 Maria Luisa Brandi1*
Maria Luisa Brandi1* Thomas O. Carpenter2
Thomas O. Carpenter2 Seiji Fukumoto3
Seiji Fukumoto3 Dieter Haffner4
Dieter Haffner4 Erik A. Imel5Masanori Kanematsu6Keith P. McCullough7
Erik A. Imel5Masanori Kanematsu6Keith P. McCullough7 Keiichi Ozono8
Keiichi Ozono8- 1Fondazione Italiana Ricerca sulle Malattie dell'Osso (FIRMO), Florence, Italy
- 2Department of Pediatrics, Section of Endocrinology, Yale School of Medicine, New Haven, CT, United States
- 3Tamaki - Aozora Hospital, Tokushima, Japan
- 4Department for Pediatric Kidney, Liver, Metabolic and Neurological Diseases, Hannover Medical School, Hannover, Germany
- 5Departments of Medicine and Pediatrics, Indiana University School of Medicine, Indianapolis, IN, United States
- 6Kyowa Kirin Corporation, Tokyo, Japan
- 7Arbor Research Collaborative for Health, Ann Arbor, MI, United States
- 8ISEIKAI International General Hospital, Osaka, Japan
X-linked hypophosphatemia (XLH) is a rare, genetic, progressive, lifelong disorder caused by pathogenic variants in the phosphate-regulating endopeptidase homolog, X-linked (PHEX) gene, resulting in excess fibroblast growth factor 23 (FGF23) and consequent renal phosphate wasting. Chronic hypophosphatemia leads to deficits of the musculoskeletal system affecting bone, muscle, joint, and dental health. XLH treatments include oral phosphate and active vitamin D—which are associated with a burdensome dosing regimen, gastrointestinal disturbances, hyperparathyroidism, and nephrocalcinosis—or burosumab, a fully human anti-FGF23 antibody. Randomized clinical trials (RCTs) demonstrated burosumab to be well tolerated and efficacious in improving serum phosphate, rickets, bone turnover, and patient-reported outcomes. However, there are limited data on the natural history of XLH or real-world comparisons of the safety, effectiveness, and long-term outcomes of XLH treatments. Advancing Patient Evidence in XLH (APEX) is a global data unification project aiming to describe the burden and lifelong progression of XLH, collect real-world data on treatment effectiveness and safety, and investigate regional differences in treatment outcomes. Participants from three observational, noninterventional, retrospective and prospective, multicenter, longitudinal (10-year) studies of patients with XLH will be included: XLH Disease Monitoring Program (NCT03651505), International XLH Registry (NCT03193476), and SUNFLOWER (NCT03745521). Data collected in the Americas, Europe, Israel, Japan, and South Korea will be processed to unify identical and similar data elements. Data unification will be an iterative process with a clinical and programming review, ensuring validity and accuracy. In this observational study, unified data involving approximately 2000 pediatric and adult participants with XLH will be analyzed to address research questions in an exploratory manner. Long-term observational studies and patient registries provide opportunities to generate real-world data and address knowledge gaps in rare diseases. APEX aims to improve clinical decision-making and practice by bridging evidence gaps that cannot be addressed by RCTs or regional registries.
1 Introduction
X-linked hypophosphatemia (XLH) is a rare, progressive and lifelong disorder caused by pathogenic variants in the phosphate-regulating endopeptidase homolog, X-linked (PHEX) gene, resulting in excess fibroblast growth factor 23 (FGF23) and consequent renal phosphate wasting (1–3) affecting approximately 1 in 20,000 to 70,000 people globally (4–7). XLH is caused by inactivating PHEX variants (1, 8) and is inherited in an X-linked dominant pattern (8). De novo variants occur in approximately 20% to 30% of patients (9, 10). Increased levels of FGF23 cause downregulation of the sodium-dependent phosphate co-transporters NPT2a and NPT2c in proximal renal tubules leading to renal phosphate wasting, decreased 1α-hydroxylase, and increased 24-hydroxylase activity, resulting in decreased serum 1,25-dihydroxyvitamin D levels, and decreased phosphate absorption in the intestines (1). Resultant chronic hypophosphatemia impairs bone and tooth mineralization resulting in rickets, osteomalacia, and odontomalacia (1, 8), amongst other multi-system consequences. In children, impaired and disproportionate growth with lower limb deformities are typical; delayed motor development and craniosynostosis may occur (1, 8, 11, 12). Adults with XLH may manifest fractures or pseudofractures, early onset osteoarthritis, enthesopathy, and less frequently, spinal stenosis (1, 8, 11). Hearing loss is not infrequent (1, 8). Dental abscesses are common across age groups as well as bone and joint pain, stiffness, and muscle weakness (1, 8, 11). The disorder severely impacts physical function, mobility, and health-related quality of life (QoL) (13, 14). In addition to the well characterized role of elevated FGF23 and hypophosphatemia in XLH, inactivating PHEX variants also modify levels of the small integrin-binding ligand, N-linked glycoproteins (SIBLING) family, such as osteopontin and the inhibitory acidic serine aspartate-rich-MEPE-associated protein (ASARM) peptides (1, 15–17). These lead to local impairment of mineralization of bone and teeth, and altered tooth morphology, contributing to osteomalacia and odontomalacia in XLH (1, 15–17). Efforts to treat XLH medically have focused primarily on modifying the influence of FGF23 on renal phosphate handling and 1,25-dihydroxyvitamin D production, or administration of conventional therapy, neither of which are likely to address aspects of the disease that are not mediated by FGF23, hypophosphatemia, or impaired vitamin D activation.
“Conventional therapy” over the past several decades has consisted of a combination of oral phosphate salts and active vitamin D analogs (8, 18). While this treatment is usually beneficial, it does not address the elevated levels of FGF23, and phosphate levels cannot be safely normalized (8, 18). Furthermore, conventional therapy does not normalize growth, nor entirely resolve bone and dental manifestations, or improve health-related QoL (13, 14, 19–21). Additionally, significant side effects, such as secondary or tertiary hyperparathyroidism, hypercalciuria, and nephrocalcinosis, may occur, and progression to chronic kidney disease has been described (18). Finally, oral phosphate supplementation is associated with poor adherence due to its unpleasant taste, the burdensome regimen of frequent dosing, and uncomfortable side effects such as diarrhea and abdominal cramping (8, 22, 23).
Burosumab is a fully human monoclonal antibody against FGF23, approved initially in 2018 for the treatment of children and adults with XLH, with varied approvals and payor coverage in children and adults across the world (24–26). Unlike conventional therapy, burosumab targets an earlier stage in the underlying pathophysiology of XLH by blocking effects of excess FGF23 and improving serum phosphate levels (24, 25, 27, 28). Clinical trials have shown burosumab to improve serum phosphate levels without hypercalciuria or elevating parathyroid hormone levels (29, 30). In children, burosumab improved rickets, and increased height and walking distance in the 6-Minute Walk Test (30). In adults, insufficiency fractures healed, walking distance in the 6-Minute Walk Test improved, and patient-reported outcomes of stiffness, pain, physical function improved (31). These benefits of burosumab treatment have been shown to be maintained long-term, for up to 144 to 184 weeks in adults (32, 33) and up to 160 weeks in children (34). Burosumab was well tolerated in clinical trials with few, if any, treatment-related serious adverse events (SAEs) (29–31, 35). Restless leg syndrome was observed as an adverse event (AE), primarily in adults (35). There were no AEs or SAEs, including deaths, that led to withdrawal of participants from the studies (29–31, 35). Neutralizing antibodies against burosumab were infrequently detected (29–31, 35, 36) and patients positive for neutralizing antibodies responded to burosumab treatment (36). To date, burosumab has not been found to increase nephrocalcinosis, ectopic myocardial mineralization, or incidence of hyperparathyroidism. However, there are important knowledge gaps regarding the long-term effects of burosumab treatment.
One such knowledge gap is the expected inability of burosumab to treat manifestations of XLH that are mediated by factors other than FGF23, such as osteopontin and ASARM peptides. To date, the effect of burosumab is not known regarding final height, degree of improvement of lower limb deformities, craniosynostosis, or the long-term effects regarding dental complications in patients with XLH (27, 30, 37–49). However, it is important to consider that most of these patients were previously treated with conventional therapy prior to burosumab initiation, and the age of burosumab initiation varied, which may influence the cumulative development of disease-related complications. Outside of randomized clinical trials (RCTs), burosumab was able to improve or prevent the worsening of some of these complications (27, 38–49), and earlier age of initiation improved outcomes relative to later age of initiation (42, 47, 49). Additionally, patients who initiate burosumab treatment tend to be those who exhibit more severe symptomatology compared with patients who persist in treatment with conventional therapy (43, 46). Therefore, information on the effect of long-term burosumab treatment and the impact of the age of initiation will be important in understanding the role burosumab can play in treating the wider symptomatology of XLH and, in addition, will also provide insights into which disease features cannot be addressed by targeting FGF23.
Due to the rarity of XLH, and with treatment expertise limited to specialized centers, significant unmet needs exist for affected patients (8). Real-world evidence is required to address knowledge gaps and to provide further information on the natural history of XLH, disease progression and burden, as well as long-term effectiveness, safety and the overall experience with burosumab outside of clinical trial conditions (2). Patient registries have been shown to be effective in the collection of large-scale, real-world patient data in rare disorders (50).
The Advancing Patient Evidence in XLH (APEX) is a ten-year global data unification project, combining data from three Kyowa Kirin-/Ultragenyx-sponsored regional observational studies to increase the overall size of the datasets and address a set of questions that would not be feasible or complete using the regional studies alone.
2 Methods
2.1 Regional study identification
Three Kyowa Kirin-/Ultragenyx-sponsored, regional observational studies in patients with XLH have been established and registered with ClinicalTrials.gov (Table 1). The XLH Disease Monitoring Program (XLH DMP, NCT03651505), initiated in July 2018 and with enrollment completed in December 2022, collects data in the Americas (51). The International XLH Registry (IXLHR, NCT03193476), collecting data from Europe and Israel, was initiated in September 2017 (52). The Study of Longitudinal Observation for Patients With X-linked Hypophosphatemic Rickets/Osteomalacia in Collaboration With Asian Partners (SUNFLOWER, NCT03745521), collecting data from participants in Japan and Korea, was initiated in April 2018 and with enrollment completed in April 2022 (53).
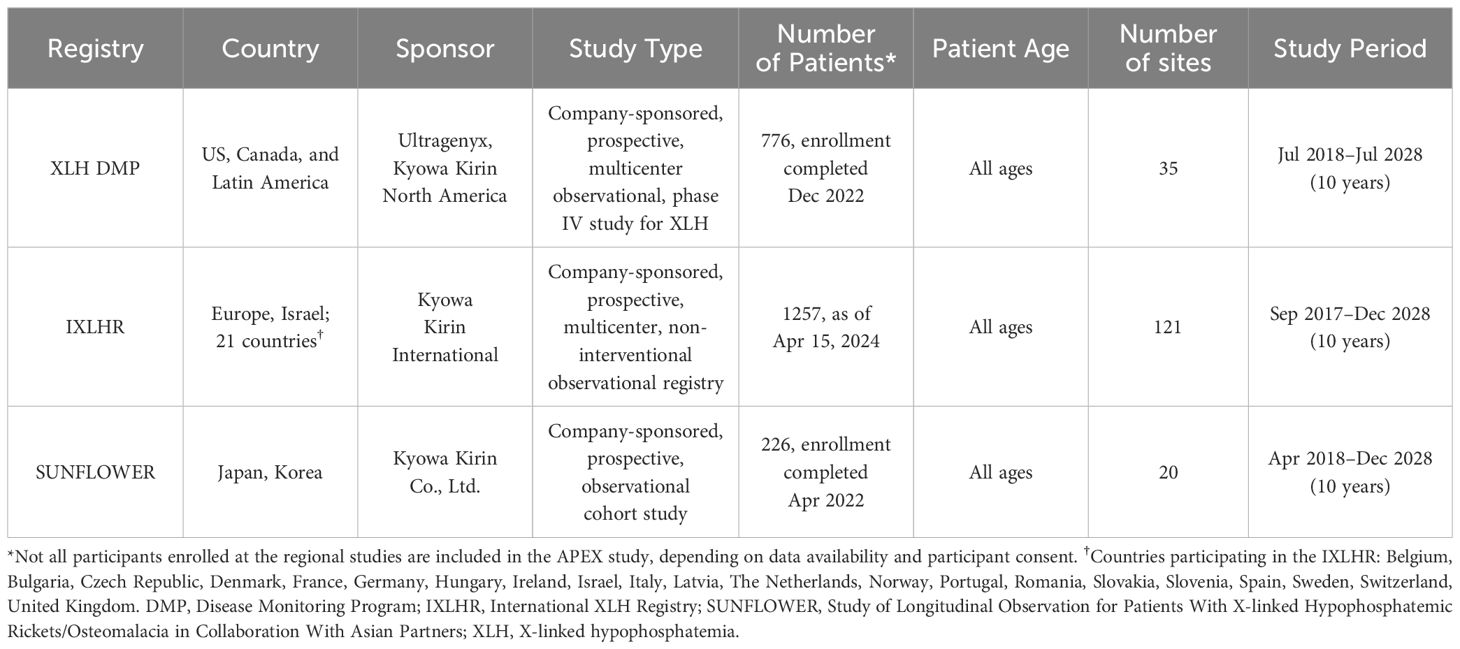
Table 1. Characteristics of Kyowa Kirin-/Ultragenyx-sponsored regional XLH studies.
2.2 Inclusion and exclusion criteria
Inclusion and exclusion criteria for the three regional studies are described in Table 2. Briefly, patients diagnosed with XLH who provided consent and were not participating in a clinical trial at the time (or who had received prior approval to do so in the case of the XLH DMP) were eligible for participation in the individual studies (2, 51–54). Patients were identified and enrolled by participating clinical sites.
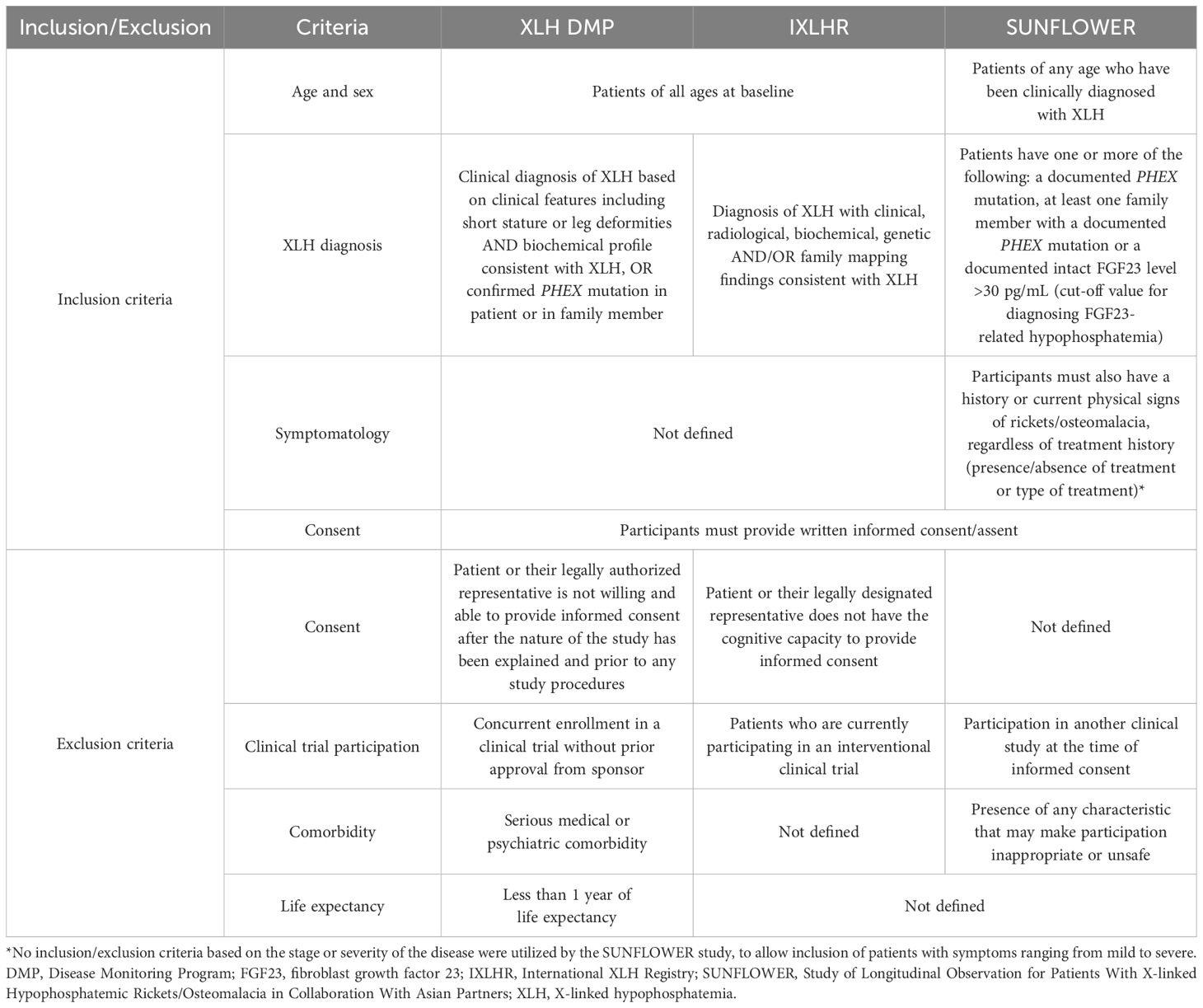
Table 2. Inclusion/exclusion criteria of Kyowa Kirin-sponsored regional XLH studies.
2.3 Study designs
Characteristics of the three regional studies are summarized in Table 1: the XLH DMP is a prospective, multicenter, longitudinal long-term outcomes program for participants with XLH (51); the IXLHR is a prospective, non-interventional observational registry (2, 52); and SUNFLOWER is a prospective, multicenter, longitudinal, long-term, observational cohort study (53, 54).
In all three studies, participants will be followed for up to ten years to capture treatment details and clinical outcome variables until withdrawal from the study or a loss to follow-up event. Participants will be treated at the discretion of the treating physician and will have access to burosumab only through authorized prescribed (commercial) use, or via early access programs in countries where burosumab is not commercially available. Where applicable and feasible, APEX will utilize these datasets through their unification (Figure 1).
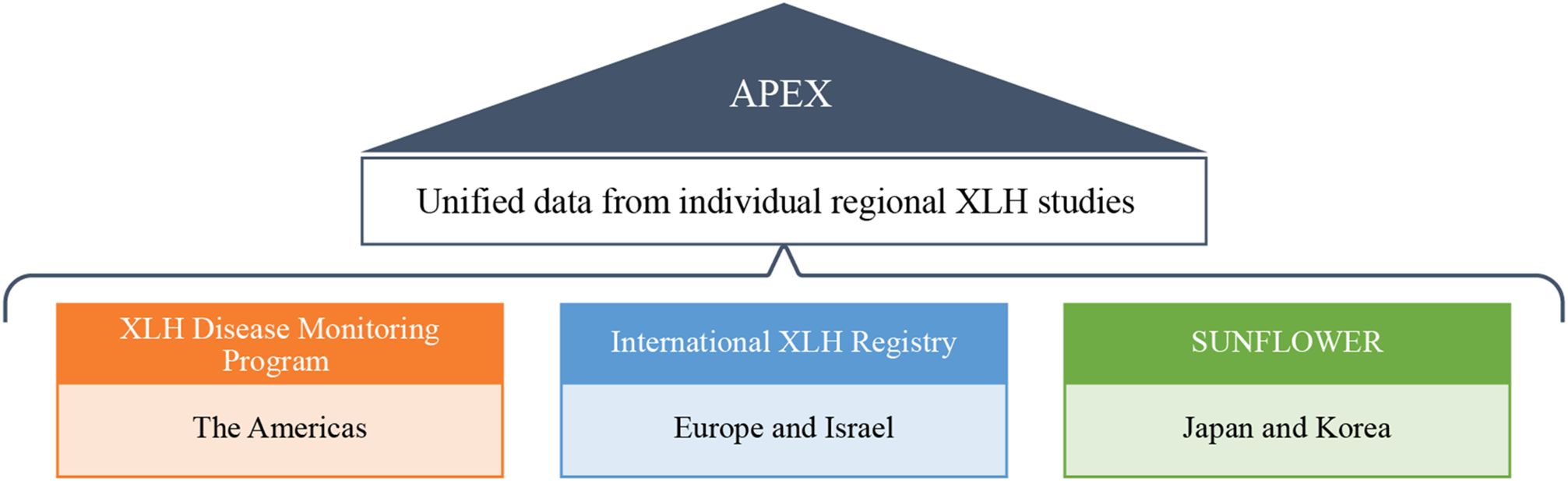
Figure 1. Data unification of XLH DMP, IXLHR, and SUNFLOWER in the APEX program. DMP, Disease Monitoring Program; IXLHR, International XLH Registry; SUNFLOWER, Study of Longitudinal Observation for Patients With X-linked Hypophosphatemic Rickets/Osteomalacia in Collaboration With Asian Partners; XLH, X-linked hypophosphatemia.
2.4 Study objectives
The overall goals of the APEX study are to:
● Describe the progressive nature of the disorder and associated burden of patients with XLH in a global real-world setting.
● Generate data on the real-world effectiveness and safety of XLH treatment options.
● Investigate regional differences in the real-world treatment of patients with XLH.
This study was planned to analyze collected information on XLH and associated clinical, patient, and disease burden data through the long-term observation of patients with XLH. APEX will enable the exploration of regional differences and generate evidence regarding the real-world effectiveness and safety of the available therapeutic options. These data will provide further context for results from RCTs to better inform clinical decision-making, and long-term consequences of XLH or its management.
2.5 Determination of sample size
Due to the rarity of XLH, statistical guidance from prior research, including the three regional studies, was limited. Sample sizes of the regional studies were determined based on feasibility and the number of potential XLH outpatients who could be enrolled in participating clinical sites (2, 54). The number of participants enrolled at the regional studies is described below; however, depending on data availability, due to the status of data processing/analysis, or whether participants in the IXLHR or SUNFLOWER studies provided additional informed consent allowing for inclusion in APEX, the number of participants in the regional studies may differ from the number available for the APEX analyses. Additional consent was not required from participants in XLH DMP. At the time of submission South American data were not available for inclusion in the APEX program but may be included at a later date.
● XLH DMP: 776 patients were enrolled.
● IXLHR: 1257 patients were enrolled as of April 15, 2024.
● SUNFLOWER: 226 patients were enrolled.
2.6 Data collection
Detailed information on the individual regional studies’ methods of data collection is available (2, 51–54). In brief, where data are available, each study collects demographic information, medical history, PHEX mutation, family history, medication history, disease and medication-related complications, laboratory assessments, functional assessments, and QoL assessments. When patients enroll into their respective regional study, both retrospective and baseline data are collected. Prospective data will be collected post-baseline at routine clinical visits and added periodically to the study databases (2, 51–54).
2.7 Data collection tools and data management
Regional study data are collected in purposely designed electronic case report forms (eCRF) via electronic data capture tools, and study databases are checked automatically using logical checks. The eCRF is completed and signed electronically for each individual participant and is completed only by qualified individuals trained in the completion and data verification of the eCRF information. Data are stored according to local regulations in order to maintain data protection.
2.8 Data quality assurance
To ensure the accuracy and robustness of the collected data, the individual regional studies have mechanisms in place to assess and, if necessary, verify the data to either confirm or correct the relevant data entry(s). These include the monitoring and auditing of study sites and automatic checks built into the electronic data capture tools (2, 51–54).
2.9 Data unification process
Data unification and the preparation of the data for analysis are separate processes and will be performed by Arbor Research Collaborative for Health (Ann Arbor, Michigan, United States).
The data will initially undergo a data checking process, including a check on the range of the values, the units used, internal consistency and the distribution of the data. Any data flagged as impossible will be queried and cross-checked, and either corrected or removed. Following the data check, the data will undergo unification.
Data unification is an iterative process with clinical and programming review to maintain accuracy. As part of the process, a consistent naming convention for variables will be applied across the study data for variables that are identical, such as age at study entry. The data will be brought into a consistent data structure to group the data that will hold all shared values, for example baseline or longitudinal data. Finally, the data will be processed to handle inconsistent variables. This will possibly include transforming variables, such as health-related QoL patient-reported outcome values, into indicators that identify values that pass clinically meaningful thresholds. Therefore, rather than trying to translate an entire incompatible scale, established diagnostic values that identify similar concepts across the different scales can be used. The unification process will also base unification schemes on clinical input and the available literature. For example, when unifying data on permanent tooth removal where XLH DMP excludes wisdom tooth removal but the other studies do not, we may restrict comparisons to specific age ranges where, according to clinical judgment and the literature, wisdom tooth removal is likely to be rare in order to have comparable data across the individual studies.
Once the data have been unified, they will be prepared for the analysis of specific research questions. This will include handling missing data and creating clinically based categories of continuous variables. When research questions require analyses of longitudinal data, regression-based methods that are robust to differences in data collection schedules will be utilized (55). Wherever possible the full combined dataset will be used to maximize statistical power; however, region-specific analyses of variables may not be available in all studies.
2.10 Statistical analyses
Appropriate to an observational study, the data will be analyzed in an exploratory manner. Continuous variables will be described by standard summary statistics (e.g. number of patients, mean and standard deviation or median and inter-quartile range, as appropriate). Categorical variables will be described by frequency and proportion. Regression modeling will be used to adjust for confounding variables between comparison groups, propensity score matching will enable the identification of comparison groups to estimate the impact of an intervention in the absence of a control group, and contrasting analyses will investigate regional differences. All analyses will attempt to identify evidence of potential biases, and diagnostic tools will be used to assess goodness of fit and appropriateness of models according to accepted statistical practices. Details of all analyses will be specified in the Statistical Analysis Plan (SAP), including how missing data will be handled.
2.11 Management and reporting of AEs
Details of AE reports associated with any treatment for XLH will be captured by the study entry for the affected patient and included in the APEX dataset. AEs will be coded at each clinical center and reported as per the local regulatory guidelines.
2.12 APEX oversight
A global medical committee (GMC) has been formed with two representatives from each regional study’s scientific steering committee, and the members of the GMC may rotate with other representatives from their study. The GMC will provide scientific oversight of the APEX program, provide their input on the data collected and the analyses performed, and author APEX reports.
3 Discussion
Multicenter, international observational studies and patient registries can be an invaluable source of real-world data in rare disorders, providing important information that would not be possible by other means, such as RCTs. It can be challenging to derive statistically robust conclusions from RCTs in rare disorders due to the limited number of patients available for inclusion. Observational studies and patient registries, in contrast, can recruit a greater number of patients as they have fewer inclusion and exclusion criteria than RCTs and typically have a longer patient follow-up. This provides invaluable information on the natural history of a disease, treatment effectiveness, additional outcomes, and safety beyond that possible in smaller RCTs (2, 50, 56, 57). Observational studies and patient registries more closely resemble real-world clinical practice compared with RCTs, and with relatively fewer exclusion criteria, they are able to provide a more generalizable insight on the impact of certain patient characteristics on disease outcomes.
APEX is an important extension of the regional XLH studies, collecting and unifying their data to form the largest XLH patient dataset in the world to date. An advantage of creating a global dataset by pooling data and increasing the sample size is in allowing analyses to be conducted that would not normally be feasible in many rare disorders. This initiative also enables a comparison of region-specific XLH populations, treatment pathways and outcomes.
There are several areas of research in which APEX may be particularly beneficial. Firstly, there is the continuing uncertainty in the management of adult patients with XLH. Clinical practice recommendations published in 2019 recommended conventional therapy for symptomatic adults (8). However, small-scale RCTs and case studies have demonstrated benefits of burosumab treatment in adults with moderate to severe XLH (31, 32, 58, 59), but real-world evidence of treatment with burosumab is lacking in this patient group. Secondly, data from APEX may also provide invaluable information on adolescent patients with XLH transitioning into adult care, as this population can be lost to follow-up and there is uncertainty regarding optimal management (60). The absence of data from this age group occurs due to their exclusion from the burosumab RCTs (29, 30), thus real-world evidence regarding their treatment and outcomes is a significant gap in our knowledge. The large patient numbers available to the APEX project and the ability to compare regional differences in practice may provide additional evidence that can be used to further refine treatment approaches in patients with XLH, and importantly, in patient groups for whom data are lacking. Finally, the analysis of the outcomes of long-term burosumab treatment and the impact of the age of initiation on outcomes may help to identify which disease complications cannot be prevented, improved, or resolved with burosumab treatment. This will hopefully lead to additional studies to characterize and address the unmet medical needs of patients with XLH.
3.1 Limitations
The limitations of APEX include inconsistencies of protocol (e.g. study visit requirements) and differences in data collection and reporting methods between the individual regional studies, which may limit the data that can be pooled or used for comparison. Therefore, it may not be possible to combine all datasets within APEX. Differences in the number of patients recruited regionally, may also result in imbalances in representation, thereby affecting the ability to describe clinical practice in underrepresented regions. In addition, there are inherent limitations of non-interventional observational studies. Unlike RCTs, there is often no standardized or mandated follow-up in registries, and this can be influenced by regional differences in data collection and clinical practice. This increases the likelihood of missing or incomplete data collection and is a potential source of bias. The XLH DMP study aims to improve follow-up, with specified visit schedules and measurements funded by the study. However, these may often be carried out in conjunction with the clinical care, leading to variability in timing and collection. SUNFLOWER has specified assessments to be collected either annually in pediatric participants or every two years in adults, while IXLHR has recommended clinical variables and collection schedule, but data are collected as per routine clinical practice. While for some registries there can be a lack of supervised enrollment, the enrollment of participants in these regional studies are supervised by the study investigators based on inclusion/exclusion criteria. The clinical diagnosis is based on the local practice of expert physicians, which ensures that the regional studies accurately reflect real-world practice. However, the lack of central definitions of diseases or diagnostic practices may result in regional variability in the diagnosis and reporting of clinical conditions.
4 Conclusion
Observational studies and patient registries offer valuable opportunities to generate real-world data and address knowledge gaps in rare diseases. The APEX program will enable the global collection and analysis of data relating to the natural history, treatment, and outcomes of patients with XLH. Analyses may help address research questions that would not be feasible in the individual regional studies, as well as allow the comparison of regional practices and outcomes. It is anticipated the findings from APEX will further increase the understanding of XLH, its treatment, and help to improve clinical practice for patients with XLH.
5 Ethics and dissemination
The XLH DMP required Institutional Review Board approvals for each participating local institution. Patients who have provided informed consent, or assent where required, with informed consent by a legally authorized representative, are included in XLH DMP (51).
The IXLHR is run in accordance with the Declaration of Helsinki and received ethical, regulatory, and institutional approvals at national, regional, and site levels for each participating country. Patient data are kept in accordance with the EU General Data Protection Regulations on the processing of personal data and the protection of privacy in the electronic communication sector (2016/679/EU). Once a patient or their legal representative provides informed consent, or assent for minors aged ≥12 years, they are enrolled in the IXLHR (2, 52).
Ethics approval for SUNFLOWER was obtained from the Ethics Committee of Osaka University, the Ethics Committee of Kyowa Kirin Co., Ltd., and the Ethics Committee of each participating medical institution. Patients or their parents/guardians are required to give informed consent before inclusion in SUNFLOWER (53, 54).
Before inclusion in the APEX project, participants in the IXLHR and SUNFLOWER are provided with information regarding the objectives and the procedures and are required to reconsent to participate.
The datasets used and analyzed that support the findings of this manuscript are available from the corresponding author and Kyowa Kirin Co., Ltd. on reasonable request.
Ethics statement
The studies involving humans were approved by the Research Ethics Review Committee of Kyowa Kirin Co., Ltd.. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants or their legal guardians.
Author contributions
MLB: Conceptualization, Investigation, Writing – original draft, Writing – review & editing. TOC: Conceptualization, Investigation, Writing – review & editing. SF: Conceptualization, Investigation, Writing – review & editing. DH: Conceptualization, Investigation, Writing – review & editing. EAI: Conceptualization, Investigation, Writing – review & editing. MK: Conceptualization, Project administration, Writing – original draft, Writing – review & editing. KPM: Conceptualization, Data curation, Methodology, Writing – original draft, Writing – review & editing. KO: Conceptualization, Investigation, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, and/or publication of this article. The authors declare that this study received funding from Kyowa Kirin Co., Ltd. The funder engaged Arbor Research Collaborative for Health Medical to conduct the data collection and analysis. The funder engaged 90TEN for writing support for this manuscript. The funders had no role in study design, data interpretation and decision to submit for publication. While Kyowa Kirin Co., Ltd funded the APEX program, the three regional studies are funded by three companies (Ultragenyx Pharmaceutical Inc, Kyowa Kirin Co., Ltd and Kyowa Kirin International plc). Ultragenyx Pharmaceutical Inc and Kyowa Kirin Co., Ltd are the sponsors of the XLH DMP study, and funded the data collection and analysis. Kyowa Kirin Co., Ltd is the sponsor of the SUNFLOWER study, and funded the data collection and analysis. Kyowa Kirin International plc is the sponsor of the IXLHR study, and funded the data collection and analysis. The funders had no role in study design or data interpretation.
Acknowledgments
The authors thank the patients/guardians who agreed to take part in the regional studies. Kyowa Kirin Co., Ltd. engaged Arbor Research Collaborative for Health Medical to conduct the data unification and analysis. Writing support for this manuscript was provided by Edward Maguire, PhD (90TEN), funded by Kyowa Kirin Co., Ltd. The authors maintained full editorial control of the paper and provided their final approval of all content.
Conflict of interest
Author MLB has received honoraria from Amgen, Ascendis, Bruno Farmaceutici, Calcilytix, Kyowa Kirin, and UCB; grants and/or speaker fees from Abiogen, Alexion, Amgen, Amolyt, Amorphical, Bruno Farmaceutici, CoGeDi, Echolight, Eli Lilly, Enterabio, Gedeon Richter, Italfarmaco, Kyowa Kirin, Menarini, Monte Rosa Therapeutics, SPA, Takeda, Theramex, and UCB; and consultancy fees from Aboca, Alexion, Amolyt, Bruno Farmaceutici, Calcilytix, Echolight, Kyowa Kirin, Personal Genomics, Smoke Free World Foundation. Author TOC has received research funding and consulting fees from Ultragenyx and Kyowa Kirin. Author SF has received consultant fees from Kyowa Kirin Co., Ltd. Author DH has received speaker and consultancy fees from Amgen, Chiesi, Horizon, Kyowa Kirin, and Ultragenyx; research grants from Amgen, Chiesi and Kyowa Kirin. Author EAI has received research funding and consulting fees from Ultragenyx and Kyowa Kirin North America. Author MK is an employee of Kyowa Kirin Co., Ltd. Author KPM has worked on projects receiving research funding from Kyowa Kirin. Author KO has received lecture fees from Kyowa Kirin Co. Ltd. and Alexion Pharma.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Beck-Nielsen SS, Mughal Z, Haffner D, Nilsson O, Levtchenko E, Ariceta G, et al. FGF23 and its role in X-linked hypophosphatemia-related morbidity. Orphanet J Rare Dis. (2019) 14:58. doi: 10.1186/s13023-019-1014-8
2. Padidela R, Nilsson O, Makitie O, Beck-Nielsen S, Ariceta G, Schnabel D, et al. The international X-linked hypophosphataemia (XLH) registry (NCT03193476): rationale for and description of an international, observational study. Orphanet J Rare Dis. (2020) 15:172. doi: 10.1186/s13023-020-01434-4
3. Javaid MK, Ward L, Pinedo-Villanueva R, Rylands AJ, Williams A, Insogna K, et al. Musculoskeletal features in adults with X-linked hypophosphatemia: an analysis of clinical trial and survey data. J Clin Endocrinol Metab. (2022) 107:e1249–e62. doi: 10.1210/clinem/dgab739
4. Beck-Nielsen SS, Brock-Jacobsen B, Gram J, Brixen K, Jensen TK. Incidence and prevalence of nutritional and hereditary rickets in southern Denmark. Eur J Endocrinol. (2009) 160:491–7. doi: 10.1530/EJE-08-0818
5. Endo I, Fukumoto S, Ozono K, Namba N, Inoue D, Okazaki R, et al. Nationwide survey of fibroblast growth factor 23 (FGF23)-related hypophosphatemic diseases in Japan: prevalence, biochemical data and treatment. Endocr J. (2015) 62:811–6. doi: 10.1507/endocrj.EJ15-0275
6. Hawley S, Shaw NJ, Delmestri A, Prieto-Alhambra D, Cooper C, Pinedo-Villanueva R, et al. Prevalence and mortality of individuals with X-linked hypophosphatemia: A United Kingdom real-world data analysis. J Clin Endocrinol Metab. (2020) 105:e871–8. doi: 10.1210/clinem/dgz203
7. Rafaelsen S, Johansson S, Raeder H, Bjerknes R. Hereditary hypophosphatemia in Norway: a retrospective population-based study of genotypes, phenotypes, and treatment complications. Eur J Endocrinol. (2016) 174:125–36. doi: 10.1530/EJE-15-0515
8. Haffner D, Emma F, Eastwood DM, Duplan MB, Bacchetta J, Schnabel D, et al. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat Rev Nephrol. (2019) 15:435–55. doi: 10.1038/s41581-019-0152-5
9. Rajah J, Thandrayen K, Pettifor JM. Clinical practice: diagnostic approach to the rachitic child. Eur J Pediatr. (2011) 170:1089–96. doi: 10.1007/s00431-011-1529-z
10. Whyte MP, Schranck FW, Armamento-Villareal R. X-linked hypophosphatemia: a search for gender, race, anticipation, or parent of origin effects on disease expression in children. J Clin Endocrinol Metab. (1996) 81:4075–80. doi: 10.1210/jcem.81.11.8923863
11. Ariceta G, Beck-Nielsen SS, Boot AM, Brandi ML, Briot K, de Lucas Collantes C, et al. The International X-Linked Hypophosphatemia (XLH) Registry: first interim analysis of baseline demographic, genetic and clinical data. Orphanet J Rare Dis. (2023) 18:304. doi: 10.1186/s13023-023-02882-4
12. Munns CF, Maguire EP, Williams A, Wood S, Biggin A. Craniosynostosis in patients with X-linked hypophosphatemia: A review. JBMR Plus. (2023) 7:e10728. doi: 10.1002/jbm4.10728
13. Cheung M, Rylands AJ, Williams A, Bailey K, Bubbear J. Patient-reported complications, symptoms, and experiences of living with X-linked hypophosphatemia across the life-course. J Endocr Soc. (2021) 5:bvab070. doi: 10.1210/jendso/bvab070
14. Yanes MIL, Diaz-Curiel M, Peris P, Vicente C, Marin S, Ramon-Krauel M, et al. Health-related quality of life of X-linked hypophosphatemia in Spain. Orphanet J Rare Dis. (2022) 17:298. doi: 10.1186/s13023-022-02452-0
15. Boukpessi T, Hoac B, Coyac BR, Leger T, Garcia C, Wicart P, et al. Osteopontin and the dento-osseous pathobiology of X-linked hypophosphatemia. Bone. (2017) 95:151–61. doi: 10.1016/j.bone.2016.11.019
16. Coyac BR, Hoac B, Chafey P, Falgayrac G, Slimani L, Rowe PS, et al. Defective mineralization in X-linked hypophosphatemia dental pulp cell cultures. J Dent Res. (2018) 97:184–91. doi: 10.1177/0022034517728497
17. El Hakam C, Parenté A, Baraige F, Magnol L, Forestier L, Di Meo F, et al. PHEX(L222P) mutation increases phex expression in a new ENU mouse model for XLH disease. Genes (Basel). (2022) 13:1356. doi: 10.3390/genes13081356
18. Carpenter TO, Imel EA, Holm IA, Jan de Beur SM, Insogna KL. A clinician's guide to X-linked hypophosphatemia. J Bone Miner Res. (2011) 26:1381–8. doi: 10.1002/jbmr.340
19. Kruse K, Hinkel GK, Griefahn B. Calcium metabolism and growth during early treatment of children with X-linked hypophosphataemic rickets. Eur J Pediatr. (1998) 157:894–900. doi: 10.1007/s004310050962
20. Mäkitie O, Doria A, Kooh SW, Cole WG, Daneman A, Sochett E. Early treatment improves growth and biochemical and radiographic outcome in X-linked hypophosphatemic rickets. J Clin Endocrinol Metab. (2003) 88:3591–7. doi: 10.1210/jc.2003-030036
21. Quinlan C, Guegan K, Offiah A, Neill RO, Hiorns MP, Ellard S, et al. Growth in PHEX-associated X-linked hypophosphatemic rickets: the importance of early treatment. Pediatr Nephrol. (2012) 27:581–8. doi: 10.1007/s00467-011-2046-z
22. Al Juraibah F, Al Amiri E, Al Dubayee M, Al Jubeh J, Al Kandari H, Al Sagheir A, et al. Diagnosis and management of X-linked hypophosphatemia in children and adolescent in the Gulf Cooperation Council countries. Arch Osteoporos. (2021) 16:52. doi: 10.1007/s11657-021-00879-9
23. Padidela R, Cheung MS, Saraff V, Dharmaraj P. Clinical guidelines for burosumab in the treatment of XLH in children and adolescents: British paediatric and adolescent bone group recommendations. Endocr Connect. (2020) 9:1051–6. doi: 10.1530/EC-20-0291
24. European Medicines Agency. CRYSVITA. Summary of Product Characteristics . Available online at: https://www.ema.europa.eu/en/documents/product-information/crysvita-epar-product-information_en.pdf (Accessed January 2024).
25. U.S. Food and Drug Administration. Crysvita Full Prescribing Information. Available online at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761068s005lbl.pdf (Accessed January 2024).
26. Kyowa Kirin Announces Approval of Crysvita® (burosumab) for the Treatment of FGF23-related Hypophosphatemic Rickets and Osteomalacia in Japan . Available online at: https://www.kyowakirin.com/media_center/news_releases/2019/e20190920_01.html (Accessed January 2024).
27. Brener A, Lebenthal Y, Cleper R, Kapusta L, Zeitlin L. Body composition and cardiometabolic health of pediatric patients with X-linked hypophosphatemia (XLH) under burosumab therapy. Ther Adv Endocrinol Metab. (2021) 12:20420188211001150. doi: 10.1177/20420188211001150
28. Saraff V, Nadar R, Hogler W. New developments in the treatment of X-linked hypophosphataemia: implications for clinical management. Paediatr Drugs. (2020) 22:113–21. doi: 10.1007/s40272-020-00381-8
29. Carpenter TO, Imel EA, Ruppe MD, Weber TJ, Klausner MA, Wooddell MM, et al. Randomized trial of the anti-FGF23 antibody KRN23 in X-linked hypophosphatemia. J Clin Invest. (2014) 124:1587–97. doi: 10.1172/JCI72829
30. Carpenter TO, Whyte MP, Imel EA, Boot AM, Hogler W, Linglart A, et al. Burosumab therapy in children with X-linked hypophosphatemia. N Engl J Med. (2018) 378:1987–98. doi: 10.1056/NEJMoa1714641
31. Portale AA, Carpenter TO, Brandi ML, Briot K, Cheong HI, Cohen-Solal M, et al. Continued beneficial effects of burosumab in adults with X-linked hypophosphatemia: results from a 24-week treatment continuation period after a 24-week double-blind placebo-controlled period. Calcif Tissue Int. (2019) 105:271–84. doi: 10.1007/s00223-019-00568-3
32. Kamenicky P, Briot K, Brandi ML, Cohen-Solal M, Crowley RK, Keen R, et al. Benefit of burosumab in adults with X-linked hypophosphataemia (XLH) is maintained with long-term treatment. RMD Open. (2023) 9:e002676. doi: 10.1136/rmdopen-2022-002676
33. Weber TJ, Imel EA, Carpenter TO, Peacock M, Portale AA, Hetzer J, et al. Long-term burosumab administration is safe and effective in adults with X-linked hypophosphatemia. J Clin Endocrinol Metab. (2022) 108:155–65. doi: 10.1210/clinem/dgac518
34. Linglart A, Imel EA, Whyte MP, Portale AA, Hogler W, Boot AM, et al. Sustained efficacy and safety of burosumab, a monoclonal antibody to FGF23, in children with X-linked hypophosphatemia. J Clin Endocrinol Metab. (2021) 107:813–24. doi: 10.1210/clinem/dgab729
35. Imel EA, Zhang X, Ruppe MD, Weber TJ, Klausner MA, Ito T, et al. Prolonged correction of serum phosphorus in adults with X-linked hypophosphatemia using monthly doses of KRN23. J Clin Endocrinol Metab. (2015) 100:2565–73. doi: 10.1210/jc.2015-1551
36. Imel EA, Glorieux FH, Whyte MP, Munns CF, Ward LM, Nilsson O, et al. Burosumab versus conventional therapy in children with X-linked hypophosphataemia: a randomised, active-controlled, open-label, phase 3 trial. Lancet. (2019) 393:2416–27. doi: 10.1016/S0140-6736(19)30654-3
37. Insogna KL, Briot K, Imel EA, Kamenicky P, Ruppe MD, Portale AA, et al. A randomized, double-blind, placebo-controlled, phase 3 trial evaluating the efficacy of burosumab, an anti-FGF23 antibody, in adults with X-linked hypophosphatemia: week 24 primary analysis. J Bone Miner Res. (2018) 33:1383–93. doi: 10.1002/jbmr.3475
38. Ertl DA, Le Lorier J, Gleiss A, Trabado S, Bensignor C, Audrain C, et al. Growth pattern in children with X-linked hypophosphatemia treated with burosumab and growth hormone. Orphanet J Rare Dis. (2022) 17:412. doi: 10.1186/s13023-022-02562-9
39. Gadion M, Hervé A, Herrou J, Rothenbuhler A, Smail-Faugeron V, Courson F, et al. Burosumab and dental abscesses in children with X-linked hypophosphatemia. JBMR Plus. (2022) 6:e10672. doi: 10.1002/jbm4.10672
40. Hervé A, Gadion M, Herrou J, Izart M, Linglart A, Cohen-Solal M, et al. Improved oral health in adults with X-linked hypophosphatemia treated with burosumab. J Clin Endocrinol Metab. (2024) dgae398. doi: 10.1210/clinem/dgae398
41. Levy-Shraga Y, Levi S, Regev R, Gal S, Brener A, Lebenthal Y, et al. Linear growth of children with X-linked hypophosphatemia treated with burosumab: a real-life observational study. Eur J Pediatr. (2023) 182:5191–202. doi: 10.1007/s00431-023-05190-y
42. Martin Ramos S, Gil-Calvo M, Roldan V, Castellano Martinez A, Santos F. Positive response to one-year treatment with burosumab in pediatric patients with X-linked hypophosphatemia. Front Pediatr. (2020) 8:48. doi: 10.3389/fped.2020.00048
43. Michigami T, Kang HG, Namba N, Ito N, Kubota T, Shintani A, et al. Burosumab treatment of X-linked hypophosphatemia patients: interim analysis of the SUNFLOWER longitudinal, observational cohort study. JBMR Plus. (2024) 8:ziae079. doi: 10.1093/jbmrpl/ziae079
44. Mindler GT, Stauffer A, Kranzl A, Penzkofer S, Ganger R, Radler C, et al. Persistent lower limb deformities despite amelioration of rickets in X-linked hypophosphatemia (XLH) - A prospective observational study. Front Endocrinol (Lausanne). (2022) 13:866170. doi: 10.3389/fendo.2022.866170
45. Paloian NJ, Nemeth B, Sharafinski M, Modaff P, Steiner RD. Real-world effectiveness of burosumab in children with X-linked hypophosphatemic rickets. Pediatr Nephrol. (2022) 37:2667–77. doi: 10.1007/s00467-022-05484-7
46. Sandy JL, Nunez C, Wheeler BJ, Jefferies C, Morris A, Siafarikas A, et al. Prevalence and characteristics of paediatric X-linked hypophosphataemia in Australia and New Zealand: Results from the Australian and the New Zealand Paediatric Surveillance Units survey. Bone. (2023) 173:116791. doi: 10.1016/j.bone.2023.116791
47. Sawamura K, Hamajima T, Izawa M, Kaneko H, Kitamura A, Kitoh H. Changes of the lower limb deformity in children with FGF23-related hypophosphatemic rickets treated with Burosumab: a single-center prospective study. J Pediatr Orthop B. (2024) 33:90–6. doi: 10.1097/bpb.0000000000001054
48. Vaisbich MH, de Cillo ACP, Silva BCC, DÁlva CB, de Carvalho ÉH, de Almeida J, et al. Real-world data of Brazilian adults with X-linked hypophosphatemia (XLH) treated with burosumab and comparison with other worldwide cohorts. Mol Genet Genomic Med. (2024) 12:e2387. doi: 10.1002/mgg3.2387
49. Walker EYX, Lindsay TAJ, Allgrove J, Marlais M, Bockenhauer D, Hayes W. Burosumab in management of X-linked hypophosphataemia: a retrospective cohort study of growth and serum phosphate levels. Arch Dis Child. (2023) 108:379–84. doi: 10.1136/archdischild-2022-324962
50. Bassanese G, Wlodkowski T, Servais A, Heidet L, Roccatello D, Emma F, et al. The European Rare Kidney Disease Registry (ERKReg): objectives, design and initial results. Orphanet J Rare Dis. (2021) 16:251. doi: 10.1186/s13023-021-01872-8
51. X-linked Hypophosphatemia Disease Monitoring Program . Available online at: https://www.clinicaltrials.gov/ct2/show/NCT03651505 (Accessed January 2024).
52. Registry for Patients With X-Linked Hypophosphatemia (International XLH Registry) . Available online at: https://clinicaltrials.gov/study/NCT03193476?term=NCT03193476&rank=1 (Accessed January 2024).
53. Study of Longitudinal Observation for Patient With X-linked Hypophosphatemic Rickets/Osteomalacia in Collaboration With Asian Partners (SUNFLOWER) . Available online at: https://clinicaltrials.gov/study/NCT03745521?term=NCT03745521&rank=1 (Accessed January 2024).
54. Kubota T, Fukumoto S, Cheong HI, Michigami T, Namba N, Ito N, et al. Long-term outcomes for Asian patients with X-linked hypophosphataemia: rationale and design of the SUNFLOWER longitudinal, observational cohort study. BMJ Open. (2020) 10:e036367. doi: 10.1136/bmjopen-2019-036367
55. Zee J, Muenz D, McCullough KP, Bieber B, Metzger M, Alencar de Pinho N, et al. Potential surrogate outcomes for kidney failure in advanced CKD: evaluation of power and predictive ability in CKDopps. Kidney Med. (2022) 4:100395. doi: 10.1016/j.xkme.2021.10.008
56. Viviani L, Zolin A, Mehta A, Olesen HV. The European Cystic Fibrosis Society Patient Registry: valuable lessons learned on how to sustain a disease registry. Orphanet J Rare Dis. (2014) 9:81. doi: 10.1186/1750-1172-9-81
57. Kempf L, Goldsmith JC, Temple R. Challenges of developing and conducting clinical trials in rare disorders. Am J Med Genet A. (2018) 176:773–83. doi: 10.1002/ajmg.a.38413
58. Aiello F, Pasquali D, Baronio F, Cassio A, Rossi C, Di Fraia R, et al. Rare PHEX intron variant causes complete and severe phenotype in a family with hypophosphatemic rickets: a case report. J Pediatr Endocrinol Metab. (2023) 36:91–5. doi: 10.1515/jpem-2022-0365
59. Zagari MC, Chiarello P, Iuliano S, D'Antona L, Rocca V, Colao E, et al. The variant p.Ala84Pro is causative of X-linked hypophosphatemic rickets: possible relationship with burosumab swinging response in adults. Genes (Basel). (2022) 14:80. doi: 10.3390/genes14010080
Keywords: X-linked hypophosphatemia (XLH), phosphate-regulating endopeptidase homologX-linked (PHEX) gene, fibroblast growth factor 23 (FGF23), musculoskeletal, rickets, osteomalacia, odontomalacia, Advancing Patient Evidence in XLH (APEX)
Citation: Brandi ML, Carpenter TO, Fukumoto S, Haffner D, Imel EA, Kanematsu M, McCullough KP and Ozono K (2025) Advancing patient evidence in XLH (APEX): rationale and design of a real-world XLH global data unification program. Front. Endocrinol. 16:1471127. doi: 10.3389/fendo.2025.1471127
Received: 26 July 2024; Accepted: 21 February 2025;
Published: 07 April 2025.
Edited by:
Mohammed S. Razzaque, The University of Texas Rio Grande Valley, United StatesReviewed by:
Khashayar Sakhaee, University of Texas Southwestern Medical Center, United StatesAvivit Brener, Dana-Dwek Children’s Hospital, Israel
Yan Jiang, Peking Union Medical College Hospital (CAMS), China
Suma Uday, Birmingham Women’s and Children’s Hospital, United Kingdom
Copyright © 2025 Brandi, Carpenter, Fukumoto, Haffner, Imel, Kanematsu, McCullough and Ozono. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Luisa Brandi, bWFyaWFsdWlzYUBtYXJpYWx1aXNhYnJhbmRpLml0