 Ashish Gupta1†‡Rajeev Kasaliwal2†‡
Ashish Gupta1†‡Rajeev Kasaliwal2†‡ Liza Das3‡Surendra Kumar Sharma2‡
Liza Das3‡Surendra Kumar Sharma2‡ Vaishali Kaur1‡Alexandre Vasiljevic4‡
Vaishali Kaur1‡Alexandre Vasiljevic4‡ Véronique Raverot5‡
Véronique Raverot5‡ Márta Korbonits6‡
Márta Korbonits6‡ Pinaki Dutta1*‡
Pinaki Dutta1*‡- 1Department of Endocrinology, Post Graduate Institute of Medical Education and Research (PGIMER), Chandigarh, India
- 2Department of Endocrinology, Mahatma Gandhi Medical College and Hospital, Jaipur, India
- 3Department of Telemedicine, Post Graduate Institute of Medical Education & Research (PGIMER), Chandigarh, India
- 4PraticienHospitalier, PharmacienBiologiste, Laboratoired’hormonologie, Hospices Civils de Lyon (Centre HospitalierUniversitaire de Lyon) CHU Lyon, Lyon, France
- 5Service d’Anatomie et CytologiePathologiques, Hospices Civils de Lyon (Centre Hospitalier Universitaire de Lyon), CHU Lyon, Lyon, France
- 6Department of Endocrinology, William Harvey Research Institute, Barts and the London School of Medicine, Queen Mary University of London, London, United Kingdom
Acromegaly due to ectopic secretion of growth hormone-releasing hormone (GHRH) is a rare disorder. The signs and symptoms of ectopic acromegaly are indistinguishable from acromegaly due to a somatotroph adenoma. A 35-year-old female presented with secondary amenorrhea for 10 years, intermittent headache, and reduced vision in both eyes for 4 years, which worsened over 4 months before presentation. Additionally, she was diagnosed with uncontrolled diabetes mellitus. On examination, she had coarse facial features, a fleshy nose, and acral enlargement. She had diminished visual acuity (left>right) and bitemporal hemianopia on perimetry. Biochemical investigations revealed elevated IGF-1 [588 ng/ml, reference range (RR) 100–242], markedly elevated basal growth hormone (>80 ng/ml; RR, 0.12–9.88), and hyperprolactinemia in the tumoral range (832 ng/ml; RR, 5–25). MRI sella demonstrated a 22×30×34mm sellar-suprasellar mass with T2 hypointensity. Chest imaging revealed a 75×87×106mm left lung mass, which was found to be a well-differentiated neuroendocrine tumor (NET) on biopsy. Plasma GHRH levels were elevated [38,088 ng/l; RR, <250–300], and a diagnosis of ectopic acromegaly secondary to lung neuroendocrine tumor was considered. During workup, the patient developed in-hospital pituitary apoplexy, which improved with medical management. After a left pneumonectomy, her clinical features of acromegaly improved, her diabetes underwent remission, and there was a marked reduction in plasma GHRH and pituitary size. Histopathology was suggestive of a neuroendocrine tumor, with immunohistochemistry positive for GHRH and negative for prolactin. Her final diagnosis was ectopic acromegaly due to GHRH secreting a lung NET with pituitary somatotroph and lactotroph pituitary hyperplasia and apoplexy in the hyperplastic pituitary.
Introduction
Acromegaly is a systemic disorder caused by growth hormone (GH) excess and is characterized by typical facial, acral, skeletal, and systemic manifestations affecting all the organ systems (1). Most often it occurs due to a GH-secreting tumor located in the sella, known as eutopic acromegaly. Ectopic acromegaly is extremely rare, comprising <1% of all acromegaly cases. The sources of ectopic growth hormone-releasing hormone (GHRH) include neuroendocrine tumors (NETs) arising from the pancreas, lung, thymus and appendix, pheochromocytomas, paragangliomas, and hypothalamic choristomas and gangliocytomas (2). Herein, we present a unique case of acromegaly due to a GHRH-secreting lung NET (bronchial carcinoid) causing enlargement of the pituitary gland, pituitary apoplexy, tumoral range hyperprolactinemia with remarkable resolution of clinical features hyperprolactinemia and IGF-1 levels after resection of the primary lung tumor.
Case presentation
A 35-year-old female presented with secondary amenorrhea for 10 years, intermittent headache, and reduced vision in both eyes for 4 years before presentation, which had worsened over the last 4 months. There was no history of galactorrhea, weight gain, striae, proximal muscle weakness, or other features suggestive of Cushing’s syndrome. She was diagnosed with diabetes mellitus 4 months before presentation to our hospital, with poor blood glucose control despite receiving four oral hypoglycemic agents [glimepiride (4 mg), metformin (1,000 mg), sitagliptin (100 mg), and dapagliflozin (10 mg) daily]. She had two children; she had breastfed the younger for 18 months 12 years ago. There was no personal or family history of pituitary or other tumors suggestive of multiple endocrine neoplasia type 1 (MEN1). On examination, her height was 140 cm (familial short stature), her BMI was 27kg/m², and she had coarse facial features, a fleshy nose, and acral enlargement. Her visual acuity was 2/60 in the right eye and 6/60 in the left eye. Fundus examination revealed bilateral moderate non-proliferative diabetic retinopathy (left eye> right eye). Visual field testing showed bitemporal hemianopia. Other aspects of systemic examination were normal. Her laboratory investigations are depicted in Table 1. She had raised baseline GH (>80 ng/ml) and IGF-1 [588 ng/ml; reference range (RR) 100–242]. She also had hyperprolactinemia [prolactin 330 and 832 ng/ml on two separate occasions (RR, 5–25ng/ml)]. Macroprolactin was normal. Further work-up revealed secondary hypocortisolism, hypogonadism, and hypothyroidism. Serum calcium (9.3 mg/dl; RR, 8.8–10.2) and phosphate (4.3 mg/dl; RR, 2.7–4.5) were normal. She was started on hormone replacement with daily prednisone (5 mg) and levothyroxine (100 mg). Contrast-enhanced dynamic MRI of the sella showed a 22×30×34mm sellar-suprasellar mass closely abutting the cavernous segment of the bilateral internal carotid artery and splaying of the optic chiasma, reported as pituitary macroadenoma (Figure 1). Initially, for headache and visual symptoms, she consulted a neurosurgeon and was scheduled for elective transsphenoidal excision for the pituitary lesion; however, as part of the pre-anesthetic check-up and evaluation of breathlessness, a chest X-ray was performed, which revealed a mass lesion in the left hemithorax (Figure 2A). Contrast-enhanced CT revealed a large heterogeneous mass in the left hilar region extending into the left upper lobe, lingula, and left lower lobe with an ipsilateral mediastinal shift (Figure 2B). The findings of the physical examination of the chest were missed by the treating doctors. Ultrasound-guided biopsy from the left lung mass revealed a well-differentiated NET with a Ki-67 of <2%, consistent with a WHO grade 1 tumor. Based on the above presentation and laboratory and radiological investigations, the possibility of an ectopic GHRH-secreting lung NET was raised. For further evaluation, an FDG PET scan was performed, which revealed avid lesions in the left lung (Figure 2C) and in the suprasellar region, without any other uptake elsewhere. Her arterial blood gas analysis was within normal limits, but the ventilation-perfusion scan suggested a severely decreased perfusion in the left lung (Figure 2D). Plasma GHRH levels, measured using a radioimmunoassay, as previously described (3), were extremely high (38,088 ng/l; RR <250–300 ng/l).
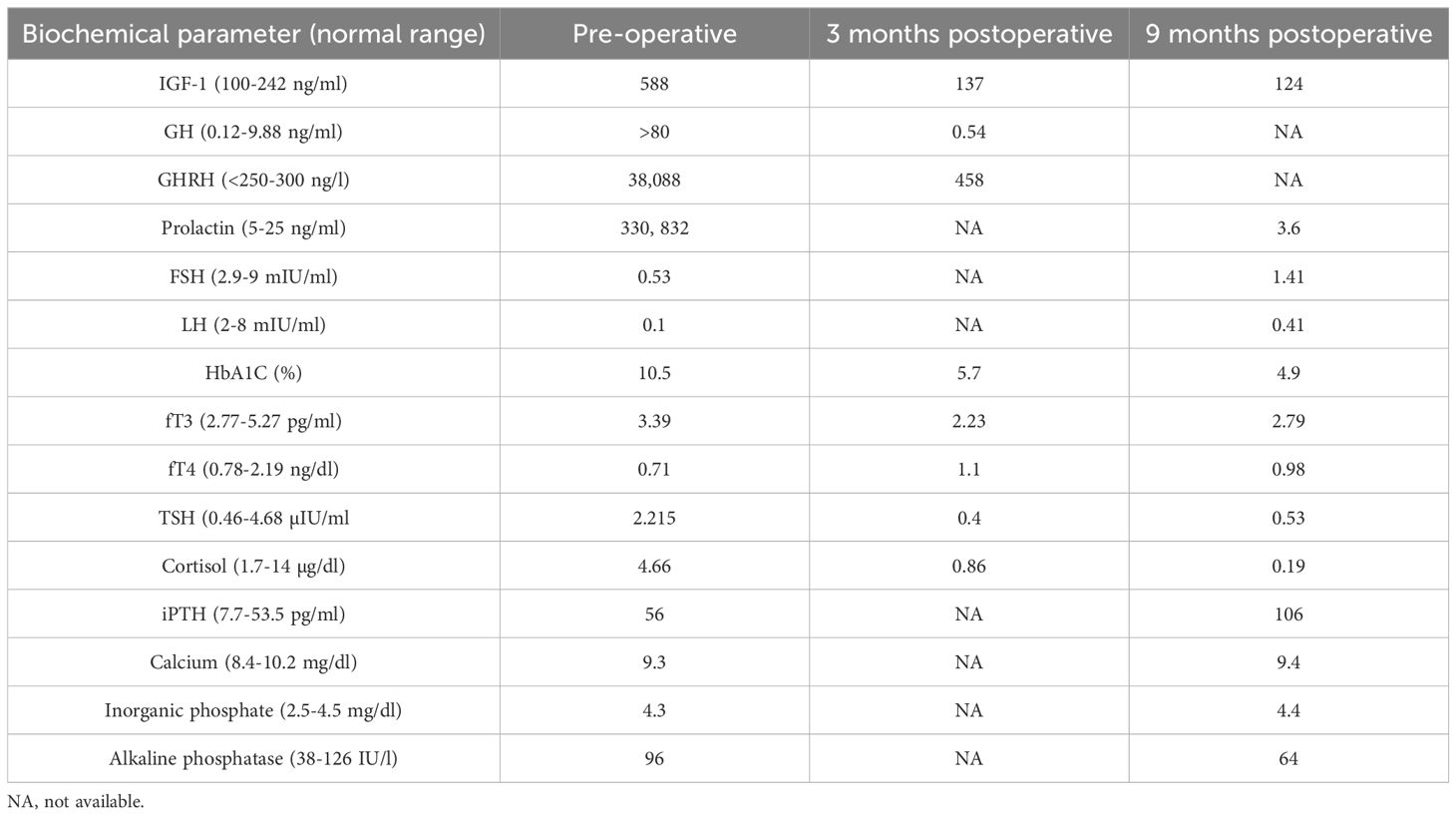
Table 1. Pre- and post-operative biochemical and hormonal investigations in the patient.
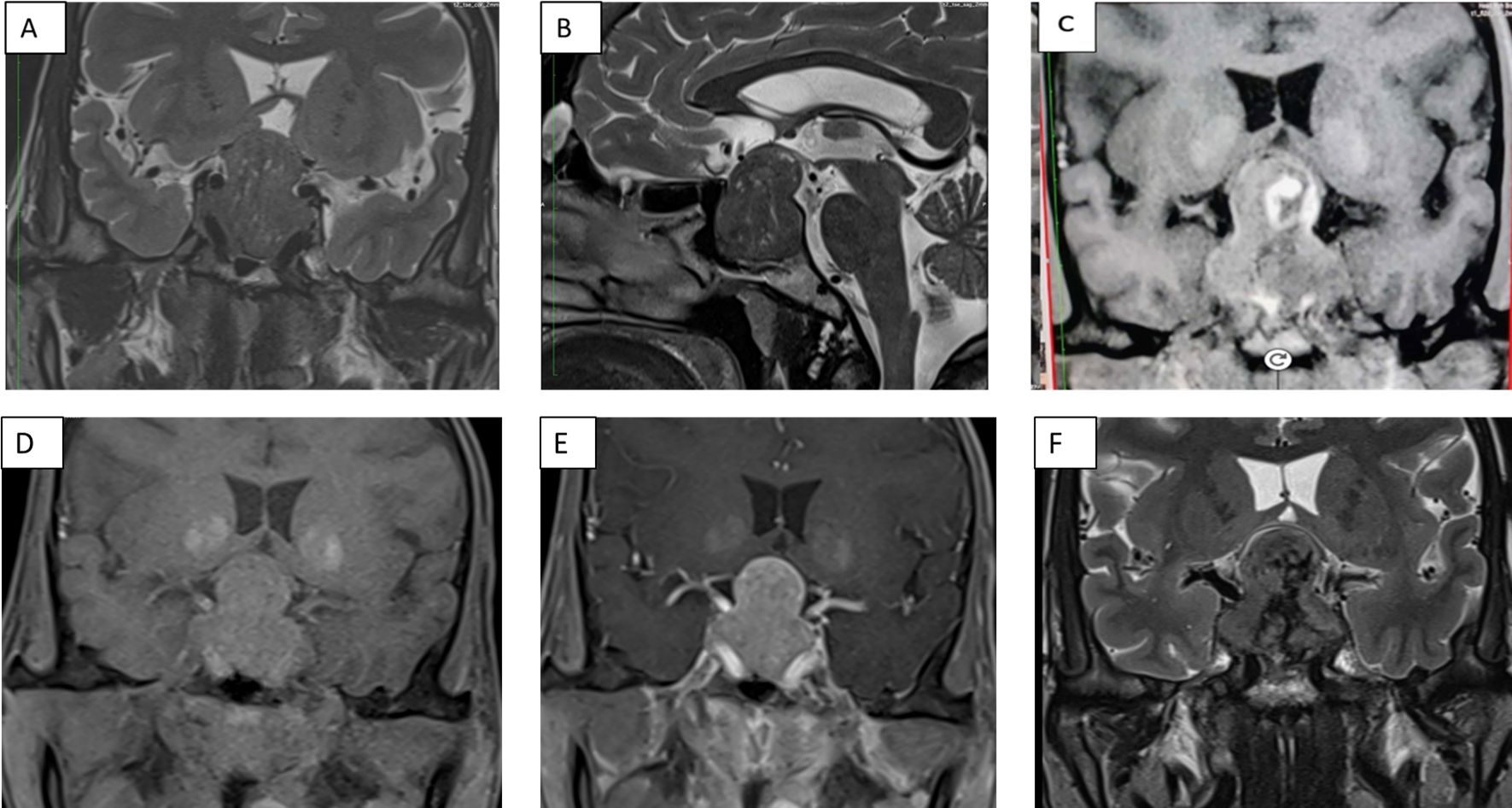
Figure 1. Panel of contrast-enhanced MRI of the sella showing (A) a 22×30×34 mm hypointense heterogenous lesion on a T2-weighted sequence coronal section (A) and sagittal section (B), and coronal T1-weighted non-contrast (D) and post-contrast (E) sections. T1-weighted non-contrast (C) and T2-weighted (F) coronal sections show hyperintensities and hypointensities, respectively, with an increase in size of the lesion to 21×35×41mm, which is suggestive of pituitary apoplexy.
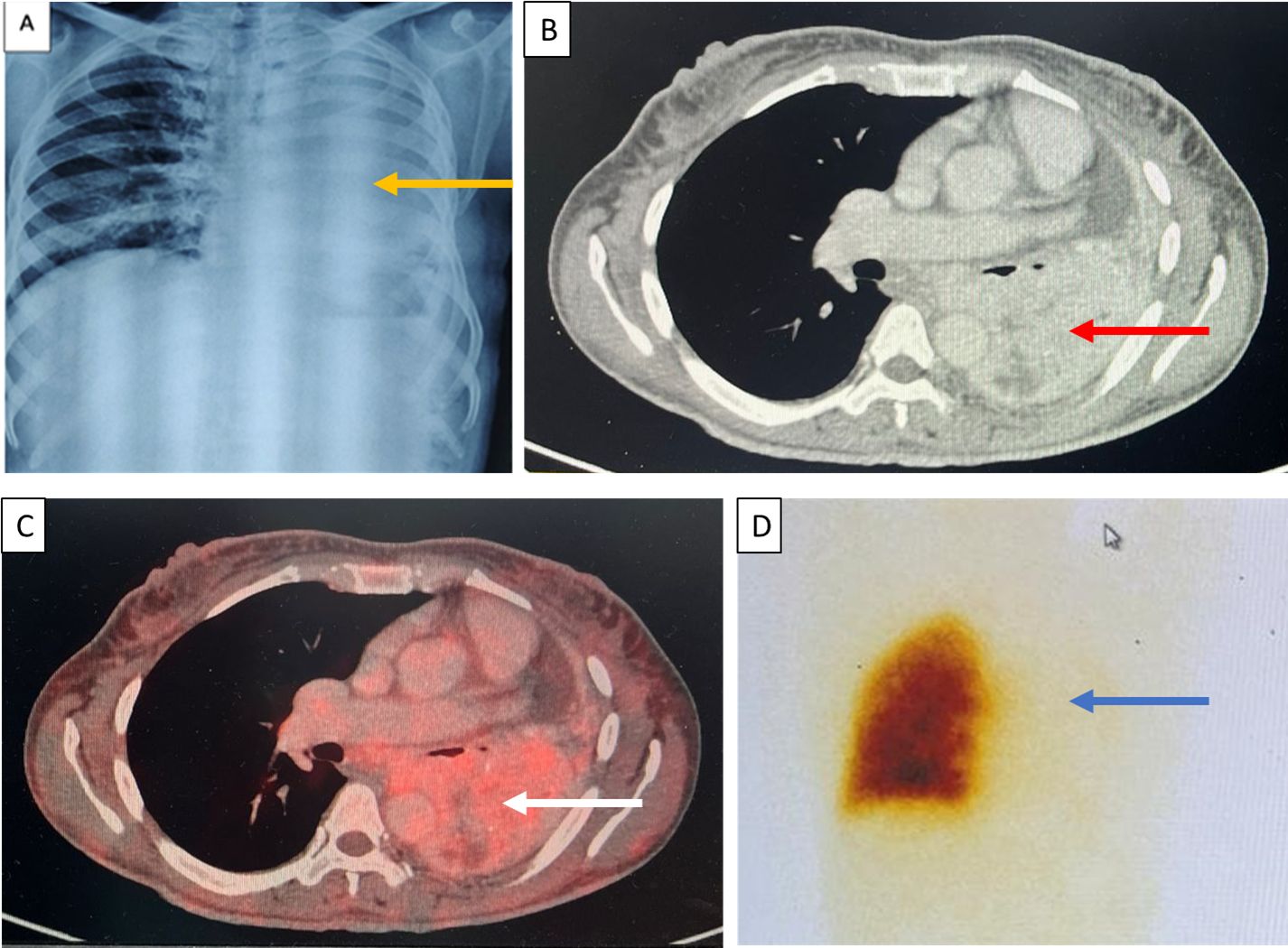
Figure 2. (A) Chest X-ray on day 4 of admission with complete opacification of the left hemithorax (yellow arrows). (B) Contrast-enhanced CT chest showing a heterogeneous mass with intense enhancement in the left lung (red arrow). (C) FDG-PET CT revealing an avid lung mass in the left lung (white arrow). (D) A ventilation perfusion scan suggestive of a severe decreased to negligible perfusion in the left lung (blue arrow).
While awaiting definitive management in the hospital, the patient developed severe headaches, multiple episodes of vomiting, drooping of the right eye (Figure 3A), and a deterioration of visual acuity (only perception of light in the right eye and finger counting at 2 m with a best corrected visual acuity of 6/60 in the left eye). MRI revealed apoplexy with an increase in size of the sellar-suprasellar mass (Figure 1C). The patient was managed with intravenous hydrocortisone (100 mg) every 8 h for 5 days followed by oral prednisone (7.5 mg) daily. Short-acting octreotide (100 mcg) administered subcutaneously every 8 h was initiated. After an improvement in her general condition, she underwent a left pneumonectomy a week later, and octreotide was discontinued. During surgery, a large yellow-colored tumor was identified. Her headache and ptosis improved within 5 days of the pneumonectomy (Figure 3B) and GH decreased to 22.4 ng/ml and prolactin decreased to 1.9 ng/ml. Histopathology revealed a NET with a Ki-67 of <3%, and immunohistochemistry was positive for synaptophysin, thyroid transcription factor 1 (TTF-1), and GHRH. Immunohistochemical analysis was negative for GH and prolactin (Figures 4A–D). The visual fields and acuity improved at 3 months (6/12 in the left eye and finger counting at 3 m in the right eye) along with a regression of soft tissue enlargement (Figure 3C). Repeat biochemical assessment at 3 months showed a normal IGF-1 (133 ng/ml; RR, 100–242) and GH (0.54 ng/ml), and a marked decrease in GHRH (458 ng/l; RR <250–300) (Table 1). Pituitary MRI at 3 months (Figure 5A, B) revealed a reduction in the size of the sellar-suprasellar mass by 48.7% (20×24×24mm). Whole- exome sequencing of peripheral blood-derived DNA showed no pathogenic variants in the MEN1, CDKN1B, and AIP genes. She continued to receive oral glucocorticoids and levothyroxine for central hypocortisolism and hypothyroidism. At 9 months follow-up, she was asymptomatic (Table 1), with significantly improved visual acuity (6/60 in the right eye and 6/12 in the left eye) and a resumption of spontaneous menstrual cycles. There was a further decrease in the size of the pituitary mass to 19×20x×17.6mm (Figures 5C, D), consistent with a 70.2% reduction in tumor volume.
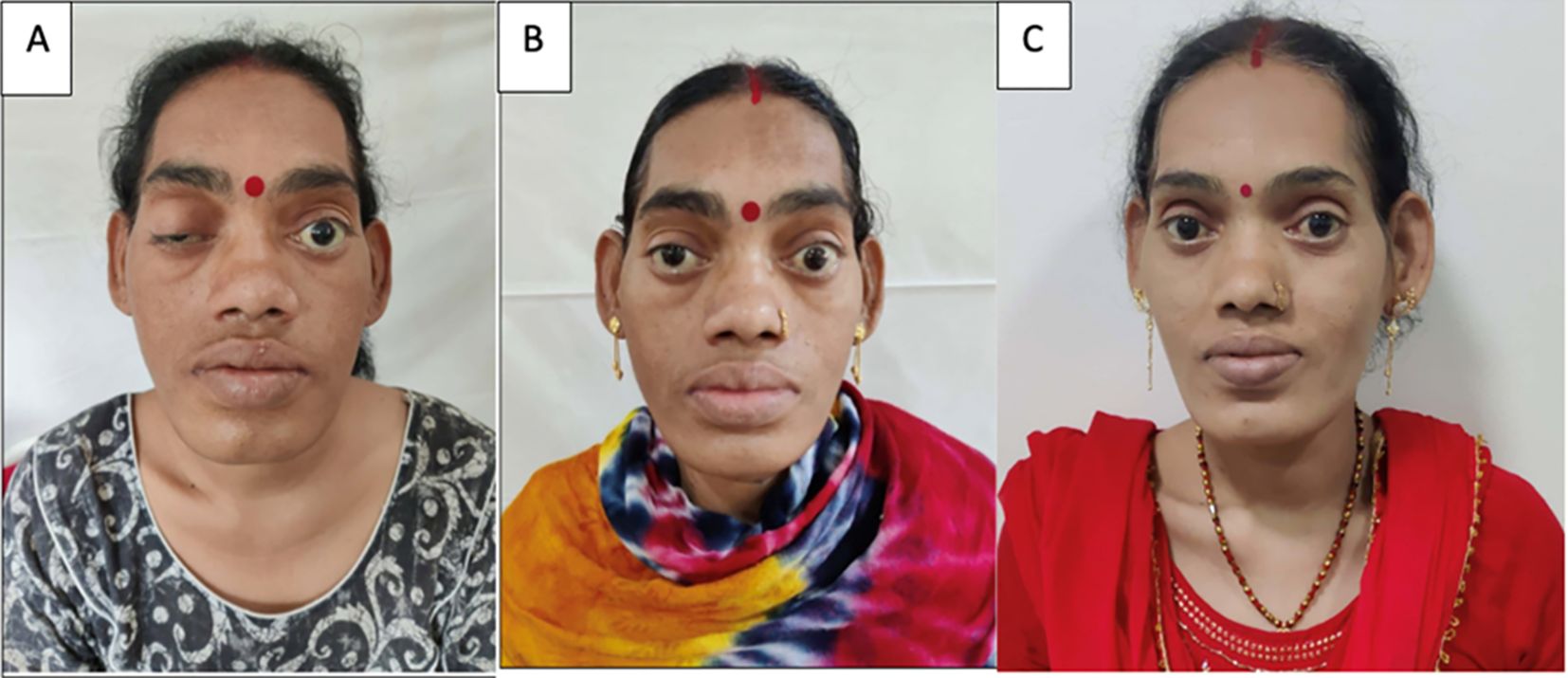
Figure 3. Clinical photographs of the patient. (A) At presentation showing ptosis of the right eye and coarse facial features, (B) resolution of the ptosis with persistent coarse features after 5 days with conservative management of apoplexy, and (C) an image after 9 months showing the resolution of the coarse facial features after surgical excision of the lung mass.
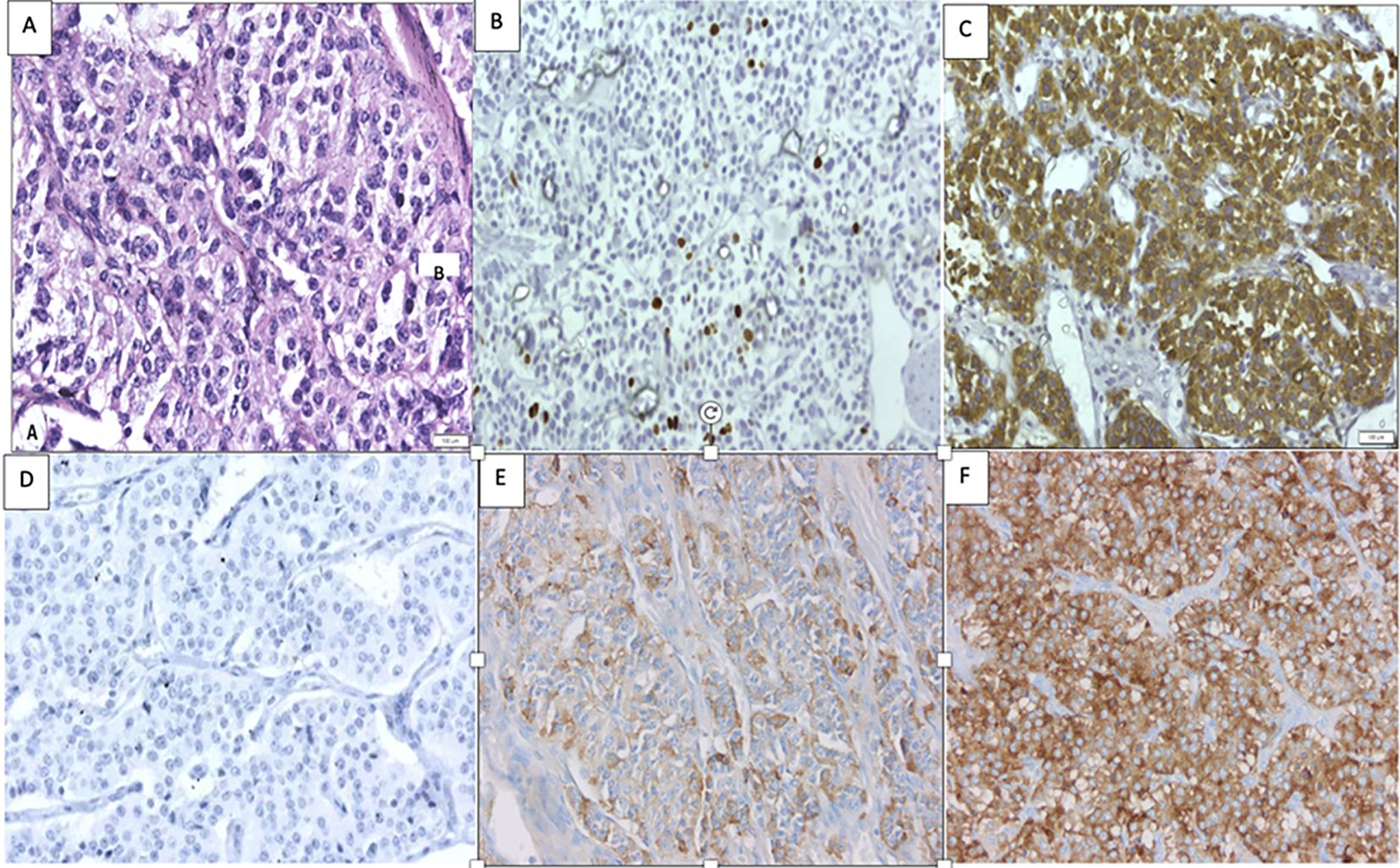
Figure 4. Histopathology of the lung mass with (A) hematoxylin and eosin staining revealing features of lung NET (400×). (B) A Ki-67 index of <3% (400×). (C) Positive for synaptophysin (400×). (D) Negative immunohistochemistry for prolactin negative (400×). (E) Immunohistochemistry for GHRH demonstrating focal positivity (5% of cells) (400×). (F) Positive GHRH control of a previously studied lung NET (400×).

Figure 5. Panel of a contrast-enhanced MRI of the sella showing the initial reduction in size of a residual lesion to 20×24×24 mm (48% reduction) 3 months after lung surgery on coronal (A) and sagittal sections (B). Further significant reduction of the lesion to 19×20×17.6 mm (70% reduction) in the coronal (C) and sagittal (D) sections 9 months after lung surgery.
Discussion
Here, we present a case of ectopic acromegaly caused by a GHRH-secreting lung NET. The unique features of the case include marked hyperplasia of the pituitary gland mimicking pituitary macroadenoma, elevated prolactin in the range usually seen in macroprolactinomas, and apoplexy in the hyperplastic pituitary gland. There was a dramatic regression of facial features and pituitary lesion volume within 3 months after the removal of the lung NET, with near-normal GHRH and prolactin levels, implicating that the GHRH-secreting lung tumor caused the acromegaly.
Ectopic acromegaly contributes to <1% of all cases of acromegaly. The clinical characteristics of ectopic acromegaly are indistinguishable from those of acromegaly resulting from GH-secreting pituitary adenomas. Lung NETs are the most common cause of ectopic acromegaly, accounting for 43% of all cases, followed by pancreatic NETs, which account for 35% of cases (2). Females are more commonly affected than males (70.1% vs. 29.9%) (2).
GHRH has been shown to be highly specific for ectopic acromegaly, as shown in a series of 177 consecutive GH-secreting adenomas in which all patients had undetectable plasma GHRH. A GHRH cutoff of 250–300 ng/l has been proposed to have a high specificity (93.8%) for the diagnosis of ectopic acromegaly, along with imaging (4, 5). However, it is not known whether GHRH levels between 30–250 ng/l are physiological or can be due to mild GHRH excess. GHRH is expressed in a variety of normal peripheral tissues, including the gastrointestinal tract, lymphocytes, uterus, ovary, testis, placenta, cerebral cortex, pituitary, kidney, prostate, liver, and lung. The major contributor to plasma GHRH is the gastrointestinal tract and the sample for GHRH needs to be taken in a fasting state (6). Our case had unequivocally high GHRH levels pre-operatively (38,088 ng/l), which decreased post-operatively to 458 ng/l (<250–300 ng/l). This was slightly above normal and could be attributed to microscopic lymph node metastases (as seen in two of four resected lymph nodes), circulating bioinactive GHRH, or secretion from other physiological sources of GHRH, such as the gastrointestinal tract, lymphocytes, uterus, ovary, cerebral cortex, pituitary, kidney, liver, or lung. A contrast-enhanced CT carried out at 9 months did not reveal any macroscopic residue, and her GH and IGF1 values were normal until the reporting of this case. Nevertheless, because of a detectable GHRH value in the post-operative period she was kept under constant surveillance to look for a residue or recurrence.
An interesting feature in this case was the markedly elevated prolactin at diagnosis, in a range that is only seen in patients with macroprolactinoma. Ectopic prolactin secretion from the lung lesion was excluded by a negative immunohistochemical analysis for prolactin in the lung NET. Ectopic prolactin production has been reported from tumors such as a leiomyoma of the uterus, a gonadoblastoma, an ovarian teratoma, a perivascular epithelioid cell tumor, a uterine cervical carcinoma, and a colorectal adenocarcinoma (7). Elevated serum prolactin was reported in 35% (44/124) or 29% (6/21) of cases in reviews of ectopic GHRH secretion (Table 2). Our case is unique in that prolactin levels were markedly elevated pre-operatively and decreased to normal levels after 9 months of follow-up (Table 1). Hyperprolactinemia is likely attributable to increased GHRH, which is known to cause high prolactin in normal subjects and in patients with acromegaly, due to lactotroph hyperplasia (15, 16). In stalk disinhibition syndrome, prolactin levels seldom exceed 100 ng/ml (17). Marked prolactin elevation should also lead to a suspicion of MEN1 syndrome (18). However, in our case, it was excluded due to a lack of clinical signs and characteristic family history and by genetics analysis.
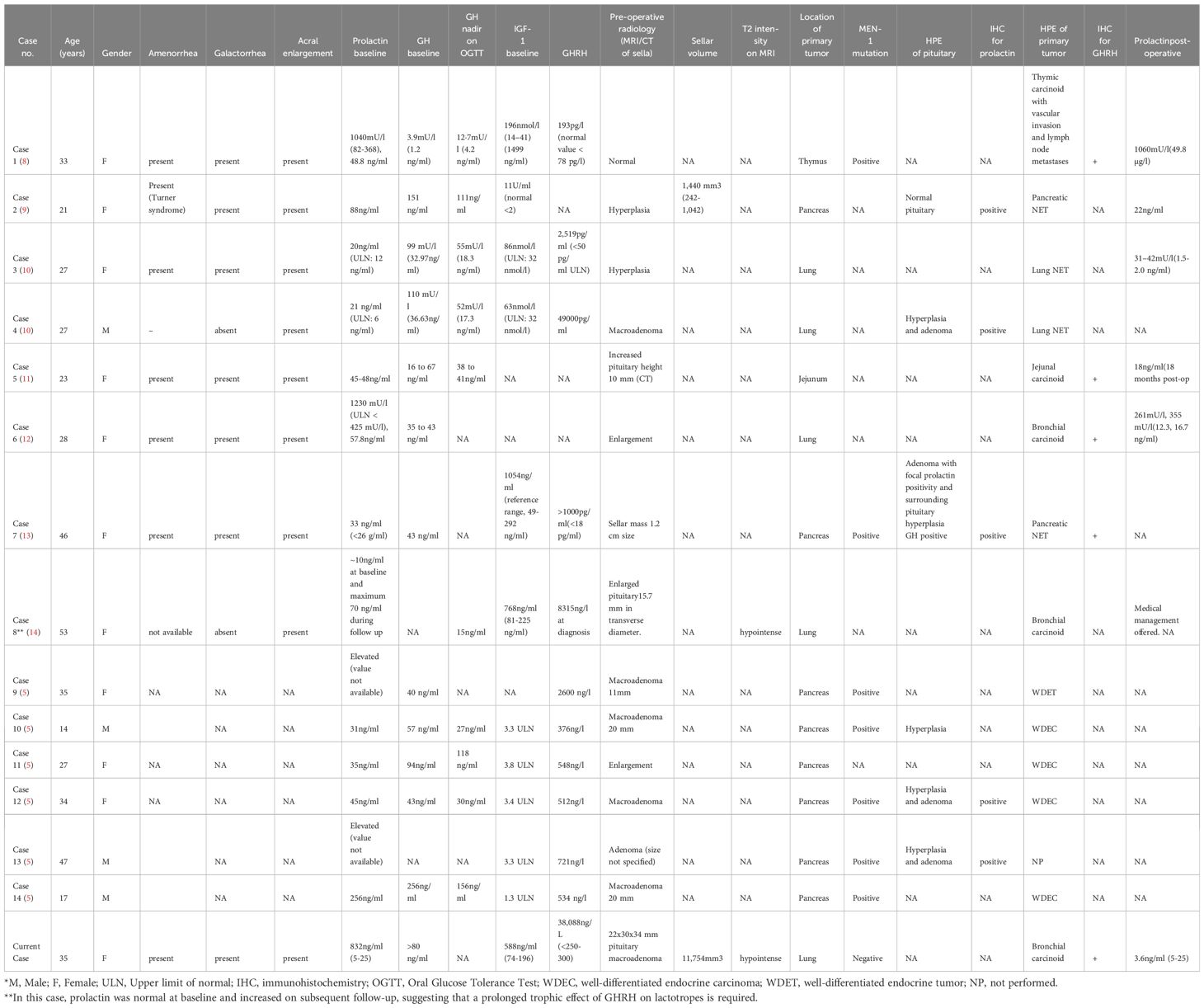
Table 2. Literature review of cases with hyperprolactinemia associated with ectopic acromegaly due to GHRH secretion.
Pituitary hyperplasia can be mistaken for pituitary macroadenoma on imaging. In a large review of ectopic GHRH secretion with acromegaly, 43 out of 96 cases were reported as pituitary enlargement, 20 as adenomas, 18 normal on imaging, 10 as unclear lesions, 3 as empty sellae, and 2 as microcystic lesions (2). Similarly, in another retrospective study comprising 30 cases of GHRH-secreting neuroendocrine tumors, radiological hyperplasia of the pituitary was reported in 80% (24/30) of cases (20). However, on histopathology, only 20/29 were hyperplasia, 3/29 cases turned out to be adenoma, 3/29 mixed adenoma, and hyperplasia, 2/29 cases there in no mention of histopathological finding and 1 out of 29 normal pituitary was found (2). In another series of 21 cases, 5/21 cases of ectopic acromegaly due to GHRH with hyperprolactinemia were reported to have adenoma on imaging (4 macroadenoma and 1 adenoma not specified) (5). Three of these cases underwent pituitary surgery, with histopathology revealing somatotroph hyperplasia in one and somatotroph hyperplasia with prolactin-GH adenoma in the other two cases (5). All the patients had ectopic acromegaly resulting from gastro-pancreatic NETs and four out of these six cases had metastases at diagnosis, mainly to the liver. Our case, however, had a lung NET and the sellar-suprasellar mass was 34 mm in maximum dimension, with the radiologist reporting it as a pituitary macroadenoma. Adenoma can be seen in areas adjacent to hyperplasia. It has been demonstrated that excessive GHRH stimulation leads to somatotroph hyperplasia and, ultimately, pituitary adenoma formation in metallothionein promoter-driven human GHRH transgenic mice (19), some of which had shown focal positivity for prolactin, further emphasizing the phenomenon of specificity spillover akin to thyro-lactotroph hyperplasia in long-standing untreated juvenile hypothyroidism (20). A characteristic radiological feature attributed to ectopic acromegaly is the presence of T2-weighted hypointensity on MRI, which was present in 83% (25/30) of cases in a previous series. Rarely, microcysts (2.1% to 9.5% in some series) have been reported (21). Although T2 hypointense signal may be present in eutopic acromegaly as well, the prevalence is much lower (52.9%), as observed in a large series of 297 patients (22).
Histopathologically, hyperplasia was present in 20, adenoma in 3, and both adenoma with surrounding areas of hyperplasia in 3, unrelated lesions in 2, and not specified for 1 respectively. The exact histopathological nature of the lesion in our patient is unknown, as she did not have pituitary surgery. Another interesting feature of the current case was the apoplexy in the pituitary hyperplasia that occurred during the course of the hospital stay. The patient presented with right eye ptosis, which improved significantly over 5 days with conservative management. The index patient had a large sellar suprasellar mass, which was possibly the only predisposition for apoplexy. She did not have any other known risk factors such as hemodynamic disturbances due to major surgery, an intervention for surgery or gamma knife radiosurgery, hypertension, a hemorrhagic pregnancy, anticoagulation use, endocrine function tests (stimulation tests), cabergoline use, or a vasculotoxic snake bite. Although histopathological analysis of the sellar-suprasellar mass was not available in the current case, the rapidity of volume reduction after removal of the primary tumor favors a diagnosis of hyperplasia over adenoma. Apoplexy has been reported in hyperplasia, with only a single case in the past in which histopathology of the pituitary revealed an adenoma adjacent to hyperplasia (19).
Despite her history of long-standing secondary amenorrhea and exertional dyspnea for the last 3 months in retrospect, her lung mass could be diagnosed only during a pre-operative assessment before pituitary surgery. The pre-operative radiological impression was that of an invasive macroadenoma that could be a hyperplasia, as the size decreased significantly and dramatically after the removal of the primary tumor, with a dramatic improvement in signs and symptoms. Moreover, histopathology also revealed positive GHRH immunostaining in the lung tissue, with a decrease in serum GHRH levels post-surgery.
Regarding the prognosis of GHRH-secreting tumors (NETs), surgical resection of the primary tumor is the treatment of choice in the majority of cases. The prognosis of these patients is excellent, with survival exceeding 80% in the majority of cases and up to 94% in lung tumors, as described previously (2, 23). This is despite the fact that up to 50% of these patients can have metastatic disease at diagnosis. Residual disease following surgery can be managed successfully by using somatostatin-receptor ligands (SRLs) or, rarely, even pegvisomant. Adjuvant medical management can lead to the successful resolution of symptoms and normalization of IGF1, with or without a significant decrease in GHRH (24).
Conclusion
Pituitary hyperplasia due to ectopic GHRH-secreting tumors can radiologically mimic pituitary adenoma, and apoplexy is extremely unusual but can lead to rapid shrinkage of the tumor, unlike eutopic somatotropinomas. Hyperprolactinemia is not uncommon but is rarely in the tumoral range in GHRH-secreting ectopic acromegaly.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
AG: Writing – original draft. RK: Methodology, Project administration, Resources, Writing – review & editing. LD: Data curation, Formal Analysis, Methodology, Project administration, Visualization, Writing – review & editing. SS: Project administration, Supervision, Writing – review & editing. VK: Data curation, Writing – review & editing. AV: Investigation, Project administration, Writing – review & editing. VR: Project administration, Resources, Supervision, Writing – review & editing. MK: Project administration, Validation, Visualization, Writing – review & editing. PD: Conceptualization, Project administration, Resources, Supervision, Validation, Visualization, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
2. Zendran I, Gut G, Kałużny M, Zawadzka K, Bolanowski M. Acromegaly caused by ectopic growth hormone releasing hormone secretion: A review. Front Endocrinol. (2022) 13:867965. doi: 10.3389/fendo.2022.867965
3. Girard P, Cohen R, Sassolas G, Harthe C, Cabrera P, Boissel JP. Pharmacokinetics of human growth hormone releasing factor (hGRF-44 NH2) in normal men after intravenous administration of a large range of doses. Eur J Clin Pharmacol. (1987) 32:507–13. doi: 10.1007/BF00637679
4. Thorner MO, Frohman LA, Leong DA, Thominet J, Downs T, Hellmann P, et al. Extrahypothalamic growth-hormone-releasing factor (GRF) secretion is a rare cause of acromegaly: plasma GRF levels in 177 acromegalic patients. J Clin Endocrinol Metab. (1984) 59:846–9. doi: 10.1210/jcem-59-5-846
5. Garby L, Caron P, Claustrat F, Chanson P, Tabarin A, Rohmer V, et al. Clinical characteristics and outcome of acromegaly induced by ectopic secretion of growth hormone-releasing hormone (GHRH): a French nationwide series of 21 cases. J Clin Endocrinol Metab. (2012) 97:2093–104. doi: 10.1210/jc.2011-2930
6. Barabutis N, Schally AV. Growth hormone-releasing hormone: Extrapituitary effects in physiology and pathology. Cell Cycle. (2010) 9:4110–6. doi: 10.4161/cc.9.20.13787
7. Sachdev S, Reyes MC, Snyder PJ. Ectopic prolactin secretion from a uterine leiomyoma. J Endocr Soc. (2020) 4:bvaa035. doi: 10.1210/jendso/bvaa035
8. Boix E, Picó A, Pinedo R, Aranda I, Kovacs K. Ectopic growth hormone-releasing hormone secretion by thymic carcinoid tumor. Clin Endocrinol (Oxf). (2002) 57:131–4. doi: 10.1046/j.1365-2265.2002.01535.x
9. Thorner MO, Perryman RL, Cronin MJ, Rogol AD, Draznin M, Johanson A, et al. Somatotroph hyperplasia. J Clin Invest. (1982) 70:965–77. doi: 10.1172/JCI110708
10. Biermasz NR, Smit JWA, Pereira AM, Frölich M, Romijn JA, Roelfsema F. Acromegaly caused by growth hormone-releasing hormone-producing tumors: long-term observational studies in three patients. Pituitary. (2007) 10:237–49. doi: 10.1007/s11102-007-0045-7
11. Spero M, White EA. Resolution of acromegaly, amenorrhea-galactorrhea syndrome, and hypergastrinemia after resection of jejunal carcinoid. J Clin Endocrinol Metab. (1985) 60:392–5. doi: 10.1210/jcem-60-2-392
12. Carroll DG, Delahunt JW, Teague CA, Cooke RR, Adams EF, Christofides ND, et al. Resolution of acromegaly after removal of a bronchial carcinoid shown to secrete growth hormone releasing factor. Aust N Z J Med. (1987) 17:63–7. doi: 10.1111/j.1445-5994.1987.tb05054.x
13. Weiss DE, Vogel H, Lopes MBS, Chang SD, Katznelson L. Ectopic acromegaly due to A pancreatic neuroendocrine tumor producing growth hormone-releasing hormone. EndocrPract. (2011) 17:79–84. doi: 10.4158/EP10165.CR
14. Rak-Makowska B, Khoo B, Sen Gupta P, Plowman PN, Grossman AB, Korbonits M. Ockham’s razor for a retinal lesion and acromegaly and breaking the vicious circle. J Endocr Soc. (2022) 6:bvac083. doi: 10.1210/jendso/bvac083
15. Goldman JA, Molitch ME, Thorner MO, Vale W, Rivier J, Reichlin S. Growth hormone and prolactin responses to bolus and sustained infusions of GRH-1-40-OH in man. J Endocrinol Invest. (1987) 10:397–406. doi: 10.1007/BF03348157
16. Serri O, Somma M, Rasio E, Brazeau P. Growth hormone-releasing factor increases serum prolactin concentrations in normal subjects and in patients with pituitary adenomas. Clin Endocrinol (Oxf). (1989) 30:65–75. doi: 10.1111/j.1365-2265.1989.tb03728.x
17. Vilar L, Vilar CF, Lyra R, Freitas M da C. Pitfalls in the diagnostic evaluation of hyperprolactinemia. Neuroendocrinology. (2019) 109:7–19. doi: 10.1159/000499694
18. Thakker RV, Newey PJ, Walls GV, Bilezikian J, Dralle H, Ebeling PR, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab. (2012) 97:2990–3011. doi: 10.1210/jc.2012-1230
19. Lamback EB, Henriques DG, Vazquez-Borrego MC, Lima CHDA, Kasuki L, Luque RM, et al. Growth hormone-releasing hormone-secreting pulmonary neuroendocrine tumor associated with pituitary hyperplasia and somatotropinoma. Arch Endocrinol Metab. (2021) 65(5):648–63.
20. Pioro EP, Scheithauer BW, Laws ER Jr., Randall RV, Kovacs KT, Horvath E. Combined thyrotroph and lactotroph cell hyperplasia simulating prolactin-secreting pituitary adenoma in long-standing primary hypothyroidism. Surg Neurol. (1988) 29:218–26. doi: 10.1016/0090-3019(88)90010-9
21. Potorac I, Bonneville JF, Daly AF, de Herder W, Fainstein-Day P, Chanson P, et al. Pituitary MRI features in acromegaly resulting from ectopic GHRH secretion from a neuroendocrine tumor: analysis of 30 cases. J Clin Endocrinol Metab. (2022) 107:e3313–20. doi: 10.1210/clinem/dgac274
22. Potorac I, Petrossians P, Daly AF, Schillo F, Ben Slama C, Nagi S, et al. Pituitary MRI characteristics in 297 acromegaly patients based on T2-weighted sequences. EndocrRelat Cancer. (2015) 22:169–77. doi: 10.1530/ERC-14-0305
23. Borson-Chazot F, Garby L, Raverot G, Claustrat F, Raverot V, Sassolas G, et al. Acromegaly induced by ectopic secretion of GHRH: a review 30 years after GHRH discovery. Ann Endocrinol (Paris). (2012) 73(6):497–502.
Keywords: ectopic acromegaly, growth hormone-releasing hormone, pituitary hyperplasia, hyperprolactinemia, apoplexy
Citation: Gupta A, Kasaliwal R, Das L, Sharma SK, Kaur V, Vasiljevic A, Raverot V, Korbonits M and Dutta P (2024) Ectopic acromegaly with tumoral range hyperprolactinemia and apoplexy with a dramatic regression of pituitary hyperplasia. Front. Endocrinol. 15:1473167. doi: 10.3389/fendo.2024.1473167
Received: 30 July 2024; Accepted: 16 September 2024;
Published: 10 October 2024.
Edited by:
Elena Varlamov, Oregon Health and Science University, United StatesReviewed by:
Mirjana Doknic, University of Belgrade, SerbiaElisa Lamback, Federal University of Rio de Janeiro, Brazil
Copyright © 2024 Gupta, Kasaliwal, Das, Sharma, Kaur, Vasiljevic, Raverot, Korbonits and Dutta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pinaki Dutta, ZHJwaW5ha2lkdXR0YTEyQGdtYWlsLmNvbQ==
†These authors share first authorship
‡ORCID: Ashish Gupta, orcid.org/0009-0004-8659-2891
Rajeev Kasaliwal, orcid.org/0000-0002-2377-6616
Liza Das, orcid.org/0000-0002-9701-9554
Surendra Kumar Sharma, orcid.org/0009-0002-2353-7049
Vaishali Kaur, orcid.org/0009-0008-3021-4544
Alexandre Vasiljevic, orcid.org/0000-0001-5954-0317
Véronique Raverot, orcid.org/0000-0003-4336-1271
Márta Korbonits, orcid.org/0000-0002-4101-9432
Pinaki Dutta, orcid.org/0000-0001-5415-1611