 Naila Rabbani
Naila Rabbani Paul J. Thornalley
Paul J. Thornalley- 1College of Medicine, Qatar University, Doha, Qatar
- 2College of Health and Life Sciences, Hamad Bin Khalifa University, Doha, Qatar
Hyperglycemia is a risk factor for the development of insulin resistance, beta-cell glucotoxicity, and vascular complications of diabetes. We propose the hypothesis, hexokinase-linked glycolytic overload and unscheduled glycolysis, in explanation. Hexokinases (HKs) catalyze the first step of glucose metabolism. Increased flux of glucose metabolism through glycolysis gated by HKs, when occurring without concomitant increased activity of glycolytic enzymes—unscheduled glycolysis—produces increased levels of glycolytic intermediates with overspill into effector pathways of cell dysfunction and pathogenesis. HK1 is saturated with glucose in euglycemia and, where it is the major HK, provides for basal glycolytic flux without glycolytic overload. HK2 has similar saturation characteristics, except that, in persistent hyperglycemia, it is stabilized to proteolysis by high intracellular glucose concentration, increasing HK activity and initiating glycolytic overload and unscheduled glycolysis. This drives the development of vascular complications of diabetes. Similar HK2-linked unscheduled glycolysis in skeletal muscle and adipose tissue in impaired fasting glucose drives the development of peripheral insulin resistance. Glucokinase (GCK or HK4)-linked glycolytic overload and unscheduled glycolysis occurs in persistent hyperglycemia in hepatocytes and beta-cells, contributing to hepatic insulin resistance and beta-cell glucotoxicity, leading to the development of type 2 diabetes. Downstream effector pathways of HK-linked unscheduled glycolysis are mitochondrial dysfunction and increased reactive oxygen species (ROS) formation; activation of hexosamine, protein kinase c, and dicarbonyl stress pathways; and increased Mlx/Mondo A signaling. Mitochondrial dysfunction and increased ROS was proposed as the initiator of metabolic dysfunction in hyperglycemia, but it is rather one of the multiple downstream effector pathways. Correction of HK2 dysregulation is proposed as a novel therapeutic target. Pharmacotherapy addressing it corrected insulin resistance in overweight and obese subjects in clinical trial. Overall, the damaging effects of hyperglycemia are a consequence of HK-gated increased flux of glucose metabolism without increased glycolytic enzyme activities to accommodate it.
Introduction
Insulin resistance, diabetes mellitus, and vascular complications of diabetes have become a major cause of morbidity and mortality in westernized societies. In 2021, there were 529 million people living with diabetes worldwide, of which 96% were patients with type 2 diabetes mellitus (T2DM) and the remaining patients with type 1 diabetes mellitus (T1DM). This is projected to increase to over 1.3 billion by 2050 (1). Prediabetes, defined by increased plasma glucose levels intermediate between those of euglycemia and diabetes, develops earlier and predisposes to the development of T2DM (2). In prediabetes, plasma glucose may be increased in fasting and postprandial phases—impaired fasting glucose (IFG) and impaired glucose tolerance (IGT), respectively (2). In 2021, the global prevalence of IFG was 298 million and IGT 464 million and are projected to increase to 414 million and 638 million by 2045, respectively (3). Insulin resistance is the primary initiating metabolic defect driving the development of prediabetes and T2DM (4, 5). It is defined as “a reduced response of target tissues to insulin, compared with subjects with normal glucose tolerance” (6). The main target tissues of insulin are skeletal muscle, liver, and adipocytes. Insulin resistance has high prevalence in Westernized countries: in the USA, the estimated prevalence of insulin resistance in the adult population is approximately 50% (7). Insulin resistance is often present many years before diabetes develops (8, 9), increasing dysglycemic stress in pancreatic beta-cells, hyperinsulinemia, and accelerating decline in beta-cell mass and function leading to the development of T2DM (6, 10).
With high prevalence of diabetes, there is a related high prevalence of vascular complications of diabetes—diabetic kidney disease (DKD), diabetic retinopathy, diabetic neuropathy, and increase risk of cardiovascular disease (CVD). Vascular complications of diabetes affect 30%–50% of patients of diabetes, producing increased morbidity, decreased quality of life, and premature mortality, the latter mainly through increased fatal CVD (11–14). There are also similar vascular complications in prediabetes of lower prevalence, as judged by their presence at diagnosis of T2DM (15).
Hyperglycemia, particularly increased fasting plasma glucose (FPG), is a risk factor for progression of insulin resistance and decline in beta-cell function leading to the development of T2DM (16, 17). It is also a risk factor for the development of vascular complications of diabetes (18, 19). Hyperglycemia-induced metabolic dysfunction of glucose metabolism underlies this relationship. There is currently no pharmacotherapy addressing this—most current drug treatment addressing rather hyperglycemia itself by hypoglycemic agents and incretin mimetics (20). Pharmacotherapy of DKD is mainly based on drugs directed to the renin–angiotensin–aldosterone system, severe diabetic retinopathy is treated by panretinal laser photocoagulation and anti-vascular endothelial growth factor therapy to decrease risk of visual loss, and diabetic neuropathy treatment involves mainly analgesics for pain relief. The risk of microvascular and macrovascular complications is addressed more generally by controlling hyperglycemia, blood pressure, and lipids (21–23), but there remains substantial risk of vascular complications development and progression to severe symptoms.
Herein, we re-examine experimental evidence of the origin of hyperglycemia-induced metabolic dysfunction, propose a new hypothesis in explanation of the initiation of metabolic dysfunction in hyperglycemia—hexokinase-linked glycolytic overload and unscheduled glycolysis—explaining its advantages of the over existing explanations and the promise for improved understanding of pathogenesis and therapeutics development.
Hexokinases—catalyzing the first committed step of glucose metabolism
Hexokinases (HKs) catalyze the first step of glucose metabolism, the conversion of glucose to glucose-6-phosphate (G6P) (24): glucose + MgATP → G6P + MgADP. They are gatekeepers at the entry of glucose into metabolism. Thereafter, G6P is the substrate for onward metabolism to pyruvate through glycolysis, to ribose-5-phosphate in the pentosephosphate pathway (PPP), and to glucose-1-phosphate for glycogen synthesis. G6P is reconverted to glucose by G6P phosphatase (G6Pase) in gluconeogenesis. G6P is mainly utilized in glycolysis in all tissues except for glycogen synthesis in the liver and skeletal muscle in the absorptive phase and gluconeogenesis in the liver and kidney in the fasting phase.
There are four major forms of HK: HK1, HK2, HK3, and HK4 or glucokinase (GCK). The molecular, kinetic, and physiological characteristics of HKs are summarized in Table 1. The cellular concentration of the ATP cofactor is typically in the range of 2–8 mM (33). Therefore, HK1, HK2, and GCK function in situ with saturating or non-limiting ATP. HK3 of red blood cells (RBCs) operates under ATP-limiting kinetics in vivo, which may be moderately enhanced by increased RBC ATP concentration in hyperglycemia (34). The conditions under which HKs function in situ with respect to glucose are more complex. From accumulated experimental and clinical evidence discussed below, generally, HK1 is saturated at the levels of intracellular concentration of glucose in all cells in euglycemia and provides a basal level of glucose metabolism (with HK3 providing a similar function in RBCs); HK2 is saturated at the levels of intracellular glucose concentration in euglycemia in some tissues and unsaturated in others with an abnormal, non-transcriptional mechanism circumventing saturation producing dysregulated glycolysis and abnormal accumulation of damaging glycolytic intermediates in persistent hyperglycemia; and GCK, while providing a glucose sensor function for control of insulin and glucagon secretion in pancreatic beta-cells and alpha cells and glycogen synthesis in the liver, also produces abnormal accumulation of damaging glycolytic intermediates and dysregulated glycolysis in persistent hyperglycemia.
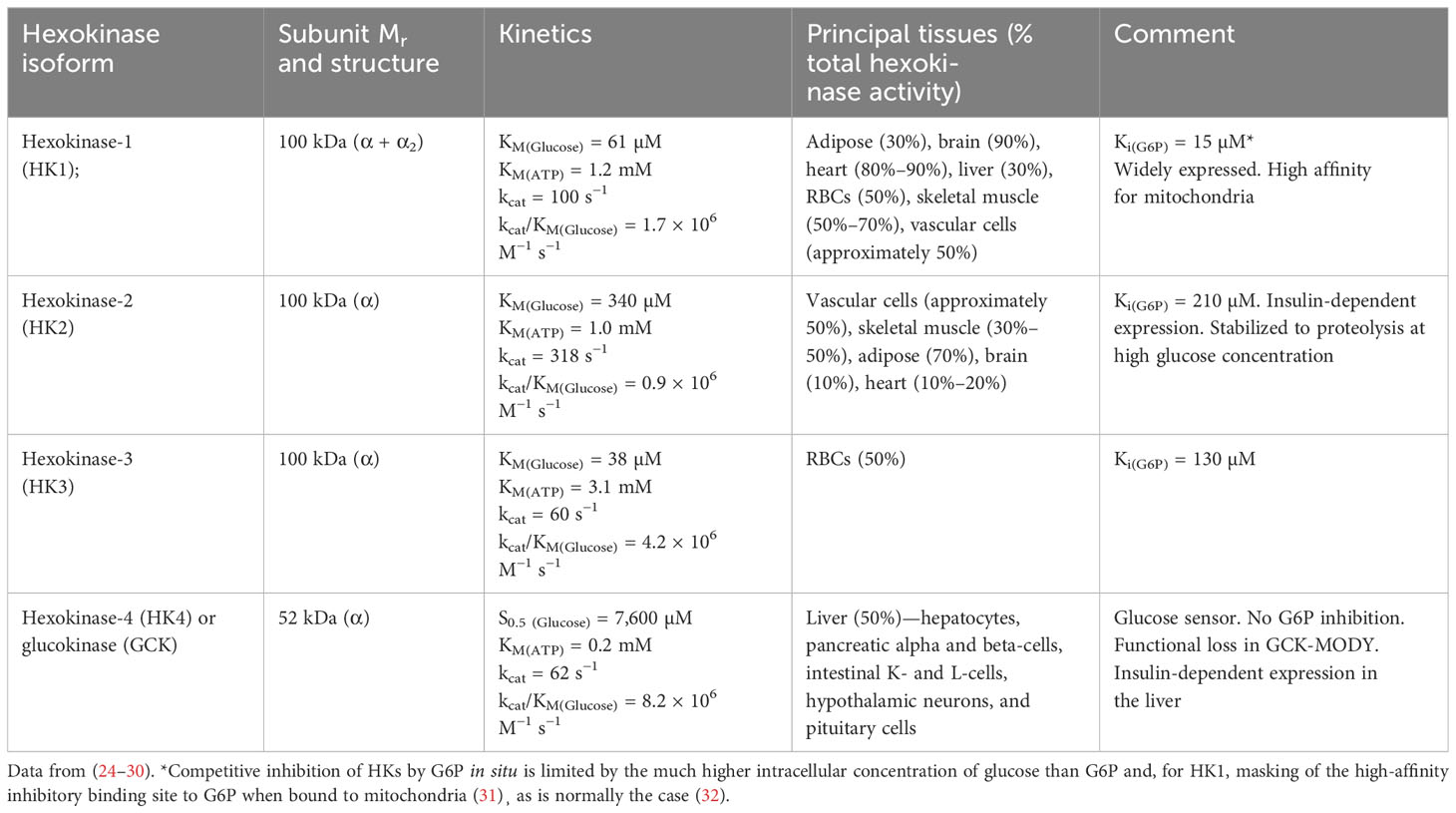
Table 1 Molecular, kinetic, and physiological characteristics of hexokinases.
There is a further minor pathway of glucose metabolism—the polyol pathway. Glucose is metabolized therein by aldose reductase (AR) to sorbitol with onward metabolism by sorbitol dehydrogenase to fructose (35). AR-catalyzed in situ kinetics of glucose metabolism are usually <1% flux of total HK activity in situ. The polyol pathway has highest activity in the renal medulla (36) where it has a role in osmoregulation, countering extracellular hypertonicity produced during anti-diuresis (37, 38).
Scheduled and unscheduled glycolysis—importance of increasing activities of glycolytic enzymes to accommodate increased flux of glucose metabolism
Early-stage glucose metabolism is regulated at three key steps: glucose uptake, hexokinase, and phosphofructokinase (PFK) (39). When increased glucose disposal occurs in the skeletal muscle in the absorptive phase, increased flux of glucose is delivered into the cytoplasm by insulin-stimulated GLUT4 (40) and enters glycolysis by HK1 and HK2 (41), with insulin stimulating increased expression of HK2 (42), producing increased flux of formation and modest increase in the concentration of G6P. G6P is converted to fructose-6-phosphate (F6P) by glucose-6-phosphate isomerase (GPI) and to glucose-1-phosphate for glycogen synthesis by phosphoglucomutase (PGM). Allosteric activation of glycogen synthase by G6P increases its consumption for glycogen synthesis (43). Insulin signaling activates phosphofructokinase-2 (PFK-2), increasing allosteric regulator fructose 2,6-bisphosphate (F26BP) (44), which increases activity of phosphofructokinase-1 (PFK-1) and converts F6P to fructose-1,6-bisphosphate (F16BP). Insulin signaling and G6P-dependent activation of transcriptional signaling of Mlx/Mondo A increases expression and activity of GPI, aldolase, triosephosphate isomerase (TPI), and glyceraldehyde-3-phosphate dehydrogenase (GA3PD) (45–48). The paralog protein, Mondo B or carbohydrate response element binding protein (ChREBP), is dominant in the liver and adipose tissue (49). This provides for regulated increased flux of glucose metabolism from G6P through and beyond glyceraldehyde-3-phosphate (GA3P) with modest increases in the steady-state levels of glycolytic intermediates—scheduled glycolysis (50) (Figure 1A). If entry of glucose into glycolysis initiated by HKs occurs without regulatory activation and increased expression of glycolytic enzymes, increased metabolic flux produces concomitant increased steady-state levels of glycolytic intermediates at least proportionate to the increase in flux of glucose metabolism—unscheduled glycolysis. Increase in some glycolytic intermediates—G6P, F6P, dihydroxyacetonephosphate (DHAP), and GA3P—beyond normal tolerable levels stimulates pathways of metabolic and cellular dysfunction (Figure 1B). The subversion of the normal gating of glucose entry into metabolism by HKs to produce glycolytic overload and unscheduled glycolysis is the origin of hyperglycemia-induced metabolic dysfunction linked to the development of insulin resistance, beta-cell glucotoxicity, and vascular complications of diabetes.
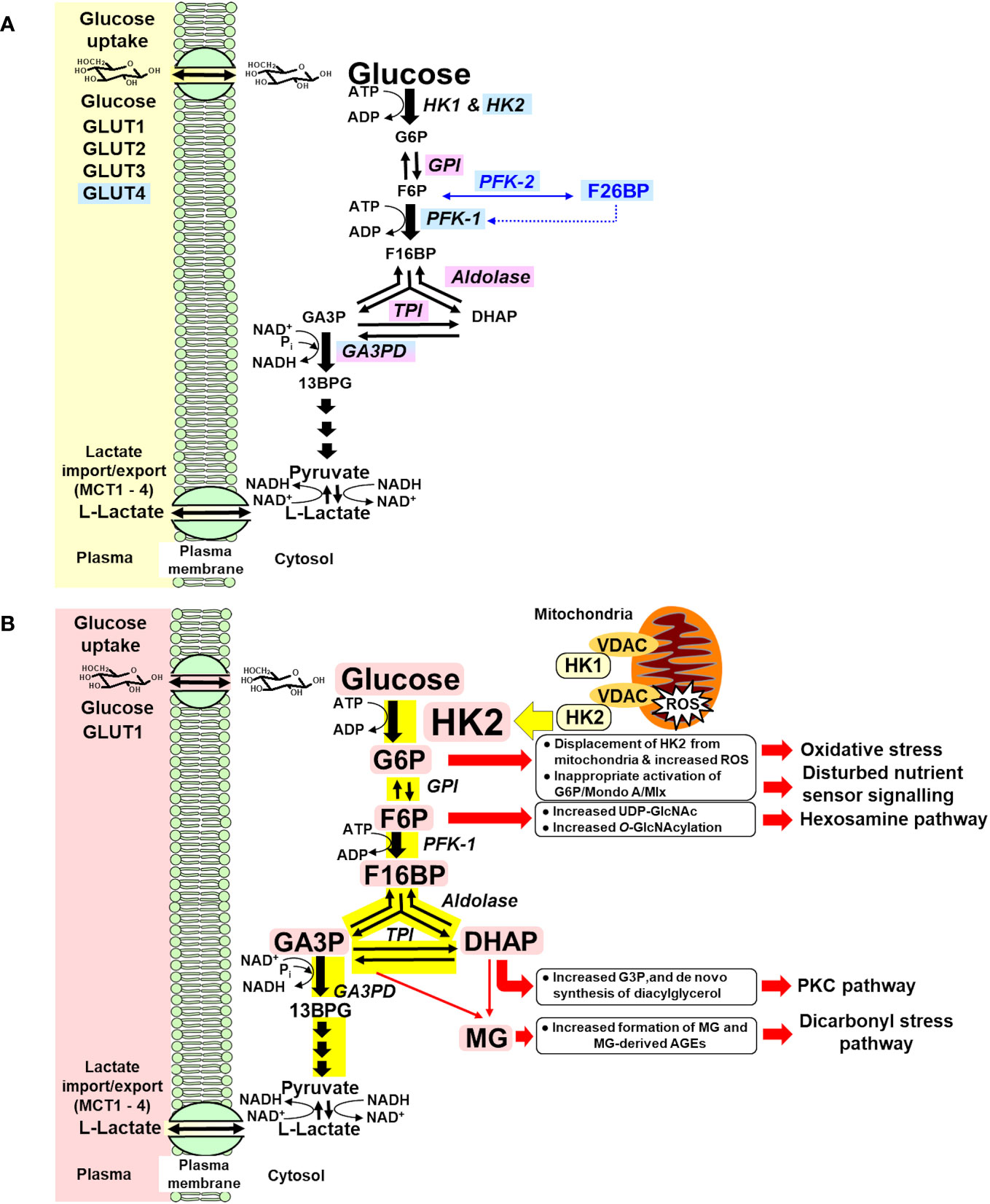
Figure 1 (A) Regulation of glycolytic enzymes by insulin and Mondo A in scheduled glycolysis. Blue highlighted text, genes with expression and/or activity regulated by insulin. Lilac highlighted text, genes with expression regulated by Mondo A/Mlx. (B) Dysregulation of glycolytic enzymes and metabolic dysfunction in hexokinase-2-linked glycolytic overload and unscheduled glycolysis hypothesis. Pink highlighted text, HK2 and metabolites increased in unscheduled glycolysis. Yellow highlight indicates increased flux in glycolytic overload.
Glucose availability and saturation status of hexokinases in cells and tissues in hyperglycemia
Glucose availability for HKs is dependent on the transport of glucose into cells by one or more facilitative glucose transport proteins (GLUTs) and sodium-dependent glucose transporters (SGLTs). The in situ activities of glucose transporters and cellular consumption of glucose by HKs usually create a negative concentration gradient of glucose from plasma and interstitial fluid to the cell cytoplasm. This is greatly diminished in cells with a glucose sensor function where intracellular glucose concentrations tracks the extracellular glucose concentration (51) (Table 2). The most relevant and important GLUTs for glucose metabolism in impaired metabolic health and glycemic disease are as follows: GLUT1—the major GLUT of vascular cells, non-parenchymal cells of liver, glucose transport of skeletal muscle and adipose tissue in the fasting phase, Schwann cells of peripheral nerve, endothelial and epithelial-like barriers of the brain, fibroblasts, and pancreatic beta-cells; GLUT2—the major GLUT of hepatocytes; GLUT3—the major GLUT of peripheral and CNS neurons; and GLUT4—the major GLUT of skeletal muscle, adipose tissue, and myocardium, with expression also in renal podocytes. In renal tubular epithelial cells, GLUT1 and GLUT2 are expressed along with high expression of sodium-dependent glucose transporter SGLT2, which normally accounts for 90% of reabsorption of glucose passing through the glomerular filter (66). GLUT1 and GLUT3 are high-affinity glucose transporters (KM(Glucose) = 0.7–3.2 mM), GLUT2 is a low-affinity glucose transporter (KM(Glucose) = 17–20 mM), and GLUT4 is a high-affinity glucose transporter (KM(Glucose) = 12.6 mM) with activity dependent on insulin. SGLT2 has high affinity for glucose (KM(Glucose) ≤ 6 mM) and relatively high capacity for glucose transport (66, 89).
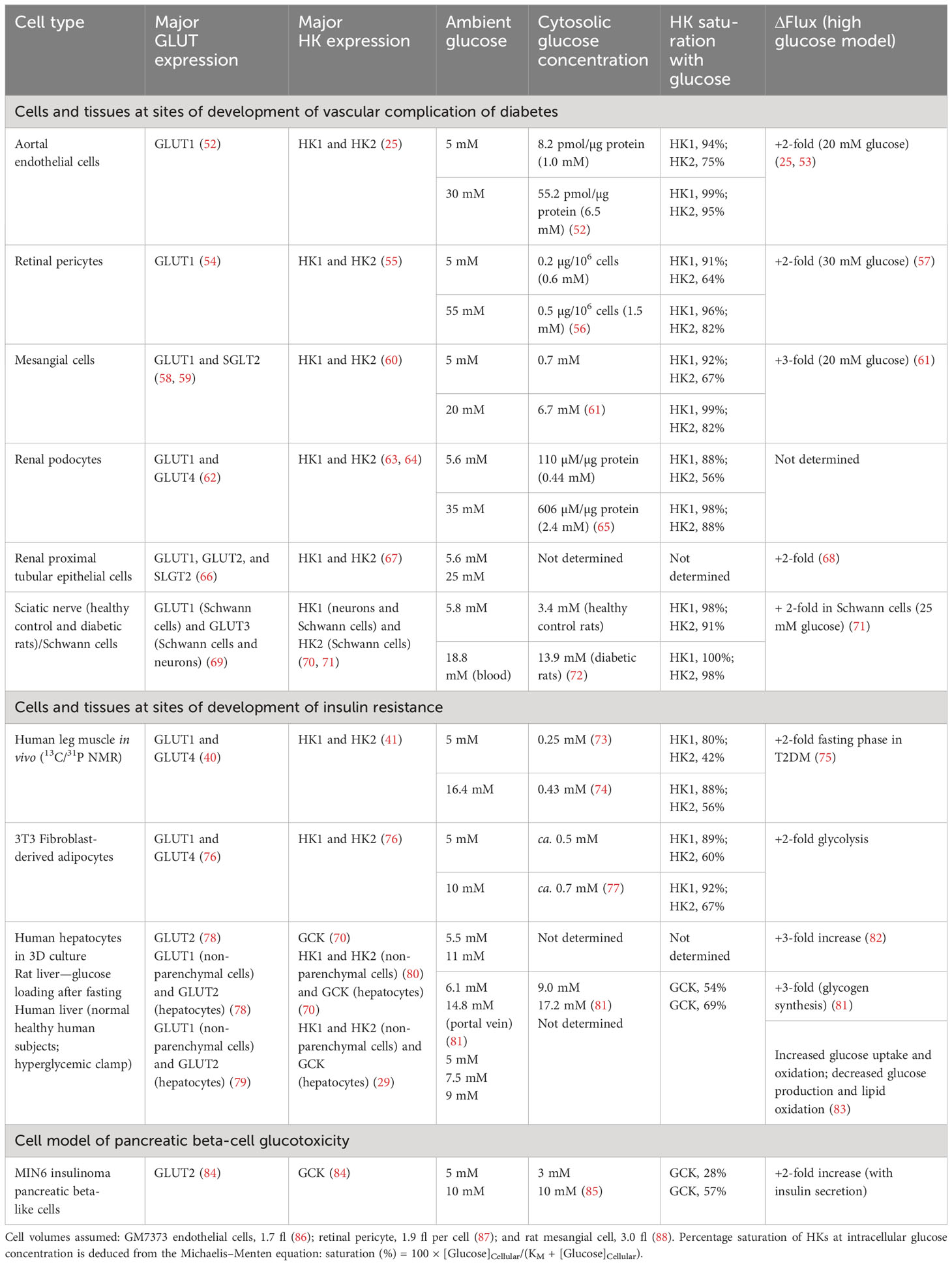
Table 2 Cytosolic glucose concentration, hexokinase saturation, and increased flux of glucose metabolism in cells and tissues suffering metabolic dysfunction in hyperglycemia.
Consider initially the glycemic status of HKs at the sites of development of vascular complications of diabetes. In vascular endothelial cells, retinal pericytes, and renal cells, glucose uptake occurs mainly by GLUT1, and hexokinases therein are HK1 and HK2 (Table 2). In vascular endothelial cells, under model euglycemic conditions with 5 mM glucose, the estimated intracellular glucose concentration is 1.0 mM producing high saturation of both HK1 and HK2 (94% and 75%, respectively). In model hyperglycemia with high glucose concentration in cell cultures, the intracellular glucose concentration was increased to 6.5 mM with both HK1 and HK2, again highly saturated (99% and 95%, respectively). Despite the high saturation of HKs with glucose, the flux of glucose metabolism in model hyperglycemia increased twofold, compared to the euglycemic control. The increased flux of glucose metabolism occurs without increased expression of HKs, as judged by HK1 and HK2 mRNA and, because of high saturation of HKs, cannot be accounted for by mass action or increased specific turnover at HKs (25). Similar saturation of HK1 and HK2 by intracellular glucose concentration in model euglycemia was found in retinal pericytes and renal mesangial cells, podocytes, and proximal tubular epithelial cells with a two- to threefold increase in flux of glucose metabolism in high glucose concentration. HK saturation by glucose is circumvented by a non-transcriptional increase in HK2 protein, which drives dysfunctional glucose metabolism and pathogenesis in the development of vascular complications of diabetes—see below.
In the peripheral nerve, glucose uptake occurs by GLUT3 in neurons and GLUT1 and GLUT3 in Schwann cells (69). HK1 is expressed in neurons and HK1 and HK2 in Schwann cells and also likely in endoneurial endothelial cells and fibroblasts (70, 71). Studies of the sciatic nerve of healthy control and streptozotocin (STZ)-induced diabetic rats indicate a high glucose content of the nerve, indicating that HK1 and HK2 are saturated in both euglycemia and hyperglycemia. Nevertheless, incubation of Schwann cells in high glucose concentration produced a twofold increase in glucose metabolism (71). This increased flux of glucose metabolism supports metabolic dysfunction in Schwann cells, which likely impairs functional support of axons implicated in the development of peripheral diabetic neuropathy (14).
In peripheral insulin resistance, there is abnormal glucose uptake and metabolism by the major target tissues of insulin, skeletal muscle, and adipose tissue. Glucose uptake by skeletal muscle cells is mediated mainly by GLUT1 in the fasting phase and GLUT4 in the absorptive phase (40). Intracellular glucose concentrations of skeletal muscle of the leg in low and high glucose were 0.25 mM and 0.43 mM, respectively (73, 74). This suggests that HK1 is saturated with glucose, whereas HK2 is not. In patients with T2DM and insulin resistance with FPG increased twofold, there was an approximately twofold increase in glucose metabolism by skeletal muscle (75). This suggests that there is increased non-insulin-mediated glucose uptake (NIMGU) by mass action effect of increased FPG, increasing glucose uptake by GLUT1 and glucose metabolism by HK2 (75, 90). This occurs without increased expression of HK1 or HK2 at the mRNA and protein level (91–94). In the fasting phase, skeletal muscle accounts for 80%–90% of glucose uptake in euglycemia, with most occurring via NIMGU (6, 90). In the absorptive phase, a larger insulin-dependent, GLUT4-mediated glucose uptake and metabolism by skeletal muscle occurs, which is decreased in insulin resistance (75, 90) (Table 2).
The glycemic status and glucose metabolism in adipocytes is similar to that of the skeletal muscle. In the adipose tissue, evidence from murine 3T3 fibroblast-derived adipocytes in low and high glucose concentration cultures indicate that HK1 is saturated by glucose in low and high glucose concentration, whereas HK2 is not (77). Under these conditions, there is a twofold increase in glucose metabolism without increase in expression of HK1 and HK2 (76, 95) (Table 2).
Hepatic insulin resistance is an important contributor to whole body insulin resistance and increased FPG (96). Glucose uptake by the liver occurs mainly by GLUT2 in hepatocytes and by GLUT1 in non-parenchymal cells (78). In hepatocytes, the metabolism of glucose by GCK accounts for approximately 95% glucose metabolism (97). GCK is not saturated by glucose but is partially inhibited by glucokinase regulatory protein (GKRP) (Table 2).
GKRP mRNA levels are high in the liver but low in most other tissues. In the liver, GKRP is expressed at the protein level: a 68-kDa polypeptide that functions as a competitive inhibitor of glucose binding to GCK. At low glucose concentrations, GCK binds to GKRP, and the inactive complex is translocated to the hepatocyte nucleus. When glucose concentration increases, GCK-GKRP complex dissociates and GCK returns to the cytosol to participate in glucose metabolism (98, 99). In human subjects, the formation of the GCK-GKRP complex is promoted by F6P and antagonized by fructose-1-phosphate (F1P), indicating that diets rich in fructose with formation of hepatic F1P can increase GCK activity.
Glucokinase has a low affinity for glucose (S0.5 = 7.6 mM) and sigmoidal kinetics with respect to glucose, reflecting positive cooperativity with a Hill coefficient of 1.7 (26). Increasing glucose levels in hepatocytes in culture produces modest increases in cellular G6P and marked increases in glycogen synthesis and glycolysis (100). Hepatic glycogen synthase is controlled by interplay between allosteric activation by G6P and reversible phosphorylation through glycogen synthase kinase-3 and protein phosphatase 1 (101). Glycogen synthesis correlates positively with G6P, increasing rapidly at 0.2–0.3 mM G6P where it is like an on–off switch for glycogen synthesis (102), increased G6P synergizing with glucose to induce activating dephosphorylation of glycogen synthase and inactivating dephosphorylation of glycogen phosphorylase (103). There is a consequent sigmoidal relationship between plasma glucose concentration and hepatic glycogen synthesis (104). Hepatic glycolytic flux correlates positively with in situ GCK activity (100). High glucose concentration induced repression of GCK and increased expression of G6Pase to decrease G6P and safeguard hepatic phosphate homeostasis (105). The induction of G6Pase is attributed to the activation of ChREBP-Mlx by increased G6P, F26BP, and xylulose 5-phosphate (49, 106). In a hepatocyte 3D primary culture model, the flux of glucose metabolism increased disproportionately (threefold) with twofold increased glucose concentration (82).
Glucose loading by the liver of normal healthy rats showed that increasing portal venous glucose approximately twofold produced a threefold increase in glucose metabolism for hepatic glycogen synthesis (81). Increased glucose uptake by the liver is associated with unchanged or decreased G6P concentration and other glycolytic intermediates, as activities of enzymes of gluconeogenesis are decreased and those of glycogen synthesis and glycolysis are increased (107). The liver responds to increased blood glucose levels in the postprandial state by uptake of glucose and conversion to glycogen (101). The formation of the GCK-GKRP complex and its stabilization by G6P limit the net hepatic glucose uptake and metabolism therein after oral ingestion of glucose (103, 108). By contrast, most fructose is readily taken up from the circulation by the liver, due to the low KM and high in situ activity of hepatic ketohexokinase (KHK) (109).
In pancreatic beta-cells, recent evidence suggests that glucose uptake by human beta-cells occurs mainly by GLUT1, whereas in rodent models, it occurs mainly by GLUT2 (110). In human and rodent beta-cells, GCK is the dominant HK (111). Intracellular glucose concentration has been reported for the murine insulinoma MIN6 cell line with glucose uptake by GLUT2 and glucose metabolism mainly by GCK (84, 112). Intracellular glucose concentration is similar to extracellular glucose concentration (85), glucose metabolism is not rate limited by GLUT2 transport (112), and flux of glucose metaboolism increases proportionately to extracellular glucose concentration through physiological range—GCK is not saturated by glucose and is coupled to the secretion of insulin (84). This is consistent with the glucose sensor function of GCK in pancreatic beta-cells (51). In subjects with normal insulin sensitivity and beta-cell function, temporary increases in plasma glucose in the absorptive phase produces increased G6P and other glycolytic intermediates, but these are corrected following insulin secretion and return of plasma glucose concentrations to the basal state in fasting phase. Sustained increased glycolytic intermediates in beta-cells is implicated in glucotoxicity (113). Although little investigated, other cells with GCK expression may be susceptible to glycolytic overload with contribution to glucotoxicity and cell dysfunction—pancreatic alpha-cells, selected neurons of the hypothalamus and brainstem, in the pituitary, and entero-endocrine K and L cells (26, 114, 115).
For other sites of critical glucose metabolism, for example, in neurons of the central nervous system (CNS) and cardiomyocytes of the heart, an abundant pool of intracellular glucose is maintained with glucose metabolism limited by dominant expression of HK1 (27, 116, 117). In the CNS, glucose enters neurons predominantly by GLUT3 (118), and the intracellular concentration of glucose in euglycemia is approximately 1 mM (119). This indicates that HK1 is 94% saturated by intracellular glucose. In cardiomyocytes, glucose uptake is mediated by GLUT1 and GLUT4 (120), intracellular glucose concentration is 1–2 mM (121), and HK1 is 94%–97% saturated by glucose. In these cells, HK1-mediated glucose metabolism prevents glycolytic overload and unscheduled glycolysis when intracellular glucose concentration is increased in hyperglycemia. Impairment of CNS and cardiac function in hyperglycemia may arise through HK2-linked glycolytic overload of vascular cells in blood vessels of the brain and heart, see below, and through the development of cardiomyocyte dysfunction, such as in atrial dilatation, which is associated with increased expression of HK2 (27), increasing the vulnerability to HK2-linked metabolic overload. This may apply particularly in ischemia reperfusion injury with G6P accumulation from glycogenolysis, as reviewed (122).
Hexokinase-2-linked glycolytic overload and unscheduled glycolysis as the initiator of metabolic dysfunction in the development of vascular complications of diabetes
Increased flux of glucose metabolism in high glucose concentration circumventing glucose saturation of HKs is the key driver of metabolic dysfunction in human endothelial cells, pericytes, renal mesangial cells, podocytes and tubular epithelial cells, Schwann cells of peripheral nerves, and fibroblasts (25, 57, 61, 68, 71, 123). Both HK1 and HK2 expessed in these cell types are saturated with glucose, and yet, the flux of glucose metabolism is increased without increased HK expression in hyperglycemia (25). The explanation for this is stabilization of HK2 protein to degradation by high intracellular glucose concentration (25). HK2 uniquely has a second glucose-binding active site in the C-terminal domain (24). This additional active site contains a degradation motif, 712QRFEK716, that binds heat shock protein cognate 70 (HSC70) and directs HK2 for proteolysis by chaperone-mediated autophagy (124). As plasma glucose concentration increases beyond the normal physiological range, there is a concomitant increase in intracellular glucose concentration. At the C-terminal active site, glucose increasingly competes with HSC70 for binding of the active site and thereby slows the degradation and increases the half-life of HK2. HK2 protein and hexokinase activity consequently increase without change in expression of other glycolytic enzymes downstream in early-stage glycolysis, producing unscheduled glycolysis (122).
Unscheduled glycolysis drives pathways of metabolic dysfunction in cells expressing HK2 in hyperglycemia with high intracellular glucose concentration. The consequences of this are:
(i) Increased levels of G6P, which displace HK2 from mitochondria, impairing disposal of ATP, producing mitochondrial membrane hyperpolarization, mitochondrial dysfunction, and increased formation of reactive oxygen species (ROS) (32, 125); and increased G6P/Mondo A/Mlx transcriptional activity, stimulating lipogenic and other gene expression with regulatory carbohydrate response elements (ChRE) (47);
(ii) Increased F6P and activation of the hexosamine pathway, increasing enzymatic protein glycosylation (126, 127);
(iii) Increased GA3P and DHAP, leading to increased formation and accumulation of the reactive dicarbonyl metabolite, methylglyoxal (MG) or dicarbonyl stress (25, 126, 127), with increased formation of MG-derived advanced glycation endproducts (AGEs) in proteins, which are misfolded proteins and activate the unfolded protein response (UPR) (25, 128);
(iv) Increased glycerol-3-phosphate (from increased DHAP), leading to increased formation of diacylglycerol de novo and abnormal activation of protein kinase c (PKC) (126, 127); and
(v) Increased metabolic channeling of G6P for glycogen synthesis as a consequence of displacement of HK2 from mitochondria (32) (Figure 1B).
Evidence of all of these effector pathways and biomarkers of HK2-linked glycolytic overload and unscheduled glycolysis has been found in diabetic endothelial dysfunction, DKD, diabetic retinopathy, and diabetic neuropathy (Table 3).
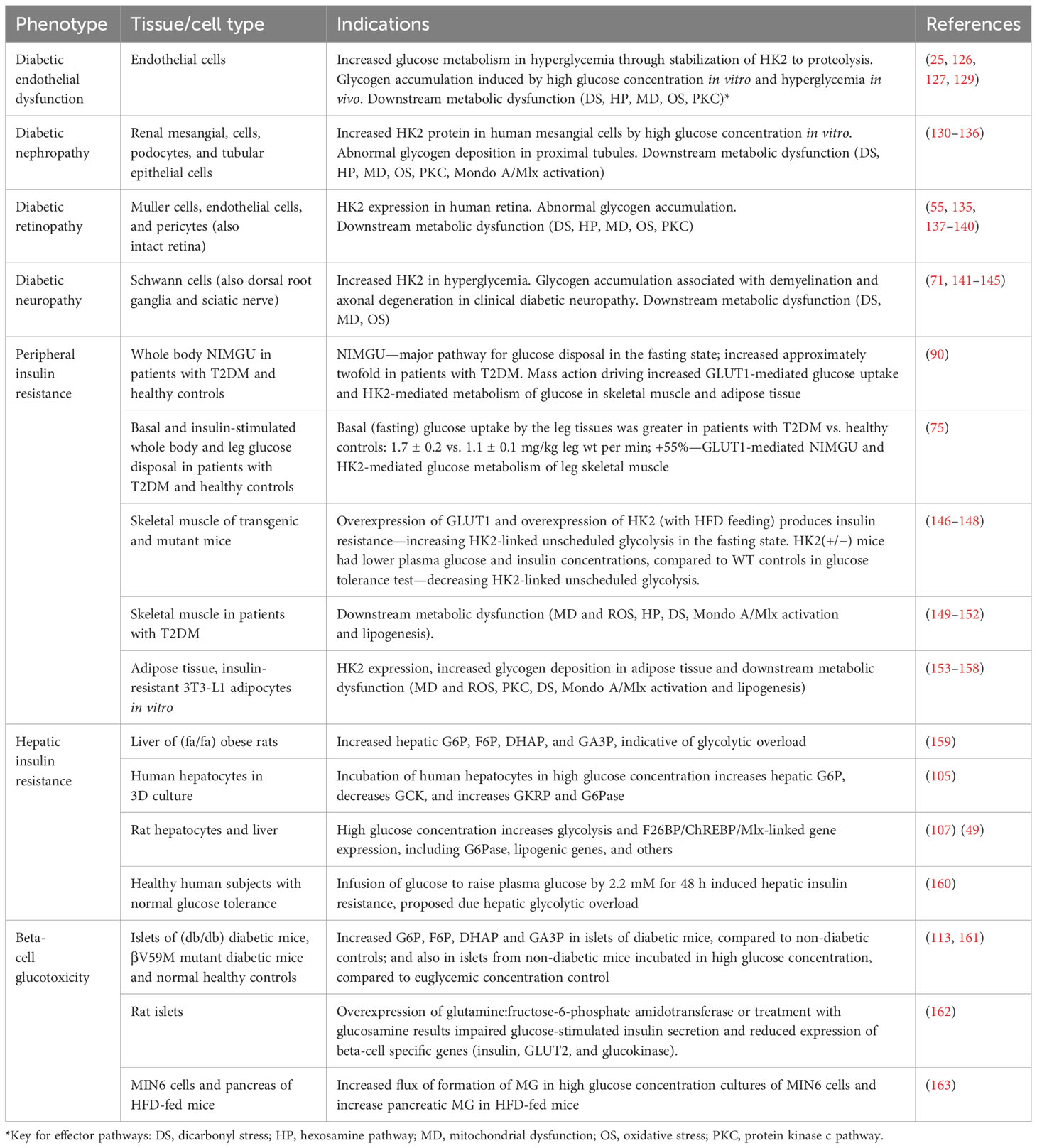
Table 3 Evidence of hexokinase-2 linked glycolytic overload and unscheduled glycolysis in glycemic disease.
The initiating event is stabilization of HK2 to proteolysis and unscheduled glycolysis. In confirmation of this, partial silencing or pharmacological treatment to maintain normal levels of HK2 in high glucose concentration prevented high glucose concentration-induced metabolic dysfunction (25, 123). A condition for the induction of glycolytic overload in hyperglycemia is that HK2 is a major contributor to HK activity at the sites of development vascular complications of diabetes. This is indeed found in vascular endothelial cells and pericytes, renal mesangial cells, podocytes and tubular epithelial cells, and Schwann cells of peripheral nerves (55, 60, 63, 64, 67, 71) (Table 3).
The extent of increase in HK2 protein by stabilization to proteolysis in hyperglycemia depends on: (i) the increase in cellular glucose concentration such that glucose competes effectively with HSC70 at the C-terminal active of HK2 and (ii) the duration of increased glucose concentration with respect to the in situ half-life t1/2 of HK2 protein. Our recent studies in endothelial cells indicate that the apparent inhibitor constant Ki value of extracellular glucose concentration for inhibition of proteolysis of HK2 is approximately 7 mM (which may vary with differing rates of cellular uptake of glucose and levels of expression of HSC70), and the half-life of HK2 protein is approximately 10 h, indicating that prolonged exposure of HK2 to increased cytosolic glucose concentration in IFG and diabetes is the condition required to stimulate HK2-linked glycolytic overload and unscheduled glycolysis. Independent estimates of the half-life of HK2 are <12 h and 11 h (164, 165). The effect of cellular glucose concentration on HK2 stability to proteolysis was identified previously by others, but the relevance to hyperglycemia and diabetes was not appreciated (124).
An interesting consequence of HK2 displacement from mitochondria in unscheduled glycolysis is increased flux of glucose metabolism for glycogen synthesis by effects of metabolic channeling (32). In human aortic endothelial cells, we found a sevenfold increase in glycogen deposition in high glucose concentration cultures (25), an effect reported previously by others without explanation (166). For in vivo translation of these findings, increased glycogen deposition was found in the aorta (129), retina (167, 168), and renal tubular epithelial cells and glomeruli of STZ rats (169). Increased glycogen deposition was also found in Schwann cells and axons of perpiheral nerve in experimental and clinical diabetes (with possible confounding effects of axonia) (141, 170). Indeed, glycogen deposition in the kidney of patients with diabetes has long been known and called “Armanni–Ebstein” diabetic nephropathy, linked to severity of disease (171). Increased glycogen deposition at the sites of vascular complications of diabetes is explained by the HK2-linked glycolytic overload and unscheduled glycolysis hypothesis.
Recent studies suggest that increased activation of Mondo A/Mlx in hyperglycemia contributes to endothelial dysfunction in diabetes and diabetic kidney disease. In vascular endothelial cells, high glucose concentration stimulated G6P/Mondo A/Mlx-induced increased expression of thioredoxin interacting protein (TXNIP) (172). Increased TXNIP synergizes with activation of the UPR, also activated in hyperglycemia (25), to stimulate inflammatory signaling via the NLRP3 inflammasome (173). Knockdown of Mondo A in podocytes decreased albuminuria, mesangial expansion, and glomerular basement membrane thickening in mutant leptin receptor (db/db) insulin-resistant, diabetic mice (174). Mondo A had increased O-GlcNAc enzymatic glycosylation through hexosamine pathway activation with increased stability to proteolysis, nuclear translocation, and transcriptional activity in mesangial cells incubated in high glucose concentration and mesangial of streptozotocin-induced diabetic rats. This was linked to increased lipid deposition and glomerulosclerosis (175). By these mechanisms, the activation of Mondo A during glycolytic overload may contribute to the damaging effects of hyperglycemia at vascular sites.
Hexokinase-2-linked unscheduled glycolysis as initiator of peripheral insulin resistance
The skeletal muscle is the major site of insulin-mediated glucose uptake in the postprandial state and the major tissue involved in direct catabolism of glucose (176, 177). Hence, the dysfunction of glucose uptake and metabolism by the skeletal muscle has a major impact on glucose homeostasis (178).
The skeletal muscle and adipose tissue have relatively high expression of HK2 (41, 76). Glucose entry into myocytes and adipocytes is mediated by GLUT1 in the fasting phase and insulin-dependent GLUT4 in the absorptive phase. A primary characteristic of insulin resistance is decreased recruitment of GLUT4 to the cell surface in response to insulin due to impaired GLUT4 trafficking, as recently reviewed (179). GLUT1 mRNA and protein in the skeletal muscle was unchanged, increased, or decreased in insulin resistance (40, 180, 181); when decreased, the decrease was more than compensated by the mass action effects of increased FPG in the in situ rate of glucose uptake (181). Unlike vascular cells, the intracellular concentration of glucose in myocytes and adipocytes is usually relatively low: HK1 is saturated by glucose euglycemia and hyperglycemia, whereas HK2 is not. Insulin increases the expression of HK2 in the skeletal muscle (42) and also binding of HK2 to mitochondria, likely by increasing phosphorylation by protein kinase B, also called Akt (182, 183). There is no indication of increased HK2 expression at the mRNA and protein level in the skeletal muscle and adipose tissues in clinical insulin resistance and some reports of decreases (91–94, 184, 185), likely due to decreased insulin-dependent expression (91). In the fasting phase, patients with insulin resistance and T2DM had higher glucose metabolism in the skeletal muscle, compared to healthy controls, mediated by mass action increased NIMGU (75, 90). As this occurs without increased insulin and other regulatory signaling, the conditions for HK2-linked unscheduled glycolysis are established. Clinical studies and experimental functional genomics studies of HK2 and GLUT1 in the skeletal muscle of mice support this, as previously discussed (186) and summarized herein in Table 3.
Overexpression of HK2 in the skeletal muscle induced insulin resistance in mice fed a high fat diet (HFD) (146). This finding was contrary to the hypotheses tested that “impaired muscle glucose uptake resulting from high fat feeding would be exposed in high flux states and could be corrected by HK2 overexpression.” In addition, heterozygous knockdown of HK2 in mice improved hyperglycemia and hyperinsulinemia (147), as we previously described (186). These findings are consistent with an inverse relationship between insulin sensitivity and HK2 expression, predicted from the hypothesis of HK2-linked unscheduled glycolysis contributing to the development of insulin resistance.
The overexpression of GLUT1 in the skeletal muscle of mice induced insulin resistance (148). We interpret this as increasing glucose uptake in the skeletal muscle in euglycemia—particularly in the fasting phase—and thereby mimicking the effect of mass action increase in glucose uptake by the skeletal muscle in increased FPG found in clinical insulin resistance (75). In the GLUT1 overexpressing mouse, there was increased intracellular glucose concentration, increased HK activity, and increased deposition of glycogen in the skeletal muscle—the latter not due to increased glycogen synthase or decreased glycogen phosphorylase activities (187). Impaired recruitment of the GLUT4 in response to insulin was linked to increased O-GlcNAcylation of membrane proteins, including GLUT4 (188). These effects are explained by HK2-linked unscheduled glycolysis.
In contrast, overexpression of GLUT4 in the skeletal muscle of mice did not induce insulin resistance (189). We interpret this as increasing insulin-stimulated GLUT4-mediated glucose uptake, occurring together with insulin- and G6P/Mondo A/Mlx-linked increase in the activity of HK2 and other glycolytic enzymes. This thereby does not produce unscheduled glycolysis and thereby does not induce insulin resistance. The transgenic mouse model with overexpression of both GLUT1 and HK2 together in the skeletal muscle was characterized by a threefold increased G6P concentration and 7.5-fold increased glycogen levels in the skeletal muscle on a standard diet, with no change in basal or insulin-stimulated whole-body glucose disposal (190). We interpret this model as establishing conditions for chronic exposure of skeletal muscle to increased G6P, inducing increased transcription of glycolytic enzymes through G6P/Mondo A/Mlx and thereby negating unscheduled glycolysis. HK2 overexpression gave markedly increased cytosolic HK2, producing increased glycogen by metabolic channeling. This may indicate that clinical HK2-linked unscheduled glycolysis is exacerbated by variability in hyperglycemia, with glycolytic intermediates accumulating most markedly with increased plasma glucose concentration after a period of lower glucose exposure and low level G6P/Mondo A/Mlx transcriptional signaling with low basal expression of glycolytic enzymes.
Evidence of activation of effector pathways of HK2-linked unscheduled glycolysis in peripheral insulin resistance has been reported: in the skeletal muscle, mitochondrial dysfunction, increased ROS formation, hexosamine pathway, MG-derived AGEs, and increased Mondo A/Mlx signaling have been found in insulin resistance (149–152) and in adipose tissue, mitochondrial dysfunction, increased ROS formation, increased PKC activity, dicarbonyl stress, and increased Mondo A/Mlx signaling (153–156) (Table 3).
Glucokinase-linked glycolytic overload and unscheduled glycolysis in hepatic insulin resistance and beta-cell glucotoxicity
GCK is the major hexokinase in hepatocytes and beta-cells, and both have efficient glucose uptake by GLUT2 and GLUT1 glucose transporters, respectively, with increased plasma glucose producing similar increases in intracellular glucose (Table 2). This reflects the role of GCK in the liver and beta-cells as glucose sensors to store or release glucose and secrete insulin in response to high glucose concentration, respectively (51). These cells are susceptible to glycolytic overload and unscheduled glycolysis because the low affinity of GCK for glucose produces increased flux of glucose metabolism with increasing intracellular glucose concentration. There is also uptake of dietary fructose by GLUT5 and metabolism by KHK to F1P in hepatocytes (191), which supplements the triosephosphate pool after cleavage to DHAP and glyceraldehyde by aldolase, with the latter phosphorylated to GA3P by triokinase, and increased formation of MG (192).
Hepatic insulin resistance is defined as raised basal hepatic glucose production in the presence of normal or raised plasma insulin levels, suggesting a failure of normal suppression of hepatic glucose production (193). In experimental obese, hyperglycemia leptin receptor mutant (fa/fa) rats with hepatic insulin resistance, there are increases in hepatic G6P, F6P, DHAP, and GA3P, indicative of glycolytic overload (159). Studies of human hepatocytes incubated in high glucose concentration in vitro suggest that glycolytic overload is countered by decreased expression of GCK and increased expression of GKRP and G6Pase, all serving to decrease the concentration of G6P (105). Increased glucose metabolism was influenced 45% by extracellular glucose concentration and 55% by insulin, the latter decreasing in an experimental model of insulin resistance (82). In normal healthy rats, increase in plasma glucose produces increased hepatic glycogen synthesis and glycolysis and decreased glucose production. Hepatic G6P was normal or lower than normal and the glycolytic regulator F26BP was increased (107). In hepatocytes, F26BP is a cofactor for ChREBP/Mlx for the induction of expression of G6Pase, lipogenic, and other ChRE target genes (49), indicating that persistent increased glucose metabolism through glycolysis in the liver stimulates increased hepatic glucose production and de novo lipogenesis. Indeed, increased hepatic de novo lipogenesis is a feature of clinical insulin resistance (194). Increased hepatic DHAP and GA3P (159) leads to increased formation of MG, accounting for the observed MG-derived AGE, hydroimidazolone MG-H1, in HFD-fed rats (195) and stimulus for the activation of the UPR (25) [which also increases hepatic G6Pase (196)]. Prolonged incubation of human hepatocytes with fructose and fatty acids decreased glucose consumption and induced G6Pase, ChREBP, and fatty acid synthase (FASN) (82). Infusion of glucose in healthy human subjects with normal glucose tolerance to raise plasma glucose by 2.2 mM for 48 h induced hepatic insulin resistance (160). We hypothesize that persistent increase in plasma glucose overwhelms the control of hepatic G6P producing hepatic glycolytic overload, simulating expression of G6Pase with increased hepatic glucose production and increased lipogenesis through activation of ChREBP/Mlx.
Activation of ChREBP by increased hepatic G6P, however, limits the increase in hepatic phosphorylated glycolytic intermediates and maintains levels of ATP during hyperglycemia (105). It may thereby be considered protective against glycolytic overload—as previously proposed (197, 198). Indeed, hepatic overexpression of ChREBP prevented the development of insulin resistance and glucose intolerance in HFD-fed mice, although hepatic steatosis increased. Overexpression of ChREBP hepatocytes in vitro induced expression of ChRE-regulated target gene, stearoyl-CoA desaturase 1 (Scd1), catalyzing the conversion of saturated fatty acids (SFAs) into monounsaturated fatty acids. SFA impairment of insulin-responsive Akt phosphorylation was thereby rescued through increased Scd1. The expression of G6Pase was also increased (199).
Studies of ChREBP silencing in hepatocytes showed decreased glucose-mediated induction of glycolytic and lipogenic genes and decreased lipid accumulation (200). Global ChREBP-deficient mice had decreased glycolytic and lipogenic gene expression and triglyceride synthesis and increased accumulation of glycogen in the liver under high starch feeding (201). Liver-specific inhibition of ChREBP by shRNA silencing in obese and insulin-resistant, leptin mutant ob/ob mice decreased hepatic steatosis, plasma triglycerides, and non-esterified fatty acids. This improved insulin signaling in the liver, skeletal muscle, and white adipose tissue, with overall improved glucose tolerance and insulin sensitivity (202). Similar effects were found in global homozygous ChREBP knockout ob/ob mice (203).
The protective effects of hepatic ChREBP are most marked in high fructose and sucrose feeding. Hepatic-specific knockout of ChREBP in mice were tolerant to high fructose diets but had increased plasma alanine aminotransferase and aspartate transaminase activities, enlarged liver, and hepatic glycogen overload. Liver G6P was increased approximately twofold, and ATP decreased approximately 40%. Hepatoxicity was rescued by overexpression of liver pyruvate kinase (204). Increased formation of F1P with activation of GCK and loss of ChREBP-induced G6Pase may synergize to increase G6P and produce damaging glycolytic overload.
In clinical studies, ChREBP expression correlates positively with glucose intolerance, insulin resistance, and steatosis, as reviewed (205). This suggests that while a protective role against hepatic glycolytic overload of ChREBP is maintained, the deleterious effects of ChREBP-mediated lipogenesis may be dominant overall during the development of insulin resistance. In the current hypothesis, we also propose a contribution to the development of insulin resistance from the activation of Mondo A in skeletal muscle in hyperglycemia.
Glycolytic overload in pancreatic beta-cells contributes to glucotoxicity—the term referring to the phenomenon of impaired β-cell function during states of elevated glucose concentration (206). In mutant leptin receptor (db/db) insulin-resistant diabetic mice, there was increased G6P, F6P, DHAP, and GA3P in islets, compared to non-diabetic controls (161). Similar effects were found in islets of βV59M mutant inactive beta-cell potassium channel diabetic mice, compared to wild-type controls, and control islets incubated in high glucose concentration (113). This suggests that glycolytic overload and unscheduled glycolysis are established in persistent hyperglycemia of diabetes and high glucose concentration in vitro. For downstream effector pathways, beta-cells are resistant to mitochondrial hyperpolarization and increased ROS formation. GCK is attached to mitochondria in a multiprotein complex with high affinity (207), despite high cellular G6P concentration (>1 mM, as judged by INS-1 insulinoma cell line) (208), and mitochondrial protein thiols of beta-cells were reduced rather than oxidized in rat islets in high glucose concentration (209). The overexpression of glutamine:fructose-6-phosphate amidotransferase or treatment with glucosamine to increase hexosamine pathway activity impaired glucose-stimulated insulin secretion and reduced the expression of beta-cell-specific genes (insulin, GLUT2, and glucokinase) (162). Recent studies have indicated that one or more glycolytic metabolites downstream of phosphofructokinase and upstream of GA3PD mediate the effects of chronic hyperglycemia (113). This is consistent with increased DHAP, GA3P, and formation of MG, which may activate the UPR by MG-modified misfolded proteins (25, 128). Indeed, the formation of MG was increased in MIN6 cells incubated in high glucose concentration, and pancreatic MG concentration was increased in HFD-fed insulin-resistant mice (163). Finally, increased Mondo A/Mlx signaling in beta-cells in persistent hyperglycemia is linked to increase in TXNIP (210), inflammation, impaired insulin secretion, and apoptosis (211) (Table 3).
In pancreatic beta-cells, there appears to be limited protection from glycolytic overload and unscheduled glycolysis. The expression of GKRP is very low (212) with no evidence for GKRP expression at the protein level, concurring with the lack of accumulation of GKRP in the nucleus of these cells (213). Protection may be afforded by downregulation of expression of GCK and increased expression of other hexokinases, thereby limiting the glycolytic response of beta-cells at high glucose concentration. Prolonged exposure to increased glucose concentration produced by 90% pancreatectomy in rats induced change in HK expression in pancreatic islets—50% decreased expression of GCK and increased expression of other HKs normally considered “forbidden” gene expression in beta-cells: HK1, +4.5-fold; HK2, +20-fold; and HK3, +47-fold (214). Similar trends were found in pancreatic islets of senescence-accelerated mice (SAM) and db/db obese mice: decreased GCK expression and increased HK1–HK3, particularly HK2; 43% and 64% decrease in GCK; and two- and sixfold increased HK2 in SAM and db/db mice, respectively (161). The downside is loss of glucose sensor function, impairing increased insulin secretion in response to increasing glucose concentration, decreasing beta-cell functionality, and progression to development of T2DM. There is also increased vulnerability to HK2-linked glycolytic overload in hyperglycemia. Mechanisms proposed for this change in islet identity are loss of transcription factor FoxO1 (215), maladaptive UPR (216, 217), and inflammation (218), which may be mediated by the UPR (211) and oxidative stress (219). In clinical translation, increasing FPG within the normal range in subjects with normal glucose tolerance or IGT correlates with decline in hyperglycemia-induced insulin secretion (220).
Hexokinase-linked glycolytic overload and unscheduled glycolysis versus mitochondrial superoxide overproduction hypotheses of metabolic dysfunction in hyperglycemia
The “hyperglycemia-induced mitochondrial superoxide overproduction” hypothesis has been proposed in explanation of hyperglycemia-induced metabolic dysfunction in the development of vascular complications of diabetes (221), with extension of this applied to insulin resistance and glucotoxicity (222). In this hypothesis, it was envisaged that “hyperglycemia-induced overproduction of superoxide by the mitochondrial electron-transport chain partially inhibits GA3PD, thereby diverting upstream metabolites from glycolysis into pathways of glucose overutilization. This results in increased flux of DHAP to DAG, an activator of PKC, and of triosephosphates to MG. Increased flux of F6P to UDP-N-acetylglucosamine increases modification of proteins by O-linked N-acetylglucosamine and increased glucose flux through the polyol pathway consumes NADPH and depletes GSH.”
A consequence of the mitochondrial superoxide overproduction hypothesis is that the proportion of the flux of glucose metabolism leading to the formation of MG is increased in high glucose concentration. However, in studies of high glucose concentration-induced metabolic dysfunction in endothelial cells and fibroblasts, the proportion of glucose metabolism accounted for as the formation of MG was unchanged (25, 123). This indicates that the in situ activity of GA3PD is not inhibited. Increased flux of formation of MG was rather due to increased flux of glucose metabolism through glycolysis by circumvention of HK saturation by glucose-induced stabilization of HK2 to proteolysis (25, 123). Increased flux of glucose metabolism and its gating by HKs was not considered in the mitochondrial superoxide overproduction hypothesis. It is the stabilization of HK2 to proteolysis and unscheduled glycolysis rather than mitochondrial superoxide overproduction that initiates metabolic dysfunction in hyperglycemia. There is also a minor increase in flux of glucose metabolism through the polyol pathway due to the low affinity of AR for glucose, but this only accounts for 1%–2% total glucose metabolism at the site of vascular complications of diabetes, insulin resistance, and beta-cell glucotoxicity.
The mitochondrial superoxide overproduction hypothesis sought to explain mitochondrial dysfunction and related increased formation of ROS as part of hyperglycemia-induced metabolic dysfunction (126), but other key aspects were not considered. The key insights and advances in the new hypothesis are as follows: (i) mitochondrial superoxide overproduction is not the initiator of metabolic dysfunction but rather one of multiple downstream effector pathways, and (ii) contribution of the displacement of HK2 from mitochondria by high cellular G6P as a cause of mitochondrial membrane hyperpolarization is recognized (25, 32, 125). Increased formation of ROS in vascular cells in hyperglycemia also occurs by activation of vascular NADPH oxidase and uncoupling of endothelial nitric oxide synthase, stimulated by activation of PKC—a further effector pathway of glycolytic overload and unscheduled glycolysis (223, 224).
The prevention of metabolic dysfunction induced by high glucose concentration in endothelial cells by the overexpression of uncoupling protein-1 (UCP1) and superoxide dismutase-2 (SOD2) appeared to provide strong support for the hyperglycemia-induced mitochondrial superoxide overproduction hypothesis (126). However, both overexpression of UCP1 and SOD2 have recently been found to decrease cellular ATP (225, 226), and this may have prevented metabolic dysfunction by decreasing in situ activity of hexokinase rather than by countering mitochondrial dysfunction and increased ROS formation.
The mitochondrial superoxide overproduction hypothesis held promise of antioxidant therapy in prediabetes and diabetes, which has been unfulfilled. Daily administration of 400 IU vitamin E for 4.5 years to middle-aged and elderly people with diabetes and CVD and diabetic nephropathy and had no effect (227). α-Lipoic acid administered for 2 years produced no benefit against endpoints of diabetic macular edema or best-corrected visual acuity in diabetic retinopathy (228). A meta-analysis concluded that there is weak evidence of benefit of α-lipoic acid treatment in diabetic neuropathy (229). Vitamin E improved insulin resistance in non-diabetic overweight subjects, but the effect appeared transient (230). A meta-analysis of studies of vitamin E and vitamin C, individually or in combination, concluded that there was no evidence for improvement of insulin resistance in diabetes by the treatments (231). Ineffectiveness of antioxidant therapy is envisaged in the HK-linked glycolytic overload and unscheduled glycolysis hypothesis as increased formation of ROS is only one of multiple effector pathways downstream of initiation by HK-gated increase in glucose metabolism.
Predictions of the hexokinase-linked glycolytic overload and unscheduled glycolysis hypothesis
From the new hypothesis, we expect to provide explanations of previously unexplainable observations and provide new strategies for improved disease management and treatment of glycemic disease. These are summarized in Table 4. Explanations may now be provided for the following: (i) susceptibility of peripheral neurons and resistance of CNS neurons to metabolic dysfunction and pathogenesis in hyperglycemia; (ii) progression of DKD and diabetic retinopathy by increased expression of HK2; (iii) increased deposition of glycogen at sites of vascular complications of diabetes linked to the progression of vascular complications; (iv) counterintuitive link of increased HK2 expression in the skeletal muscle to the development of insulin resistance and heterozygous HK2 knockdown to improved insulin sensitivity in mice; (v) FPG being a risk predictor for the progression of insulin resistance; and (vi) patients with T1DM who may also exhibit insulin resistance (Table 4).
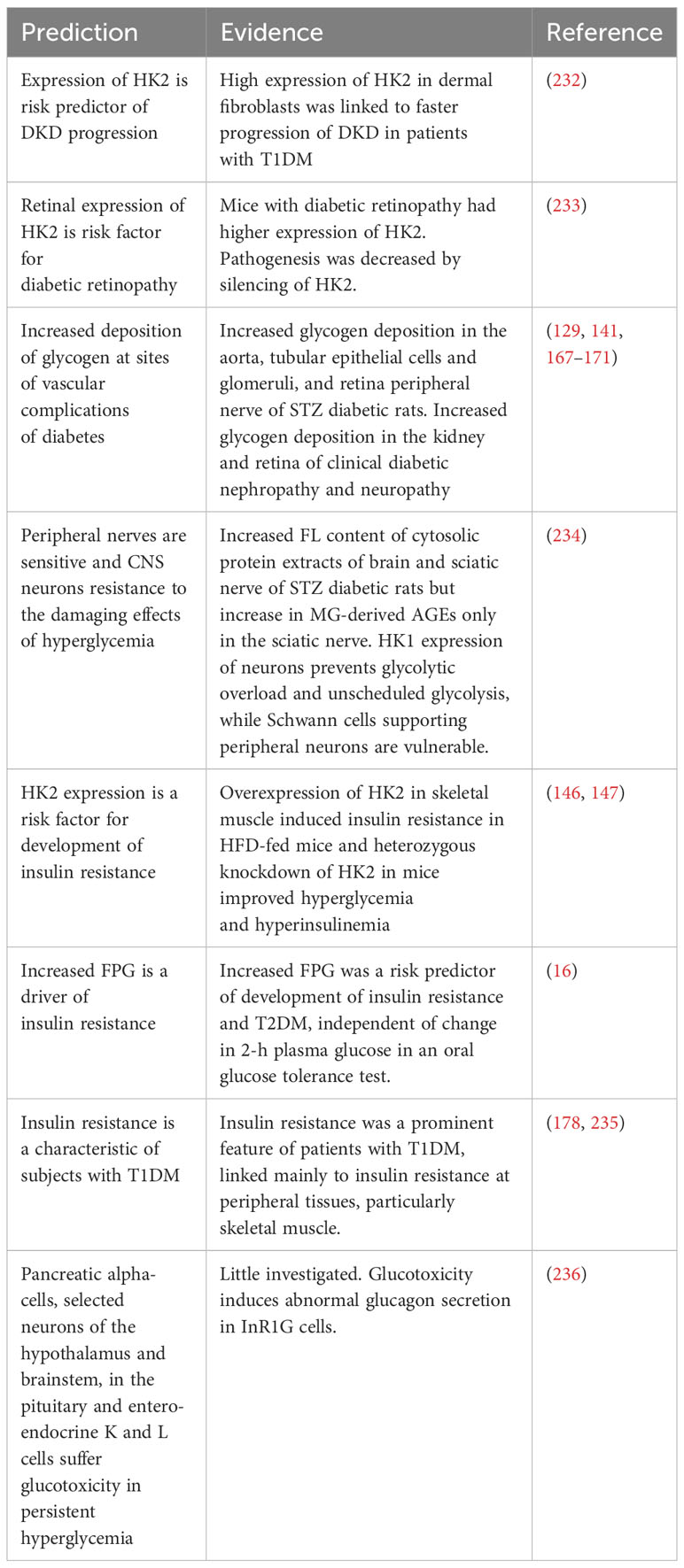
Table 4 Predictions of the hexokinase-linked glycolytic overload and unscheduled glycolysis hypothesis.
Predictions to be explored in the future are (i) the positive association of HK2 expression with clinical insulin resistance and (ii) glycolytic overload in cells other than hepatocytes and beta-cells with dominant GCK expression and increased intracellular glucose concentration in hyperglycemia.
For therapeutics development, correcting the primary initiating step of dysfunctional glucose metabolism in hyperglycemia is likely the most effective pharmacological target, that is, dysregulation of HK2 expression. In vascular complications of diabetes, the pharmacological intervention required is to normalize HK2 protein levels to those of euglycemic control. To counter peripheral insulin resistance, we seek to inhibit HK2-mediated glucose metabolism in the skeletal muscle and adipose tissue in the fasting phase but not in the absorptive phase; therefore, direct chemical inhibitors of HK2 are undesirable. We rather need to correct the dysfunctional regulation of HK2 expression. Correcting peripheral resistance is expected to increase glucose disposal, decreasing plasma glucose concentration and thereby indirectly relieving glycolytic overload linked to hepatic insulin resistance and beta-cell glucotoxicity.
An example of a therapeutic agent that corrects HK2-linked metabolic dysfunction in hyperglycemia is the glyoxalase 1 (Glo1) inducer, optimized to a combination of trans-resveratrol (tRES) and hesperetin (HESP), or tRES-HESP (237). The combination synergizes at the receptor, transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2), and HESP improves bioavailability of tRES by inhibition of intestinal glucuronosyl-transferases (238). Glo1 catalyzes the detoxification of MG and is the optimum strategy to counter dicarbonyl stress (239). The GLO1 gene has a functional antioxidant response element (ARE) where Nrf2 binds to induce increased expression of Glo1 (240). We developed a therapeutic strategy to optimize compounds for the induction of expression of Glo1 by the activation of Nrf2 with a functional GLO1-ARE reporter assay (237). A further ARE-linked, Nrf2-regulated gene is glucose-6-phosphate dehydrogenase (G6PD). Increasing expression of G6PD decreases the steady-state concentration of G6P, decreasing transcriptional activity of G6P/Mondo A/Mlx and thereby decreasing expression of HK2. The decrease in G6P also decreases displacement of HK2 from mitochondria and related mitochondrial dysfunction. There is emerging evidence that tRES-HESP corrects HK2-linked glycolytic overload and potently prevents vascular cell dysfunction in high glucose concentration.
Incubation of human aortal endothelial cells with tRES-HESP in high glucose concentration decreased HK2 protein levels and corrected the flux of glucose metabolism to those of low glucose concentration control cultures and all metabolic dysfunction (25). Similar responses have been found in other cell types (123). tRES-HESP also prevented bone-marrow-derived progenitor cell dysfunction and improved wound healing in experimental diabetes (241).
tRES-HESP was evaluated clinically in non-diabetic overweight and obese subjects with insulin resistance in a double-blind, placebo-controlled crossover study—Healthy Aging Through Functional Food (HATFF) study. tRES-HESP (90 mg tRES, 120 mg HESP), once daily given by oral capsule, for 8 weeks improved insulin sensitivity to that of lean, healthy controls, improving FPG, vascular inflammation, and dicarbonyl stress (237). Insulin resistance was measured by the oral glucose insulin sensitivity (OGIS) index, which correlates well with the reference hyperinsulinemic–euglycemic clamp technique (242). The magnitude of change in OGIS with tRES-HESP treatment, 42–58 ml min−1 m−2, was comparable to that achieved with pharmaceutical treatment of patients with T2DM (for example, 1.7 g metformin per day, ΔOGIS = +54 ml min−1 m−2) (243) and extreme weight loss with gastric band surgery in morbid obesity (ΔOGIS = +62 ml min−1 m−2) (244). There were also links to two established mediators of insulin resistance: TXNIP and tumor necrosis factor-α (TNFα). TXNIP is a mediator of insulin resistance in the liver, skeletal muscle, and adipose tissue and impaired pancreatic beta-cell insulin secretion (245–247). It has expression induced by G6P/Mondo A/Mlx (248) and therefore has expression-induced downstream of glycolytic overload. It also has expression increased by the activation of the UPR sensor IRE1α, increasing expression of XBP1 and cleavage of miR-17, the latter stabilizing TXNIP mRNA (249). TNFα decreases insulin receptor signaling in the adipose tissue and skeletal muscle, particularly prior to development of T2DM (250–252). tRES-HESP treatment produced a decrease in PBMC TNFα expression in the obese subject subgroup of the HATFF study (237). TNFα expression is likely increased through the activation of the UPR in glycolytic overload with TXNIP linked to the activation of the NLRP3 inflammasome (249). Reversal of insulin resistance by tRES-HESP was not achieved by tRES and HESP individually in clinical evaluation (253, 254), suggesting pharmacological synergism of tRES-HESP.
The correction of insulin resistance by tRES-HESP treatment in the HATFF study is consistent with both improvement of insulin resistance and glucotoxicity (237). The overweight and obese subjects treated likely have reversible impairment of beta-cell function (10). Treatment with tRES-HESP corrected peripheral insulin resistance, relieved dysglycemic stress, and thereby provided conditions for normal beta-cell function to be restored.
Targeting activation of Nrf2 to correct insulin resistance is supported by studies of tissue-specific genetic activation of Nrf2 in HFD-fed mice by partial knockdown of Nrf2 antagonist, Keap1. The activation of Nrf2 in the skeletal muscle prevented insulin resistance, and the activation of Nrf2 in the liver prevented hyperglycemia (255). A similar activation of Nrf2 in all tissues prevented the onset of diabetes in db/db mice (256). Research on the development of activators of Nrf2 for the treatment of vascular complications of diabetes has been advanced (257). In future studies, the development of Nrf2 activators to correct HK2-linked glycolytic overload and unscheduled glycolysis treatments may prove effective where optimizations for the induction of G6PD and Glo1 expression are key ARE-linked gene targets. Countering dicarbonyl stress is linked to the prevention of vascular complications of diabetes and treatment of insulin resistance, as reviewed (239). Increasing G6PD expression is expected to improve beta-cell function and glucose tolerance (258) and decrease progression of vascular complications of diabetes (259–261). Previous considerations have linked G6PD activity to countering oxidative stress but suppression of cellular G6P, Mondo A/ChREBP activation, HK2 expression, and glycolytic overload are likely also involved. Nrf2 activators have differing abilities to induce ARE-linked gene expression by mechanisms not yet fully understood (262), so functional screens of activators for the induction of target gene expression are currently required.
Concluding remarks
The hexokinase-linked glycolytic overload and unscheduled glycolysis hypothesis recognizes the crucial role of HKs in the initiation of dysfunctional metabolism and pathogenesis in hyperglycemia. When there is persistent increased flux of glucose entering glycolysis without concomitant increase in glycolytic enzyme activities to catalyze its onward metabolism, steady-state levels of glycolytic intermediates exceed tolerable limits and metabolic dysfunction leading to a decline in insulin sensitivity and beta-cell function, and glycemic disease may ensue.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Author contributions
NR: Writing – review & editing. PT: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Writing – original draft.
Funding
The author(s) declare financial support was received for the research and authorship of this article. Funding for publication costs was from Hamad bin Khalifa University and Qatar University, Doha, Qatar.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ong KL, Stafford LK, McLaughlin SA, Boyko EJ, Vollset SE, Smith AE, et al. Global, regional, and national burden of diabetes from 1990 to 2021, with projections of prevalence to 2050: a systematic analysis for the Global Burden of Disease Study 2021. Lancet (2023) 402(10397):203–34. doi: 10.1016/S0140-6736(23)01301-6
2. Echouffo-Tcheugui JB, Selvin E. Prediabetes and what it means: the epidemiological evidence. Annu Rev Public Health (2021) 42:59–77. doi: 10.1146/annurev-publhealth-090419-102644
3. Rooney MR, Fang M, Ogurtsova K, Ozkan B, Echouffo-Tcheugui JB, Boyko EJ, et al. Global prevalence of prediabetes. Diabetes Care (2023) 46(7):1388–94. doi: 10.2337/dc22-2376
4. Ruijgrok C, Dekker JM, Beulens JW, Brouwer IA, Coupé VMH, Heymans MW, et al. Size and shape of the associations of glucose, HbA1c, insulin and HOMA-IR with incident type 2 diabetes: the Hoorn Study. Diabetologia (2018) 61(1):93–100. doi: 10.1007/s00125-017-4452-7
5. Welsh P, Preiss D, Lloyd SM, de Craen AJ, Jukema JW, Westendorp RG, et al. Contrasting associations of insulin resistance with diabetes, cardiovascular disease and all-cause mortality in the elderly: PROSPER long-term follow-up. Diabetologia (2014) 57(12):2513–20. doi: 10.1007/s00125-014-3383-9
6. DeFronzo RA. and Tripathy D Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care (2009) 32 Suppl 2(Suppl 2):S157–63. doi: 10.2337/dc09-S302
7. Parcha V, Heindl B, Kalra R, Li P, Gower B, Arora G, et al. Insulin resistance and cardiometabolic risk profile among nondiabetic american young adults: insights from NHANES. J Clin Endocrinol Metab (2021) 107(1):e25–37. doi: 10.1210/clinem/dgab645
8. Lillioja S, Mott DM, Howard BV, Bennett PH, Yki-Järvinen H, Freymond D, et al. Impaired glucose tolerance as a disorder of insulin action. Longitudinal and cross-sectional studies in Pima Indians. New Engl J Med (1988) 318(19):1217–25. doi: 10.1056/nejm198805123181901
9. Warram JH, Martin BC, Krolewski AS, Soeldner JS, Kahn CR. Slow glucose removal rate and hyperinsulinemia precede the development of type II diabetes in the offspring of diabetic parents. Ann Intern Med (1990) 113(12):909–15. doi: 10.7326/0003-4819-113-12-909
10. Esser N, Utzschneider KM, Kahn SE. Early beta cell dysfunction vs insulin hypersecretion as the primary event in the pathogenesis of dysglycaemia. Diabetologia (2020) 63(10):2007–21. doi: 10.1007/s00125-020-05245-x
11. Einarson TR, Acs A, Ludwig C, Panton UH. Prevalence of cardiovascular disease in type 2 diabetes: a systematic literature review of scientific evidence from across the world in 2007–2017. Cardiovasc Diabetol (2018) 17(1):83. doi: 10.1186/s12933-018-0728-6
12. Wu B, Bell K, Stanford A, Kern DM, Tunceli O, Vupputuri S, et al. Understanding CKD among patients with T2DM: prevalence, temporal trends, and treatment patterns—NHANES 2007–2012. BMJ Open Diabetes Res Care (2016) 4(1):e000154. doi: 10.1136/bmjdrc-2015-000154
13. Thomas RL, Halim S, Gurudas S, Sivaprasad S, Owens DR. IDF Diabetes Atlas: A review of studies utilising retinal photography on the global prevalence of diabetes related retinopathy between 2015 and 2018. Diabetes Res Clin Pract (2019) 157:107840. doi: 10.1016/j.diabres.2019.107840
14. Feldman EL, Callaghan BC, Pop-Busui R, Zochodne DW, Wright DE, Bennett DL, et al. Diabetic neuropathy. Nat Rev Dis Primers (2019) 5(1):41. doi: 10.1038/s41572-019-0092-1
15. Aikaeli F, Njim T, Gissing S, Moyo F, Alam U, Mfinanga SG, et al. Prevalence of microvascular and macrovascular complications of diabetes in newly diagnosed type 2 diabetes in low-and-middle-income countries: A systematic review and meta-analysis. PloS Global Public Health (2022) 2(6):e0000599. doi: 10.1371/journal.pgph.0000599
16. Mozaffary A, Asgari S, Tohidi M, Kazempour-Ardebili S, Azizi F. and Hadaegh F Change in fasting plasma glucose and incident type 2 diabetes mellitus: results from a prospective cohort study. BMJ Open (2016) 6(5):e010889. doi: 10.1136/bmjopen-2015-010889
17. Godsland IF, Jeffs JAR, Johnston DG. Loss of beta cell function as fasting glucose increases in the non-diabetic range. Diabetologia (2004) 47(7):1157–66. doi: 10.1007/s00125-004-1454-z
18. Diabetes-Control-Complications-Trial-Research-Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. New Engl J Med (1993), 327:977–986. doi: 10.1056/NEJM199309303291401
19. Stratton IM, Adler AI, Neil HAW, Matthews DR, Manley SE, Cull CA, et al. Association of glycaemic with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ (2002) 321(7258):405–12. doi: 10.1136/bmj.321.7258.405
20. ElSayed NA, Aleppo G, Aroda VR, Bannuru RR, Brown FM, Bruemmer D, et al. 9. Pharmacologic approaches to glycemic treatment: standards of care in diabetes—2023. Diabetes Care (2022) 46(Supplement 1):S140–57. doi: 10.2337/dc23-S009
21. ElSayed NA, Aleppo G, Aroda VR, Bannuru RR, Brown FM, Bruemmer D, et al. 10. Cardiovascular disease and risk management: standards of care in diabetes—2023. Diabetes Care (2022) 46(Supplement 1):S158–90. doi: 10.2337/dc23-S010
22. ElSayed NA, Aleppo G, Aroda VR, Bannuru RR, Brown FM, Bruemmer D, et al. 11. Chronic kidney disease and risk management: standards of care in diabetes—2023. Diabetes Care (2022) 46(Supplement 1):S191–202. doi: 10.2337/dc23-S011
23. ElSayed NA, Aleppo G, Aroda VR, Bannuru RR, Brown FM, Bruemmer D, et al. 12. Retinopathy, neuropathy, and foot care: standards of care in diabetes—2023. Diabetes Care (2022) 46(Supplement 1):S203–15. doi: 10.2337/dc23-S012
25. Irshad Z, Xue M, Ashour A, Larkin JR, Thornalley PJ, Rabbani N. Activation of the unfolded protein response in high glucose treated endothelial cells is mediated by methylglyoxal. Sci Rep (2019) 9:7889. doi: 10.1155/2018/5642148
26. Matschinsky FM, Magnuson MA, Zelent D, Jetton TL, Doliba N, Han Y, et al. The network of glucokinase-expressing cells in glucose homeostasis and the potential of glucokinase activators for diabetes therapy. Diabetes (2006) 55(1):1–12. doi: 10.2337/diabetes.55.01.06.db05-0926
27. Roosimaa M, Põdramägi T, Kadaja L, Ruusalepp A, Paju K, Puhke R, et al. Dilation of human atria: increased diffusion restrictions for ADP, overexpression of hexokinase 2 and its coupling to oxidative phosphorylation in cardiomyocytes. Mitochondrion (2013) 13(5):399–409. doi: 10.1016/j.mito.2012.12.005
28. Mandarino LJ, Printz RL, Cusi KA, Kinchington P, O’Doherty RM, Osawa H, et al. Regulation of hexokinase II and glycogen synthase mRNA, protein, and activity in human muscle. Amer J Physiol - Endocrinol Metab (1995) 269(4):E701–8. doi: 10.1152/ajpendo.1995.269.4.E701
29. Lowes W, Walker M, Alberti KG, Agius L. Hexokinase isoenzymes in normal and cirrhotic human liver: suppression of glucokinase in cirrhosis. Biochim Biophys Acta (1998) 1379(1):134–42. doi: 10.1016/s0304-4165(97)00092-5
30. Heimberg H, De Vos A, Moens K, Quartier E, Bouwens L, Pipeleers D, et al. The glucose sensor protein glucokinase is expressed in glucagon-producing alpha-cells. Proc Natl Acad Sci USA (1996) 93(14):7036–41. doi: 10.1073/pnas.93.14.7036
31. Liu X, Kim CS, Kurbanov FT, Honzatko RB, Fromm HJ. Dual mechanisms for glucose 6-phosphate inhibition of human brain hexokinase. J Biol Chem (1999) 274(44):31155–9. doi: 10.1074/jbc.274.44.31155
32. John S, Weiss JN, Ribalet B. Subcellular localization of hexokinases I and II directs the metabolic fate of glucose. PloS One (2011) 6(3):e17674. doi: 10.1371/journal.pone.0017674
33. Greiner JV. and glonek T intracellular ATP concentration and implication for cellular evolution. Biol (Basel) (2021) 10(11):1166. doi: 10.3390/biology10111166
34. Standl E, Kolb HJ. 2,3-Diphosphoglycerate fluctuations in erythrocytes reflecting pronounced blood glucose variation. In-vivo and in-vitro studies in normal, diabetic and hypoglycaemic subjects. Diabetologia (1973) 9(6):461–6. doi: 10.1007/bf00461689
35. Gabbay KH. The sorbitol pathway and the complications of diabetes. New Engl J Med (1973) 288(16):831–6. doi: 10.1056/nejm197304192881609
36. Nishimura C, Furue M, Ito T, Ohmori Y, Tanimoto T. Quantitative determination of human aldose reductase by enzyme-linked immunosorbent assay. Biochem Pharmacol (1993) 46(1):21–8. doi: 10.1016/0006-2952(93)90343-u
37. Grunewald RW, Weber II, Kinne RKH. Renal inner medullary sorbitol metabolism. Am J Physiol - Renal Fluid Electrolyte Physiol (1995) 269(5):F696–701. doi: 10.1152/ajprenal.1995.269.5.F696
38. Moriyama T, Garcia-Perez A. and Burg MB Osmotic regulation of aldose reductase protein synthesis in renal medullary cells. J Biol Chem (1989) 264(28):16810–4. doi: 10.1016/S0021-9258(19)84778-6
39. Tanner LB, Goglia AG, Wei MH, Sehgal T, Parsons LR, Park JO, et al. Four key steps control glycolytic flux in mammalian cells. Cell Syst (2018) 7(1):49–62.e8. doi: 10.1016/j.cels.2018.06.003
40. Pedersen O, Bak JF, Andersen PH, Lund S, Moller DE, Flier JS, et al. Evidence against altered expression of GLUT1 or GLUT4 in skeletal muscle of patients with obesity or NIDDM. Diabetes (1990) 39(7):865–70. doi: 10.2337/diab.39.7.865
41. Ritov VB. and kelley DE hexokinase isozyme distribution in human skeletal muscle. Diabetes (2001) 50(6):1253–62. doi: 10.2337/diabetes.50.6.1253
42. Vogt C, Ardehali H, Iozzo P, Yki-Järvinen H, Koval J, Maezono K, et al. Regulation of hexokinase II expression in human skeletal muscle in vivo. Metab - Clin Exp (2000) 49(6):814–8. doi: 10.1053/meta.2000.6245
43. Viijlar-Palasí C. and Guinovart JJ The role of glucose 6-phosphate in the control of glycogen synthase. FASEB J (1997) 11(7):544–58. doi: 10.1096/fasebj.11.7.9212078
44. Hue L, Blackmore PF, Shikama H, Robinson-Steiner A, Exton JH. Regulation of fructose-2,6-bisphosphate content in rat hepatocytes, perfused hearts, and perfused hindlimbs. J Biol Chem (1982) 257(8):4308–13. doi: 10.1016/S0021-9258(18)34722-7
45. Nasrin N, Ercolani L, Denaro M, Kong XF, Kang I, Alexander M. An insulin response element in the glyceraldehyde-3-phosphate dehydrogenase gene binds a nuclear protein induced by insulin in cultured cells and by nutritional manipulations in vivo. Proc Natl Acad Sci USA (1990) 87(14):5273–7. doi: 10.1073/pnas.87.14.5273
46. Sans CL, Satterwhite DJ, Stoltzman CA, Breen KT, Ayer DE. MondoA-mlx heterodimers are candidate sensors of cellular energy status: mitochondrial localization and direct regulation of glycolysis. Molec Cell Biol (2006) 26(13):4863–71. doi: 10.1128/mcb.00657-05
47. Jeong YS, Kim D, Lee YS, Kim HJ, Han JY, Im SS, et al. Integrated expression profiling and genome-wide analysis of chREBP targets reveals the dual role for chREBP in glucose-regulated gene expression. PloS One (2011) 6(7):e22544. doi: 10.1371/journal.pone.0022544
48. Ma L, Robinson LN, Towle HC. ChREBP•Mlx is the principal mediator of glucose-induced gene expression in the liver. J Biol Chem (2006) 281(39):28721–30. doi: 10.1074/jbc.M601576200
49. Arden C, Tudhope SJ, Petrie JL, Al-Oanzi ZH, Cullen KS, Lange AJ, et al. Fructose 2,6-bisphosphate is essential for glucose-regulated gene transcription of glucose-6-phosphatase and other ChREBP target genes in hepatocytes. Biochem J (2012) 443(1):111–23. doi: 10.1042/BJ20111280
50. Noguchi R, Kubota H, Yugi K, Toyoshima Y, Komori Y, Soga T, et al. The selective control of glycolysis, gluconeogenesis and glycogenesis by temporal insulin patterns. Mol Syst Biol (2013) 9(1):664. doi: 10.1038/msb.2013.19
51. Matschinsky FM, Wilson DF. The central role of glucokinase in glucose homeostasis: A perspective 50 years after demonstrating the presence of the enzyme in islets of langerhans. Front Physiol (2019) 10148:148. doi: 10.3389/fphys.2019.00148
52. Giardino I, Edelstein D, Brownlee M. Nonenzymatic glycosylation in-vitro and in bovine endothelial cells alters basic fibroblast growth-factor activity - a model for intracellular glycosylation in diabetes. J Clin Invest (1994) 94(1):110–7. doi: 10.1172/JCI117296
53. Quijano C, Castro L, Peluffo G, Valez V, Radi R. Enhanced mitochondrial superoxide in hyperglycemic endothelial cells: direct measurements and formation of hydrogen peroxide and peroxynitrite. Amer J Physiol - Heart Circ Physiol (2007) 293(6):H3404–14. doi: 10.1152/ajpheart.00761.2007
54. Mandarino LJ, Finlayson J, Hassell JR. High glucose downregulates glucose transport activity in retinal capillary pericytes but not endothelial cells. Invest Ophthalmol Vis Sci (1994) 35(3):964–72.
55. Beemer FA, Vlug AMC, Rijksen G, Hamburg A, Staal GEJ. Characterization of some glycolytic enzymes from human retina and retinoblastoma. Cancer Res (1982) 42(10):4228–32.
56. Chibber R, Molinatti GM, Wong JSK, Mirrlees D, Kohner EM. The effect of aminoguanidine and Tolrestat on glucose toxicity in bovine retinal capillary pericytes. Diabetes (1994) 42:758–763. doi: 10.2337/diab.43.6.758
57. Wakisaka M, Yoshinari M, Yamamoto M, Nakamura S, Asano T, Himeno T, et al. Na+-dependent glucose uptake and collagen synthesis by cultured bovine retinal pericytes. Biochim Biophys Acta (1997) 1362(1):87–96. doi: 10.1016/S0925-4439(97)00071-9
58. Heilig CW, Concepcion LA, Riser BL, Freytag SO, Zhu M, Cortes P. Overexpression of glucose transporters in rat mesangial cells cultured in a normal glucose milieu mimics the diabetic phenotype. J Clin Invest (1995) 96:1802–1814. doi: 10.1172/JCI118226
59. Wakisaka M, He Q, Spiro MJ, Spiro RG. Glucose entry into rat mesangial cells is mediated by both Na+-coupled and facilitative transporters. Diabetologia (1995) 38(3):291–7. doi: 10.1007/BF00400633
60. Robey RB, Ma J, Santos AVP, Noboa OA, Coy PE, Bryson JM. Regulation of mesangial cell hexokinase activity and expression by heparin-binding epidermal growth factor-like growth factor. J Biol Chem (2002) 277(17):14370–8. doi: 10.1074/jbc.M111722200
61. Asano T, Wakisaka M, Yoshinari M, Nakamura S, Doi Y, Fujishima M. Troglitazone enhances glycolysis and improves intracellular glucose metabolism in rat mesangial cells. Metabolism (2000) 49(3):308–13. doi: 10.1016/S0026-0495(00)90088-X
62. Coward RJM, Welsh GI, Yang J, Tasman C, Lennon R, Koziell A, et al. The human glomerular podocyte is a novel target for insulin action. Diabetes (2005) 54(11):3095–102. doi: 10.2337/diabetes.54.11.3095
63. Srivastava T, Joshi T, Jiang Y, Heruth DP, Rezaiekhaligh MH, Novak J, et al. Upregulated proteoglycan-related signaling pathways in fluid flow shear stress-treated podocytes. Am J Physiol Renal Physiol (2020) 319(2):F312–f322. doi: 10.1152/ajprenal.00183.2020
64. Li J, Sun YBY, Chen W, Fan J, Li S, Qu X, et al. Smad4 promotes diabetic nephropathy by modulating glycolysis and OXPHOS. EMBO Rep (2020) 21(2):e48781. doi: 10.15252/embr.201948781
65. Kaul K, Chopra M, De Angelis P, Kohner EM, Chibber R. The novel link between inflammatory enzyme C2GNT and the shedding of syndecan-1 in podocyte dysfunction. Endocrinol Stud (2012) 2(2):e9. doi: 10.4081/es.2012.e9
66. Szablewski L. Distribution of glucose transporters in renal diseases. J BioMed Sci (2017) 24(1):64. doi: 10.1186/s12929-017-0371-7
67. Riwanto M, Kapoor S, Rodriguez D, Edenhofer I, Segerer S, Wüthrich RP. Inhibition of aerobic glycolysis attenuates disease progression in polycystic kidney disease. PloS One (2016) 11(1):e0146654. doi: 10.1371/journal.pone.0146654
68. Eleftheriadis T, Pissas G, Tsogka K, Nikolaou E, Liakopoulos V, Stefanidis I. A unifying model of glucotoxicity in human renal proximal tubular epithelial cells and the effect of the SGLT2 inhibitor dapagliflozin. Internat Urol Nephrol (2020) 52(6):1179–89. doi: 10.1007/s11255-020-02481-3
69. Magnani P, Cherian PV, Gould GW, Greene DA, Sima AAF, Brosius FC 3rd. Glucose transporters in rat peripheral nerve: paradonal expression of GLUT1 and GLUT3. Metabolism (1996) 45(12):1466–73. doi: 10.1016/s0026-0495(96)90174-2
70. Gardiner NJ, Wang Z, Luke C, Gott A, Price SA, Fernyhough P. Expression of hexokinase isoforms in the dorsal root ganglion of the adult rat and effect of experimental diabetes. Brain Res (2007) Supplement C):143–54:1175. doi: 10.1016/j.brainres.2007.08.015
71. Kim ES, Isoda F, Kurland I, Mobbs CV. Glucose-induced metabolic memory in schwann cells: prevention by PPAR agonists. Endocrinology (2013) 154(9):3054–66. doi: 10.1210/en.2013-1097
72. Obrosova IG, Pacher P, Szabo C, Zsengeller Z, Hirooka H, Stevens MJ, et al. Aldose reductase inhibition counteracts oxidative-nitrosative stress and poly(ADP-ribose) polymerase activation in tissue sites for diabetes complications. Diabetes (2005) 54(1):234–42. doi: 10.2337/diabetes.54.1.234
73. Dresner A, Laurent D, Marcucci M, Griffin ME, Dufour S, Cline GW, et al. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J Clin Invest (1999) 103(2):253–9. doi: 10.1172/jci5001
74. Roussel R, Carlier PG, Robert JJ, Velho G, Bloch G. 13C/31P NMR studies of glucose transport in human skeletal muscle. Proc Natl Acad Sci USA (1998) 95(3):1313–8. doi: 10.1073/pnas.95.3.1313
75. DeFronzo RA, Gunnarsson R, Björkman O, Olsson M, Wahren J. Effects of insulin on peripheral and splanchnic glucose metabolism in noninsulin-dependent (type II) diabetes mellitus. J Clin Invest (1985) 76(1):149–55. doi: 10.1172/jci111938
76. Romero Mdel M, Sabater D, Fernández-López JA, Remesar X, Alemany M. Glycerol production from glucose and fructose by 3T3-L1 cells: A mechanism of adipocyte defense from excess substrate. PloS One (2015) 10(10):e0139502. doi: 10.1371/journal.pone.0139502
77. Kovacic PB, Chowdhury HH, Velebit J, Kreft M, Jensen J, Zorec R. New Insights into Cytosolic Glucose Levels during Differentiation of 3T3-L1 Fibroblasts into Adipocytes. J Biol Chem (2011) 286(15):13370–81. doi: 10.1074/jbc.M110.200980
78. Hacker HJ, Thorens B, Grobholz R. Expression of facilitative glucose transporter in rat liver and choroid plexus. Histochemistry (1991) 96(5):435–9. doi: 10.1007/BF00316001
79. Karim S, Adams DH, Lalor PF. Hepatic expression and cellular distribution of the glucose transporter family. World J Gastroenterol (2012) 18(46):6771–81. doi: 10.3748/wjg.v18.i46.6771
80. Mayer D, Klimek F, Rempel A, Bannasch P. Hexokinase expression in liver preneoplasia and neoplasia. Biochem Soc Trans (1997) 25(1):122–7. doi: 10.1042/bst0250122
81. Niewoehner CB, Nuttall FQ. Relationship of hepatic glucose uptake to intrahepatic glucose concentration in fasted rats after glucose load. Diabetes (1988) 37(11):1559–66. doi: 10.2337/diab.37.11.1559
82. Kemas AM, Youhanna S, Zandi Shafagh R, Lauschke VM. Insulin-dependent glucose consumption dynamics in 3D primary human liver cultures measured by a sensitive and specific glucose sensor with nanoliter input volume. FASEB J (2021) 35(3):e21305. doi: 10.1096/fj.202001989RR
83. Nicod N, Giusti V, Besse C, Tappy L. Metabolic adaptations to dexamethasone-induced insulin resistance in healthy volunteers. Obes Res (2003) 11(5):625–31. doi: 10.1038/oby.2003.90
84. Ishihara H, Asano T, Tsukuda K, Katagiri H, Inukai K, Anai M, et al. Pancreatic beta cell line MIN6 exhibits characteristics of glucose metabolism and glucose-stimulated insulin secretion similar to those of normal islets. Diabetologia (1993) 36(11):1139–45. doi: 10.1007/BF00401058
85. Kaminski MT, Lenzen S, Baltrusch S. Real-time analysis of intracellular glucose and calcium in pancreatic beta cells by fluorescence microscopy. Biochim Biophys Acta (2012) 1823(10):1697–707. doi: 10.1016/j.bbamcr.2012.06.022
86. Fels J, Jeggle P, Kusche-Vihrog K, Oberleithner H. Cortical actin nanodynamics determines nitric oxide release in vascular endothelium. PloS One (2012) 7(7):e41520. doi: 10.1371/journal.pone.0041520
87. Sugihara K, Sasaki S, Uemura A, Kidoaki S, Miura T. Mechanisms of endothelial cell coverage by pericytes: computational modelling of cell wrapping and in vitro experiments. J R Soc Interface (2020) 17(162):20190739. doi: 10.1098/rsif.2019.0739
88. Ling H, Ardjomand P, Samvakas S, Simm A, Busch GL, Lang F, et al. Mesangial cell hypertrophy induced by NH4Cl: Role of depressed activities of cathepsins due to elevated lysosomal pH. Kidney Internat (1998) 53(6):1706–12. doi: 10.1046/j.1523-1755.1998.00952.x
89. Koepsell H. Glucose transporters in brain in health and disease. Pflugers Arch (2020) 472(9):1299–343. doi: 10.1007/s00424-020-02441-x
90. Baron AD, Kolterman OG, Bell J, Mandarino LJ, Olefsky JM. Rates of noninsulin-mediated glucose uptake are elevated in type II diabetic subjects. J Clin Invest (1985) 76(5):1782–8. doi: 10.1172/JCI112169
91. Pendergrass M, Koval J, Vogt C, Yki-Jarvinen H, Iozzo P, Pipek R, et al. Insulin-induced hexokinase II expression is reduced in obesity and NIDDM. Diabetes (1998) 47(3):387–94. doi: 10.2337/diabetes.47.3.387
92. Vestergaard H, Bjørbaek C, Hansen T, Larsen FS, Granner DK, Pedersen O. Impaired activity and gene expression of hexokinase II in muscle from non-insulin-dependent diabetes mellitus patients. J Clin Invest (1995) 96(6):2639–45. doi: 10.1172/JCI118329
93. Braithwaite SS, Palazuk B, Colca JR, Edwards CW III, Hofmann C. Reduced expression of hexokinase II in insulin-resistant diabetes. Diabetes (1995) 44(1):43–8. doi: 10.2337/diab.44.1.43
94. Cusi KJ, Pratipanawatr T, Koval J, Printz R, Ardehali H, Granner DK, et al. Exercise increases hexokinase II mRNA, but not activity in obesity and type 2 diabetes. Metabolism (2001) 50(5):602–6. doi: 10.1053/meta.2001.22568
95. Sabater D, Arriarán S, del Mar Romero M, Agnelli S, Remesar X, Fernández-López JA, et al. Cultured 3T3L1 adipocytes dispose of excess medium glucose as lactate under abundant oxygen availability. Sci Rep (2014) 4(1):3663. doi: 10.1038/srep03663
96. Bock G, Chittilapilly E, Basu R, Toffolo G, Cobelli C, Chandramouli V, et al. Contribution of hepatic and extrahepatic insulin resistance to the pathogenesis of impaired fasting glucose. Diabetes (2007) 56(6):1703–11. doi: 10.2337/db06-1776
97. Lynedjian PB, Marie S, Gjinovci A, Genin B, Deng SP, Buhler L, et al. Glucokinase and cytosolic phosphoenolpyruvate carboxykinase (GTP) in the human liver. Regulation of gene expression in cultured hepatocytes. J Clin Invest (1995) 95(5):1966–73. doi: 10.1172/jci117880
98. Agius L, Peak M, Newgard CB, Gomez-Foix AM, Guinovart JJ. Evidence for a role of glucose-induced translocation of glucokinase in the control of hepatic glycogen synthesis. J Biol Chem (1996) 271(48):30479–86. doi: 10.1074/jbc.271.48.30479
99. de la Iglesia N, Veiga-da-Cunha M, Van Schaftingen E, Guinovart JJ, Ferrer JC. Glucokinase regulatory protein is essential for the proper subcellular localisation of liver glucokinase. FEBS Lett (1999) 456(2):332–8. doi: 10.1016/s0014-5793(99)00971-0
100. Härndahl L, Schmoll D, Herling AW, Agius L. The role of glucose 6-phosphate in mediating the effects of glucokinase overexpression on hepatic glucose metabolism. FEBS J (2006) 273(2):336–46. doi: 10.1111/j.1742-4658.2005.05067.x
101. von Wilamowitz-Moellendorff A, Hunter RW, García-Rocha M, Kang L, López-Soldado I, Lantier L, et al. Glucose-6-phosphate–mediated activation of liver glycogen synthase plays a key role in hepatic glycogen synthesis. Diabetes (2013) 62(12):4070–82. doi: 10.2337/db13-0880
102. Fernández-Novell JM, Roca A, Bellido D, Vilaró S, Guinovart JJ. Translocation and aggregation of hepatic glycogen synthase during the fasted-to-refed transition in rats. Eur J Biochem (1996) 238(2):570–5. doi: 10.1111/j.1432-1033.1996.0570z.x
103. Agius L. Glucokinase and molecular aspects of liver glycogen metabolism. Biochem J (2008) 414(1):1–18. doi: 10.1042/bj20080595
104. Nozaki Y, Petersen MC, Zhang D, Vatner DF, Perry RJ, Abulizi A, et al. Metabolic control analysis of hepatic glycogen synthesis in vivo. Proc Natl Acad Sci U.S.A. (2020) 117(14):8166–76. doi: 10.1073/pnas.1921694117
105. Arden C, Petrie JL, Tudhope SJ, Al-Oanzi Z, Claydon AJ, Beynon RJ, et al. Elevated glucose represses liver glucokinase and induces its regulatory protein to safeguard hepatic phosphate homeostasis. Diabetes (2011) 60(12):3110–20. doi: 10.2337/db11-0061
106. Diaz-Moralli S, Ramos-Montoya A, Marin S, Fernandez-Alvarez A, Casado M, Cascante M. Target metabolomics revealed complementary roles of hexose- and pentose-phosphates in the regulation of carbohydrate-dependent gene expression. Amer J Physiol - Endocrinol Metab (2012) 303(2):E234–42. doi: 10.1152/ajpendo.00675.2011
107. Halimi S, Assimacopoulos-Jeannet F, Terrettaz J, Jeanrenaud B. Differential effect of steady-state hyperinsulinaemia and hyperglycaemia on hepatic glycogenolysis and glycolysis in rats. Diabetologia (1987) 30(4):268–72. doi: 10.1007/BF00270426
108. Van Schaftingen E. Short-term regulation of glucokinase. Diabetologia (1994) 37(2):S43–7. doi: 10.1007/BF00400825
109. Diggle CP, Shires M, Leitch D, Brooke D, Carr IM, Markham AF, et al. Ketohexokinase: expression and localization of the principal fructose-metabolizing enzyme. J Histochem Cytochem (2009) 57(8):763–74. doi: 10.1369/jhc.2009.953190
110. Berger C, Zdzieblo D. Glucose transporters in pancreatic islets. Pflugers Archiv (2020) 472(9):1249–72. doi: 10.1007/s00424-020-02383-4
111. De Vos A, Heimberg H, Quartier E, Huypens P, Bouwens L, Pipeleers D, et al. Human and rat beta cells differ in glucose transporter but not in glucokinase gene expression. J Clin Invest (1995) 96(5):2489–95. doi: 10.1172/jci118308
112. Ishihara H, Asano T, Tsukuda K, Katagiri H, Inukai K, Anai M, et al. Human GLUT-2 overexpression does not affect glucose-stimulated insulin secretion in MIN6 cells. Amer J Physiol - Endocrinol Metab (1995) 269(5):E897–902. doi: 10.1152/ajpendo.1995.269.5.E897
113. Haythorne E, Lloyd M, Walsby-Tickle J, Tarasov AI, Sandbrink J, Portillo I, et al. Altered glycolysis triggers impaired mitochondrial metabolism and mTORC1 activation in diabetic β-cells. Nat Commun (2022) 13(1):6754. doi: 10.1038/s41467-022-34095-x
114. Bosi E, Marchetti P, Rutter GA, Eizirik DL. Human alpha cell transcriptomic signatures of types 1 and 2 diabetes highlight disease-specific dysfunction pathways. iScience (2022) 25(10):105056. doi: 10.1016/j.isci.2022.105056
115. Filippello A, Di Mauro S, Scamporrino A, Malaguarnera R, Torrisi SA, Leggio GM, et al. High glucose exposure impairs L-cell differentiation in intestinal organoids: molecular mechanisms and clinical implications. Internat J Molec Sci (2021) 22(13):6660. doi: 10.3390/ijms22136660
116. Butt UJ, Steixner-Kumar AA, Depp C, Sun T, Hassouna I, Wüstefeld L, et al. Hippocampal neurons respond to brain activity with functional hypoxia. Mol Psychiatry (2021) 26(6):1790–807. doi: 10.1038/s41380-020-00988-w
117. De Backer I, Hussain SS, Bloom SR, Gardiner JV. Insights into the role of neuronal glucokinase. Amer J Physiol - Endocrinol Metab (2016) 311(1):E42–55. doi: 10.1152/ajpendo.00034.2016
118. Mantych GJ, James DE, Chung HD, Devaskar SU. Cellular localization and characterization of Glut 3 glucose transporter isoform in human brain. Endocrinology (1992) 131(3):1270–8. doi: 10.1210/endo.131.3.1505464
119. Gruetter R, Novotny EJ, Boulware SD, Rothman DL, Mason GF, Shulman GI, et al. Direct measurement of brain glucose concentrations in humans by 13C NMR spectroscopy. Proc Natl Acad Sci USA (1992) 89(3):1109–12. doi: 10.1073/pnas.89.3.1109
120. Szablewski L. Glucose transporters in healthy heart and in cardiac disease. Internat J Cardiol (2017) 230:70–75. doi: 10.1016/j.ijcard.2016.12.083
121. Mustroph J, Lücht CM, Wagemann O, Sowa T, Hammer KP, Sag CM, et al. Empagliflozin enhances human and murine cardiomyocyte glucose uptake by increased expression of GLUT1. Diabetologia (2019) 62(4):726–9. doi: 10.1007/s00125-019-4819-z
122. Rabbani N, Thornalley PJ. Hexokinase-2 glycolytic overload in diabetes and ischemia–reperfusion injury. Trends Endocrinol Metab (2019) 30(7):419–31. doi: 10.1016/j.tem.2019.04.011
123. Ashour A, Xue M, Al-Motawa M, Thornalley PJ, Rabbani N. Glycolytic overload-driven dysfunction of periodontal ligament fibroblasts in high glucose concentration, corrected by glyoxalase 1 inducer. BMJ Open Diabetes Res Care (2020) 8(2):e001458. doi: 10.1136/bmjdrc-2020-001458
124. Xia H-g, Najafov A, Geng J, Galan-Acosta L, Han X, Guo Y, et al. Degradation of HK2 by chaperone-mediated autophagy promotes metabolic catastrophe and cell death. J Cell Biol (2015) 210(5):705–16. doi: 10.1083/jcb.201503044
125. Viticchiè G, Agostini M, Lena AM, Mancini M, Zhou H, Zolla L, et al. p63 supports aerobic respiration through hexokinase II. Proc Natl Acad Sci USA (2015) 112(37):11577–82. doi: 10.1073/pnas.1508871112
126. Nishikawa T, Edelstein D, Liang Du X, Yamagishi S, Matsumura T, Kaneda Y, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemia damage. Nature (2000) 404:787–790. doi: 10.1038/35008121
127. Xue M, Qian Q, Adaikalakoteswari A, Rabbani N, Babaei-Jadidi R, Thornalley PJ. Activation of NF-E2-related factor-2 reverses biochemical dysfunction of endothelial cells induced by hyperglycemia linked to vascular disease. Diabetes (2008) 57(10):2809–17. doi: 10.2337/db06-1003
128. Rabbani N, Xue M, Thornalley PJ. Dicarbonyl stress, protein glycation and the unfolded protein response. Glycoconj J (2021) 38(3):331–34. doi: 10.1007/s10719-021-09980-0
129. Komolafe OA, Ofusori DA, Adewole OS, Ayoka AO, Bejide R. Histological and histochemical studies of the aorta and pulmonary trunk in STZ-induced diabetic wistar rats treated with momordica charantia. Internat J Morphol (2013) 31(2):716–23. doi: 10.4067/S0717-95022013000200060
130. Isoe T, Makino Y, Mizumoto K, Sakagami H, Fujita Y, Honjo J, et al. High glucose activates HIF-1-mediated signal transduction in glomerular mesangial cells through a carbohydrate response element binding protein. Kidney Internat (2010) 78(1):48–59. doi: 10.1038/ki.2010.99
131. Kang J, Dai X-S, Yu T-B, Wen B, Yang Z-W. Glycogen accumulation in renal tubules, a key morphological change in the diabetic rat kidney. Acta Diabetol (2005) 42(2):110–6. doi: 10.1007/s00592-005-0188-9
132. Geoffrion M, Du X, Irshad Z, Vanderhyden BC, Courville K, Sui G, et al. Differential effects of glyoxalase 1 overexpression on diabetic atherosclerosis and renal dysfunction in streptozotocin-treated, apolipoprotein E-deficient mice. Physiol Rep (2014) 2(6):e12043. doi: 10.14814/phy2.12043
133. Fantus IG, Goldberg HJ, Whiteside CI, Topic D. The hexosamine biosynthesis pathway. In: Cortes P, Mogensen CE, editors. The diabetic kidney. Totowa, NJ, USA: Humana Press (2006). p. 117–33.
134. Kiritoshi S, Nishikawa T, Sonoda K, Kukidome D, Senokuchi T, Matsuo T, et al. Reactive oxygen species from mitochondria Induce cyclooxygenase-2 gene expression in human mesangial cells: potential role in diabetic nephropathy. Diabetes (2003) 52(10):2570–7. doi: 10.2337/diabetes.52.10.2570
135. Shiba T, Inoguchi T, Sportsman JR, Heath WF, Bursell S, King GL. Correlation of diacylglycerol level and protein kinase C activity in rat retina to retinal circulation. Amer J Physiol (1993) 265(5):E783–93. doi: 10.1152/ajpendo.1993.265.5.E783
136. Suzuki S, Yokoyama A, Noro E, Aoki S, Shimizu K, Shimada H, et al. Expression and pathophysiological significance of carbohydrate response element binding protein (ChREBP) in the renal tubules of diabetic kidney. Endocrine J (2020) 67(3):335–45. doi: 10.1507/endocrj.EJ19-0133
137. Schellini SA, Gregorio EA, Spadella CT, MaChado JLM, Demoraessilva MA. Muller cells and diabetic-retinopathy. Braz J Med Biol Res (1995) 28(9):977–80.
138. Yao D, Taguchi T, Matsumura T, Pestell R, Edelstein D, Giardino I, et al. High glucose increases angiopoietin-2 transcription in microvascular endothelial cells through methylglyoxal modification of mSin3A. J Biol Chem (2007) 282(42):31038–45. doi: 10.1074/jbc.M704703200
139. Tien T, Zhang J, Muto T, Kim D, Sarthy VP, Roy S. High glucose induces mitochondrial dysfunction in retinal müller cells: implications for diabetic retinopathy. Invest Ophthalmol Vis Sci (2017) 58(7):2915–21. doi: 10.1167/iovs.16-21355
140. Semba RD, Huang H, Lutty GA, Van Eyk JE, Hart GW. The role of O-GlcNAc signaling in the pathogenesis of diabetic retinopathy. Proteomics - Clin App (2014) 8(3-4):218–31. doi: 10.1002/prca.201300076
141. Powell HC, Rosoff J, Myers RR. Microangiopathy in human diabetic neuropathy. Acta Neuropathol (1985) 68(4):295–305. doi: 10.1007/bf00690832
142. Bierhaus A, Fleming T, Stoyanov S, Leffler A, Babes A, Neacsu C, et al. Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nat Med (2012) 18(6):926–33. doi: 10.1038/nm.2750
143. Zhang L, Yu C, Vasquez FE, Galeva N, Onyango I, Swerdlow RH, et al. Hyperglycemia alters the schwann cell mitochondrial proteome and decreases coupled respiration in the absence of superoxide production. J Proteome Res (2010) 9(1):458–71. doi: 10.1021/pr900818g
144. Russell JW, Berent-Spillson A, Vincent AM, Freimann CL, Sullivan KA, Feldman EL. Oxidative injury and neuropathy in diabetes and impaired glucose tolerance. Neurobiol Dis (2008) 30(3):420–9. doi: 10.1016/j.nbd.2008.02.013
145. Duran-Jimenez B, Dobler D, Moffatt S, Rabbani N, Streuli CH, Thornalley PJ, et al. Advanced Glycation Endproducts in extracellular matrix proteins contribute to the failure of sensory nerve regeneration in diabetes. Diabetes (2009) 58:2893–2903. doi: 10.2337/db09-0320
146. Fueger PT, Bracy DP, Malabanan CM, Pencek RR, Granner DK, Wasserman DH. Hexokinase II overexpression improves exercise-stimulated but not insulin-stimulated muscle glucose uptake in high-fat-fed C57BL/6J mice. Diabetes (2004) 53(2):306–14. doi: 10.2337/diabetes.53.2.306
147. Heikkinen S, Pietilä M, Halmekytö M, Suppola S, Pirinen E, Deeb SS, et al. Hexokinase II-deficient mice: prenatal death of homozygotes without disturbances in glucose tolerance in heterozygotes. J Biol Chem (1999) 274(32):22517–23. doi: 10.1074/jbc.274.32.22517
148. Marshall BA, Ren JM, Johnson DW, Gibbs EM, Lillquist JS, Soeller WC, et al. Germline manipulation of glucose homeostasis via alteration of glucose transporter levels in skeletal muscle. J Biol Chem (1993) 268(25):18442–5. doi: 10.1016/S0021-9258(17)46645-2
149. Lefort N, Glancy B, Bowen B, Willis WT, Bailowitz Z, De Filippis EA, et al. Increased reactive oxygen species production and lower abundance of complex I subunits and carnitine palmitoyltransferase 1B protein despite normal mitochondrial respiration in insulin-resistant human skeletal muscle. Diabetes (2010) 59(10):2444–52. doi: 10.2337/db10-0174
150. Yki-Järvinen H, Daniels MC, Virkamäki A, Mäkimattila S, DeFronzo RA, McClain D. Increased glutamine : fructose-6-phosphate amidotransferase activity in skeletal muscle of patients with NIDDM. Diabetes (1996) 45(3):302–7. doi: 10.2337/diab.45.3.302
151. Mey JT, Blackburn BK, Miranda ER, Chaves AB, Briller J, Bonini MG, et al. Dicarbonyl stress and glyoxalase enzyme system regulation in human skeletal muscle. Amer J Physiol - Regulat Integrat Compar Physiol (2018) 314(2):R181–90. doi: 10.1152/ajpregu.00159.2017
152. Ahn B, Wan S, Jaiswal N, Vega RB, Ayer DE, Titchenell PM, et al. MondoA drives muscle lipid accumulation and insulin resistance. J CIin Invest Insight (2019) 5(15):e129119. doi: 10.1172/jci.insight.129119
153. Fazakerley DJ, Minard AY, Krycer JR, Thomas KC, Stöckli J, Harney DJ, et al. Mitochondrial oxidative stress causes insulin resistance without disrupting oxidative phosphorylation. J Biol Chem (2018) 293(19):7315–28. doi: 10.1074/jbc.RA117.001254
154. Brandon AE, Liao BM, Diakanastasis B, Parker BL, Raddatz K, McManus SA, et al. Protein kinase C epsilon deletion in adipose tissue, but not in liver, improves glucose tolerance. Cell Metab (2019) 29(1):183–191.e7. doi: 10.1016/j.cmet.2018.09.013
155. Masania J, Malczewska-Malec M, Razny U, Goralska J, Zdzienicka A, Kiec-Wilk B, et al. Dicarbonyl stress in clinical obesity. Glycoconj J (2016) 33:581–589. doi: 10.1007/s10719-016-9692-0
156. Herman MA, Peroni OD, Villoria J, Schon MR, Abumrad NA, Bluher M, et al. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature (2012) 484(7394):333–8. doi: 10.1038/nature10986
157. Lavis VR. Hexokinase isozymes of normal human subcutaneous adipose tissue. Metabolism (1978) 27(9):1101–8. doi: 10.1016/0026-0495(78)90156-7
158. Morton NM, Nelson YB, Michailidou Z, Di Rollo EM, Ramage L, Hadoke PWF, et al. A stratified transcriptomics analysis of polygenic fat and lean mouse adipose tissues identifies novel candidate obesity genes. PloS One (2011) 6(9):e23944–4. doi: 10.1371/journal.pone.0023944
159. Sugiyama Y, Shimura Y, Ikeda H. Pathogenesis of hyperglycemia in genetically obese-hyperglycemic rats, Wistar fatty: presence of hepatic insulin resistance. Endocrinol Jpn (1989) 36(1):65–73. doi: 10.1507/endocrj1954.36.65
160. Tripathy D, Merovci A, Basu R, Abdul-Ghani M, DeFronzo RA. Mild physiologic hyperglycemia induces hepatic insulin resistance in healthy normal glucose-tolerant participants. J Clin Endocrinol Metab (2019) 104(7):2842–50. doi: 10.1210/jc.2018-02304
161. Murao N, Yokoi N, Takahashi H, Hayami T, Minami Y, Seino S. Increased glycolysis affects β-cell function and identity in aging and diabetes. Mol Metab (2022) 55:101414. doi: 10.1016/j.molmet.2021.101414
162. Kaneto H, Xu G, Song KH, Suzuma K, Bonner-Weir S, Sharma A, et al. Activation of the hexosamine pathway leads to deterioration of pancreatic beta-cell function through the induction of oxidative stress. J Biol Chem (2001) 276(33):31099–104. doi: 10.1074/jbc.M104115200
163. Tym A, Jin T, Rossmeisl M, Kopecky J, Squires P, Rabbani N, et al. Increased pancreatic methylglyoxal in insulin resistance and beta cell detachment from the extracellular matrix. Diabetes (2014) 63(Suppl 1):A553–4.
164. Frank SK, Fromm HJ. Effect of streptozotocin-induced diabetes on the turnover of hexokinase II in the rat. Biochem Biophys Res Commun (1982) 106(2):379–84. doi: 10.1016/0006-291X(82)91120-2
165. Wolf A, Agnihotri S, Micallef J, Mukherjee J, Sabha N, Cairns R, et al. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J Exp Med (2011) 208(2):313–26. doi: 10.1084/jem.20101470
166. Artwohl M, Brunmair B, Fürnsinn C, Hölzenbein T, Rainer G, Freudenthaler A, et al. Insulin does not regulate glucose transport and metabolism in human endothelium. Eur J Clin Invest (2007) 37(8):643–50. doi: 10.1111/j.1365-2362.2007.01838.x
167. Osorio-Paz I, Sánchez-Chávez G, Salceda R. Control of glycogen content in retina: allosteric regulation of glycogen synthase. PloS One (2012) 7(2):e30822. doi: 10.1371/journal.pone.0030822
168. Sanchez-Chavez G, Hernandez-Berrones J, Luna-Ulloa LB, Coffe V, Salceda R. Effect of diabetes on glycogen metabolism in rat retina. Neurochem Res (2008) 33(7):1301–8. doi: 10.1007/s11064-007-9583-7
169. Sullivan MA, Forbes JM. Glucose and glycogen in the diabetic kidney: Heroes or villains? EBioMedicine (2019) 47:590–597. doi: 10.1016/j.ebiom.2019.07.067
170. Punsoni M, Drexler S, Palaia T, Stevenson M, Stecker MM. Acute anoxic changes in peripheral nerve: anatomic and physiologic correlations. Brain Behav (2015) 5(7):e00347. doi: 10.1002/brb3.347
171. Ritchie S, Waugh D. The pathology of Armanni-Ebstein diabetic nephropathy. Am J Pathol (1957) 33(6):1035–57.
172. Li X, Kover KL, Heruth DP, Watkins DJ, Moore WV, Jackson K, et al. New insight into metformin action: regulation of chREBP and FOXO1 activities in endothelial cells. Mol Endocrinol (2015) 29(8):1184–94. doi: 10.1210/me.2015-1090
173. Mohamed I, Hafez S, FaIraq A, Ergul A, Imig J, El-Remessy A. Thioredoxin-interacting protein is required for endothelial NLRP3 inflammasome activation and cell death in a rat model of high-fat diet. Diabetologia (2014) 57(2):413–23. doi: 10.1007/s00125-013-3101-z
174. Li L, Long J, Mise K, Poungavrin N, Lorenzi PL, Mahmud I, et al. The transcription factor ChREBP links mitochondrial lipidomes to mitochondrial morphology and progression of diabetic kidney disease. J Biol Chem (2023) 299(9):105185. doi: 10.1016/j.jbc.2023.105185
175. Park M-J, Kim D-I, Lim S-K, Choi J-H, Han H-J, Yoon K-C, et al. High glucose-induced O-GlcNacylated carbohydrate response element-binding protein (ChREBP) mediates mesangial cell lipogenesis and fibrosis. J Biol Chem (2014) 289(19):13519–30. doi: 10.1074/jbc.M113.530139
176. Wasserman DH, Kang L, Ayala JE, Fueger PT, Lee-Young RS. The physiological regulation of glucose flux into muscle in vivo. J Exp Biol (2011) 214(2):254–62. doi: 10.1242/jeb.048041
177. TeSlaa T, Bartman CR, Jankowski CSR, Zhang Z, Xu X, Xing X, et al. The source of glycolytic intermediates in mammalian tissues. Cell Metab (2021) 33(2):367–378.e5. doi: 10.1016/j.cmet.2020.12.020
178. DeFronzo RA, Hendler R, Simonson D. Insulin resistance is a prominent feature of insulin-dependent diabetes. Diabetes (1982) 31(9):795–801. doi: 10.2337/diab.31.9.795
179. van Gerwen J, Shun-Shion AS, Fazakerley DJ. Insulin signalling and GLUT4 trafficking in insulin resistance. Biochem Soc Trans (2023) 51:1057–1069. doi: 10.1042/bst20221066
180. Miele C, Formisano P, Condorelli G, Caruso M, Oriente F, Andreozzi F, et al. Abnormal glucose transport and GLUT1 cell-surface content in fibroblasts and skeletal muscle from NIDDM and obese subjects. Diabetologia (1997) 40(4):421–9. doi: 10.1007/s001250050696
181. Ciaraldi TP, Mudaliar S, Barzin A, Macievic JA, Edelman SV, Park KS, et al. Skeletal muscle GLUT1 transporter protein expression and basal leg glucose uptake are reduced in type 2 diabetes. J Clin Endocrinol Metab (2005) 90(1):352–8. doi: 10.1210/jc.2004-0516
182. Vogt C, Yki-Jarvinen H, Iozzo P, Pipek R, Pendergrass M, Koval J, et al. Effects of insulin on subcellular localization of hexokinase II in human skeletal muscle in vivo. J Clin Endocrinol Metab (1998) 83(1):230–4. doi: 10.1210/jcem.83.1.4476
183. Roberts DJ, Tan-Sah VP, Smith JM, Miyamoto S. Akt phosphorylates HK-II at Thr-473 and increases mitochondrial HK-II association to protect cardiomyocytes. J Biol Chem (2013) 288(33):23798–806. doi: 10.1074/jbc.M113.482026
184. Galton DJ, Wilson JPD. The effect of starvation and diabetes on glycolytic enzymes in human adipose tissue. Clin Sci (1971) 41(6):545–53. doi: 10.1042/cs0410545
185. Ducluzeau P-H, Perretti N, Laville M, Andreelli F, Vega N, Riou J-P, et al. Regulation by insulin of gene expression in human skeletal muscle and adipose tissue: evidence for specific defects in type 2 diabetes. Diabetes (2001) 50(5):1134–42. doi: 10.2337/diabetes.50.5.1134
186. Rabbani N, Xue M, Thornalley PJ. Hexokinase-2-linked glycolytic overload and unscheduled glycolysis - driver of insulin resistance and development of vascular complications of diabetes. Internat J Molec Sci (2022) 23(4):2165. doi: 10.3390/ijms23042165
187. Ren JM, Marshall BA, Gulve EA, Gao J, Johnson DW, Holloszy JO, et al. Evidence from transgenic mice that glucose transport is rate-limiting for glycogen deposition and glycolysis in skeletal muscle. J Biol Chem (1993) 268(22):16113–5. doi: 10.1016/S0021-9258(19)85395-4
188. Buse MG, Robinson KA, Marshall BA, Hresko RC, Mueckler MM. Enhanced O-GlcNAc protein modification is associated with insulin resistance in GLUT1-overexpressing muscles. Am J Physiol Endocrinol Metab (2002) 283(2):E241–50. doi: 10.1152/ajpendo.00060.2002
189. Ren JM, Marshall BA, Mueckler MM, McCaleb M, Amatruda JM, Shulman GI. Overexpression of Glut4 protein in muscle increases basal and insulin-stimulated whole body glucose disposal in conscious mice. J Clin Invest (1995) 95(1):429–32. doi: 10.1172/JCI117673
190. Hansen PA, Marshall BA, Chen M, Holloszy JO, Mueckler M. Transgenic overexpression of hexokinase II in skeletal muscle does not increase glucose disposal in wild-type or GLUT1-overexpressing mice. J Biol Chem (2000) 275(29):22381–6. doi: 10.1074/jbc.M001946200
191. Herman MA, Birnbaum MJ. Molecular aspects of fructose metabolism and metabolic disease. Cell Metab (2021) 33(12):2329–54. doi: 10.1016/j.cmet.2021.09.010
192. Erkin-Cakmak A, Bains Y, Caccavello R, Noworolski SM, Schwarz J-M, Mulligan K, et al. Isocaloric fructose restriction reduces serum d-lactate concentration in children with obesity and metabolic syndrome. J Clin Endocrinol Metab (2019) 104(7):3003–11. doi: 10.1210/jc.2018-02772
193. DeFronzo RA, Simonson D, Ferrannini E. Hepatic and peripheral insulin resistance: a common feature of type 2 (non-insulin-dependent) and type 1 (insulin-dependent) diabetes mellitus. Diabetologia (1982) 23(4):313–9. doi: 10.1007/bf00253736
194. Smith GI, Shankaran M, Yoshino M, Schweitzer GG, Chondronikola M, Beals JW, et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J Clin Invest (2020) 130(3):1453–60. doi: 10.1172/jci134165
195. Li SY, Liu Y, Sigmon VK, McCort A, Ren J. High-fat diet enhances visceral advanced glycation end products, nuclear O-Glc-Nac modification, p38 mitogen-activated protein kinase activation and apoptosis. Diabetes Obes Metab (2005) 7(4):448–54. doi: 10.1111/j.1463-1326.2004.00387.x
196. Wang D, Wei Y, Schmoll D, Maclean KN, Pagliassotti MJ. Endoplasmic reticulum stress increases glucose-6-phosphatase and glucose cycling in liver cells. Endocrinology (2006) 147(1):350–8. doi: 10.1210/en.2005-1014
197. Agius L. High-carbohydrate diets induce hepatic insulin resistance to protect the liver from substrate overload. Biochem Pharmacol (2013) 85(3):306–12. doi: 10.1016/j.bcp.2012.09.019
198. Agius L. Dietary carbohydrate and control of hepatic gene expression: mechanistic links from ATP and phosphate ester homeostasis to the carbohydrate-response element-binding protein. Proc Nutrit. Soc (2016) 75(1):10–8. doi: 10.1017/S0029665115002451
199. Benhamed F, Denechaud PD, Lemoine M, Robichon C, Moldes M, Bertrand-Michel J, et al. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J Clin Invest (2012) 122(6):2176–94. doi: 10.1172/jci41636
200. Dentin R, Pégorier J-P, Benhamed F, Foufelle F, Ferré P, Fauveau V, et al. Hepatic glucokinase is required for the synergistic action of ChREBP and SREBP-1c on glycolytic and lipogenic gene expression. J Biol Chem (2004) 279(19):20314–26. doi: 10.1074/jbc.M312475200
201. Iizuka K, Bruick RK, Liang G, Horton JD, Uyeda K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci USA (2004) 101(19):7281–6. doi: 10.1073/pnas.0401516101
202. Dentin R, Benhamed F, Hainault I, Fauveau V, Foufelle F, Dyck JRB, et al. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes (2006) 55(8):2159–70. doi: 10.2337/db06-0200
203. Iizuka K, Miller B, Uyeda K. Deficiency of carbohydrate-activated transcription factor ChREBP prevents obesity and improves plasma glucose control in leptin-deficient (ob/ob) mice. Amer J Physiol - Endocrinol Metab (2006) 291(2):E358–64. doi: 10.1152/ajpendo.00027.2006
204. Shi J-H, Lu J-Y, Chen H-Y, Wei C-C, Xu X, Li H, et al. Liver ChREBP protects against fructose-induced glycogenic hepatotoxicity by regulating L-type pyruvate kinase. Diabetes (2020) 69(4):591–602. doi: 10.2337/db19-0388
205. Abdul-Wahed A, Guilmeau S, Postic C. Sweet sixteenth for ChREBP: established roles and future goals. Cell Metab (2017) 26(2):324–41. doi: 10.1016/j.cmet.2017.07.004
206. Rossetti L, Giaccari A, DeFronzo RA. Glucose toxicity. Diabetes Care (1990) 13(6):610–30. doi: 10.2337/diacare.13.6.610
207. Danial NN, Gramm CF, Scorrano L, Zhang CY, Krauss S, Ranger AM, et al. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature (2003) 424(6951):952–6. doi: 10.1038/nature01825
208. Han J-H, Kim S, Kim S, Lee H, Park S-Y, Woo C-H. FMK, an Inhibitor of p90RSK, Inhibits High Glucose-Induced TXNIP Expression via Regulation of ChREBP in Pancreatic β Cells. Internat J Molec Sci (2019) 20(18):4424. doi: 10.3390/ijms20184424
209. Roma LP, Duprez J, Takahashi HK, Gilon P, Wiederkehr A, Jonas JC. Dynamic measurements of mitochondrial hydrogen peroxide concentration and glutathione redox state in rat pancreatic β-cells using ratiometric fluorescent proteins: confounding effects of pH with HyPer but not roGFP1. Biochem J (2012) 441(3):971–8. doi: 10.1042/bj20111770
210. Richards P, Rachdi L, Oshima M, Marchetti P, Bugliani M, Armanet M, et al. MondoA is an essential glucose-responsive transcription factor in human pancreatic β-cells. Diabetes (2018) 67(3):461–72. doi: 10.2337/db17-0595
211. Oslowski CM, Hara T, O’Sullivan-Murphy B, Kanekura K, Lu S, Hara M, et al. Thioredoxin-interacting protein mediates ER stress-induced β cell death through initiation of the inflammasome. Cell Metab (2012) 16(2):265–73. doi: 10.1016/j.cmet.2012.07.005
212. Beer NL, Tribble ND, McCulloch LJ, Roos C, Johnson PR, Orho-Melander M, et al. The P446L variant in GCKR associated with fasting plasma glucose and triglyceride levels exerts its effect through increased glucokinase activity in liver. Hum Mol Genet (2009) 18(21):4081–8. doi: 10.1093/hmg/ddp357
213. Ashcroft FM, Lloyd M, Haythorne EA. Glucokinase activity in diabetes: too much of a good thing? Trends Endocrinol Metab (2023) 34(2):119–30. doi: 10.1016/j.tem.2022.12.007
214. Ebrahimi AG, Hollister-Lock J, Sullivan BA, Tsuchida R, Bonner-Weir S, Weir GC. Beta cell identity changes with mild hyperglycemia: Implications for function, growth, and vulnerability. Molec Metab (2020) 35:100959. doi: 10.1016/j.molmet.2020.02.002
215. Talchai C, Xuan S, Lin Hua V, Sussel L, Accili D. Pancreatic beta-Cell Dedifferentiation as a Mechanism of Diabetic beta-Cell Failure. Cell (2012) 150(6):1223–34. doi: 10.1016/j.cell.2012.07.029
216. Back SH, Scheuner D, Han J, Song B, Ribick M, Wang J, et al. Translation attenuation through eIF2alpha phosphorylation prevents oxidative stress and maintains the differentiated state in beta cells. Cell Metab (2009) 10(1):13–26. doi: 10.1016/j.cmet.2009.06.002
217. Zhang W, Feng D, Li Y, Iida K, McGrath B, Cavener DR. PERK EIF2AK3 control of pancreatic beta cell differentiation and proliferation is required for postnatal glucose homeostasis. Cell Metab (2006) 4(6):491–7. doi: 10.1016/j.cmet.2006.11.002
218. Nordmann TM, Dror E, Schulze F, Traub S, Berishvili E, Barbieux C, et al. The role of inflammation in β-cell dedifferentiation. Sci Rep (2017) 7(1):6285. doi: 10.1038/s41598-017-06731-w
219. Guo S, Dai C, Guo M, Taylor B, Harmon JS, Sander M, et al. Inactivation of specific β cell transcription factors in type 2 diabetes. J Clin Invest (2013) 123(8):3305–16. doi: 10.1172/JCI65390
220. Abdul-Ghani MA, Matsuda M, Jani R, Jenkinson CP, Coletta DK, Kaku K, et al. The relationship between fasting hyperglycemia and insulin secretion in subjects with normal or impaired glucose tolerance. Am J Physiol Endocrinol Metab (2008) 295(2):E401–6. doi: 10.1152/ajpendo.00674.2007
221. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature (2001) 414(6865):813–20. doi: 10.1038/414813a
222. Kim J-a, Wei Y. and sowers JR role of mitochondrial dysfunction in insulin resistance. Circ Res (2008) 102(4):401–14. doi: 10.1161/CIRCRESAHA.107.165472
223. Hink U, Li H, Mollnau H, Oelze M, Matheis E, Hartmann M, et al. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ Res (2001) 88:14–22. doi: 10.1161/01.res.88.2.e14
224. Guzik TJ, Mussa S, Gastaldi D, Sadowski J, Ratnatunga C, Pillai R, et al. Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation (2002) 105(14):1656–62. doi: 10.1161/01.cir.0000012748.58444.08
225. González-Muniesa P, Milagro FI, Campión J, Martínez JA. Ectopic UCP1 gene expression in HepG2 cells affects ATP production. J Physiol Biochem (2005) 61(2):389–93. doi: 10.1007/BF03167056
226. Wang X, Zhang X, Wu D, Huang Z, Hou T, Jian C, et al. Mitochondrial flashes regulate ATP homeostasis in the heart. Elife (2017) 6:e23908. doi: 10.7554/eLife.23908
227. Lonn E, Yusuf S, Hoogwerf B, Pogue J, Yi QL, Zinman B, et al. Effects of vitamin E on cardiovascular and microvascular outcomes in high-risk patients with diabetes - Results of the HOPE study and MICRO-HOPE substudy. Diabetes Care (2002) 25(11):1919–27. doi: 10.2337/diacare.25.11.1919
228. Haritoglou C, Gerss J, Hammes HP, Kampik A, Ulbig MW. Alpha-lipoic acid for the prevention of diabetic macular edema. Ophthalmologica (2011) 226(3):127–37. doi: 10.1159/000329470
229. Han T, Bai J, Liu W, Hu Y. A systematic review and meta-analysis of α-lipoic acid in the treatment of diabetic peripheral neuropathy. Eur J Endocrinol (2012) 167(4):465–71. doi: 10.1530/eje-12-0555
230. Manning PJ, Sutherland WHF, Walker RJ, Williams SM, de Jong SA, Ryalls AR, et al. Effect of high-dose vitamin E on insulin resistance and associated parameters in overweight subjects. Diabetes Care (2004) 27(9):2166–71. doi: 10.2337/diacare.27.9.2166
231. Khodaeian M, Tabatabaei-Malazy O, Qorbani M, Farzadfar F, Amini P, Larijani B. Effect of vitamins C and E on insulin resistance in diabetes: a meta-analysis study. Eur J Clin Invest (2015) 45(11):1161–74. doi: 10.1111/eci.12534
232. Iori E, Millioni R, Puricelli L, Arrigoni G, Lenzini L, Trevisan R, et al. Glycolytic enzyme expression and pyruvate kinase activity in cultured fibroblasts from type 1 diabetic patients with and without nephropathy. Biochim Biophys Acta (2008) 1782(11):627–33. doi: 10.1016/j.bbadis.2008.08.012
233. Xia F, Sun J-J, Jiang Y-Q, Li C-F. MicroRNA-384-3p inhibits retinal neovascularization through targeting hexokinase 2 in mice with diabetic retinopathy. J Cell Physiol (2019) 234(1):721–30. doi: 10.1002/jcp.26871
234. Karachalias N, Babaei-Jadidi R, Rabbani N, Thornalley PJ. Increased protein damage in renal glomeruli, retina, nerve, plasma and urine and its prevention by thiamine and benfotiamine therapy in a rat model of diabetes. Diabetologia (2010) 53(7):1506–16. doi: 10.1007/s00125-010-1722-z
235. Kaul K, Apostolopoulou M, Roden M. Insulin resistance in type 1 diabetes mellitus. Metabolism (2015) 64(12):1629–39. doi: 10.1016/j.metabol.2015.09.002
236. Katsura T, Kawamori D, Aida E, Matsuoka TA, Shimomura I. Glucotoxicity induces abnormal glucagon secretion through impaired insulin signaling in InR1G cells. PloS One (2017) 12(4):e0176271. doi: 10.1371/journal.pone.0176271
237. Xue M, Weickert MO, Qureshi S, Ngianga-Bakwin K, Anwar A, Waldron M, et al. Improved glycemic control and vascular function in overweight and obese subjects by glyoxalase 1 inducer formulation. Diabetes (2016) 65(8):2282–94. doi: 10.2337/db16-0153
238. Rabbani N, Thornalley PJ. Glyoxalase 1 modulation in obesity and diabetes. Antioxidants Redox Signaling (2018)30:354–374. doi: 10.1089/ars.2017.7424
239. Rabbani N, Xue M, Thornalley PJ. Methylglyoxal-induced dicarbonyl stress in aging and disease: first steps towards glyoxalase 1-based treatments. Clin Sci (2016) 130:1677–1696. doi: 10.1042/CS20160025
240. Xue M, Rabbani N, Momiji H, Imbasi P, Anwar MM, Kitteringham NR, et al. Transcriptional control of glyoxalase 1 by Nrf2 provides a stress responsive defence against dicarbonyl glycation. Biochem J (2012) 443(1):213–22. doi: 10.1042/BJ20111648
241. Li H, O’Meara M, Zhang X, Zhang K, Seyoum B, Yi Z, et al. Ameliorating methylglyoxal-induced progenitor cell dysfunction for tissue repair in diabetes. Diabetes (2019) 68(6):1287–302. doi: 10.2337/db18-0933
242. Mari A, Pacini G, Murphy E, Ludvik B, Nolan JJ. A model-based method for assessing insulin sensitivity from the oral glucose tolerance test. Diabetes Care (2001) 24(3):539–48. doi: 10.2337/diacare.24.3.539
243. Kautzky-Willer A, Tura A, Winzer C, Wagner OF, Ludvik B, Hanusch-Enserer U, et al. Insulin sensitivity during oral glucose tolerance test and its relations to parameters of glucose metabolism and endothelial function in type 2 diabetic subjects under metformin and thiazolidinedione. Diabetes Obes Metab (2006) 8(5):561–7. doi: 10.1111/j.1463-1326.2005.00568.x
244. Hanusch-Enserer U, Cauza E, Spak M, Endler G, Dunky A, Tura A, et al. Improvement of insulin resistance and early atherosclerosis in patients after gastric banding. Obes Res (2004) 12(2):284–91. doi: 10.1038/oby.2004.36
245. Jo SH, Kim MY, Park JM, Kim TH, Ahn YH. Txnip contributes to impaired glucose tolerance by upregulating the expression of genes involved in hepatic gluconeogenesis in mice. Diabetologia (2013) 56(12):2723–32. doi: 10.1007/s00125-013-3050-6
246. Xu G, Chen J, Jing G, Shalev A. Thioredoxin-interacting protein regulates insulin transcription through microRNA-204. Nat Med (2013) 19(9):1141–6. doi: 10.1038/nm.3287
247. Parikh H, Carlsson E, Chutkow WA, Johansson LE, Storgaard H, Poulsen P, et al. TXNIP regulates peripheral glucose metabolism in humans. PloS Med (2007) 4(5):e158. doi: 10.1371/journal.pmed.0040158
248. Stoltzman CA, Peterson CW, Breen KT, Muoio DM, Billin AN, Ayer DE. Glucose sensing by MondoA : Mlx complexes: A role for hexokinases and direct regulation of thioredoxin-interacting protein expression. Proc Natl Acad Sci USA (2008) 105(19):6912–7. doi: 10.1073/pnas.0712199105
249. Lerner AG, Upton J-P, Praveen PVK, Ghosh R, Nakagawa Y, Igbaria A, et al. IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab (2012) 16(2):250–64. doi: 10.1016/j.cmet.2012.07.007
250. Hotamisligil GS, Murray DL, Choy LN, Spiegelman BM. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc Natl Acad Sci USA (1994) 91(11):4854–8. doi: 10.1073/pnas.91.11.4854
251. Miyazaki Y, Pipek R, Mandarino LJ, DeFronzo RA. Tumor necrosis factor-alpha and insulin resistance in obese type 2 diabetic patients. Int J Obes Relat Metab Disord (2003) 27(1):88–94. doi: 10.1038/sj.ijo.0802187
252. Plomgaard P, Bouzakri K, Krogh-Madsen R, Mittendorfer B, Zierath JR, Pedersen BK. Tumor necrosis factor-α Induces skeletal muscle insulin resistance in healthy human subjects via inhibition of akt substrate 160 phosphorylation. Diabetes (2005) 54(10):2939–45. doi: 10.2337/diabetes.54.10.2939
253. Liu K, Zhou R, Wang B, Mi M-T. Effect of resveratrol on glucose control and insulin sensitivity: a meta-analysis of 11 randomized controlled trials. Amer J Clin Nutrit (2014) 99(6):1510–9. doi: 10.3945/ajcn.113.082024
254. Rizza S, Muniyappa R, Iantorno M, Kim J-a, Chen H, Pullikotil P, et al. Citrus polyphenol hesperidin stimulates production of nitric oxide in endothelial cells while improving endothelial function and reducing inflammatory markers in patients with metabolic syndrome. J Clin Endocrinol Metab (2011) 96(5):E782–92. doi: 10.1210/jc.2010-2879
255. Uruno A, Yagishita Y, Katsuoka F, Kitajima Y, Nunomiya A, Nagatomi R, et al. Nrf2-mediated regulation of skeletal muscle glycogen metabolism. Molec Cell Biol (2016) 36(11):1655–72. doi: 10.1128/MCB.01095-15
256. Uruno A, Furusawa Y, Yagishita Y, Fukutomi T, Muramatsu H, Negishi T, et al. The keap1-nrf2 system prevents onset of diabetes mellitus. Molec Cell Biol (2013) 33(15):2996–3010. doi: 10.1128/mcb.00225-13
257. Dodson M, Shakya A, Anandhan A, Chen J, Garcia JGN, Zhang DD. NRF2 and diabetes: the good, the bad, and the complex. Diabetes (2022) 71(12):2463–76. doi: 10.2337/db22-0623
258. De la Rosa A, Gomez-Cabrera MC, Vinue A, Gonzalez-Navarro H, Sanchez-Andres JV, Viña J. Overexpression of glucose 6 phosphate dehydrogenase preserves mouse pancreatic beta cells function until late in life. Free Radical Biol Med (2021) 164:149–153. doi: 10.1016/j.freeradbiomed.2020.12.439
259. Xu Y, Zhang Z, Hu J, Stillman IE, Leopold JA, Handy DE, et al. Glucose-6-phosphate dehydrogenase-deficient mice have increased renal oxidative stress and increased albuminuria. FASEB J (2010) 24(2):609–16. doi: 10.1096/fj.09-135731
260. Cappai G, Songini M, Doria A, Cavallerano JD, Lorenzi M. Increased prevalence of proliferative retinopathy in patients with type 1 diabetes who are deficient in glucose-6-phosphate dehydrogenase. Diabetologia (2011) 54(6):1539–42. doi: 10.1007/s00125-011-2099-3
261. Sun Q, Zhang BY, Zhang PA, Hu J, Zhang HH, Xu GY. Downregulation of glucose-6-phosphate dehydrogenase contributes to diabetic neuropathic pain through upregulation of toll-like receptor 4 in rats. Molec Pain (2019) 15:1744806919838659. doi: 10.1177/1744806919838659
Keywords: hyperglycermia, glucose metabolism, insulin resistance, glucotoxicity, diabetes, vascular complications
Citation: Rabbani N and Thornalley PJ (2024) Hexokinase-linked glycolytic overload and unscheduled glycolysis in hyperglycemia-induced pathogenesis of insulin resistance, beta-cell glucotoxicity, and diabetic vascular complications. Front. Endocrinol. 14:1268308. doi: 10.3389/fendo.2023.1268308
Received: 27 July 2023; Accepted: 12 December 2023;
Published: 16 January 2024.
Edited by:
Guy A. Rutter, Imperial College London, United KingdomReviewed by:
Loranne Agius, Newcastle University, United KingdomHirotake Komatsu, Beckman Research Institute of City of Hope, United States
Copyright © 2024 Rabbani and Thornalley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul J. Thornalley, cGp0aG9ybmFsbGV5QGdtYWlsLmNvbQ==; Naila Rabbani, bi5yYWJiYW5pQHF1LmVkdS5xYQ==