 Lucas S. Santana
Lucas S. Santana Augusto G. Guimaraes
Augusto G. Guimaraes Madson Q. Almeida
Madson Q. Almeida- 1Unidade de Adrenal, Laboratório de Hormônios e Genética Molecular Laboratório de Investigação Médica 42 (LIM/42), Serviço de Endocrinologia e Metabologia, Hospital das Clínicas, Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil
- 2Divisão de Oncologia Endócrina, Instituto do Câncer do Estado de São Paulo (ICESP), Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil
Primary aldosteronism (PA) is the most common form of secondary arterial hypertension, with a prevalence of approximately 20% in patients with resistant hypertension. In the last decade, somatic pathogenic variants in KCNJ5, CACNA1D, ATP1A1 and ATP2B3 genes, which are involved in maintaining intracellular ionic homeostasis and cell membrane potential, were described in aldosterone-producing adenomas (aldosteronomas). All variants in these genes lead to the activation of calcium signaling, the major trigger for aldosterone production. Genetic causes of familial hyperaldosteronism have been expanded through the report of germline pathogenic variants in KCNJ5, CACNA1H and CLCN2 genes. Moreover, PDE2A and PDE3B variants were associated with bilateral PA and increased the spectrum of genetic etiologies of PA. Of great importance, the genetic investigation of adrenal lesions guided by the CYP11B2 staining strongly changed the landscape of somatic genetic findings of PA. Furthermore, CYP11B2 staining allowed the better characterization of the aldosterone-producing adrenal lesions in unilateral PA. Aldosterone production may occur from multiple sources, such as solitary aldosteronoma or aldosterone-producing nodule (classical histopathology) or clusters of autonomous aldosterone-producing cells without apparent neoplasia denominated aldosterone-producing micronodules (non-classical histopathology). Interestingly, KCNJ5 mutational status and classical histopathology of unilateral PA (aldosteronoma) have emerged as relevant predictors of clinical and biochemical outcome, respectively. In this review, we summarize the most recent advances in the pathogenesis of PA and discuss their impact on clinical outcome.
Introduction
Arterial hypertension (AH) represents one of the main risk factors for premature death, affecting about 10 to 40% of the world population (1, 2). Primary aldosteronism (PA) is the most frequent cause of endocrine AH, with a prevalence of around 4% and 10% in hypertensive patients treated in primary and tertiary care services, respectively, reaching around 20% of patients with resistant AH (3–6).
PA is characterized by autonomous production of aldosterone, independent of the renin-angiotensin system. As a consequence, sodium retention, plasma renin suppression, blood pressure (BP) elevation and K+ excretion increase occur, with consequent cardiovascular damage (7). The latter is due to the fact that excess of aldosterone exerts its deleterious cardiovascular effects independent of blood pressure levels, resulting in higher cardiovascular morbidity and mortality in patients with PA when compared with patients with essential AH (8, 9).
Aldosterone is a mineralocorticoid hormone, which is synthesized by the zona glomerulosa (ZG) of the adrenal cortex. Its play a major role in electrolyte regulation through sodium and water renal reabsorption (10, 11). Aldosterone is synthetized from cholesterol and its biosynthesis is under the control of two principal factors: angiotensin II (Ang II) and extracellular potassium concentration (K+) (10).
Stimulation of ZG cells by Ang II or an increase in plasma K+ concentration leads to cell membrane depolarization and increase in intracellular Ca2+, by opening of voltage-gated Ca2+ channels and inositol triphosphate-dependent Ca2+ release from the endoplasmic reticulum. The increase of intracellular Ca2+ leads to activation of a phosphorylation cascade that positively regulate aldosterone synthesis and cell proliferation, specifically by increasing the CYP11B2 gene transcription (10, 12, 13).
Effects of aldosterone are mediated through the mineralocorticoid receptor (MR), a hormone dependent transcription factor that is expressed in non-epithelial tissues, such as the heart and vessels, and in epithelial tissues such as the salivary glands and kidney distal tubule, where aldosterone regulates sodium/water reabsorption and potassium excretion (10).
The main causes of PA are bilateral cortical adrenal hyperplasia (idiopathic hyperaldosteronism) and aldosteronomas (14). Idiopathic hyperaldosteronism is caused by bilateral nodular hyperplasia originating from the cortical zona glomerulosa, whereas aldosteronomas are aldosterone-producing adenomas usually measuring between 1-3 cm (but can even measure less than 1 cm). Each of these accounts for about 50-60% and 40-50% of PA cases, respectively (7, 14).
The two major causes of PA account for more than 95% of cases, with approximately 5% of bilateral hyperplasia occurring in a familial context. Thus, bilateral hyperplasia remains without a defined genetic etiology in most cases. Although somatic allelic variants are identified in about 90% of aldosteronomas, few advances have been made in the genetic elucidation of bilateral PA (10, 15).
Several genes that encode ion channels that modulate zona glomerulosa cell depolarization and aldosterone synthesis pathways have already been associated with the pathogenesis of PA (Figure 1), differing in prevalence among aldosteronomas and familial PA cohorts (12). The aim of this review is to discuss the most recent discoveries about the PA pathogenesis, as well as the clinical and prognostic impact of the genetic characterization of this very prevalent disorder, associated with a high cardiovascular morbidity.
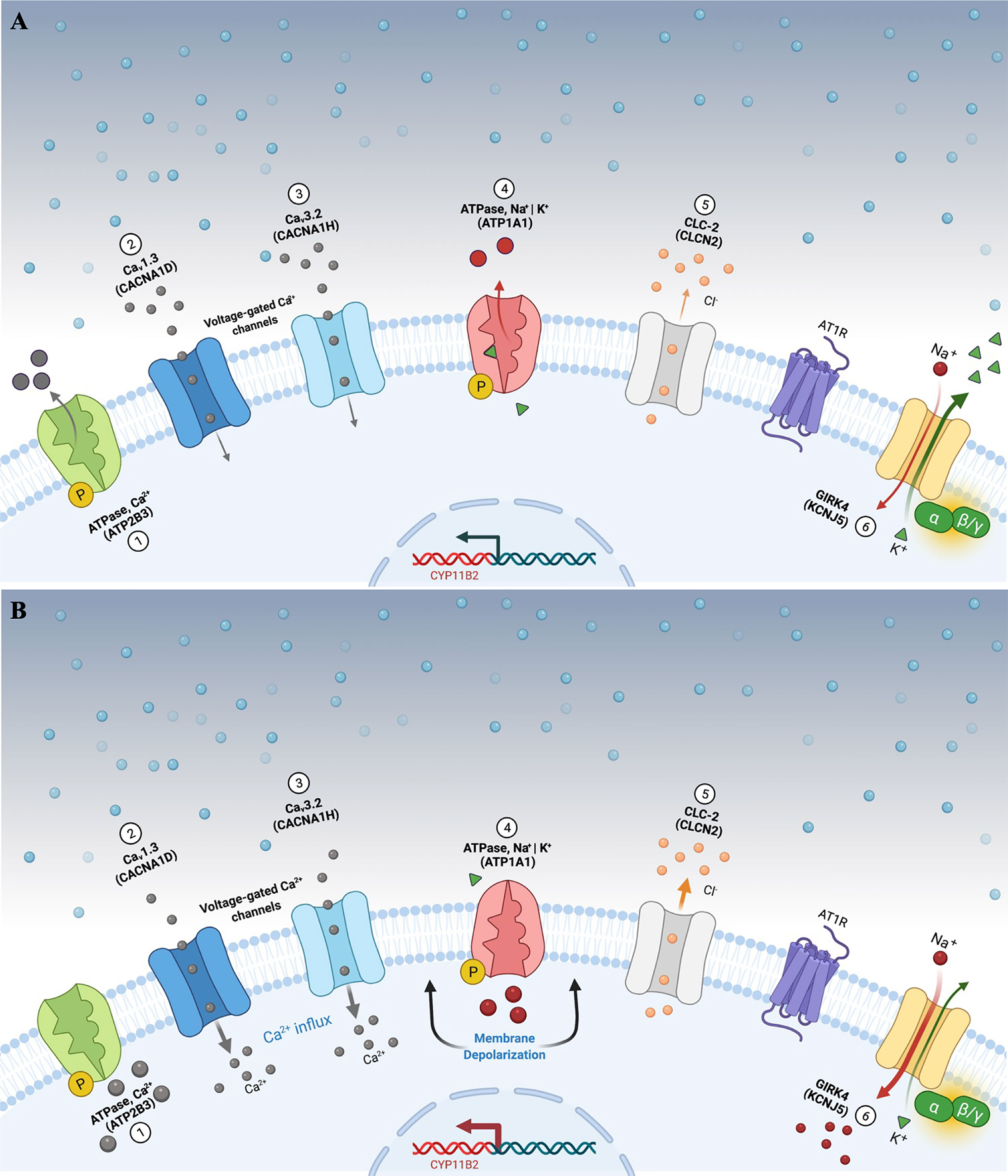
Figure 1 Aldosterone secretion in adrenal zona glomerulosa cells under physiological (A) and pathological (B) conditions. (A) Adrenal glomerulosa cell resting. The normal resting potential of zona glomerulosa cells is hyperpolarized (highly negative membrane potential). Activation of the angiotensin receptor (AT1R) by angiotensin II or extracellular hyperkalemia results in membrane depolarization and calcium influx via activated voltage-gated calcium channels. Calcium influx activates signaling to increase expression of aldosterone synthase (CYP11B2) and ultimately aldosterone production. (B) Genetic alterations leading to cell membrane depolarization, intracellular ionic modification, and autonomous aldosterone secretion in PA. Pathogenic variants in the KCNJ5 gene (G-protein-activated inward rectifier potassium channel GIRK4) [6] promote loss of channel K+ selectivity and increase permeability to Na+, leading to membrane depolarization and calcium influx via voltage-gated calcium channels. Similarly, impaired ATPase, Ca2+ (ATP2B3) [1]; Cav1.3 (CACNA1D) [2]; Cav3.2 (CACNA1H) [3]; ATPase, Na+ | K+ (ATP1A1) [4], and CLC-2 (CLCN2) [5] function results in cell membrane depolarization, calcium influx and autonomous aldosterone secretion. PA, primary aldosteronism.
Diagnosis and Clinical Management
According to the American Endocrine Society (The Endocrine Society), the following scenarios are indicated for PA screening in hypertensive patients: I) AH and hypokalemia (spontaneous or induced by diuretic therapy); II) AH and adrenal incidentaloma; III) Blood pressure >150x100 mmHg on three different occasions; IV) AH not controlled (≥140/90 mmHg) on three or more antihypertensive drugs (resistant AH); V) controlled AH (<140x90 mmHg) on four antihypertensive drugs (resistant AH); VI) AH associated with obstructive sleep apnea; VII) AH and family history of AH or cerebrovascular disease of the young (<40 years); VIII) AH in first-degree relatives of patients with PH (7).
PA screening should be performed with plasma aldosterone (A) and renin (R) measurements, with hypokalemia correction before the test. To avoid false negative results, diuretics and spironolactone should be withheld for at least 4 to 6 weeks before the test. Aldosterone concentration >10 ng/dL and an A/PRA ratio (plasma renin activity) ≥ 30 ng/dL/ng/mL/h or A/R ≥2.0 confer a sensitivity and specificity greater than 90% for PH diagnosis (7, 16, 17). It should be emphasized that A/PRA or A/R ratio should be calculated only for patients with suppressed or very low renin levels.
After laboratorial PA confirmation, patients should undergo adrenal computed tomography (CT) for etiologic characterization and exclusion of adrenal cortical carcinoma. Adrenal CT has limited accuracy (around 60-70%), especially for detection of small (<1 cm) aldosteronomas (7) or for patients with bilateral nodules (to differentiate non-functioning or cortisol producing-adenomas). Therefore, adrenal vein sampling (AVS) is indicated for the majority of patients with PA for the proper characterization of the lateralization of aldosterone production (7, 16). Although AVS is the gold standard approach to define aldosterone lateralization, it should be carried out only in centers with expertise for this procedure and by a very experienced interventional radiologist. In addition, AVS should be considered only if laparoscopic surgery is a treatment option. A detailed description of PA work-up investigation is beyond the scope of this review.
Treatment of unilateral PA consists of laparoscopic adrenalectomy. The bilateral hyperplasia is treated with a mineralocorticoid antagonist (spironolactone or eplerenone). Both approaches are associated with reduced cardiovascular morbidity caused by excess of aldosterone (18, 19). The reduction of cardiovascular risk after medical treatment for PA is associated with normalization of renin levels (PRA >1 ng/mL/h) (20).
Familial Hyperaldosteronism
Familial hyperaldosteronism (FH) is rare, but likely a highly underdiagnosed entity due to lack of routine screening (Table 1). Therefore, there is a lack of prevalence data for most of pathogenic variants listed in Table 1. The first report of FH occurred in 1966 (30), with subsequent characterization of its molecular etiology (21). This autosomal dominant form of PH was caused by a chimeric gene consisting of the 11β-hydroxylase promoter (CYP11B1) and aldosterone synthase (CYP11B2) coding region, resulting from a non-homologous pairing during crossing-over. Therefore, aldosterone synthesis becomes now regulated by adrenocorticotropic hormone (ACTH) instead of Ang II (21). This presentation of familial PH was then termed FH type 1 (OMIM #103900), or glucocorticoid-suppressible hyperaldosteronism.

Table 1 Genetic causes of familial hyperaldosteronism.
A diagnosis of FH 1 is highly suggestive if aldosterone suppression (<4 ng/dL) occurs after a dexamethasone suppression test (0.5 mg each 6h for 48h). However, the FH 1 diagnosis should be confirmed by the presence of the chimeric gene in a long range PCR (31). The treatment of FH 1 consists of low dose dexamethasone administration in adults (0.125–0.25mg/d) to suppress ACTH and block aldosterone synthesis (32, 33). If additional blood pressure control is required, a mineralocorticoid antagonist can be added.
The molecular pathogenesis of Type 2 FH (OMIM #605635) consists of gain-of-function heterozygous germline variants in the CLCN2 gene (Table 1). Type 2 FH is characterized by autosomal dominant inheritance, incomplete penetrance and a family history of aldosteronoma or bilateral PA (34, 35). CLCN2 was mapped as a FH gene in 2018 and encodes an inwardly rectifying chloride channel (ClC-2), a member of the CLC voltage-gated Cl– channels family which is expressed in the cortical zona glomerulosa (28, 29).
So far, 6 missense pathogenic variants in CLCN2 have been reported in the literature associated with FH 2 (Table 2) (48). The presence of these alleles causes an increase in Cl- conductance through the channel, leading to a continuous depolarization of the plasma membrane, resulting in an increase of CYP11B2 expression and consequent stimulus for aldosterone synthesis (Figure 1B) (28, 29).
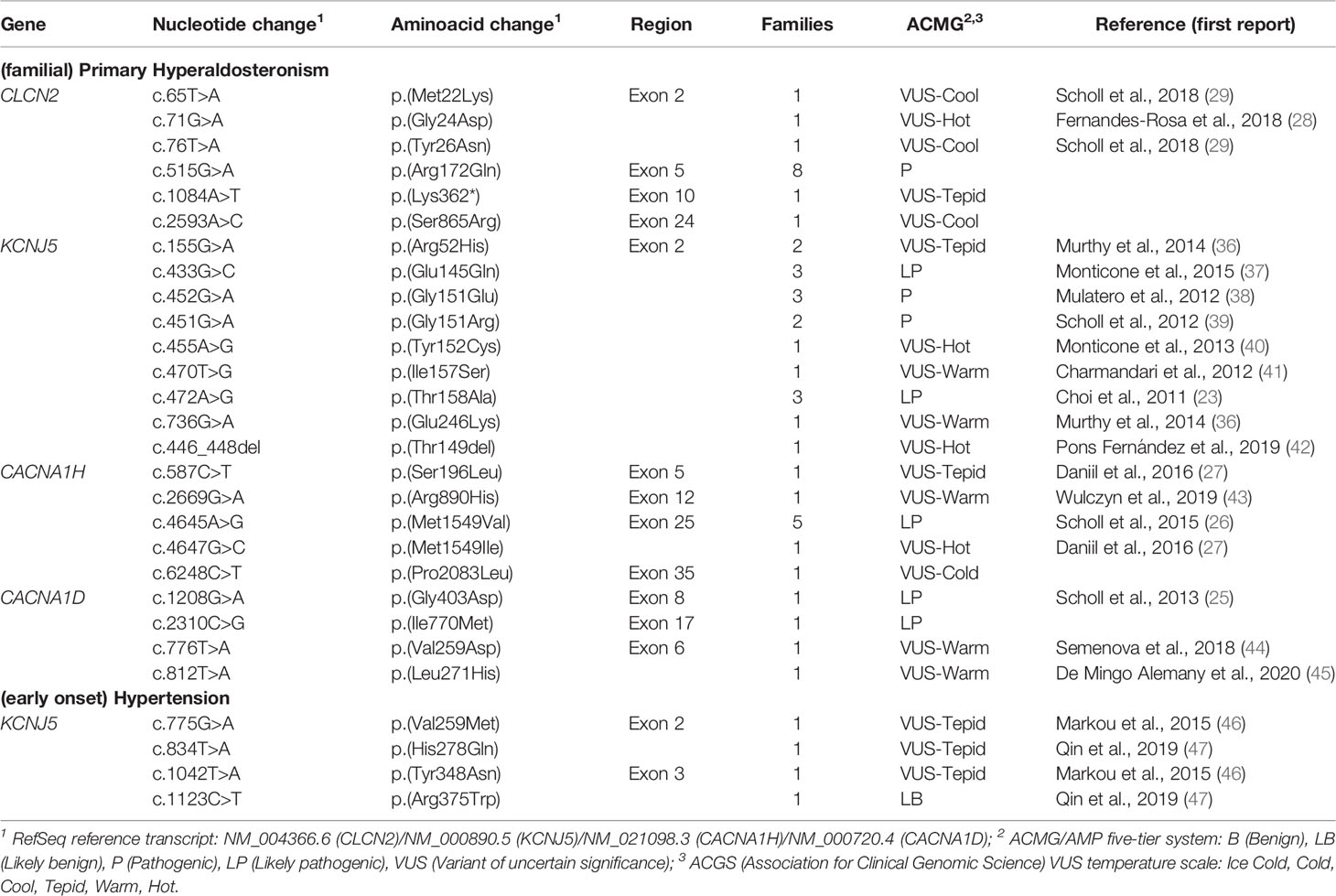
Table 2 Germline allelic variants identified in probands with (familial) primary hyperaldosteronism/(early onset) hypertension.
In 2008, individuals with childhood-onset PA, resistant AH, hypokalemia and bilateral macronodular adrenal hyperplasia were reported (31). In 2011, an inactivating germline variant in the KCNJ5 gene was identified in a case with a similar clinical presentation. Named FH 3 (OMIM #613677), this autosomal dominant PA subtype is caused by an impaired function of K+ GIRK4 (Kir3.4) potassium channel, which is encoded by KCNJ5 gene (23).
The molecular defect in the K+ GIRK4 potassium channel leads to the loss of its ionic selectivity, with a consequent increase in sodium conductance (Figure 1B). Naturally responsible for maintaining the zona glomerulosa membrane potential, it starts to act as a channel in favor of sodium influx, promoting continuous membrane depolarization and subsequent activation of voltage-dependent Ca+ channels. These increased intracellular calcium concentrations trigger CYP11B2 overexpression and aldosterone synthesis (39).
The genetic study of numerous PA cohorts and the consequent mapping of new KCNJ5 pathogenic variants allowed, over the years, to expand the phenotypic heterogeneity of this PA subtype (39–41) (Table 2). Certain alleles between amino acids residues 151-158 of the K+ GIRK4 potassium channel, more specifically p.(Gly151Arg), p.(Ile157Ser), and p.(Thr158Ala), are correlated with a more severe PA clinical presentation, with early-onset hypertension, more resistant to drug treatment and with a frequent need for bilateral adrenalectomy (24). On the other hand, some substitutions in this same region, namely p.(Gly151Glu) and p.(Tyr152Cys), result in a mild clinical presentation, with an adequate blood pressure control with aldosterone antagonists and without evidence of adrenal hyperplasia in CT evaluation (13, 39). Interestingly, in vitro experiments showed that mutant KCNJ5 channels can be undermined with the use of macrolide antibiotics such as roxithromycin and clarithromycin, suppressing CYP11B2 expression and aldosterone production (49).
Four KCNJ5 germline variants were reported in cohorts of patients with AH without a typical familial and biochemical diagnosis of PA (Table 2) (46, 47). The p.(His278Gln) variant, for example, was reported in a patient with resistant AH with normal serum K+ levels, plasma renin activity and aldosterone levels. The allele was inherited from his father who had essential AH without PA (47). None of the other reported cases had a phenotype similar to FH 3 patients, with early-onset medication-resistant hypertension, hypokalemia and bilateral adrenal macronodular hyperplasia (24).
Type 4 FH (OMIM #617027), the rarest subtype of PA, is caused by gain-of-function germline variants in CACNA1H gene (autosomal dominant inheritance), which encodes calcium voltage-gated channel subunit α1 H (Cav3.2) (26) (Tables 1, 2). Scholl et al. identified a recurrent heterozygous variant in the CACNA1H gene in five patients with early-onset PA (26). In silico studies with the identified p.(Met1549Val) mutant demonstrated an increase in calcium influx into zona glomerulosa cells, resulting in continual stimulation of aldosterone synthesis (50). Later studies demonstrated a late and incomplete penetrance of this PA subtype (27).
In 2013, Scholl et al. sequenced the candidate CACNA1D gene in 100 unrelated individuals with early-onset PA and identified two de novo heterozygous alleles in two girls with an undescribed syndrome featuring PA, AH, seizures and neuromuscular abnormalities (OMIM #615474) (25) (Table 2). This gene encodes the α 1D subunit of the L-type voltage-gated Ca2+ channel Cav1.3. The identified variants promote an activation of the Ca+2 channel at lower depolarization potentials, resulting in increased Ca+2 influx (25). Subsequently, two more cases were reported with de novo heterozygous CACNA1D variants, leading to a severe developmental disorder also associated with developmental delay, intellectual impairment, neurological symptoms (including seizures), and endocrine symptoms, evident as PA and/or congenital hyperinsulinemic hypoglycemia (44, 45).
Recently, rare heterozygous missense germline variants in the phosphodiesterase 2A (PDE2A) and 3B (PDE3B) genes were identified in 3 out of 11 patients with PA caused by bilateral hyperplasia (51). In addition, PDE2A was a marker of zona glomerulosa and aldosterone-producing hyperplastic areas and micronodules. In vitro functional studies supported the involvement of PDE2A and PDE3B in the pathogenesis of bilateral PA. PKA activity in frozen tissue was significantly higher in adrenals from patients harboring PDE2A and PDE3B variants. Interestingly, inactivating PDE2A and PDE3B variants increased SGK1 and SCNN1G/ENaCg at mRNA or protein levels (51).
SGK1 (serum and glucocorticoid inducible kinase-isoform 1) belongs to a large family of serine-threonine kinases. SGK1 is expressed in numerous tissues and plays a major role in transmembrane ionic transport, being established as an important regulator of Na+ transporters (52). Aldosterone is the most notorious hormonal regulator of SGK1 expression. After binding to the cytosolic mineralocorticoid receptor, aldosterone promotes the transcription of SGK1, which regulates a variety of ion transporters, such as ENaC (epithelial sodium channel). SGK1 reduces ENaC ubiquitination and degradation, as well as its cellular internalization (53). Therefore, PDE2A and PDE3B variants can induce aldosterone signaling by increasing SGK1/SCNN1G(ENag) (51). In addition, an increase in SGK1 activity also stimulates hypercoagulability, fibrosis and inflammation processes (54).
Unilateral Primary Aldosteronism
Aldosteronomas are a major cause of unilateral PA, associated with somatic variants in KCNJ5, CACNA1D, ATP1A1, ATP2B3, CLCN2, CACNA1H and CTNNB1 genes (Table 3). These genes drive autonomous aldosterone production and/or directly contribute for tumorigenesis (68). In 2011, Choi et al. identified recurrent KCNJ5 gain-of-function variants in aldosteronomas, namely p.(Gly151Arg) and p.(Leu168Arg), that affects residues at the channel ion selectivity filter (23) (Table 4 and Figure 1B).
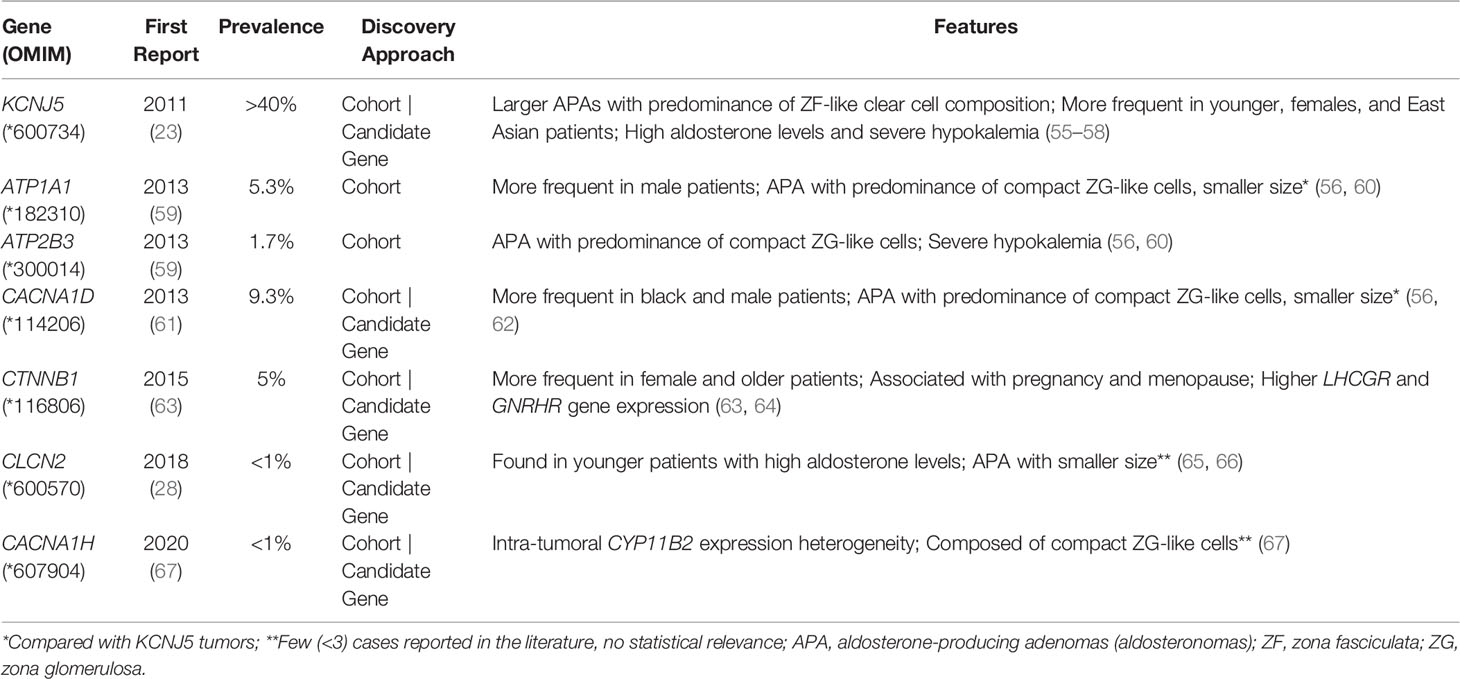
Table 3 Genetic causes of unilateral primary aldosteronism.
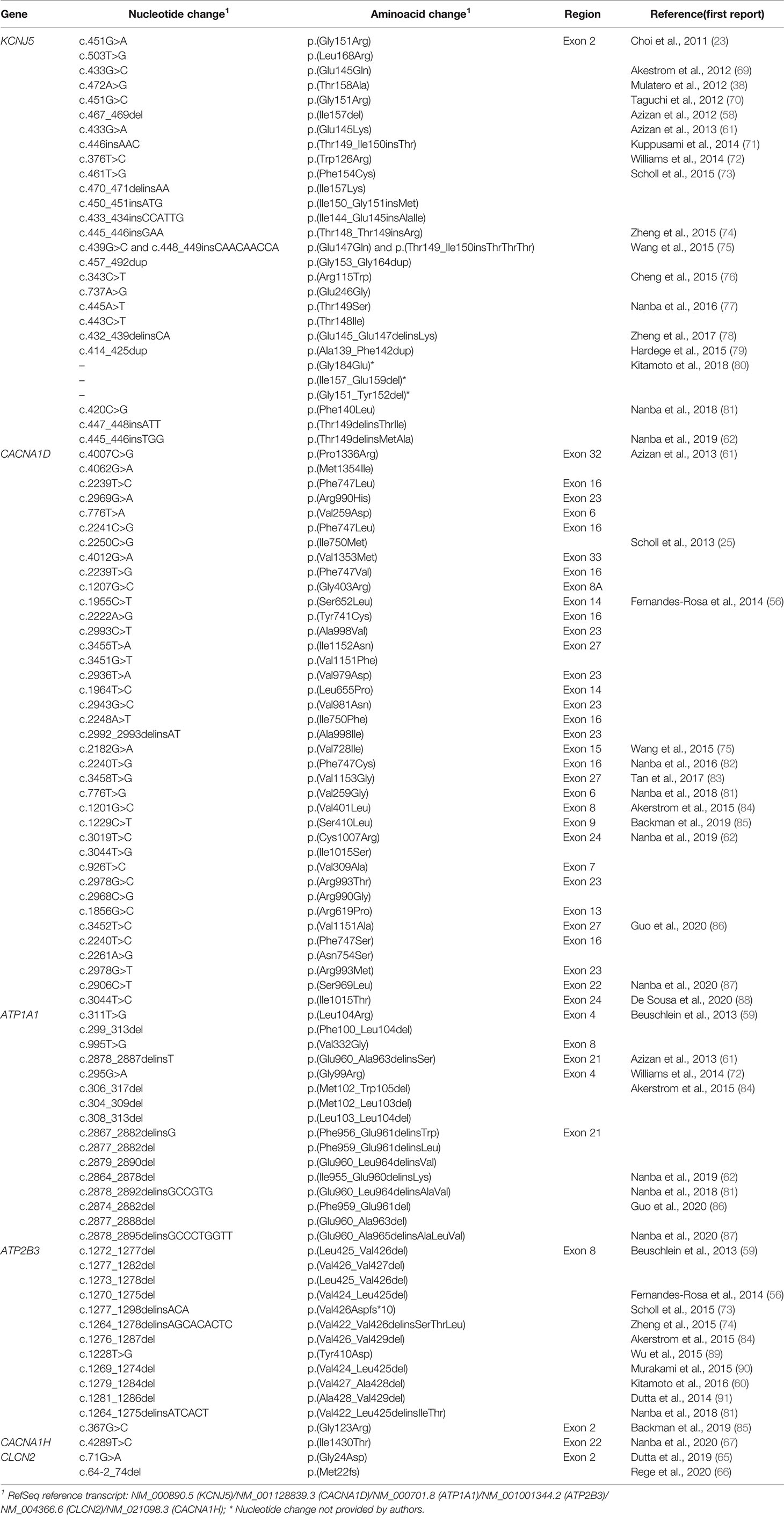
Table 4 Somatic variants identified in adrenal lesions associated with unilateral primary aldosteronism.
KCNJ5 is the most frequently affected gene in aldosteronomas (>40%), with even higher prevalence in Japanese and/or Eastern Asian cohorts (65-69% approximately). Characteristically, KCNJ5 mutant aldosteronomas are more frequent in female (>70%) and younger patients, with larger tumor size. Higher preoperative aldosterone and reduced potassium levels were also identified in these patients, which could contribute to early-onset disease, severity and earlier diagnosis (23, 24, 55, 74, 92).
In 2013 after KCNJ5 discovery, somatic CACNA1D gain-of-function variants were reported in aldosteronomas, with a prevalence of around 10%. CACNA1D encodes the α1 subunit Cav1.3 of a voltage dependent L-type (long-lasting) calcium channel and its pathogenic variants affect conserved residues within the channel activation gate (Table 3). Compared to wild-type, mutated Cav1.3 reaches activation in less depolarized membrane potentials, causing abnormal Ca+ influx, CYP11B2 expression, and aldosterone production (Table 4 and Figure 1B). In contrast with KCNJ5 related aldosteronomas, CACNA1D tumors are significantly smaller and more frequent in older male patients (25, 56, 61).
In 2013, Beuschlein et al. identified somatic variants in genes encoding ATPases, ATP1A1 and ATP2B3 in aldosteronomas (59). Missense and in-frame deletion variants in ATP1A1, which encodes Na+/K+ ATPase α subunit, impair pump activity and significantly reduce affinity for potassium, resulting in inappropriate membrane depolarization (Table 4 and Figure 1B). ATP2B3 encodes a Ca+ ATPase in which loss-of-function alleles (in-frame deletions) lead to a loss of physiological pump function, responsible for sodium and possibly calcium ions leaking into the cell, inducing membrane depolarization, and contributing to increased calcium concentrations. The combined prevalence of somatic variants in ATPases is around 7% and, until now, no ATPase pathogenic variants were found as germline or surrounding aldosteronoma tissue. Additionally, ATPase mutant aldosteronomas showed a high prevalence among older male patients (61, 93).
As found in other adrenocortical tumors, somatic gain of function variants in CTNNB1 gene, encoding β catenin, also have been reported in around 5% of aldosteronomas (Tables 3, 4). Affected adrenals had an aberrant β catenin accumulation in the Wnt cell-differentiation pathway and overexpression of luteinizing hormone/choriogonadotropin receptor (LHCGR) and gonadotropin-releasing hormone receptor (GnRHR) (63, 94, 95). Patients harboring aldosteronomas with CTNNB1 variants are more frequently females (60-70%) and older individuals, with no significant differences in preoperative aldosterone levels, tumor size and frequency familial hypertension compared with those with KCNJ5 variants (64). Unfortunately, the underlying mechanism leading to CYP11B2 overexpression due to CTNNB1 mutations remains unclear. Berthon et al. (96) showed that β-catenin plays an essential role in the control of basal and Angi II-induced aldosterone secretion, by activating AT1R, CYP21 and CYP11B2 transcription (96).
Due to recent advances in high throughput sequencing, few somatic variants have been recently identified in 2 genes only so far related to FH (CLCN2 and CACNA1H): the missense p.(Gly24Asp) (CLCN2), previously reported in FH 2 (28, 65); the splice junction loss c.64-2_74del (CLCN2) (65), and more recently, the missense p.(Ile1430Thr) (CACNA1H) (Table 4) (67).
The knowledge about adrenal lesions associated with PA and the detection rate of somatic variant have been significantly changed since the development of highly specific monoclonal antibodies against CYP11B1 and CYP11B2 (97). Under normal conditions, CYP11B2 was sporadically detected in the zona glomerulosa, whereas CYP11B1 was entirely detected in the zonae fasciculata-reticularis (98). In younger individuals, immunohistochemistry from normal adrenals reveals a continuous CYP11B2 expression throughout the ZG layer, but this pattern changes in adults and CYP11B2 expression becomes discontinuing in ZG (98, 99). Next, Nanba et al. demonstrated that CYP11B2 immunostaining was a powerful tool for histopathological identification of adrenal lesions associated with aldosterone overproduction (100).
Fernandes Rosa et al. performed the most comprehensive study in a cohort of 474 aldosteronomas from the European Network for the Study of Adrenal Tumors, reaching a detection rate of somatic variants of 54%, although CTNNB1 sequencing was not included in this study (56). Two other studies, which included CTNNB1 sequencing, demonstrated similar findings: Wu et al. studied 219 aldosteronomas, detecting somatic variants in 58.4% of them (101), and Vilela et al. reported a discovery rate of approximately 50% (102).
Recent studies using immunohistochemistry-guided approach to determine the exact source of abnormal aldosterone production led to the identification of pathogenic somatic variants in around 90% of screened aldosteronomas (81, 82, 88, 103). The lower prevalence of somatic variants found in aldosteronomas in previous studies using conventional approaches, not taking in account CYP11B2 expression, is explained due to the macroscopical selection of non-aldosterone-producing adrenal lesions (81). A recent review confirmed these previous findings, showing a higher detection rate of somatic variants with CYP11B2-guided extraction (85%) when compared to the classical approach with DNA extraction from fresh frozen tissue (54%) (57). Overall, the variant-negative ratio decreased from 46% to 15%. Gene-specific detection rate also increased from 34% to 56% in KCNJ5, 8% to 10% in CACNA1D, 8% to 12% in ATP1A1 and 4% to 5% in ATP2B3 (57).
Moreover, the CYP11B2-guided high throughput sequencing method has revealed a wide complexity of aldosterone-producing lesions in patients with PA (81, 82, 88, 103, 104). In multinodular cases, tumors from the same adrenal might harbor different recurrent somatic variants, suggesting independent triggers for the somatic events (82, 105). Interestingly, aldosterone production may occur from multiple sources: multiple aldosteronomas in the same adrenal gland, dominant non-producing adenoma with satellite CYP11B2 positive non-dominant nodules, and clusters of autonomous aldosterone-producing cells (APCCs) without apparent neoplasia (55, 81, 88, 106, 107).
APCCs are common in normal adrenals and accumulate with age, becoming more often detectable in morphologically normal adult adrenals (108). Somatic pathogenic variants in CACNA1D, ATP1A1 and ATP2B3 were found in 35% to 76% of the APCCs, with CACNA1D being the most mutated gene (108, 109). Interestingly, the spectrum of affected gene in APCCs is different from aldosteronomas. APCCs may a key player to the understanding of the physiology and pathophysiology of aldosterone production. It has been hypothesized that aldosteronomas can derive from APCCs with autonomous aldosterone production (harboring somatic in aldosterone-driver genes) (15, 99, 108).
Recently, the international histopathology consensus for unilateral PA (HISTALDO) classified the aldosterone-producing lesions (110). (Table 5). Aldosteronoma was defined as a well circumscribed CYP11B2-positive solitary neoplasm (≥ 10 mm diameter) composed of clear or compact eosinophilic cells or both cell types. Aldosterone-producing nodule is a CYP11B2-positive lesion (<10 mm diameter) morphologically visible with hematoxylin-eosin staining (“microaldosteronoma”). In this consensus, the nomenclature for APPC was changed to aldosterone-producing micronodules (APMs). APMs are defined as CYP11B2-positive lesion (<10 mm diameter) composed of ZG cells located beneath adrenal capsule. APMs are indistinguishable from normal zona glomerulosa (ZG) cells in hematoxylin-eosin staining (108, 110). In CYP11B2 staining, APMs have a strong uniform immunoreactivity for CYP11B2, without evident neoplasia or hyperplasia (108, 110).

Table 5 New histopathological nomenclature (HISTALDO) of aldosterone-producing adrenal lesions in patients with unilateral primary aldosteronism.
These advances in PA histopathology allowed the definition of classical and non-classical histopathological features associated with PA (Table 5) (110). The classical histology is defined by the presence of a solitary aldosteronoma or APN. In contrast, “non-classical” histology is characterized by adrenals with multiple APNs or APMs (or multiple APMs and multiple APNs together) or aldosterone-producing diffuse hyperplasia (110, 111). In summary, non-classical histology is defined by the absence of a dominant aldosterone-producing lesion (such as a solitary aldosteronoma or APN). Interestingly, the mutational spectrum is different between classical and non-classical histology. KCNJ5 somatic variants are predominant among aldosteronomas (classical histology), whereas CACNA1D is the most frequent mutated gene in APMs (81, 111).
Impact on Clinical Outcome
The impact of genetic and clinical variables in outcome in PA patients have been more properly evaluated after the Primary Aldosteronism Surgical Outcome (PASO) study, which established criteria for clinical and biochemical success in unilateral PA patients after adrenalectomy (112). PASO criteria classified PA patients after adrenalectomy according to the biochemical outcome and clinical success. Complete biochemical success is defined by correction of hypokalemia when present pre-surgery and normalization of the aldosterone-to-renin ratio, and partial biochemical success as a correction of hypokalemia when present pre-surgery and a raised aldosterone-to-renin ratio, but with at least 50% decrease in baseline plasma aldosterone concentration compared to pre-surgical levels. Regarding blood pressure control, complete clinical success is defined as blood pressure <140x90 mmHg without anti-hypertensive medications after 6 months of follow-up, whereas partial clinical success as a reduction in the number or dose of anti-hypertensive medications when compared to pre-surgery (98).
Recently, non-classical histopathological lesions associated with aldosterone excess were found in 25% of the cases in a German cohort of unilateral PA (111). On the other side, APMs were found in only 5% (7 out of 137) of the cases in a Chinese cohort of unilateral PA (113). Therefore, additional studies from patients with different genetic backgrounds are essential to define the prevalence of classical and non-classical unilateral PA among different cohorts.
Of great importance in clinical practice, postsurgical complete biochemical success after adrenalectomy was correlated with histological features in a German cohort of unilateral PA. The rate of biochemical cure of PA was 98% in patients with the classical histopathology (solitary aldosteronoma or APN) compared with 67% in the patients with unilateral PA caused by non-classical histopathology (111). These findings suggested the presence of a baseline abnormal aldosterone production from the contralateral gland in patients with non-classical unilateral PA (Table 5).
KCNJ5 somatic pathogenic variants have been associated with complete clinical success in cohorts of unilateral PA from Australia, West Norway, Japan and Brazil (80, 102, 114, 115). In a Brazilian cohort of PA, complete clinical success based in PASO criteria was more frequent in patients with aldosteronomas harboring KCNJ5 pathogenic variants than in those with pathogenic variants in other driver genes (102). However, it should be emphasized that these previous studies did not conduct a genetic investigation based on CYP11B2 staining.
Interestingly, KCNJ5 pathogenic variants have been more frequently detected in aldosteronomas (classical histopathology), which is associated with a higher chance of postsurgical complete biochemical success (57). Recently, somatic KCNJ5 pathogenic variants were not associated with clinical and biochemical outcome in a small group of 38 aldosteronomas with genetic investigation guided by CYP11B2 staining. However, the influence of KCNJ5 status in the outcome of PA patients cannot be ruled out and should be further evaluated in larger cohorts of unilateral PA with genetic investigation guided by CYP11B2 staining. Furthermore, the impact of somatic KCNJ5 pathogenic variants on clinical outcome might depend on the frequency of classical histopathology among unilateral PA cases.
Perspectives
Genetics of unilateral PA has remarkably improved in the last decade. However, most cases of bilateral hyperplasia remain without genetic etiology (15). Of great importance, a new histopathological classification has been recently proposed for aldosterone-producing lesions in unilateral PA (110). Besides the impact on the comprehension of PA pathophysiology, the histopathological features have influence in the outcome after unilateral adrenalectomy. KCNJ5 mutational status and classical histopathology of unilateral PA (aldosteronoma) have emerged as relevant predictors of clinical and biochemical outcome, respectively (102, 111). Further studies will be important to characterize the spectrum of classical and non-classical unilateral PA among cohorts from different genetic backgrounds.
Author Contributions
LS and AG participated on acquisition, analysis and interpretation of data, and drafted the manuscript. MA designed, drafted and revising critically the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Sao Paulo Research Foundation (FAPESP) grant 2019/15873-6 (to MA).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Murray CJL, Lopez AD. Measuring the Global Burden of Disease. N Engl J Med (2013) 369(5):448–57. doi: 10.1056/NEJMra1201534
2. Egan BM. US Trends in Prevalence, Awareness, Treatment, and Control of Hypertension, 1988-2008. JAMA (2010) 303(20):2043. doi: 10.1001/jama.2010.650
3. Hannemann A, Wallaschofski H. Prevalence of Primary Aldosteronism in Patient’s Cohorts and in Population-Based Studies - A Review of the Current Literature. Horm Metab Res (2012) 44(03):157–62. doi: 10.1055/s-0031-1295438
4. Plouin P-F, Amar L, Chatellier G. Trends in the Prevalence of Primary Aldosteronism, Aldosterone-Producing Adenomas, and Surgically Correctable Aldosterone-Dependent Hypertension. Nephrol Dial Transplant (2004) 19(4):774–7. doi: 10.1093/ndt/gfh112
5. Douma S, Petidis K, Doumas M, Papaefthimiou P, Triantafyllou A, Kartali N, et al. Prevalence of Primary Hyperaldosteronism in Resistant Hypertension: A Retrospective Observational Study. Lancet (2008) 371(9628):1921–6. doi: 10.1016/S0140-6736(08)60834-X
6. Calhoun DA, Nishizaka MK, Zaman MA, Thakkar RB, Weissmann P. Hyperaldosteronism Among Black and White Subjects With Resistant Hypertension. Hypertension (2002) 40(6):892–6. doi: 10.1161/01.HYP.0000040261.30455.B6
7. Funder JW, Carey RM, Mantero F, Murad MH, Reincke M, Shibata H, et al. The Management of Primary Aldosteronism: Case Detection, Diagnosis, and Treatment: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab (2016) 101(5):1889–916. doi: 10.1210/jc.2015-4061
8. Milliez P, Girerd X, Plouin P-F, Blacher J, Safar ME, Mourad J-J. Evidence for an Increased Rate of Cardiovascular Events in Patients With Primary Aldosteronism. J Am Coll Cardiol (2005) 45(8):1243–8. doi: 10.1016/j.jacc.2005.01.015
9. Stowasser M, Sharman J, Leano R, Gordon RD, Ward G, Cowley D, et al. Evidence for Abnormal Left Ventricular Structure and Function in Normotensive Individuals With Familial Hyperaldosteronism Type I. J Clin Endocrinol Metab (2005) 90(9):5070–6. doi: 10.1210/jc.2005-0681
10. Boulkroun S, Fernandes-Rosa FL, Zennaro M-C. Old and New Genes in Primary Aldosteronism. Best Pract Res Clin Endocrinol Metab (2020) 34(2):101375. doi: 10.1016/j.beem.2020.101375
11. Zennaro M-C, Fernandes-Rosa FL, Boulkroun S. Overview of Aldosterone-Related Genetic Syndromes and Recent Advances. Curr Opin Endocrinol Diabetes Obes (2018) 25(3):147–54. doi: 10.1097/MED.0000000000000409
12. Zennaro M-C, Rickard AJ, Boulkroun S. Genetics in Endocrinology: Genetics of Mineralocorticoid Excess: An Update for Clinicians. Eur J Endocrinol (2013) 169(1):R15–25. doi: 10.1530/EJE-12-0813
13. Zennaro M-C, Boulkroun S, Fernandes-Rosa F. An Update on Novel Mechanisms of Primary Aldosteronism. J Endocrinol (2015) 224(2):R63–77. doi: 10.1530/JOE-14-0597
14. Rossi GP, Bernini G, Caliumi C, Desideri G, Fabris B, Ferri C, et al. A Prospective Study of the Prevalence of Primary Aldosteronism in 1,125 Hypertensive Patients. J Am Coll Cardiol (2006) 48(11):2293–300. doi: 10.1016/j.jacc.2006.07.059
15. Zennaro M-C, Boulkroun S, Fernandes-Rosa F. Genetic Causes of Functional Adrenocortical Adenomas. Endocr Rev (2017) 38(6):516–37. doi: 10.1210/er.2017-00189
16. Vilela LAP, Almeida MQ. Diagnosis and Management of Primary Aldosteronism. Arch Endocrinol Metab (2017) 61(3):305–12. doi: 10.1590/2359-3997000000274
17. Rossi GP, Ceolotto G, Rossitto G, Seccia TM, Maiolino G, Berton C, et al. Prospective Validation of an Automated Chemiluminescence-Based Assay of Renin and Aldosterone for the Work-Up of Arterial Hypertension. Clin Chem Lab Med (2016) 54(9):1441–50. doi: 10.1515/cclm-2015-1094
18. Rossi GP, Bolognesi M, Rizzoni D, Seccia TM, Piva A, Porteri E, et al. Vascular Remodeling and Duration of Hypertension Predict Outcome of Adrenalectomy in Primary Aldosteronism Patients. Hypertension (2008) 51(5):1366–71. doi: 10.1161/HYPERTENSIONAHA.108.111369
19. Catena C, Colussi G, Lapenna R, Nadalini E, Chiuch A, Gianfagna P, et al. Long-Term Cardiac Effects of Adrenalectomy or Mineralocorticoid Antagonists in Patients With Primary Aldosteronism. Hypertension (2007) 50(5):911–8. doi: 10.1161/HYPERTENSIONAHA.107.095448
20. Hundemer GL, Curhan GC, Yozamp N, Wang M, Vaidya A. Cardiometabolic Outcomes and Mortality in Medically Treated Primary Aldosteronism: A Retrospective Cohort Study. Lancet Diabetes Endocrinol (2018) 6(1):51–9. doi: 10.1016/S2213-8587(17)30367-4
21. Lifton RP, Dluhy RG, Powers M, Rich GM, Cook S, Ulick S, et al. A Chimaeric Llβ-Hydroxylase/Aldosterone Synthase Gene Causes Glucocorticoid-Remediable Aldosteronism and Human Hypertension. Nature (1992) 355(6357):262–5. doi: 10.1038/355262a0
22. Stowasser M, Bachmann AW, Huggard PR, Rossetti TR, Gordon RD. Severity of Hypertension in Familial Hyperaldosteronism Type I: Relationship to Gender and Degree of Biochemical Disturbance 1. J Clin Endocrinol Metab (2000) 85(6):2160–6. doi: 10.1210/jc.85.6.2160
23. Choi M, Scholl UI, Yue P, Björklund P, Zhao B, Nelson-Williams C, et al. K + Channel Mutations in Adrenal Aldosterone-Producing Adenomas and Hereditary Hypertension. Science (80- ) (2011) 331(6018):768–72. doi: 10.1126/science.1198785
24. Mulatero P, Monticone S, Rainey WE, Veglio F, Williams TA. Role of KCNJ5 in Familial and Sporadic Primary Aldosteronism. Nat Rev Endocrinol (2013) 9(2):104–12. doi: 10.1038/nrendo.2012.230
25. Scholl UI, Goh G, Stölting G, de Oliveira RC, Choi M, Overton JD, et al. Somatic and Germline CACNA1D Calcium Channel Mutations in Aldosterone-Producing Adenomas and Primary Aldosteronism. Nat Genet (2013) 45(9):1050–4. doi: 10.1038/ng.2695
26. Scholl UI, Stölting G, Nelson-Williams C, Vichot AA, Choi M, Loring E, et al. Recurrent Gain of Function Mutation in Calcium Channel CACNA1H Causes Early-Onset Hypertension With Primary Aldosteronism. Elife (2015) 4:e06315. doi: 10.7554/eLife.06315.019
27. Daniil G, Fernandes-Rosa FL, Chemin J, Blesneac I, Beltrand J, Polak M, et al. CACNA1H Mutations Are Associated With Different Forms of Primary Aldosteronism. EBioMedicine (2016) 13:225–36. doi: 10.1016/j.ebiom.2016.10.002
28. Fernandes-Rosa FL, Daniil G, Orozco IJ, Göppner C, El Zein R, Jain V, et al. A Gain-of-Function Mutation in the CLCN2 Chloride Channel Gene Causes Primary Aldosteronism. Nat Genet (2018) 50(3):355–61. doi: 10.1038/s41588-018-0053-8
29. Scholl UI, Stölting G, Schewe J, Thiel A, Tan H, Nelson-Williams C, et al. CLCN2 Chloride Channel Mutations in Familial Hyperaldosteronism Type II. Nat Genet (2018) 50(3):349–54. doi: 10.1038/s41588-018-0048-5
30. Sutherland DJ, Ruse JL, Laidlaw JC. Hypertension, Increased Aldosterone Secretion and Low Plasma Renin Activity Relieved by Dexamethasone. Can Med Assoc J (1966) 95(22):1109–19.
31. Geller DS, Zhang J, Wisgerhof MV, Shackleton C, Kashgarian M, Lifton RP. A Novel Form of Human Mendelian Hypertension Featuring Nonglucocorticoid-Remediable Aldosteronism. J Clin Endocrinol Metab (2008) 93(8):3117–23. doi: 10.1210/jc.2008-0594
32. Dluhy RG, Anderson B, Harlin B, Ingelfinger J, Lifton R. Glucocorticoid-Remediable Aldosteronism is Associated With Severe Hypertension in Early Childhood. J Pediatr (2001) 138(5):715–20. doi: 10.1067/mpd.2001.112648
33. Dluhy RG, Lifton RP. Glucocorticoid-Remediable Aldosteronism. J Clin Endocrinol Metab (1999) 84(12):4341–4. doi: 10.1210/jcem.84.12.6256
34. Pallauf A, Schirpenbach C, Zwermann O, Fischer E, Morak M, Holinski-Feder E, et al. The Prevalence of Familial Hyperaldosteronism in Apparently Sporadic Primary Aldosteronism in Germany: A Single Center Experience. Horm Metab Res (2012) 44(03):215–20. doi: 10.1055/s-0031-1299730
35. Lafferty AR. A Novel Genetic Locus for Low Renin Hypertension: Familial Hyperaldosteronism Type II Maps to Chromosome 7 (7p22). J Med Genet (2000) 37(11):831–5. doi: 10.1136/jmg.37.11.831
36. Murthy M, Xu S, Massimo G, Wolley M, Gordon RD, Stowasser M, et al. Role for Germline Mutations and a Rare Coding Single Nucleotide Polymorphism Within the KCNJ5 Potassium Channel in a Large Cohort of Sporadic Cases of Primary Aldosteronism. Hypertension (2014) 63(4):783–9. doi: 10.1161/HYPERTENSIONAHA.113.02234
37. Monticone S, Bandulik S, Stindl J, Zilbermint M, Dedov I, Mulatero P, et al. A Case of Severe Hyperaldosteronism Caused by a De Novo Mutation Affecting a Critical Salt Bridge Kir3.4 Residue. J Clin Endocrinol Metab (2015) 100(1):E114–8. doi: 10.1210/jc.2014-3636
38. Mulatero P, Tauber P, Zennaro M-C, Monticone S, Lang K, Beuschlein F, et al. KCNJ5 Mutations in European Families With Nonglucocorticoid Remediable Familial Hyperaldosteronism. Hypertension (2012) 59(2):235–40. doi: 10.1161/HYPERTENSIONAHA.111.183996
39. Scholl UI, Nelson-Williams C, Yue P, Grekin R, Wyatt RJ, Dillon MJ, et al. Hypertension With or Without Adrenal Hyperplasia Due to Different Inherited Mutations in the Potassium Channel KCNJ5. Proc Natl Acad Sci (2012) 109(7):2533–8. doi: 10.1073/pnas.1121407109
40. Monticone S, Hattangady NG, Penton D, Isales CM, Edwards MA, Williams TA, et al. A Novel Y152C KCNJ5 Mutation Responsible for Familial Hyperaldosteronism Type III. J Clin Endocrinol Metab (2013) 98(11):E1861–5. doi: 10.1210/jc.2013-2428
41. Charmandari E, Sertedaki A, Kino T, Merakou C, Hoffman DA, Hatch MM, et al. A Novel Point Mutation in the KCNJ5 Gene Causing Primary Hyperaldosteronism and Early-Onset Autosomal Dominant Hypertension. J Clin Endocrinol Metab (2012) 97(8):E1532–9. doi: 10.1210/jc.2012-1334
42. Pons Fernández N, Moreno F, Morata J, Moriano A, León S, De Mingo C, et al. Familial Hyperaldosteronism Type III a Novel Case and Review of Literature. Rev Endocr Metab Disord (2019) 20(1):27–36. doi: 10.1007/s11154-018-9481-0
43. Wulczyn K, Perez-Reyes E, Nussbaum RL, Park M. Primary Aldosteronism Associated With a Germline Variant in CACNA1H. BMJ Case Rep (2019) 12(5):e229031. doi: 10.1136/bcr-2018-229031
44. Semenova NA, Ryzhkova OR, Strokova TV, Taran NN. The Third Case Report a Patient With Primary Aldosteronism, Seizures, and Neurologic Abnormalities (PASNA) Syndrome De Novo Variant Mutations in the CACNA1D Gene. Zh Nevrol Psikhiatr Im S S Korsakova (2018) 118(12):49. doi: 10.17116/jnevro201811812149
45. De Mingo Alemany MC, Mifsud Grau L, Moreno Macián F, Ferrer Lorente B, León Cariñena SA. De Novo CACNA1D Missense Mutation in a Patient With Congenital Hyperinsulinism, Primary Hyperaldosteronism and Hypotonia. Channels (2020) 14(1):175–80. doi: 10.1080/19336950.2020.1761171
46. Markou A, Sertedaki A, Kaltsas G, Androulakis II, Marakaki C, Pappa T, et al. Stress-Induced Aldosterone Hyper-Secretion in a Substantial Subset of Patients With Essential Hypertension. J Clin Endocrinol Metab (2015) 100(8):2857–64. doi: 10.1210/jc.2015-1268
47. Qin F, Liu K, Zhang C, Sun X, Zhang Y, Wu Y, et al. Steroid Metabolism Gene Variants and Their Genotype-Phenotype Correlations in Chinese Early-Onset Hypertension Patients. Hypertens Res (2019) 42(10):1536–43. doi: 10.1038/s41440-019-0306-7
48. Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, et al. The Human Gene Mutation Database (HGMD®): Optimizing Its Use in a Clinical Diagnostic or Research Setting. Hum Genet (2020) 139(10):1197–207. doi: 10.1007/s00439-020-02199-3
49. Scholl UI, Abriola L, Zhang C, Reimer EN, Plummer M, Kazmierczak BI, et al. Macrolides Selectively Inhibit Mutant KCNJ5 Potassium Channels That Cause Aldosterone-Producing Adenoma. J Clin Invest (2017) 127(7):2739–50. doi: 10.1172/JCI91733
50. Reimer EN, Walenda G, Seidel E, Scholl UI. CACNA1HM1549V Mutant Calcium Channel Causes Autonomous Aldosterone Production in HAC15 Cells and Is Inhibited by Mibefradil. Endocrinology (2016) 157(8):3016–22. doi: 10.1210/en.2016-1170
51. Rassi-Cruz M, Maria AG, Faucz FR, London E, Vilela LAP, Santana LS, et al. Phosphodiesterase 2A and 3B Variants Are Associated With Primary Aldosteronism. Endocr Relat Cancer (2021) 28(1):1–13. doi: 10.1530/ERC-20-0384
52. Pearce D. SGK1 Regulation of Epithelial Sodium Transport. Cell Physiol Biochem (2003) 13(1):13–20. doi: 10.1159/000070245
53. Staub O, Dho S, Henry P, Correa J, Ishikawa T, McGlade J, et al. WW Domains of Nedd4 Bind to the Proline-Rich PY Motifs in the Epithelial Na+ Channel Deleted in Liddle’s Syndrome. EMBO J (1996) 15(10):2371–80. doi: 10.1002/j.1460-2075.1996.tb00593.x
54. Terada Y, Kuwana H, Kobayashi T, Okado T, Suzuki N, Yoshimoto T, et al. Aldosterone-Stimulated SGK1 Activity Mediates Profibrotic Signaling in the Mesangium. J Am Soc Nephrol (2008) 19(2):298–309. doi: 10.1681/ASN.2007050531
55. Nanba K, Rainey WE. GENETICS IN ENDOCRINOLOGY: Impact of Race and Sex on Genetic Causes of Aldosterone-Producing Adenomas. Eur J Endocrinol (2021) 185(1):R1–11. doi: 10.1530/EJE-21-0031
56. Fernandes-Rosa FL, Williams TA, Riester A, Steichen O, Beuschlein F, Boulkroun S, et al. Genetic Spectrum and Clinical Correlates of Somatic Mutations in Aldosterone-Producing Adenoma. Hypertension (2014) 64(2):354–61. doi: 10.1161/HYPERTENSIONAHA.114.03419
57. Williams TA, Reincke M. Pathophysiology and Histopathology of Primary Aldosteronism. Trends Endocrinol Metab (2022) 33(1):36–49. doi: 10.1016/j.tem.2021.10.002
58. Azizan EAB, Lam BYH, Newhouse SJ, Zhou J, Kuc RE, Clarke J, et al. Microarray, qPCR, and KCNJ5 Sequencing of Aldosterone-Producing Adenomas Reveal Differences in Genotype and Phenotype Between Zona Glomerulosa- and Zona Fasciculata-Like Tumors. J Clin Endocrinol Metab (2012) 97(5):E819–29. doi: 10.1210/jc.2011-2965
59. Beuschlein F, Boulkroun S, Osswald A, Wieland T, Nielsen HN, Lichtenauer UD, et al. Somatic Mutations in ATP1A1 and ATP2B3 Lead to Aldosterone-Producing Adenomas and Secondary Hypertension. Nat Genet (2013) 45(4):440–4. doi: 10.1038/ng.2550
60. Kitamoto T, Suematsu S, Yamazaki Y, Nakamura Y, Sasano H, Matsuzawa Y, et al. Clinical and Steroidogenic Characteristics of Aldosterone-Producing Adenomas With ATPase or CACNA1D Gene Mutations. J Clin Endocrinol Metab (2016) 101(2):494–503. doi: 10.1210/jc.2015-3284
61. Azizan EAB, Poulsen H, Tuluc P, Zhou J, Clausen MV, Lieb A, et al. Somatic Mutations in ATP1A1 and CACNA1D Underlie a Common Subtype of Adrenal Hypertension. Nat Genet (2013) 45(9):1055–60. doi: 10.1038/ng.2716
62. Nanba K, Omata K, Gomez-Sanchez CE, Stratakis CA, Demidowich AP, Suzuki M, et al. Genetic Characteristics of Aldosterone-Producing Adenomas in Blacks. Hypertension (2019) 73(4):885–92. doi: 10.1161/HYPERTENSIONAHA.118.12070
63. Teo AED, Garg S, Haris Shaikh L, Zhou J, Karet Frankl FE, Gurnell M, et al. Pregnancy, Primary Aldosteronism, and Adrenal CTNNB1 Mutations. N Engl J Med (2015) 373(15):1429–36. doi: 10.1056/NEJMoa1504869
64. Wang J-J, Peng K-Y, Wu V-C, Tseng F-Y, Wu K-D. CTNNB1 Mutation in Aldosterone Producing Adenoma. Endocrinol Metab (2017) 32(3):332. doi: 10.3803/EnM.2017.32.3.332
65. Dutta RK, Arnesen T, Heie A, Walz M, Alesina P, Söderkvist P, et al. A Somatic Mutation in CLCN2 Identified in a Sporadic Aldosterone-Producing Adenoma. Eur J Endocrinol (2019) 181(5):K37–41. doi: 10.1530/EJE-19-0377
66. Rege J, Nanba K, Blinder AR, Plaska S, Udager AM, Vats P, et al. Identification of Somatic Mutations in CLCN2 in Aldosterone-Producing Adenomas. J Endocr Soc (2020) 4(10):bvaa123. doi: 10.1210/jendso/bvaa123
67. Nanba K, Blinder AR, Rege J, Hattangady NG, Else T, Liu C-J, et al. Somatic CACNA1H Mutation As a Cause of Aldosterone-Producing Adenoma. Hypertension (2020) 75(3):645–9. doi: 10.1161/HYPERTENSIONAHA.119.14349
68. Oki K, Gomez-Sanchez CE. The Landscape of Molecular Mechanism for Aldosterone Production in Aldosterone-Producing Adenoma. Endocr J (2020) 67(10):989–95. doi: 10.1507/endocrj.EJ20-0478
69. Åkerström T, Crona J, Delgado Verdugo A, Starker LF, Cupisti K, Willenberg HS, et al. Comprehensive Re-Sequencing of Adrenal Aldosterone Producing Lesions Reveal Three Somatic Mutations Near the KCNJ5 Potassium Channel Selectivity Filter. PloS One (2012) 7(7):e41926. doi: 10.1371/journal.pone.0041926
70. Taguchi R, Yamada M, Nakajima Y, Satoh T, Hashimoto K, Shibusawa N, et al. Expression and Mutations of KCNJ5 mRNA in Japanese Patients With Aldosterone-Producing Adenomas. J Clin Endocrinol Metab (2012) 97(4):1311–9. doi: 10.1210/jc.2011-2885
71. Kuppusamy M, Caroccia B, Stindl J, Bandulik S, Lenzini L, Gioco F, et al. A Novel KCNJ5-Inst149 Somatic Mutation Close to, But Outside, the Selectivity Filter Causes Resistant Hypertension by Loss of Selectivity for Potassium. J Clin Endocrinol Metab (2014) 99(9):E1765–73. doi: 10.1210/jc.2014-1927
72. Williams TA, Monticone S, Schack VR, Stindl J, Burrello J, Buffolo F, et al. Somatic ATP1A1 , ATP2B3 , and KCNJ5 Mutations in Aldosterone-Producing Adenomas. Hypertension (2014) 63(1):188–95. doi: 10.1161/HYPERTENSIONAHA.113.01733
73. Scholl UI, Healy JM, Thiel A, Fonseca AL, Brown TC, Kunstman JW, et al. Novel Somatic Mutations in Primary Hyperaldosteronism are Related to the Clinical, Radiological and Pathological Phenotype. Clin Endocrinol (Oxf) (2015) 83(6):779–89. doi: 10.1111/cen.12873
74. Zheng F-F, Zhu L-M, Nie A-F, Li X-Y, Lin J-R, Zhang K, et al. Clinical Characteristics of Somatic Mutations in Chinese Patients With Aldosterone-Producing Adenoma. Hypertension (2015) 65(3):622–8. doi: 10.1161/HYPERTENSIONAHA.114.03346
75. Wang B, Li X, Zhang X, Ma X, Chen L, Zhang Y, et al. Prevalence and Characterization of Somatic Mutations in Chinese Aldosterone-Producing Adenoma Patients. Medicine (Baltimore) (2015) 94(16):e708. doi: 10.1097/MD.0000000000000708
76. Cheng C-J, Sung C-C, Wu S-T, Lin Y-C, Sytwu H-K, Huang C-L, et al. Novel KCNJ5 Mutations in Sporadic Aldosterone-Producing Adenoma Reduce Kir3.4 Membrane Abundance. J Clin Endocrinol Metab (2015) 100(1):E155–63. doi: 10.1210/jc.2014-3009
77. Nanba K, Omata K, Tomlins SA, Giordano TJ, Hammer GD, Rainey WE, et al. Double Adrenocortical Adenomas Harboring Independent KCNJ5 and PRKACA Somatic Mutations. Eur J Endocrinol (2016) 175(2):K1–6. doi: 10.1530/EJE-16-0262
78. Zheng F-F, Zhu L-M, Zhou W-L, Zhang Y, Li M-Y, Zhu Y-C, et al. A Novel Somatic Mutation 145–147deleteinsk in KCNJ5 Increases Aldosterone Production. J Hum Hypertens (2017) 31(11):756–9. doi: 10.1038/jhh.2017.50
79. Hardege I, Xu S, Gordon RD, Thompson AJ, Figg N, Stowasser M, et al. Novel Insertion Mutation in KCNJ5 Channel Produces Constitutive Aldosterone Release From H295R Cells. Mol Endocrinol (2015) 29(10):1522–30. doi: 10.1210/me.2015-1195
80. Kitamoto T, Omura M, Suematsu S, Saito J, Nishikawa T. KCNJ5 Mutation as a Predictor for Resolution of Hypertension After Surgical Treatment of Aldosterone-Producing Adenoma. J Hypertens (2018) 36(3):619–27. doi: 10.1097/HJH.0000000000001578
81. Nanba K, Omata K, Else T, Beck PCC, Nanba AT, Turcu AF, et al. Targeted Molecular Characterization of Aldosterone-Producing Adenomas in White Americans. J Clin Endocrinol Metab (2018) 103(10):3869–76. doi: 10.1210/jc.2018-01004
82. Nanba K, Chen AX, Omata K, Vinco M, Giordano TJ, Else T, et al. Molecular Heterogeneity in Aldosterone-Producing Adenomas. J Clin Endocrinol Metab (2016) 101(3):999–1007. doi: 10.1210/jc.2015-3239
83. Tan GC, Negro G, Pinggera A, Tizen Laim NMS, Mohamed Rose I, Ceral J, et al. Aldosterone-Producing Adenomas. Hypertension (2017) 70(1):129–36. doi: 10.1161/HYPERTENSIONAHA.117.09057
84. Åkerström T, Willenberg HS, Cupisti K, Ip J, Backman S, Moser A, et al. Novel Somatic Mutations and Distinct Molecular Signature in Aldosterone-Producing Adenomas. Endocr Relat Cancer (2015) 22(5):735–44. doi: 10.1530/ERC-15-0321
85. Backman S, Åkerström T, Maharjan R, Cupisti K, Willenberg HS, Hellman P, et al. RNA Sequencing Provides Novel Insights Into the Transcriptome of Aldosterone Producing Adenomas. Sci Rep (2019) 9(1):6269. doi: 10.1038/s41598-019-41525-2
86. Guo Z, Nanba K, Udager A, McWhinney BC, Ungerer JPJ, Wolley M, et al. Biochemical, Histopathological, and Genetic Characterization of Posture-Responsive and Unresponsive APAs. J Clin Endocrinol Metab (2020) 105(9):e3224–35. doi: 10.1210/clinem/dgaa367
87. Nanba K, Yamazaki Y, Bick N, Onodera K, Tezuka Y, Omata K, et al. Prevalence of Somatic Mutations in Aldosterone-Producing Adenomas in Japanese Patients. J Clin Endocrinol Metab (2020) 105(11):e4066–73. doi: 10.1210/clinem/dgaa595
88. De Sousa K, Boulkroun S, Baron S, Nanba K, Wack M, Rainey WE, et al. Genetic, Cellular, and Molecular Heterogeneity in Adrenals With Aldosterone-Producing Adenoma. Hypertension (2020) 75(4):1034–44. doi: 10.1161/HYPERTENSIONAHA.119.14177
89. Wu V-C, Huang K-H, Peng K-Y, Tsai Y-C, Wu C-H, Wang S-M, et al. Prevalence and Clinical Correlates of Somatic Mutation in Aldosterone Producing Adenoma-Taiwanese Population. Sci Rep (2015) 5(1):11396. doi: 10.1038/srep11396
90. Murakami M, Yoshimoto T, Minami I, Bouchi R, Tsuchiya K, Hashimoto K, et al. A Novel Somatic Deletion Mutation of ATP2B3 in Aldosterone-Producing Adenoma. Endocr Pathol (2015) 26(4):328–33. doi: 10.1007/s12022-015-9400-9
91. Dutta RK, Welander J, Brauckhoff M, Walz M, Alesina P, Arnesen T, et al. Complementary Somatic Mutations of KCNJ5, ATP1A1, and ATP2B3 in Sporadic Aldosterone Producing Adrenal Adenomas. Endocr Relat Cancer (2014) 21(1):L1–4. doi: 10.1530/ERC-13-0466
92. Kitamoto T, Suematsu S, Matsuzawa Y, Saito J, Omura M, Nishikawa T. Comparison of Cardiovascular Complications in Patients With and Without KCNJ5 Gene Mutations Harboring Aldosterone-Producing Adenomas. J Atheroscler Thromb (2015) 22(2):191–200. doi: 10.5551/jat.24455
93. Tauber P, Aichinger B, Christ C, Stindl J, Rhayem Y, Beuschlein F, et al. Cellular Pathophysiology of an Adrenal Adenoma-Associated Mutant of the Plasma Membrane Ca2+-ATPase Atp2b3. Endocrinology (2016) 157(6):2489–99. doi: 10.1210/en.2015-2029
94. Berthon A, Drelon C, Val P. Pregnancy, Primary Aldosteronism, and Somatic CTNNB1 Mutations. N Engl J Med (2016) 374(15):1492–4. doi: 10.1056/NEJMc1514508
95. Åkerström T, Maharjan R, Sven Willenberg H, Cupisti K, Ip J, Moser A, et al. Activating Mutations in CTNNB1 in Aldosterone Producing Adenomas. Sci Rep (2016) 6(1):19546. doi: 10.1038/srep19546
96. Berthon A, Drelon C, Ragazzon B, Boulkroun S, Tissier F, Amar L, et al. WNT/β-Catenin Signalling Is Activated in Aldosterone-Producing Adenomas and Controls Aldosterone Production. Hum Mol Genet (2014) 23(4):889–905. doi: 10.1093/hmg/ddt484
97. Gomez-Sanchez CE, Qi X, Velarde-Miranda C, Plonczynski MW, Parker CR, Rainey W, et al. Development of Monoclonal Antibodies Against Human CYP11B1 and CYP11B2. Mol Cell Endocrinol (2014) 383(1–2):111–7. doi: 10.1016/j.mce.2013.11.022
98. Nishimoto K, Nakagawa K, Li D, Kosaka T, Oya M, Mikami S, et al. Adrenocortical Zonation in Humans Under Normal and Pathological Conditions. J Clin Endocrinol Metab (2010) 95(5):2296–305. doi: 10.1210/jc.2009-2010
99. Nishimoto K, Koga M, Seki T, Oki K, Gomez-Sanchez EP, Gomez-Sanchez CE, et al. Immunohistochemistry of Aldosterone Synthase Leads the Way to the Pathogenesis of Primary Aldosteronism. Mol Cell Endocrinol (2017) 441:124–33. doi: 10.1016/j.mce.2016.10.014
100. Nanba K, Tsuiki M, Sawai K, Mukai K, Nishimoto K, Usui T, et al. Histopathological Diagnosis of Primary Aldosteronism Using CYP11B2 Immunohistochemistry. J Clin Endocrinol Metab (2013) 98(4):1567–74. doi: 10.1210/jc.2012-3726
101. Wu V-C, Wang S-M, Chueh S-CJ, Yang S-Y, Huang K-H, Lin Y-H, et al. The Prevalence of CTNNB1 Mutations in Primary Aldosteronism and Consequences for Clinical Outcomes. Sci Rep (2017) 7(1):39121. doi: 10.1038/srep39121
102. Vilela LAP, Rassi-Cruz M, Guimaraes AG, Moises CCS, Freitas TC, Alencar NP, et al. KCNJ5 Somatic Mutation Is a Predictor of Hypertension Remission After Adrenalectomy for Unilateral Primary Aldosteronism. J Clin Endocrinol Metab (2019) 104(10):4695–702. doi: 10.1210/jc.2019-00531
103. Ono Y, Yamazaki Y, Omata K, Else T, Tomlins SA, Rhayem Y, et al. Histological Characterization of Aldosterone-Producing Adrenocortical Adenomas With Different Somatic Mutations. J Clin Endocrinol Metab (2020) 105(3):e282–9. doi: 10.1210/clinem/dgz235
104. Omata K, Yamazaki Y, Nakamura Y, Anand SK, Barletta JA, Sasano H, et al. Genetic and Histopathologic Intertumor Heterogeneity in Primary Aldosteronism. J Clin Endocrinol Metab (2017) 102(6):1792–6. doi: 10.1210/jc.2016-4007
105. Fernandes-Rosa FL, Giscos-Douriez I, Amar L, Gomez-Sanchez CE, Meatchi T, Boulkroun S, et al. Different Somatic Mutations in Multinodular Adrenals With Aldosterone-Producing Adenoma. Hypertension (2015) 66(5):1014–22. doi: 10.1161/HYPERTENSIONAHA.115.05993
106. Zennaro M-C, Boulkroun S, Fernandes-Rosa FL. Pathogenesis and Treatment of Primary Aldosteronism. Nat Rev Endocrinol (2020) 16(10):578–89. doi: 10.1038/s41574-020-0382-4
107. Prada ETA, Burrello J, Reincke M, Williams TA. Old and New Concepts in the Molecular Pathogenesis of Primary Aldosteronism. Hypertension (2017) 70(5):875–81. doi: 10.1161/HYPERTENSIONAHA.117.10111
108. Nishimoto K, Tomlins SA, Kuick R, Cani AK, Giordano TJ, Hovelson DH, et al. Aldosterone-Stimulating Somatic Gene Mutations are Common in Normal Adrenal Glands. Proc Natl Acad Sci (2015) 112(33):E4591–9. doi: 10.1073/pnas.1505529112
109. Omata K, Anand SK, Hovelson DH, Liu C-J, Yamazaki Y, Nakamura Y, et al. Aldosterone-Producing Cell Clusters Frequently Harbor Somatic Mutations and Accumulate With Age in Normal Adrenals. J Endocr Soc (2017) 1(7):787–99. doi: 10.1210/js.2017-00134
110. Williams TA, Gomez-Sanchez CE, Rainey WE, Giordano TJ, Lam AK, Marker A, et al. International Histopathology Consensus for Unilateral Primary Aldosteronism. J Clin Endocrinol Metab (2021) 106(1):42–54. doi: 10.1210/clinem/dgaa484
111. Meyer LS, Handgriff L, Lim JS, Udager AM, Kinker I-S, Ladurner R, et al. Single-Center Prospective Cohort Study on the Histopathology, Genotype, and Postsurgical Outcomes of Patients With Primary Aldosteronism. Hypertension (2021) 78(3):738–46. doi: 10.1161/HYPERTENSIONAHA.121.17348
112. Williams TA, Lenders JWM, Mulatero P, Burrello J, Rottenkolber M, Adolf C, et al. Outcomes After Adrenalectomy for Unilateral Primary Aldosteronism: An International Consensus on Outcome Measures and Analysis of Remission Rates in an International Cohort. Lancet Diabetes Endocrinol (2017) 5(9):689–99. doi: 10.1016/S2213-8587(17)30135-3
113. Wang H, Wang F, Zhang Y, Wen J, Dong D, Chang X, et al. Surgical Outcomes of Aldosterone-Producing Adenoma on the Basis of the Histopathological Findings. Front Endocrinol (Lausanne) (2021) 12:663096. doi: 10.3389/fendo.2021.663096
114. Ip JCY, Pang TCY, Pon CK, Zhao JT, Sywak MS, Gill AJ, et al. Mutations in KCNJ5 Determines Presentation and Likelihood of Cure in Primary Hyperaldosteronism. ANZ J Surg (2015) 85(4):279–83. doi: 10.1111/ans.12470
Keywords: primary aldosteronism, aldosterone, aldosterone synthase, genetics, outcome
Citation: Santana LS, Guimaraes AG and Almeida MQ (2022) Pathogenesis of Primary Aldosteronism: Impact on Clinical Outcome. Front. Endocrinol. 13:927669. doi: 10.3389/fendo.2022.927669
Received: 24 April 2022; Accepted: 23 May 2022;
Published: 23 June 2022.
Edited by:
Ricardo Correa, University of Arizona, United StatesReviewed by:
Avinaash Vickram Maharaj, Queen Mary University of London, United KingdomEmanuele Pignatti, University of Bern, Switzerland
Copyright © 2022 Santana, Guimaraes and Almeida. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Madson Q. Almeida, bWFkc29uLmFAaGMuZm0udXNwLmJy