 Cristina Franco
Cristina Franco Lorella Maria Teresa Canzoniero
Lorella Maria Teresa Canzoniero- Department of Science and Technology, University of Sannio, Benevento, Italy
Impairment of both cellular zinc and redox homeostasis is a feature of several chronic diseases, including obesity. A significant two-way interaction exists between redox metabolism and the relatively redox-inert zinc ion. Redox metabolism critically influences zinc homeostasis and controls its cellular availability for various cellular functions by regulating zinc exchange from/to zinc-binding proteins. Zinc can regulate redox metabolism and exhibits multiple pro-antioxidant properties. On the other hand, even minor disturbances in zinc status and zinc homeostasis affect systemic and cellular redox homeostasis. At the cellular level, zinc homeostasis is regulated by a multi-layered machinery consisting of zinc-binding molecules, zinc sensors, and two selective families of zinc transporters, the Zinc Transporter (ZnT) and Zrt, Irt-like protein (ZIP). In the present review, we summarize the current state of knowledge on the role of the mutual interaction between zinc and redox homeostasis in physiology and pathophysiology, pointing to the role of zinc in the alterations responsible for redox stress in obesity. Since zinc transporters primarily control zinc homeostasis, we describe how changes in the expression and activity of these zinc-regulating proteins are associated with obesity.
1 Introduction
It has been widely documented that oxidative stress (OS) occurs in overweight and obesity and plays a central role in obesity-related comorbidities (1). Several conditions underlie OS in obesity, including hyperglycemia, hyperlipidemia, chronic inflammation, and inadequate antioxidant defenses, which are closely linked, although some contribute more than others.
Impairment in antioxidant defenses in obesity has also been associated with a deficiency in various microelements and vitamins. A combined vitamin and trace element deficiency has been demonstrated in overweight individuals, which worsens with increasing obesity (2). In fact, obese individuals frequently experience low levels of carotenoids, vitamins A, B6, C, D, and E (3, 4), as well as deficiency in selenium, magnesium, iron, and zinc microelements (4, 5).
Here we review the current state of knowledge on the interplay between zinc and redox homeostasis in obesity. Since zinc transporters primarily control zinc homeostasis, we also describe how changes in the expression and activity of these zinc-regulating proteins are associated with obesity.
2 Redox signaling and homeostasis: two faced role in obesity
OS relies on the excessive production of Reactive Oxygen Species (ROS) and Reactive Nitrogen Species (RNS) (RO(N)S) (6). Recently the concept of RO(N)S as harmful molecules has been reconsidered. It is now known that ROS are not always “evil” and antioxidants are not always “good,” but that the extent and contest in which ROS are produced determine whether it can cause beneficial or harmful effects on living systems. This “two-faced” nature of RO(N)S is supported by their functional role as signaling molecules (second messengers) in numerous redox-regulated biological processes such as cell division, differentiation, death, host defense, and metabolic regulation (7). RO(N)S are an integral part of normal cell signaling, responsible for reversible redox-based (oxidation/reduction) post-translational modifications (redox PTMs) of reactive and redox-sensitive sulfur-containing amino acid residues of cell signaling pathway components in a highly selective manner (7). Hence, as with other signaling molecules, the generation of RO(N)S is regulated to avoid RO(N)S concentrations higher than those tolerated by cells (8). Paradoxically, they also act as sensors of changes in the cellular redox state and contribute to maintain redox homeostasis (9).
In this regard, the close relationship between ROS and glucose-lipid metabolism is not surprising, considering that the cell’s redox status is related to glucose and lipid metabolism (10, 11). However, the production of ROS is not only the consequence of glucose and lipid usage, but it also controls glucose and lipid metabolism. Indeed, a close relationship exists between ROS and insulin signaling in its target cells (12, 13). Following cellular insulin stimulation, a transient burst of hydrogen peroxide (H2O2) is essential for fine-tuning insulin signal transduction (14, 15). Namely, H2O2 can trigger biochemical modifications through oxidation of the reduced cysteine thiol side chains of protein tyrosine phosphatases (PTPs) that negatively regulate insulin signal transduction, leading to their inactivation (14, 16). In addition, ROS promotes glucose uptake by positively regulating gene expression of genes encoding glucose transporters (GLUTs) and signaling pathways responsible for translocating GLUTs from intracellular vesicles to the plasma membrane (10).
In contrast, chronic malnutrition and consumption of high-fat and high-carbohydrate meals deliver an excessive amount of energy substrates to the metabolic pathway in adipose and non-adipose cells, which, in turn, can increase the production of ROS, mainly via the mitochondrial electron transport chain (17). Greater availability of reducing equivalents from increased fatty acid and glucose loading results in less efficient oxidative phosphorylation in mitochondria that yields relatively large amounts of superoxide anion (O2-·). Impaired glucose utilization favors the occurrence of hyperglycemia, which enhances OS by oxidative degradation of glucose through autoxidation processes (18). Similarly, accumulated lipids are themselves targets of oxidation, causing an increase in lipid peroxidation (19). Lipid peroxidation products in turn can be released from adipose tissue and enter the liver, where they can alter the respiratory chain, forming more ROS and starting a vicious cycle (20).
Concomitantly, expanded adipose tissue synthesizes and secretes huge amounts of cytokines and chemokines, collectively defined adipochemokines or adipokines, such as leptin, that promote infiltration of adipose tissue by macrophages with subsequent overproduction of RO(N)S and inflammatory cytokines, leading to adipose tissue inflammation (21, 22). Such a process is triggered by OS, which leads to the activation of transcription factors that control the expression of proinflammatory cytokines, which further increase RO(N)S production (23).
Finally, the lack of antioxidant defenses, both in the form of enzymatic and nonenzymatic molecules, can contribute to OS in obesity. Indeed, reduced expression of superoxide dismutase (SOD), catalase, and glutathione peroxidase (GPX) enzymes has been described in obesity (19). It is conceivable that reduced activity of nuclear factor E2-related factor 2 (Nrf2) that controls the expression of diverse antioxidant enzymes contributes to weakening enzymatic antioxidant defenses in insulin resistance and obesity, thereby worsening OS (24).
Several studies have addressed the relationship between zinc status and the changes in obesity in animal models and obese individuals (25). Interestingly, zinc participates in all the primary metabolic processes contributing to OS in obesity (1). Although zinc is relatively redox-inert in living organisms, it can regulate redox metabolism and exhibits several pro-antioxidant properties (26). Similarly, redox metabolism regulates zinc exchange from/to zinc-binding proteins and controls zinc availability for various cellular functions. This two-way interaction between redox metabolism and zinc ion entails that even small perturbations in zinc status and zinc homeostasis can affect cellular and systemic redox homeostasis (27). Hence, it is conceivable that changes in dietary zinc status may exacerbate OS in obesity.
3 Zinc: an essential micronutrient
The importance of zinc to human health has been increasingly appreciated since the first evidence by Prasad (28), and much has been learned about the molecular basis of its indispensability. However, many aspects of zinc biology remain to be further explored. Zinc is the second most abundant transition metal in living organisms after iron and the most abundant intracellular metal. Albeit zinc is considered a dietary microelement, intracellularly, it reaches very high concentrations (29). Indeed, of all total 2-3 g of body zinc, only 0.1% is present in the plasma, most of which is bound to proteins, whereas the remaining 99.9% is confined within cells (30). In plasma, zinc is mainly bound to albumin, α-macroglobulin, and transferrin (31–33), while only a very small fraction, less than 2%, is present as free zinc (33). It is present throughout the body, especially in skeletal muscle (~60%) and bone (~30%), followed by skin and liver. The remaining fraction is distributed among the other tissues and organs, including prostate, pancreas, heart, kidney, and brain (34, 35). Zinc is critically involved in cell proliferation, differentiation, survival, apoptosis, and neurotransmission (36, 37). Estimates place overall intracellular zinc concentration between 200-300 μM, depending on cell type (38). Such high intracellular concentration is substantiated by the fact that a huge number of proteins requires zinc: about 1 in 10 proteins (~3000 proteins) contains a zinc-binding motif (39). In about 90% of the overall zinc-dependent proteins, zinc is required as a catalytic cofactor by 300 enzymes of all classes and a structural component of thousands of protein domains, such as the “zinc finger” domain of numerous transcription factors (35). Such a large number of proteins that require zinc to fulfill their function stimulates reflection on how complex the entire zinc proteome is and how sophisticated is the mechanism by which proteins gain access to zinc ions within a cell. Maintaining cellular zinc levels within an appropriate range is critical, as even a slight deficiency or excess can significantly affect human growth, health, and well-being (40, 41) (Figure 1).
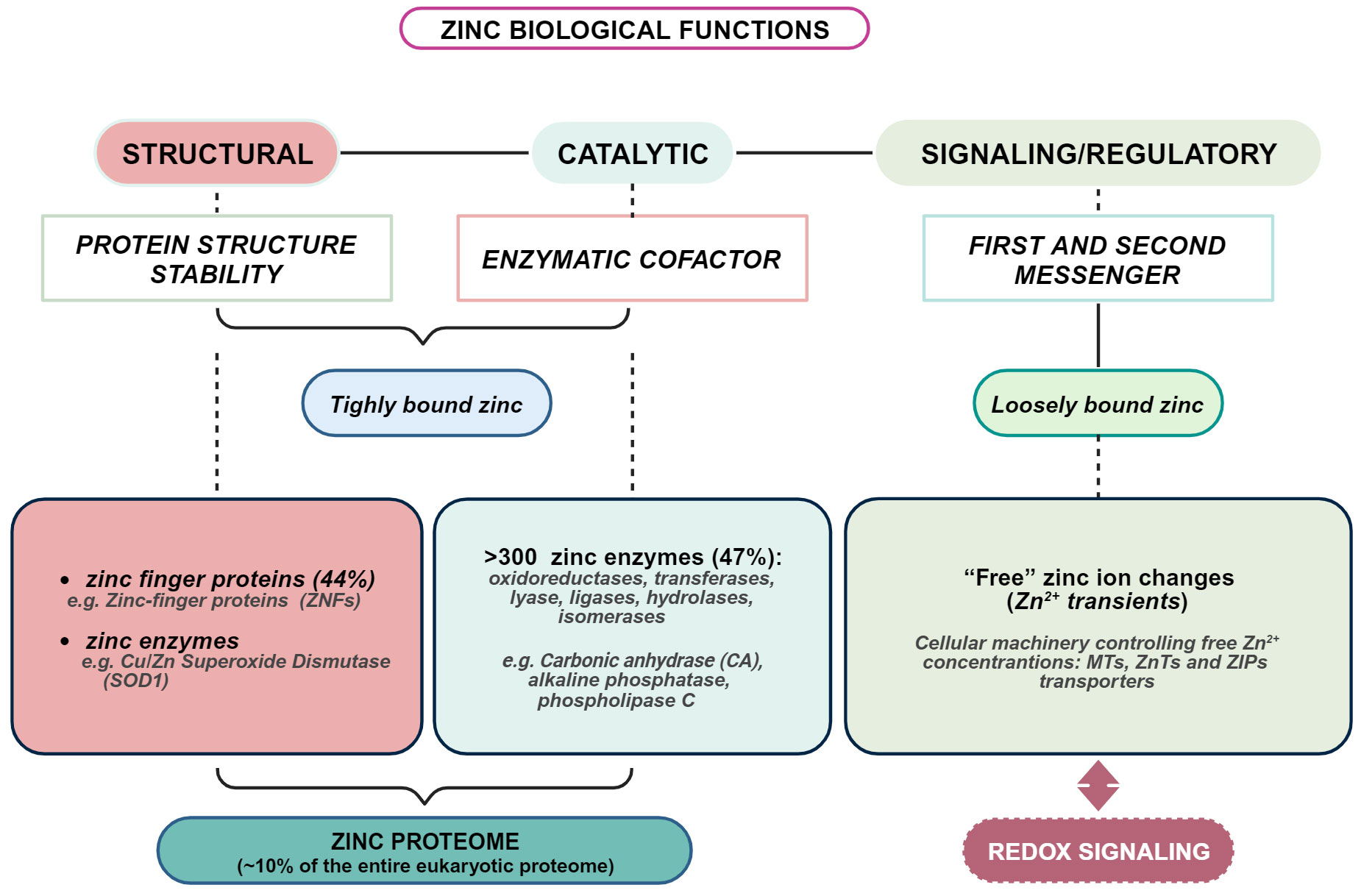
Figure 1 Overview of biological functions of zinc in living organisms. Created with BioRender.com.
The average daily zinc intake (Recommended Dietary Allowance, RDA) sufficient to meet the nutrient requirements of healthy adults is 11 mg for men and 8 mg for women. Depending on age, children require 2-8 mg for both sexes (42). The tolerable upper limit (UL) for zinc has been set at 40 mg/day (43). Zinc is naturally present in a wide variety of foods. The major dietary sources of zinc are shellfish (oysters, crabs, lobsters), red meat, poultry, pork, eggs, dairy products, legumes, nuts, seeds, whole grains, and vegetables (44, 45). However, zinc from plant sources is less bioavailable than from animal foods because they contain large amounts of phytic acid and some other indigestible zinc-binding ligands that make zinc unavailable for absorption (45–49).
Unfortunately, zinc deficiency in humans remains a significant global public health problem. In low- and middle-income countries, an estimated 4% of childhood morbidity and mortality is due to severe zinc deficiency (50, 51). However, slightly inadequate zinc intake is also observed in young children and the elderly over 69 years of age in developed countries (42, 52), suggesting that many factors interact to cause zinc deficiency (53). In particular, dietary habits and food preferences, such as high consumption of phytate-containing foods, e.g., corn, cereals, rice, and legumes, reduce zinc absorption (48). In addition, chronic diseases such as diabetes, gastrointestinal, liver, and kidney diseases, or infections are considered important risk factors for zinc deficiency (54–56).
The body responds to insufficient intake with rapid metabolic adaptations aimed at reducing endogenous losses, primarily by lowering zinc-dependent processes, such as growth and immune system functions. In fact, zinc deficiency mainly affects the immune, skeletal, gastrointestinal, epidermal, nervous, and reproductive systems (49, 57, 58). As a result, severe zinc deficiency leads to serious complications such as immune system dysfunction with recurrent infections, growth retardation, weight loss, alopecia, diarrhea, dermatitis, hypogonadism, hematologic abnormalities, mental disorders, and increased oxidative stress (55, 59–61). However, mild/moderate zinc deficiency is more common than severe zinc deficiency and, in humans, is usually associated with growth retardation, male hypogonadism, skin changes, loss of appetite, loss of taste, mild weight loss, mental lethargy, abnormal dark adaptation, and delayed wound healing (59, 62). Therefore, an adequate supply of zinc is critical for both nutritional status and treatment of various diseases. In this regard, zinc supplementation is potentially beneficial and may serve to correct intercurrent deficiency, such as in preterm infants (63), or as adjunct therapy for various acute and chronic diseases (59, 64).
However, zinc deficiency is not the only health problem associated with zinc intake. Although in healthy individuals the risk of zinc accumulation is much lower than that of zinc deficiency and is a relatively rare event, long-term exposure to elevated zinc concentrations well above the UL results in clinical manifestations of zinc toxicity (65, 66). Zinc toxicity mainly results from dietary supplements, including multivitamins, or the overuse of denture adhesive creams (67). Conversely, even if they contain high amounts of the mineral, excess zinc from food sources alone is relatively harmless. In this context, healthy infants are often unnecessarily exposed to the risk of zinc poisoning, primarily through consumption of supplements, foods, and beverages used to meet needs during growth and to prevent possible inadequate intakes in early childhood (68). Adverse effects of high zinc intake include nausea, dizziness, headache, upset stomach, vomiting, loss of appetite, and lower immunity (65). It is also known that intake of very high doses of zinc supplements can impair the HDL: LDL ratio and copper absorption, resulting in decreased serum levels of HDL and copper, respectively (69–71).
In addition, zinc status in humans and other organisms is of particular importance for the optimal maintenance of a normal gut microbiota composition (72). Zinc shapes host–pathogen interactions and actively protects the host from pathogen invasion. In addition, zinc contributes to maintaining the integrity of the intestinal barrier (73) by regulating the activity of alkaline phosphatase (74). On the other hand, competition for zinc between the immune system and the pathogen and zinc accumulation in immune cells represents the first line of defense against host colonization to limit bacterial infection, a process known as nutritional immunity (29, 74). On the other hand, zinc also serves as an essential micronutrient in prokaryotic cells (29) and is involved in many aspects of their biology. Therefore, it is not surprising that both zinc deficiency and excess can significantly affect the gut microbiota by altering microbial diversity and changing susceptibility to bacterial infections (68, 75–77). This aspect is of great interest considering that dysbiosis of the gut microbiome is causally associated with obesity in humans (78–80).
Even in severe deficiency, a slight increase in dietary zinc intake can rapidly improve clinical symptoms and restore the small amount of zinc loss. An effective homeostatic mechanism prevents variations in tissue zinc and meets tissue requirements over a wide range of zinc intakes (81). The daily zinc requirement is relatively low due to the slow turnover of body zinc (1/1000 per day); however, a correct daily dietary intake is essential because it influences the size of a small but significant endogenous zinc pool, the so-called “exchangeable zinc pool” (EZP), which is probably the most important zinc reservoir (61). EZP accounts for approximately 10% of total body zinc (51) and is localized mainly in plasma and liver (82, 83). The metal ion that makes up this pool is readily mobilized and exchanged with tissues, making zinc available for all functions that require this metal (84).
Consequently, an increase in tissue uptake could decrease serum zinc if it is not rapidly absorbed, transferred to plasma, and replaced within the EZP. The availability of zinc controls the rate of EZP turnover (81). EZP turnover increases markedly (~35) when zinc intake is deficient (85) and immediately decreases when zinc intake is restored (85). Interestingly, intake of glucose and fat results in a significant decrease in plasma zinc, albeit slowest with fat meals, likely due to the slower absorption rate (81). Most importantly, the extent of the decline in plasma zinc induced by glucose and fat depends on the energy intake rather than the meal composition (81, 86).
4 Zinc status and obesity
The propensity to develop obesity varies between the sexes. Indeed, obesity is a sexually dimorphic trait and is more common in boys than in girls (6% of girls and 8% of boys). Conversely, it is more frequent in adult women than in age- and weight-matched men (World Health Organization. Obesity and overweight. Available at: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight). Similarly, it is generally observed that women, both normal weight and obese, have slightly higher serum zinc levels than men, although not significantly (87, 88). Nevertheless, changes in zinc status occur in obese children and adults regardless of gender. In particular, obese patients of both sexes frequently exhibit alterations in the metabolism of this trace element, as demonstrated by the significantly lower serum zinc concentrations that they exhibit compared with lean control subjects (87–101). In the same individuals, zinc balance is worsened by increased hyperzincuria due to increased urinary zinc excretion (87). The latter aspect contrasts sharply with conditions characterized by dietary zinc deficiency, in which the body attempts to conserve zinc and reduce its excretion (102).
In addition, hypozincemia in obese individuals is negatively associated with anthropometric parameters such as body mass index (BMI) and waist circumference, as well as with biochemical parameters such as fasting blood glucose, insulin and leptin (88, 96, 98, 103, 104). Also, women with poorer zinc status had higher body weight, waist circumference, and plasma glucose levels than women with normal levels of this trace mineral (101). The association between zinc and obesity has been underscored in genetically obese (ob/ob) mice that exhibited hypozincemia and hyperzincuria (95) and in null mice for zinc-binding proteins metallothioneins (MTs) that spontaneously develop obesity (105).
Alterations of zinc distribution occur in obese mice. For instance, zinc levels were significantly lower in the pancreas, whereas higher levels were found in muscle, brown and white adipose tissue, and liver (106, 107). Alterations in the sequestration and zinc transport mechanisms in the liver and adipose tissue may be responsible for the hypozincemia observed in obesity (106). Such a decrease in serum zinc would affect the zinc supply to all tissues, reducing the amounts needed to maintain EZP.
However, even though hypozincemia seems to be a common feature of obesity, it is not a direct cause of the disease (108). No significant differences in body weight, adipose tissue mass, or fat distribution are observed in obese individuals on a low zinc diet. In contrast, even modest weight loss in obese individuals following a hypocaloric diet increased circulating zinc to normal levels (89, 101, 109). Most important, obesity-induced hypozincemia is inversely correlated with OS markers (59). Weight loss following moderate caloric restriction while restoring normal zinc levels improved obesity-related OS (110).
5 Zinc and obesity-related metabolic abnormalities
Numerous observations emphasize the role of nutritional zinc in all the significant features of obesity. From a mechanistic point of view, the biological functions of zinc are closely associated with all the major metabolic mechanisms that generate OS in obesity, especially with a lack of antioxidant defenses, but also with insulin resistance, hyperleptinemia, and chronic low-grade inflammation (1, 111).
5.1 Zinc and obesity-related insulin resistance and hyperleptinemia
Zinc has insulin-mimetic activity and is involved in the synthesis, storage, and release of insulin in pancreatic β-cells (112). In particular, zinc is stoichiometrically associated with insulin molecules in the secretory granules of pancreatic β-cells to maintain the crystalline structure of insulin and prevent its degradation by the action of proteolytic enzymes (113, 114). Furthermore, upon glucose stimulation, zinc is released along with insulin into the extracellular space, exerting autocrine and paracrine effects within the pancreas (113). It controls insulin secretion by positively regulating the ATP-activated K+ channel (KATP) activity and potentiating KATP currents, ultimately reducing cellular excitability and, thus, insulin release (115). Moreover, zinc promotes the action of insulin (116, 117) by lowering blood glucose levels and sensitizing target cells in muscle, liver, and adipose tissue to insulin signaling and exerting a regulatory function on various signaling cascades (118–122). Specifically, changes in intracellular zinc appear to be crucial in modulating insulin response through crosstalk with phosphorylation signaling pathways. The mechanisms underlying these effects include the inhibition of protein tyrosine phosphatase 1B (PTP1B), which is physiologically responsible for insulin receptor inhibition through dephosphorylation and attenuates insulin signal transduction (123, 124).
Resistance to the cellular effects of insulin is a significant contributor to obesity (125) and gradually leads to a failure of pancreatic β-cell function, which inevitably results in impaired insulin secretion due to a progressive loss of insulin sensitivity of peripheral target tissues. Initially, pancreatic β-cells attempt to compensate for decreased sensitivity to insulin action by increasing the rate of insulin synthesis and release through hypertrophic and hyperplastic processes in pancreatic β-islets to improve glucose sensing until the compensatory potential of the pancreas is exhausted (126, 127).
Zinc deficiency has been shown to decrease the total number of insulin granules in pancreatic β-cells and to impair insulin sensitivity and glucose tolerance in obese rats and mice (128, 129). As in rodents, obese individuals with lower dietary zinc intakes have higher insulin levels than obese ones with normal dietary zinc intakes (97). In ob/ob mice, zinc supplementation markedly attenuates glucose-induced insulin secretion and lessens fasting plasma glucose levels (107). Similarly, zinc treatment leads to a change in metabolic profile with beneficial effects on insulin sensitivity in both obese children and adults (130–134).
Zinc can improve glucose utilization in insulin-resistant individuals through its insulin-like effects, increasing glucose uptake in insulin-resistant muscle cells by upregulating critical components of the insulin signaling cascade (122). These results are also supported by the blood glucose-lowering effects of zinc compounds observed in several preclinical studies (135–138). Concerning the potential risk of insulin resistance resulting from impaired zinc status in obesity, Cruz and his coworkers (131) systematically reviewed the results of several clinical trials conducted to determine the efficacy or inefficacy of zinc supplementation in obese men and women. The available evidence supports the notion that improving zinc status helps relieve insulin resistance (131, 139).
Similarly, zinc’s regulation of leptin production has been a matter of intense research. Leptin functions as a hormone to give information about the status of body fat depots by acting on the hypothalamus to diminish food intake and increase energy expenditure to maintain constant adipose tissue mass. Brain leptin controls the hypothalamic production of a central appetite-regulating neuropeptide called neuropeptide Y (NPY), which, in turn, stimulates appetite (140, 141). During fasting, NPY levels are high while leptin levels are low, stimulating appetite. It is now apparent that, among other biological functions, zinc regulates appetite, modulating leptin production (55, 104, 142, 143). Interestingly, in healthy humans and rodents, dietary zinc deficiency decreased circulating leptin levels (104, 143–145), whereas zinc supplementation increased leptin levels proportionally to zinc adjustments (104). Thus, in non-obese individuals, zinc provides a regulatory signal for food intake via leptin that can explain the decreased appetite and anorexia observed in zinc deficiency (105,145. 144). Thus, in healthy individuals, zinc deficiency caused a general reduction of NPY, which may ultimately lead to weight loss and anorexia (146, 147).
Consequently, changes in the leptin-NPY axis may occur in obesity, but paradoxically, the vast majority of obese individuals do not have reduced but higher circulating leptin concentrations that do not lead to a reduction in appetite, indicating leptin resistance (148, 149). In obesity, a low zinc diet alters leptin production, although in this case zinc deficiency leads to increased rather than decreased serum leptin levels. Indeed, high blood and adipose leptin levels are associated with low serum zinc levels in obese mice (108, 150). As in mice, an inverse association between dietary zinc intake and leptinemia has also been observed in young obese women (151). Zinc deficiency may also exacerbate hyperleptinemia in obesity by other mechanisms. In this context, as in the case of insulin, PTP1B directly regulated leptin by controlling the phosphorylation state of the leptin receptor and negatively affecting leptin sensitivity (152). Zinc deficiency could contribute to leptin insensitivity in non-adipose tissues such as liver and muscle through loss of the inhibitory effect of zinc on PTP1B. In addition, a positive association between hyperinsulinemia and hyperleptinemia in obesity has been observed. Specifically, higher blood leptin levels reflect higher insulin resistance (153). Mechanistically, chronic hyperinsulinemia resulting from decreased responsiveness to insulin promotes excessive synthesis and release of leptin from adipose tissue (91, 154).
5.2 Zinc and obesity-related inflammation
Obesity is an inflammatory disease (155). Indeed, inflammation begins in adipose tissue and gradually spreads to adjacent sites such as skeletal muscle and liver until a systemic, low-grade inflammatory state, also known as metabolic inflammation, develops and fails to resolve (156). Adipose tissue is able to synthesize and release locally and systemically pro-inflammatory and anti-inflammatory cytokines, such as leptin but also TNF-α, IL-6, adiponectin, and resistin. All fat depots are composed of adipocytes, resident innate and adaptive immune cells, represented mainly by macrophages, but also including neutrophils, dendritic cells, eosinophils, natural killer cells, innate lymphoid cells (ILCs), and B and T cells distributed in the stromal vascular fraction surrounded by a dense blood network (157). Excessive fat accumulation inflames adipose tissue during obesity and favors adipose tissue macrophages (ATM) polarization from an anti-inflammatory to a pro-inflammatory phenotype, leading to further recruitment and infiltration of macrophages and other immune cells. The latter, in turn, massively produce and release pro-inflammatory cytokines, leading to an inflammatory cycle that exacerbates adipose tissue inflammation and triggers a systemic inflammatory cascade through immune cell infiltration of other insulin-dependent tissues (22).
In this regard, the dual function exhibited by leptin, namely as hormone and cytokine, is paradigmatic of the close and evolutionary conserved tie between the metabolic and immune system. Leptin acts as a modulator of the innate and adaptive immune response and has immunological activity functioning as cytokine (158–160). As such, leptin, like many immune system mediators, functions as signaling molecules of both the immune and metabolic systems and is considered the cornerstone signal that links these two systems, as through which they regulate and influence each other (158). In fact, leptin acts as a chemoattractant for monocytes/macrophages and is required for macrophage activation and cytokines expression of TNF-α, IL-6, and IL-12 (161). It derives that, in obesity, hyperleptinemia may therefore contribute to macrophage accumulation in tissues (162–164). An adequate dietary zinc intake is pivotal in the maintenance of the delicate balance between inflammatory and metabolic responses. In fact, in zinc deficient-obesity, leptin rise promoted the increase in macrophage infiltration in adipose tissue of mice fed a high-fat diet (108).
Yet, the influence of zinc on the immune response is not limited to its regulatory role on leptin levels but refers to the specific role that zinc plays in immunity compared to all other microelements. Indeed, zinc availability supports and influences both humoral and cell-mediated immune responses, affects many aspects of the immune system, and contributes to the maintenance of immune system functions, including the inflammatory response (165–168). Accordingly, both severe and mild zinc deficiency can weaken the immune system (61). More interesting is the fact that zinc is an anti-inflammatory agent that is able to reduce cytokine production (169) and zinc deficiency is not only associated with immune deficiencies but can also cause systemic inflammation (97). Therefore, zinc deficiency may directly contribute to the inflammatory state of adipose tissue in obese individuals, and it may also affect the amount of leptin produced and released by this tissue which ultimately has a proinflammatory effect itself and promotes the secretion of other cytokines. Accordingly, obese subjects with a lower zinc status displayed a sustained inflammation and activation of the immune response (97). Likewise, lower dietary zinc intake negatively associates with levels of IL-6 and leptin in young obese women with respect to normal zinc intake counterparts (151). Importantly, sex-related differences in the inverse relationship between zinc status and inflammatory markers have been found (170), which, in turn, may influence susceptibility to the development of diseases, particularly in women. Regarding the effects of zinc on obesity-related inflammation and its complications, zinc supplementation combined or not with restricted caloric diets has showed to modify inflammatory markers, reducing the levels of IL-6 and C-reactive protein (CRP) in obese individuals (100, 151). Of note, CRP levels are directly related to those of IL-6 (171), and IL-6 together with TNF-α concurs to reduce the sensitivity to insulin action (172, 173). While experimental and clinical data consistently indicate that long-term zinc supplementation directly improves inflammatory markers (100), clinical studies showed conflicting results on the effects of zinc on anthropometric measurements (e.g., weight, BMI, waist and hip circumference) (143, 150, 174, 175). Of relevance, the improvement in the inflammatory process observed following zinc supplementation is comparable to that obtained in case of body weight reduction. Moreover, weight loss alone produces the normalization of reduced zinc levels (109).
Cytokines themselves may, at least in part, cause the redistribution of zinc under inflammatory conditions (176). Remarkably, IL-6 is able to regulate metallothionein expression (177). Even more interestingly a polymorphism in the gene encoding metallothionein 2A (MT2A) is associated with higher plasma levels of IL-6, hyperglycemia, and marked zinc deficiency in patients with the AA genotype than in carriers of the AG allele, suggesting the presence of a specific genetic background that may influence susceptibility to the development of zinc deficiency associated with inflammation (177).
6 Zinc homeostasis
Eukaryotic cells have evolved a complex machinery consisting of importer/exporter proteins, sensors, and MTs to ensure that free zinc ion levels are in the femto/pico-molar range.
However, even in a buffered environment such as the cytoplasm, short-term intracellular increases in free zinc occur, typically in a narrow concentration range from picomolar to low nanomolar, referred to as “zinc transients” or “zinc signals”. In contrast to structural and catalytic functions, such zinc changes are due to a very small intracellular zinc pool that is not stably bound to proteins and is therefore referred to as ‘labile’ or “rapidly exchangeable”. It can be transiently released within and from cells (178), allowing zinc to exert regulatory functions and act as a first and second messenger.
Zinc signals originate from extracellular zinc translocation and intracellular release of zinc from intracellular stores or MTs (116). When zinc is released into the surrounding milieu, it can be taken up by neighboring cells through transport mechanisms responsible for translocating zinc into the cell (179). Extracellularly, zinc concentrations are extremely low, and their increase is probably due to the release of zinc stored in membrane-enclosed vesicles and released by exocytosis. Well known examples are the zinc released with glutamate at the synaptic level in the central nervous system (180) and zinc delivered with insulin to secretory vesicles released by pancreatic β-cells in response to stimulation with glucose (181).
Alternatively, zinc signals may originate from ions stored intracellularly. Similar to calcium, eukaryotic cells store zinc ions in some cellular compartments where they perform important functions, and they can be released in response to various stimuli (181–186). Remarkably, extracellular and intracellular zinc signals trigger a zinc burst, but the time scale of zinc release from intracellular stores is slightly slower than that of extracellular zinc flux (116, 187).
Finally, cellular zinc signals may result from its oxidative release from the thiol groups of MTs. In humans, at least 11 functional isoforms of MTs are known, divided into four classes (MT-1 to MT-4), all of which contain twenty conserved cysteine residues that give them the ability to coordinate seven zinc ions (188, 189). The unique intramolecular arrangement by which zinc ions are coordinated in MTs gives them the ability to bind zinc tightly but at the same time mobilize it readily without altering its valence. Indeed, in biological systems, the zinc ion is relatively redox-inert, exhibiting only one valence (Zn2+). In contrast, the thiol groups of the cysteine residues of MTs can be oxidized and reduced, which confers redox activity to the zinc clusters. Once oxidized or reduced, the thiol groups release or bind zinc (188). Because of the coupling of zinc binding/release with the ionization state of thiol groups of MTs display two major functions, namely zinc acceptor and zinc donor (190, 191). Certainly, such coupling links cellular zinc to the cellular redox state, as a shift to more oxidizing conditions results in the release of zinc, whereas a shift to more reducing conditions results in its binding (192). This is particularly interesting as there is growing evidence that redox signaling is critical for various cellular functions (6, 193).
The ionization state of MTs can be altered by cellular oxidants such as glutathione disulfide (GSSG) or ROS. Oxidation of the thiol groups causes the release of zinc from its binding sites on the MTs and the formation of disulfide bonds that can be reduced by glutathione (GSH). In contrast, cysteine-sulfur reduction leads to binding of zinc to MTs. Zinc released by MTs promotes further expression of MTs via the transcription factor Metal Regulatory Transcription Factor 1 (MTF1), which acts as a sensor of labile zinc levels (194, 195), to reduce zinc availability.
6.1 Pro-antioxidant actions of zinc
Although intimate relationship between zinc release from MTs and cellular redox state, zinc cannot be considered an antioxidant because it does not interact directly with oxidants to scavenge them. In contrast, it exerts this effect indirectly by interfering with or suppressing oxidative reactions and protecting cellular components from oxidative damage. Therefore, it should be referred to zinc as a pro-antioxidant (196).
Zinc acts as a pro-antioxidant with multiple actions integrated into the cellular system to defend against oxidants (197). First, zinc generally binds the negative charge of cell membrane phospholipids and, together with nonenzymatic antioxidants, protects membrane lipids from peroxidative damage caused by heavy metals. In particular, it competes with redox-active metals such as iron and copper, preventing the formation of highly oxidant lipid peroxides (198–200) (Figure 2A).
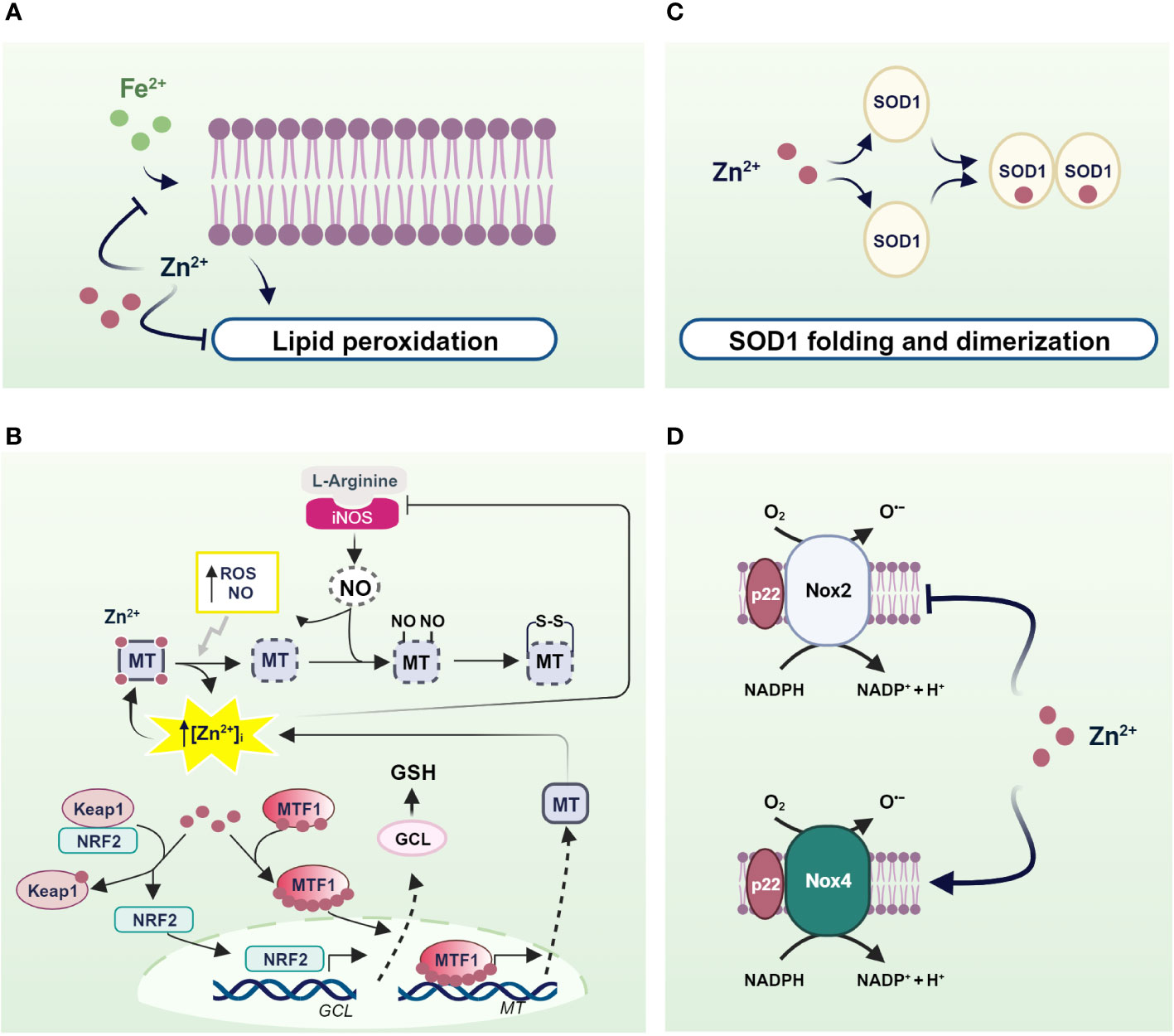
Figure 2 Mechanisms of pro-antioxidant actions of zinc in living organisms. (A) Zinc stabilizes the cell membrane by competing with redox-active metals and preventing the formation of highly oxidant lipid peroxides; (B) NO- and ROS-induced zinc release from MTs helps to counteract OS through the translocation of MTF1 and NRF2 to the nucleus, which in turn activate the transcription of genes encoding MTs and antioxidant defenses; (C) SOD1 and (D) NOXs as examples of enzymes in which zinc is an essential structural component and a regulator of the activity of ROS-producing enzymes. Created with BioRender.com.
Besides to reduced free zinc levels, zinc binding to MTs directly protects the sulfhydryl groups of MTs from oxidation. In addition, the oxidation of MT causes zinc release and is coupled to MTs expression to confer protection against OS. In fact, zinc release from MTs contributes to counteract OS inducing MT expression by activating MTF1. A similar mechanism is observed after MTs thiol oxidation by nitric oxide (NO), H2O2, and GSSG (201–204) (Figure 2B).
Similarly, zinc released by MTs is capable of modulating the expression of antioxidant enzymes under the control of other zinc-modulated transcription factors. Interestingly, Nrf2 is emerging as an important regulator of cellular resistance to oxidants and OS (205). Under basal conditions, Nrf2 signaling is suppressed as it is sequestered in the cytosol by interacting with zinc metalloprotein Keap1 (Kelch-like erythroid cell-derived protein with CNC homology-associated protein 1) (206), which promotes its degradation via a ubiquitination proteasome system. As a result of various stimuli, the Nrf2/Keap1 complex can be dissociated and Nrf2 is free to migrate to the nucleus (207). In both healthy and stressed cells, released zinc from MTs can alter the conformation of Keap1 by binding to cysteine residues on Keap1, thereby reducing its affinity for Nrf2. As a result, Nrf2 migrates to the nucleus where it promotes the expression of genes involved in antioxidant defense (208) (Figure 2B).
In addition to MTs, zinc can be bound by other cellular components containing cysteine residues such as GSH (209). As with MTs, zinc binding to thiol groups protects them from oxidation. Furthermore, zinc/GSH interaction confers to the cell the capability to counteract redox changes, controlling zinc release from MTs. In fact, GSH binding to MTs provokes an increase in labile zinc levels affecting MT conformation and displacing zinc from its binding sites (210). In turn, zinc positively modulates the de novo synthesis of GSH through induction of Nrf2 transcription factor, which is responsible for the upregulation of the gene coding for the rate-limiting enzyme of GSH synthesis, glutamate cysteine ligase (GCL) (211). In addition, zinc stabilizes GCL preventing its cleavage by caspase 3 (212, 213) (Figure 2B).
Similarly, zinc exerts pro-antioxidant actions also as a cofactor of antioxidant enzymes. A well-known example of a zinc metalloenzyme is the copper/zinc superoxide dismutase (Cu/Zn SOD, SOD1), which rids the cells from superoxide radicals turning it into water and H2O2. Notably, zinc binding is needed for the proper functioning of SOD1. In fact, zinc stabilizes the native structure of each SOD1 monomer, accelerating its folding and promoting its dimerization. In contrast, zinc mis-metalation alters SOD1 folding and indirectly affects its catalytic activities (214) (Figure 2C).
There is convincing evidence that zinc also controls the expression and activity of NADPH oxidase (Nox), a superoxide-producing enzyme. Seven Nox catalytic components have been identified, namely Nox 1-5, Duox1, and Duox2. Interestingly, Nox 1, 2, 3, and 5 produce superoxide anions, whereas Nox4, Duox1, and 2 mainly generate hydrogen peroxide and release it into the extracellular space. However, the superoxide anion is rapidly disproportionated to the more stable product H2O2 by SOD. Moreover, unlike other members of the Nox family, Nox4 is constitutively activated, suggesting that it is actively involved in generating ROS, which plays a second messenger role in numerous physiological and biochemical processes (215). Interestingly, several lines of evidence suggest that Nox2- and Nox4-containing NADPH oxidase are differentially regulated by zinc. Specifically, intracellular zinc exerted an inhibitory effect on Nox2, as demonstrated by increased expression, activation, and activity of Nox2 under zinc deficiency, which was greatly attenuated by zinc enrichment (216). Accordingly, silencing of Nox2 attenuates the OS observed under zinc deficiency, suggesting that Nox2 is an essential regulator of OS under reduced zinc availability due to its ability to produce superoxide. Of note, by contrast, Nox4 is downregulated in zinc deficiency (216). This evidence is undoubtedly important considering the endogenous mechanism of coupling oxidative signaling via Nox4 and the insulin signaling cascade (15). In this context, the downregulation of Nox4 observed in zinc deficiency could promote depression of the insulin cascade (Figure 2D).
Moreover, zinc is not only a critical structural component of all isoforms of nitric oxide synthase (NOS) (217, 218) but also a key player in regulating both their expression and activity. More deeply, the ability of zinc to inhibit the production of NO through inducible (iNOS) and constitutive NOS (cNOS) is responsible for the reported anti-inflammatory effect of zinc (219). This zinc effect is based on the central role of MTs in affecting NO-mediated changes in labile zinc (203, 220). Indeed, iNOS-derived NO can nitrosate the cysteine thiol groups of MTs and other zinc-containing intracellular proteins, releasing bound zinc and increasing labile zinc (221).
In turn, the increase in intracellular zinc may have a cytoprotective effect by limiting the production of NO through the inhibition of iNOS (222). Spahl et al. (223) demonstrated that iNOS-derived NO causes a transient increase in free zinc concentration in the nucleus that correlates with the translocation of MTs to the nucleus, where they rapidly exchange zinc with zinc-finger transcription factors and regulate gene expression. Indeed, under the influence of NO, zinc released from MTs suppresses the expression of iNOS, through inhibition of NF-κB transactivation (222, 224), whereas it promotes the expression of MTs, which, in turn, can scavenge NO through covalent binding to form S-nitrothiols (223) (Figure 2B). In addition, Berendji et al. (225) demonstrated that labile zinc increased in the nucleus after exposure to NO. The local release of zinc appears to be associated with the release of the metal from zinc-containing domains of transcription factors as a result of nitrosylation of zinc thiolate clusters and consequent disruption of these domains, which affects gene expression. These results suggest that the cytotoxicity of excessive and long-term NO exposure, as in inflammation, directly targets zinc clusters in transcription factors that destroy DNA-binding activity (202). Prolonged exposure to NO disrupts zinc homeostasis in pancreatic islet cells and reduces the pool of labile zinc (226). In addition, NO could S-nitrosate the thiols of the MTs and cause dissociation of the zinc from the sulfur ligands of the MTs. As indicated by the reduction in zinc levels, it is conceivable that the zinc released from MTs is not properly handled during nitrosative stress, suggesting that pancreatic islet cells lose the ability to efficiently complex and store zinc under these conditions (226).
On the other hand, NO as well as other free radicals have been shown to play a key role in the dysfunction and damage of pancreatic β-cells in diabetes, in part due to the intrinsic susceptibility of β-cells to OS (227). The role of NO in insulin secretion is less clear and sometimes contradictory. Indeed, physiologically low levels of NO, produced by cNOS, stimulate insulin secretion. In contrast, cytokines induce iNOS expression in islet β-cells in response to inflammatory stimuli, resulting in the production of an excessive amount of NO (228), which is directly involved in cytokine-mediated inhibition of insulin secretion and islet degeneration (229). In this regard, zinc dyshomeostasis induced by excessive amounts of NO could have an important impact on the ability of pancreatic β-cells to release insulin because zinc is involved in the crystallization and storage of insulin in secretory granules (113).
Although the role of NO in regulating adipocyte differentiation is still unclear, a similar mechanism has been proposed to be responsible for the upregulation of adipocyte differentiation leading to the development of obesity. Specifically, abnormal production of NO could trigger intracellular zinc mobilization that positively affects adipocyte differentiation (230). However, the role of isoforms of NOS in this process remains to be elucidated (231, 232).
6.2 Zinc muffling and buffering
The short lifetime of zinc signals themselves implies that when cytosolic zinc concentration increases, cells are able to control it, i.e., they generate zinc signals of different amplitudes, minimize cytosolic zinc fluctuations, and eventually restore the equilibrium state of zinc concentration. As for calcium (233), all processes involved in the control of zinc transients are referred to as zinc “muffling”. Together, the buffering and muffling processes not only ensure that the free cytosolic zinc concentration remains low, thereby protecting the cell from the toxic effects of zinc overload but also modulate the availability of zinc ions for binding to proteins that require them. The molecular basis of the buffering and muffling processes is based on the interplay between MTs, and specific zinc-transporting proteins involved in zinc distribution (116).
In this context, in addition to their frequently cited function as zinc buffers, MTs also function as zinc attenuators. Because MT-bound zinc can be mobilized, they are not only zinc scavengers but also able to dynamically release zinc ions to or accept them from other metal-binding proteins and make the metal ion available to other proteins. Although these two functions appear quite different at first glance, they are closely related and depend on the coordination of zinc binding sites on MTs and thiol reactivity. The partial saturation of the zinc-binding sites of MTs in the resting state allows them to bind additional zinc ions in the event of an increased concentration of free zinc (178, 234). Although important, either the binding affinity or even cellular expression of MTs alone is sufficient to account for total cellular zinc buffering while ensuring maintenance of zinc concentration at a critical level and subsequent return to baseline (235). On the other hand, excessive cytosolic zinc buffering capacity prevents zinc fluctuations rapidly extinguishing any zinc signals. In this context, several studies have shown that MTs play a role in controlling the intracellular free zinc pool by zinc muffling. It is based on the ability of MT to attenuate the increase in cytosolic zinc by translocating into cells and supplying zinc to their specific transporters located at the plasma membrane or the membrane of subcellular compartments when the concentration of free zinc is elevated (236, 237). Indeed, in addition to the cytosol, MTs have been found in various cellular compartments such as the nucleus and the intermembrane space of mitochondria (238–240).
As for MTs, the activity of zinc transporters, which transport zinc from the cytosol to the extracellular space or sequester zinc in intracellular compartments, contributes to zinc buffering, allowing the cell to store zinc and rapidly release it when needed temporarily. The zinc buffering capacity of MTs, in conjunction with their translocation within cells and the activity of zinc transporters, are collectively responsible for zinc muffling.
Hitherto, twenty-four mammalian transporters have been described. They belong to two complementary protein families: the Zinc Transporter (ZnT) and the Zrt, Irt-like Protein (ZIP), which in humans are encoded by the Slc30a1-10 and Slc39a1-14 genes, respectively (241). In addition to the plasma membrane, zinc-transporting proteins have been found on ER membranes, in mitochondria, in the Golgi apparatus, intracellular vesicles, and lysosomes; in contrast, the nuclear membrane appears to be devoid of specific zinc transporters (237). Although both protein families share selectivity for zinc binding, ZnT and ZIP transporters move zinc in opposite directions. ZnTs are responsible for removing excess zinc from the cytoplasm and transporting it out of cells or into the lumen of intracellular compartments, whereas ZIPs promote the influx of zinc from the extracellular space or intracellular stores into the cytoplasm. Thus, ZnTs prevent cellular overaccumulation of zinc, whereas ZIPs replenish cytosolic zinc. While most ZIPs are localized at the plasma membrane, most ZnTs are localized in the intracellular compartments, except ZnT1, which is on the cell surface, where it functions as the major pathway of zinc efflux and provides control of metal ion levels (242–244).
In addition to maintaining zinc homeostasis, ZnTs and ZIPs enable the compartmentalization of zinc and play an important role in zinc movement across the compartment membrane in which they are localized (245). They promote zinc entry into the lumens of subcellular compartments, where it is required (for zinc proteins, e.g., zinc-containing enzymes (246); on the other hand, they mediate the local release of zinc in the cell, which is accompanied by zinc transients through which the zinc ion affects gene expression and cell signaling (247). The cooperative regulation of MTs and ZnT transporters, whose expression is tissue- and cell-specific, is essential for zinc homeostasis (35).
The expression and cellular distribution of several physiological mediators regulating MTs and specific ZnT and ZIP proteins are strictly regulated by zinc availability (248, 249). For instance, excessive zinc boost increases ZnT1 surface expression, whereas zinc deficiency causes ZnT1 internalization and degradation (250).
7 Zinc transporters and obesity
Because zinc homeostasis is primarily controlled by zinc transporters, a possible explanation for the different zinc levels and distribution in obesity could be changes in the expression and activity of these zinc-regulating proteins. Changes in the expression of zinc transporters may be due, at least in part, to the inflammatory state characteristic of obesity. Indeed, an inverse relationship between inflammatory markers, BMI, and body fat percentage and the expression of various zinc transporters such as ZnT4, ZnT5, ZnT9, ZIP1, ZIP4, and ZIP6 was found in obese women (251). Similarly, the expression of ZnT1 and ZnT5 was upregulated in leukocytes in another zinc intervention study in obese subjects. Most importantly, an increase in the expression of both zinc transporters was positively correlated with zinc intake in both females and males, although it reached a higher statistical significance in females (251). This suggests that changes in expression levels, mainly of ZnT1, are a more sensitive marker of zinc status than circulating zinc concentrations. ZnT1 upregulation was also observed in the blood samples of obese women, followed by ZIP1 (252). Not surprisingly, the expression of ZnT1, the only transporter responsible for the efflux of zinc from cells, is directly controlled by the availability of zinc and its expression decreases with low zinc intake to protect cells from excessive zinc loss (253). Similarly, the upregulation of ZnT5 serves to restore zinc levels in cellular secretory pathways, particularly in the Golgi apparatus, where luminal zinc is loaded onto secreted proteins that require zinc for their catalytic activity (254).
Since the first report on the expression pattern of ZIPs and ZnTs in adipose tissue, it has become apparent that the biology of the different fat depots, namely subcutaneous and visceral fat (VAT), correlate to different expression levels of zinc-transporting proteins (255). Further changes observed in these depots from lean and obese individuals have reinforced the notion that zinc differentially affects lipid metabolism according to metabolic contest, ultimately reflecting the differential expression of zinc transporters.
In this regard, alterations in the expression of zinc transporters were found in the subcutaneous adipose tissue (SAT) of obese patients. In particular, the expression of ZIP14 showed a significant and reversible reduction in these fat depots of obese individuals, which was restored after a period of weight loss (256). Interestingly, ZIP14 expression increased sharply during the early differentiation of preadipocytes into mature adipocytes, suggesting a role for this transporter in adipogenesis and not during lipogenesis (257). It is likely that the localization of ZIP14 to the plasma membrane is responsible for zinc influx into preadipocytes and controls the intracellular zinc increase that regulates the final differentiation of preadipocytes into mature adipocytes. Therefore, reduction or deletion of ZIP14 negatively affects adipose function, impairs late adipocyte differentiation, and promotes the acquisition of a hypertrophic phenotype often associated with insulin resistance. Notably, Troche and coworkers (258) elegantly demonstrated that deletion of ZIP14 alters the metabolism of white adipose tissue (WAT), rendering it insulin insensitive and increases the expression of cytokines such as leptin and IL-6 by disinhibiting NK-kB and JAK2/STAT3 signaling pathways (Figure 3A a, b). The reduction of ZIP14 in obese patients negatively correlated with both leptinemia and adipose tissue leptin levels in obesity, as in ZIP14 knockout mice, which exhibited higher levels of leptin. Altogether, knockout of ZIP14 mimics a state of zinc deficiency similar to that observed in obese individuals, and the occurrence of metabolic changes similar to those observed in adipose tissue of obese individuals demonstrates that ZIP14 is critical for controlling zinc availability in metabolism and in inhibiting inflammatory processes. Remarkably, ZIP14 knockout mice showed hyperinsulinemia and body fat accumulation, two major features of type 2 diabetes and obesity (259). Moreover, in obese adipose tissue, ZIP14 downregulation has been associated with the increased expression of several cytokines, such as TNF-a and IL-10 (256).
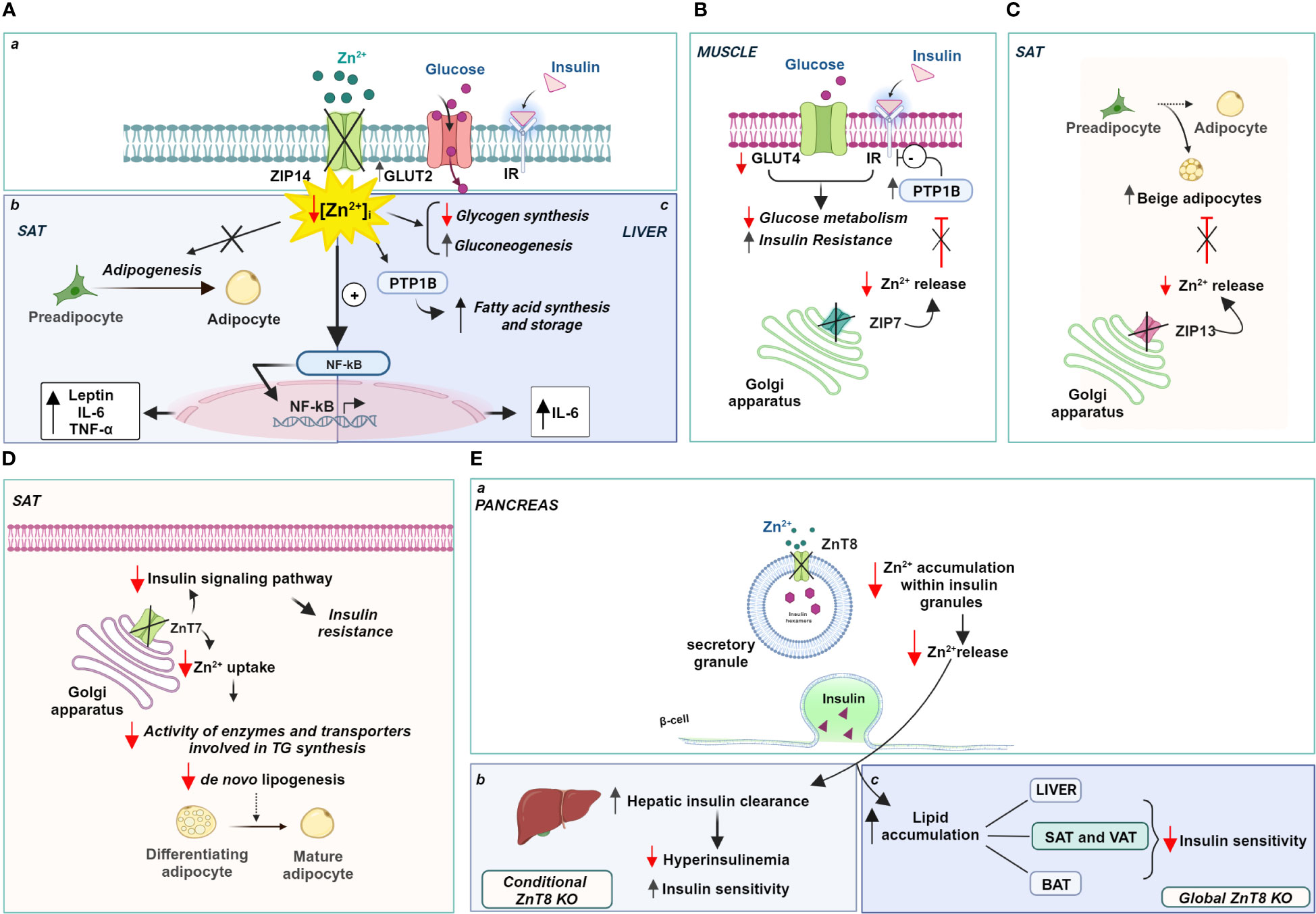
Figure 3 Zinc-dependent pathways in obesity: role of ZIPs and ZnTs. (A) Localization (a) and role of Zip14 in the SAT (b) and liver (c); (B) Zip7 functional role in the Golgi apparatus of muscle, (C) Zip13 and (D) ZnT7 functional role in the Golgi apparatus of SAT; (E) ZnT8 role in the regulation of insulin release (a) and insulin sensitivity (b, c). Created with BioRender.com.
Similarly, ZIP14 expression has functional significance in hepatocytes under physiological conditions. Up-regulation of ZIP14 is responsible for an increase in liver zinc content and a concomitant decrease in serum zinc, which, in turn, is directly related to zinc transport into hepatocytes. Accordingly, downregulation of ZIP14 and consequent zinc deficiency was observed in a mouse model of alcoholic liver disease (260), Moreover, the lack of ZIP14 upregulation in both IL-6 and iNOS knockout mice clearly suggests that the expression of this zinc transporter is under the control of pro-inflammatory cytokines, namely IL-1 β and IL-6 (261, 262). Interestingly, inflammation-induced increase in zinc levels regulates inflammatory response inhibiting further IL-6 release and interfering with NF-kB activation (262) (Figure 3A a, c).
ZIP14 is also involved in regulating hepatic glucose metabolism with opposite effects compared to adipose tissue. The abundance of ZIP14 on the plasma membrane of hepatocytes is strongly regulated by postprandial glucose metabolism. At this stage, higher ZIP14 expression regulated the intensity and duration of insulin action. Indeed, ZIP14 directly controlled insulin signaling through the activation of the two endosomal enzymes, cathepsin D and insulin-degrading enzyme (IDE), which are responsible for dissociating insulin from its receptor (263). ZIP14 KO mice had a higher glucose transport rate and higher glucose concentration in the liver than WT mice, due to a stronger expression of the glucose transporter GLUT2 at the plasma membrane. Interestingly, decreased expression of ZIP14 not only did not affect the insulin sensitivity of hepatocytes but also potentiated hepatic glucose metabolism and promoted glycogen synthesis. Hence, it is conceivable that ZIP14-mediated zinc influx is essential for fine-tuning insulin signaling in hepatocytes and positively regulates glucose metabolism by promoting both glycolysis and gluconeogenesis (263) (Figure 3A a, c).
Consistent with the role played by ZIP14 in regulating glucose-lipid metabolism, induction of ZIP14 in hepatocytes also assumes a relevant role in adaptation to ER stress induced by high-fat diet (HFD). Indeed, ZIP14 increased cellular zinc availability and reduces the risk of liver disease (264). Mechanistically, ZIP14 mediated the influx of zinc that inhibits PTP1B activity in the liver and negatively affects the hepatic synthesis of fatty acids and storage of triglycerides during HFD feeding (265). In contrast, the ablation of ZIP14 has a negative effect favoring triglyceride accumulation because PTP1B activity is not inhibited due to the reduced availability of zinc (264) (Figure 3A a, c).
Similarly, studies recapitulating high-fat diets indicate the involvement of the zinc transporter ZIP7 in insulin signaling and glucose metabolism in skeletal muscle cells (266). ZIP7 is responsible for the zinc homeostasis of the Golgi apparatus transporting zinc out of the lumen of this subcellular compartment and contributing to an increase in intracytoplasmic zinc concentration (267). Physiologically, the function of ZIP7 is associated with glycemic control through a positive modulation of the molecular components of the insulin signaling pathway. ZIP7 thus supports glucose uptake and its use for glycogen synthesis in skeletal muscle. Interestingly, mice fed a high-fat diet exhibited lower expression of ZIP7, consistent with the onset of insulin resistance in muscle (266) (Figure 3B). The exact mechanism by which ZIP7 is involved in insulin resistance and, more importantly, how its zinc transport activity relates to its physiological effects in skeletal muscle requires further investigation, as opposite changes in ZIP7 expression have been found in diabetic cardiomyocytes (268).
Recently, ZIP13, a transporter localized in the Golgi apparatus that mediates zinc transport into the cytoplasm (269), has been linked to the biogenesis of the so-called “beige adipocytes”. They derive from the browning of white adipose cells in visceral tissue and display functional properties intermediate between brown (BAT) and white adipocytes (270). Indeed, beige adipocytes accumulate lipids like white adipocytes and produce heat like brown adipocytes, improving insulin sensitivity and glucose metabolism. They are a potential new therapeutic target for treating metabolic disorders such as obesity, which is known to be associated with decreased thermogenesis. In this context, ZIP13 has attracted interest because it is highly expressed in pancreatic β-cells and the gene encoding this zinc transporter is one of the genes involved in glucose homeostasis during fasting (271). A loss-of-function mutation in the ZIP13 gene has been associated with Ehlers-Danlos syndrome, which is characterized by decreased white adipose tissue mass, among other pathological manifestations (269). Interestingly, ZIP13-deficient mice show increased biogenesis of beige adipocytes due to accelerated differentiation of preadipocytes into beige cells, suggesting that ZIP13 physiologically acts as a negative regulator of adipocyte browning. ZIP13 likely provides zinc ions for modulating the activity of enzymes responsible for degradation of the key components of the adipocyte browning process (272) (Figure 3C).
Further evidence supporting the role of zinc transporters in adipose tissue metabolism was provided by ZnT7 KO mice. ZnT7 is ubiquitously expressed and localized in the Golgi apparatus, promoting zinc influx into this subcellular compartment’s lumen (273). Huang and colleagues (274) have observed that ZnT7 KO mice display a lower body fat percentage than their counterparts WT. These results were subsequently confirmed by Tepaamorndech and coworkers (275), which elegantly demonstrated that adipose tissue is the only tissue affected by ablation of ZnT7, with no relevant differences in other body tissues. Accordingly, since the synthesis and release of leptin are directly dependent on the extent of fat mass, in parallel with a reduction in fat depots, ZnT7 KO mice exhibit decreased circulating leptin levels (276). The main effect on lipid metabolism observed in knockout mice can be attributed to the reduced availability of zinc in the Golgi apparatus which is required for the proper activity of enzymes and transporters involved in triglyceride synthesis.
In addition, ZnT7 is differentially expressed in subcutaneous and visceral fat pads, with a higher expression of ZnT7 in SAT. As a result, the main effects of ZnT7 deficiency have been observed in SAT fat cells, showing a size reduction, probably due to reduced lipid accumulation. Importantly, ZnT7 expression within SAT is controlled and occurs only when adipocyte lipogenesis and not differentiation is induced (275). Genetic ablation of ZnT7 impaired the ability of fat cells to synthesize lipids resulting in insulin insensitivity and glucose intolerance in SAT adipocytes (Figure 3D). Reduction of ZnT7 expression impaired insulin signaling pathway activity and decreased glucose uptake by quantitatively reducing insulin-stimulated activation of Akt. These changes decrease the glycolysis rate and thus the availability of metabolic intermediates necessary for fatty acid production, altering lipogenesis within SAT. From a whole-body perspective, the reduced sensitivity of SAT to insulin action results in a significantly poorer ability of the body to store excess glucose and lipids. In light of these findings, it can be argued that the higher expression of ZnT7 in SAT appears to confer unique properties to this fat depot that predispose it to the accumulation of excess fat, reducing the risk of developing metabolic abnormalities associated with obesity (277). It is worth noting that ZnT7 deficiency is associated with a negative zinc status in the body that cannot be corrected by zinc supplementation. This suggests that ZnT7 expression is not dependent on and cannot be modulated by dietary zinc intake.
Significant alterations in body fat homeostasis and glucose tolerance have been observed in ZnT8 knockout mouse strains. ZnT8 was first identified on insulin granules in pancreatic β-cells, where ZnT8 operates by accumulating cytoplasmic zinc inside the granules (278). The identification of polymorphisms in the gene encoding ZnT8 associated with type 2 diabetes in nonobese individuals (279, 280) and the presence of autoantibodies to ZnT8 in patients with type 1 diabetes has generated considerable interest in the specific role of this transporter in obesity. However, it has been shown that specific deletion of ZnT8 in pancreatic β-cells does not increase the risk of developing obesity but, on the contrary, protects against insulin resistance induced by a high-fat diet (281). Indeed, ZnT8 deficiency prevents the hyperinsulinemia often observed with high fat intake and maintains insulin sensitivity. It is conceivable that reduced zinc accumulation in insulin granules due to specific ZnT8 deficiency of β-cells does not lead to hyperinsulinemia because hepatic clearance of pancreatic hormone is increased (282). Zinc contained in insulin granules and released along with insulin reduces hepatic degradation of the hormone by inhibiting its endocytosis and subsequent degradation, thus ensuring proper insulin delivery to target tissues. Hence, the loss of this endocrine effect of zinc in ZnT8 KO mice affects the rate of hepatic insulin excretion (Figure 3E a, b).
The scenario changes dramatically in global ZnT8 knockout mice exhibiting severe insulin resistance and obesity. Indeed, Mao and colleagues (283) found that global KO mice, in contrast to conditional β-cell ZnT8 KO mice, exhibited adipocyte hypertrophy due to lipid accumulation in all major white adipose depots (VAT and SAT), accumulation of lipids in the liver, and increased expression of genes related to fatty acid synthesis and uptake. Notably, BAT also accumulated more lipids upon ZnT8 ablation, although the expression of genes controlling energy expenditure through heat production was not affected. In this contest, ZnT8 deficiency combined with a high-fat diet significantly exacerbated the effects caused by the absence of this zinc store alone and contributes to increased obesity (283) (Figure 3E a, c).
8 Conclusions
Zinc abnormalities are considered a common feature of obesity, and zinc supplementation strategies are attracting considerable interest as a potential strategy to improve body weight management, inflammatory biomarkers, and insulin resistance in obese individuals. However, zinc supplementation does not produce consistent results and is not always successful. Zinc deficiency likely exacerbates the general state of micronutrient deficiency characteristic of obese individuals. At the same time, the diversity of mechanisms affected by zinc deficiency underscores the indispensable role that zinc plays physiologically and, more importantly, in pathological conditions.
In recent years, new insights have been gained into the function of various zinc transporters. The strict interplay between MTs and members of the ZIPs and ZnTs families is critical for zinc buffering and muffling. In addition, there is a close functional relationship with redox metabolism. Changes in zinc buffering and muffling capacity assume a central role under physiological conditions, but even more so under conditions characterized by oxidative stress, as in obesity. We are beginning to determine how individual zinc transporters may be involved in obesity. However, a more comprehensive view of the full spectrum of alterations in zinc homeostasis can improve the understanding of the mechanisms underlying obesity and its associated comorbidities and develop novel therapeutic strategies aimed at reducing the impact of obesity.
Author contributions
CF: Conceptualization, Writing – original draft, Writing – review & editing. LC: Writing – review & editing, Conceptualization, Funding acquisition, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This manuscript was financially supported by the University of Sannio (Fondi FRA).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Savini I, Catani MV, Evangelista D, Gasperi V, Avigliano L. Obesity-associated oxidative stress: strategies finalized to improve redox state. Int J Mol Sci (2013) 14:10497–538. doi: 10.3390/ijms140510497
2. Vincent HK, Innes KE, Vincent KR. Oxidative stress and potential interventions to reduce oxidative stress in overweight and obesity. Diabetes Obes Metab (2007) 9:813–39. doi: 10.1111/j.1463-1326.2007.00692.x
3. Aasheim ET, Hofsø D, Hjelmesaeth J, Birkeland KI, Bøhmer T. Vitamin status in morbidly obese patients: a cross-sectional study. Am J Clin Nutr (2008) 87:362–9. doi: 10.1093/ajcn/87.2.362
4. Kaidar-Person O, Person B, Szomstein S, Rosenthal RJ. Nutritional deficiencies in morbidly obese patients: a new form of malnutrition? Part A: vitamins. Obes Surg (2008) 18:870–6. doi: 10.1007/s11695-007-9349-y
5. Lapik IA, Galchenko AV, Gapparova KM. Micronutrient status in obese patients: A narrative review. Obes Med (2020) 18. doi: 10.1016/j.obmed.2020.100224
6. Sies H, Jones DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol (2020) 21:363–83. doi: 10.1038/s41580-020-0230-3
7. Sies H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol (2017) 11:613–9. doi: 10.1016/j.redox.2016.12.035
8. Reczek CR, Chandel NS. ROS-dependent signal transduction. Curr Opin Cell Biol (2015) 33:8–13. doi: 10.1016/j.ceb.2014.09.010
9. Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol (2007) 39:44–84. doi: 10.1016/j.biocel.2006.07.001
10. Liemburg-Apers DC, Willems PH, Koopman WJ, Grefte S. Interactions between mitochondrial reactive oxygen species and cellular glucose metabolism. Arch Toxicol (2015) 89:1209–26. doi: 10.1007/s00204-015-1520-y
11. Duchen MR. Roles of mitochondria in health and disease. Diabetes (2004) 53 Suppl 1:S96–102. doi: 10.2337/diabetes.53.2007.s96
12. May JM, de Haën C. Insulin-stimulated intracellular hydrogen peroxide production in rat epididymal fat cells. J Biol Chem (1979) 254:2214–20. doi: 10.1016/S0021-9258(17)30209-0
13. Loh K, Deng H, Fukushima A, Cai X, Boivin B, Galic S, et al. Reactive oxygen species enhance insulin sensitivity. Cell Metab (2009) 10:260–72. doi: 10.1016/j.cmet.2009.08.009
14. Mahadev K, Zilbering A, Zhu L, Goldstein BJ. Insulin-stimulated hydrogen peroxide reversibly inhibits protein-tyrosine phosphatase 1b in vivo and enhances the early insulin action cascade. J Biol Chem (2001) 276:21938–42. doi: 10.1074/jbc.C100109200
15. Mahadev K, Motoshima H, Wu X, Ruddy JM, Arnold RS, Cheng G, et al. The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol Cell Biol (2004) 24:1844 –1854. doi: 10.1128/MCB.24.5.1844-1854.2004
16. Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell (2002) 9:387–99. doi: 10.1016/s1097-2765(02)00445-8
17. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature (2001) 414:813–20. doi: 10.1038/414813a
18. Wolff SP. Diabetes mellitus and free radicals. Free radicals, transition metals and oxidative stress in the aetiology of diabetes mellitus and complications. Br Med Bull (1993) 49:642–52. doi: 10.1093/oxfordjournals.bmb.a072637
19. Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. (2004) 114:1752–61. doi: 10.1172/JCI21625
20. Milagro FI, Campión J, Martínez JA. Weight gain induced by high-fat feeding involves increased liver oxidative stress. Obes (Silver Spring). (2006) 14:1118–23. doi: 10.1038/oby.2006.128
21. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. (2007) 117:175–84. doi: 10.1172/JCI29881
22. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. (2003) 112:1796–808. doi: 10.1172/JCI19246
23. Shoelson SE, Herrero L, Naaz A. Obesity, inflammation, and insulin resistance. Gastroenterology (2007) 132:2169–80. doi: 10.1053/j.gastro.2007.03.059
24. Yu Z, Shao W, Chiang Y, Foltz W, Zhang Z, Ling W, et al. Oltipraz upregulates the nuclear factor (erythroid-derived 2)-like 2 [corrected](NRF2) antioxidant system and prevents insulin resistance and obesity induced by a high-fat diet in C57BL/6J mice. Diabetologia (2011) 54:922–34. doi: 10.1007/s00125-010-2001-8
25. García OP, Long KZ, Rosado JL. Impact of micronutrient deficiencies on obesity. Nutr Rev (2009) 67:559–72. doi: 10.1111/j.1753-4887.2009.00228.x
26. Lee SR. Critical role of zinc as either an antioxidant or a prooxidant in cellular systems. Oxid Med Cell Longev (2018) 18:9156285. doi: 10.1155/2018/9156285
27. Maret W. The redox biology of redox-inert zinc ions. Free Radic Biol Med (2019) 134:311–26. doi: 10.1016/j.freeradbiomed.2019.01.006
28. Prasad AS, Miale A, Farid Z, Sandstead HH, Schulert AR. Zinc metabolism in patients with the syndrome of iron deficiency anemia, hepatosplenomegaly, dwarfism, and hypogonadism. J Lab Clin Med (1963) 61:537–49.
29. Cuajungco MP, Ramirez MS, Tolmasky ME. Zinc: multidimensional effects on living organisms. Biomedicines (2021) 9:208. doi: 10.3390/biomedicines9020208
30. Haase H, Rink L. Multiple impacts of zinc on immune function. Metallomics (2014) 6:1175–80. doi: 10.1039/c3mt00353a
31. Prasad AS, Oberleas D. Binding of zinc to amino acids and serum proteins in vitro. J Lab Clin Med (1970) 76:416–25.
32. Foote JW, Delves HT. Albumin bound and alpha 2-macroglobulin bound zinc concentrations in the sera of healthy adults. J Clin Pathol (1984) 37(9):1050–4. doi: 10.1136/jcp.37.9.1050
33. Craig GM, Evans SJ, Brayshaw BJ, Raina SK. A study of serum zinc, albumin, alpha-2-macroglobulin and transferrin levels in acute and long stay elderly hospital patients. Postgrad Med J (1990) 66:205–9. doi: 10.1136/pgmj.66.773.205
34. Takagishi T, Hara T, Fukada T. Recent advances in the role of SLC39A/ZIP zinc transporters in vivo. Int J Mol Sci (2017) 18(12):2708. doi: 10.3390/ijms18122708
35. Kambe T, Tsuji T, Hashimoto A, Itsumura N. The physiological, biochemical, and molecular roles of zinc transporters in zinc homeostasis and metabolism. Physiol Rev (2015) 95:749–84. doi: 10.1152/physrev.00035.2014
36. Beyersmann D, Haase H. Functions of zinc in signaling, proliferation and differentiation of mammalian cells. Biometals (2001) 14:331–41. doi: 10.1023/a:1012905406548
37. Sensi SL, Paoletti P, Bush AI, Sekler I. Zinc in the physiology and pathology of the CNS. Nat Rev Neurosci (2009) 10:780–91. doi: 10.1038/nrn2734
38. Maret W. Analyzing free zinc(II) ion concentrations in cell biology with fluorescent chelating molecules. Metallomics (2015) 7:202–11. doi: 10.1039/c4mt00230j
39. Andreini C, Banci L, Bertini I, Rosato A. Counting the zinc-proteins encoded in the human genome. J Proteome Res (2006) 5:196–201. doi: 10.1021/pr050361j
40. MacDonald RS. The role of zinc in growth and cell proliferation. J Nutr (2000) 130:1500S–8S. doi: 10.1093/jn/130.5.1500S
41. Canzoniero LM, Turetsky DM, Choi DW. Measurement of intracellular free zinc concentrations accompanying zinc-induced neuronal death. J Neurosci (1999) 19:RC31. doi: 10.1523/JNEUROSCI.19-19-j0005.1999
42. Maret W, Sandstead HH. Zinc requirements and the risks and benefits of zinc supplementation. J Trace Elem Med Biol (2006) 20:3–18. doi: 10.1016/j.jtemb.2006.01.006
43. Institute of Medicine (US) Panel on Micronutrients. Dietary reference intakes for vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc Vol. 12. . Washington (DC: National Academies Press (US (2001). Available at: https://www.ncbi.nlm.nih.gov/books/NBK222317/.
44. Foster M, Samman S. Implications of a plant-based diet on zinc requirements and nutritional status. In: Mariotti F, editor. Vegetarian and plant-based diets in health and disease prevention. Academic Press. (2017). p. 683–713. doi: 10.1016/B978-0-12-803968-7.00038-1
45. Solomons NW. Dietary sources of zinc and factors affecting its bioavailability. Food Nutr Bulletin. (2001) 22:138–54. doi: 10.1177/156482650102200204
46. Foster M, Chu A, Petocz P, Samman S. Effect of vegetarian diets on zinc status: a systematic review and meta-analysis of studies in humans. J Sci Food Agric (2013) 93:2362–71. doi: 10.1002/jsfa.6179
47. Hunt JR. Bioavailability of iron, zinc, and other trace minerals from vegetarian diets. Am J Clin Nutr (2003) 78(3 Suppl):633S–9S. doi: 10.1093/ajcn/78.3.633S
48. Lönnerdal B. Dietary factors influencing zinc absorption. J Nutr (2000) 130(5S Suppl):1378S–83S. doi: 10.1093/jn/130.5.1378S
49. Roohani N, Hurrell R, Kelishadi R, Schulin R. Zinc and its importance for human health: An integrative review. J Res Med Sci (2013) 18(2):144–57.
50. Caulfield LE, Richard SA, Black RE. Undernutrition as an underlying cause of malaria morbidity and mortality in children less than five years old. Am J Trop Med Hyg (2004) 71:55–63. doi: 10.4269/ajtmh.2004.71.55
51. Walker FCL, Fontaine O, Young MW, Black RE. Zinc and low osmolarity oral rehydration salts for diarrhoea: a renewed call to action. Bull World Health Organ. (2009) 87:780–6. doi: 10.2471/blt.08.058990
52. Yasuda H, Tsutsui T. Infants and elderlies are susceptible to zinc deficiency. Sci Rep (2016) 6:21850. doi: 10.1038/srep21850
53. Krebs NF. Update on zinc deficiency and excess in clinical pediatric practice. Ann Nutr Metab (2013) 62 Suppl 1:19–29. doi: 10.1159/000348261
54. Chausmer AB. Zinc, insulin and diabetes. J Am Coll Nutr (1998) 17:109–15. doi: 10.1080/07315724.1998.10718735
55. Prasad AS. Discovery of human zinc deficiency: its impact on human health and disease. Adv Nutr (2013) 4:176–90. doi: 10.3945/an.112.003210
56. Wessels I, Rolles B, Slusarenko AJ, Rink L. Zinc deficiency as a possible risk factor for increased susceptibility and severe progression of Corona Virus Disease 19. Br J Nutr (2022) 127:214–32. doi: 10.1017/S0007114521000738
57. Chasapis CT, Ntoupa PA, Spiliopoulou CA, Stefanidou ME. Recent aspects of the effects of zinc on human health. Arch Toxicol (2020) 94:1443–60. doi: 10.1007/s00204-020-02702-9
58. Corona C, Masciopinto F, Silvestri E, Viscovo AD, Lattanzio R, Sorda RL, et al. Dietary zinc supplementation of 3xTg-AD mice increases BDNF levels and prevents cognitive deficits as well as mitochondrial dysfunction. Cell Death Dis (2010) 1:e91. doi: 10.1038/cddis.2010.73
59. Prasad AS. Clinical manifestations of zinc deficiency. Annu Rev Nutr (1985) 5:341–63. doi: 10.1146/annurev.nu.05.070185.002013
60. Rink L, Kirchner H. Zinc-altered immune function and cytokine production. J Nutr (2000) 130:1407S–11S. doi: 10.1093/jn/130.5.1407S
61. Wessels I, Maywald M, Rink L. Zinc as a gatekeeper of immune function. Nutrients (2017) 9(12):1286. doi: 10.3390/nu9121286
62. Prasad AS. Lessons learned from experimental human model of zinc deficiency. J Immunol Res (2020) 2020:9207279. doi: 10.1155/2020/9207279
63. Willoughby JL, Bowen CN. Zinc deficiency and toxicity in pediatric practice. Curr Opin Pediatr (2014) 26:579–84. doi: 10.1097/MOP.0000000000000132
64. Berni Canani R, Secondo A, Passariello A, Buccigrossi V, Canzoniero LM, Ruotolo S, et al. Zinc inhibits calcium-mediated and nitric oxide-mediated ion secretion in human enterocytes. Eur J Pharmacol (2010) 626:266–70. doi: 10.1016/j.ejphar.2009.09.042
65. Plum LM, Rink L, Haase H. The essential toxin: impact of zinc on human health. Int J Environ Res Public Health (2010) 7:1342–65. doi: 10.3390/ijerph704134
66. Haase H, Overbeck S, Rink L. Zinc supplementation for the treatment or prevention of disease: current status and future perspectives. Exp Gerontol. (2008) 43:394–408. doi: 10.1016/j.exger.2007.12.002
67. Doherty K, Connor M, Cruickshank R. Zinc-containing denture adhesive: a potential source of excess zinc resulting in copper deficiency myelopathy. Br Dent J (2011) 210:523–5. doi: 10.1038/sj.bdj.2011.428
68. Podany A, Rauchut J, Wu T, Kawasawa YI, Wright J, Lamendella R, et al. Excess dietary zinc intake in neonatal mice causes oxidative stress and alters intestinal host-microbe interactions. Mol Nutr Food Res (2019) 63:e1800947. doi: 10.1002/mnfr.201800947
69. Hooper PL, Visconti L, Garry PJ, Johnson GE. Zinc lowers high-density lipoprotein-cholesterol levels. JAMA (1980) 244(17):1960–1. doi: 10.1001/jama.1980.03310170058030
70. Ogiso T, Moriyama K, Sasaki S, Ishimura Y, Minato A. Inhibitory effect of high dietary zinc on copper absorption in rats. Chem Pharm Bull (Tokyo) (1974) 22:55–60. doi: 10.1248/cpb.22.55
71. Ogiso T, Ogawa N, Miura T. Inhibitory effect of high dietary zinc on copper absorption in rats. II. Binding of copper and zinc to cytosol proteins in the intestinal mucosa. Chem Pharm Bull (1979) 27:515–21. doi: 10.1248/cpb.27.515
72. Southon S, Gee JM, Bayliss CE, Wyatt GM, Horn N, Johnson IT. Intestinal microflora, morphology and enzyme activity in zinc-deficient and Zn-supplemented rats. Br J Nutr (1986) 55:603–11. doi: 10.1079/bjn19860065
73. Islam T, Albracht-Schulte K, Ramalingam L, Schlabritz-Lutsevich N, Park OH, Zabet-Moghaddam M, et al. Anti-inflammatory mechanisms of polyphenols in adipose tissue: role of gut microbiota, intestinal barrier integrity and zinc homeostasis. J Nutr Biochem (2023) 115:109242. doi: 10.1016/j.jnutbio.2022.109242
74. Lopez CA, Skaar EP. The impact of dietary transition metals on host-bacterial interactions. Cell Host Microbe (2018) 23:737–48. doi: 10.1016/j.chom.2018.05.00
75. Chen X, Jiang Y, Wang Z, Chen Y, Tang S, Wang S, et al. Alteration in gut microbiota associated with zinc deficiency in school-age children. Nutrients (2022) 14:2895. doi: 10.3390/nu14142895
76. Chen L, Wang Z, Wang P, Yu X, Ding H, Wang Z, et al. Effect of long-term and short-term imbalanced zn manipulation on gut microbiota and screening for microbial markers sensitive to zinc status. Microbiol Spectr. (2021) 9:e0048321. doi: 10.1128/Spectrum.00483-21
77. Skalny AV, Aschner M, Lei XG, Gritsenko VA, Santamaria A, Alekseenko SI, et al. Gut microbiota as a mediator of essential and toxic effects of zinc in the intestines and other tissues. Int J Mol Sci (2021) 22:13074. doi: 10.3390/ijms222313074
78. Liu R, Hong J, Xu X. Gut microbiome and serum metabolome alterations in obesity and after weight-loss intervention. Nat Med (2017) 23:859–68. doi: 10.1038/nm.4358
79. Koren O, Tako E. Chronic dietary zinc deficiency alters gut microbiota composition and function. Proceedings (2020) 61:16. doi: 10.3390/IECN2020-06993
80. Saad MJ, Santos A, Prada PO. Linking gut microbiota and inflammation to obesity and insulin resistance. Physiol (Bethesda). (2016) 31:283–93. doi: 10.1152/physiol.00041.201
81. Goode HF, Robertson DA, Kelleher J, Walker BE. Effect of fasting, self-selected and isocaloric glucose and fat meals and intravenous feeding on plasma zinc concentrations. Ann Clin Biochem (1991) 28:442–5. doi: 10.1177/000456329102800503
82. Wastney ME, Aamodt RL, Rumble WF, Henkin RI. Kinetic analysis of zinc metabolism and its regulation in normal humans. Am J Physiol (1986) 251:R398–408. doi: 10.1152/ajpregu.1986.251.2.R398
83. Miller LV, Hambidge KM, Naake VL, Hong Z, Westcott JL, Fennessey PV. Size of the zinc pools that exchange rapidly with plasma zinc in humans: alternative techniques for measuring and relation to dietary zinc intake. J Nutr (1994) 124:268–76. doi: 10.1093/jn/124.2.268
84. Long JM, Khandaker AM, Sthity RA, Westcott JE, Matveev A, Black RE, et al. Exchangeable zinc pool size reflects form of zinc supplementation in young children and is not associated with markers of inflammation. Nutrients (2022) 14(3):481. doi: 10.3390/nu14030481
85. King JC. Yet again, serum zinc concentrations are unrelated to zinc intakes. J Nutr (2018) 148:1399–401. doi: 10.1093/jn/nxy190
86. Sweeney MA, Dinsmore WW, McKee CM, Love AH. The effect of food composition on serum zinc. Acta Pharmacol Toxicol (Copenh). (1986) 7:170–3. doi: 10.1111/j.1600-0773.1986.tb02735.x
87. Marreiro ND, Fisberg M, Cozzolino SMF. Zinc nutritional status in obese children and adolescents. Biol Trace Elem Res (2002) 86:107–22. doi: 10.1385/bter:86:2:107
88. Rios-Lugo MJ, Madrigal-Arellano C, Gaytán-Hernández D, Hernández-Mendoza H, Teresita Romero-Guzmán ET. Association of serum zinc levels in overweight and obesity. Biol Trace Elem Res (2020) 198:51–7. doi: 10.1007/s12011-020-02060-8
89. Di Martino G, Matera MG, De Martino B, Vacca C, Di Martino S, Rossi F. Relationship between zinc and obesity. J Med (1993) 24(2-3):177–83.
90. Ozata M, Mergen M, Oktenli C, Aydin A, Sanisoglu SY, Bolu E, et al. Increased oxidative stress and hypozincemia in male obesity. Clin Biochem (2002) 35:627–31. doi: 10.1016/s0009-9120(02)00363-6
91. Konukoglu D, Turhan MS, Ercan M, Serin O. Relationship between plasma leptin and zinc levels and the effect of insulin and oxidative stress on leptin levels in obese diabetic patients. J Nutr Biochem (2004) 15:757–60. doi: 10.1016/j.jnutbio.2004.07.007
92. Perrone L, Gialanella G, Moro R, Feng SL, Boccia E, Palombo G, et al. Zinc, copper, and iron in obese children and adolescents. Nutr Res (1998) 18:183–9. doi: 10.1016/S0271-5317(98)00011-6
93. Chen MD, Lin PY, Lin WH, Cheng V. Zinc in hair and serum of obese individuals in Taiwan. Am J Clin Nutr (1988) 48:1307–9. doi: 10.1093/ajcn/48.5.1307
94. Chen MD, Lin PY, Sheu WHH. Zinc status in plasma of obese individuals during glucose administration. Biol Trace Elem Res (1997) 60:123–9. doi: 10.1007/BF02783315
95. Chen MD, Liou SJ, Lin PY, Yang VC, Alexander P, Lin WH. Effects of zinc supplementation on the plasma glucose level and insulin activity in genetically obese (ob/ob) mice. Biol Trace Elem Res (1998) 61:303–11. doi: 10.1007/BF02789090
96. Marreiro DDN, Fisberg M, Cozzolino SMF. Zinc nutritional status and its relationships with hyperinsulinemia in obese children and adolescents. Biol Trace Elem Res (2004) 100:137–49. doi: 10.1385/bter:100:2:137
97. Costarelli L, Muti E, Malavolta M, Cipriano C, Giacconi R, Tesei S, et al. Distinctive modulation of inflammatory and metabolic parameters in relation to zinc nutritional status in adult overweight/obese subjects. J Nutr Biochem (2010) 21:432–7. doi: 10.1016/j.jnutbio.2009.02.001
98. Habib SA, Saad EA, Elsharkawy AA, Attia ZR. Pro-inflammatory adipocytokines, oxidative stress, insulin, Zn and Cu: Interrelations with obesity in Egyptian non-diabetic obese children and adolescents. Adv Med Sci (2015) 60:179–85. doi: 10.1016/j.advms.2015.02.002
99. Gu K, Xiang W, Zhang Y, Sun K, Jiang X. The association between serum zinc level and overweight/obesity: a meta-analysis. Eur J Nutr (2019) 58:2971–82. doi: 10.1007/s00394-018-1876-x
100. Khorsandi H, Nikpayam O, Yousefi R, Parandoosh M, Hosseinzadeh N, Saidpour A, et al. Zinc supplementation improves body weight management, inflammatory biomarkers and insulin resistance in individuals with obesity: a randomized, placebo-controlled, double-blind trial. Diabetol Metab Syndr (2019) 11:101. doi: 10.1186/s13098-019-0497-8
101. Voruganti VS, Cai G, Klohe DM, Jordan KC, Lane MA, Freeland-Graves JH. Short-term weight loss in overweight/obese low-income women improves plasma zinc and metabolic syndrome risk factors. J Trace Elem Med Biol (2010) 24:271–6. doi: 10.1016/j.jtemb.2010.05.001
102. King JC, Shames DM, Lowe NM, Woodhouse LR, Sutherland B, Abrams SA, et al. Effect of acute zinc depletion on zinc homeostasis and plasma zinc kinetics in men. Am J Clin Nutr (2001) 74:116–24. doi: 10.1093/ajcn/74.1.116
103. Chen MD, Lin PY. Zinc-induced hyperleptinemia relates to the amelioration of sucrose-induced obesity with zinc repletion. Obes Res (2000) 8:525–9. doi: 10.1038/oby.2000.65
104. Mantzoros CS, Prasad AS, Beck FW, Grabowski S, Kaplan J, Adair C, et al. Zinc may regulate serum leptin concentrations in humans. J Am Coll Nutr (1998) 17:270–5. doi: 10.1080/07315724.1998.10718758
105. Beattie JH, Wood AM, Newman AM, Bremner I, Choo KH, Michalska AE, et al. Obesity and hyperleptinemia in metallothionein (-I and -II) null mice. Proc Natl Acad Sci U.S.A. (1998) 95:358–63. doi: 10.1073/pnas.95.1.358
106. Feitosa MC, Lima VB, Moita Neto JM, Marreiro Ddo N. Plasma concentration of IL-6 and TNF-α and its relationship with zincemia in obese women. Rev Assoc Med Bras 1992 (2013) 59:429–34. doi: 10.1016/j.ramb.2013.03.003
107. Begin-Heick N, Dalpe-Scott M, Rowe J, Heick HM. Zinc supplementation attenuates insulin secretory activity in pancreatic islets of the ob/ob mouse. Diabetes (1985) 34:179–84. doi: 10.2337/diab.34.2.179
108. Liu MJ, Bao S, Bolin ER, Burris DL, Xu X, Sun Q, et al. Zinc deficiency augments leptin production and exacerbates macrophage infiltration into adipose tissue in mice fed a high-fat diet. J Nutr (2013) 143:1036–45. doi: 10.3945/jn.113.175158
109. Di Toro A, Marotta A, Todisco N, Ponticiello E, Collini R, Di Lascio R, et al. Unchanged iron and copper and increased zinc in the blood of obese children after two hypocaloric diets. Biol Trace Elem Res (1997) 57:97–104. doi: 10.1007/BF02778192
110. Buchowski MS, Hongu N, Acra S, Wang L, Warolin J, Roberts LJ 2nd. Effect of modest caloric restriction on oxidative stress in women, a randomized trial. PloS One (2012) 7:e47079. doi: 10.1371/journal.pone.0047079
111. Olechnowicz J, Tinkov A, Skalny A, Suliburska J. Zinc status is associated with inflammation, oxidative stress, lipid, and glucose metabolism. J Physiol Sci (2018) 68:19–31. doi: 10.1007/s12576-017-0571-7
112. Chimienti F. Zinc, pancreatic islet cell function and diabetes: new insights into an old story. Nutr Res Rev (2013) 26(1):1–11. doi: 10.1017/S0954422412000212
113. Emdin SO, Dodson GG, Cutfield JM, Cutfield SM. Role of zinc in insulin biosynthesis. Some possible zinc-insulin interactions in the pancreatic B-cell. Diabetologia (1980) 19:174–82. doi: 10.1007/BF00275265
114. Dodson G, Steiner D. The role of assembly in insulin’s biosynthesis. Curr Opin Struct Biol (1998) 8:189–94. doi: 10.1016/s0959-440x(98)80037-7
115. Prost AL, Bloc A, Hussy N, Derand R, Vivaudou M. Zinc is both an intracellular and extracellular regulator of KATP channel function. J Physiol (2004) 559:157–67. doi: 10.1113/jphysiol.2004.065094
116. Maret W. Zinc in cellular regulation: the nature and significance of “Zinc signals”. Int J Mol Sci (2017) 18:2285. doi: 10.3390/ijms18112285
117. May JM, Contoreggi CS. The mechanism of the insulin-like effects of ionic zinc. J Biol Chem (1982) 257(8):4362–8. doi: 10.1016/S0021-9258(18)34730-6
118. Tang X, Shay NF. Zinc has an insulin-like effect on glucose transport mediated by phosphoinositol-3-kinase and Akt in 3T3-L1 fibroblasts and adipocytes. J Nutr (2001) 131:1414–20. doi: 10.1093/jn/131.5.1414
119. Norouzi S, Adulcikas J, Sohal SS, Myers S. Zinc stimulates glucose oxidation and glycemic control by modulating the insulin signaling pathway in human and mouse skeletal muscle cell lines. PloS One (2018) 13:e0191727. doi: 10.1371/journal.pone.0191727
120. Myers SA, Nield A, Chew GS, Myers MA. The zinc transporter, Slc39a7 (Zip7) is implicated in glycaemic control in skeletal muscle cells. PloS One (2013) 8:e79316. doi: 10.1371/journal.pone.0079316
121. Miranda ER, Dey CS. Effect of chromium and zinc on insulin signaling in skeletal muscle cells. Biol Trace Elem Res (2004) 101:19–36. doi: 10.1385/BTER:101:1:19
122. Wu Y, Lu H, Yang H, Li C, Sang Q, Liu X, et al. Zinc stimulates glucose consumption by modulating the insulin signaling pathway in L6 myotubes: essential roles of Akt-GLUT4, GSK3β and mTOR-S6K1. J Nutr Biochem (2016) 34:126–35. doi: 10.1016/j.jnutbio.2016.05.008
123. Myers SA. Zinc transporters and zinc signaling: new insights into their role in type 2 diabetes. Int J Endocrinol (2015) 2015:167503. doi: 10.1155/2015/167503
124. Haase H, Maret W. Intracellular zinc fluctuations modulate protein tyrosine phosphatase activity in insulin/insulin-like growth factor-1 signaling. Exp Cell Res (2003) 291:289–98. doi: 10.1016/s0014-4827(03)00406-3
125. Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes (1988) 37:1595–607. doi: 10.2337/diab.37.12.1595
126. Kasuga M. Insulin resistance and pancreatic beta cell failure. J Clin Invest. (2006) 116:1756–60. doi: 10.1172/JCI29189
127. Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature (2006) 444(7121):840–6. doi: 10.1038/nature05482
128. Boquist L, Lernmark A. Effects on the endocrine pancreas in Chinese hamsters fed zinc deficient diets. Acta Pathol Microbiol Scand (1969) 76:215–28. doi: 10.1111/j.1699-0463.1969.tb03252.x
129. Huber AM, Gershoff SN. Effect of zinc deficiency in rats on insulin release from the pancreas. J Nutr (1973) 103:1739–44. doi: 10.1093/jn/103.12.1739
130. Kelishadi R, Hashemipour M, Adeli K, Tavakoli N, Movahedian-Attar A, Shapouri J, et al. Effect of zinc supplementation on markers of insulin resistance, oxidative stress, and inflammation among prepubescent children with metabolic syndrome. Metab Syndr Relat Disord (2010) 8:505–10. doi: 10.1089/met.2010.0020
131. Cruz KJ, Morais JB, de Oliveira AR, Severo JS, Marreiro DD. The effect of zinc supplementation on insulin resistance in obese subjects: a systematic review. Biol Trace Elem Res (2017) 176:239–43. doi: 10.1007/s12011-016-0835-8
132. Kim J, Lee S. Effect of zinc supplementation on insulin resistance and metabolic risk factors in obese Korean women. Nutr Res Pract (2012) 6:221–5. doi: 10.4162/nrp.2012.6.3.221
133. Marreiro DN, Geloneze B, Tambascia MA, Lerário AC, Halpern A, Cozzolino SM. Effect of zinc supplementation on serum leptin levels and insulin resistance of obese women. Biol Trace Elem Res (2006) 112:109–18. doi: 10.1385/bter:112:2:109
134. Hashemipour M, Kelishadi R, Shapouri J, Sarrafzadegan N, Amini M, Tavakoli N, et al. Effect of zinc supplementation on insulin resistance and components of the metabolic syndrome in prepubertal obese children. Hormones (Athens) (2009) 8:279–85. doi: 10.14310/horm.2002.1244
135. Adachi Y, Yoshida J, Kodera Y, Kato A, Yoshikawa Y, Kojima Y, et al. A new insulin-mimetic bis(allixinato)zinc(II) complex: structure-activity relationship of zinc(II) complexes. J Biol Inorg Chem (2004) 9:885–93. doi: 10.1007/s00775-004-0590-8
136. Sakurai H, Adachi Y. The pharmacology of the insulinomimetic effect of zinc complexes. Biometals (2005) 184:319–23. doi: 10.1007/s10534-005-3688-8
137. Vardatsikos G, Pandey NR, Srivastava AK. Insulino-mimetic and anti-diabetic effects of zinc. J Inorg Biochem (2013) 120:8–17. doi: 10.1016/j.jinorgbio.2012.11.006
138. Chukwuma CI, Mashele SS, Eze KC, Matowane GR, Islam SM, Bonnet SL, et al. A comprehensive review on zinc(II) complexes as anti-diabetic agents: The advances, scientific gaps and prospects. Pharmacol Res (2020) 155:104744. doi: 10.1016/j.phrs.2020.104744
139. Thoen RU, Barther NN, Schemitt E, Bona S, Fernandes S, Coral G, et al. Zinc supplementation reduces diet-induced obesity and improves insulin sensitivity in rats. Appl Physiol Nutr Metab (2019) 44:580–6. doi: 10.1139/apnm-2018-0519
140. Kalra SP, Dube MG, Sahu A, Phelps CP, Kalra PS. Neuropeptide Y secretion increases in the paraventricular nucleus in association with increased appetite for food. Proc Natl Acad Sci U S A. (1991) 88:10931–5. doi: 10.1073/pnas.88.23.10931
141. Sahu A, Kalra PS, Kalra SP. Food deprivation and ingestion induce reciprocal changes in neuropeptide Y concentrations in the paraventricular nucleus. Peptides (1988) 9:83–6. doi: 10.1016/0196-9781(88)90013-7
142. Lee RG, Rains TM, Tovar-Palacio C, Beverly JL, Shay NF. Zinc deficiency increases hypothalamic neuropeptide Yand neuropeptide Y mRNA levels and does not block neuropeptide Y-induced feeding in rats. J Nutr (1998) 128:1218– 23. doi: 10.1093/jn/128.7.1218
143. Mangian H, Lee R, Paul G, Emmert, Shay N. Zinc deficiency suppresses plasma leptin concentrations in rats. Nutr Biochem (1998) 9:47–51. doi: 10.1016/S0955-2863(97)00165-4
144. Baltaci AK, Mogulkoc R, Halifeoglu I. Effects of zinc deficiency and supplementation on plasma leptin levels in rats. Biol Trace Elem Res (2005) 104:41–6. doi: 10.1385/BTER:104:1:041
145. Gaetke LM, Frederich RC, Oz HS, McClain CJ. Decreased food intake rather than zinc deficiency is associated with changes in plasma leptin, metabolic rate, and activity levels in zinc deficient rats. J Nutr Biochem (2002) 13:237–44. doi: 10.1016/s0955-2863(01)00220-0
146. Levenson CW. Zinc regulation of food intake: new insights on the role of neuropeptide Y. Nutr Rev (2003) 61:247–9. doi: 10.1301/nr.2003.jul.247-249
147. Huntington CE, Shay NF, Grouzmann E, Arseneau LM, Beverly JL. Zinc status affects neurotransmitter activity in the paraventricular nucleus of rats. J Nutr (2002) 132:270 –275. doi: 10.1093/jn/132.2.270
148. Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med (1996) 334:292–5. doi: 10.1056/NEJM199602013340503
149. Gruzdeva O, Borodkina D, Uchasova E, Dyleva Y, Barbarash O. Leptin resistance: underlying mechanisms and diagnosis. Diabetes Metab Syndr Obes (2019) 12:191–8. doi: 10.2147/DMSO.S182406
150. Tallman DL, Taylor CG. Effects of dietary fat and zinc on adiposity, serum leptin and adipose fatty acid composition in C57BL/6J mice. J Nutr Biochem (2003) 14:17–23. doi: 10.1016/s0955-2863(02)00228-0
151. Kim J, Ahn J. Effect of zinc supplementation on inflammatory markers and adipokines in young obese women. Biol Trace Elem Res (2014) 157:101–6. doi: 10.1007/s12011-013-9885-3
152. Cook WS, Unger RH. Protein tyrosine phosphatase 1B: a potential leptin resistance factor of obesity. Dev Cell (2002) 2:385–7. doi: 10.1016/s1534-5807(02)00158-2
153. Carantoni M, Abbasi F, Azhar S, Chen YD, Klebanov M, Wang PW, et al. Plasma leptin concentrations do not appear to decrease insulin-mediated glucose disposal or glucose-stimulated insulin secretion in women with normal glucose tolerance. Diabetes (1998) 47:244–7. doi: 10.2337/diab.47.2.244
154. Fischer S, Hanefeld M, Haffner SM, Fusch C, Schwanebeck U, Köhler C, et al. Insulin-resistant patients with type 2 diabetes mellitus have higher serum leptin levels independently of body fat mass. Acta Diabetol (2002) 39:105–10. doi: 10.1007/s005920200027
155. Hildebrandt X, Ibrahim M, Peltzer N. Cell death and inflammation during obesity: “Know my methods, WAT(son)”. Cell Death Differ (2023) 30:279–92. doi: 10.1038/s41418-022-01062-4
156. Hotamisligil GS. Inflammation and metabolic disorders. Nature (2006) 444:860–7. doi: 10.1038/nature05485
157. Curat CA, Miranville A, Sengenès C, Diehl M, Tonus C, Busse R, et al. From blood monocytes to adipose tissue-resident macrophages: induction of diapedesis by human mature adipocytes. Diabetes (2004) 53:1285–92. doi: 10.2337/diabetes.53.5.1285
158. La Cava A. Leptin in inflammation and autoimmunity. Cytokine (2017) 98:51–8. doi: 10.1016/j.cyto.2016.10.011
159. Pérez-Pérez A, Vilariño-García T, Fernández-Riejos P, Martín-González J, Segura-Egea JJ, Sánchez-Margalet V. Role of leptin as a link between metabolism and the immune system. Cytokine Growth Factor Rev (2017) 35:71–84. doi: 10.1016/j.cytogfr.2017.03.001
160. Francisco V, Pino J, Campos-Cabaleiro V, Ruiz-Fernández C, Mera A, Gonzalez-Gay MA, et al. Obesity, fat mass and immune system: role for leptin. Front Physiol (2018) 9:640. doi: 10.3389/fphys.2018.00640
161. Loffreda S, Yang SQ, Lin HZ, Karp CL, Brengman ML, Wang DJ, et al. Leptin regulates proinflammatory immune responses. FASEB J (1998) 12:57–65. doi: 10.1096/fsb2fasebj.12.1.57
162. Gruen ML, Hao M, Piston DW, Hasty AH. Leptin requires canonical migratory signaling pathways for induction of monocyte and macrophage chemotaxis. Am J Physiol Cell Physiol (2007) 293:C1481–8. doi: 10.1152/ajpcell.00062.2007
163. Yao J, Wu D, Qiu Y. Adipose tissue macrophage in obesity-associated metabolic diseases. Front Immunol (2022) 13:977485. doi: 10.3389/fimmu.2022.977485
164. de Candia P, Prattichizzo F, Garavelli S, Alviggi C, La Cava A, Matarese G. The pleiotropic roles of leptin in metabolism, immunity, and cancer. J Exp Med (2021) 218:e20191593. doi: 10.1084/jem.20191593
165. Prasad AS. Zinc in human health: effect of zinc on immune cells. Mol Med (2008) 14:353–7. doi: 10.2119/2008-00033.Prasad
166. Rink L, Haase H. Zinc homeostasis and immunity. Trends Immunol (2007) 28:1–4. doi: 10.1016/j.it.2006.11.005
167. Haase H, Rink L. Functional significance of zinc-related signaling pathways in immune cells. Annu Rev Nutr (2009) 29:133–52. doi: 10.1146/annurev-nutr-080508-141119
168. Bonaventura P, Benedetti G, Albarède F, Miossec P. Zinc and its role in immunity and inflammation. Autoimmun Rev (2015) 14:277–85. doi: 10.1016/j.autrev.2014.11.008
169. Prasad AS. Zinc is an antioxidant and anti-inflammatory agent: its role in human health. Front Nutr (2014) 1:14. doi: 10.3389/fnut.2014.00014
170. Jung S, Kim MK, Choi BY. The relationship between zinc status and inflammatory marker levels in rural korean adults aged 40 and older. PloS One (2015) 10:e0130016. doi: 10.1371/journal.pone.0130016
171. Castell JV, Gómez-Lechón MJ, David M, Hirano T, Kishimoto T, Heinrich PC. Recombinant human interleukin-6 (IL-6/BSF-2/HSF) regulates the synthesis of acute phase proteins in human hepatocytes. FEBS Lett (1988) 232:347–50. doi: 10.1016/0014-5793(88)80766-x
172. Kroder G, Bossenmaier B, Kellerer M, Capp E, Stoyanov B, Mühlhöfer A, et al. Tumor necrosis factor-alpha- and hyperglycemia-induced insulin resistance. Evidence for different mechanisms and different effects on insulin signaling. J Clin Invest. (1996) 97:1471–7. doi: 10.1172/JCI118569
173. Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science (1996) 271:665–8. doi: 10.1126/science.271.5249.665
174. Payahoo L, Ostadrahimi A, Mobasseri M, Khaje Bishak Y, Farrin N, Asghari Jafarabadi M, et al. Effects of zinc supplementation on the anthropometric measurements, lipid profiles and fasting blood glucose in the healthy obese adults. Adv Pharm Bull (2013) 3:161–5. doi: 10.5681/apb.2013.027
175. Abdollahi S, Toupchian O, Jayedi A, Meyre D, Tam V, Soltani S. Zinc supplementation and body weight: A systematic review and dose-response meta-analysis of randomized controlled trials. Adv Nutr (2020) 11:398–411. doi: 10.1093/advances/nmz084
176. Gaetke LM, McClain CJ, Talwalkar RT, Shedlofsky SI. Effects of endotoxin on zinc metabolism in human volunteers. Am J Physiol (1997) 272:E952–6. doi: 10.1152/ajpendo.1997.272.6.E952
177. Mocchegiani E, Giacconi R, Cipriano C, Muti E, Gasparini N, Malavolta M. Are zinc-bound metallothionein isoforms (I+II and III) involved in impaired thymulin production and thymic involution during ageing? Immun Ageing (2004) 1:5. doi: 10.1186/1742-4933-1-5
178. Krezel A, Maret W. Dual nanomolar and picomolar Zn(II) binding properties of metallothionein. J Am Chem Soc (2007) 129:10911–21. doi: 10.1021/ja071979s
179. Reyes JG. Zinc transport in mammalian cells. Am J Physiol (1996) 270:C401–10. doi: 10.1152/ajpcell.1996.270.2.C401
180. Frederickson CJ, Koh JY, Bush AI. The neurobiology of zinc in health and disease. Nat Rev Neurosci (2005) 6:449–62. doi: 10.1038/nrn1671
181. Ishihara H, Maechler P, Gjinovci A, Herrera PL, Wollheim CB. Islet beta-cell secretion determines glucagon release from neighbouring alpha-cells. Nat Cell Biol (2003) 5:330–5. doi: 10.1038/ncb951
182. Yamasaki S, Sakata-Sogawa K, Hasegawa A, Suzuki T, Kabu K, Sato E, et al. Zinc is a novel intracellular second messenger. J Cell Biol (2007) 177:637–45. doi: 10.1083/jcb.200702081
183. Taylor KM, Hiscox S, Nicholson RI, Hogstrand C, Kille P. Protein kinase CK2 triggers cytosolic zinc signaling pathways by phosphorylation of zinc channel ZIP7. Sci Signal (2012). doi: 10.1126/scisignal.2002585
184. Nishida K, Yamasaki S. Zinc signaling by “Zinc wave”. In: Fukada T, Kambe T, editors. Zinc signals in cellular functions and disorders. Tokyo: Springer (2014) 5(210):ra11. doi: 10.1007/978-4-431-55114-0_5
185. Maywald M, Wessels I, Rink L. Zinc signals and immunity. Int J Mol Sci (2017) 18:2222. doi: 10.3390/ijms18102222
186. Woodruff G, Bouwkamp CG, de Vrij FM, Lovenberg T, Bonaventure P, Kushner SA, et al. The zinc transporter SLC39A7 (ZIP7) is essential for regulation of cytosolic zinc levels. Mol Pharmacol (2018) 94:1092–100. doi: 10.1124/mol.118.112557
188. Bell SG, Vallee BL. The metallothionein/thionein system: an oxidoreductive metabolic zinc link. Chembiochem (2009) 10:55–62. doi: 10.1002/cbic.200800511
189. Kimura T, Kambe T. The functions of metallothionein and ZIP and znT transporters: an overview and perspective. Int J Mol Sci (2016) 17:336. doi: 10.3390/ijms17030336
190. Maret W. The function of zinc metallothionein: a link between cellular zinc and redox state. J Nutr (2000) 30:1455S–8S. doi: 10.1093/jn/130.5.1455S
191. Jacob C, Maret W, Vallee BL. Control of zinc transfer between thionein, metallothionein, and zinc proteins. Proc Natl Acad Sci U S A. (1998) 95:3489–94. doi: 10.1073/pnas.95.7.3489
192. Hübner C, Haase H. Interactions of zinc- and redox-signaling pathways. Redox Biol (2021) 41:101916. doi: 10.1016/j.redox.2021.101916
193. Marinho HS, Real C, Cyrne L, Soares H, Antunes F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol (2014) 2:535–62. doi: 10.1016/j.redox.2014.02.006
194. Andrews GK. Cellular zinc sensors: MTF-1 regulation of gene expression. Biometals (2001) 14:223–37. doi: 10.1023/a:1012932712483
195. Hardyman JE, Tyson J, Jackson KA, Aldridge C, Cockell SJ, Wakeling LA, et al. Zinc sensing by metal-responsive transcription factor 1 (MTF1) controls metallothionein and ZnT1 expression to buffer the sensitivity of the transcriptome response to zinc. Metallomics (2016) 8:337–43. doi: 10.1039/C5MT00305A
196. Maret W. Zinc coordination environments in proteins as redox sensors and signal transducers. Antioxid Redox Signal (2006) 8:1419–41. doi: 10.1089/ars.2006.8.1419
197. Bettger WJ. Zinc and selenium, site-specific versus general antioxidation. Can J Physiol Pharmacol (1993) 71:721–4. doi: 10.1139/y93-108
198. Bettger WJ, O’Dell BL. A critical physiological role of zinc in the structure and function of biomembranes. Life Sci (1981) 28:1425–38. doi: 10.1016/0024-3205(81)90374-x
199. Girotti AW, Thomas JP, Jordan JE. Inhibitory effect of zinc on free radical lipid peroxidation in erythrocyte membranes. J Free Radic Biol Med (1985) 1:395–401. doi: 10.1016/0748-5514(85)90152
200. Zago MP, Oteiza PI. The antioxidant properties of zinc: interactions with iron and antioxidants. Free Radic Biol Med (2001) 31:266–74. doi: 10.1016/s0891-5849(01)00583-4
201. Stitt MS, Wasserloos KJ, Tang X, Liu X, Pitt BR, Croix St CM. Nitric oxide-induced nuclear translocation of the metal responsive transcription factor, MTF-1 is mediated by zinc release from metallothionein. Vascul Pharmacol (2006) 44:149–55. doi: 10.1016/j.vph.2005.10.004
202. Kroncke KD, Fehsel K, Schmidt T, Zenke FT, Dasting I, Wesener JR, et al. Nitric oxide destroys zinc-sulfur clusters inducing zinc release from metallothionein and inhibition of the zinc finger-type yeast transcription activator LAC9. Biochem Biophys Res Commun (1994) 200:1105–10. doi: 10.1006/bbrc.1994.1564
203. Pearce LL, Wasserloos K, St Croix CM, Gandley R, Levitan ES, Pitt BR. Metallothionein, nitric oxide and zinc homeostasis in vascular endothelial cells. J Nutr (2000) 130:1467S–70S. doi: 10.1093/jn/130.5.1467S
204. Quesada AR, Byrnes RW, Krezoski SO, Petering DH. Direct reaction of H2O2 with sulfhydryl groups in HL-60 cells: zinc-metallothionein and other sites. Arch Biochem Biophys (1996) 334(2):241–50. doi: 10.1006/abbi.1996.0452
205. Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol (2013) 53:401–26. doi: 10.1146/annurev-pharmtox-011112-140320
206. Kopacz A, Kloska D, Forman HJ, Jozkowicz A, Grochot-Przeczek A. Beyond repression of Nrf2: An update on Keap1. Free Radic Biol Med (2020) 157:63–74. doi: 10.1016/j.freeradbiomed.2020.03.023
207. McMahon M, Lamont DJ, Beattie KA, Hayes JD. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proc Natl Acad Sci U.S.A. (2010) 107:18838–43. doi: 10.1073/pnas.1007387107
208. Kang M-I, Kobayashi A, Wakabayashi N, Kim S-G, Yamamoto M. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc Natl Acad Sci U S A. (2004) 101:2046–51. doi: 10.1073/pnas.0308347100
209. Krezel A, Bal W. Studies of zinc(II) and nickel(II) complexes of GSH, GSSG and their analogs shed more light on their biological relevance. Bioinorg Chem Appl (2004) 2:293–305. doi: 10.1155/S1565363304000172
210. Jiang LJ, Maret W, Vallee BL. The glutathione redox couple modulates zinc transfer from metallothionein to zinc-depleted sorbitol dehydrogenase. Proc Natl Acad Sci U.S.A. (1998) 95:3483–8. doi: 10.1073/pnas.95.7.3483
211. Ha KN, Chen Y, Cai J, Sternberg P Jr. Increased glutathione synthesis through an ARE-Nrf2-dependent pathway by zinc in the RPE: implication for protection against oxidative stress. Invest Ophthalmol Vis Sci (2006) 47:2709–15. doi: 10.1167/iovs.05-1322
212. Clegg MS, Hanna LA, Niles BJ, Momma TY, Keen CL. Zinc deficiency-induced cell death. IUBMB Life (2005) 57:661–9. doi: 10.1080/15216540500264554
213. Omata Y, Salvador GA, Supasai S, Keenan AH, Oteiza PI. Decreased zinc availability affects glutathione metabolism in neuronal cells and in the developing brain. Toxicol Sci (2013) 133:90–100. doi: 10.1093/toxsci/kft022
214. Lelie HL, Liba A, Bourassa MW, Chattopadhyay M, Chan PK, Gralla EB, et al. Copper and zinc metallation status of copper-zinc superoxide dismutase from amyotrophic lateral sclerosis transgenic mice. J Biol Chem (2011) 286:2795–806. doi: 10.1074/jbc.M110.186999
215. Martyn KD, Frederick LM, von Loehneysen K, Dinauer MC, Knaus UG. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal (2006) 18:69–82. doi: 10.1016/j.cellsig.2005.03.023
216. Li MS, Adesina SE, Ellis CL, Gooch JL, Hoover RS, Williams CR. NADPH oxidase-2 mediates zinc deficiency-induced oxidative stress and kidney damage. Am J Physiol Cell Physiol (2017) 312:C47–55. doi: 10.1152/ajpcell.00208.2016
217. Hemmens B, Goessler W, Schmidt K, Mayer B. Role of bound zinc in dimer stabilization but not enzyme activity of neuronal nitric-oxide synthase. J Biol Chem (2000) 275:35786–91. doi: 10.1074/jbc.M005976200
218. Li H, Raman CS, Glaser CB, Blasko E, Young TA, Parkinson JF, et al. Crystal structures of zinc-free and -bound heme domain of human inducible nitric-oxide synthase. Implications for dimer stability and comparison with endothelial nitric-oxide synthase. J Biol Chem (1999) 274:21276 –21284. doi: 10.1074/jbc.274.30.21276
219. Abou-Mohamed G, Papapetropoulos A, Catravas JD, Caldwell RW. Zn2+ inhibits nitric oxide formation in response to lipopolysaccharides: implication in its anti-inflammatory activity. Eur J Pharmacol (1998) 341(2-3):265–72. doi: 10.1016/s0014-2999(97)01416-7
220. St Croix CM, Wasserloos KJ, Dineley KE, Reynolds IJ, Levitan ES, Pitt BR. Nitric oxide-induced changes in intracellular zinc homeostasis are mediated by metallothionein/thionein. Am J Physiol Lung Cell Mol Physiol (2002) 282:L185–92. doi: 10.1152/ajplung.00267.2001
221. Aravindakumar CT, Ceulemans J, De Ley M. Nitric oxide induces Zn2+ release from metallothionein by destroying zinc-sulphur clusters without concomitant formation of S-nitrosothiol. Biochem J (1999) 344:253–8. doi: 10.1042/0264-6021:3440253
222. Cortese-Krott MM, Kulakov L, Opländer C, Kolb-Bachofen V, Kröncke K-D, Suschek CV. Zinc regulates iNOS-derived nitric oxide formation in endothelial cells. Redox Biol (2014) 2:945–54. doi: 10.1016/j.redox.2014.06.011
223. Spahl DU, Berendji-Grün D, Suschek CV, Kolb-Bachofen V, Kröncke K-D. Regulation of zinc homeostasis by inducible NO synthase-derived NO: nuclear metallothionein translocation and intranuclear Zn2+ release. Proc Natl Acad Sci U S A. (2003) 100(24):13952–7. doi: 10.1073/pnas.2335190100
224. Shumilla JA, Wetterhahn KE, Barchowsky A. Inhibition of NF-kappa B binding to DNA by chromium, cadmium, mercury, zinc, and arsenite in vitro: evidence of a thiol mechanism. Arch Biochem Biophys (1998) 349:356–62. doi: 10.1006/abbi.1997.0470
225. Berendji D, Kolb-Bachofen V, Meyer KL, Grapenthin O, Weber H, Wahn V, et al. Nitric oxide mediates intracytoplasmic and intranuclear zinc release. FEBS Lett (1997) 405:37–41. doi: 10.1016/s0014-5793(97)00150-6
226. Tartler U, Kröncke KD, Meyer KL, Suschek CV, Kolb-Bachofen V. Nitric oxide interferes with islet cell zinc homeostasis. Nitric Oxide (2000) 4:609–14. doi: 10.1006/niox.2000.0314
227. Wang J, Wang H. Oxidative stress in pancreatic beta cell regeneration. Oxid Med Cell Longev (2017) 2017:1930261. doi: 10.1155/2017/1930261
228. Darville MI, Eizirik DL. Regulation by cytokines of the inducible nitric oxide synthase promoter in insulin-producing cells. Diabetologia (1998) 41:1101–8. doi: 10.1007/s001250051036
229. Loweth AC, Williams GT, Scarpello JH, Morgan NG. Evidence for the involvement of cGMP and protein kinase G in nitric oxide-induced apoptosis in the pancreatic B-cell line, HIT-T15. FEBS Lett (1997) 400:285–8. doi: 10.1016/s0014-5793(96)01392-0
230. Chen C-W, Chen L-K, Huang T-Y, Yang D-M, Liu S-Y, Tsai P-J, et al. Nitric oxide mobilizes intracellular zn 2+ via the GC/cGMP/PKG signaling pathway and stimulates adipocyte differentiation. Int J Mol Sci (2022) 23:5488. doi: 10.3390/ijms23105488
231. Sansbury BE, Cummins TD, Tang Y, Hellmann J, Holden CR, Harbeson MA, et al. Overexpression of endothelial nitric oxide synthase prevents diet-induced obesity and regulates adipocyte phenotype. Circ Res (2012) 111:1176–89. doi: 10.1161/CIRCRESAHA.112.266395
232. Engeli S, Janke J, Gorzelniak K, Böhnke J, Ghose N, Lindschau C, et al. Regulation of the nitric oxide system in human adipose tissue. J Lipid Res (2004) 45:1640–8. doi: 10.1194/jlr.M300322-JLR200
233. Thomas RC, Coles JA, Deitmer JW. Homeostatic muffling. Nature (1991) 350:564. doi: 10.1038/350564b0
234. Haase H, Maret W. Partial oxidation and oxidative polymerization of metallothionein. Electrophoresis (2008) 29:4169–76. doi: 10.1002/elps.200700922
235. Kägi JH. Overview of metallothionein. Methods Enzymol (1991) 205:613–26. doi: 10.1016/0076-6879(91)05145-l
236. Maret W. Metals on the move: zinc ions in cellular regulation and in the coordination dynamics of zinc proteins. Biometals (2011) 24:411–8. doi: 10.1007/s10534-010-9406-1
237. Colvin RA, Holmes WR, Fontaine CP, Maret W. Cytosolic zinc buffering and muffling: their role in intracellular zinc homeostasis. Metallomics (2010) 2:306–17. doi: 10.1039/b926662c
238. Zhou Z, Kang YJ. Immunocytochemical localization of metallothionein and its relation to doxorubicin toxicity in transgenic mouse heart. Am J Pathol (2000) 156:1653–62. doi: 10.1016/S0002-9440(10)65036-5
239. Ye B, Maret W, Vallee BL. Zinc metallothionein imported into liver mitochondria modulates respiration. Proc Natl Acad Sci U.S.A. (2001) 98:2317–22. doi: 10.1073/pnas.041619198
240. Hennigar SR, Kelleher SL. Zinc networks: the cell-specific compartmentalization of zinc for specialized functions. Biol Chem (2012) 393:565–78. doi: 10.1515/hsz-2012-0128
241. Kambe T, Taylor KM, Fu D. Zinc transporters and their functional integration in mammalian cells. J Biol Chem (2021) 296:100320. doi: 10.1016/j.jbc.2021.100320
242. Palmiter RD. Constitutive expression of metallothionein-III (MT-III), but not MT-I, inhibits growth when cells become zinc deficient. Toxicol Appl Pharmacol (1995) 135:139–46. doi: 10.1006/taap.1995.1216
243. Palmiter RD. Protection against zinc toxicity by metallothionein and zinc transporter 1. Proc Natl Acad Sci U S A. (2004) 101:4918–23. doi: 10.1073/pnas.0401022101
244. Sankavaram K, Freake HC. The effects of transformation and ZnT-1 silencing on zinc homeostasis in cultured cells. J Nutr Biochem (2012) 23:629–34. doi: 10.1016/j.jnutbio.2011.03.006
245. Lu Q, Haragopal H, Slepchenko KG, Stork C, Li YV. Intracellular zinc distribution in mitochondria, ER and the Golgi apparatus. Int J Physiol Pathophysiol Pharmacol (2016) 8:35–43.
246. Suzuki T, Ishihara K, Migaki H, Matsuura W, Kohda A, Okumura K, et al. Zinc transporters, ZnT5 and ZnT7, are required for the activation of alkaline phosphatases, zinc-requiring enzymes that are glycosylphosphatidylinositol-anchored to the cytoplasmic membrane. J Biol Chem (2005) 280:637–43. doi: 10.1074/jbc.M411247200
247. Sanford L, Carpenter MC, Palmer AE. Intracellular Zn2+ transients modulate global gene expression in dissociated rat hippocampal neurons. Sci Rep (2019) 9:9411. doi: 10.1038/s41598-019-45844-2
248. Langmade SJ, Ravindra R, Daniels PJ, Andrews GK. The transcription factor MTF-1 mediates metal regulation of the mouse ZnT1 gene. J Biol Chem (2000) 275:34803–9. doi: 10.1074/jbc.M007339200
249. Laity JH, Andrews GK. Understanding the mechanisms of zinc-sensing by metal-response element binding transcription factor-1 (MTF-1) Arch. Biochem Biophys (2007) 463:201–10. doi: 10.1016/j.abb.2007.03.019
250. Nishito Y, Kambe T. Zinc transporter 1 (ZNT1) expression on the cell surface is elaborately controlled by cellular zinc levels. J Biol Chem (2019) 294:15686–97. doi: 10.1074/jbc.RA119.010227
251. Noh H, Paik HY, Kim J, Chung J. The changes of zinc transporter ZnT gene expression in response to zinc supplementation in obese women. Biol Trace Elem Res (2014) 162:38–45. doi: 10.1007/s12011-014-0128-z
252. dos Santos Rocha PB, de Castro Amorim A, de Sousa AF, do Monte SJ, da Mata Sousa LC, do Nascimento Nogueira N, et al. Expression of the zinc transporters genes and metallothionein in obese women. Biol Trace Elem Res (2011) 143:603–11. doi: 10.1007/s12011-010-8887-7
253. McMahon RJ, Cousins RJ. Regulation of the zinc transporter ZnT-1 by dietary zinc. Proc Natl Acad Sci U.S.A. (1998) 95:4841–6. doi: 10.1073/pnas.95.9.4841
254. Kambe T, Narita H, Yamaguchi-Iwai Y, Hirose J, Amano T, Sugiura N, et al. Cloning and characterization of a novel mammalian zinc transporter, zinc transporter 5, abundantly expressed in pancreatic beta cells. J Biol Chem (2002) 277:19049–55. doi: 10.1074/jbc.M200910200
255. Smidt K, Pedersen SB, Brock B, Schmitz O, Fisker S, Bendix J, et al. Zinc-transporter genes in human visceral and subcutaneous adipocytes: lean versus obese. Mol Cell Endocrinol (2007) 264:68–73. doi: 10.1016/j.mce.2006.10.010
256. Maxel T, Smidt K, Larsen A, Bennetzen M, Cullberg K, Fjeldborg K, et al. Gene expression of the zinc transporter ZIP14 (SLC39a14) is affected by weight loss and metabolic status and associates with PPARγ in human adipose tissue and 3T3-L1 pre-adipocytes. BMC Obes (2015) 2:46. doi: 10.1186/s40608-015-0076-y
257. Tominaga K, Kagata T, Johmura Y, Hishida T, Nishizuka M, Imagawa M, et al. SLC39A14, a LZT protein, is induced in adipogenesis and transports zinc. FEBS J (2005) 272:1590–9. doi: 10.1111/j.1742-4658.2005.04580.x
258. Troche C, Aydemir TB, Cousins RJ. Zinc transporter Slc39a14 regulates inflammatory signaling associated with hypertrophic adiposity. Am J Physiol Endocrinol Metab (2016) 310:E258–68. doi: 10.1152/ajpendo.00421.2015
259. Aydemir TB, Chang SM, Guthrie GJ, Maki AB, Ryu MS, Karabiyik A, et al. Zinc transporter ZIP14 functions in hepatic zinc, iron and glucose homeostasis during the innate immune response (endotoxemia). PloS One (2012) 7:e48679. doi: 10.1371/journal.pone.0048679
260. Sun Q, Li Q, Zhong W, Zhang J, Sun X, Tan X, et al. Dysregulation of hepatic zinc transporters in a mouse model of alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol (2014) 307:G313–22. doi: 10.1152/ajpgi.00081.2014
261. Lichten LA, Liuzzi JP, Cousins RJ. Interleukin-1beta contributes via nitric oxide to the upregulation and functional activity of the zinc transporter Zip14 (Slc39a14) in murine hepatocytes. Am J Physiol Gastrointest Liver Physiol (2009) 296:G860–7. doi: 10.1152/ajpgi.90676.2008
262. Liuzzi JP, Lichten LA, Rivera S, Blanchard RK, Aydemir TB, Knutson MD, et al. Interleukin-6 regulates the zinc transporter Zip14 in liver and contributes to the hypozincemia of the acute-phase response. Proc Natl Acad Sci U.S.A. (2005) 102:6843–8. doi: 10.1073/pnas.0502257102
263. Aydemir TB, Troche C, Kim MH, Cousins RJ. Hepatic ZIP14-mediated zinc transport contributes to endosomal insulin receptor trafficking and glucose metabolism. J Biol Chem (2016) 291:23939–51. doi: 10.1074/jbc.M116.748632
264. Kim MH, Aydemir TB, Kim J, Cousins RJ. Hepatic ZIP14-mediated zinc transport is required for adaptation to endoplasmic reticulum stress. Proc Natl Acad Sci USA (2017) 114(29):E5805–14. doi: 10.1073/pnas.1704012114
265. Delibegovic M, Zimmer D, Kauffman C, Rak K, Hong EG, Cho YR, et al. Liver-specific deletion of protein-tyrosine phosphatase 1B (PTP1B) improves metabolic syndrome and attenuates diet-induced endoplasmic reticulum stress. Diabetes (2009) 58:590–9. doi: 10.2337/db08-0913
266. Norouzi S, Adulcikas J, Henstridge DC, Sonda S, Sohal SS, Myers S. The zinc transporter zip7 is downregulated in skeletal muscle of insulin-resistant cells and in mice fed a high-fat diet. Cells (2019) 8:663. doi: 10.3390/cells8070663
267. Taylor KM, Vichova P, Jordan N, Hiscox S, Hendley R, Nicholson RI. ZIP7-mediated intracellular zinc transport contributes to aberrant growth factor signaling in antihormone-resistant breast cancer Cells. Endocrinology (2008) 149:4912–20. doi: 10.1210/en.2008-0351
268. Tuncay E, Bitirim VC, Durak A, Carrat GRJ, Taylor KM, Rutter GA, et al. Hyperglycemia-induced changes in ZIP7 and znT7 expression cause zn2+ Release from the sarco(endo)plasmic reticulum and mediate ER stress in the heart. Diabetes (2017) 66:1346–58. doi: 10.2337/db16-1099
269. Bin BH, Fukada T, Hosaka T, Yamasaki S, Ohashi W, Hojyo S, et al. Biochemical characterization of human ZIP13 protein: a homo-dimerized zinc transporter involved in the spondylocheiro dysplastic Ehlers-Danlos syndrome. J Biol Chem (2011) 286:40255–65. doi: 10.1074/jbc.M111.256784
270. Ikeda K, Maretich P, Kajimura S. The common and distinct features of brown and beige adipocytes. Trends Endocrinol Metab (2018) 29:191–200. doi: 10.1016/j.tem.2018.01.001
271. Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, Jackson AU, et al. New genetic loci implicated in fasting glucose 1. homeostasis and their impact on type 2 diabetes risk. Nat Genet (2010) 42:105–16. doi: 10.1038/ng.520
272. Fukunaka A, Fukada T, Bhin J, Suzuki L, Tsuzuki T, Takamine Y, et al. Zinc transporter ZIP13 suppresses beige adipocyte biogenesis and energy expenditure by regulating C/EBP-β expression. PloS Genet (2017) 13:e1006950. doi: 10.1371/journal.pgen.1006950
273. Kirschke CP, Huang L. ZnT7, a novel mammalian zinc transporter, accumulates zinc in the Golgi apparatus. J Biol Chem (2003) 278:4096–102. doi: 10.1074/jbc.M207644200
274. Huang L, Yu YY, Kirschke CP, Gertz ER, Lloyd KK. Znt7 (Slc30a7)-deficient mice display reduced body zinc status and body fat accumulation. J Biol Chem (2007) 282:37053–63. doi: 10.1074/jbc.M706631200
275. Tepaamorndech S, Kirschke CP, Pedersen TL, Keyes WR, Newman JW, Huang L. Zinc transporter 7 deficiency affects lipid synthesis in adipocytes by inhibiting insulin-dependent Akt activation and glucose uptake. FEBS J (2016) 283:378–94. doi: 10.1111/febs.1358
276. Huang L, Kirschke CP, Lay YA, Levy LB, Lamirande DE, Zhang PH. Znt7-null mice are more susceptible to diet-induced glucose intolerance and insulin resistance. J Biol Chem (2012) 287:33883–96. doi: 10.1074/jbc.M111.309666
277. Porter SA, Massaro JM, Hoffmann U, Vasan RS, O’Donnel CJ, Fox CS. Abdominal subcutaneous adipose tissue: a protective fat depot? Diabetes Care (2009) 32:1068–75. doi: 10.2337/dc08-2280
278. Nicolson TJ, Bellomo EA, Wijesekara N, Loder MK, Baldwin JM, Gyulkhandanyan AV, et al. Insulin storage and glucose homeostasis in mice null for the granule zinc transporter ZnT8 and studies of the type 2 diabetes-associated variants. Diabetes (2009) 58:2070–83. doi: 10.2337/db09-055
279. Cauchi S, Nead KT, Choquet H, Horber F, Potoczna N, Balkau B, et al. The genetic susceptibility to type 2 diabetes may be modulated by obesity status: implications for association studies. BMC Med Genet (2008) 9:45. doi: 10.1186/1471-2350-9-45
280. Wu Y, Li H, Loos RJ, Yu Z, Ye X, Chen L, et al. Common variants in CDKAL1, CDKN2A/B, IGF2BP2, SLC30A8, and HHEX/IDE genes are associated with type 2 diabetes and impaired fasting glucose in a Chinese Han population. Diabetes (2008) 57:2834–42. doi: 10.2337/db08-0047
281. Hardy AB, Wijesekara N, Genkin I, Prentice KJ, Bhattacharjee A, Kong D, et al. Effects of high-fat diet feeding on Znt8-null mice: differences between β-cell and global knockout of Znt8. Am J Physiol Endocrinol Metab (2012) 302:E1084–96. doi: 10.1152/ajpendo.00448.2011
282. Tamaki M, Fujitani Y, Hara A, Uchida T, Tamura Y, Takeno K, et al. The diabetes-susceptible gene SLC30A8/ZnT8 regulates hepatic insulin clearance. J Clin Invest. (2013) 123:4513–24. doi: 10.1172/JCI68807
Keywords: zinc status, oxidative stress, buffering, ZnT, ZIP, metallothioneins
Citation: Franco C and Canzoniero LMT (2024) Zinc homeostasis and redox alterations in obesity. Front. Endocrinol. 14:1273177. doi: 10.3389/fendo.2023.1273177
Received: 05 August 2023; Accepted: 18 December 2023;
Published: 08 January 2024.
Edited by:
Cristoforo Silvestri, Laval University, CanadaReviewed by:
Fredy Alexander Guevara Agudelo, Laval University, CanadaPaola Lanuti, G. D’Annunzio University of Chieti-Pescara, Italy
Copyright © 2024 Franco and Canzoniero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lorella Maria Teresa Canzoniero, Y2Fuem9uaWVyb0B1bmlzYW5uaW8uaXQ=