 Sachi Matsubayashi
Sachi Matsubayashi Saburo Ito
Saburo Ito Jun Araya
Jun Araya Kazuyoshi Kuwano
Kazuyoshi Kuwano- Division of Respiratory Diseases, Department of Internal Medicine, The Jikei University School of Medicine, Tokyo, Japan
Recent advances in aging research have provided novel insights for the development of senotherapy, which utilizes cellular senescence as a therapeutic target. Cellular senescence is involved in the pathogenesis of various chronic diseases, including metabolic and respiratory diseases. Senotherapy is a potential therapeutic strategy for aging-related pathologies. Senotherapy can be classified into senolytics (induce cell death in senescent cells) and senomorphics (ameliorate the adverse effects of senescent cells represented by the senescence-associated secretory phenotype). Although the precise mechanism has not been elucidated, various drugs against metabolic diseases may function as senotherapeutics, which has piqued the interest of the scientific community. Cellular senescence is involved in the pathogenesis of chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF), which are aging-related respiratory diseases. Large-scale observational studies have reported that several drugs, such as metformin and statins, may ameliorate the progression of COPD and IPF. Recent studies have reported that drugs against metabolic diseases may exert a pharmacological effect on aging-related respiratory diseases that can be different from their original effect on metabolic diseases. However, high non-physiological concentrations are needed to determine the efficacy of these drugs under experimental conditions. Inhalation therapy may increase the local concentration of drugs in the lungs without exerting systemic adverse effects. Thus, the clinical application of drugs against metabolic diseases, especially through an inhalation treatment modality, can be a novel therapeutic approach for aging-related respiratory diseases. This review summarizes and discusses accumulating evidence on the mechanisms of aging, as well as on cellular senescence and senotherapeutics, including drugs against metabolic diseases. We propose a developmental strategy for a senotherapeutic approach for aging-related respiratory diseases with a special focus on COPD and IPF.
Introduction
The global population is aging at an unprecedented rate. Aging adversely affects physiological functions and consequently increases the susceptibility of most organs to various pathological conditions. In the respiratory system, aging can induce pathological changes, such as deterioration of respiratory function, increased susceptibility to infection, and malignancy (1, 2). The incidence of chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF), which are representative aging-related respiratory diseases, increases with age (3). COPD and IPF are a major socioeconomic burden due to the cumulative cost and effort associated with medical events. Hence, there is an urgent need to understand the pathogenesis of aging-related respiratory diseases and develop novel therapies. In recent years, basic research on aging mechanisms has markedly advanced. The identification of the lifespan-extending effect of caloric restriction in model organisms and the subsequent discovery of lifespan-related genes (represented by Sirtuin-encoding genes) provide clues for understanding the molecular mechanisms of aging (4–7). Furthermore, mouse studies involving genetic manipulation of cyclin-dependent kinase (CDK) inhibitory proteins, which regulate the cell cycle, have demonstrated that the elimination of senescent cells can extend lifespan and mitigate the development of various aging-related diseases (8, 9). Thus, aging research is now rapidly developing.
In addition to their effects on the primary target diseases, drugs against metabolic diseases, such as diabetes and dyslipidemia, may exert therapeutic effects on various aging-related diseases (10). Metabolic diseases are common complications in patients with aging-related respiratory diseases. Hence, the anti-aging effects of drugs against metabolic diseases are attracting attention in the field of respiratory diseases. Retrospective studies on metabolic disorders have delineated several interesting findings. For example, statins are reported to reduce the incidence of COPD exacerbation episodes and mitigate the decline in respiratory function (11, 12). The anti-diabetic drug metformin is also expected to suppress decline of lung function in COPD patients (13). In patients with both COPD and type 2 diabetes, the mortality rate among metformin users is lower than that of the control cohort with similar backgrounds (14). Further to attributing their therapeutic effects on metabolic diseases, recent advances indicate that these drugs may directly suppress cellular senescence.
Cellular senescence is phenotypically characterized by irreversible cell cycle arrest and apoptosis resistance, which are adaptive responses to various intrinsic and extrinsic stresses (15, 16). Senescent cells are also characterized by a senescence-associated secretory phenotype (SASP) that produces various cytokines and growth factors. Excessive SASP caused by the accumulation of senescent cells has been implicated in chronic inflammation, aberrant tissue repair, and fibrotic tissue remodeling. Therefore, the regulation of cellular senescence is proposed to be a promising approach to prevent aging-related diseases. Many researchers are focusing on an anti-senescence modality of treatment, namely senotherapy (17). Although various new agents are being developed, repositioning pre-existing drugs with potential clinical efficacy as a senotherapeutic can be a reasonable strategy. If drug repositioning, which is also called repurposing, is possible without safety concerns, it will enable the immediate clinical application of the drug. In this review, we discuss the repositioning of drugs against metabolic diseases as potential senotherapeutics for aging-related respiratory diseases with a special focus on COPD and IPF.
Mechanism of aging
Organisms employ dynamic defense mechanisms, which are called homeostasis and robustness, to maintain stability against internal and external disturbances. Homeostasis refers to short-term mechanisms, whereas robustness refers to systemic long-term protective mechanisms, including defense mechanisms against perturbation, dynamic responses to environmental changes, and tissue regeneration in response to injury or defect (18–20). Organismal aging can be, at least partly, assumed to be the dysregulation of robustness (21). Therefore, the organism becomes fragile, exhibiting a reduced ability to respond to changes and insults and a low ability to regenerate. Aging-induced impaired robustness, which is associated with the accumulation of senescent cells, is believed to result from a decline in organ function. The hallmarks of aging include genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, disabled macroautophagy, deregulated nutrient-sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, altered intercellular communication, chronic inflammation, and dysbiosis (22). These pathological changes are interdependent and can drive the progression of cellular senescence. Additionally, aging is a heterogenous process even at the cellular level. The aging process is not equal in all cells with some and not all cells exhibiting senescence (23, 24), indicating the presence of complex mechanisms for regulating organismal aging. To explore a druggable approach against this complex aging process, the molecular mechanisms of aging have been widely investigated. Recent studies have suggested that the accumulated senescent cells can be a promising therapeutic target. Senotherapeutics, which target cellular senescence, are classified into the following two types: senolytics, which induce cell death in senescent cells, and senomorphics, which suppress the SASP-inducing effect and prevent cellular senescence. In the following section, we provide an overview of cellular senescence and a recent understanding of senotherapy.
Cellular senescence and concept of senotherapeutics (senolytics and senomorphics)
Cellular senescence, first reported by Hayflick, is a phenomenon that repeatedly divided human fibroblasts in vitro cannot proliferate beyond certain limits and have finite proliferative capacity (25). Initially, this phenomenon was attributed to an artificial change that occurs only under cell culture conditions. However, accumulated evidence revealed that cellular senescence has physiological roles (26–30) and is involving in aging (31–34). Internal and external stimuli, such as DNA damage stress (induced by radiation, chemotherapy, and reactive oxygen species (ROS)), inflammation, mechanical stress, repeated cell growth signals (growth factors and insulin-like growth factor-1 (IGF-1) signaling), metabolic aberrations, mitochondrial dysfunction, accumulation of unfolded proteins, certain oncogenes, and nuclear membrane dysfunction, can induce cellular senescence (15). These stimuli activate several signaling pathways and can converge on IGF-1/Akt/mammalian target of rapamycin (mTOR) signaling, which regulates transcription factor cascades, including the cell cycle inhibitors p16INK4A/RB and p53/p21CIP1, resulting in sustained irreversible cell cycle arrest (35). Cellular senescence plays physiological roles in diverse conditions and the number of senescent cells increases with aging. Excessive and disorganized senescent cells promote chronic inflammation and fibrotic tissue remodeling, exert paracrine effects on distant organs, and consequently drive the pathogenesis of many aging-related diseases (16, 36).
Novel treatment strategies targeting cellular senescence have been investigated based on these molecular mechanisms. Senolytics have been validated using mouse models. Mice have been genetically engineered to selectively eliminate p16-expressing senescent cells. These mice exhibit enhanced lifespan and delayed onset of aging-related pathologies (8). Senolytics have been explored using the STRING database (functional protein association networks) based on the biological differences between healthy and senescent cells. Dasatinib (a pan-tyrosine kinase inhibitor originally developed as an anti-cancer drug) and quercetin (a flavonoid) were selected as novel senolytic drugs (37). Subsequently, the Bcl-2 family inhibitor navitoclax (ABT-263) and selective Bcl-xL inhibitors A1331852 and A1155463 have been developed (38, 39). Fisetin, a polyphenol, exhibits senolytic activity by inhibiting the phosphatidylinositol-3 kinase-mTOR pathway (38). The administration of these senolytic agents decreases the accumulation of senescent cells and inflammatory cytokines, improves physical activity, and enhances the lifespan in aged mice (40). Other compounds, such as cardiac glycosides (ouabain) and HSP90 inhibitors (geldanamycin derivatives) also exhibit senolytic activity (41, 42).
Senomorphics attenuate the pathological effect of SASP without inducing cell death. Rapamycin, resveratrol, and metformin are representative senomorphics (43). Promising senomorphics include aspirin, NF-κB inhibitors, p38MAPK inhibitors, JAK/STAT inhibitors, ATM inhibitors, and statins (17). The molecular mechanisms of senomorphics are more complex than those of senolytics. Based on the complex mechanism and physiological role of cellular senescence, most senomorphics target SASP. Cellular senescence is initially induced in a small number of stressed cells and subsequently induced in neighboring and distant cells through the paracrine/autocrine effect of SASP. Hence, senomorphics targeting SASP may block the induction of cellular senescence at multiple sites (17). Senomorphics not only suppress senescence expansion by targeting the initial few senescent cells but also inhibit a vicious cycle that promotes further accumulation of senescent cells. Senotherapeutics can be promising therapeutic agents and may prevent the progression of aging-related pathology.
To develop a senotherapeutic strategy for aging-related respiratory diseases, one attractive approach is the repositioning of drugs as potential senomorphics. In contrast to de novo drug discovery, drug repositioning has several advantages, including low discovery costs and clinically established safety. A recent successful drug repositioning model is the application of an SGLT2 inhibitor, an anti-diabetic drug, for the treatment of heart failure (44, 45). In respiratory disease, another potential approach is changing the treatment modality to inhalation therapy, which may achieve a high local drug concentration in the lungs without exerting systemic adverse effects. Although several drugs may exert a senotherapeutic effect, high non-physiological concentrations are required under experimental conditions. Thus, the potential repositioning of drugs can be explored by changing the treatment modality to inhalation therapy for aging-related respiratory diseases (Figure 1). In the following section, we explain the involvement of cellular senescence in metabolic diseases and aging-related respiratory diseases and describe the potential therapeutic use of drugs against metabolic diseases as senotherapeutics.
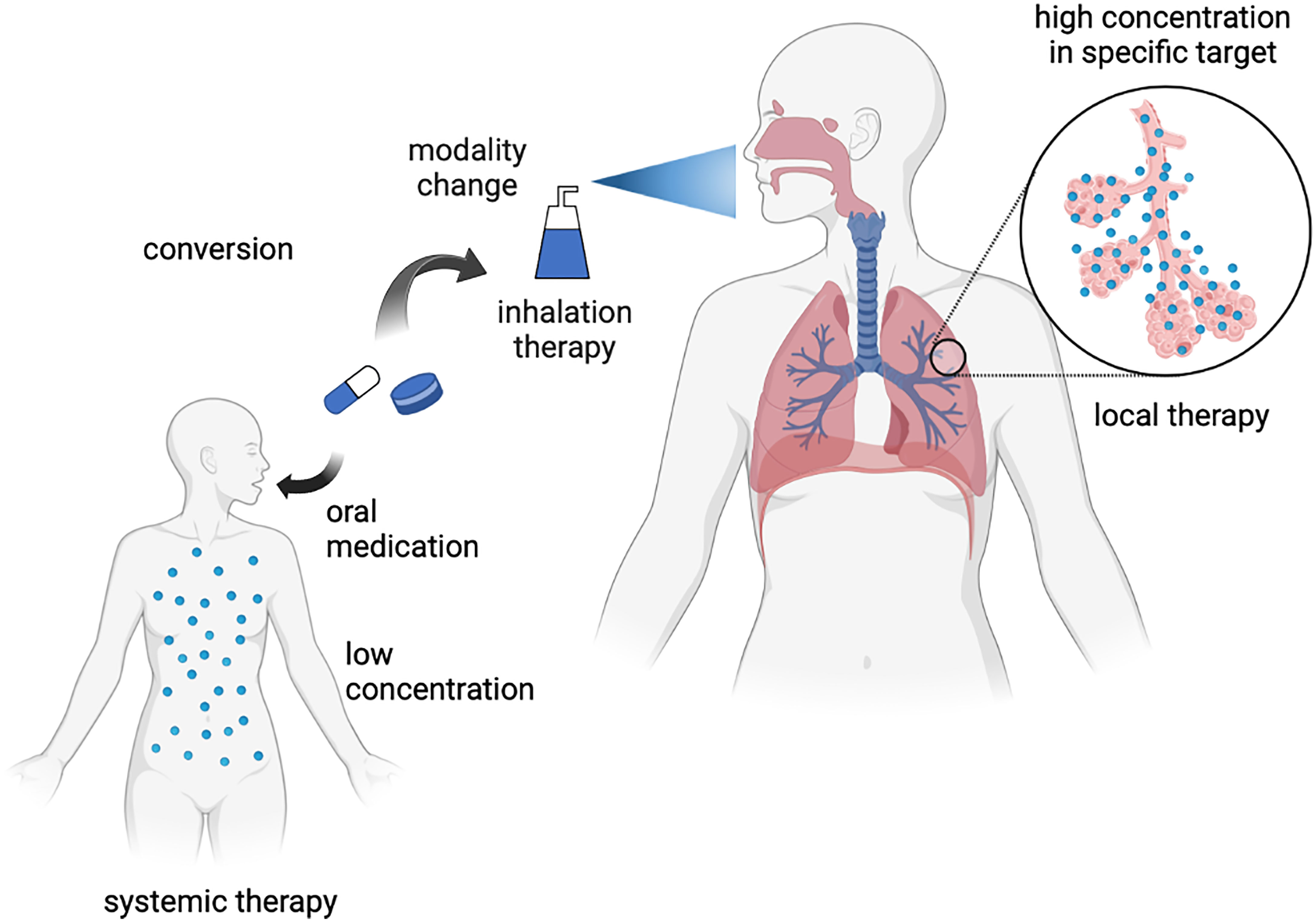
Figure 1 Concept of inhalation therapy with drugs against metabolic diseases for respiratory diseases as drug repositioning. Created with Biorender.
Metabolic diseases and cellular senescence
Cellular senescence is involved in the pathogenesis of various metabolic diseases (10), including metabolic syndrome, type 2 diabetes, and osteoporosis. Aging and obesity are the major risk factors for the development of type 2 diabetes (46). Senescent adipocytes accumulate in both mouse models and human cases of type 2 diabetes and obesity (47). Additionally, senescent adipocytes with p53 activation and enhanced ROS production are closely related to insulin resistance (48), glucose intolerance, and systemic inflammation (49), indicating the presence of a vicious cycle between adipocyte senescence and progression of type 2 diabetes. Furthermore, hyperglycemia and increased serum lipids induce adipocyte senescence and cellular senescence in various organs.
The efficacy of senolytics against metabolic diseases has been previously investigated. Senolytic therapy with dasatinib and quercetin (D + Q) is demonstrated to reduce the number of senescent adipocytes and restore insulin sensitivity in diet-induced obese mice (50). D + Q may also reduce the accumulation of senescent adipocytes in humans. The administration of D + Q for 3 days decreases the counts of macrophages and senescent adipocytes in adipose tissue and circulating SASP in patients with diabetic nephropathy (51). Thus, these results indicate that senolytics can be a promising approach for treating type 2 diabetes, which should be examined in future clinical trials. Additionally, the number of senescent cells is reported to increase with aging in the bone tissue in both mouse models and human samples (52, 53). D + Q, or the SASP inhibitor ruxolitinib alleviate osteoporosis in the mouse models (54).
Local organ aging may affect systemic aging in most metabolic diseases by inducing metabolic disturbances and upregulating SASP-related factors (10). Hence, the senotherapeutic effect of drugs against metabolic diseases is conferred through the improvement of metabolic conditions or the systemic anti-senescence properties during systemic administration. The therapeutic effect of drugs on metabolic diseases can be attributed to the accumulation of these pleiotropic effects. Moreover, this complex interaction between focal and systemic cellular senescence may be involved in other aging-related diseases. Several studies have demonstrated that drugs against metabolic disorders can exert a senotherapeutic effect on various cell types and tissues (targets other than the original therapeutic targets), including respiratory cells and tissues.
Aging-related respiratory diseases and cellular senescence
COPD and cellular senescence
Aging is closely associated with COPD development (55). Cellular senescence is observed in lung epithelial cells, vascular endothelial cells, and fibroblasts of patients with COPD (56, 57). Stimulation with cigarette smoke extract (CSE) induces senescence in the lung epithelial cells and lung fibroblasts in vitro (58–61). Additionally, cellular senescence and DNA damage in vascular endothelial progenitor cells and type II alveolar epithelial cells of patients with COPD are upregulated when compared with those cells of healthy controls, which may lead to the depletion of progenitor cells and stem cells required for regeneration of the damaged lung (62). In addition to increased numbers of senescent cells, elevated levels of SASP-related factors have been reported in the lungs of patients with COPD. In terms of COPD pathogenesis, the accumulation of senescent cells induced by repeated cigarette smoke exposure and chronological aging contribute to SASP-mediated inflammation, aberrant tissue repair, and loss of regenerative capacity, resulting in airway wall thickening and emphysema (3). Differential effects of cigarette smoke exposure on cellular senescence progression may be a critical determinant of COPD development. A recent study reported a negative correlation between epigenetic aging regulated by DNA methylation and respiratory function, suggesting a pivotal role of epigenetic modification in cellular senescence during COPD pathogenesis (63).
The expression of cellular senescence markers of CDK inhibitors (CDKI), such as p21CIP1 and p16INK4A, is upregulated in the airway epithelium of patients with COPD (58, 64). Several studies have demonstrated the potential efficacy of a senotherapeutic approach for preventing COPD development by regulating CDKI levels and CDKI-expressing cell numbers. Genetic deletion of p16INK4A may suppress smoking-stimulated respiratory function decline, emphysema, and cellular senescence of the airway epithelium in mouse models (65). In contrast, other studies have reported that genetic deletion of p16INK4A alone is not sufficient to suppress cellular senescence and emphysema (66). p14ARF (p19ARF in mice), encoded by the INK4a-ARF locus (also encodes p16INK4A; CDKN2A in humans), regulates cell cycle arrest and is used as a cellular senescence marker (67). The removal of p19ARF-expressing senescent cells by specifically inducing apoptosis with diphtheria toxin suppressed emphysematous changes induced by both cigarette smoke exposure and elastase in mouse models (68–70). Thus, the use of senolytics may be a promising approach for COPD; however, the safety and clinical application of senolytics for COPD treatment remain obscure. Moreover, the beneficial or adverse effects of a senolytic approach on organs with a high senescence burden, such as the lungs of patients with COPD have not been evaluated (71). A senomorphic approach using drugs against metabolic diseases for COPD is discussed in the following sections.
IPF and cellular senescence
Aging is a known risk factor for IPF development. The incidence of IPF increases with age (72). Telomere shortening, mainly reflecting replicative cellular senescence, is reported to be closely involved in IPF pathogenesis. Mutations in the telomerase-encoding gene (TERT) are detected in 8%–15% of patients with familial pulmonary fibrosis (73, 74). The frequency of telomere shortening is high in patients with sporadic pulmonary fibrosis (75). A study on three large cohorts of patients with IPF reported that shorter telomere length is associated with poor prognosis (76). This indicated that telomere attrition-induced cellular senescence is closely associated with IPF pathogenesis. Previously, we have detected senescence-associated β-galactosidase (SA-β-Gal), a representative marker of cellular senescence, in the lungs of patients with IPF. Epithelial cells covering fibroblastic foci and cuboidal metaplasia in the active fibrosing area are mainly stained with SA-β-Gal, whereas no positive senescent cells are detected in the healthy lung (77), which further indicated that cellular senescence is at least partly involved in the pathogenesis of lung fibrosis in IPF.
The p16INK4A expression levels in the lungs are positively correlated with the severity of pulmonary dysfunctions in patients with IPF (78). The removal of p16INK4A-positive cells mitigates the bleomycin-induced deterioration of pulmonary function without markedly altering lung fibrosis in INK-ATTAC mice. A similar benefit was achieved by administering D + Q (78). One pilot clinical trial examined the safety and efficacy of administering D + Q thrice a week for 3 weeks in patients with IPF, and D + Q improved performances in physical tests, such as 6-minute walk distance, 4-meter gait speed, and chair stand time (79). Other senotherapeutics examined for IPF include NADPH oxidase (NOX) 1/4 dual inhibitors. An imbalance between the levels of NOX4 and nuclear factor-erythroid 2-related factor 2 (NRF2) promotes the pathogenesis of lung fibrosis by enhancing cellular senescence in myofibroblasts, resulting in resistance to apoptosis and persistent fibrosis during IPF pathogenesis (80, 81). The NOX1/4 dual inhibitor GKT137831 attenuates bleomycin-induced lung fibrosis development in an aged mouse model (82). A phase II trial with GKT137831 involving patients with IPF is currently ongoing (NCT03865927).
Other anti-aging therapies, including rapamycin, nicotinamide riboside, nicotinamide mononucleotide, sirtuin activators are widely implicated in the treatment of COPD and IPF, and those anti-aging therapies are well summarized in recent reviews (3, 71). In the following sections, we summarize and focus on the therapeutic mechanisms and effects of representative drugs (metformin, statins, fibrates, and thiazolidinedione derivatives) against metabolic diseases on the pathogenesis of COPD and IPF.
Metformin
Metformin, a biguanide anti-diabetic agent, has been used as a first-line drug for diabetes. The orally administered metformin is absorbed from the intestine and transported to the hepatocytes where it inhibits the mitochondrial respiratory chain complex I (83, 84). Subsequently, the intracellular adenosine triphosphate (ATP)/adenosine monophosphate (AMP) ratio decreases, resulting in the activation of AMP-activated protein kinase (AMPK). Activated AMPK suppresses gluconeogenesis in the liver and downregulates serum glucose levels (85). In addition to its anti-diabetic activity, several studies have demonstrated the senomorphic effect of metformin. The mechanism underlying the senomorphic effect of metformin has not been completely elucidated and is assumed to be diverse (86, 87). Metformin attenuates IGF-1 signaling by decreasing the blood insulin levels, resulting in the inhibition of mTORC1 signaling, which is postulated to be a major systemic senomorphic mechanism (88–91). Additionally, metformin may exert a systemic senotherapeutic effect and directly exhibit senomorphic activity. Metformin imported into the cell via organic cationic transporter 1 exerts its senomorphic effect through several mechanisms (92). AMPK activation by metformin inhibits the downstream mTORC1 signaling, which is accompanied by the enhancement of nutrition sensing and autophagy. Additionally, AMPK improves mitochondrial biogenesis by activating peroxisome proliferator-activated receptor γ coactivator-1α (92). Furthermore, AMPK contributes to epigenetic transcriptional regulation via histone modification and microRNA (92, 93). Metformin-mediated inhibition of mitochondrial complex I suppresses ROS with concomitant production of advanced glycation end-products, resulting in a decreased accumulation of macromolecular damage (94). Moreover, metformin downregulates the inflammatory cytokine levels via NF-κB inhibition, activates NRF2, and consequently mitigates SASP secretion (95, 96). Thus, metformin exerts its senomorphic effect through the above-mentioned pleiotropic mechanisms. Several studies have reported that metformin extends the lifespan of model organisms (95, 97–99).
The lifespan-extending effects of metformin have been reported in patients with diabetes (100–102). In addition to its contribution to reducing cardiovascular disease risk, observational studies have demonstrated that metformin treatment decreases the incidence of malignancy (103–106). Furthermore, metformin mitigates age-related cognitive function decline (107, 108). The Food and Drug Administration and National Institute of Health are conducting the Targeting Aging with Metformin study, which aims to demonstrate the effects of metformin in non-diabetic populations on various physiological and pathological conditions, such as cancer, dementia, cardiovascular disease, lifespan, and other age-related diseases (86). However, some groups argue that the therapeutic effect of metformin is not a universal phenomenon in non-diabetic patients and healthy elderly individuals (109–111).
Stimulation of cultured human bronchial epithelial cells (HBECs) with CSE promotes mitochondrial injury, resulting in a decreased oxygen consumption rate (OCR). Metformin concentration-dependently reverses the CSE-induced depletion of OCR (112). The same study also examined the effect of a metformin-containing diet on the COPD mouse model exposed to cigarette smoke for 6 months. Lung inflammation, emphysematous change, and airway remodeling in the metformin-containing diet-fed group were lower than those in the routine diet-fed control group. Additionally, oxidative stress, cell death, telomere attrition, and cellular senescence were suppressed in the metformin-containing diet-fed group (112). Other researchers have reported that the AMPK-activating drug adenosine analog 5-aminoimidazole-4-carboxamide riboside suppresses CSE-induced cellular senescence and SASP-related factors in BEAS-2B, a human bronchial epithelial cell line (113). Additionally, the metformin-containing diet prevented the progression of elastase-induced emphysematous changes in a mouse model (114). Furthermore, metformin activated the epithelial sodium ion channel ENaC in lung epithelial cells, which may be involved in COPD prevention (115). Previously, we reported that metformin attenuates CSE-induced cellular senescence by suppressing mTOR signaling through the upregulation of DEPTOR expression in HBECs (116). Thus, local lung administration may be sufficient for metformin to exert a senomorphic effect, especially under cigarette smoke exposure conditions.
Several clinical studies have reported favorable effects of metformin in patients with COPD. The clinical outcomes of drugs against metabolic diseases in patients with COPD and IPF are summarized in Table 1. The COPDGene study reported that in patients with COPD, lung function decline with age among metformin users is slower than that among non-metformin users (112). Another COPDGene analysis demonstrated that metformin reduced the frequency of disease exacerbations in patients with both COPD and asthma and that metformin use was associated with an improved respiratory symptom score (117). In contrast, another study revealed no apparent clinical benefit of metformin administration in patients with COPD who were hospitalized for disease exacerbation although this study evaluated a small number of patients for a short duration (118). A large retrospective analysis of patients with both COPD and type 2 diabetes reported that all-cause mortality in the metformin-treated group was lower than that in the matched control group (14). Furthermore, a recent study demonstrated that metformin prevents the decline in pulmonary diffusion capacity in patients with both COPD and diabetes (13).
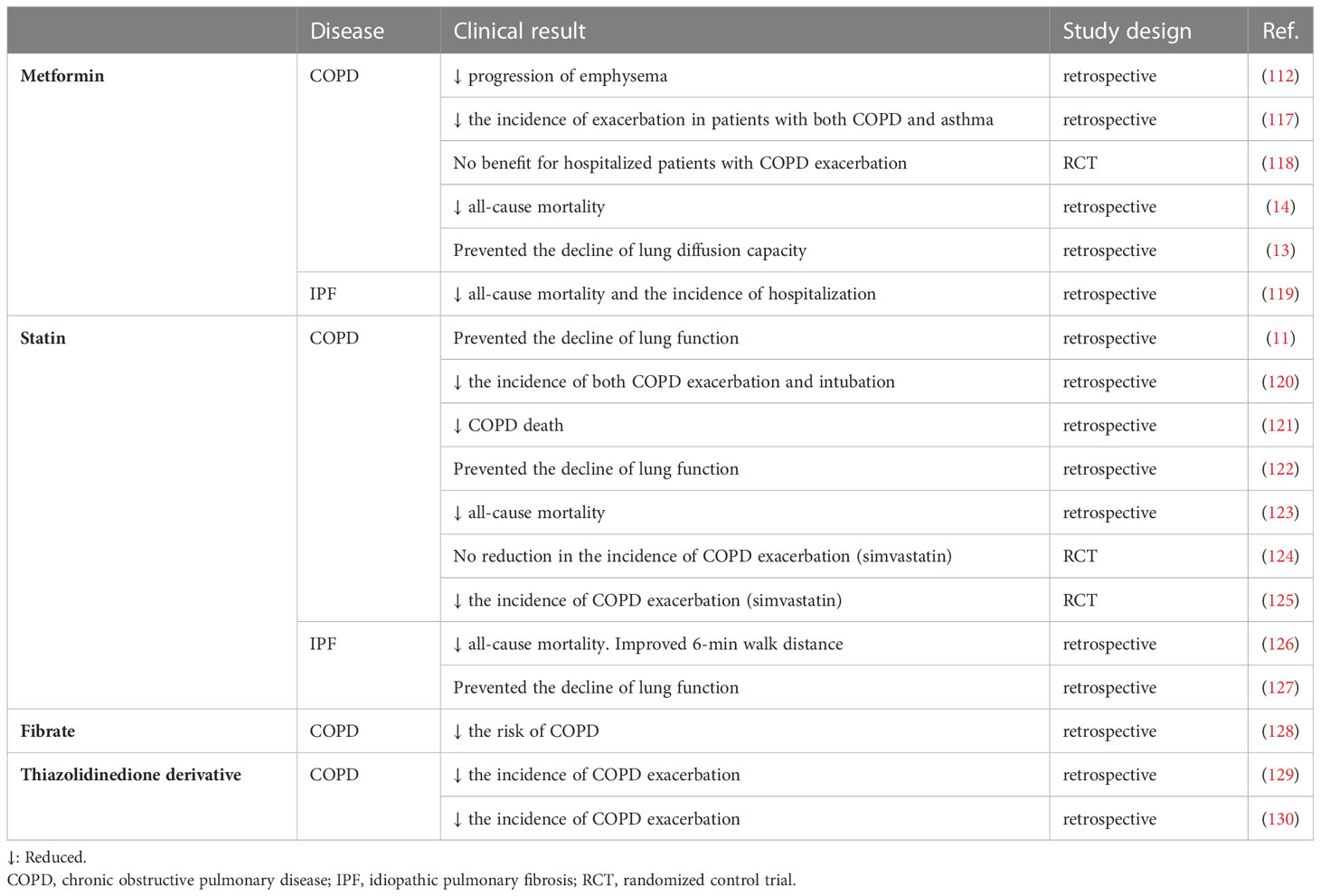
Table 1 Clinical outcomes of drugs against metabolic disease in COPD and IPF.
Several studies have demonstrated the potential efficacy of metformin in treating IPF. We previously reported that metformin suppresses TGF-β-induced myofibroblast differentiation of lung fibroblasts (131). Metformin activates AMPK, which inhibits TGF-β-induced NOX4 expression and concomitantly enhanced ROS production in lung fibroblasts. In a bleomycin-induced mouse model, metformin attenuated NOX4 upregulation and SMAD phosphorylation, resulting in the amelioration of lung fibrosis development (131). This inhibitory effect of metformin on pulmonary fibrosis has been further confirmed by several groups with one study suggesting that metformin may reverse the established pulmonary fibrosis (132, 133). A clinical retrospective study of patients with both IPF and diabetes demonstrated that the all-cause mortality and hospitalization in the metformin-treated group were lower than those in the non-metformin-treated group (119).
The potential application of metformin in the treatment of aging-related respiratory diseases has been demonstrated in both COPD and IPF. However, the drug concentration used under in vitro conditions is higher than the maximum blood concentration (Cmax) required for clinical use. The clinical and experimental dosages of the drugs against metabolic diseases are summarized in Table 2. Clinical dose and clinical Cmax were mainly based on the information from the National Library of Medicine in the USA, the electronic medicines compendium in the UK, and the Kyoto Encyclopedia of Genes and Genomes (KEGG) MEDICUS in Japan. The Cmax level of metformin for clinical use is 6.2–12.1 μM, whereas the concentrations used under in vitro experimental conditions are in the range of 0.5–10 mM. Therefore, the previously elucidated in vitro anti-senescent mechanisms may not precisely explain the potential use of metformin in patients with COPD and IPF. Furthermore, the oral administration of metformin at a clinically available dosage may not be sufficient to obtain appropriate clinical benefits as a senotherapeutic agent. We speculate that inhalation therapy with a high local concentration may address this issue associated with the clinical application of these agents in the treatment of aging-associated respiratory diseases.
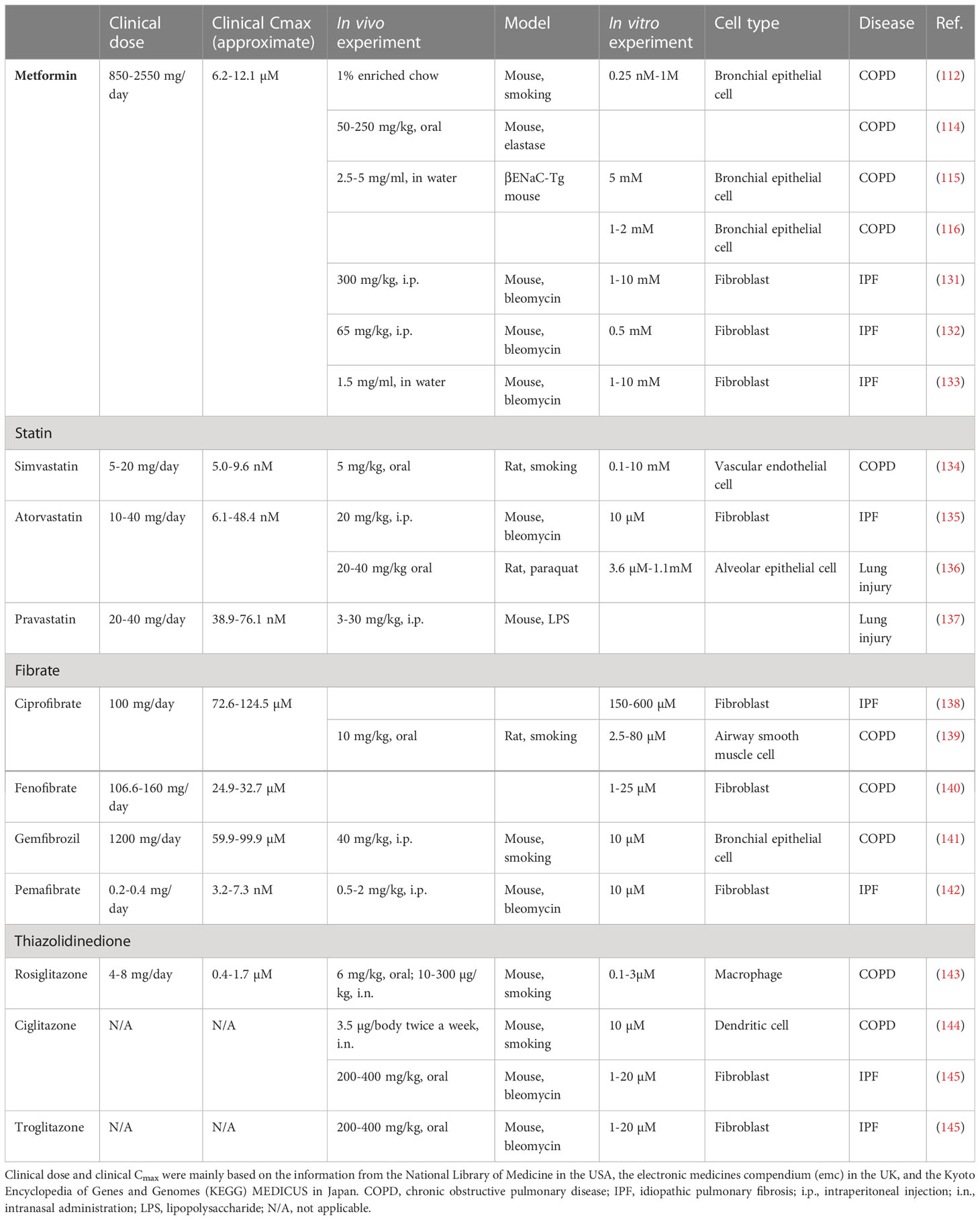
Table 2 Concentration of drugs against metabolic diseases in clinical and experimental setting.
Statins
Statins reduce cholesterol levels in hepatocytes by inhibiting the conversion of hydroxymethylglutaryl-CoA to mevalonate, which is the rate-limiting step in cholesterol biosynthesis in hepatocytes (146). The downregulation of cholesterol in hepatocytes promotes the nuclear translocation of sterol regulatory element-binding protein 2 (SREBP2) from the cytoplasm. In the nucleus, SREBP2 binds to the low-density lipoprotein (LDL) receptor-encoding gene promoter and upregulates the expression of LDL receptor. Subsequently, LDL in the serum is transferred to hepatocytes, leading to the downregulation of serum LDL levels (146, 147). In addition to cholesterol-lowering effects, statins exert other beneficial effects (also called pleiotropic effects) (148), and several studies have demonstrated the senotherapeutic property of statins. Hydrogen peroxide-induced cellular senescence is ameliorated by atorvastatin, pravastatin, and pitavastatin in vitro using human umbilical vein endothelial cells (149). Statin-mediated activation of Akt, through phosphorylation of Ser 473, led to expression of endothelial nitric oxide synthase, SIRT1, and catalase, all of which were implicated in the attenuation of cellular senescence in this study (149). In addition, fluvastatin inhibits the onset of endothelial progenitor cell senescence induced by ex vivo culture conditions, and its effect is independent of nitric oxide, ROS, and Rho kinase, but dependent on geranylgeranylpyrophosphate (150). Statins inhibit the prenylation of various proteins by blocking the formation of isoprenoid intermediates, which are essential for protein prenylation (151). This is postulated to be part of the mechanism for the suppression of cellular senescence because it leads to the upregulation of cell cycle-related proteins and downregulation of the expression of cell cycle inhibitor p27Kip1 (150). Another report showed that the administration of low-dose fluvastatin and valsartan increased the expression of longevity genes, including SIRT1, PRKAA, and KL in human subjects (152). Accordingly, statins act through various mechanisms to inhibit cellular senescence and have potential as senotherapeutic agents.
Large-scale retrospective studies conducted in several countries have reported the potential benefits of statins for patients with COPD in COPD-related hospitalization, cardiovascular complications, and mortality (11, 120–123). One study reported that statins may prevent the decline in pulmonary functions (11). Large-scale follow-up studies have been performed focusing on the role of statins in patients with COPD. The STATCOPE trial is a multicenter, placebo-controlled, randomized prospective study that focuses on the effect of statins on COPD exacerbation (124). In this trial, the primary outcome of the incidence of COPD exacerbation was not significantly different between the placebo and simvastatin-treated groups. Additionally, the period to first exacerbation was not significantly different between the placebo and simvastatin-treated groups, indicating no clinical benefit of simvastatin in preventing COPD exacerbation. However, this trial enrolled advanced high-risk patients with a decreased percentage of forced expiratory volume in one second and a history of emergency visits or hospitalization due to COPD exacerbation within one year, which may have affected the results. Although conclusive studies examining the benefit of statins in patients with mild to moderate COPD are not available, a recent study has attempted to clarify this question. A prospective double-blind study revealed that simvastatin significantly decreased COPD exacerbation (125). The beneficial effects of statins were also examined in COPD animal model. Oral administration of simvastatin ameliorated cigarette smoke-induced emphysema and pulmonary hypertension in rats exposed to cigarette smoke for 16 weeks, although the involvement of cellular senescence was not evaluated (134).
The therapeutic benefits of statins have also been reported in patients with IPF. Combined retrospective analysis of large-scale IPF trials for the anti-fibrotic drug pirfenidone (CAPACITY study and ASCEND study) revealed that statin users were associated with lower mortality and higher 6-minute walking distance than non-statin users (126). Additionally, overall hospitalizations, respiratory disease-related hospitalizations, and IPF-related deaths were low among statin users. A similar analysis was performed in another IPF trial for the anti-fibrotic drug nintedanib (INPULSIS study). Irrespective of nintedanib use, significant suppression of the decline in forced vital capacity/year was observed among statin users (127). The effects of statins were also examined in a bleomycin-induced lung fibrosis mouse model. Myofibroblast differentiation and pulmonary fibrosis in the group intraperitoneally administered with atorvastatin at a dose of 20 mg/kg bodyweight were attenuated when compared with those in the control group (135). Pravastatin attenuated lipopolysaccharide (LPS)-induced acute lung injury in mice (137), while atorvastatin inhibited paraquat-induced epithelial-mesenchymal transition (136).
Therefore, statins may alleviate the pathogenesis of both COPD and IPF through various mechanisms. However, their direct senotherapeutic effects on respiratory diseases are still unclear, and future studies are required to reveal the precise mechanism.
Fibrates
Fibrates, which are peroxisome proliferator-activated receptor (PPAR) α agonists, are used for the clinical treatment of dyslipidemia (153, 154). PPARα is expressed mostly in the liver, adipocytes, and skeletal muscles although its expression is also detected in other organs and immune cells (155). Fibrates promote fatty acid metabolism by activating PPARα and its transcripts. Additionally, activated PPARα upregulates the expression of the lipid enzymes medium-chain acyl-coenzyme A dehydrogenase and long-chain acyl-coenzyme A dehydrogenase and regulates mitochondrial β-oxidation (156, 157). In addition to their effects on lipid metabolism, several studies have demonstrated the senotherapeutic potential of fibrates. Fenofibrate protects against aging-related renal damage and dysfunction by improving proteinuria, tissue remodeling, inflammation, and apoptosis through the activation of AMPK and SIRT1 signaling in aged mice (158). One study reported that fenofibrate decreased the accumulation of senescent cells and inhibited cartilage degradation by inducing apoptosis and autophagic flux (159). Fenofibrate-induced PPARα upregulation reverses the aging effect on monocytes as evidenced by the restoration of fatty acid oxidation accompanied by high levels of lipid droplet formation (160). PPARα downregulation is implicated in Paneth cell senescence in the intestinal epithelial niche (161).
Peroxisome biosynthesis and metabolism are markedly downregulated in the lungs of patients with IPF. Treatment with ciprofibrate or pemafibrate promotes peroxisome proliferation and downregulates myofibroblast differentiation of fibroblasts (138, 142). Additionally, fenofibrate suppresses TGF-β-induced myofibroblast differentiation independent of PPARα activation by downregulating mitochondrial respiration (140). Fibrates suppress neutrophil infiltration, increase vascular permeability, and promote inflammatory cytokine production in bleomycin-induced or LPS-induced lung injury models and reduce lung compliance (162–164).
Ciprofibrate suppresses cytokines involved in smoking-induced airway remodeling and smooth muscle hyperplasia (139). The induction of TFEB by gemfibrozil mitigates CSE-induced autophagy impairment in airway epithelial cells, resulting in the suppression of ROS production and cellular senescence (141). This suggests the potential efficacy of ciprofibrates in activating autophagy by modulating TFEB through PPARα activation during COPD pathogenesis. A population-based retrospective cohort study reported that in patients with dyslipidemia, the incidence of COPD development among fibrate users was lower than that among non-fibrate users within the 6-year observation period (128). These findings indicate that fibrate is a promising therapeutic for aging-related lung diseases and that it can function as a senotherapeutic by activating PPARα and its transcripts. In contrast to metformin, oral administration of fibrates at a clinical dosage may sufficiently achieve high drug concentrations to exert a senomorphic effect based on in vitro experiments (Table 2), suggesting that fibrates are potential candidates that can be repositioned as senotherapeutics.
Thiazolidinedione derivatives
In addition to metformin, statins, and fibrates, thiazolidinedione derivatives (such as rosiglitazone and pioglitazone) have been reported to exert senotherapeutic effects. Rosiglitazone and pioglitazone function as PPARγ agonists and are used as oral hypoglycemic agents (165). Cellular senescence induced by angiotensin II is inhibited by pioglitazone in endothelial progenitor cells (166). In this study, pioglitazone prevents cellular senescence by downregulating angiotensin type 1 receptor. Pioglitazone also restores telomerase activity, which may also be involved in its anti-senescence activity. Age-related functional decline in renal mesenchymal stem cells contributes to the pathogenesis of chronic kidney disease. Indoxyl sulfate, a uremia-related toxin, evokes cellular senescence in renal mesenchymal stem cells, which is inhibited by pioglitazone (167). This effect of pioglitazone is, at least in part, attributable to the activation of PPARγ, which leads to suppression of prion protein gene expression. The senotherapeutic properties of thiazolidinediones have also been demonstrated in UV irradiation-induced cellular senescence in murine skin fibroblasts. Pioglitazone inhibits UV-induced cellular senescence through attenuating ROS production with concomitant suppression of cell cycle arrest-associated proteins such as p53 and p21 (168). The senotherapeutic potential of thiazolidinediones is also demonstrated using in vivo models. Pioglitazone ameliorates age-related renal dysfunction in 24-months old rats, which is attributed to increased klotho expression and decreased oxidative stress and mitochondrial injury (169). Long-term treatment with low-dose rosiglitazone extended the lifespan of aged mice (170). Additionally, rosiglitazone mitigated inflammation and tissue atrophy, improved cognitive ability, and alleviated depression-like symptoms (170). Further, patients with diabetes treated with pioglitazone had a lower mortality rate compared to non-PPARγ agonists users (170).
Several studies have demonstrated the efficacy of PPARγ agonists in treating respiratory diseases. A retrospective study on veterans with both diabetes and COPD revealed that the risk of COPD exacerbations in patients receiving PPARγ agonists was significantly lower than that in patients receiving other diabetes medications (129). Another retrospective study reported similar results with PPARγ agonists (130). The effects of other thiazolidinedione derivatives have been examined in mouse models. Rosiglitazone prevents the upregulation of neutrophil counts in the bronchoalveolar lavage fluid of mice exposed to cigarette smoke for a short duration (5 days) (143). Ciglitazone is reported to attenuate lung emphysema in mice chronically exposed to cigarette smoke (3–5 months) (144). The therapeutic efficacies of troglitazone and ciglitazone have also been demonstrated in the bleomycin-induced lung fibrosis mouse model (145).
Compared with those on other drugs, studies on senotherapeutic potential of thiazolidinedione derivatives and their application for treating aging-related respiratory diseases are at a nascent stage. Hence, further studies on thiazolidinedione derivatives are needed.
Inhalation therapy
As we have described in this review, the drugs against metabolic diseases exhibit senotherapeutic properties and may be beneficial for the treatment of aging-related respiratory diseases. However, the usefulness and clinical application of these agents to respiratory disease remains uncertain, because of the high drug concentrations used in the experimental models compared to the expected drug concentrations in the lungs based on their approved oral doses. We speculate that a possible solution to these issues is the development of an inhalation treatment modality. Repurposing pre-existing drugs with potential efficacy for respiratory disease to suit inhalation therapy may save time and cost compared with developing a new drug from scratch. Currently, the clinically available inhaled drugs for respiratory diseases are mainly composed of inhaled steroids and bronchodilators. However, under experimental conditions, several drugs against metabolic diseases, such as statins and thiazolidinediones, have been investigated in asthma and pulmonary hypertension models. In comparison to the distribution during intraperitoneal or forced oral administration, better lung-localized drug distribution is demonstrated during inhalation and intratracheal administration of simvastatin in OVA-induced asthma mouse model (171). Simvastatin inhalation suppresses airway inflammation and remodeling in a dose-dependent manner. Intratracheal administration of pravastatin is shown to suppress bronchial goblet cell hyperplasia and reduce TNF-α and KC expression in bronchoalveolar lavage fluid, but not the expression of other chemokines or airway irritability in OVA-induced asthma mouse model (172). In a rat model of monocrotaline-induced pulmonary arterial hypertension (PAH), intratracheal administration of nanoparticulated pitavastatin attenuates the progression of PAH accompanied by a reduction in inflammation and pulmonary artery remodeling (173). In another study, the effects of rosiglitazone on PAH were examined. While no obvious effect of oral rosiglitazone administration on pulmonary hemodynamics is demonstrated, intratracheal administration of poly(lactic-co-glycolic) acid-based particles of rosiglitazone induces selective pulmonary vasodilation and reduces the proliferation of vascular endothelial cells and smooth muscle cells in rats with PAH (174). Furthermore, combined inhalation of rosiglitazone and sildenafil leads to improvement in cardiac function, delayed right heart remodeling, and inhibition of arterial muscularization in rats with PAH (175). Nebulized pioglitazone in combination with synthetic lung surfactant promotes lung maturation and attenuates the development of neonatal hyperoxia-induced lung injury (176). Interestingly, inhalation therapy using resveratrol as a senomorphic to treat respiratory disease shows that intratracheal administration preserves lung compliance and structure and prevents DNA damage in prematurely aging telomerase null (terc-/-) mice (177). In addition, resveratrol-β-cyclodextrin inclusion complexes significantly suppresses ZnCl2 smoke-induced acute lung injury through anti-inflammatory and anti-apoptotic mechanism (178).
Accordingly, we speculate that the development of inhalation therapy using drugs against metabolic diseases can be a promising approach for potential senotherapy; however, several concerns must be noted. High drug concentrations in the lungs during inhalation may evoke previously unrecognized toxicity. In addition, the conversion of the drug delivery system from oral to inhalation could cause critical pharmacological alterations based on drug properties and pharmacokinetics which should be carefully examined. Overcoming drug insolubility in water and low stability in solution should be an urgent task in inhalation therapy development. Furthermore, the development of efficient drug delivery devices is a critical problem. In future studies, these issues should be addressed for each drug before adopting them for clinical applications.
Conclusions
Cellular metabolism is closely related to the mechanisms of cellular senescence. Hence, drugs against metabolic diseases may have senotherapeutic potential. Cellular senescence plays a pivotal role in the pathogenesis of various aging-related disorders. Hence, senotherapy can be a promising approach to develop efficient treatments for refractory aging-related respiratory diseases, including COPD and IPF. Drugs against metabolic diseases can be potentially repositioned as senotherapeutics. However, based on the experimental results, concentrations higher than those achieved by oral administration may be necessary to determine the clinical efficacy of some promising senotherapeutics in treating COPD and IPF. In addition to the oral administration modality, the development of an inhalation modality for the treatment of metabolic diseases can be an attractive approach to achieve high and effective local concentrations of drugs in the lungs without inducing systemic adverse events. Future studies should focus on determining appropriate drugs, optimal drug concentrations, and effective treatment modalities to develop clinically applicable therapeutics for aging-related respiratory diseases using drugs against metabolic diseases.
Author contributions
SM, SI, and JA prepared the manuscript. All authors have made substantial contributions to the manuscript and have read and approved the final manuscript.
Funding
This work was supported by JSPS KAKENHI (Grant numbers: 21K16123, 22H03082, and 21K08213).
Acknowledgments
We would like to thank Editage (www.editage.com) for English language editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AMP, adenosine monophosphate; AMPK, AMP-activated protein kinase; ATP, adenosine triphosphate; CDK, cyclin-dependent kinase; CDKI, cyclin-dependent kinase inhibitor; COPD, chronic obstructive pulmonary disease; CSE, cigarette smoke extract; HBEC,human bronchial epithelial cell; IGF-1, insulin growth factor-1; IPF, idiopathic pulmonary fibrosis; LDL, low-density lipoprotein; LPS, lipopolysaccharide; mTOR, mammalian target of rapamycin; NRF2, nuclear factor-erythroid 2-related factor 2; OCR, oxygen consumption rate; PAH, pulmonary arterial hypertension; PPAR, peroxisome proliferator-activated receptor; ROS, reactive oxygen species; SA-β-gal, senescence-associated β-galactosidase; SASP, senescence-associated secretory phenotype; SREBP2, sterol regulatory element-binding protein 2; TFEB, transcriptional factor EB.
References
1. Bowdish DME. The aging lung: Is lung health good health for older adults? Chest (2019) 155:391–400. doi: 10.1016/j.chest.2018.09.003
2. Janssens JP, Pache JC, Nicod LP. Physiological changes in respiratory function associated with ageing. Eur Respir J (1999) 13:197–205. doi: 10.1183/09031936.99.14614549
3. Barnes PJ, Baker J, Donnelly LE. Cellular senescence as a mechanism and target in chronic lung diseases. Am J Respir Crit Care Med (2019) 200:556–64. doi: 10.1164/rccm.201810-1975TR
4. Weindruch R, Walford RL, Fligiel S, Guthrie D. The retardation of aging in mice by dietary restriction: longevity, cancer, immunity and lifetime energy intake. J Nutr (1986) 116:641–54. doi: 10.1093/jn/116.4.641
5. Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature (1993) 366:461–4. doi: 10.1038/366461a0
6. Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in saccharomyces cerevisiae by two different mechanisms. Genes Dev (1999) 13:2570–80. doi: 10.1101/gad.13.19.2570
7. Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature (2000) 403:795–800. doi: 10.1038/35001622
8. Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature (2011) 479:232–6. doi: 10.1038/nature10600
9. Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med (2015) 21:1424–35. doi: 10.1038/nm.4000
10. Khosla S, Farr JN, Tchkonia T, Kirkland JL. The role of cellular senescence in ageing and endocrine disease. Nat Rev Endocrinol (2020) 16:263–75. doi: 10.1038/s41574-020-0335-y
11. Alexeeff SE, Litonjua AA, Sparrow D, Vokonas PS, Schwartz J. Statin use reduces decline in lung function: VA normative aging study. Am J Respir Crit Care Med (2007) 176:742–7. doi: 10.1164/rccm.200705-656OC
12. Ingebrigtsen TS, Marott JL, Nordestgaard BG, Lange P, Hallas J, Vestbo J. Statin use and exacerbations in individuals with chronic obstructive pulmonary disease. Thorax (2015) 70:33–40. doi: 10.1136/thoraxjnl-2014-205795
13. Kahnert K, Andreas S, Kellerer C, Lutter JI, Lucke T, Yildirim Ö, et al. Reduced decline of lung diffusing capacity in COPD patients with diabetes and metformin treatment. Sci Rep (2022) 12:1435. doi: 10.1038/s41598-022-05276-x
14. Yen F-S, Chen W, Wei JC-C, Hsu C-C, Hwu C-M. Effects of metformin use on total mortality in patients with type 2 diabetes and chronic obstructive pulmonary disease: A matched-subject design. PloS One (2018) 13:e0204859. doi: 10.1371/journal.pone.0204859
15. Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol (2014) 15:482–96. doi: 10.1038/nrm3823
16. Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol (2007) 8:729–40. doi: 10.1038/nrm2233
17. Di Micco R, Krizhanovsky V, Baker D, d’Adda di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol (2021) 22:75–95. doi: 10.1038/s41580-020-00314-w
18. Heinrich R, Schuster S. The regulation of cellular systems. New York: Springer Science & Business Media (2012).
19. Kriete A. Robustness and aging–a systems-level perspective. Biosystems (2013) 112:37–48. doi: 10.1016/j.biosystems.2013.03.014
21. Barth E, Sieber P, Stark H, Schuster S. Robustness during aging-molecular biological and physiological aspects. Cells (2020) 9:1862. doi: 10.3390/cells9081862
22. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. Hallmarks of aging: An expanding universe. Cell (2023) 186(2):243–78. doi: 10.1016/j.cell.2022.11.001
23. Uyar B, Palmer D, Kowald A, Murua Escobar H, Barrantes I, Möller S, et al. Single-cell analyses of aging, inflammation and senescence. Ageing Res Rev (2020) 64:101156. doi: 10.1016/j.arr.2020.101156
24. Cohn RL, Gasek NS, Kuchel GA, Xu M. The heterogeneity of cellular senescence: insights at the single-cell level. Trends Cell Biol (2023) 33:9–17. doi: 10.1016/j.tcb.2022.04.011
25. Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res (1965) 37:614–36. doi: 10.1016/0014-4827(65)90211-9
26. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell (1997) 88:593–602. doi: 10.1016/S0092-8674(00)81902-9
27. Muñoz-Espín D, Cañamero M, Maraver A, Gómez-López G, Contreras J, Murillo-Cuesta S, et al. Programmed cell senescence during mammalian embryonic development. Cell (2013) 155:1104–18. doi: 10.1016/j.cell.2013.10.019
28. Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell (2013) 155:1119–30. doi: 10.1016/j.cell.2013.10.041
29. Jun J-I, Lau LF. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol (2010) 12:676–85. doi: 10.1038/ncb2070
30. Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, et al. Senescence of activated stellate cells limits liver fibrosis. Cell (2008) 134:657–67. doi: 10.1016/j.cell.2008.06.049
31. Baker DJ, Perez-Terzic C, Jin F, Pitel KS, Niederländer NJ, Jeganathan K, et al. Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat Cell Biol (2008) 10:825–36. doi: 10.1038/ncb1744
32. Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM. Cellular senescence in aging primates. Science (2006) 311:1257. doi: 10.1126/science.1122446
33. Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, von Zglinicki T. DNA Damage response and cellular senescence in tissues of aging mice. Aging Cell (2009) 8:311–23. doi: 10.1111/j.1474-9726.2009.00481.x
34. Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, et al. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature (2006) 443:453–7. doi: 10.1038/nature05092
35. Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, et al. Cellular senescence: Defining a path forward. Cell (2019) 179:813–27. doi: 10.1016/j.cell.2019.10.005
36. van Deursen JM. The role of senescent cells in ageing. Nature (2014) 509:439–46. doi: 10.1038/nature13193
37. Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, et al. The achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell (2015) 14:644–58. doi: 10.1111/acel.12344
38. Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, Dai HM, Ling YY, Stout MB, et al. Identification of a novel senolytic agent, navitoclax, targeting the bcl-2 family of anti-apoptotic factors. Aging Cell (2016) 15:428–35. doi: 10.1111/acel.12445
39. Zhu Y, Doornebal EJ, Pirtskhalava T, Giorgadze N, Wentworth M, Fuhrmann-Stroissnigg H, et al. New agents that target senescent cells: the flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging (2017) 9:955–63. doi: 10.18632/aging.101202
40. Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, et al. Senolytics improve physical function and increase lifespan in old age. Nat Med (2018) 24:1246–56. doi: 10.1038/s41591-018-0092-9
41. Guerrero A, Herranz N, Sun B, Wagner V, Gallage S, Guiho R, et al. Cardiac glycosides are broad-spectrum senolytics. Nat Metab (2019) 1:1074–88. doi: 10.1038/s42255-019-0122-z
42. Fuhrmann-Stroissnigg H, Ling YY, Zhao J, McGowan SJ, Zhu Y, Brooks RW, et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat Commun (2017) 8:422. doi: 10.1038/s41467-017-00314-z
43. Zhang L, Pitcher LE, Prahalad V, Niedernhofer LJ, Robbins PD. Targeting cellular senescence with senotherapeutics: senolytics and senomorphics. FEBS J (2022) 290:1362–83. doi: 10.1111/febs.16350
44. Nassif ME, Windsor SL, Borlaug BA, Kitzman DW, Shah SJ, Tang F, et al. The SGLT2 inhibitor dapagliflozin in heart failure with preserved ejection fraction: a multicenter randomized trial. Nat Med (2021) 27:1954–60. doi: 10.1038/s41591-021-01536-x
45. Zannad F, Ferreira JP, Pocock SJ, Anker SD, Butler J, Filippatos G, et al. SGLT2 inhibitors in patients with heart failure with reduced ejection fraction: a meta-analysis of the EMPEROR-reduced and DAPA-HF trials. Lancet (2020) 396:819–29. doi: 10.1016/S0140-6736(20)31824-9
46. Palmer AK, Gustafson B, Kirkland JL, Smith U. Cellular senescence: at the nexus between ageing and diabetes. Diabetologia (2019) 62:1835–41. doi: 10.1007/s00125-019-4934-x
47. Xu M, Palmer AK, Ding H, Weivoda MM, Pirtskhalava T, White TA, et al. Targeting senescent cells enhances adipogenesis and metabolic function in old age. Elife (2015) 4:e12997. doi: 10.7554/eLife.12997
48. Minamino T, Orimo M, Shimizu I, Kunieda T, Yokoyama M, Ito T, et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat Med (2009) 15:1082–7. doi: 10.1038/nm.2014
49. Vergoni B, Cornejo P-J, Gilleron J, Djedaini M, Ceppo F, Jacquel A, et al. DNA Damage and the activation of the p53 pathway mediate alterations in metabolic and secretory functions of adipocytes. Diabetes (2016) 65:3062–74. doi: 10.2337/db16-0014
50. Palmer AK, Xu M, Zhu Y, Pirtskhalava T, Weivoda MM, Hachfeld CM, et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell (2019) 18:e12950. doi: 10.1111/acel.12950
51. Hickson LJ, Langhi Prata LG, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of dasatinib plus quercetin in individuals with diabetic kidney disease. EBioMedicine (2019) 47:446–56. doi: 10.1016/j.ebiom.2019.08.069
52. Piemontese M, Almeida M, Robling AG, Kim H-N, Xiong J, Thostenson JD, et al. Old age causes de novo intracortical bone remodeling and porosity in mice. JCI Insight (2017) 2:e93771. doi: 10.1172/jci.insight.93771
53. Farr JN, Fraser DG, Wang H, Jaehn K, Ogrodnik MB, Weivoda MM, et al. Identification of senescent cells in the bone microenvironment. J Bone Miner Res (2016) 31:1920–9. doi: 10.1002/jbmr.2892
54. Farr JN, Xu M, Weivoda MM, Monroe DG, Fraser DG, Onken JL, et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat Med (2017) 23:1072–9. doi: 10.1038/nm.4385
55. Barnes PJ. Senescence in COPD and its comorbidities. Annu Rev Physiol (2017) 79:517–39. doi: 10.1146/annurev-physiol-022516-034314
56. Tsuji T, Aoshiba K, Nagai A. Alveolar cell senescence in patients with pulmonary emphysema. Am J Respir Crit Care Med (2006) 174:886–93. doi: 10.1164/rccm.200509-1374OC
57. Müller K-C, Welker L, Paasch K, Feindt B, Erpenbeck VJ, Hohlfeld JM, et al. Lung fibroblasts from patients with emphysema show markers of senescence in vitro. Respir Res (2006) 7:32. doi: 10.1186/1465-9921-7-32
58. Tsuji T, Aoshiba K, Nagai A. Cigarette smoke induces senescence in alveolar epithelial cells. Am J Respir Cell Mol Biol (2004) 31:643–9. doi: 10.1165/rcmb.2003-0290OC
59. Nyunoya T, Monick MM, Klingelhutz A, Yarovinsky TO, Cagley JR, Hunninghake GW. Cigarette smoke induces cellular senescence. Am J Respir Cell Mol Biol (2006) 35:681–8. doi: 10.1165/rcmb.2006-0169OC
60. Fujii S, Hara H, Araya J, Takasaka N, Kojima J, Ito S, et al. Insufficient autophagy promotes bronchial epithelial cell senescence in chronic obstructive pulmonary disease. Oncoimmunology (2012) 1:630–41. doi: 10.4161/onci.20297
61. Ito S, Araya J, Kurita Y, Kobayashi K, Takasaka N, Yoshida M, et al. PARK2-mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy (2015) 11:547–59. doi: 10.1080/15548627.2015.1017190
62. Paschalaki KE, Starke RD, Hu Y, Mercado N, Margariti A, Gorgoulis VG, et al. Dysfunction of endothelial progenitor cells from smokers and chronic obstructive pulmonary disease patients due to increased DNA damage and senescence. Stem Cells (2013) 31:2813–26. doi: 10.1002/stem.1488
63. Breen M, Nwanaji-Enwerem JC, Karrasch S, Flexeder C, Schulz H, Waldenberger M, et al. Accelerated epigenetic aging as a risk factor for chronic obstructive pulmonary disease and decreased lung function in two prospective cohort studies. Aging (2020) 12:16539–54. doi: 10.18632/aging.103784
64. Aoshiba K, Zhou F, Tsuji T, Nagai A. DNA Damage as a molecular link in the pathogenesis of COPD in smokers. Eur Respir J (2012) 39:1368–76. doi: 10.1183/09031936.00050211
65. Cottage CT, Peterson N, Kearley J, Berlin A, Xiong X, Huntley A, et al. Targeting p16-induced senescence prevents cigarette smoke-induced emphysema by promoting IGF1/Akt1 signaling in mice. Commun Biol (2019) 2:307. doi: 10.1038/s42003-019-0532-1
66. Sundar IK, Rashid K, Gerloff J, Li D, Rahman I. Genetic ablation of p16INK4a does not protect against cellular senescence in mouse models of chronic obstructive pulmonary Disease/Emphysema. Am J Respir Cell Mol Biol (2018) 59:189–99. doi: 10.1165/rcmb.2017-0390OC
67. Sherr CJ. Divorcing ARF. And p53: an unsettled case. Nat Rev Cancer (2006) 6:663–73. doi: 10.1038/nrc1954
68. Hashimoto M, Asai A, Kawagishi H, Mikawa R, Iwashita Y, Kanayama K, et al. Elimination of p19ARF-expressing cells enhances pulmonary function in mice. JCI Insight (2016) 1:e87732. doi: 10.1172/jci.insight.87732
69. Mikawa R, Sato T, Suzuki Y, Baskoro H, Kawaguchi K, Sugimoto M. p19Arf exacerbates cigarette smoke-induced pulmonary dysfunction. Biomolecules (2020) 10:462. doi: 10.3390/biom10030462
70. Mikawa R, Suzuki Y, Baskoro H, Kanayama K, Sugimoto K, Sato T, et al. Elimination of p19ARF -expressing cells protects against pulmonary emphysema in mice. Aging Cell (2018) 17:e12827. doi: 10.1111/acel.12827
71. Araya J, Kuwano K. Cellular senescence-an aging hallmark in chronic obstructive pulmonary disease pathogenesis. Respir Investig (2022) 60:33–44. doi: 10.1016/j.resinv.2021.09.003
72. Raghu G, Chen S-Y, Yeh W-S, Maroni B, Li Q, Lee Y-C, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med (2014) 2:566–72. doi: 10.1016/S2213-2600(14)70101-8
73. Armanios MY, Chen JJ-L, Cogan JD, Alder JK, Ingersoll RG, Markin C, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med (2007) 356:1317–26. doi: 10.1056/NEJMoa066157
74. Tsakiri KD, Cronkhite JT, Kuan PJ, Xing C, Raghu G, Weissler JC, et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci USA (2007) 104:7552–7. doi: 10.1073/pnas.0701009104
75. Alder JK, Chen JJ-L, Lancaster L, Danoff S, Su S-C, Cogan JD, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci USA (2008) 105:13051–6. doi: 10.1073/pnas.0804280105
76. Stuart BD, Lee JS, Kozlitina J, Noth I, Devine MS, Glazer CS, et al. Effect of telomere length on survival in patients with idiopathic pulmonary fibrosis: an observational cohort study with independent validation. Lancet Respir Med (2014) 2:557–65. doi: 10.1016/S2213-2600(14)70124-9
77. Minagawa S, Araya J, Numata T, Nojiri S, Hara H, Yumino Y, et al. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-β-induced senescence of human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol (2011) 300:L391–401. doi: 10.1152/ajplung.00097.2010
78. Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun (2017) 8:14532. doi: 10.1038/ncomms14532
79. Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK, et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine (2019) 40:554–63. doi: 10.1016/j.ebiom.2018.12.052
80. Hecker L, Vittal R, Jones T, Jagirdar R, Luckhardt TR, Horowitz JC, et al. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med (2009) 15:1077–81. doi: 10.1038/nm.2005
81. Amara N, Goven D, Prost F, Muloway R, Crestani B, Boczkowski J. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGF 1-induced fibroblast differentiation into myofibroblasts. Thorax (2010) pp:733–8. doi: 10.1136/thx.2009.113456
82. Hecker L, Logsdon NJ, Kurundkar D, Kurundkar A, Bernard K, Hock T, et al. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci Transl Med (2014) 6:231ra47. doi: 10.1126/scitranslmed.3008182
83. El-Mir MY, Nogueira V, Fontaine E, Avéret N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem (2000) 275:223–8. doi: 10.1074/jbc.275.1.223
84. Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J (2000) 348(Pt 3):607–14. doi: 10.1042/bj3480607
85. Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia (2017) 60:1577–85. doi: 10.1007/s00125-017-4342-z
86. Barzilai N, Crandall JP, Kritchevsky SB, Espeland MA. Metformin as a tool to target aging. Cell Metab (2016) 23:1060–5. doi: 10.1016/j.cmet.2016.05.011
87. Kulkarni AS, Gubbi S, Barzilai N. Benefits of metformin in attenuating the hallmarks of aging. Cell Metab (2020) 32:15–30. doi: 10.1016/j.cmet.2020.04.001
88. Liu B, Fan Z, Edgerton SM, Yang X, Lind SE, Thor AD. Potent anti-proliferative effects of metformin on trastuzumab-resistant breast cancer cells via inhibition of erbB2/IGF-1 receptor interactions. Cell Cycle (2011) 10:2959–66. doi: 10.4161/cc.10.17.16359
89. Kickstein E, Krauss S, Thornhill P, Rutschow D, Zeller R, Sharkey J, et al. Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc Natl Acad Sci USA (2010) 107:21830–5. doi: 10.1073/pnas.0912793107
90. Nair V, Sreevalsan S, Basha R, Abdelrahim M. Mechanism of metformin-dependent inhibition of mammalian target of rapamycin (mTOR) and ras activity in pancreatic cancer: role of specificity protein (Sp) transcription factors. J Biol Chem (2014) 289:27692–701. doi: 10.1074/jbc.M114.592576
91. Pérez-Revuelta BI, Hettich MM, Ciociaro A, Rotermund C, Kahle PJ, Krauss S, et al. Metformin lowers ser-129 phosphorylated α-synuclein levels via mTOR-dependent protein phosphatase 2A activation. Cell Death Dis (2014) 5:e1209. doi: 10.1038/cddis.2014.175
92. Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B. Metformin: from mechanisms of action to therapies. Cell Metab (2014) 20:953–66. doi: 10.1016/j.cmet.2014.09.018
93. Wang Y, An H, Liu T, Qin C, Sesaki H, Guo S, et al. Metformin improves mitochondrial respiratory activity through activation of AMPK. Cell Rep (2019) 29:1511–1523.e5. doi: 10.1016/j.celrep.2019.09.070
94. Ishibashi Y, Matsui T, Takeuchi M, Yamagishi S. Metformin inhibits advanced glycation end products (AGEs)-induced renal tubular cell injury by suppressing reactive oxygen species generation via reducing receptor for AGEs (RAGE) expression. Horm Metab Res (2012) 44:891–5. doi: 10.1055/s-0032-1321878
95. Moiseeva O, Deschênes-Simard X, St-Germain E, Igelmann S, Huot G, Cadar AE, et al. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κB activation. Aging Cell (2013) 12:489–98. doi: 10.1111/acel.12075
96. Hu Q, Peng J, Jiang L, Li W, Su Q, Zhang J, et al. Metformin as a senostatic drug enhances the anticancer efficacy of CDK4/6 inhibitor in head and neck squamous cell carcinoma. Cell Death Dis (2020) 11:925. doi: 10.1038/s41419-020-03126-0
97. Anisimov VN, Berstein LM, Popovich IG, Zabezhinski MA, Egormin PA, Piskunova TS, et al. If started early in life, metformin treatment increases life span and postpones tumors in female SHR mice. Aging (2011) 3:148–57. doi: 10.18632/aging.100273
98. Cabreiro F, Au C, Leung K-Y, Vergara-Irigaray N, Cochemé HM, Noori T, et al. Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell (2013) 153:228–39. doi: 10.1016/j.cell.2013.02.035
99. De Haes W, Frooninckx L, Van Assche R, Smolders A, Depuydt G, Billen J, et al. Metformin promotes lifespan through mitohormesis via the peroxiredoxin PRDX-2. Proc Natl Acad Sci USA (2014) 111:E2501–9. doi: 10.1073/pnas.1321776111
100. UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet (1998) 352:854–65. doi: 10.1016/S0140-6736(98)07037-8
101. Campbell JM, Bellman SM, Stephenson MD, Lisy K. Metformin reduces all-cause mortality and diseases of ageing independent of its effect on diabetes control: A systematic review and meta-analysis. Ageing Res Rev (2017) 40:31–44. doi: 10.1016/j.arr.2017.08.003
102. Bannister CA, Holden SE, Jenkins-Jones S, Morgan CL, Halcox JP, Schernthaner G, et al. Can people with type 2 diabetes live longer than those without? a comparison of mortality in people initiated with metformin or sulphonylurea monotherapy and matched, non-diabetic controls. Diabetes Obes Metab (2014) 16:1165–73. doi: 10.1111/dom.12354
103. Landman GWD, Kleefstra N, van Hateren KJJ, Groenier KH, Gans ROB, Bilo HJG. Metformin associated with lower cancer mortality in type 2 diabetes: ZODIAC-16. Diabetes Care (2010) 33:322–6. doi: 10.2337/dc09-1380
104. Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JMM. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care (2009) 32:1620–5. doi: 10.2337/dc08-2175
105. Monami M, Colombi C, Balzi D, Dicembrini I, Giannini S, Melani C, et al. Metformin and cancer occurrence in insulin-treated type 2 diabetic patients. Diabetes Care (2011) 34:129–31. doi: 10.2337/dc10-1287
106. Gandini S, Puntoni M, Heckman-Stoddard BM, Dunn BK, Ford L, DeCensi A, et al. Metformin and cancer risk and mortality: a systematic review and meta-analysis taking into account biases and confounders. Cancer Prev Res (2014) 7:867–85. doi: 10.1158/1940-6207.CAPR-13-0424
107. Ng TP, Feng L, Yap KB, Lee TS, Tan CH, Winblad B. Long-term metformin usage and cognitive function among older adults with diabetes. J Alzheimers Dis (2014) 41:61–8. doi: 10.3233/JAD-131901
108. Cheng C, Lin C-H, Tsai Y-W, Tsai C-J, Chou P-H, Lan T-H. Type 2 diabetes and antidiabetic medications in relation to dementia diagnosis. J Gerontol A Biol Sci Med Sci (2014) 69:1299–305. doi: 10.1093/gerona/glu073
109. Mohammed I, Hollenberg MD, Ding H, Triggle CR. A critical review of the evidence that metformin is a putative anti-aging drug that enhances healthspan and extends lifespan. Front Endocrinol (2021) 12:718942. doi: 10.3389/fendo.2021.718942
110. Soukas AA, Hao H, Wu L. Metformin as anti-aging therapy: Is it for everyone? Trends Endocrinol Metab (2019) 30:745–55. doi: 10.1016/j.tem.2019.07.015
111. Wang C, Chen B, Feng Q, Nie C, Li T. Clinical perspectives and concerns of metformin as an anti-aging drug. Aging Med (2020) 3:266–75. doi: 10.1002/agm2.12135
112. Polverino F, Wu TD, Rojas-Quintero J, Wang X, Mayo J, Tomchaney M, et al. Metformin: Experimental and clinical evidence for a potential role in emphysema treatment. Am J Respir Crit Care Med (2021) 204:651–66. doi: 10.1164/rccm.202012-4510OC
113. Park CS, Bang B-R, Kwon H-S, Moon K-A, Kim T-B, Lee K-Y, et al. Metformin reduces airway inflammation and remodeling via activation of AMP-activated protein kinase. Biochem Pharmacol (2012) 84:1660–70. doi: 10.1016/j.bcp.2012.09.025
114. Cheng X-Y, Li Y-Y, Huang C, Li J, Yao H-W. AMP-activated protein kinase reduces inflammatory responses and cellular senescence in pulmonary emphysema. Oncotarget (2017) 8:22513–23. doi: 10.18632/oncotarget.15116
115. Nakashima R, Nohara H, Takahashi N, Nasu A, Hayashi M, Kishimoto T, et al. Metformin suppresses epithelial sodium channel hyperactivation and its associated phenotypes in a mouse model of obstructive lung diseases. J Pharmacol Sci (2022) 149:37–45. doi: 10.1016/j.jphs.2022.03.002
116. Saito N, Araya J, Ito S, Tsubouchi K, Minagawa S, Hara H, et al. Involvement of lamin B1 reduction in accelerated cellular senescence during chronic obstructive pulmonary disease pathogenesis. J Immunol (2019) 202:1428–40. doi: 10.4049/jimmunol.1801293
117. Wu TD, Fawzy A, Kinney GL, Bon J, Neupane M, Tejwani V, et al. Metformin use and respiratory outcomes in asthma-COPD overlap. Respir Res (2021) 22:70. doi: 10.1186/s12931-021-01658-3
118. Hitchings AW, Lai D, Jones PW, Baker EH. Metformin in COPD trial team. metformin in severe exacerbations of chronic obstructive pulmonary disease: a randomised controlled trial. Thorax (2016) 71:587–93. doi: 10.1136/thoraxjnl-2015-208035
119. Teague TT, Payne SR, Kelly BT, Dempsey TM, McCoy RG, Sangaralingham LR, et al. Evaluation for clinical benefit of metformin in patients with idiopathic pulmonary fibrosis and type 2 diabetes mellitus: a national claims-based cohort analysis. Respir Res (2022) 23:91. doi: 10.1186/s12931-022-02001-0
120. Blamoun AI, Batty GN, DeBari VA, Rashid AO, Sheikh M, Khan MA. Statins may reduce episodes of exacerbation and the requirement for intubation in patients with COPD: evidence from a retrospective cohort study. Int J Clin Pract (2008) 62:1373–8. doi: 10.1111/j.1742-1241.2008.01731.x
121. Frost FJ, Petersen H, Tollestrup K, Skipper B. Influenza and COPD mortality protection as pleiotropic, dose-dependent effects of statins. Chest (2007) 131:1006–12. doi: 10.1378/chest.06-1997
122. Keddissi JI, Younis WG, Chbeir EA, Daher NN, Dernaika TA, Kinasewitz GT. The use of statins and lung function in current and former smokers. Chest (2007) 132:1764–71. doi: 10.1378/chest.07-0298
123. Søyseth V, Brekke PH, Smith P, Omland T. Statin use is associated with reduced mortality in COPD. Eur Respir J (2007) 29:279–83. doi: 10.1183/09031936.00106406
124. Criner GJ, Connett JE, Aaron SD, Albert RK, Bailey WC, Casaburi R, et al. Simvastatin for the prevention of exacerbations in moderate-to-severe COPD. N Engl J Med (2014) 370:2201–10. doi: 10.1056/NEJMoa1403086
125. Schenk P, Spiel AO, Hüttinger F, Gmeiner M, Fugger J, Pichler M, et al. Can simvastatin reduce COPD exacerbations? A randomised double-blind controlled study. Eur Respir J (2021) 58:2001798. doi: 10.1183/13993003.01798-2020
126. Kreuter M, Bonella F, Maher TM, Costabel U, Spagnolo P, Weycker D, et al. Effect of statins on disease-related outcomes in patients with idiopathic pulmonary fibrosis. Thorax (2017) 72:148–53. doi: 10.1136/thoraxjnl-2016-208819
127. Kreuter M, Costabel U, Richeldi L, Cottin V, Wijsenbeek M, Bonella F, et al. Statin therapy and outcomes in trials of nintedanib in idiopathic pulmonary fibrosis. Respiration (2018) 95:317–26. doi: 10.1159/000486286
128. Lei Y-F, Lin H-C, Lin H-L, Uang Y-S, Cheng H-W, Wang L-H. Association between use of antihyperlipidemic agents and chronic obstructive pulmonary disease in patients with hyperlipidemia: A population-based retrospective cohort study. Int J Chron Obstruct Pulmon Dis (2020) 15:2573–81. doi: 10.2147/COPD.S267017
129. Rinne ST, Liu C-F, Feemster LC, Collins BF, Bryson CL, O’Riordan TG, et al. Thiazolidinediones are associated with a reduced risk of COPD exacerbations. Int J Chron Obstruct Pulmon Dis (2015) 10:1591–7. doi: 10.2147/COPD.S82643
130. Chen K-Y, Wu S-M, Tseng C-H, Lee K-Y, Lin Y-H, Liu H-Y, et al. Combination therapies with thiazolidinediones are associated with a lower risk of acute exacerbations in new-onset COPD patients with advanced diabetic mellitus: a cohort-based case-control study. BMC Pulm Med (2021) 21:141. doi: 10.1186/s12890-021-01505-7
131. Sato N, Takasaka N, Yoshida M, Tsubouchi K, Minagawa S, Araya J, et al. Metformin attenuates lung fibrosis development via NOX4 suppression. Respir Res (2016) 17:107. doi: 10.1186/s12931-016-0420-x
132. Rangarajan S, Bone NB, Zmijewska AA, Jiang S, Park DW, Bernard K, et al. Metformin reverses established lung fibrosis in a bleomycin model. Nat Med (2018) 24:1121–7. doi: 10.1038/s41591-018-0087-6
133. Kheirollahi V, Wasnick RM, Biasin V, Vazquez-Armendariz AI, Chu X, Moiseenko A, et al. Metformin induces lipogenic differentiation in myofibroblasts to reverse lung fibrosis. Nat Commun (2019) 10:2987. doi: 10.1038/s41467-019-10839-0
134. Lee J-H, Lee D-S, Kim E-K, Choe K-H, Oh Y-M, Shim T-S, et al. Simvastatin inhibits cigarette smoking-induced emphysema and pulmonary hypertension in rat lungs. Am J Respir Crit Care Med (2005) 172:987–93. doi: 10.1164/rccm.200501-041OC
135. Yildirim M, Kayalar O, Atahan E, Oztay F. Atorvastatin attenuates pulmonary fibrosis in mice and human lung fibroblasts, by the regulation of myofibroblast differentiation and apoptosis. J Biochem Mol Toxicol (2022) 36:e23074. doi: 10.1002/jbt.23074
136. Du J, Zhu Y, Meng X, Xie H, Wang J, Zhou Z, et al. Atorvastatin attenuates paraquat poisoning-induced epithelial-mesenchymal transition via downregulating hypoxia-inducible factor-1 alpha. Life Sci (2018) 213:126–33. doi: 10.1016/j.lfs.2018.10.026
137. Yao H-W, Mao L-G, Zhu J-P. Protective effects of pravastatin in murine lipopolysaccharide-induced acute lung injury. Clin Exp Pharmacol Physiol (2006) 33:793–7. doi: 10.1111/j.1440-1681.2006.04440.x
138. Oruqaj G, Karnati S, Vijayan V, Kotarkonda LK, Boateng E, Zhang W, et al. Compromised peroxisomes in idiopathic pulmonary fibrosis, a vicious cycle inducing a higher fibrotic response via TGF-β signaling. Proc Natl Acad Sci USA (2015) 112:E2048–57. doi: 10.1073/pnas.1415111112
139. Ke Q, Yang L, Cui Q, Diao W, Zhang Y, Xu M, et al. Ciprofibrate attenuates airway remodeling in cigarette smoke-exposed rats. Respir Physiol Neurobiol (2020) 271:103290. doi: 10.1016/j.resp.2019.103290
140. Kikuchi R, Maeda Y, Tsuji T, Yamaguchi K, Abe S, Nakamura H, et al. Fenofibrate inhibits TGF-β-induced myofibroblast differentiation and activation in human lung fibroblasts in vitro. FEBS Open Bio (2021) 11:2340–9. doi: 10.1002/2211-5463.13247
141. Bodas M, Patel N, Silverberg D, Walworth K, Vij N. Master autophagy regulator transcription factor EB regulates cigarette smoke-induced autophagy impairment and chronic obstructive pulmonary disease-emphysema pathogenesis. Antioxid Redox Signal (2017) 27:150–67. doi: 10.1089/ars.2016.6842
142. Liu Y, Chen S, Yu L, Deng Y, Li D, Yu X, et al. Pemafibrate attenuates pulmonary fibrosis by inhibiting myofibroblast differentiation. Int Immunopharmacol (2022) 108:108728. doi: 10.1016/j.intimp.2022.108728
143. Lea S, Plumb J, Metcalfe H, Spicer D, Woodman P, Fox JC, et al. The effect of peroxisome proliferator-activated receptor-γ ligands on in vitro and in vivo models of COPD. Eur Respir J (2014) 43:409–20. doi: 10.1183/09031936.00187812
144. Shan M, You R, Yuan X, Frazier MV, Porter P, Seryshev A, et al. Agonistic induction of PPARγ reverses cigarette smoke–induced emphysema. J Clin Invest 3 (2014) 124:1371–81. doi: 10.1172/JCI70587
145. Milam JE, Keshamouni VG, Phan SH, Hu B, Gangireddy SR, Hogaboam CM, et al. PPAR-gamma agonists inhibit profibrotic phenotypes in human lung fibroblasts and bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol (2008) 294:L891–901. doi: 10.1152/ajplung.00333.2007
146. Sirtori CR. The pharmacology of statins. Pharmacol Res (2014) 88:3–11. doi: 10.1016/j.phrs.2014.03.002
147. Istvan ES, Deisenhofer J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science (2001) 292:1160–4. doi: 10.1126/science.1059344
148. Wang C-Y, Liu P-Y, Liao JK. Pleiotropic effects of statin therapy: molecular mechanisms and clinical results. Trends Mol Med (2008) 14:37–44. doi: 10.1016/j.molmed.2007.11.004
149. Ota H, Eto M, Kano MR, Kahyo T, Setou M, Ogawa S, et al. Induction of endothelial nitric oxide synthase, SIRT1, and catalase by statins inhibits endothelial senescence through the akt pathway. Arterioscler Thromb Vasc Biol (2010) 30:2205–11. doi: 10.1161/ATVBAHA.110.210500
150. Assmus B, Urbich C, Aicher A, Hofmann WK, Haendeler J, Rössig L, et al. HMG-CoA reductase inhibitors reduce senescence and increase proliferation of endothelial progenitor cells via regulation of cell cycle regulatory genes. Circ Res (2003) 92:1049–55. doi: 10.1161/01.RES.0000070067.64040.7C
151. Palsuledesai CC, Distefano MD. Protein prenylation: enzymes, therapeutics, and biotechnology applications. ACS Chem Biol (2015) 10:51–62. doi: 10.1021/cb500791f
152. Janić M, Lunder M, Novaković S, Škerl P, Šabovič M. Expression of longevity genes induced by a low-dose fluvastatin and valsartan combination with the potential to Prevent/Treat “Aging-related disorders”. Int J Mol Sci (2019) 20:1844. doi: 10.3390/ijms20081844
153. Staels B, Dallongeville J, Auwerx J, Schoonjans K, Leitersdorf E, Fruchart JC. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation (1998) 98:2088–93. doi: 10.1161/01.CIR.98.19.2088
154. Berglund L, Brunzell JD, Goldberg AC, Goldberg IJ, Sacks F, Murad MH, et al. Evaluation and treatment of hypertriglyceridemia: an endocrine society clinical practice guideline. J Clin Endocrinol Metab (2012) 97:2969–89. doi: 10.1210/jc.2011-3213
155. Lefebvre P, Chinetti G, Fruchart J-C, Staels B. Sorting out the roles of PPAR alpha in energy metabolism and vascular homeostasis. J Clin Invest (2006) 116:571–80. doi: 10.1172/JCI27989
156. Aoyama T, Peters JM, Iritani N, Nakajima T, Furihata K, Hashimoto T, et al. Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor alpha (PPARalpha). J Biol Chem (1998) 273:5678–84. doi: 10.1074/jbc.273.10.5678
157. Gulick T, Cresci S, Caira T, Moore DD, Kelly DP. The peroxisome proliferator-activated receptor regulates mitochondrial fatty acid oxidative enzyme gene expression. Proc Natl Acad Sci USA (1994) 91:11012–6. doi: 10.1073/pnas.91.23.11012
158. Kim EN, Lim JH, Kim MY, Kim HW, Park CW, Chang YS, et al. PPARα agonist, fenofibrate, ameliorates age-related renal injury. Exp Gerontol (2016) 81:42–50. doi: 10.1016/j.exger.2016.04.021
159. Nogueira-Recalde U, Lorenzo-Gómez I, Blanco FJ, Loza MI, Grassi D, Shirinsky V, et al. Fibrates as drugs with senolytic and autophagic activity for osteoarthritis therapy. EBioMedicine (2019) 45:588–605. doi: 10.1016/j.ebiom.2019.06.049
160. Wang M, Yan Y, Zhang Z, Yao X, Duan X, Jiang Z, et al. Programmed PPAR-α downregulation induces inflammaging by suppressing fatty acid catabolism in monocytes. iScience (2021) 24:102766. doi: 10.1016/j.isci.2021.102766
161. Pentinmikko N, Iqbal S, Mana M, Andersson S, Cognetta AB 3rd, Suciu RM, et al. Notum produced by paneth cells attenuates regeneration of aged intestinal epithelium. Nature (2019) 571:398–402. doi: 10.1038/s41586-019-1383-0
162. Genovese T, Mazzon E, Di Paola R, Muià C, Crisafulli C, Caputi AP, et al. Role of endogenous and exogenous ligands for the peroxisome proliferator-activated receptor alpha in the development of bleomycin-induced lung injury. Shock (2005) 24:547–55. doi: 10.1097/01.shk.0000190825.28783.a4
163. Hecker M, Behnk A, Morty RE, Sommer N, Vadász I, Herold S, et al. PPAR-α activation reduced LPS-induced inflammation in alveolar epithelial cells. Exp Lung Res (2015) 41:393–403. doi: 10.3109/01902148.2015.1046200
164. Delayre-Orthez C, Becker J, Guenon I, Lagente V, Auwerx J, Frossard N, et al. PPARα downregulates airway inflammation induced by lipopolysaccharide in the mouse. Respir Res (2005) 6:1–10. doi: 10.1186/1465-9921-6-91
165. American Diabetes Association Professional Practice Committee. Classification and diagnosis of diabetes: Standards of medical care in diabetes–2022. Diabetes Care (2022) 45:S17–38. doi: 10.2337/dc22-S002
166. Imanishi T, Kobayashi K, Kuroi A, Ikejima H, Akasaka T. Pioglitazone inhibits angiotensin II-induced senescence of endothelial progenitor cell. Hypertens Res (2008) 31:757–65. doi: 10.1291/hypres.31.757
167. Han YS, Kim SM, Lee JH, Lee SH. Co-Administration of melatonin effectively enhances the therapeutic effects of pioglitazone on mesenchymal stem cells undergoing indoxyl sulfate-induced senescence through modulation of cellular prion protein expression. Int J Mol Sci (2018) 19(5):1367. doi: 10.3390/ijms19051367
168. Chen L, Bi B, Zeng J, Zhou Y, Yang P, Guo Y, et al. Rosiglitazone ameliorates senescence-like phenotypes in a cellular photoaging model. J Dermatol Sci (2015) 77:173–81. doi: 10.1016/j.jdermsci.2015.01.007
169. Yang H-C, Deleuze S, Zuo Y, Potthoff SA, Ma L-J, Fogo AB. The PPARgamma agonist pioglitazone ameliorates aging-related progressive renal injury. J Am Soc Nephrol (2009) 20:2380–8. doi: 10.1681/ASN.2008111138
170. Xu L, Ma X, Verma N, Perie L, Pendse J, Shamloo S, et al. PPARγ agonists delay age-associated metabolic disease and extend longevity. Aging Cell (2020) 19:e13267. doi: 10.1111/acel.13267
171. Xu L, Dong X-W, Shen L-L, Li F-F, Jiang J-X, Cao R, et al. Simvastatin delivery via inhalation attenuates airway inflammation in a murine model of asthma. Int Immunopharmacol (2012) 12:556–64. doi: 10.1016/j.intimp.2012.01.012
172. Zeki AA, Bratt JM, Chang KY, Franzi LM, Ott S, Silveria M, et al. Intratracheal instillation of pravastatin for the treatment of murine allergic asthma: a lung-targeted approach to deliver statins. Physiol Rep (2015) 3:e12352. doi: 10.14814/phy2.12352
173. Chen L, Nakano K, Kimura S, Matoba T, Iwata E, Miyagawa M, et al. Nanoparticle-mediated delivery of pitavastatin into lungs ameliorates the development and induces regression of monocrotaline-induced pulmonary artery hypertension. Hypertension (2011) 57:343–50. doi: 10.1161/HYPERTENSIONAHA.110.157032
174. Rashid J, Alobaida A, Al-Hilal TA, Hammouda S, McMurtry IF, Nozik-Grayck E, et al. Repurposing rosiglitazone, a PPAR-γ agonist and oral antidiabetic, as an inhaled formulation, for the treatment of PAH. J Control Release (2018) 280:113–23. doi: 10.1016/j.jconrel.2018.04.049
175. Rashid J, Nozik-Grayck E, McMurtry IF, Stenmark KR, Ahsan F. Inhaled combination of sildenafil and rosiglitazone improves pulmonary hemodynamics, cardiac function, and arterial remodeling. Am J Physiol Lung Cell Mol Physiol (2019) 316:L119–30. doi: 10.1152/ajplung.00381.2018
176. Sakurai R, Lee C, Shen H, Waring AJ, Walther FJ, Rehan VK. A combination of the aerosolized PPAR-γ agonist pioglitazone and a synthetic surfactant protein b peptide mimic prevents hyperoxia-induced neonatal lung injury in rats. Neonatology (2018) 113:296–304. doi: 10.1159/000486188
177. Navarro S, Reddy R, Lee J, Warburton D, Driscoll B. Inhaled resveratrol treatments slow ageing-related degenerative changes in mouse lung. Thorax (2017) 72:451–9. doi: 10.1136/thoraxjnl-2016-208964
Keywords: senotherapy, senolytics, senomorphics, drugs against metabolic diseases, aging-related respiratory disease, chronic obstructive pulmonary disease (COPD), idiopathic pulmonary fibrosis (IPF), inhalation therapy
Citation: Matsubayashi S, Ito S, Araya J and Kuwano K (2023) Drugs against metabolic diseases as potential senotherapeutics for aging-related respiratory diseases. Front. Endocrinol. 14:1079626. doi: 10.3389/fendo.2023.1079626
Received: 25 October 2022; Accepted: 22 March 2023;
Published: 03 April 2023.
Edited by:
Ritesh Kumar Baboota, University of Gothenburg, SwedenReviewed by:
Brenna Osborne, University of Copenhagen, DenmarkAimo Kannt, Fraunhofer Institute for Translational Medicine and Pharmacology ITMP, Germany
Copyright © 2023 Matsubayashi, Ito, Araya and Kuwano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saburo Ito, c2FidS1zNTVAamlrZWkuYWMuanA=
†These authors have contributed equally to this work