivo studies have shown that GPR30 plays an important role in adipogenesis by reducing the fat mass and adipocyte size (77). Compared to BAT, GPR30 is highly expressed in WAT (77). Deletion of GPR30 by reducing plasma insulin and leptin levels protects female mice from developing obesity, glucose intolerance and insulin resistance after nutritional challenge (77). How GPR30-mediated estrogen signaling interacts with mechanisms of epigenomic regulation remains to be determined.
Adipose Tissue Gene Regulation by Estrogen Receptors
ERs can bind directly or indirectly to promoters of target genes to repress or activate their expression (26). Manipulation of estrogen levels or ERs have provided insights into adipocyte target genes and thereby the mechanisms of ER-mediated gene regulation. For example, loss of estrogens by ovariectomizing reduced WAT expression of glutathione peroxidase 3 (Gpx3) (41), a gene important for the protection of cells from oxidative stress in the form of reactive oxygen species (ROS) (78). Furthermore, E2 reduced ROS levels and enhanced browning in female mouse SAT through promoting macrophage heme oxygenase-1 (Hmox1, also known as HO-1) expression (79). Similarly, E2 treatment of 3T3-L1 adipocytes increased expression of genes encoding the ROS reducing antioxidants HO-1, NAD(P)H:quinone oxidoreductase 1 (NQO1) and glutamate-cysteine ligase (GCL), directly in the adipocytes (80). High levels of ROS have previously been linked to decreased mitochondrial respiration (81) and increased fat storage (82), which are typical hallmarks of adipocyte dysfunction (83). Correspondingly, postmenopausal females showed increased VAT ROS levels compared to premenopausal individuals (80). Together, these data may suggest that intact E2 signaling could, through regulation of genes involved in antioxidant processes, play a role in increasing the resilience to nutritional/metabolic stress and prevent adipose dysfunction, a key contributor of obesity and metabolic syndrome (84).
In support of this theory, adipose-specific deletion of Estrogen receptor 1 (Esr1, gene encoding ERα) in both female and male mice have recently been shown to decrease mitochondrial DNA (mtDNA) copy number in both WAT and BAT (34) (Figure 1). The investigators demonstrated that ERα binds directly to the nuclear-encoded mtDNA polymerase subunit γ (Polg1), thereby controlling mtDNA replication in WAT (34). Moreover, loss of ERα was further accompanied by reduced expression of key genes involved in mitochondrial biogenesis (Pgc1b, Nrf1), and transcription (Polrmt) (34). Other studies have previously shown that NRF1 is under control of E2-mediated ERα and ERβ activities in other tissues such as breast cancer, mammary glands, and the uterus (85, 86). In female mice BAT, ERα is necessary for mitochondrial remodeling through Dynamin-related protein 1 (Drp1) (34), and thermogenesis through Ucp1 (34) and Cidea (41). Overall, these data suggest that estrogen signaling is important for maintaining mitochondrial function in females, an important prerequisite for preventing adipocyte dysfunction and metabolic complications (87).
A number of microRNAs (miRNAs) have been found to play crucial roles in both white and beige/brown adipocyte development and function (reviewed in (88)). Knockdown of ERα in rat bone marrow-derived mesenchymal stem cells (BMSCs) has been found to alter the expression of almost 200 miRNAs, including downregulation of miR-210-3p, accompanied with increased Pparg protein levels and reduced expression of the osteogenic regulator Runx2 (89). Conversely, overexpression of miR-210-3p was found to increase Wnt signaling and promoted osteogenesis over adipogenesis (89). Interestingly, endometriosis is an estrogen-driven inflammatory disease characterized by reduced BMI and abnormal levels of circulating miRNAs (90), including miR-342 (91) and Let-7b (92, 93). Overexpression or inhibition of these miRNAs in primary preadipocytes from healthy donors altered the expression of C/ebpa, C/ebpb and Pparg (94). Of note, miRNAs may affect gene expression not only in the cells they are produced, but also in distant cells and tissues through secreted extracellular vesicles, including exosomes (95). Importantly, small motifs in the miRNAs have recently been found to dictate their retention or secretion, with white adipocytes demonstrating by far the highest production and secretion rates per cell compared to several other cell types (96). Thus, future studies should be better equipped to predict and assess local and systemic effects of ER-regulated miRNAs.
Estrogen-Mediated Epigenetic Regulation in Adipocytes
Epigenetics plays a causal role in the development of obesity (97), and adipogenesis is extensively regulated by DNA methylation and demethylation, histone tail modifications and chromatin remodeling (97, 98). Strikingly, E2-bound ERs have been shown to be involved in these epigenetic processes in various tissues through recruitment of co-regulators and epigenetic remodeling enzymes (99, 100). We will here review general known mechanisms of epigenetic regulation via estrogens and highlight known aspects in adipocytes (Figure 2).
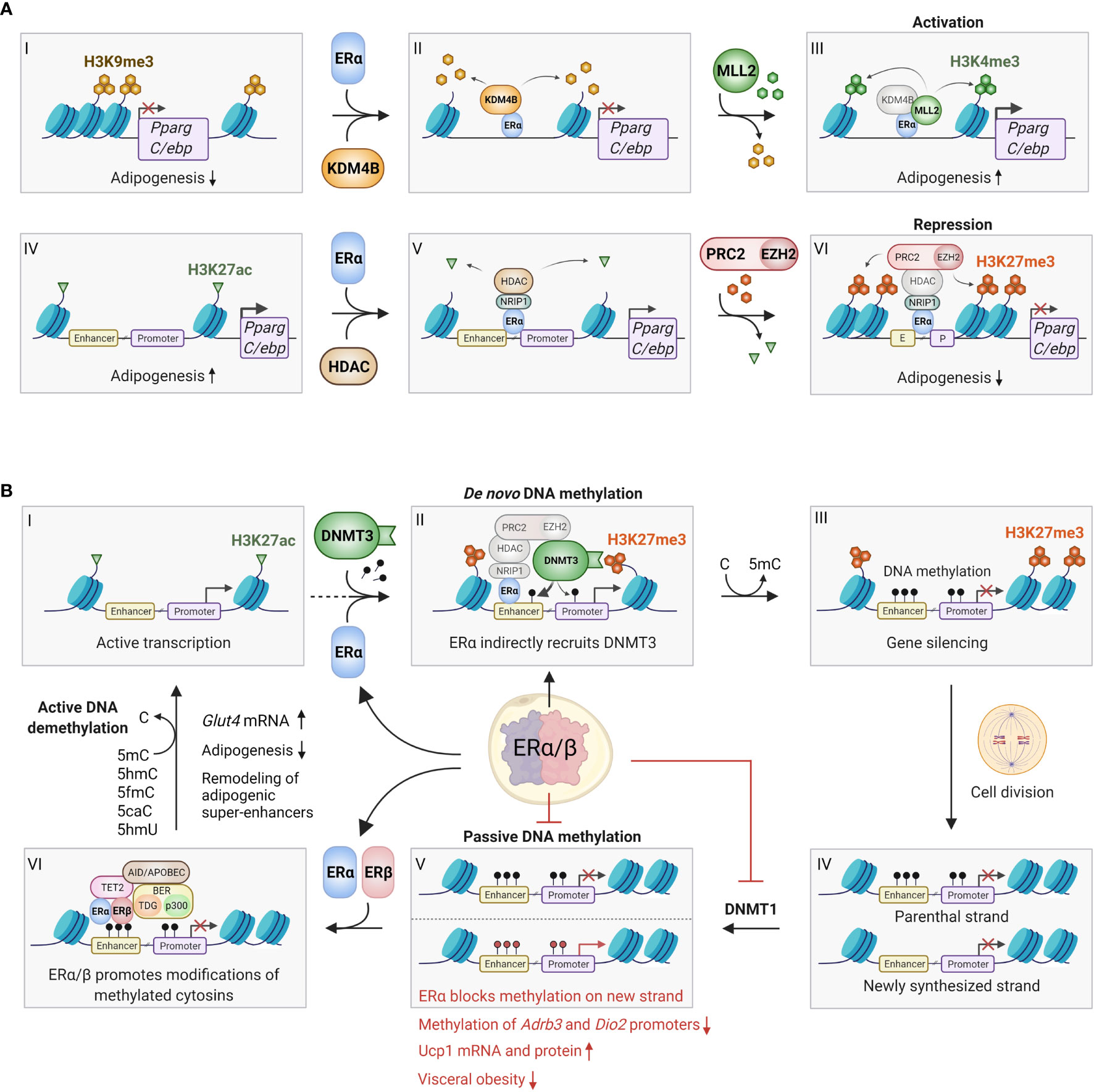
Figure 2 Epigenetic effects of ERα/β in adipocytes. (A) ERα can bind to promoter regions with repressive H3K9me3 marks (I-VI), where it recruits the histone demethylase KDM4B (also known as JMJD2B), which specifically removes these marks (II). This enables the recruitment and activity of the methyl transferase MLL2, which trimethylates lysine 4 on histone 3, forming activating H3K4me3 marks, which promotes gene expression (III). This process may occur on the promoters of Pparg and C/ebp, promoting adipogenesis. Conversely, ERα can also bind to actively transcribed genes characterized by H3K27ac marks (IV), where it binds various coregulators, including NRIP1, that enables binding of histone deacetylases (HDACs), which remove the acetyl groups on H3K27 (V). Finally, the ERα/NRIP1/HDAC complex can further bind the PRC2/EZH2 polycomb complex, which adds methyl groups to form repressive H3K27me3 marks (VI). This process can also occur on the Pparg and C/ebp promoter/enhancers, inhibiting adipogenesis. Although the repressive pathway appears most predominant, further studies should investigate whether the activating pathway indeed plays a role in certain preadipocyte/mesenchymal stem cell subpopulations. (B) ERα and ERβ affects DNA methylation through several mechanisms. ERα promotes de novo methylation and gene silencing by binding to actively transcribed regions (I), where the ERα/HDAC/PRC2/EZH2 complex first converts activating H3K27ac marks to repressive H3K27me3 marks (see Figure 2A IV-VI for details). The DNA methyl transferase DNMT3 recognizes the H3K27me3 marks, and stabilized by the ERα/HDAC/PRC2/EZH2 complex it adds a methyl group to cytosine residues on the surrounding DNA, leading to stable gene silencing (II-III). Conversely, ERα can inhibit passive DNA methylation after cell division. This occurs by transcriptional inhibition of DNMT1, which copies the DNA methylation pattern of the old DNA strand onto the newly synthesized DNA (IV-V). Red methyl groups (bottom panel V) represent hypomethylated regions in response to ERα-mediated repression of DNMT1, leading to increased beiging. ERα and/or ERβ can also promote active demethylation by recruitment of TET2, AID/APOBEC/BER complexes, which alter methylated cytosines in numerous ways that ultimately restores unmodified cytosine (VI-I). Active demethylation likely remodels adipogenic super-enhancers, and has been found to inhibit adipogenesis and increase Glut4 expression. C, Cytosine; 5mC, 5-methylcytosine; 5hmC, 5-hydroxymethylcytosine; 5fmC, 5-formylcytosine; 5caC, 5-carboxylcytosine; 5hmU, 5-hydroxymethyluracyl. Figure created in BioRender.com.
Histone Modifications by ERα
Cellular DNA is wrapped around histone proteins to form nucleosomes and higher-order chromatin structures (101, 102), which constitutes a major layer in transcriptional regulation (101, 102). H2, H3 and H4 histone family members, with tails of various lengths, are subjected to extensive post-translational modifications, including methylation and acetylation (101, 102). ERα interacts with, and promotes the activity of MLL2 (99, 103), a specific H3K4 histone methyltransferase (HMT) that confers epigenetic activation of gene expression (104) (Figure 2A). Mutations in MLL2 lead to insulin resistance and reduced glucose tolerance in mice (105) and humans (98, 106). Because MLL2-dependent H3K4me3 activating marks are mutually exclusive with repressing H3K9me3 marks, the MLL2/ERα complex also includes KDM4B, a H3K9 demethylase that coordinates the conversion from repressive to activating marks (103). In preadipocytes, KDM4B is known to act on the promoters of Pparg and C/ebp and promote adipogenesis (107). Taken together, ERα may promote adipogenesis through KDM4B/MLL2 (Figure 2A), but this remains to be confirmed. Of particular interest would be whether this mechanism exists predominantly in subcutaneous (gluteal and femoral) WAT.
In contrast, as detailed further above, estrogen and/or ERα has mainly been found to inhibit adipogenesis (108–111). ERα mediates epigenetic silencing by recruiting histone deacetylase HDAC1 and HMTs like EZH2 to convert activating H3K27ac marks to repressive H3K27me3 marks (99) (Figure 2A). In rats, E2 treatment increased the binding of ERα/EZH2 to the promoters of Pparg, C/ebpa and Cfd (encoding Adipsin) in mesenchymal stem cells (MSCs), leading to increased H3K27 methylation and repression of these genes (112). These data support a predominantly inhibitory effect of estrogen on adipogenesis, and this effect is at least partly due to epigenetic silencing of adipogenic master regulators.
DNA Methylation and Demethylation by ERα
DNA methylation on CpG islands (99), which are present in most promoters (113), has a repressive effect on gene expression (114). This reaction can be catalyzed by two types of DNA methyltransferases (DNMTs) depending on the purpose of the methylation. While DNMT1 is active during cell division where it copies the parental DNA methylation pattern, DNMT3 can establish new methylation patterns, also known as de novo DNA methylation (115). ERα promotes the latter by indirect recruitment and activation of DNMT3 (99) to EREs (Figure 2B). Thus, mapping the genomic binding pattern of ERα in different adipose tissues at different developmental stages is critical to understand its epigenetic effects. Strikingly, ERα has shown a strong preference for binding to intergenic regions (116). Interestingly, about half of the CpG islands are also found in intergenic regions, and have recently been shown to be an essential part of poised enhancers, acting as anchors between the enhancer and target promoters (113). Consequently, methylation of CpG islands plays a crucial role in determining enhancer-promoter selectivity. Importantly, there are significant changes in enhancer interactions during adipocyte differentiation (117). Collectively, ERα may be involved in methylation-dependent regulation of enhancer-promoter interactions during adipogenesis. However, future studies are needed to test this hypothesis.
ERα and ERβ are also involved in demethylation, both passively during cell division by transcriptional inhibition of DNMT1, and actively by interacting with a range of enzymes that modify and remove the methyl group (99, 118) (Figure 2B). ERβ promotes active demethylation and increased expression of Glut4 in mouse embryonic fibroblasts by recruiting the demethylation machinery to the Glut4 promoter (119, 120). This ER-bound demethylation machinery includes TET2, which has been shown to inhibit adipogenesis (121), and p300, a known master epigenetic writer of enhancers during adipogenesis (122). It is therefore plausible that ERα and/or ERβ-dependent DNA demethylation is involved in the epigenetic regulation of adipogenesis, although this was not directly investigated. However, E2 has been shown to epigenetically promote beiging in mice by promoting demethylation of the Adrb3 and Dio2 promoters, leading to increased Ucp1 expression (39). These changes were accompanied by reduced visceral lipogenic gene expression, improved fatty acid utilization, which reversed diet-induced visceral obesity and glucose intolerance (39). Moreover, activation of both ERα (70) and ERβ (123) has been shown by others to activate WAT browning (124). Taken together, ERα may promote thermogenesis by relieving repressive methylation marks on key positive regulators of beiging and mitochondrial uncoupling.
Discussion
At the time morphological differences between individuals with obesity were first described (3), the direct influence of sex hormones on adipocytes had not been explored. Since then, much has been learned about how metabolic processes differ by sex and how estrogen affects developmental, metabolic and epigenetic processes, including adipogenesis and the fate of adipocyte progenitor cells towards thermogenic brown/beige or white fat cells.
In the research performed by Pedersen et al., Santos et al. and Zhou et al. (34, 64, 70), an effort has been made to differentiate the mechanism of estrogen signaling in different subtypes of adipocytes. However, despite technological advances allowing improved distinction of the metabolic properties of subcutaneous and visceral adipose depots, the effects of estrogen on distinct subtypes of fat cells in different depots remains to be described. More detailed insight into the role of estrogen signaling in adipocyte subtypes may be critical, as different adipocytes possess unique metabolic and endocrine profiles regardless of adipogenic capacities (125, 126).
The first evidence for epigenetic control of adipogenesis by estrogen was provided by the study of Rüegg et al. in 2011 (119), and progress has since been made in this field of research. However, more research is needed to fully understand estrogen-dependent mechanisms in different adipose tissue depots and adipocyte subtypes, and to what extent these mechanisms are distinct in males and females. New detailed insight into estrogen-mediated epigenetic changes may also help to assess health effects of environmental xenoestrogens, which partly act via epigenetic changes (127). Furthermore, it will be important to clarify functional differences and similarities between ERα and Erβ in metabolic and epigenetic regulation in different adipose cell types and depots.
At the same time, we must consider that estrogen effects on adipocytes are not limited to the classical types of ERs. For example, Wang et al. revealed that the relatively recently described non-genomic estrogen receptor GPR30 regulates adiposity in mice in a sex-specific manner (77). A relevant challenge is therefore also to evaluate whether GPR30-mediated estrogen signaling might interact with mechanisms of epigenomic regulation.
In conclusion, the emerging knowledge of estrogen-mediated metabolic and epigenetic regulation in different adipocytes provides a deeper understanding of how cellular programming regulates metabolic health. Further research in this area may uncover new molecular targets for improving body composition, insulin resistance and reducing the risk of lifestyle-related diseases.
Author Contributions
Conceptualization and literature investigation: SD, PPS, J-IB, RÅJ, and GM. Original draft preparation: PPS, J-IB, RÅJ, and SD. Writing and editing: SD, J-IB, RÅJ, PPS, and GM. Figures preparation: RÅJ and J-IB. Supervision: SD and GM. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by grants received from the Trond Mohn Foundation (BFS2017NUTRITIONLAB) and the Western Norway Regional Health Authority (912010).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Wells J. Sexual Dimorphism of Body Composition. Best Pract Res Clin Endocrinol Metab (2007) 21:415–30. doi: 10.1016/J.BEEM.2007.04.007
2. Goossens GH, Jocken JWE, Blaak EE. Sexual Dimorphism in Cardiometabolic Health: The Role of Adipose Tissue, Muscle and Liver. Nat Rev Endocrinol (2021) 17:47–66. doi: 10.1038/S41574-020-00431-8
3. Vague J. Sexual Differentiation. A Determinant Factor of the Forms of Obesity. 1947. Obes Res (1996) 4:201–3. doi: 10.1002/J.1550-8528.1996.TB00535.X
4. Despres JP, Nadeau A, Tremblay A, Ferland M, Moorjani S, Lupien PJ, et al. Role of Deep Abdominal Fat in the Association Between Regional Adipose Tissue Distribution and Glucose Tolerance in Obese Women. Diabetes (1989) 38:304–9. doi: 10.2337/diabetes.38.3.304
5. Pouliot MC, Després JP, Nadeau A, Moorjani S, Prud’Homme D, Lupien PJ, et al. Visceral Obesity in Men: Associations With Glucose Tolerance, Plasma Insulin, and Lipoprotein Levels. Diabetes (1992) 41:826–34. doi: 10.2337/diab.41.7.826
6. Wajchenberg BL. Subcutaneous and Visceral Adipose Tissue: Their Relation to the Metabolic Syndrome. Endocr Rev (2000) 21:697–738. doi: 10.1210/edrv.21.6.0415
7. Janssen I, Katzmarzyk PT, Ross R. Waist Circumference and Not Body Mass Index Explains Obesity-Related Health Risk. Am J Clin Nutr (2004) 79:379–84. doi: 10.1093/ajcn/79.3.379
8. Zhang C, Rexrode KM, Van Dam RM, Li TY, Hu FB. Abdominal Obesity and the Risk of All-Cause, Cardiovascular, and Cancer Mortality: Sixteen Years of Follow-Up in US Women. Circulation (2008) 117:1658–67. doi: 10.1161/CIRCULATIONAHA.107.739714
9. Després JP, Lemieux I. Abdominal Obesity and Metabolic Syndrome. Nature (2006) 444:881–7. doi: 10.1038/nature05488
10. Balkau B, Deanfield JE, Després JP, Bassand JP, Fox KAA, Smith SC, et al. International Day for the Evaluation of Abdominal Obesity (IDEA): A Study of Waist Circumference, Cardiovascular Disease, and Diabetes Mellitus in 168 000 Primary Care Patients in 63 Countries. Circulation (2007) 116:1942–51. doi: 10.1161/CIRCULATIONAHA.106.676379
11. Krotkiewski M, Bjorntorp P, Sjostrom L, Smith U. Impact of Obesity on Metabolism in Men and Women. Importance of Regional Adipose Tissue Distribution. J Clin Invest (1983) 72:1150–62. doi: 10.1172/JCI111040
12. Lee MJ, Wu Y, Fried SK. Adipose Tissue Heterogeneity: Implication of Depot Differences in Adipose Tissue for Obesity Complications. Mol Aspects Med (2013) 34:1–11. doi: 10.1016/j.mam.2012.10.001
13. Walker GE, Marzullo P, Ricotti R, Bona G, Prodam F. The Pathophysiology of Abdominal Adipose Tissue Depots in Health and Disease. Horm Mol Biol Clin Investig (2014) 19:57–74. doi: 10.1515/HMBCI-2014-0023/PDF
14. Ilich JZ, Kelly OJ, Inglis JE, Panton LB, Duque G, Ormsbee MJ. Interrelationship Among Muscle, Fat, and Bone: Connecting the Dots on Cellular, Hormonal, and Whole Body Levels. Ageing Res Rev (2014) 15:51–60. doi: 10.1016/J.ARR.2014.02.007
15. Kalyani RR, Corriere M, Ferrucci L. Age-Related and Disease-Related Muscle Loss: The Effect of Diabetes, Obesity, and Other Diseases. Lancet Diabetes Endocrinol (2014) 2:819–29. doi: 10.1016/S2213-8587(14)70034-8
16. Mayes JS, Watson GH. Direct Effects of Sex Steroid Hormones on Adipose Tissues and Obesity. Obes Rev (2004) 5:197–216. doi: 10.1111/J.1467-789X.2004.00152.X
17. Law J, Bloor I, Budge H, Symonds ME. The Influence of Sex Steroids on Adipose Tissue Growth and Function. Horm Mol Biol Clin Investig (2014) 19:13–24. doi: 10.1515/HMBCI-2014-0015/PDF
18. Chang E, Varghese M, Singer K. Gender and Sex Differences in Adipose Tissue. Curr Diabetes Rep (2018) 18:69. doi: 10.1007/S11892-018-1031-3
19. Shi H, Seeley RJ, Clegg DJ. Sexual Differences in the Control of Energy Homeostasis. Front Neuroendocrinol (2009) 30:396–404. doi: 10.1016/J.YFRNE.2009.03.004
20. Zamboni M. Effects of Age on Body Fat Distribution and Cardiovascular Risk Factors in Women. Am J Clin Nutr (1997) 66:111–5. doi: 10.1093/AJCN/66.1.111
21. Rosen ED, Spiegelman BM. What We Talk About When We Talk About Fat. Cell (2014) 156:20–44. doi: 10.1016/j.cell.2013.12.012
22. Bao TZ, Han GZ, Shim JY, Wen Y, Jiang XR. Quantitative Structure-Activity Relationship of Various Endogenous Estrogen Metabolites for Human Estrogen Receptor Alpha and Beta Subtypes: Insights Into the Structural Determinants Favoring a Differential Subtype Binding. Endocrinology (2006) 147:4132–50. doi: 10.1210/EN.2006-0113
23. Ryan KJ. Biochemistry of Aromatase: Significance to Female Reproductive Physiology. Cancer Res (1982) 42:3342s–4s.
24. Cui J, Shen Y, Li R. Estrogen Synthesis and Signaling Pathways During Aging: From Periphery to Brain. Trends Mol Med (2013) 19:197–209. doi: 10.1016/J.MOLMED.2012.12.007
25. Millier SG, Whitelaw PF, Smyth CD. Follicular Oestrogen Synthesis: The “Two-Cell, Two-Gonadotrophin” Model Revisited. Mol Cell Endocrinol (1994) 100:51–4. doi: 10.1016/0303-7207(94)90278-X
26. Nilsson S, Mäkelä S, Treuter E, Tujague M, Thomsen J, Andersson G, et al. Mechanisms of Estrogen Action. Physiol Rev (2001) 81:1535–65. doi: 10.1152/PHYSREV.2001.81.4.1535
27. Kuiper GGJM, Carlsson B, Grandien K, Enmark E, Häggblad J, Nilsson S, et al. Comparison of the Ligand Binding Specificity and Transcript Tissue Distribution of Estrogen Receptors Alpha and Beta. Endocrinology (1997) 138:863–70. doi: 10.1210/ENDO.138.3.4979
28. Jadhav RR, Ye Z, Huang RL, Liu J, Hsu PY, Huang YW, et al. Genome-Wide DNA Methylation Analysis Reveals Estrogen-Mediated Epigenetic Repression of Metallothionein-1 Gene Cluster in Breast Cancer. Clin Epigenet (2015) 7:13. doi: 10.1186/S13148-015-0045-9
29. Fortress AM, Frick KM. Epigenetic Regulation of Estrogen-Dependent Memory. Front Neuroendocrinol (2014) 35:530–49. doi: 10.1016/J.YFRNE.2014.05.001
30. Sklias A, Halaburkova A, Vanzan L, Jimenez NF, Cuenin C, Bouaoun L, et al. Epigenetic Remodelling of Enhancers in Response to Estrogen Deprivation and Re-Stimulation. Nucleic Acids Res (2021) 49:9738–54. doi: 10.1093/NAR/GKAB697
31. Jeffery E, Wing A, Holtrup B, Sebo Z, Kaplan JL, Saavedra-Peña R, et al. The Adipose Tissue Microenvironment Regulates Depot-Specific Adipogenesis in Obesity. Cell Metab (2016) 24:142–50. doi: 10.1016/J.CMET.2016.05.012
32. Min SY, Desai A, Yang Z, Sharma A, DeSouza T, Genga RMJ, et al. Diverse Repertoire of Human Adipocyte Subtypes Develops From Transcriptionally Distinct Mesenchymal Progenitor Cells. Proc Natl Acad Sci USA (2019) 116:17970–9. doi: 10.1073/PNAS.1906512116
33. Nilsson M, Dahlman I, Rydén M, Nordström EA, Gustafsson J-A, Arner P, et al. Oestrogen Receptor Alpha Gene Expression Levels are Reduced in Obese Compared to Normal Weight Females. Int J Obes (Lond) (2007) 31:900–7. doi: 10.1038/SJ.IJO.0803528
34. Zhou Z, Moore TM, Drew BG, Ribas V, Wanagat J, Civelek M, et al. Estrogen Receptor α Controls Metabolism in White and Brown Adipocytes by Regulating Polg1 and Mitochondrial Remodeling. Sci Transl Med (2020) 12:eaax8096. doi: 10.1126/SCITRANSLMED.AAX8096
35. Babaei P, Dastras A, Tehrani BS, Pourali Roudbaneh S. The Effect of Estrogen Replacement Therapy on Visceral Fat, Serum Glucose, Lipid Profiles and Apelin Level in Ovariectomized Rats. J menopausal Med (2017) 23:182. doi: 10.6118/JMM.2017.23.3.182
36. Dam TV, Dalgaard LB, Thomsen CB, Hjortebjerg R, Ringgaard S, Johansen FT, et al. Estrogen Modulates Metabolic Risk Profile After Resistance Training in Early Postmenopausal Women: A Randomized Controlled Trial. Menopause (2021) 28:1214–24. doi: 10.1097/GME.0000000000001841
37. Haarbo J, Marslew U, Gotfredsen A, Christiansen C. Postmenopausal Hormone Replacement Therapy Prevents Central Distribution of Body Fat After Menopause. Metabolism (1991) 40:1323–6. doi: 10.1016/0026-0495(91)90037-W
38. Gambacciani M, Ciaponi M, Cappagli B, Piaggesi L, De Simone L, Orlandi R, et al. Body Weight, Body Fat Distribution, and Hormonal Replacement Therapy in Early Postmenopausal Women. J Clin Endocrinol Metab (1997) 82:414–7. doi: 10.1210/JCEM.82.2.3735
39. Al-Qahtani SM, Bryzgalova G, Valladolid-Acebes I, Korach-André M, Dahlman-Wright K, Efendić S, et al. 17β-Estradiol Suppresses Visceral Adipogenesis and Activates Brown Adipose Tissue-Specific Gene Expression. Horm Mol Biol Clin Investig (2017) 29:13–26. doi: 10.1515/HMBCI-2016-0031
40. Mauvais-Jarvis F. Estrogen and Androgen Receptors: Regulators of Fuel Homeostasis and Emerging Targets for Diabetes and Obesity. Trends Endocrinol Metab (2011) 22:24–33. doi: 10.1016/J.TEM.2010.10.002
41. Lundholm L, Putnik M, Otsuki M, Andersson S, Ohlsson C, Gustafsson JÅ, et al. Effects of Estrogen on Gene Expression Profiles in Mouse Hypothalamus and White Adipose Tissue: Target Genes Include Glutathione Peroxidase 3 and Cell Death-Inducing DNA Fragmentation Factor, Alpha-Subunit-Like Effector A. J Endocrinol (2008) 196:547–57. doi: 10.1677/JOE-07-0277
42. D’Eon TM, Souza SC, Aronovitz M, Obin MS, Fried SK, Greenberg AS. Estrogen Regulation of Adiposity and Fuel Partitioning. Evidence of Genomic and non-Genomic Regulation of Lipogenic and Oxidative Pathways. J Biol Chem (2005) 280:35983–91. doi: 10.1074/JBC.M507339200
43. Bryzgalova G, Lundholm L, Portwood N, Gustafsson JÅ, Khan A, Efendic S, et al. Mechanisms of Antidiabetogenic and Body Weight-Lowering Effects of Estrogen in High-Fat Diet-Fed Mice. Am J Physiol - Endocrinol Metab (2008) 295:904–12. doi: 10.1152/AJPENDO.90248.2008
44. Rodriguez-Cuenca S, Monjo M, Proenza AM, Roca P. Depot Differences in Steroid Receptor Expression in Adipose Tissue: Possible Role of the Local Steroid Milieu. Am J Physiol Endocrinol Metab (2005) 288:200–7. doi: 10.1152/ajpendo.00270.2004.-Sex
45. Bélanger C, Luu-The V, Dupont P, Tchernof A. Adipose Tissue Intracrinology: Potential Importance of Local Androgen/Estrogen Metabolism in the Regulation of Adiposity. Horm Metab Res (2002) 34:737–45. doi: 10.1055/S-2002-38265
46. Clegg DJ, Brown LM, Woods SC, Benoit SC. Gonadal Hormones Determine Sensitivity to Central Leptin and Insulin. Diabetes (2006) 55:978–87. doi: 10.2337/DIABETES.55.04.06.DB05-1339
47. Gao Q, Mezei G, Nie Y, Rao Y, Choi CS, Bechmann I, et al. Anorectic Estrogen Mimics Leptin’s Effect on the Rewiring of Melanocortin Cells and Stat3 Signaling in Obese Animals. Nat Med 2006 131 (2006) 13:89–94. doi: 10.1038/nm1525
48. Schneider G, Kirschner MA, Berkowitz R, Ertel NH. Increased Estrogen Production in Obese Men. J Clin Endocrinol Metab (1979) 48:633–8. doi: 10.1210/JCEM-48-4-633
49. Tchernof A, Després JP, Dupont A, Bélanger A, Nadeau A, Prud’homme D, et al. Relation of Steroid Hormones to Glucose Tolerance and Plasma Insulin Levels in Men. Importance of Visceral Adipose Tissue. Diabetes Care (1995) 18:292–9. doi: 10.2337/DIACARE.18.3.292
50. Simpson ER, Mahendroo MS, Means GD, Kilgore MW, Hinshelwood MM, Graham-Lorence S, et al. Aromatase Cytochrome P450, the Enzyme Responsible for Estrogen Biosynthesis. Endocr Rev (1994) 15:342–55. doi: 10.1210/EDRV-15-3-342
51. Brodsky IG, Balagopal P, Nair KS. Effects of Testosterone Replacement on Muscle Mass and Muscle Protein Synthesis in Hypogonadal Men–a Clinical Research Center Study. J Clin Endocrinol Metab (1996) 81:3469–75. doi: 10.1210/JCEM.81.10.8855787
52. Cohe PG. The Hypogonadal-Obesity Cycle: Role of Aromatase in Modulating the Testosterone-Estradiol Shunt–a Major Factor in the Genesis of Morbid Obesity. Med Hypotheses (1999) 52:49–51. doi: 10.1054/MEHY.1997.0624
53. Ohlsson C, Hammarstedt A, Vandenput L, Saarinen N, Ryberg H, Windahl SH, et al. Increased Adipose Tissue Aromatase Activity Improves Insulin Sensitivity and Reduces Adipose Tissue Inflammation in Male Mice. Am J Physiol - Endocrinol Metab (2017) 313:E450–62. doi: 10.1152/AJPENDO.00093.2017
54. Pedersen SB, Fuglsig S, Sjøgren P, Richelsen B. Identification of Steroid Receptors in Human Adipose Tissue. Eur J Clin Invest (1996) 26:1051–6. doi: 10.1046/J.1365-2362.1996.380603.X
55. Mizutani T, Nishikawa Y, Adachi H, Enomoto T, Ikegami H, Kurachi H, et al. Identification of Estrogen Receptor in Human Adipose Tissue and Adipocytes. J Clin Endocrinol Metab (1994) 78:950–4. doi: 10.1210/JCEM.78.4.8157726
56. Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS. Increased Adipose Tissue in Male and Female Estrogen Receptor-Alpha Knockout Mice. Proc Natl Acad Sci USA (2000) 97:12729–34. doi: 10.1073/PNAS.97.23.12729
57. Shi H, Kumar SPDS. Liu X. G Protein-Coupled Estrogen Receptor in Energy Homeostasis and Obesity Pathogenesis. Prog Mol Biol Transl Sci (2013) 114:193. doi: 10.1016/B978-0-12-386933-3.00006-6
58. Giudicelli Y, Dieudonne MN, Lacasa D, Pasquier YN, Pecquery R. Modulation by Sex Hormones of the Membranous Transducing System Regulating Fatty Acid Mobilization in Adipose Tissue. Prostaglandins Leukot Essent Fatty Acids (1993) 48:91–100. doi: 10.1016/0952-3278(93)90015-O
59. Cooke PS, Naaz A. Role of Estrogens in Adipocyte Development and Function. Exp Biol Med (Maywood) (2004) 229:1127–35. doi: 10.1177/153537020422901107
60. Yepuru M, Eswaraka J, Kearbey JD, Barrett CM, Raghow S, Veverka KA, et al. Estrogen Receptor-β-Selective Ligands Alleviate High-Fat Diet- and Ovariectomy-Induced Obesity in Mice. J Biol Chem (2010) 285:31292–303. doi: 10.1074/JBC.M110.147850/ATTACHMENT/357C29FE-9F5F-43EC-B0AC-C4C20266C48D/MMC1.PDF
61. Foryst-Ludwig A, Clemenz M, Hohmann S, Hartge M, Sprang C, Frost N, et al. Metabolic Actions of Estrogen Receptor Beta (Erβ) are Mediated by a Negative Cross-Talk With Pparγ. PloS Genet (2008) 4:e1000108. doi: 10.1371/JOURNAL.PGEN.1000108
62. Sutjarit N, Thongon N, Weerachayaphorn J, Piyachaturawat P, Suksamrarn A, Suksen K, et al. Inhibition of Adipogenic Differentiation of Human Bone Marrow-Derived Mesenchymal Stem Cells by a Phytoestrogen Diarylheptanoid From Curcuma Comosa. J Agric Food Chem (2020) 68:9993–10002. doi: 10.1021/ACS.JAFC.0C04063
63. Tao Z, Zheng LD, Smith C, Luo J, Robinson A, Almeida FA, et al. Estradiol Signaling Mediates Gender Difference in Visceral Adiposity. via autophagy Cell Death Dis (2018) 9:309. doi: 10.1038/S41419-018-0372-9
64. Pedersen SB, Kristensen K, Hermann PA, Katzenellenbogen JA, Richelsen B. Estrogen Controls Lipolysis by Up-Regulating Alpha2a-Adrenergic Receptors Directly in Human Adipose Tissue Through the Estrogen Receptor Alpha. Implications for the Female Fat Distribution. J Clin Endocrinol Metab (2004) 89:1869–78. doi: 10.1210/JC.2003-031327
65. Shungin D, Winkler TW, Croteau-Chonka DC, Ferreira T, Locke AE, Mägi R, et al. New Genetic Loci Link Adipose and Insulin Biology to Body Fat Distribution. Nature (2015) 518:187–96. doi: 10.1038/nature14132
66. Lovejoy JC, Champagne CM, De Jonge L, Xie H, Smith SR. Increased Visceral Fat and Decreased Energy Expenditure During the Menopausal Transition. Int J Obes 2008 326 (2008) 32:949–58. doi: 10.1038/ijo.2008.25
67. Anderson LA, McTernan PG, Barnett AH, Kumar S. The Effects of Androgens and Estrogens on Preadipocyte Proliferation in Human Adipose Tissue: Influence of Gender and Site. J Clin Endocrinol Metab (2001) 86:5045–51. doi: 10.1210/JCEM.86.10.7955
68. Davis KE, Neinast MD, Sun K, Skiles WM, Bills JD, Zehr JA, et al. The Sexually Dimorphic Role of Adipose and Adipocyte Estrogen Receptors in Modulating Adipose Tissue Expansion, Inflammation, and Fibrosis. Mol Metab (2013) 2:227. doi: 10.1016/J.MOLMET.2013.05.006
69. Ribas V, Nguyen MTA, Henstridge DC, Nguyen A-K, Beaven SW, Watt MJ, et al. Impaired Oxidative Metabolism and Inflammation are Associated With Insulin Resistance in Erα-Deficient Mice. Am J Physiol - Endocrinol Metab (2010) 298:E304. doi: 10.1152/AJPENDO.00504.2009
70. Santos RS, Frank AP, Fátima LA, Palmer BF, Öz OK, Clegg DJ. Activation of Estrogen Receptor Alpha Induces Beiging of Adipocytes. Mol Metab (2018) 18:51. doi: 10.1016/J.MOLMET.2018.09.002
71. Liang YQ, Akishita M, Kim S, Ako J, Hashimoto M, Iijima K, et al. Estrogen Receptor Beta is Involved in the Anorectic Action of Estrogen. Int J Obes Relat Metab Disord (2002) 26:1103–9. doi: 10.1038/SJ.IJO.0802054
72. Levin ER. Plasma Membrane Estrogen Receptors. Trends Endocrinol Metab (2009) 20:477–82. doi: 10.1016/J.TEM.2009.06.009
73. Faure A, Vergnaud -Th M, Sutter-Dub Th M, Sutter Ch BJ. Immediate, Short- and Long-Term Opposite Effects of Oestradiol-17 Beta on Glucose Metabolism in Rat Adipocytes: Relationship With the Biphasic Changes in Body Weight and Food Intake. J Endocrinol (1984) 101:13–9. doi: 10.1677/JOE.0.1010013
74. Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an Estrogen Membrane Receptor Coupled to a G Protein in Human Breast Cancer Cells. Endocrinology (2005) 146:624–32. doi: 10.1210/EN.2004-1064
75. Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A Transmembrane Intracellular Estrogen Receptor Mediates Rapid Cell Signaling. Science (2005) 307:1625–30. doi: 10.1126/SCIENCE.1106943
76. Liu S, Mauvais-Jarvis F. Minireview: Estrogenic Protection of β-Cell Failure in Metabolic Diseases. Endocrinology (2010) 151:859–64. doi: 10.1210/EN.2009-1107
77. Wang A, Luo J, Moore W, Alkhalidy H, Wu L, Zhang J, et al. GPR30 Regulates Diet-Induced Adiposity in Female Mice and Adipogenesis In Vitro. Sci Rep (2016) 6:34302. doi: 10.1038/SREP34302
78. Yun SL, YK A, Jin WC, Kim M, Yasue S, Hee JS, et al. Dysregulation of Adipose Glutathione Peroxidase 3 in Obesity Contributes to Local and Systemic Oxidative Stress. Mol Endocrinol (2008) 22:2176–89. doi: 10.1210/ME.2008-0023
79. Sul OJ, Hyun HJ, Rajasekaran M, Suh JH, Choi HS. Estrogen Enhances Browning in Adipose Tissue by M2 Macrophage Polarization. via heme oxygenase-1 J Cell Physiol (2021) 236:1875–88. doi: 10.1002/JCP.29971
80. Narumi M, Takahashi K, Yamatani H, Seino M, Yamanouchi K, Ohta T, et al. Oxidative Stress in the Visceral Fat Is Elevated in Postmenopausal Women With Gynecologic Cancer. J Womens Health (Larchmt) (2018) 27:99–106. doi: 10.1089/JWH.2016.6301.
81. Wang T, Si Y, Shirihai OS, Si H, Schultz V, Corkey RF, et al. Respiration in Adipocytes is Inhibited by Reactive Oxygen Species. Obesity (2010) 18:1493–502. doi: 10.1038/oby.2009.456
82. Jones AR IV, Meshulam T, Oliveira MF, Burritt N, Corkey BE. Extracellular Redox Regulation of Intracellular Reactive Oxygen Generation, Mitochondrial Function and Lipid Turnover in Cultured Human Adipocytes. PloS One (2016) 11:e0164011. doi: 10.1371/journal.pone.0164011
83. Castro JP, Grune T, Speckmann B. The Two Faces of Reactive Oxygen Species (ROS) in Adipocyte Function and Dysfunction. Biol Chem (2016) 397:709–24. doi: 10.1515/hsz-2015-0305
84. Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte Dysfunctions Linking Obesity to Insulin Resistance and Type 2 Diabetes. Nat Rev Mol Cell Biol (2008) 9:367–77. doi: 10.1038/nrm2391
85. Ivanova MM, Radde BN, Son J, Mehta FF, Chung SH, Klinge CM. Estradiol and Tamoxifen Regulate NRF-1 and Mitochondrial Function in Mouse Mammary Gland and Uterus. J Mol Endocrinol (2013) 51:233–46. doi: 10.1530/JME-13-0051
86. Mattingly KA, Ivanova MM, Riggs KA, Wickramasinghe NS, Barch MJ, Klinge CM. Estradiol Stimulates Transcription of Nuclear Respiratory Factor-1 and Increases Mitochondrial Biogenesis. Mol Endocrinol (2008) 22:609–22. doi: 10.1210/ME.2007-0029
87. Longo M, Zatterale F, Naderi J, Parrillo L, Formisano P, Raciti GA, et al. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. Int J Mol Sci (2019) 20:2358. doi: 10.3390/ijms20092358
88. Heyn GS, Corrêa LH, Magalhães KG. The Impact of Adipose Tissue-Derived miRNAs in Metabolic Syndrome, Obesity, and Cancer. Front Endocrinol (Lausanne) (2020) 11:563816. doi: 10.3389/FENDO.2020.563816
89. Li X, Peng B, Zhu X, Wang P, Sun K, Lei X, et al. MiR-210-3p Inhibits Osteogenic Differentiation and Promotes Adipogenic Differentiation Correlated With Wnt Signaling in Erα-Deficient rBMSCs. J Cell Physiol (2019) 234:23475–84. doi: 10.1002/JCP.28916
90. Bjorkman S, Taylor HS. MicroRNAs in Endometriosis: Biological Function and Emerging Biomarker Candidates†. Biol Reprod (2019) 100:1135–46. doi: 10.1093/BIOLRE/IOZ014
91. Cosar E, Mamillapalli R, Ersoy GS, Cho SY, Seifer B, Taylor HS. Serum microRNAs as Diagnostic Markers of Endometriosis: A Comprehensive Array-Based Analysis. Fertil Steril (2016) 106:402–9. doi: 10.1016/J.FERTNSTERT.2016.04.013
92. Cho S, Mutlu L, Grechukhina O, Taylor HS. Circulating microRNAs as Potential Biomarkers for Endometriosis. Fertil Steril (2015) 103:1252–1260.e1. doi: 10.1016/J.FERTNSTERT.2015.02.013
93. Grechukhina O, Petracco R, Popkhadze S, Massasa E, Paranjape T, Chan E, et al. A Polymorphism in a Let-7 microRNA Binding Site of KRAS in Women With Endometriosis. EMBO Mol Med (2012) 4:206–17. doi: 10.1002/EMMM.201100200
94. Zolbin MM, Mamillapalli R, Nematian SE, Goetz L, Taylor HS. Adipocyte Alterations in Endometriosis: Reduced Numbers of Stem Cells and microRNA Induced Alterations in Adipocyte Metabolic Gene Expression. Reprod Biol Endocrinol (2019) 17:36. doi: 10.1186/S12958-019-0480-0
95. Mori MA, Ludwig RG, Garcia-Martin R, Brandão BB, Kahn CR. Extracellular miRNAs: From Biomarkers to Mediators of Physiology and Disease. Cell Metab (2019) 30:656–73. doi: 10.1016/J.CMET.2019.07.011
96. Garcia-Martin R, Wang G, Brandão BB, Zanotto TM, Shah S, Kumar Patel S, et al. MicroRNA Sequence Codes for Small Extracellular Vesicle Release and Cellular Retention. Nat 2021 (2021) 601:446–51. doi: 10.1038/s41586-021-04234-3
97. Gao W, Liu J-L, Lu X, Yang Q. Epigenetic Regulation of Energy Metabolism in Obesity. J Mol Cell Biol (2021) 13:480–99. doi: 10.1093/JMCB/MJAB043
98. Pant R, Firmal P, Shah VK, Alam A, Chattopadhyay S. Epigenetic Regulation of Adipogenesis in Development of Metabolic Syndrome. Front Cell Dev Biol (2021) 8:619888. doi: 10.3389/FCELL.2020.619888
99. Kovács T, Szabó-Meleg E, Ábrahám IM. Estradiol-Induced Epigenetically Mediated Mechanisms and Regulation of Gene Expression. Int J Mol Sci 2020 (2020) 2121:3177. doi: 10.3390/IJMS21093177
100. Gadaleta RM, Magnani L. Nuclear Receptors and Chromatin: An Inducible Couple. J Mol Endocrinol (2014) 52:R137–49. doi: 10.1530/JME-13-0170
101. Bannister AJ, Kouzarides T. Regulation of Chromatin by Histone Modifications. Cell Res (2011) 21:381–95. doi: 10.1038/cr.2011.22
102. Zhao S, Allis CD, Wang GG. The Language of Chromatin Modification in Human Cancers. Nat Rev Cancer (2021) 21:413–30. doi: 10.1038/S41568-021-00357-X
103. Shi L, Sun L, Li Q, Liang J, Yu W, Yi X, et al. Histone Demethylase JMJD2B Coordinates H3K4/H3K9 Methylation and Promotes Hormonally Responsive Breast Carcinogenesis. Proc Natl Acad Sci USA (2011) 108:7541–6. doi: 10.1073/PNAS.1017374108
104. Gole B, Wiesmüller L. Leukemogenic Rearrangements at the Mixed Lineage Leukemia Gene (MLL)—multiple Rather Than a Single Mechanism. Front Cell Dev Biol (2015) 3:41. doi: 10.3389/FCELL.2015.00041
105. Goldsworthy M, Absalom NL, Schröter D, Matthews HC, Bogani D, Moir L, et al. Mutations in Mll2, an H3K4 Methyltransferase, Result in Insulin Resistance and Impaired Glucose Tolerance in Mice. PloS One (2013) 8:e61870. doi: 10.1371/JOURNAL.PONE.0061870
106. Yap KL, Johnson AEK, Fischer D, Kandikatla P, Deml J, Nelakuditi V, et al. Congenital Hyperinsulinism as the Presenting Feature of Kabuki Syndrome: Clinical and Molecular Characterization of 9 Affected Individuals. Genet Med (2019) 21:233–42. doi: 10.1038/S41436-018-0013-9
107. Jang MK, Kim JH, Jung MH. Histone H3K9 Demethylase JMJD2B Activates Adipogenesis by Regulating H3K9 Methylation on Pparγ and C/Ebpα During Adipogenesis. PloS One (2017) 12:e0168185. doi: 10.1371/JOURNAL.PONE.0168185
108. Dang ZC, Löwik CWGM. The Balance Between Concurrent Activation of ERs and PPARs Determines Daidzein-Induced Osteogenesis and Adipogenesis. J Bone Miner Res (2004) 19:853–61. doi: 10.1359/JBMR.040120
109. Foryst-Ludwig A, Clemenz M, Hohmann S, Hartge M, Sprang C, Frost N, et al. Metabolic Actions of Estrogen Receptor Beta (ERbeta) are Mediated by a Negative Cross-Talk With PPARgamma. PloS Genet (2008) 4:e1000108. doi: 10.1371/JOURNAL.PGEN.1000108
110. Jeong S, Yoon M. 17β-Estradiol Inhibition of Pparγ-Induced Adipogenesis and Adipocyte-Specific Gene Expression. Acta Pharmacol Sin (2011) 32:230–8. doi: 10.1038/APS.2010.198
111. Hong L, Colpan A, Peptan IA. Modulations of 17-Beta Estradiol on Osteogenic and Adipogenic Differentiations of Human Mesenchymal Stem Cells. Tissue Eng (2006) 12:2747–53. doi: 10.1089/TEN.2006.12.2747
112. Bitirim CV, Ozer ZB, Akcali KC. Estrogen Receptor Alpha Regulates the Expression of Adipogenic Genes Genetically and Epigenetically in Rat Bone Marrow-Derived Mesenchymal Stem Cells. PeerJ (2021) 9:e12071. doi: 10.7717/PEERJ.12071
113. Pachano T, Sánchez-Gaya V, Ealo T, Mariner-Faulí M, Bleckwehl T, Asenjo HG, et al. Orphan CpG Islands Amplify Poised Enhancer Regulatory Activity and Determine Target Gene Responsiveness. Nat Genet 2021 537 (2021) 53:1036–49. doi: 10.1038/s41588-021-00888-x
114. Mohn F, Weber M, Rebhan M, Roloff TC, Richter J, Stadler MB, et al. Lineage-Specific Polycomb Targets and De Novo DNA Methylation Define Restriction and Potential of Neuronal Progenitors. Mol Cell (2008) 30:755–66. doi: 10.1016/J.MOLCEL.2008.05.007
115. Okano M, Bell DW, Haber DA, Li E. DNA Methyltransferases Dnmt3a and Dnmt3b Are Essential for De Novo Methylation and Mammalian Development. Cell (1999) 99:247–57. doi: 10.1016/S0092-8674(00)81656-6
116. Kittler R, Zhou J, Hua S, Ma L, Liu Y, Pendleton E, et al. A Comprehensive Nuclear Receptor Network for Breast Cancer Cells. Cell Rep (2013) 3:538–51. doi: 10.1016/J.CELREP.2013.01.004
117. Madsen JGS, Madsen MS, Rauch A, Traynor S, Van Hauwaert EL, Haakonsson AK, et al. Highly Interconnected Enhancer Communities Control Lineage-Determining Genes in Human Mesenchymal Stem Cells. Nat Genet (2020) 52:1227–38. doi: 10.1038/S41588-020-0709-Z
118. Jin X, Li Y, Guo Y, Jia Y, Qu H, Lu Y, et al. Erα is Required for Suppressing OCT4-Induced Proliferation of Breast Cancer Cells via DNMT1/ISL1/ERK axis. Cell Prolif (2019) 52:e12612. doi: 10.1111/CPR.12612
119. Rüegg J, Cai W, Karimi M, Kiss NB, Swedenborg E, Larsson C, et al. Epigenetic Regulation of Glucose Transporter 4 by Estrogen Receptor β. Mol Endocrinol (2011) 25:2017–28. doi: 10.1210/ME.2011-1054
120. Liu Y, Duong W, Krawczyk C, Bretschneider N, Borbély G, Varshney M, et al. Oestrogen Receptor β Regulates Epigenetic Patterns at Specific Genomic Loci Through Interaction With Thymine DNA Glycosylase. Epigenet Chromatin (2016) 9:7. doi: 10.1186/S13072-016-0055-7
121. Hou Y, Zhang Z, Wang Y, Gao T, Liu X, Tang T, et al. 5mc Profiling Characterized TET2 as an Anti-Adipogenic Demethylase. Gene (2020) 733:144265. doi: 10.1016/J.GENE.2019.144265
122. Lai B, Lee JE, Jang Y, Lifeng W, Peng W, Ge K. MLL3/MLL4 are Required for CBP/p300 Binding on Enhancers and Super-Enhancer Formation in Brown Adipogenesis. Nucleic Acids Res (2017) 45:6388–403. doi: 10.1093/NAR/GKX234
123. Miao YF, Su W, Dai YB, Wu WF, Huang B, Barros RPA, et al. An Erβ Agonist Induces Browning of Subcutaneous Abdominal Fat Pad in Obese Female Mice. Sci Rep (2016) 6:38579. doi: 10.1038/SREP38579
124. Nanduri R. Epigenetic Regulators of White Adipocyte Browning. Epigenomes 2021 (2021) 55:3. doi: 10.3390/EPIGENOMES5010003
125. Raajendiran A, Ooi G, Bayliss J, O’Brien PE, Schittenhelm RB, Clark AK, et al. Identification of Metabolically Distinct Adipocyte Progenitor Cells in Human Adipose Tissues. Cell Rep (2019) 27:1528–1540.e7. doi: 10.1016/J.CELREP.2019.04.010
126. Biagi CAO, Cury SS, Alves CP, Rabhi N, Silva WA, Farmer SR, et al. Multidimensional Single-Nuclei RNA-Seq Reconstruction of Adipose Tissue Reveals Adipocyte Plasticity Underlying Thermogenic Response. Cells (2021) 10:3073. doi: 10.3390/CELLS10113073
Keywords: sexual dimorphism, steroids, estrogen, adipocyte, epigenetic
Citation: Bjune J-I, Strømland PP, Jersin RÅ, Mellgren G and Dankel SN (2022) Metabolic and Epigenetic Regulation by Estrogen in Adipocytes. Front. Endocrinol. 13:828780. doi: 10.3389/fendo.2022.828780
Received: 03 December 2021; Accepted: 24 January 2022;
Published: 22 February 2022.
Edited by:
Dwight J. Klemm, University of Colorado Anschutz Medical Campus, United StatesReviewed by:
Deborah Clegg, Cedars Sinai Medical Center, United StatesMaria Marino, Roma Tre University, Italy
Copyright © 2022 Bjune, Strømland, Jersin, Mellgren and Dankel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simon Nitter Dankel, c2ltb24uZGFua2VsQHVpYi5ubw==
†ORCID: Jan-Inge Bjune, orcid.org/0000-0001-8255-4282
Pouda Panahandeh Strømland, orcid.org/0000-0003-4733-801X
‡These authors have contributed equally to this work