 Grace E. Shryack1,2,3
Grace E. Shryack1,2,3 Alexa A. Krause1,2,3Simone Hernandez Ruano4Laura C. Schulz5Kathleen A. Pennington4*
Alexa A. Krause1,2,3Simone Hernandez Ruano4Laura C. Schulz5Kathleen A. Pennington4* R. Scott Rector1,2,3*
R. Scott Rector1,2,3*- 1Research Service, Harry S Truman Memorial Veterans Medical Center, Columbia, MO, United States
- 2NextGen Precision Health, University of Missouri, Columbia, MO, United States
- 3Department of Nutrition and Exercise Physiology, University of Missouri, Columbia, MO, United States
- 4Department of Obstetrics and Gynecology, Baylor College of Medicine, Houston, TX, United States
- 5Deparment of Obstetrics, Gynecology, and Women’s Health, University of Missouri, Columbia, MO, United States
Introduction: Gestational Diabetes Mellitus (GDM) impacts roughly 1 in 7 pregnancies and results in metabolic dysfunction-associated steatotic liver disease (MASLD) in 30% of these women. Nonetheless, there exists a dearth of investigation into the relationship between GDM and MASLD. Here, we sought to investigate the potential role of hepatic mitochondrial function in GDM and MASLD.
Methods: One week prior to conception and throughout pregnancy, mice were fed either a low-fat control diet (CD) or a high-fat, high-sucrose (HFHS) diet to induce an established model of GDM. Maternal livers were collected at day 0, 6.5, 13.5 and 17.5 of pregnancy. Hepatic markers (via mRNA and western blot analyses) of mitochondrial biogenesis, autophagy, mitophagy, activity, and function were assessed, as well as markers of inflammation and antioxidant status were evaluated.
Results: Progressing gestation in both CD and GDM dams significantly decreased protein and mRNA markers of hepatic mitochondrial biogenesis (Pgc1-α, Tfam), autophagy (Atg5, Sqstm1), mitophagy (Pink1, Bnip3) and lipid handling (Ampk, pAMPK/AMPK, FAS, ACC, pACC, Mttp) with a main effect for time (P<0.05). HFHS-induced model of GDM lead to significant elevations in liver triglycerides and NAFLD Activity Score (NAS) (P<0.0001, P<0.0001) independent of body weight gain during gestation. MASLD development in the GDM mice occurred in conjunction with significant reductions in hepatic mitochondrial activity at day 6.5 (citrate synthase, p<0.01) and day 17.5 (β-HAD, citrate synthase, P<0.001) compared to CD mice. However, GDM lead to elevated protein and/or mRNA markers of mitochondrial biogenesis (Tfam), mitophagy (BNIP3, Bnip3, Sqstm1, Pink1), lipid handling (Mttp), inflammation (Il-1β, Tnf-α, Tgf-β) and antioxidant defense (Gxp1, Nfe2l2, Sod2) (P<0.05).
Discussion: Pregnancy, independent of diet, decreased markers of liver mitochondrial biogenesis, autophagy, and mitophagy in dams. The GDM mouse model exhibited elevated hepatic TG and NAS, as well as decreased liver mitochondrial activity. These findings demonstrate that pregnancy and GDM significantly impact maternal liver mitochondrial metabolism and unveil new insight on the potential relationship between MASLD and GDM.
Introduction
The diagnosis of glucose intolerance during pregnancy or Gestational Diabetes Mellitus (GDM), affects up to 14% of pregnancies globally (1, 2). Although pregnancy is a natural state of insulin resistance, women with GDM face a worsened level of insulin resistance coupled with glucose intolerance (3–8). This disease can then further be exacerbated by a lack of beta cell expansion in the pancreas, leading to reduced insulin secretion (9–11). This failure to adapt to the metabolic demands of pregnancy is associated with an increased risk of type 2 diabetes, GDM in subsequent pregnancies, hypertension, cardiovascular disease, increased body mass index, and cesarean delivery (12). Moreover, GDM induces adverse outcomes in the fetus, including increased risk for preterm birth, macrosomia, and type 2 diabetes (12).
Recently, studies have found that women with GDM have a heightened risk for developing metabolic dysfunction-associated steatotic liver disease (MASLD), formerly known as nonalcoholic fatty liver disease (NAFLD), during pregnancy and post-partum (13–15). Women with MASLD were three times more likely to develop GDM (16, 17). Furthermore, compared to their healthy, pregnant counterparts, women with GDM display alterations in hepatic serum metabolites such as lysophosphatidylcholine, glycerophospholipids, monoacylglycerol, serine, proline, leucine, and isoleucine (18, 19). These metabolites are involved in lipid and amino acid metabolism (19, 20), and such alterations of these metabolites may help explain the metabolic impact of GDM (21–23), but a mechanistic link between GDM and MASLD has yet to be determined.
MASLD, characterized by a greater than 5% of hepatocytes containing lipid, is the most common form of chronic liver disease (24, 25). MASLD is linked to several metabolic disorders including type 2 diabetes, cardiovascular disease, and obesity (24, 26). Although the specific cause of MASLD is unknown, there are a number of processes that influence this disease, such as alterations in lipid metabolism and mitochondrial function (27–30). The hepatic mitochondrion is responsible for the breakdown of different substrates via glycolysis and fatty acid oxidation which then feed into the TCA cycle and activates ATP synthesis via the electron transport chain, a process known as respiration (31–34). Further, functional mitochondria are in a constant cycle of turnover via biogenesis, autophagy, and mitophagy. During MASLD, mitochondrial respiration, metabolism, activity, and turnover are impaired (35–37). This creates a pool of poorly functioning mitochondria that exhibits inflammation, increases reactive oxygen species generation, and decreases in antioxidant capacity (35). The health of a hepatic mitochondria depends on the interaction between these processes and when impaired significantly contributes to the development and progression of MASLD (32, 37).
As stated previously, there is limited investigation into the mechanistic links between GDM and MASLD. Previous murine models of GDM have shown alterations in maternal hepatic electron transport chain and UCP2, a marker of ROS (38). Offspring of mice with GDM display impaired mitochondrial function and hepatic lipid accumulation (38, 39). The purpose of this study was to evaluate the impact of GDM on hepatic lipid metabolism, mitochondrial function, and inflammation in the maternal liver. It is imperative to understand how GDM and MASLD are connected. By evaluating the liver at varying time points during gestation, we can further understand the natural changes that occur in the liver in a healthy vs. GDM pregnancy. We hypothesized that GDM will significantly increase maternal hepatocellular injury.
Materials and methods
Experimental animals
The Baylor College of Medicine Institutional Animal Care and Use Committee approved all animal procedures. All methods performed were in accordance with the Guide for the Care and Use of Laboratory animals. Seven-week-old female and 12-week-old male wild type C57BL/6J were purchased from JAX (Bar Harbor, ME). Mice were kept at 23°C, 40-60% humidity, and a 14-hour light/10-hour dark cycle. Mice had ab libitum access to food and water. Female mice were mated to C57BL/6J breeder males for five days. Following, observation of a copulatory plug was identified and marked as day 0 of pregnancy. A total of 60 mice were included in this study. CD: day 0 n=8, day 6.5 n=8, day 13.5 n=4, and day 17.5 n=10. GDM: day 0 n=8, day 6.5 n=5, day 13.5 n=7, and day 17.5 n=10.
Diet and timeline
Mice were randomized and fed either a high-fat, high-sucrose (HFHS) diet (D12451, Research Diets Inc., New Brunswick, NJ) or control diet (CD) (D12450K, Research Diets Inc.). Maternal feeding started 1-week prior to conception and continued throughout pregnancy. This feeding timeline has been verified to confirm a model of GDM in C57BL/6J mice (40–42). Livers were collected on days 0, 6.5, 13.5, and 17.5 of pregnancy. After a 6-hour fast, mice were euthanized via CO2 inhalation and cardiac exsanguination.
Histology
Liver tissues fixed in 10% formalin for 24 hours were embedded in paraffin, sectioned, and stained with hematoxylin-eosin (H&E) by IDEXX BioAnalytics (Columbia, MO, USA) for histological evaluation. Steatosis was scored by direct observation of the percentage of overall surface area covered by lipid accumulation within cells on a low to medium power and scored as a 0 (<5%), 1 (5-33%), 2 (33-66%), or 3 (>66%). Lobular inflammation refers to the number of foci with white blood cell infiltrate present per 200x powered field, scored as 0 (no foci), 1 (<2 foci), 2 (2-4 foci), or 3 (>4 foci). Hepatocyte ballooning refers to the presence of hepatocytes that have pathologically swollen with or without the presence of lipid vacuoles, indicating a trend towards apoptosis and necrosis of the cell, and is scored as 0 (no ballooned hepatocytes), 1 (rare but definite ballooned cells), or 2 (many/most cells are prominent with ballooning). NAS (NAFLD Activity Score) assessment consists of histologically examining the liver for steatosis, lobular inflammation, and hepatocyte ballooning utilizing H&E staining, and is indicated as a sum of these scores (0–8) (43).
Serum triglycerides and insulin
Triglycerides were isolated from the liver utilizing previous methods (44). Once extracted, liver and serum triglycerides were measured using the Serum Triglyceride Determination Ket (Sigma-Aldrich). Serum insulin was measured after a 6-hour fast using a Rat/Mouse Insulin ELISA kit from EMD Millipore according to the manufacturer’s instructions. Serum insulin and triglycerides were part of previous studies that have been published (40–42).
Mitochondrial activity
β-hydroxyacyl-CoA dehydrogenase (β-HAD) and citrate synthase activities were measured utilizing methods from Srere et al. (45) and Bass et al. (46) as previously described by our lab (28, 47, 48). For citrate synthase, liver homogenates were incubated with acetyl-CoA, DTNB, and oxaloacetate. Following, detection of reduced DTNB at a wavelength of 412nm served as an index of enzymatic activity. For β-HAD, liver homogenate was placed in an assay buffer of EDTA, triethanolamine-HCl, and NADH at a pH of 7.0. After baseline reading, acetoacetyl-CoA was added, and the rates of NADH disappearance to NAD appearance ratio were measured every 10 seconds for 5 minutes at 340 nm.
RNA extraction and quantitative PCR
Gene expression was completed from whole liver tissue samples. RNA and complementary DNA were isolated based on previous protocols reported from our lab (48, 49). Quantitative Real-time PCR (qPCR) was conducted using iTAQ Universal SYBR Green Supermix (Bio-Rad). Results are displayed as RQ (Relative Quantification) and was calculated using the Ct of the target gene, Ct of the housekeeping gene, and the average control group (day 0, CD). Results were calculated using the delta Ct methods and values are expressed relative to the control group (day 0, CD). Primer sequences are shown in Supplementary Table 1.
Western blotting
Whole liver homogenates were prepared for Western blot analysis as previously described (28, 47, 49, 50). Primary antibodies were used at 1:1,000 dilutions, and secondary antibodies at 1:5,000 dilutions. Primary antibodies used are listed in Supplementary Table 1. Blots were analyzed via densiometric analysis (Image Lab v5.1, Bio-Rad Laboratories Inc., Hercules, CA). Total protein was assessed with Amido black (0.1%; Millipore Sigma) to control differences in protein loading and transfer, as previously described (28, 47, 49, 50).
Statistical analysis
Statistical analyses were completed using GraphPad Prism 10.0.2. All data was analyzed via two-way ANOVA (diet, time) with Tukey’s multiple comparison post-hoc test employed when necessary. Differences were considered statistically significant when P < 0.05. Data are shown as mean ± SEM.
Results
Animal characteristics and MASLD development
As we have previously reported for this mouse model of GDM, a significant increase in body weight across pregnancy stages occurred in both groups with a main effect for time (Figure 1A, P<0.0001). There were significant main effects for diet and time for serum insulin, serum triglycerides, and liver triglycerides (P<0.01). On days 13.5 and 17.5, serum insulin increased in CD, whereas GDM caused insulin secretion to be blunted (P=0.01, P=0.004, Figure 1B). This model, employed by our group, also exhibited systemic insulin resistance compared to control by day 13.5, as confirmed by glucose tolerance test and euglycemic-hyperinsulinemic clamp studies (41, 42). GDM mice compared to control mice displayed a non-significant elevation in serum triglycerides at day 0, but both groups were decreased by day 17.5 (Figure 1C). Liver triglycerides were markedly elevated in the GDM mice compared to control at day 17.5 (P<0.0001, Figure 1D). NAFLD activity score (NAS) and its three main components were evaluated to determine MASLD progression (Figure 1E). GDM displayed elevations in hepatic steatosis and significant increases in total NAS for day 0 (P<0.01), 6.5 (P<0.0001), and 17.5 (P<0.0001) compared to control mice. Hepatic inflammation and ballooning were also increased in GDM mice compared to CD at day 6.5 and 17.5 (P<0.05). There was a main effect for time and diet for all three components and NAS (P<0.01).
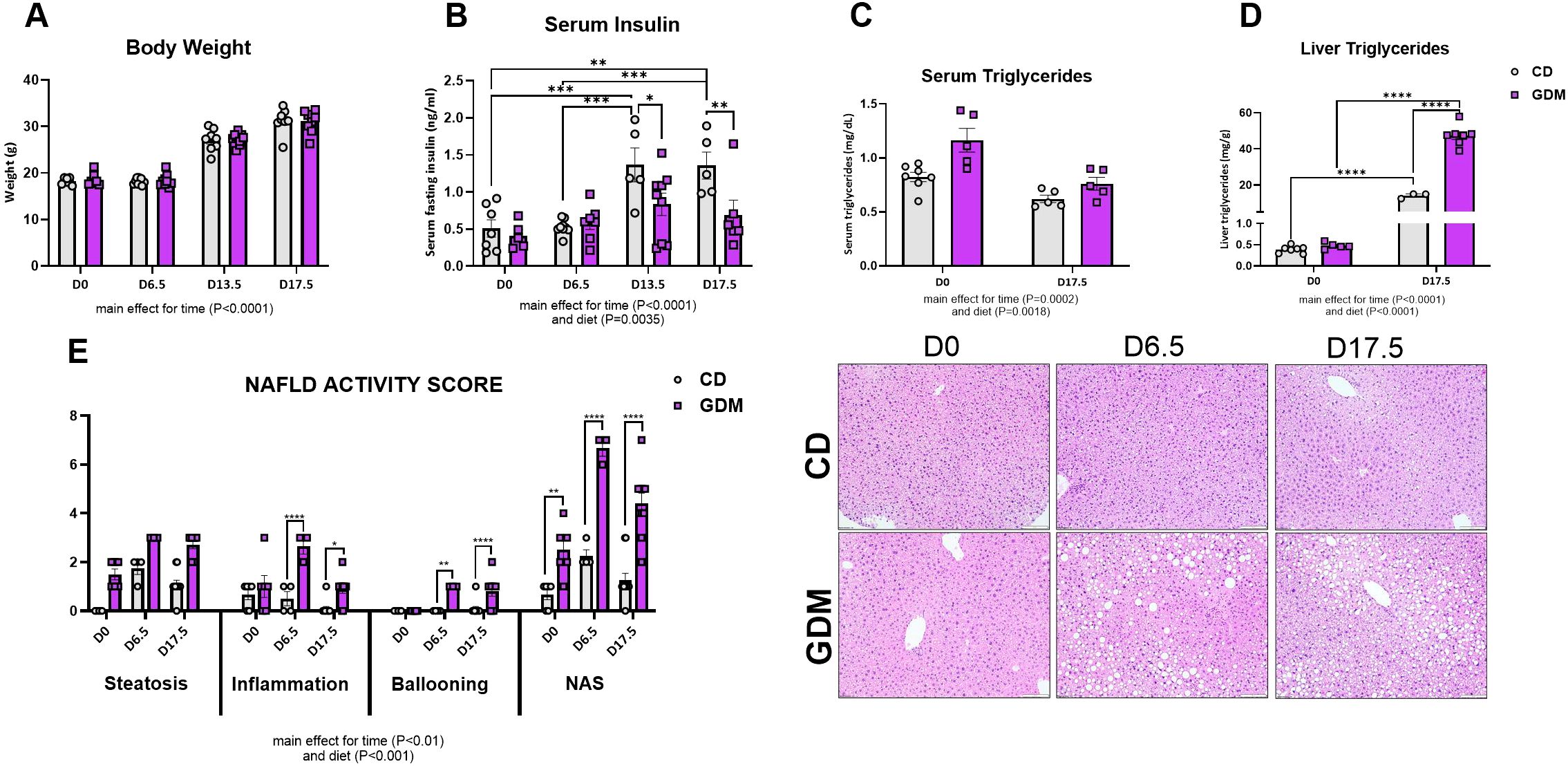
Figure 1. Animal characteristics. (A) Body weight of dams throughout stages of pregnancy (g). (B) Fasting serum insulin concentrations (ng/ml). (C) Serum triglycerides (mg/dL). Data were previously reported (40–42). (D) Liver triglycerides (mg/dL). (E) NAFLD Activity Score and Liver H&E Representative images. D0 = day 0, D6.5 = day 6.5, D13.5 = day 13.5, D17.5 = day 17.5 of pregnancy. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 all denote an interaction. Main effect for time and/or diet indicated under each graph. Error bars represent SEM. N=6-10. CD, Control Diet; GDM, Gestational Diabetes Mellitus.
Markers of hepatic lipid metabolism decreased throughout gestation
Markers of hepatic lipid metabolism were evaluated throughout gestational development. mRNA expression of Ampk (AMP-activated protein kinase) decreased as pregnancy progressed (main effect for time, P<0.0001, Figure 2A) while protein expression of phospho-AMPK to AMPK decreased at day 6.5 but slightly increased by day 17.5 (main effect for time, P=0.0373, Figure 2B). Other major markers of lipid handling [fatty acid synthase (FAS), acetyl-CoA carboxylase (ACC), phospho-ACC, microsomal triglyceride transfer protein (Mttp)] except ACC (acetyl-CoA Carboxylase) protein were significantly decreased with gestational duration (P<0.05, Figure 2). GDM significantly decreased hepatic de novo lipogenesis marker FAS (fatty acid synthase) protein at day 0 (P=0.024), day 6.5 (P<0.0001), and 17.5 (P=0.0001) compared to CD (Figure 2D) and there were main effects for GDM for ACC and phospho-ACC protein content (P<0.01, Figures 2E, F). Mttp, which aids in transporting lipids across cell membranes, was elevated at day 0 (P=0.007) and then decreased at day 6.5 (P=0.035) in GDM compared to control mice (Figure 2G).
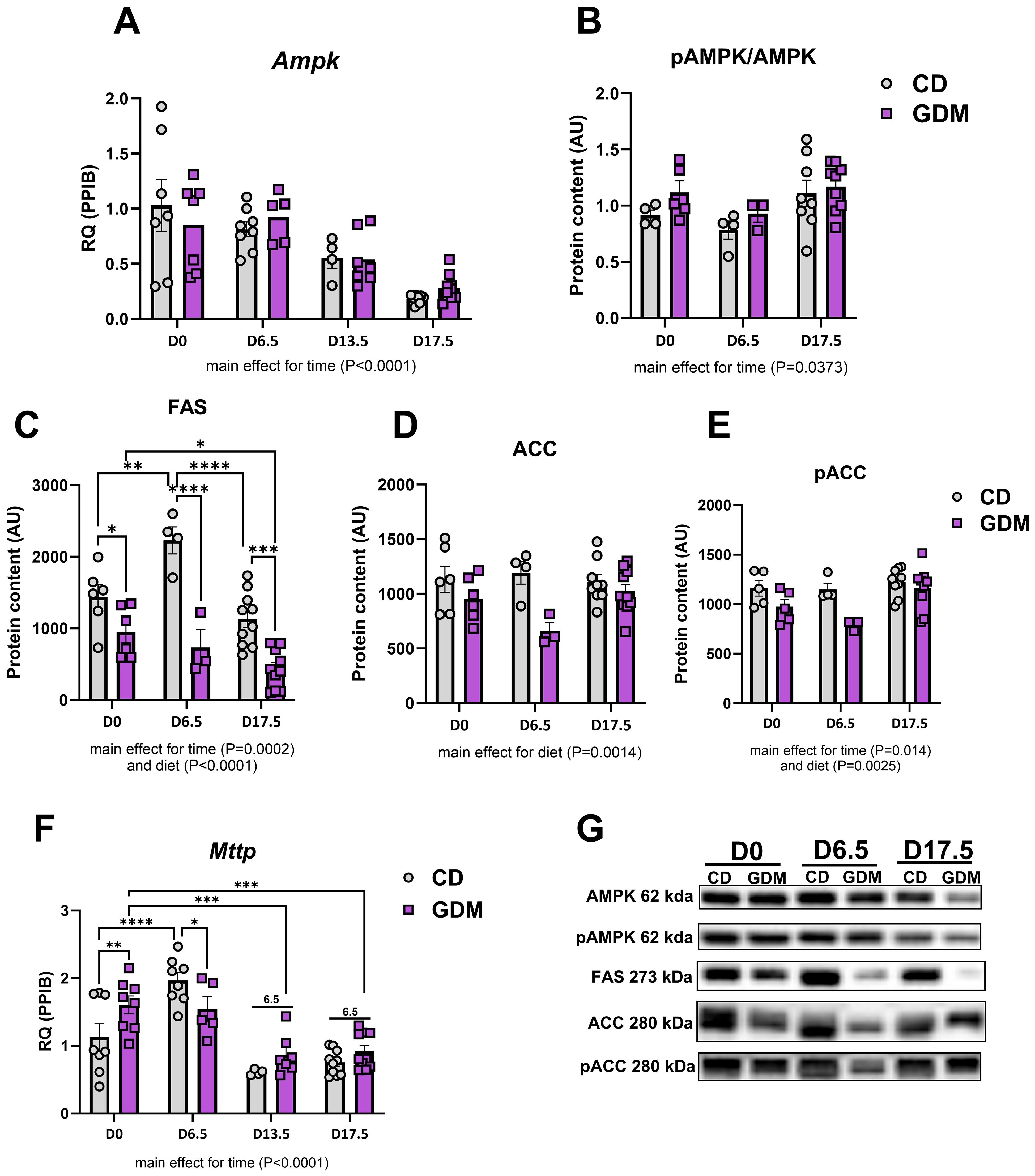
Figure 2. Hepatic lipid handling. (A) Ampk mRNA expression. Protein expression of: (B) pAMPK/AMPK, (C) FAS, (D) ACC, (E) pACC. (F) Mttp mRNA expression. (G) Representative protein bands from western blot for: AMPK, pAMPK, FAS, ACC, pACC. D0 = day 0, D6.5 = day 6.5, D13.5 = day 13.5, D17.5 = day 17.5 of pregnancy (H). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, all denote an interaction. 6.5: an interaction of both respective groups compared to day 6.5 with P<0.05. Main effect for time and/or diet indicated under each graph. Error bars represent SEM. N=8-10. CD, Control Diet; GDM, Gestational Diabetes Mellitus; RQ, Relative Quantification; AU, Arbitrary Units; PPIB, Peptidylprolyl Isomerase B, housekeeping gene.
Alterations in markers of mitochondrial turnover in GDM and gestation
Measures of hepatic mitochondrial function, citrate synthase and β-HAD activities, significantly decreased in dams with GDM at days 6.5 (P=0.001), 13.5 (P=0.019) and 17.5 (P=0.003) (Figure 3A). Markers of mitochondrial biogenesis, Pgc1-α (peroxisome proliferator-activated receptor gamma coactivator 1-alpha), and Tfam (mitochondrial transcription factor A) exhibited parallel declines as pregnancy progresses (main effect of time, P<0.0001, Figure 3B). Markers of autophagy (Atg5: Autophagy-related protein 5, Sqstm1: Sequestosome-1) and mitophagy (Pink1: PTEN Induced Kinase 1, Bnip3: BCL2 Interacting Protein 3) in both groups were significantly attenuated as gestation progressed to day 17.5 (main effect for time, P<0.0001, Figures 3C, D). GDM significantly increased BNIP3 protein and mRNA expression at day 0 compared with CD mice (P=0.004 and P<0.001, respectively, Figure 3D) but had similar expression to CD by day 17.5. Interestingly, there was a main effect for diet with GDM mice exhibiting elevated protein and/or mRNA markers of mitochondrial biogenesis (Tfam), autophagy (Sqstm1), mitophagy (BNIP3, Bnip3, Pink1) (P<0.05).
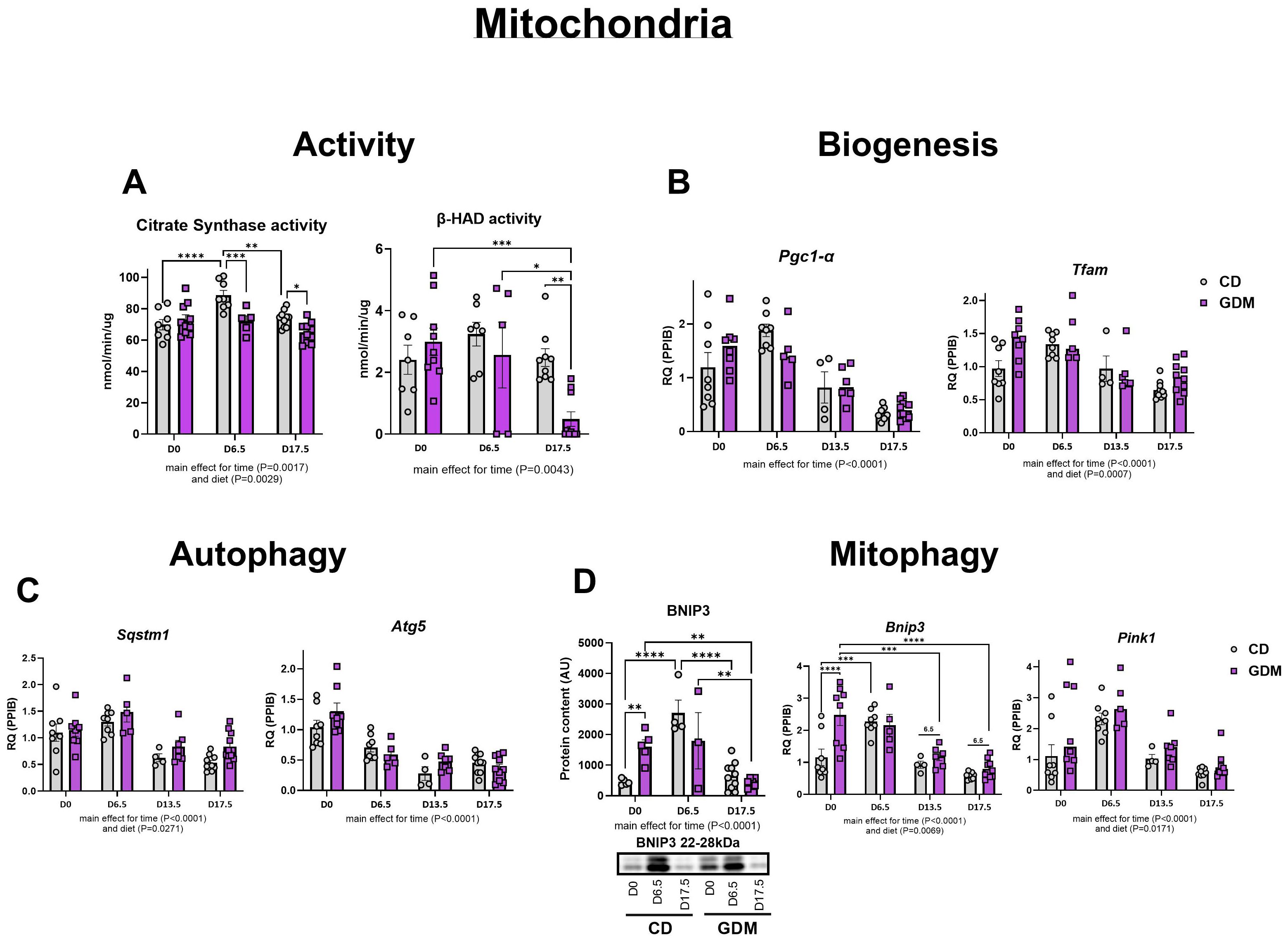
Figure 3. Hepatic mitochondrial activity, biogenesis, autophagy, and mitophagy. (A) Citrate synthase and (nmol/min/ug) and β-had activity (nmol/min/ug). (B) Biogenesis: mRNA expression of Pgc1-α, and Tfam. (C) Autophagy: mRNA expression of Atg5 and Sqstm1. (D) Mitophagy: Protein expression of BNIP3 with its representative protein bands from western blot and mRNA expression of Pink1 and Bnip3. D0 = day 0, D6.5 = day 6.5, D13.5 = day 13.5, D17.5 = day 17.5 of pregnancy. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 all denote an interaction. 6.5: an interaction of both respective groups compared to day 6.5 with P<0.05. Main effect for time and/or diet indicated under each graph. Error bars represent SEM. N=4-10. CD, Control Diet; GDM, Gestational Diabetes Mellitus; RQ, Relative Quantification; AU, Arbitrary Units; PPIB, Peptidylprolyl Isomerase B, housekeeping gene.
GDM elevates markers of inflammation and antioxidant defense
GDM had no effect on the protein expression of hepatic CD68 (cluster of differentiation 68), a marker of macrophage infiltration, but there was a main effect for time (P=0.0077, Figure 4A). Time during gestation also significantly decreased other markers of inflammation [Il-1β (interleukin-1 beta), Tnf-α (tumor necrosis factor-alpha), and Tgf-β (transforming growth factor β)] and hepatic markers of antioxidant defense [Nfe2l2 (nuclear-factor erythroid-derived 2-like 2), Sod2 (superoxide dismutase 2), Gpx1 (glutathione peroxidase 1)] (Figure 4B). GDM markedly upregulated Il-1β, Tnf-α, and Gpx1 at day 0 (P<0.0001, Figure 4A). There was also a main effect for diet with increases in hepatic Tnf-α (P<0.001), Tgf-β (P=0.0009), Nfe2l (P=0.0287), Sod2 (P=0.0207), and Gpx1 (P=0.0003) (Figure 4B).
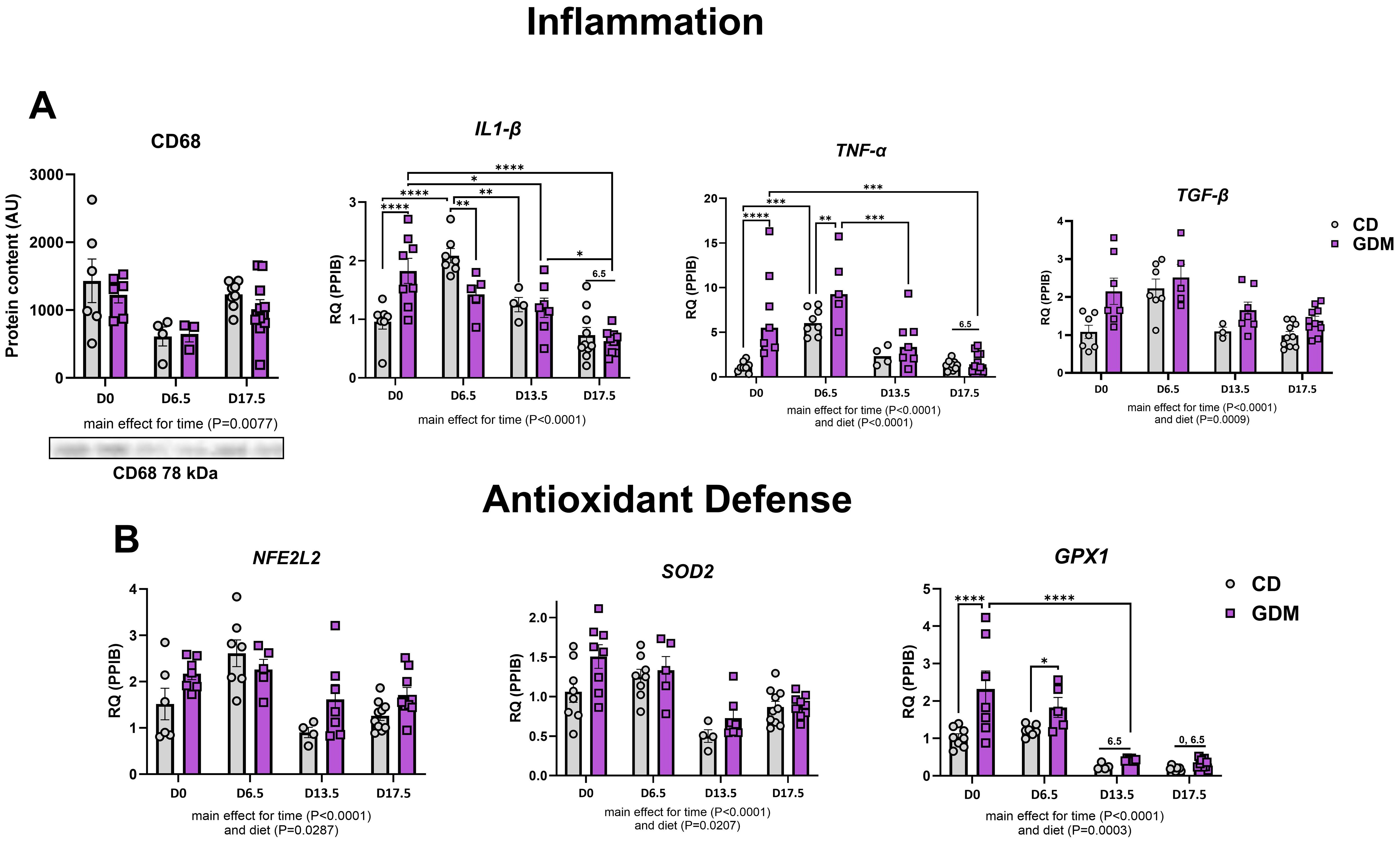
Figure 4. Markers of hepatic inflammation and antioxidant defense. (A) Inflammation: Protein expression of CD68 with its representative protein bands from western blot, and mRNA expression of Il1-β, Tnf-α, and Tgf-β. (B) Antioxidant defense: mRNA expression of Nfe2l2, Sod2 and Gpx1. D0 = day 0, D6.5 = day 6.5, D13.5 = day 13.5, D17.5 = day 17.5 of pregnancy. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 all denote an interaction. 6.5: an interaction of both respective groups compared to day 6.5 with P<0.05. 0: an interaction of both respective groups compared to day 0. Main effect for time and/or diet indicated under each graph. Error bars represent SEM. N=4-10. CD, Control Diet; GDM, Gestational Diabetes Mellitus; RQ, Relative Quantification; AU, Arbitrary Units; PPIB, Peptidylprolyl Isomerase B, housekeeping gene.
Discussion
Gestational diabetes mellitus is associated with an increased risk of MASLD, but there is a paucity of data mechanistically linking these two conditions. Here we provide novel evidence in an established high fat, high sucrose-fed mouse model of GDM (40–42) that changes in maternal liver lipid metabolism may play an important role in GDM pathology. GDM mice developed hepatic steatosis, the accumulation of lipid in the liver, with increases in hepatocellular inflammation and ballooning degeneration, consistent with MASH (35). The GDM mice also exhibited global downregulation in markers of lipid metabolism, and mitochondrial function, biogenesis, mitophagy, and autophagy with increasing gestation time.
Heightened insulin resistance is the distinguishing feature of GDM that is commonly accompanied by inadequate pancreatic beta cell expansion (51). Obesity is a risk factor for the development of GDM (and MASLD), but it has been found that up to one third of women with GDM are considered lean (BMI < 18.5 kg/m2) (52). To better characterize the condition, GDM is now being classified into subtypes dependent on insulin metabolism. Approximately 50% of women with GDM will have systemic insulin resistance, 35% will display insufficient insulin secretion, and 15% will exhibit variations of altered insulin metabolism (53–55). These data highlight the importance of understanding and investigating the varying pathophysiology of GDM. The mouse model used in the current study exhibited systemic insulin resistance and impaired insulin secretion with attenuated beta cell expansion, making it a preferred translational model to investigate GDM (40–42).
During healthy gestation, several metabolic adaptations occur such as elevated glucose utilization, decreases in fatty acid oxidation in the liver, and subsequent systemic insulin resistance (56, 57). As pregnancy progresses into its later stages, adipose tissue undergoes increased lipolysis to ensure sufficient circulating free fatty acids (58). GDM appears to extend these adaptations beyond what is metabolically necessary, aligning with MASLD development. Here, we found in our model of GDM that maternal liver triglycerides were significantly elevated at day 17.5 compared to control diet fed mice, while serum triglycerides were markedly decreased (Figure 1). Similarly, NAS, which includes steatosis, was markedly increased at day 0, 6.5, and 17.5 in GDM compared to CD with elevations occurring as gestation processed. Our data showed that markers of lipid breakdown (Ampk, pAMPK/AMPK) and export (Mttp) decreased as gestation progresses, in both groups. These data suggest that increased hepatic lipid uptake occurs in GDM, concomitant with significant decreases in lipid breakdown and export. This outcome was accompanied with decreased levels of FAS and ACC, markers of hepatic de novo lipogenesis (DNL), which is the formation of fats in the liver from other sources such as sugar (59). Decreased markers of hepatic DNL correspond with other short-term high fat diet feedings studies that show similar reductions (60–64). Overall, these data suggest GDM significantly decreases in lipid breakdown and export, eliciting the formation of lipid accumulation in the liver.
The mitochondrion is the main residence for lipid import and break down. The dysfunction of this organelle is an important feature of MASLD development (27, 28). With our findings of altered lipid storage and export, we hypothesized that our model of GDM would display significant alterations in hepatic mitochondrial function, aligning with MASLD progression. Citrate synthase and β-HAD activities, enzymes that catalyze the TCA cycle and fatty acid β-oxidation within the mitochondria, respectively, were both significantly decreased in GDM mice compared to control mice by day 17.5 (65). Interestingly, early in GDM pregnancy, markers of mitochondrial activity (citrate synthase, β-HAD), biogenesis (Tfam), autophagy (Atg5), and mitophagy (BNIP3, Bnip3, Pink1) were either significantly elevated or trending. Similar outcomes were seen with inflammation (Il1-β, Tnf-α, Tgf-β) and antioxidant defense (Sod2, Gpx1). This information suggests that early pregnancy is a critical window where transcriptional hepatic adaptations to GDM first emerge. In line with these findings, work from our group have previously shown that mitochondrial dysfunction actually precedes MASLD development (28), and findings from others suggest that alterations in plasma lipidomics in early pregnacy predict GDM development (21–23). It is known that pregnancy requires great metabolic flexibility, and substrate and additional energy requirements by the fetus, placenta, and uterus take priority (66–68). Regardless of GDM or control conditions, we found that all measures of mitochondrial turnover, lipid metabolism, inflammation, and antioxidant defense were attenuated by day 17.5 of pregnancy. Early pregnancy displays the largest alterations in hepatic mRNA and protein markers between GDM and CD, but it is not until later in pregnancy that the liver cannot compensate, and it develops overt metabolic characteristics of MASLD exhibited by increased liver triglycerides, elevated NAS (steatosis, inflammation, and ballooning), and attenuated mitochondrial activity.
One of the strengths of this study is the use of an established diet-fed mouse model to develop GDM. This model closely mimics key metabolic features of human GDM, such as insulin resistance and impaired insulin secretion. Furthermore, this is the first study to evaluate the impact of GDM on hepatic markers of mitochondrial activity, biogenesis, mitophagy, autophagy, inflammation, antioxidant defense, and lipid metabolism throughout gestation. Despite these strengths, there are limitations. While the HFHS-fed mouse model does recapitulate GDM pathophysiology, it is important to note that pregnancy in a human is much lengthier and likely more complex, limiting the translation of these findings from rodent GDM to human GDM. Secondly, the present study does not include any non-pregnant mice as a control group. Although previous literature suggests that control-diet fed non-pregnant mice would not display any hepatic metabolic alterations over time (69–72), direct comparisons with this experimental setting would help strengthen the conclusions.
In conclusion, our data demonstrates that HFHS feeding in a GDM model leads to increased maternal hepatic triglyceride accumulation, elevated NAS, and reduced mitochondrial activity, accompanied by increases in hepatic inflammation and antioxidant defense. Pregnancy itself acts as a powerful metabolic switch, reprogramming nutrient utilization and energy balance to support fetal development. This metabolic shift appears strong enough to override liver adaptations to high-fat diet-induced GDM, ensuring continued development of the fetus and support organs but leading to hepatic lipid increases. To our knowledge, this is the first study to evaluate and characterize alterations in hepatic mitochondrial and lipid metabolism during both normal gestation and in a GDM model. These findings highlight the significant impact of gestation and GDM on liver health and MASLD risk.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Ethics statement
The animal study was approved by Baylor College of Medicine Institutional Animal Care and Use Committee. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
GS: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. AK: Methodology, Visualization, Writing – review & editing. SR: Methodology, Writing – review & editing. LS: Conceptualization, Methodology, Writing – review & editing. KP: Conceptualization, Data curation, Investigation, Methodology, Project administration, Resources, Visualization, Writing – review & editing. RR: Conceptualization, Funding acquisition, Methodology, Project administration, Software, Supervision, Writing – review & editing, Data curation, Formal analysis, Investigation, Resources, Validation, Visualization, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The work was supported in part by VA-Merit Grant I01BX003271 (Salary support for RR) and NIH grant R03HD105831 (LS). This work was supported by resources and the use of facilities at the University of Missouri and Harry S. Truman Memorial Veterans Hospital in Columbia, MO.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2025.1498764/full#supplementary-material
Glossary
GDM: Gestational Diabetes Mellitus
MASLD: Metabolic Dysfunction-Associated Steatotic Liver Disease
MASH: Metabolic-Dysfunction Associated Steatohepatitis
CD68: Cluster of Differentiation 68
ACC: Acetyl-CoA Carboxylase
pACC: phosphorylated Acetyl-CoA Carboxylase
FAS: Fatty acid synthase
TGF-β: Transforming growth factor beta
NFE2L2/NRF2: Nuclear factor, erythroid 2-like 2
β-HAD: β-hydroxyacyl-CoA dehydrogenase
AMPK: AMP-activated protein kinase
PPIB: Peptidylprolyl Isomerase B
PGC1-α: Peroxisome proliferator-activated receptor gamma coactivator 1-alpha
TFAM: Mitochondrial transcription factor A
PINK1: PTEN Induced Kinase 1
SQSTM1: Sequestosome 1
BNIP3: BCL2 interacting protein 3
ATG5: Autophagy protein 5
SOD2: Superoxide dismutase 2
GPX1: Glutathione peroxidase 1
TNF-α: Tumor necrosis factor alpha
IL1-β: Interleukin-1 beta
MTTP: Microsomal triglyceride transfer protein
References
1. American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. (2010) 33 Suppl 1:S62–69. doi: 10.2337/dc10-S062
2. Sweeting A, Hannah W, Backman H, Catalano P, Feghali M, Herman WH, et al. Epidemiology and management of gestational diabetes. Lancet. (2024) 404:175–92. doi: 10.1016/S0140-6736(24)00825-0
3. Catalano PM, Tyzbir ED, Wolfe RR, Calles J, Roman NM, Amini SB, et al. Carbohydrate metabolism during pregnancy in control subjects and women with gestational diabetes. Am J Physiol. (1993) 264:E60–67. doi: 10.1152/ajpendo.1993.264.1.E60
4. Yang SJ, Kim TN, Baik SH, Kim TS, Lee KW, Nam M, et al. Insulin secretion and insulin resistance in Korean women with gestational diabetes mellitus and impaired glucose tolerance. Korean J Intern Med. (2013) 28:306–13. doi: 10.3904/kjim.2013.28.3.306
5. Homko C, Sivan E, Chen X, Reece EA, and Boden G. Insulin secretion during and after pregnancy in patients with gestational diabetes mellitus. J Clin Endocrinol Metab. (2001) 86:568–73. doi: 10.1210/jc.86.2.568
6. McIntyre HD, Catalano P, Zhang C, Desoye G, Mathiesen ER, and Damm P. Gestational diabetes mellitus. Nat Rev Dis Primers. (2019) 5:47. doi: 10.1038/s41572-019-0098-8
7. American Diabetes Association. 2. Classification and diagnosis of diabetes: standards of medical care in diabetes-2019. Diabetes Care. (2019) 42:S13–s28. doi: 10.2337/dc19-S002
8. Baz B, Riveline JP, and Gautier JF. ENDOCRINOLOGY OF PREGNANCY: Gestational diabetes mellitus: definition, aetiological and clinical aspects. Eur J Endocrinol. (2016) 174:R43–51. doi: 10.1530/EJE-15-0378
9. Salazar-Petres ER and Sferruzzi-Perri AN. Pregnancy-induced changes in β-cell function: what are the key players? J Physiol. (2022) 600:1089–117. doi: 10.1113/JP281082
10. Lee K, Kuang A, Bain JR, Hayes MG, Muehlbauer MJ, Ilkayeva OR, et al. and Lowe WL, Jr. Metabolomic and genetic architecture of gestational diabetes subtypes. Diabetologia. (2024) 67:895–907. doi: 10.1007/s00125-024-06110-x
11. Mittal R, Prasad K, Lemos JRN, Arevalo G, and Hirani K. Unveiling gestational diabetes: an overview of pathophysiology and management. Int J Mol Sci. (2025) 26. doi: 10.3390/ijms26052320
12. Gregory EC and Ely DM. Trends and characteristics in gestational diabetes: United States, 2016-2020. Natl Vital Stat Rep. (2022) 71:1–15.
13. Ajmera VH, Gunderson EP, VanWagner LB, Lewis CE, Carr JJ, and Terrault NA. Gestational diabetes mellitus is strongly associated with non-alcoholic fatty liver disease. Am J Gastroenterol. (2016) 111:658–64. doi: 10.1038/ajg.2016.57
14. Lavrentaki A, Thomas T, Subramanian A, Valsamakis G, Thomas N, Toulis KA, et al. Increased risk of non-alcoholic fatty liver disease in women with gestational diabetes mellitus: A population-based cohort study, systematic review and meta-analysis. J Diabetes Complications. (2019) 33:107401. doi: 10.1016/j.jdiacomp.2019.06.006
15. Cho Y, Chang Y, Ryu S, Kim C, Wild SH, and Byrne CD. History of gestational diabetes and incident nonalcoholic fatty liver disease: the Kangbuk Samsung health study. Am J Gastroenterol. (2023) 118:1980–8. doi: 10.14309/ajg.0000000000002250
16. Song S, Duo Y, Zhang Y, Qiao X, Xu J, Zhang J, et al. The predictive ability of hepatic steatosis index for gestational diabetes mellitus and large for gestational age infant compared with other noninvasive indices among Chinese pregnancies: A preliminary double-center cohort study. Diabetes Metab Syndr Obes. (2021) 14:4791–800. doi: 10.2147/DMSO.S335364
17. Retnakaran R, Luo J, and Shah BR. Gestational diabetes in young women predicts future risk of serious liver disease. Diabetologia. (2019) 62:306–10. doi: 10.1007/s00125-018-4775-z
18. Zhan Y, Wang J, He X, Huang M, Yang X, He L, et al. Plasma metabolites, especially lipid metabolites, are altered in pregnant women with gestational diabetes mellitus. Clin Chim Acta. (2021) 517:139–48. doi: 10.1016/j.cca.2021.02.023
19. Zhao H, Li H, Chung ACK, Xiang L, Li X, Zheng Y, et al. Large-scale longitudinal metabolomics study reveals different trimester-specific alterations of metabolites in relation to gestational diabetes mellitus. J Proteome Res. (2019) 18:292–300. doi: 10.1021/acs.jproteome.8b00602
20. Wang Y, Pan XF, and Pan A. Lipidomics in gestational diabetes mellitus. Curr Opin Lipidol. (2023) 34:1–11. doi: 10.1097/MOL.0000000000000858
21. Chen X, Chen H, Zhang Y, Jiang Y, Wang Y, Huang X, et al. Maternal liver dysfunction in early pregnancy predisposes to gestational diabetes mellitus independent of preconception overweight: A prospective cohort study. Bjog. (2022) 129:1695–703. doi: 10.1111/1471-0528.17117
22. Wang Y, Huang Y, Wu P, Ye Y, Sun F, Yang X, et al. Plasma lipidomics in early pregnancy and risk of gestational diabetes mellitus: a prospective nested case-control study in Chinese women. Am J Clin Nutr. (2021) 114:1763–73. doi: 10.1093/ajcn/nqab242
23. Zhang Z, Zhou Z, and Li H. The role of lipid dysregulation in gestational diabetes mellitus: Early prediction and postpartum prognosis. J Diabetes Investig. (2024) 15:15–25. doi: 10.1111/jdi.14119
24. Cotter TG and Rinella M. Nonalcoholic fatty liver disease 2020: the state of the disease. Gastroenterology. (2020) 158:1851–64. doi: 10.1053/j.gastro.2020.01.052
25. Perumpail BJ, Khan MA, Yoo ER, Cholankeril G, Kim D, and Ahmed A. Clinical epidemiology and disease burden of nonalcoholic fatty liver disease. World J Gastroenterol. (2017) 23:8263–76. doi: 10.3748/wjg.v23.i47.8263
26. Pouwels S, Sakran N, Graham Y, Leal A, Pintar T, Yang W, et al. Non-alcoholic fatty liver disease (NAFLD): a review of pathophysiology, clinical management and effects of weight loss. BMC Endocr Disord. (2022) 22:63. doi: 10.1186/s12902-022-00980-1
27. Wei Y, Rector RS, Thyfault JP, and Ibdah JA. Nonalcoholic fatty liver disease and mitochondrial dysfunction. World J Gastroenterol. (2008) 14:193–9. doi: 10.3748/wjg.14.193
28. Rector RS, Thyfault JP, Uptergrove GM, Morris EM, Naples SP, Borengasser SJ, et al. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J Hepatol. (2010) 52:727–36. doi: 10.1016/j.jhep.2009.11.030
29. Prasun P, Ginevic I, and Oishi K. Mitochondrial dysfunction in nonalcoholic fatty liver disease and alcohol related liver disease. Transl Gastroenterol Hepatol. (2021) 6:4. doi: 10.21037/tgh-20-125
30. Ramanathan R, Ali AH, and Ibdah JA. Mitochondrial dysfunction plays central role in nonalcoholic fatty liver disease. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms23137280
31. Javadov S, Kozlov AV, and Camara AKS. Mitochondria in health and diseases. Cells. (2020) 9. doi: 10.3390/cells9051177
32. Morio B, Panthu B, Bassot A, and Rieusset J. Role of mitochondria in liver metabolic health and diseases. Cell Calcium. (2021) 94:102336. doi: 10.1016/j.ceca.2020.102336
33. Ajaz S, McPhail MJ, Gnudi L, Trovato FM, Mujib S, Napoli S, et al. Mitochondrial dysfunction as a mechanistic biomarker in patients with non-alcoholic fatty liver disease (NAFLD). Mitochondrion. (2021) 57:119–30. doi: 10.1016/j.mito.2020.12.010
34. Fromenty B and Roden M. Mitochondrial alterations in fatty liver diseases. J Hepatol. (2023) 78:415–29. doi: 10.1016/j.jhep.2022.09.020
35. Pierantonelli I and Svegliati-Baroni G. Nonalcoholic fatty liver disease: basic pathogenetic mechanisms in the progression from NAFLD to NASH. Transplantation. (2019) 103:e1–e13. doi: 10.1097/TP.0000000000002480
36. Longo M, Meroni M, Paolini E, Macchi C, and Dongiovanni P. Mitochondrial dynamics and nonalcoholic fatty liver disease (NAFLD): new perspectives for a fairy-tale ending? Metabolism. (2021) 117:154708. doi: 10.1016/j.metabol.2021.154708
37. Legaki AI, Moustakas II, Sikorska M, Papadopoulos G, Velliou RI, and Chatzigeorgiou A. Hepatocyte mitochondrial dynamics and bioenergetics in obesity-related non-alcoholic fatty liver disease. Curr Obes Rep. (2022) 11:126–43. doi: 10.1007/s13679-022-00473-1
38. Stevanović-Silva J, Beleza J, Coxito P, Pereira S, Rocha H, Gaspar TB, et al. Maternal high-fat high-sucrose diet and gestational exercise modulate hepatic fat accumulation and liver mitochondrial respiratory capacity in mothers and male offspring. Metabolism. (2021) 116:154704. doi: 10.1016/j.metabol.2021.154704
39. Pereira TJ, Fonseca MA, Campbell KE, Moyce BL, Cole LK, Hatch GM, et al. Maternal obesity characterized by gestational diabetes increases the susceptibility of rat offspring to hepatic steatosis via a disrupted liver metabolome. J Physiol. (2015) 593:3181–97. doi: 10.1113/jphysiol.2015.593.issue-14
40. Pennington KA, Dong Y, Ruano SH, van der Walt N, Sangi-Haghpeykar H, and Yallampalli C. Brief high fat high sugar diet results in altered energy and fat metabolism during pregnancy in mice. Sci Rep. (2020) 10:20866. doi: 10.1038/s41598-020-77529-6
41. Pennington KA, van der Walt N, Pollock KE, Talton OO, and Schulz LC. Effects of acute exposure to a high-fat, high-sucrose diet on gestational glucose tolerance and subsequent maternal health in mice. Biol Reprod. (2017) 96:435–45. doi: 10.1095/biolreprod.116.144543
42. Mishra A, Ruano SH, Saha PK, and Pennington KA. A novel model of gestational diabetes: Acute high fat high sugar diet results in insulin resistance and beta cell dysfunction during pregnancy in mice. PloS One. (2022) 17:e0279041. doi: 10.1371/journal.pone.0279041
43. Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, and Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol. (1999) 94:2467–74. doi: 10.1111/j.1572-0241.1999.01377.x
44. Folch J, Lees M, and Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. (1957) 226:497–509. doi: 10.1016/S0021-9258(18)64849-5
45. Srere PA. Citrate synthase: [EC 4.1.3.7. Citrate oxaloacetate-lyase (CoA-acetylating). Methods Enzymol. (1969) 13:3–11. doi: 10.1016/0076-6879(69)13005-0
46. Bass A, Brdiczka D, Eyer P, Hofer S, and Pette D. Metabolic differentiation of distinct muscle types at the level of enzymatic organization. Eur J Biochem. (1969) 10:198–206. doi: 10.1111/j.1432-1033.1969.tb00674.x
47. Rector RS, Thyfault JP, Morris RT, Laye MJ, Borengasser SJ, Booth FW, et al. Daily exercise increases hepatic fatty acid oxidation and prevents steatosis in Otsuka Long-Evans Tokushima Fatty rats. Am J Physiol Gastrointest Liver Physiol. (2008) 294:G619–626. doi: 10.1152/ajpgi.00428.2007
48. Linden MA, Fletcher JA, Meers GM, Thyfault JP, and Laughlin MH. and Rector RS. A return to ad libitum feeding following caloric restriction promotes hepatic steatosis in hyperphagic OLETF rats. Am J Physiol Gastrointest Liver Physiol. (2016) 311:G387–395. doi: 10.1152/ajpgi.00089.2016
49. Moore MP, Cunningham RP, Meers GM, Johnson SA, Wheeler AA, Ganga RR, et al. Compromised hepatic mitochondrial fatty acid oxidation and reduced markers of mitochondrial turnover in human NAFLD. Hepatology. (2022) 76:1452–65. doi: 10.1002/hep.32324
50. Moore MP, Shryack G, Alessi I, Wieschhaus N, Meers GM, Johnson SA, et al. Relationship between serum β-hydroxybutyrate and hepatic fatty acid oxidation in individuals with obesity and NAFLD. Am J Physiol Endocrinol Metab. (2024) 326:E493–e502. doi: 10.1152/ajpendo.00336.2023
51. Moyce BL and Dolinsky VW. Maternal β-cell adaptations in pregnancy and placental signalling: implications for gestational diabetes. Int J Mol Sci. (2018) 19. doi: 10.3390/ijms19113467
52. Wadivkar P and Hawkins M. Is gestational diabetes mellitus in lean women a distinct entity warranting a modified management approach? Front Clin Diabetes Healthc. (2024) 5:1338597. doi: 10.3389/fcdhc.2024.1338597
53. Kotzaeridi G, Blätter J, Eppel D, Rosicky I, Linder T, Geissler F, et al. Characteristics of gestational diabetes subtypes classified by oral glucose tolerance test values. Eur J Clin Invest. (2021) 51:e13628. doi: 10.1111/eci.13628
54. Ortega-Montiel J, Martinez-Juarez LA, Montoya A, Morales-Juárez L, Gallardo-Rincón H, Galicia-Hernández V, et al. Gestational diabetes mellitus subtypes classified by oral glucose tolerance test and maternal and perinatal outcomes: results of a Mexican multicenter prospective cohort study “Cuido Mi Embarazo. Diabetes Metab Syndr Obes. (2024) 17:1491–502. doi: 10.2147/DMSO.S450939
55. Powe CE, Allard C, Battista MC, Doyon M, Bouchard L, Ecker JL, et al. Heterogeneous contribution of insulin sensitivity and secretion defects to gestational diabetes mellitus. Diabetes Care. (2016) 39:1052–5. doi: 10.2337/dc15-2672
56. Fang H, Li Q, Wang H, Ren Y, Zhang L, and Yang L. Maternal nutrient metabolism in the liver during pregnancy. Front Endocrinol (Lausanne). (2024) 15:1295677. doi: 10.3389/fendo.2024.1295677
57. Wild R and Feingold KR. Effect of pregnancy on lipid metabolism and lipoprotein levels. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, et al, editors. Endotext. MDText.com, Inc. Copyright © 2000-2024, MDText.com, Inc., South Dartmouth (MA (2000).
58. Chavan-Gautam P, Rani A, and Freeman DJ. Distribution of fatty acids and lipids during pregnancy. Adv Clin Chem. (2018) 84:209–39. doi: 10.1016/bs.acc.2017.12.006
59. Kawano Y and Cohen DE. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J Gastroenterol. (2013) 48:434–41. doi: 10.1007/s00535-013-0758-5
60. Costabile G, Della Pepa G, Salamone D, Luongo D, Naviglio D, Brancato V, et al. Reduction of de novo lipogenesis mediates beneficial effects of isoenergetic diets on fatty liver: mechanistic insights from the MEDEA randomized clinical trial. Nutrients. (2022) 14. doi: 10.3390/nu14102178
61. Schwarz JM, Linfoot P, Dare D, and Aghajanian K. Hepatic de novo lipogenesis in normoinsulinemic and hyperinsulinemic subjects consuming high-fat, low-carbohydrate and low-fat, high-carbohydrate isoenergetic diets. Am J Clin Nutr. (2003) 77:43–50. doi: 10.1093/ajcn/77.1.43
62. Duarte JA, Carvalho F, Pearson M, Horton JD, Browning JD, and Jones JG. and Burgess SC. A high-fat diet suppresses de novo lipogenesis and desaturation but not elongation and triglyceride synthesis in mice. J Lipid Res. (2014) 55:2541–53. doi: 10.1194/jlr.M052308
63. Reis-Costa A, Belew GD, Viegas I, Tavares LC, Meneses MJ, Patrício B, et al. The effects of long-term high fat and/or high sugar feeding on sources of postprandial hepatic glycogen and triglyceride synthesis in mice. Nutrients. (2024) 16. doi: 10.3390/nu16142186
64. Goedeke L, Strober JW, Suh R, Paolella LM, Li X, Rogers JC, et al. High-fat-diet-induced hepatic insulin resistance per se attenuates murine de novo lipogenesis. iScience. (2024) 27:111175. doi: 10.1016/j.isci.2024.111175
65. Nassir F and Ibdah JA. Role of mitochondria in nonalcoholic fatty liver disease. Int J Mol Sci. (2014) 15:8713–42. doi: 10.3390/ijms15058713
66. Lain KY and Catalano PM. Metabolic changes in pregnancy. Clin Obstet Gynecol. (2007) 50:938–48. doi: 10.1097/GRF.0b013e31815a5494
67. Prentice AM and Goldberg GR. Energy adaptations in human pregnancy: limits and long-term consequences. Am J Clin Nutr. (2000) 71:1226s–32s. doi: 10.1093/ajcn/71.5.1226s
68. Armistead B, Johnson E, VanderKamp R, Kula-Eversole E, Kadam L, Drewlo S, et al. Placental regulation of energy homeostasis during human pregnancy. Endocrinology. (2020) 161. doi: 10.1210/endocr/bqaa076
69. Poussin C, Ibberson M, Hall D, Ding J, Soto J, Abel ED, et al. Oxidative phosphorylation flexibility in the liver of mice resistant to high-fat diet-induced hepatic steatosis. Diabetes. (2011) 60:2216–24. doi: 10.2337/db11-0338
70. Ciapaite J, van den Broek NM, Te Brinke H, Nicolay K, Jeneson JA, Houten SM, et al. Differential effects of short- and long-term high-fat diet feeding on hepatic fatty acid metabolism in rats. Biochim Biophys Acta. (2011) 1811:441–51. doi: 10.1016/j.bbalip.2011.05.005
71. Velázquez KT, Enos RT, Bader JE, Sougiannis AT, Carson MS, Chatzistamou I, et al. Prolonged high-fat-diet feeding promotes non-alcoholic fatty liver disease and alters gut microbiota in mice. World J Hepatol. (2019) 11:619–37. doi: 10.4254/wjh.v11.i8.619
Keywords: GDM, MASLD, NAFLD, mitochondria, liver
Citation: Shryack GE, Krause AA, Hernandez Ruano S, Schulz LC, Pennington KA and Rector RS (2025) A murine model of gestational diabetes reveals MASLD risk and alterations in markers of hepatic mitochondrial metabolism. Front. Endocrinol. 16:1498764. doi: 10.3389/fendo.2025.1498764
Received: 19 September 2024; Accepted: 29 May 2025;
Published: 25 June 2025.
Edited by:
Adriana Laura Burgueño, National Scientific and Technical Research Council (CONICET), ArgentinaReviewed by:
Maria Luisa Lazo De La Vega Monroy, University of Guanajuato, MexicoLuz Andreone, Universidad Austral, Argentina
Copyright © 2025 Shryack, Krause, Hernandez Ruano, Schulz, Pennington and Rector. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kathleen A. Pennington, a2F0aGxlZW4ucGVubmluZ3RvbkBiY20uZWR1; R. Scott Rector, cmVjdG9yc0BoZWFsdGgubWlzc291cmkuZWR1