 Bin Liu1*
Bin Liu1* Qing Jin2Yi Kang Sun2Zhi Ming Yang1Ping Meng3Xi Zhang2Qiu Chen2,3Pin Gan2Tao Zhao2Jia Ji He2
Qing Jin2Yi Kang Sun2Zhi Ming Yang1Ping Meng3Xi Zhang2Qiu Chen2,3Pin Gan2Tao Zhao2Jia Ji He2 Gui Ping He2
Gui Ping He2 Qiang Xue2*
Qiang Xue2*- 1Department of Cardiology, The Fifth Affiliated Hospital of Kunming Medical University, Gejiu People’s Hospital, Gejiu, Yunnan, China
- 2Department of Cardiology, Yan’an Hospital Affiliated to Kunming Medical University, Key Laboratory of Cardiovascular Disease of Yunnan Province, Kun Min, Yunnan, China
- 3Yan’an Hospital Affiliated to Kunming Medical University, Key Laboratory of Cardiovascular Disease of Yunnan Province, Kun Min, Yunnan, China
Diabetic cardiomyopathy (DCM) is a common and fatal cardiac complication caused by diabetes, with its pathogenesis involving various forms of cell death and mitochondrial dysfunction, particularly ferroptosis and mitochondrial injury. Recent studies have indicated that ferroptosis and mitochondrial damage play crucial roles in the onset and progression of DCM, though their precise regulatory mechanisms remain unclear. Of particular interest is the interaction between ferroptosis and mitochondrial damage, as well as their synergistic effects, which are not fully understood. This review summarizes the roles of ferroptosis and mitochondrial injury in the progression of DCM and explores the molecular mechanisms involved, with an emphasis on the interplay between these two processes. Additionally, the article offers an overview of targeted drugs shown to be effective in cellular experiments, animal models, and clinical trials, analyzing their mechanisms of action and potential side effects. The goal is to provide insights for future drug development and clinical applications. Moreover, the review explores the challenges and prospects of multi-target combination therapies and personalized medicine interventions in clinical practice to offer strategic guidance for the comprehensive prevention and management of DCM.
1 Introduction
Diabetic cardiomyopathy (DCM) is a unique form of myocardial injury that occurs in patients with diabetes. It is not only closely linked to the systemic complications of diabetes but also involves specific pathological changes in myocardial metabolism and oxidative stress (1). Key factors contributing to DCM pathogenesis include disruptions in iron metabolism and mitochondrial dysfunction. The characteristic pathological features of DCM include lipid accumulation in myocardial cells, myocardial hypertrophy, myocardial fibrosis, mitochondrial damage, and myocardial ischemia (2). These alterations ultimately contribute to heart failure, which is a leading cause of mortality among patients with diabetes (Figure 1).
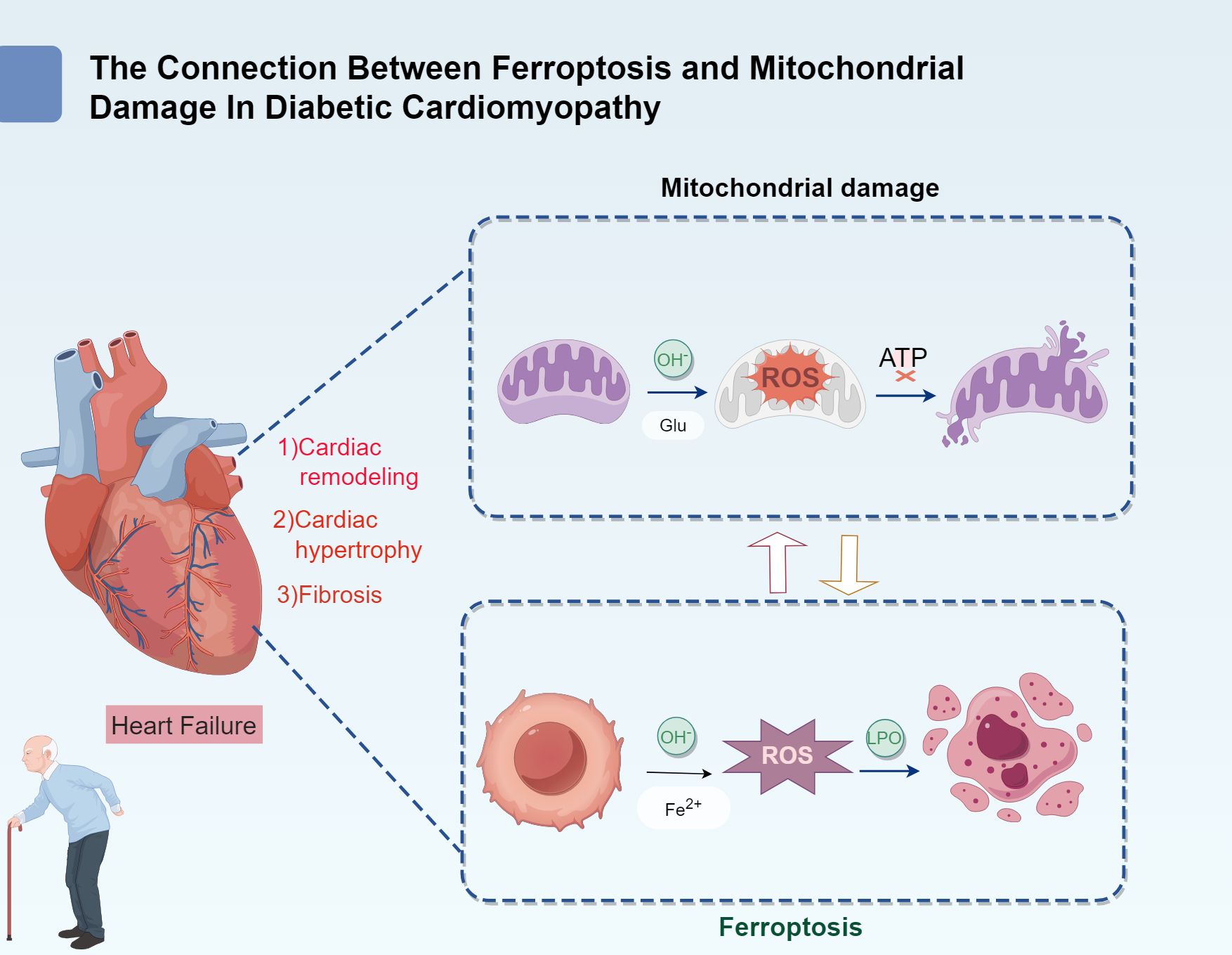
Figure 1. The connection between ferroptosis and mitochondrial damage in diabetic cardiomyopathy.
The increasing global prevalence of diabetes presents significant challenges to the prevention and treatment of DCM. According to the latest report by the International Diabetes Federation (IDF) (3, 4), the global prevalence of diabetes among adults aged 20–79 years had reached 14% by 2022, doubling the rate 30 years ago. The number of individuals with diabetes now exceeds 800 million, and this figure is projected to grow further in the coming decades. Early-stage cardiac damage in diabetic patients often goes unnoticed due to a lack of clear symptoms. Diagnosis typically occurs only when symptoms such as fatigue, shortness of breath, or edema appear after excluding other heart diseases (5). Due to its chronic and progressive nature, and the absence of specific early symptoms, early detection and intervention for DCM remain challenging. Research indicates that 20%–30% of diabetic patients may eventually develop DCM, and the risk of heart failure among these patients exceeds 70% (6, 7).
The pathogenesis of DCM is exceptionally complex, involving dysregulation of glucose and lipid metabolism, insulin resistance, oxidative stress, inflammatory responses, mitochondrial dysfunction, and ferroptosis (1, 8, 9). Among these, ferroptosis and mitochondrial damage have emerged as new areas of focus in recent years and are considered potential new therapeutic targets for DCM treatment. Ferroptosis is a form of programmed cell death induced by iron-dependent lipid peroxidation (10). Its core mechanisms include iron overload, lipid peroxidation, and disruption of the antioxidant defense system (11). Meanwhile, mitochondria, the cellular energy powerhouses, are directly involved in myocardial energy supply, and their dysfunction exacerbates myocardial impairment (12). The pathophysiological mechanisms of DCM are highly complex, involving factors such as metabolic disturbances in glucose and lipid homeostasis, insulin resistance, oxidative stress, inflammatory responses, mitochondrial dysfunction, and ferroptosis (1, 8, 9). Among these factors, ferroptosis and mitochondrial injury have emerged as key areas of focus and are considered potential therapeutic targets for DCM treatment (13).
Ferroptosis is a form of programmed cell death induced by iron-mediated lipid peroxidation (14). Its primary mechanisms involve iron overload, lipid peroxidation, imbalance in the antioxidant system, and the resulting cellular dysfunction (15). At the same time, mitochondria, which serve as the energy powerhouse of cells, are essential for maintaining cellular homeostasis. Mitochondrial dysfunction directly impairs myocardial energy supply, thereby exacerbating myocardial dysfunction (16). Ferroptosis has been observed in various organs, including the heart, liver, kidneys, brain, and retina of patients with diabetes (17–19). However, cardiac tissue is particularly vulnerable due to its high energy demands, unique metabolic characteristics (such as high mitochondrial density and continuous oxidative stress), and its critical role in physiological functions (20). This heightened vulnerability becomes more evident when there is damage to the diabetic heart, ventricular remodeling, and heart failure, where therapeutic strategies targeting ferroptosis could offer promising benefits. However, current studies on ferroptosis in diabetic cardiomyopathy are primarily restricted to limited clinical observations and animal experiments (21), underscoring the need for more comprehensive and systematic research.
This review will focus on exploring the role of ferroptosis and mitochondrial injury in cardiac cells exposed to chronic diabetic conditions, their impact on patients with diabetes, and the associated pathophysiological mechanisms. The main focus is to provide new strategies and recommendations for interventions targeting these mechanisms, as well as for personalized treatment approaches, combination therapies, and the development of innovative drug therapies.
2 Ferroptosis and its link to DCM
Ferroptosis is a form of programmed cell death driven by the excessive accumulation of iron ions within cells, triggering the Fenton reaction, activating oxidative stress, and disrupting lipid metabolism, ultimately leading to cellular demise (22, 23). The primary mechanisms underlying ferroptosis involve imbalances in iron metabolism and dysfunction of the antioxidant defense system. Glutathione (GSH), present in the cytoplasm of normal cells, is one of the most important low-molecular-weight antioxidants (24). It directly participates in scavenging reactive oxygen species (ROS) and reducing oxidative products, thus preventing oxidative stress-induced cellular damage (25). During ferroptosis, GPX4 binds to GSH to reduce lipid peroxides (such as PE-PUFA-OOH) to their corresponding alcohols, preventing lipid peroxide accumulation and protecting cellular membranes and structures from oxidative damage (20, 26).
Acyl-CoA synthetase long-chain family member 4 (ACSL4) is a key enzyme involved in the synthesis of polyunsaturated fatty acid phosphatidylethanolamine (PUFA-PE) (27). It catalyzes the acylation of polyunsaturated fatty acids (PUFAs) to form polyunsaturated fatty acid-CoA (PUFA-CoA), which is subsequently esterified by lysophosphatidylcholine acyltransferase 3 (LPCAT3) to form PUFA-CoA (28). The esterified PUFA-CoA then reacts with phosphatidylethanolamine (PE) to form PUFA-PEs. These PUFA-PEs play a fundamental role in maintaining cell membrane integrity; however, their oxidation leads to excessive lipid peroxide accumulation, ultimately triggering ferroptosis (29). In the context of DCM, chronic hyperglycemia disrupts fatty acid metabolism, leading to ACSL4 overexpression, increased PUFA-CoA production, and accelerated lipid peroxidation. This cascade not only elevates lipid peroxide levels but also intensifies iron accumulation in myocardial cells, exacerbating ferroptosis and contributing to myocardial cell death and fibrosis (30).
In patients with DCM, the GSH-dependent antioxidant mechanism is suppressed, whereas ACSL4 activation promotes ferroptosis in myocardial cells (31). The combined effects of oxidative stress and fatty acid metabolism disorder further exacerbate myocardial cell damage and promote ferroptosis (1, 8, 9). Unlike apoptosis, necrosis, or autophagy, ferroptosis exhibits distinct pathological features, including cell membrane rupture, mitochondrial dysfunction, and significant oxidative stress (32). Ferroptosis is particularly prominent in metabolic diseases, with its manifestation being more pronounced in DCM.
In the development of DCM, sustained hyperglycemia acts as a key driver of ferroptosis. Hyperglycemia disrupts myocardial fatty acid metabolism and reduces the capacity of myocardial cells to tolerate iron overload (33). Excessive accumulation of iron ions intensifies the production of reactive oxygen species (ROS) through the Fenton reaction, which, in turn, exacerbates lipid peroxidation. This process leads to a decrease in the activity of antioxidant enzymes such as glutathione peroxidase 4 (GPX4), superoxide dismutase 1 (SOD1), and NAD(P)H quinone oxidoreductase 1 (NQO1) (34). This decline in antioxidant enzyme activity further exacerbates ferroptosis (23, 33, 35), leading to uncontrolled lipid peroxidation, accelerating myocardial cell death, and triggering myocardial fibrosis and heart dysfunction (36). Among these, the inactivation of GPX4—a crucial inhibitor of ferroptosis—plays a significant role in the pathogenesis of myocardial injury in DCM.
Clinical and pathological studies have shown that patients with DCM exhibit significantly increased iron content in myocardial tissue, along with expansion of the cardiac chambers (37). In these patients, oxidative stress markers such as ACSL4, malondialdehyde (MDA), and NOX4 are generally elevated, whereas the expression of antioxidant enzymes like GPX4 and GSH is significantly reduced (36, 38). Furthermore, mitochondrial damage in myocardial cells of patients with DCM manifests as mitochondrial swelling, cristae disruption, and functional disturbances (32, 39). These pathological changes closely overlap with the characteristics of ferroptosis. These phenomena may be interrelated, further exacerbating myocardial damage and promoting the onset and progression of DCM.
2.1 Impact of glycemic disorder on ferroptosis in DCM and molecular regulatory mechanisms
In recent years, the dysregulation of glucose metabolism has emerged as a pivotal focus in research on DCM. Chronic fluctuations in blood glucose not only trigger oxidative stress but also promote the accumulation of advanced glycation end products (AGEs), exacerbating iron overload and lipid peroxidation (40). In DCM patients, impaired oxidative phosphorylation and the accumulation of AGEs in myocardial cells result in reduced ATP production. This energy deficiency makes cardiac cells more vulnerable to oxidative damage, particularly in high-glucose environments where iron overload is prevalent (41). Elevated blood glucose levels further increase the production of ROS, activating intracellular oxidative stress responses and further facilitating iron accumulation (42). Additionally, hyperglycemia promotes fatty acid synthesis pathways, enhancing the production of lipid peroxides, which accelerates ferroptosis in cardiac cells (33, 43).
Hyperglycemia promotes ferroptosis in myocardial cells through several interrelated mechanisms, which collectively contribute to the progression of DCM. These mechanisms include: (1) hyperglycemia induces an imbalance in the redox state of iron ions, promoting free radical generation and accelerating ferroptosis (44); (2) glucose dysregulation triggers iron ion accumulation, activating lipid peroxidation via the Fenton reaction, thereby promoting ferroptosis (23, 45); (3) chronic inflammation in diabetes exacerbates iron transport, storage, and utilization disorders, leading to the accumulation of free iron and inducing ferroptosis and myocardial fibrosis (46); (4) hyperglycemia disrupts mitochondrial energy supply, increasing ROS generation, further aggravating iron metabolic imbalance and eventually causing myocardial damage and ventricular remodeling (42, 47); (5) glucose dysregulation suppresses the expression of antioxidant genes via the Nrf2/ARE pathway, weakening the antioxidant defense and DNA repair capacity of the p53 pathway (48, 49); (6) insulin resistance, along with hyperglycemia, inhibits GPX4 activity, promotes AGEs formation, and disrupts fatty acid metabolism, ultimately accelerating the onset of ferroptosis (40, 50, 51).
2.2 Impact of iron homeostasis imbalance on ferroptosis in DCM and molecular regulatory mechanisms
Iron homeostasis refers is the regulation of iron intake, transport, storage, and excretion, which directly impacts cellular energy metabolism, antioxidant defense, and immune responses (46, 52). Hepcidin, a key hormone synthesized by the liver, regulates iron absorption and storage (52). Under normal physiological conditions, hepcidin reduces the expression of iron transport proteins like ferroportin, thereby limiting iron release and absorption to maintain iron homeostasis (53). Ferritin, a vital intracellular storage protein, sequesters excess iron to prevent its presence as free iron, thus reducing the risk of oxidative stress (54). Ferroportin facilitates iron transport by transferring intracellular iron into the bloodstream, thereby preventing iron accumulation in cells (55). When iron levels are high, ferroportin function is suppressed, leading to reduced iron release and its accumulation in cells. Additionally, transferrin receptor 1 (TfR1) mediates iron uptake by binding transferrin-bound iron and internalizing the complex via receptor-mediated endocytosis (56).
In DCM, blood glucose dysregulation triggers excessive oxidative stress and chronic inflammation, both of which are major contributors to ferroptosis (23). Hyperglycemia and insulin resistance promote oxidative stress and the release of inflammatory mediators, directly influencing the expression of hepcidin (13, 57). The upregulation of hepcidin inhibits the function of ferroportin, leading to intracellular iron accumulation (58), an increase in intracellular free iron levels, and subsequent lipid peroxidation and ferroptosis (59). Furthermore, chronic inflammation induces an increase in ferritin expression as a protective mechanism to store excess iron and reduce toxicity. However, excessive iron storage is insufficient to prevent iron overload, ultimately exacerbating ferroptosis.
Iron regulatory proteins (IRPs) are critical for maintaining iron homeostasis, as they control the expression of genes involved in iron absorption, storage, and export by interacting with iron response elements (IREs) (60). For instance, IRPs promote iron uptake by regulating TfR1 expression and increase iron storage by modulating ferritin synthesis (61). However, in diabetes, oxidative stress and chronic inflammation alter IRP activity, leading to iron metabolism dysregulation, iron overload, and increased ferroptosis (52). Thus, IRPs play a dual role in DCM: while they help maintain iron balance under normal conditions, their dysregulation in diabetes may contribute to cardiomyocyte ferroptosis and mitochondrial dysfunction (54, 55).
2.3 Role of oxidative stress in diabetic cardiomyopathy
In DCM, the disruption of iron homeostasis and persistent oxidative stress are key drivers of ferroptosis (52). These factors collectively contribute to lipid peroxidation, mitochondrial impairment, and ultimately, cardiomyocyte death (62). Several studies have shown that oxidative stress exacerbates ferroptosis through multiple signaling pathways, including System Xc-, Nrf2/KEAP1 pathway, and NOX4 (63).
System Xc- is an essential amino acid antiporter composed of two subunits: SLC7A11 (solute carrier family 7 member 11) and SLC3A2 (solute carrier family 3 member 2) (64–66). This system imports cysteine, a precursor for GSH. GSH, a crucial antioxidant, effectively scavenges ROS to prevent lipid peroxidation and ferroptosis (31). A dysfunction in System Xc- results in reduced cysteine availability, which limits GSH synthesis, weakens antioxidant defenses, and increases susceptibility to lipid peroxidation, thereby accelerating ferroptosis (67). In DCM, System Xc- dysfunction further promotes ferroptosis, making it a critical molecular marker (68). The accumulation of advanced glycation end-products (AGEs) and oxidative stress caused by hyperglycemia further disrupts System Xc-, leading to iron deposition and exacerbated oxidative stress. This cascade directly promotes lipid peroxidation and ferroptosis (40, 67, 69) (Figure 2).
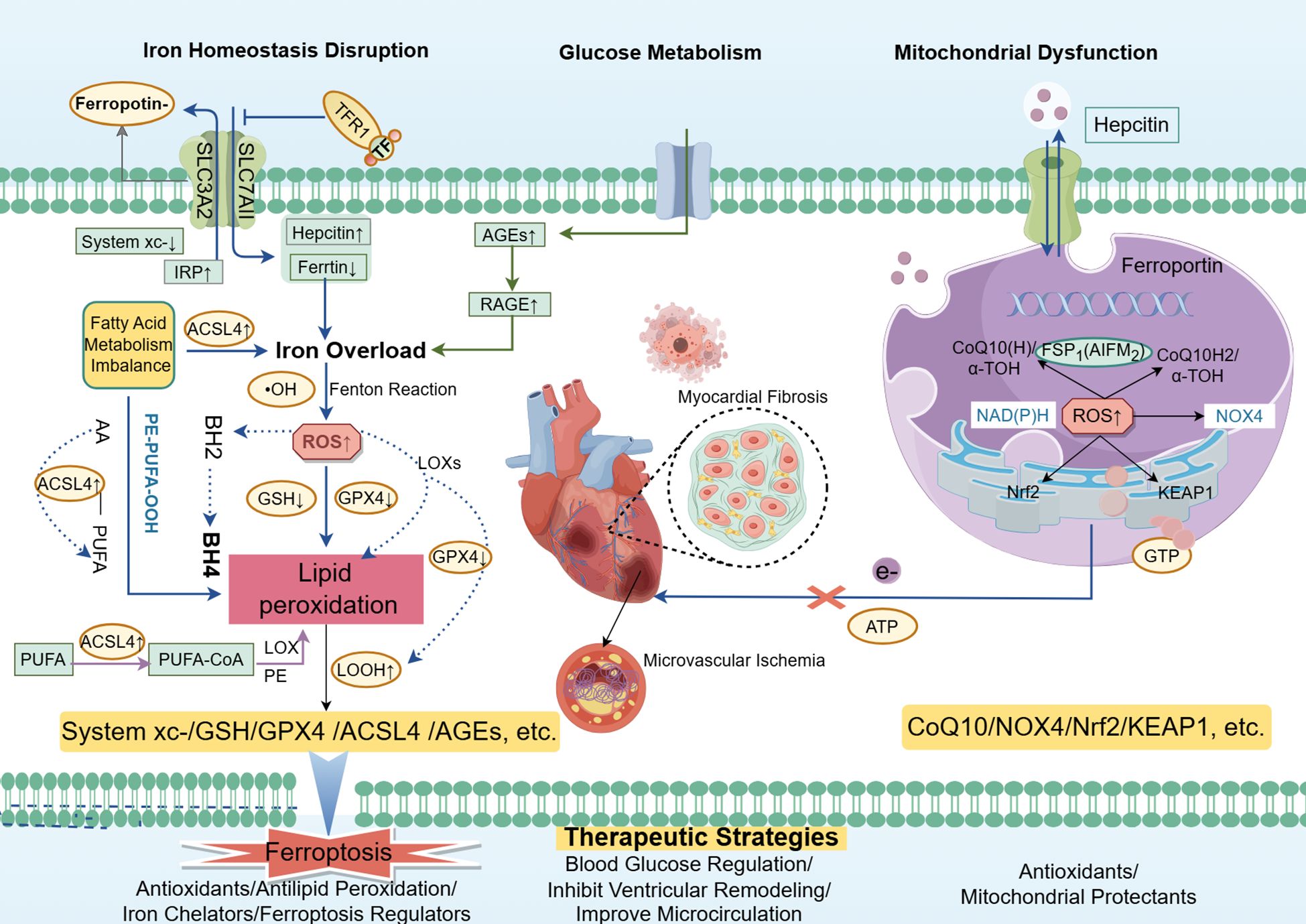
Figure 2. Ferroptosis and mitochondrial damage in diabetic cardiomyopathy (DCM) are driven by dysregulated glucose metabolism and insulin resistance, leading to iron overload and disturbed fatty acid metabolism in cardiomyocytes. This cascade results in excessive ROS generation and lipid peroxidation, activating pathways such as ACSL4 and hepcidin while inhibiting antioxidant systems like System Xc-, GSH, and GPX4. Iron overload and lipid peroxidation exacerbate mitochondrial dysfunction, impairing ATP synthesis and energy metabolism, ultimately contributing to cardiac fibrosis, heart failure, and the progression of DCM. Targeting ferroptosis and mitochondrial damage offers potential therapeutic strategies for managing DCM Courtesy of reference (70).
The nuclear factor erythroid-2-related factor 2 (Nrf2)/Kelch-like epichlorohydrin (ECH)-associated protein 1 (KEAP1) pathway plays a crucial role in DCM. Nrf2, a key transcription factor in oxidative stress response, is typically degraded by KEAP1 under normal conditions (71). However, under oxidative stress or pathological conditions like AGEs (72), Nrf2 is activated and translocated to the nucleus, initiating the expression of antioxidant genes such as GPX4, which are essential for reducing lipid peroxidation and delaying ferroptosis (73). Additionally, emerging research suggests that ferroptosis plays a significant role in diabetic retinopathy (DR) (74). Glucose dysregulation promotes upregulates long non-coding RNA ZFAS1, which enhances the expression of miR-7-5p. This cascade subsequently activates ACSL4, leading to increased lipid peroxidation and exacerbating oxidative damage and ferroptosis in retinal endothelial cells (75).
NOX4, a member of the NADPH oxidase family, interacts with Nrf2 and plays a significant role in oxidative stress (76). It catalyzes the production of superoxide and hydrogen peroxide, which increase lipid peroxidation, iron accumulation, and mitochondrial damage, thus driving ferroptosis (77). Under hyperglycemic conditions, NOX4 expression is significantly upregulated, further exacerbating ferroptosis (78). ROS generated by NOX4 also damage crucial components of the mitochondrial respiratory chain, further promoting mitochondrial dysfunction, which is a characteristic feature of DCM (79, 80).
2.3.1 Summary
Disruption in iron homeostasis, coupled with oxidative stress and the involvement of critical signaling pathways such as System Xc-, Nrf2/KEAP1, and NOX4, plays a significant role in the induction of ferroptosis in DCM. Dysregulated iron metabolism further exacerbates myocardial damage, while the ZFAS1/miR-7-5p/ACSL4 axis promotes lipid peroxidation in DR, highlighting potential therapeutic targets.
3 Mitochondrial damage and its role in DCM
Disturbances in myocardial energy metabolism are a crucial factor in the development of DCM. Mitochondria, as the primary energy producers in cells, are central to cardiac energy production and function (47, 79). Mitochondrial dysfunction in DCM is often characterized by a loss of membrane potential, which inhibits ATP synthesis and triggers the release of cytochrome C and apoptosis-related factors, thereby promoting cardiomyocyte apoptosis or necrosis (81). Furthermore, insulin resistance (IR) and chronic hyperglycemia (CH) associated with diabetes exacerbate mitochondrial damage by increasing the production of reactive oxygen species (ROS) within mitochondria. This oxidative stress intensifies mitochondrial dysfunction, disrupts energy metabolism, and further promotes oxidative damage (82).
In a hyperglycemic environment, non-enzymatic glycation reactions are enhanced, leading to the formation of AGEs (40, 51, 83). AGEs not only react with biomolecules such as proteins and lipids but also exacerbate oxidative stress, impairing the energy metabolism of cardiomyocytes and ultimately leading to cardiac dysfunction (84). Oxidative stress can directly damage the mitochondrial structure, particularly the mitochondrial respiratory chain, severely limiting ATP synthesis (85, 86). This metabolic dysregulation reduces mitochondrial energy production efficiency, adversely affecting cardiac contraction and relaxation functions (87). Furthermore, these processes accelerate cardiomyocyte aging, compounding the risk of heart failure (50, 88). Coenzyme Q10 (CoQ10) plays a crucial role in maintaining cellular energy metabolism and ensuring normal electron transfer in the mitochondrial respiratory chain (89, 90). As a potent antioxidant, CoQ10 effectively scavenges excess ROS, thereby protecting cells from oxidative stress-induced damage (91). Moreover, CoQ10 shows considerable promise in inhibiting ferroptosis and reducing oxidative harm in cardiomyocytes. Ferroptosis Suppressor Protein 1 (FSP1) is a newly identified inhibitor of ferroptosis, functioning by catalyzing the reduction of membrane phospholipids to prevent the oxidation of PUFA-PE (92, 93). This action reduces lipid peroxidation, thereby slowing the progression of ferroptosis. The synergistic action between FSP1 and CoQ10 significantly suppresses oxidative stress, further inhibiting ferroptosis (94).
Overall, the metabolic disturbances associated with diabetes not only worsen mitochondrial dysfunction but also intensify oxidative damage to cardiomyocytes through ferroptosis. The interplay between these processes is a critical driver of DCM progression, ultimately leading to cardiac dysfunction (10). CoQ10, FSP1, and other mitochondrial protectants, with their antioxidant properties and ability to reduce lipid peroxidation, offer a promising therapeutic strategy to safeguard mitochondria, inhibit ferroptosis, and mitigate oxidative damage (91–93). This approach could provide new perspectives for the treatment of DCM.
3.1 Interaction between ferroptosis and mitochondrial damage in DCM
The interaction between ferroptosis and mitochondrial damage in DCM is a complex pathological process involving iron overload, mitochondrial energy metabolism disturbances, and oxidative stress. Studies have suggested that these factors promote myocardial cell damage and drive the pathological progression of DCM.
Iron overload is a key trigger of ferroptosis. In the diabetic environment, disruptions in glucose and lipid metabolism result in abnormal iron uptake and storage, particularly within cardiomyocytes, where iron accumulation is markedly increased (45, 79, 95). The imbalance in iron transporters and export proteins leads to intracellular iron buildup, with some of the excess iron entering the mitochondria. Once inside, iron participates in Fenton reactions, generating highly reactive free radicals, such as hydroxyl radicals. These radicals inflict severe cellular damage by disrupting membranes, proteins, and DNA, ultimately causing myocardial cell injury and death (96).
Iron accumulation not only directly damages cardiomyocytes but also exacerbates mitochondrial damage through the promotion of lipid peroxidation (13). Lipid peroxidation, part of the oxidative stress response, disrupts the lipid bilayer of cell membranes, impairing the integrity and function of mitochondrial membranes (97). These damages, in turn, promote the generation of additional free radicals, further exacerbating the oxidative damage to cardiomyocytes, thus creating a vicious cycle. Moreover, oxidative stress amplifies the ferroptosis process. Elevated levels of ROS damage membrane structures and regulate iron metabolism-related signaling pathways, including those involving hepcidin, ferritin, and iron transporters. This regulation increases iron release and facilitates further iron entry into cells (70). As ROS levels increase, mitochondrial function progressively deteriorates, leading to energy depletion and, ultimately, cardiomyocyte death.
In summary, the interaction between ferroptosis and mitochondrial damage in DCM forms a vicious cycle: from iron overload to oxidative damage and ultimately to cell death. This process not only aggravates myocardial cell injury but also accelerates the pathological progression of DCM (64). Therefore, targeting iron metabolism and mitochondrial protection may provide a novel strategy for the prevention and treatment of DCM.
3.2 Impact of ferroptosis and mitochondrial damage on cardiac structure and function in DCM
Ferroptosis and mitochondrial damage play a pivotal role in the pathogenesis of DCM, and their interaction exacerbates the metabolic crisis in the heart, significantly affecting cardiac structure and function (98). Research has shown that iron overload not only promotes the deposition of extracellular matrix (ECM) components such as collagen (99) but also activates signaling pathways like transforming growth factor-beta (TGF-β), triggering cardiac remodeling (100). Additionally, iron overload can activate inflammatory responses, initiating cell death pathways such as apoptosis and autophagy (32, 101, 102), further accelerating myocardial cell damage and death.
The oxidative stress induced by ferroptosis exacerbates mitochondrial damage, particularly by disrupting mitochondrial structure and function (85, 86, 103). This disruption severely impairs ATP synthesis in cardiomyocytes, leading to energy depletion. The subsequent energy metabolic disorder contributes to cardiac dysfunction, which may ultimately result in heart failure. Therefore, the interaction between ferroptosis and mitochondrial damage provides a mechanistic basis for the deterioration of cardiac function.
In patients with DCM, the interplay between ferroptosis and mitochondrial dysfunction significantly disrupts myocardial energy metabolism, exacerbates myocardial fibrosis, and accelerates the progression of heart failure (104). Studies conducted on animal models have demonstrated that treatment with iron chelators or antioxidants can effectively reduce iron buildup and ROS levels, leading to an improvement in cardiac function (105). These results highlight the importance of restoring iron balance and repairing mitochondrial function as key therapeutic strategies for enhancing cardiac health in DCM patients.
Given the critical roles of ferroptosis and mitochondrial dysfunction in the pathogenesis of DCM, developing targeted therapeutic strategies to restore iron balance and repair mitochondrial function may offer a promising approach for treating DCM. Such interventions could potentially improve cardiac function and provide new avenues for effective clinical management in the future.
4 Ferroptosis and mitochondrial injury interventions in DCM
4.1 Current treatment strategies for DCM and their limitations
Currently, treatment strategies for DCM primarily aim at controlling blood glucose levels and alleviating heart failure symptoms. Key therapeutic approaches include regulating blood glucose, managing blood lipids, improving insulin resistance, and preventing or treating cardiovascular and cerebrovascular complications induced by diabetes (106). Commonly used drugs in DCM and their mechanisms of action, as along with potential side effects are listed in Table 1. For example, metformin, a widely used antidiabetic drug, not only exerts its classic hypoglycemic effects but has also been shown to possess antioxidant and ferroptosis-inhibiting properties in recent studies (124). Metformin reduces the levels of lipid peroxidation products such as MDA and 4-hydroxy-2-nonenal (HNE), decreases intracellular ROS levels, and improves mitochondrial function, thereby inhibiting ferroptosis and alleviating the progression of DCM (125). Studies have demonstrated that metformin activates the AMP-activated protein kinase (AMPK) signaling pathway, improves iron homeostasis, enhances cellular antioxidant capacity, and helps mitigate cardiac damage associated with DCM (126, 127).
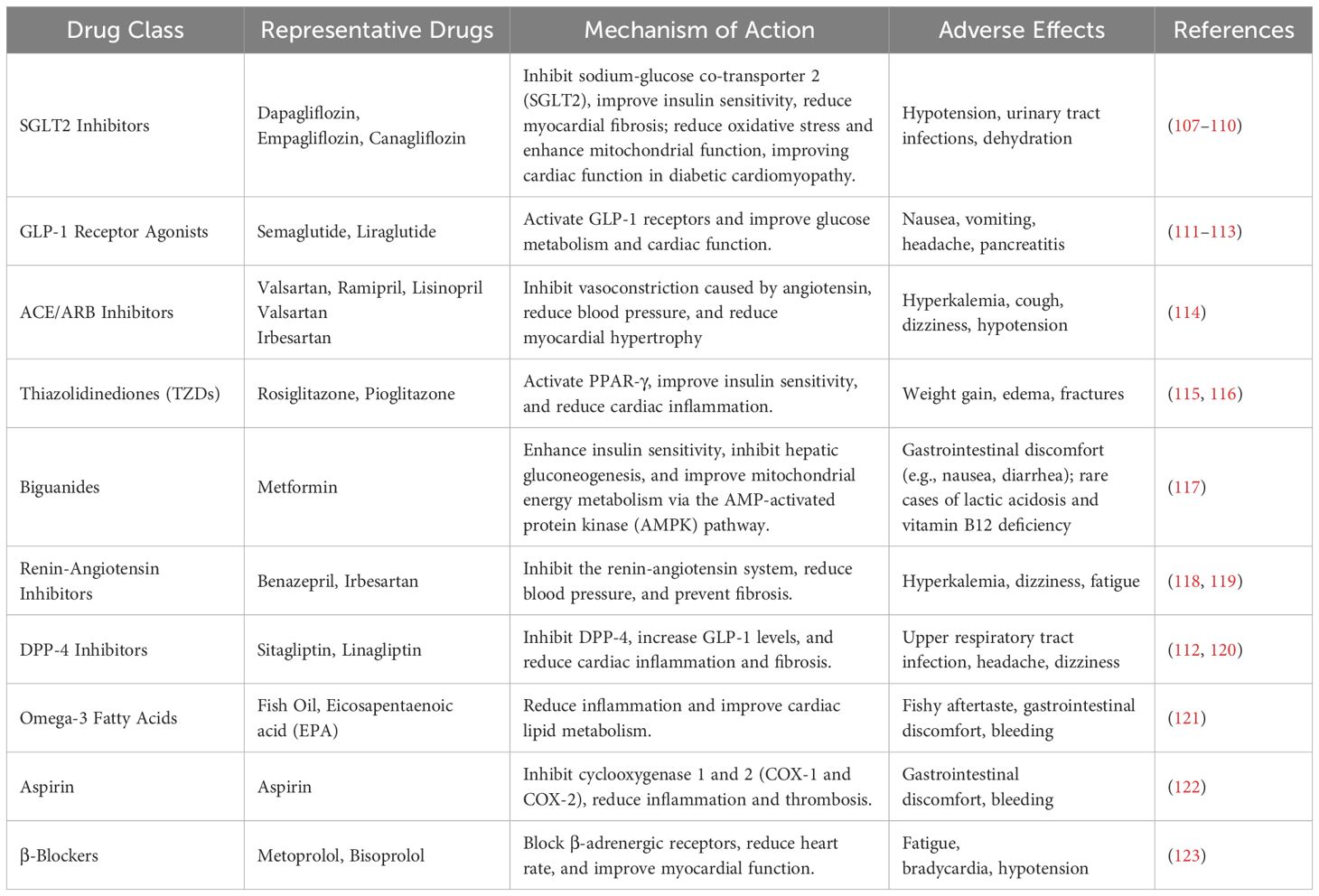
Table 1. Drugs for improving heart failure in DCM and alleviating cardiovascular and cerebrovascular complications.
Sodium-glucose cotransporter 2 (SGLT-2) inhibitors, such as dapagliflozin and empagliflozin (107–110), work by inhibiting SGLT-2 in the kidneys, which decreases glucose reabsorption and lowers blood glucose levels. These drugs also exert diuretic effects, reduce cardiac inflammation, enhance myocardial energy metabolism, and alleviate oxidative stress, offering cardiac protection in DCM.
Glucagon-like peptide-1 receptor (GLP-1 receptor) agonists), such as liraglutide and semaglutide (111), mimic the action of endogenous GLP-1 to increase insulin secretion, suppress glucagon secretion, delay gastric emptying, and promote weight loss. In addition to improving blood glucose control, these drugs have been shown to improve cardiac structure, reduce myocardial hypertrophy, decrease cardiac fibrosis, and enhance heart function (112). They also help reduce vascular inflammation and oxidative stress.
Dipeptidyl peptidase-4 (DPP-4) inhibitors enhance insulin secretion and reduce glucose production by inhibiting DPP-4 enzyme activity (128). They also improve endothelial function and reduce cardiovascular burden (129). These medications represent a range of therapeutic options for preventing and treating DCM. However, current treatment strategies primarily focus on alleviating symptoms, such as controlling blood glucose and managing heart failure (130). A comprehensive treatment approach that targets the core pathological mechanisms of DCM, including myocardial energy metabolism dysregulation, mitochondrial dysfunction, and ferroptosis inhibition, has yet to be established.
While current treatments delay the progression of DCM to some extent, their effectiveness and safety remain limited. Notably, no specific medications have been identified that can significantly improve cardiac energy metabolism or repair mitochondrial function in DCM (131). Some studies suggest that certain drugs may have positive effects on cardiac metabolism and reduce myocardial injury, but their long-term efficacy and safety require further investigation (132). For example, the GLP-1 receptor agonist semaglutide has been shown to promote insulin secretion, improve cardiac metabolism, and reduce myocardial injury, thus delaying the progression of DCM (111). While preliminary results are promising, clinical data on the long-term effects and potential side effects of this drug remain insufficient, warranting further research and validation.
Moreover, new glucose level-lowering medications such as DPP-4 inhibitors (e.g., sitagliptin and vildagliptin) are widely used in diabetes treatment. In vitro and animal studies suggest that DPP-4 inhibitors can exert positive effects on DCM through mechanisms such as reducing cardiac inflammation and improving heart function (112, 120). However, the findings from relevant studies have been inconsistent. Some clinical trials report that DPP-4 inhibitors improve heart failure symptoms (133), while other studies suggest that their cardiac protective effects are limited (134). These discrepancies may be attributed to variations in study design, sample size, and trial standards. Therefore, future research should focus on prospective, multicenter, randomized controlled trials to further verify the efficacy and safety of DPP-4 inhibitors in DCM.
In conclusion, while current therapeutic approaches provide some relief in managing DCM, interventions targeting the disease’s core pathological mechanisms require further investigation. Future treatment strategies should place greater emphasis on addressing disorders in iron metabolism and mitochondrial damage to develop targeted therapies that offer more effective treatment options for patients with DCM.
4.2 Current Status of Targeted Ferroptosis and Mitochondrial Injury Treatment Strategies in DCM
As research into the pathogenesis of DCM progresses, ferroptosis and mitochondrial injury are increasingly recognized as key pathological contributors to the disease. Current targeted treatment strategies focus on iron chelators, antioxidants, mitochondrial protectants, and nanoparticle-based therapies (Table 2). These therapeutic approaches aim to reduce myocardial damage and prevent or delay the progression of DCM by targeting ferroptosis, repairing mitochondrial damage, and counteracting oxidative stress (85, 88, 103, 140). Ferroptosis inhibitors, particularly Ferrostatin-1, have been shown to effectively suppress ferroptosis under various pathological conditions (152, 176). Ferrostatin-1 exerts its action by inhibiting lipid peroxidation, particularly through its action on membrane lipids, thereby slowing the progression of ferroptosis (177). Additionally, Ferrostatin-1 exerts anti-inflammatory and antioxidant effects, further alleviating oxidative stress by inhibiting the Nrf2/ARE pathway and other antioxidant signaling pathways (178).
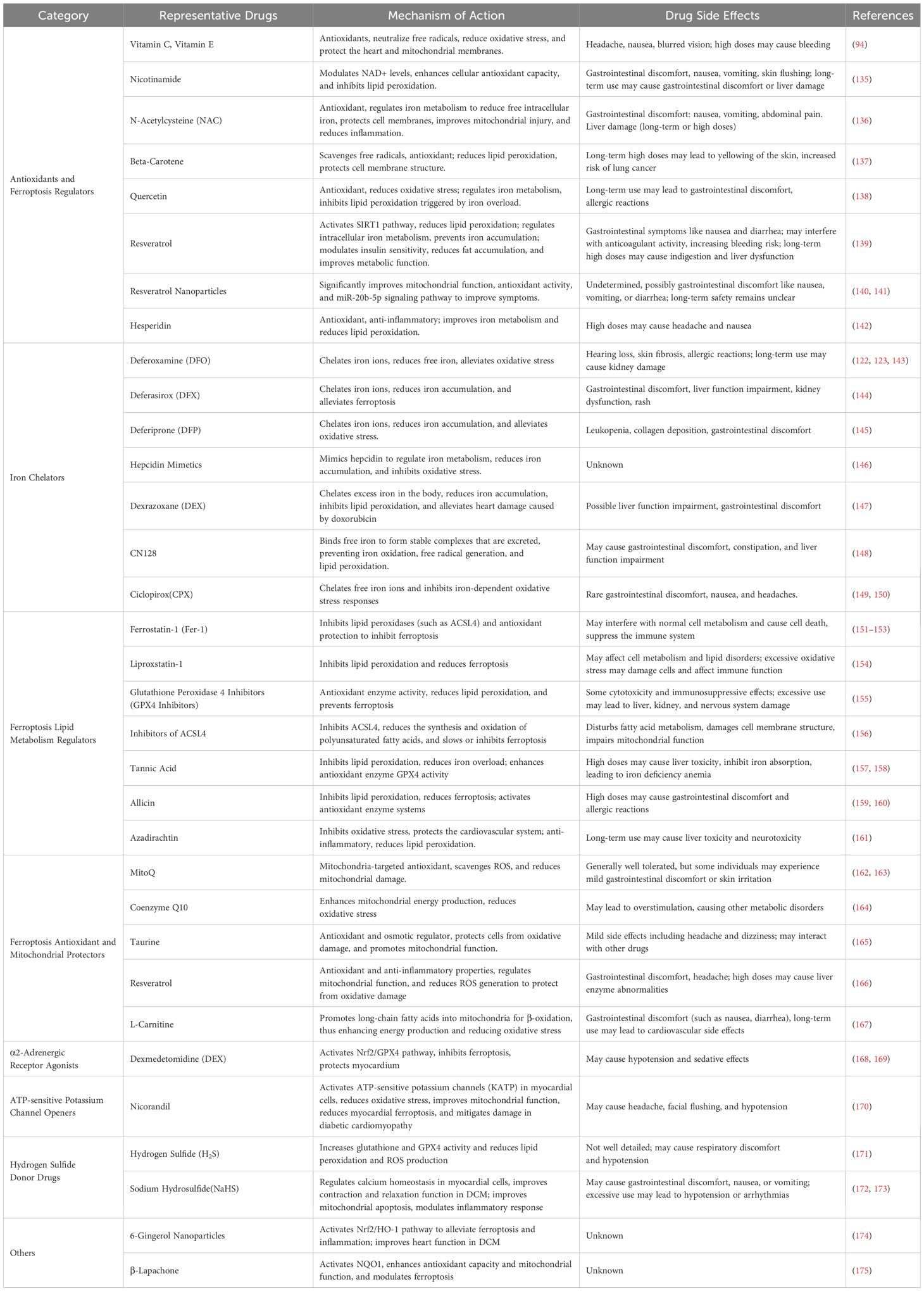
Table 2. Representative drugs for the improvement of ferroptosis and mitochondrial injury.
Iron chelators are also essential in managing DCM. Deferoxamine, a widely used iron chelator, has proven effective in treating iron overload-related disorders but carries significant side effects, particularly affecting renal function and the immune system (179, 180). Deferiprone, another iron chelator, offers better oral bioavailability but still poses risks, including kidney toxicity. On the other hand, the novel oral iron chelator CN128 has shown superior iron chelation and antioxidant properties in clinical trials (148). It also has a longer half-life, fewer side effects, and is able to effectively reduce body iron overload without causing kidney damage (181). Additionally, other iron chelators, such as Ciclopirox (CPX) (149), have demonstrated the potential to inhibit iron-dependent oxidative stress, reduce lipid peroxidation, and improve myocardial cell health, offering enhanced myocardial protection in DCM. Furthermore, resveratrol nanoparticles have shown promising effects in protecting the heart by mitigating mitochondrial damage and inhibiting lipid peroxidation reactions (89).
Additionally, resveratrol nanoparticles have demonstrated protective effects on the heart by alleviating mitochondrial damage and inhibiting lipid peroxidation (182). These therapies function through multiple mechanisms, including modulating intracellular redox balance and improving mitochondrial function, offering new possibilities for the treatment of DCM.
Although these therapeutic strategies have shown promising results in laboratory settings, most studies are still limited to cell experiments, animal models, and a few clinical case reports. Comprehensive experimental data on drug mechanisms of action, pharmacodynamics, toxicology, and pharmacokinetics remain scarce (183). Furthermore, there is a lack of large-scale, multicenter, prospective preclinical studies and randomized clinical trials, meaning the effectiveness and safety of these treatment strategies have not been fully validated in clinical practice (151).
Therefore, while ferroptosis and mitochondrial injury-targeted therapies show great promise in the treatment of DCM, further in-depth exploration of the drugs’ mechanisms of action, efficacy, and safety is crucial to advance the clinical translation of these therapeutic approaches.
5 Prospects for basic research and clinical applications of therapeutic strategies for DCM
The pathogenesis of DCM is multifaceted, involving ferroptosis, oxidative stress, mitochondrial injury, and other factors. Advancements in understanding these pathological processes are paving the way for innovative therapeutic strategies, particularly those centered on multi-target combination therapies and personalized medicine. Future therapeutic approaches may combine ferroptosis-targeted agents with mitochondrial protectants to achieve synergistic effects in alleviating myocardial injury and improving cardiac function (Figure 3).
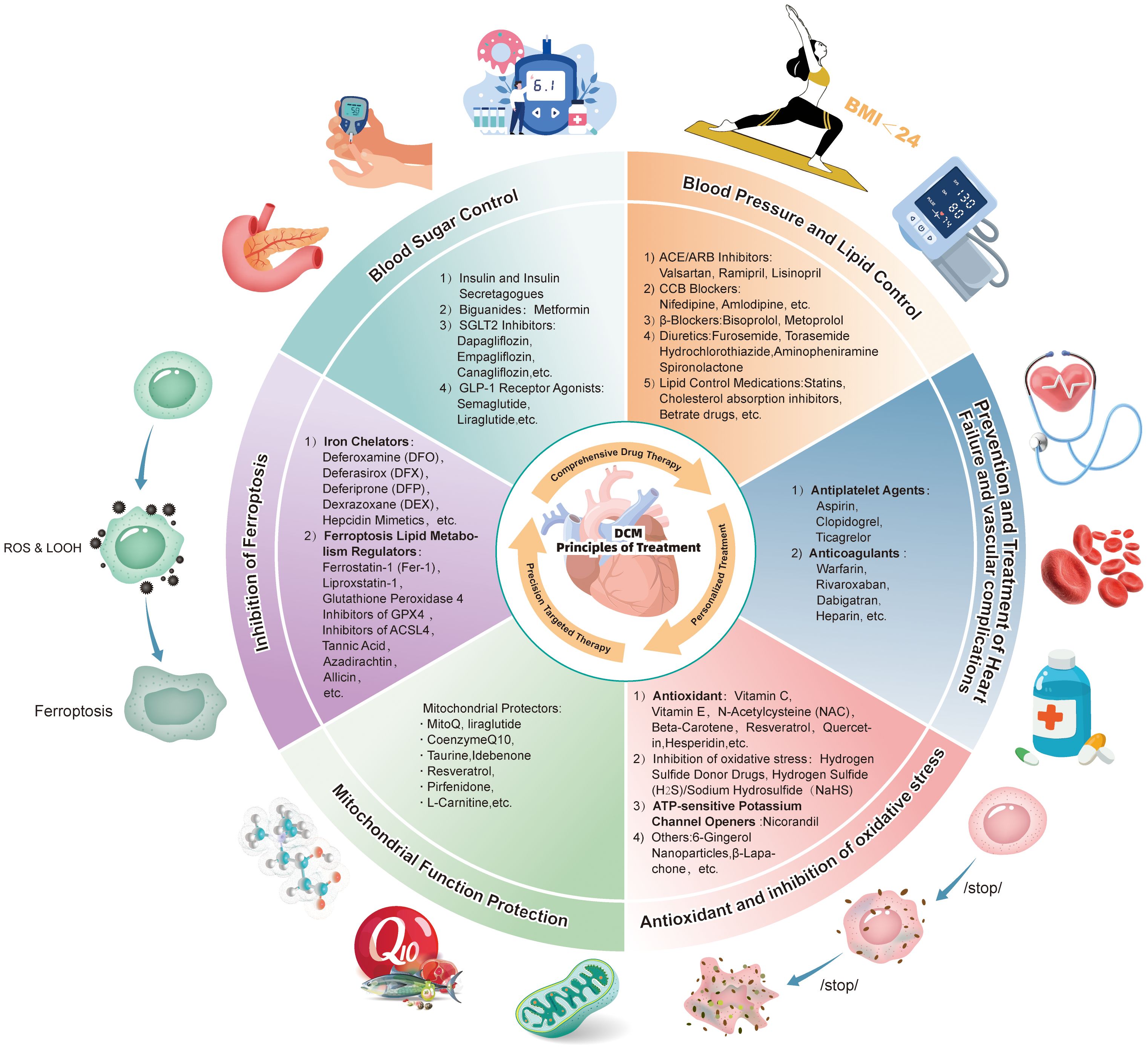
Figure 3. Comprehensive prevention and treatment strategies for diabetic cardiomyopathy: A schematic diagram of drug regimens based on combined pharmacological therapy, individualized treatment, and precision-targeted therapy targeting ferroptosis and mitochondrial damage.
5.1 Potential of combination therapy
The intricate pathological mechanisms of DCM present significant challenges for single-treatment approaches to address all aspects of the disease comprehensively. The interaction between ferroptosis and mitochondrial injury creates a synergistic effect, which exacerbates cardiac dysfunction during DCM progression (184). Consequently, combination therapies that target both ferroptosis and mitochondrial dysfunction have emerged as a promising direction for future clinical treatment. For example, the combined use of iron chelators and antioxidants has shown potential to effectively reduce iron overload while inhibiting oxidative stress, thereby offering enhanced protection for myocardial cells (136, 137, 143, 145). Similarly, combining GPX4 agonists with sodium-calcium exchanger inhibitors could suppress lipid peroxidation and safeguard mitochondrial integrity, thereby delaying myocardial cell death (185, 186). These combination strategies demonstrate strong synergistic effects, laying a robust theoretical foundation for the development of personalized treatment strategies. By tailoring treatment regimens based on an individual’s pathological state, these approaches could improve therapeutic efficacy while minimizing side effects.
With advancements in precision medicine, identifying specific pathological mechanisms in patients will be instrumental in guiding the development of targeted therapies. This approach would ensure that interventions are both effective and safe, addressing the variability in drug responses among patients (187). Ultimately, combination therapies grounded in multi-target and personalized strategies are poised to play a transformative role in the clinical management of DCM, offering hope for more effective and patient-specific treatments in the future.
5.2 Challenges and progress in clinical research
Although novel therapies such as ferroptosis inhibitors and mitochondrial protectants have shown promising efficacy in preclinical studies (164, 166, 188), their clinical translation still faces numerous challenges. Firstly, long-term drug efficacy evaluation and drug resistance are major difficulties in clinical trial design (189). The slow progression of DCM necessitates extended trial durations, thereby escalating costs and adding complexity to the research process (190). Furthermore, variability among patients, including genetic factors and individual pathological conditions, can profoundly influence drug responses, presenting a considerable hurdle in developing personalized treatment strategies (191).
Moreover, the combination of iron chelators and mitochondrial protectants has shown promising results in some studies (192), but several challenges remain, including issues related to drug formulation, safety, and potential interactions between medications. For instance, iron chelators such as deferoxamine and deferasirox effectively reduce iron overload and improve iron-related damage. However, prolonged or high-dose usage can lead to adverse effects such as liver and kidney toxicity or allergic reactions (122, 123, 179, 180). Therefore, the clinical use of these agents requires careful management, with dosages and treatment durations tailored to the patient’s specific condition.
Additionally, while drugs like SGLT2 inhibitors and GLP-1 receptor agonists have demonstrated benefits in improving cardiac function and glucose metabolism in patients with DCM (107, 108, 111, 113), their side effects present significant limitations in clinical practice. For example, SGLT2 inhibitors may cause hypoglycemia and urinary tract infections, whereas GLP-1 receptor agonists may trigger nausea and pancreatitis (193). Thiazolidinediones (e.g., rosiglitazone and pioglitazone) enhance insulin sensitivity and reduce cardiac inflammation but are linked to side effects such as weight gain, edema, and bone fractures with long-term use (115, 116, 194). These issues are particularly concerning for elderly patients and those with osteoporosis, who require careful evaluation of the risks and benefits of such medications (195). These concerns are particularly relevant for elderly patients and those with osteoporosis, necessitating a thorough assessment of risks and benefits.
5.3 Future research directions
As the pathogenesis of DCM continues to be explored, future research is likely to focus on several key areas:
5.3.1 Development of precision-targeted drugs
Current pharmacological treatments for DCM and its complications largely rely on non-specific approaches, focusing on symptom management rather than addressing the underlying pathological mechanisms. Moving forward, research will prioritize the development of drugs that specifically target key processes such as ferroptosis and oxidative stress (14, 196). For example, therapeutics that modulate iron metabolism by targeting proteins like hepcidin or ferritin are anticipated to gain prominence in future studies. Additionally, compounds aimed at restoring mitochondrial function, particularly those capable of enhancing ATP synthesis or mitigating the generation of ROS, represent promising clinical advancements (197). These targeted therapeutic strategies have the potential to not only improve treatment outcomes but also pave the way for personalized therapeutic approaches tailored to the unique pathological conditions of individual patients.
5.3.2 Development of novel drugs
Mitochondrial dysfunction is a central pathological feature of DCM, and developing drugs that restore mitochondrial function holds significant therapeutic potential (198). Agents that enhance ATP synthesis or reduce the generation of ROS are among the promising candidates for addressing this dysfunction (199). In addition to mitochondrial protectants, anti-inflammatory drugs may also offer new treatment avenues for DCM. Specifically, drugs that target inflammation pathways unique to the heart, such as those modulating inflammatory responses associated with myocardial fibrosis or cardiomyocyte (200).
5.3.3 Drug combination strategies
Many monotherapeutic strategies have failed to yield the desired outcomes when used alone, prompting increasing interest in combination therapies. Current research indicates that combining SGLT2 inhibitors with GLP-1 receptor agonists has shown synergistic effects (67–69, 76). Future studies are likely to explore additional combination therapies, particularly those involving drugs with distinct mechanisms of action, such as pairing iron chelators with antioxidants or mitochondrial protectants (64). These combinations could improve therapeutic efficacy while minimizing side effects (201, 202). By targeting multiple pathological processes simultaneously, drug combinations offer a more comprehensive approach to treating DCM.
5.3.4 Gene and stem cell therapies
With the ongoing progress in gene editing and stem cell research, gene therapy and cell-based treatments hold the potential to deliver groundbreaking therapeutic options for DCM (203). Gene editing technologies that repair mitochondrial function or stem cell therapies aimed at repairing myocardial damage could offer innovative solutions in the future. Gene therapy can address the molecular root causes of DCM, while stem cell therapy shows promise in promoting myocardial regeneration and functional recovery. These approaches could provide new avenues for the treatment of DCM, offering hope for improved outcomes for patients (204).
5.3.5 Personalized medicine and precision healthcare
Future research should focus on personalized treatment strategies based on genomics, metabolomics, and other technologies. The pathological mechanisms may vary from patient to patient, and thus, precision medicine will play a key role in developing individualized therapeutic approaches. This will ensure that drug treatments are tailored to a patient’s specific pathological state, thereby improving efficacy and reducing side effects (205). Personalized medicine will not only enhance treatment outcomes but also reduce unwanted side effects, optimizing the overall treatment outcome for patients.
5.4 Clinical translation and challenges
Despite significant advancements in basic research for DCM treatment, translating these findings into clinical applications remains fraught with challenges. Firstly, although many drugs show promising results in animal models, there is a lack of sufficient large-scale clinical trial data to support their efficacy and safety across diverse patient populations (206). Thus, future research will require multi-center, large-scale randomized controlled trials to validate the feasibility and effectiveness of these drugs in clinical settings. Moreover, the combination of novel drugs with existing treatment modalities (e.g., SGLT2 inhibitors with ferroptosis inhibitors, antioxidants like selenocysteine with Ferrostatin-1 or Liproxstatin-1, and GLP-1 receptor agonists with ferroptosis inhibitors) (207–209) will be an important area of future research. Investigating the potential synergistic effects and interactions between these drugs will help optimize treatment regimens and improve overall efficacy (205, 210). Combination therapies may not only enhance therapeutic outcomes but also provide more treatment options to meet individual patient needs.
6 Conclusion
In conclusion, treating diabetic cardiomyopathy remains a complex challenge. While current medications can alleviate symptoms and slow the progression of the disease, their efficacy and safety still require further enhancement. Future research should prioritize precision medicine, aiming to minimize drug side effects while optimizing the effectiveness of multi-target combination therapies. With technological advances, the development of drugs targeting ferroptosis and mitochondrial dysfunction, personalized treatment strategies, and the integration of stem cell and gene therapies could pave the way for significant breakthroughs in the clinical management of diabetic cardiomyopathy.
Author contributions
BL: Conceptualization, Data curation, Writing – original draft, Writing – review & editing. QJ: Resources, Writing – review & editing. YS: Conceptualization, Writing – review & editing. ZY: Conceptualization, Project administration, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Resources, Software, Supervision, Validation, Visualization, Writing – review & editing. PM: Conceptualization, Resources, Writing – review & editing. XZ: Formal Analysis, Writing – review & editing. QC: Conceptualization, Writing – review & editing. PG: Conceptualization, Resources, Writing – review & editing. TZ: Conceptualization, Writing – review & editing. JH: Resources, Validation, Writing – review & editing. GH: Conceptualization, Resources, Writing – review & editing. QX: Conceptualization, Funding acquisition, Resources, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was supported by the Scientific Research Fund of the Yunnan Provincial Department of Education (Project No. K132191027) and by the Key Laboratory of Cardiovascular Disease of Yunnan Province (Project No. 2018DG008).
Acknowledgments
The authors thank the academic editor and reviewers for their hard work and dedication. Some of the schematic figures in this article were sourced from Figdraw.com, and we would like to express our gratitude.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
DCM: Diabetic cardiomyopathy
IDF: International Diabetes Federation
ROS: Reactive oxygen species
GSH: Glutathione
GPX4: Glutathione peroxidase 4
AGEs: Advanced glycation end products
Nrf2: Nuclear factor erythroid 2-related factor 2
ARE: Antioxidant response element
Tf: Transferrin
Ft: Ferritin
Fpn: Ferroportin
IRPs: Iron regulatory proteins
DMT1: Divalent Metal Transporter 1
System Xc-: Cystine/Glutamate Antiporter System Xc-
TGF-β: Transforming growth factor-beta
IR: Insulin resistance
CH: Chronic hyperglycemia
ATP: Adenosine triphosphate
ECM: Extracellular matrix
SGLT-2: Sodium-glucose cotransporter 2
GLP-1: Glucagon-like peptide-1
DPP-4: Dipeptidyl peptidase-4
ACE: Angiotensin-converting enzyme
ARB: Angiotensin receptor blockers
TZDs: Thiazolidinediones
AMPK: Adenosine monophosphate-activated protein kinase
RAS: Renin-Angiotensin Inhibitors
EPA: Eicosapentaenoic acid
COX-1 and COX-2: Cyclooxygenase 1 and 2
NAC: N-Acetylcysteine
DFO: Deferoxamine
DFX: Deferasirox
DFP: Deferiprone
DEX: Dexrazoxane
Fer-1: Ferrostatin-1
GPX4 Inhibitors: Glutathione Peroxidase 4 Inhibitors
DEX: Dexmedetomidine
H₂S: Hydrogen Sulfide
NaHS: Sodium Hydrosulfide
NAD(P)H: Nicotinamide Adenine Dinucleotide (Phosphate)
NQO1: Quinone Oxidoreductase 1
GPT: Glutamate Pyruvate Transaminase
NOX4: NADPH Oxidase 4
REAP1: Receptor Interacting Protein 1
ACSL4: Acyl-CoA Synthetase Long Chain Family Member 4
SLC3A2: Solute Carrier Family 3 Member 2
SLC7A11: Solute Carrier Family 7 Member 11
PUFA: Polyunsaturated Fatty Acids
PE-PUFA-OOH: Phosphatidylethanolamine Polyunsaturated Fatty Acids
SOD1: Superoxide Dismutase 1
Nrf2: Nuclear factor erythroid-2-related factor 2
KEAP1: Kelch-like epichlorohydrin (ECH)-associated protein 1
References
1. Quaiyoom A, Kumar R. An overview of diabetic cardiomyopathy. Curr Diabetes Rev. (2024) 20:97–109. doi: 10.2174/0115733998255538231001122639
2. Heather LC, Gopal K, Srnic N, Ussher JR. Redefining diabetic cardiomyopathy: perturbations in substrate metabolism at the heart of its pathology. Diabetes. (2024) 73:659–70. doi: 10.2337/dbi23-0019
3. Federation ID. New estimates show that over 800 million people are living with diabetes. (2024). Available online at: https://idf.org/news/new-diabetes-estimates.
4. Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract. (2022) 183:109119. doi: 10.1016/j.diabres.2021.109119
5. Bhatt DL, Szarek M, Steg PG, Cannon CP, Leiter LA, McGuire DK, et al. Sotagliflozin in patients with diabetes and recent worsening heart failure. New Engl J Med. (2021) 384:117–28. doi: 10.1056/NEJMoa2030183
6. Ritchie RH, Abel ED. Basic mechanisms of diabetic heart disease. Circ Res. (2020) 126:1501–25. doi: 10.1161/CIRCRESAHA.120.315913
7. Nakamura K, Miyoshi T, Yoshida M, Akagi S, Saito Y, Ejiri K, et al. Pathophysiology and treatment of diabetic cardiomyopathy and heart failure in patients with diabetes mellitus. Int J Mol Sci. (2022) 23:3587. doi: 10.3390/ijms23073587
8. Lou X, Zhang Y, Guo J, Gao L, Ding Y, Zhuo X, et al. What is the impact of ferroptosis on diabetic cardiomyopathy: a systematic review. Heart Failure Rev. (2024) 29:1–11. doi: 10.1007/s10741-023-10336-z
9. Tan Y, Zhang Z, Zheng C, Wintergerst KA, Keller BB, Cai L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: preclinical and clinical evidence. Nat Rev Cardiol. (2020) 17:585–607. doi: 10.1038/s41569-020-0339-2
10. Fang X, Ardehali H, Min J, Wang F. The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat Rev Cardiol. (2023) 20:7–23. doi: 10.1038/s41569-022-00735-4
11. Liu P, Zhang Z, Cai Y, Li Z, Zhou Q, Chen Q. Ferroptosis: mechanisms and role in diabetes mellitus and its complications. Ageing Res Rev. (2024) 94(000):102201. doi: 10.1016/j.arr.2024.102201
12. Chen Z, Li S, Liu M, Yin M, Chen J, Li Y, et al. Nicorandil alleviates cardiac microvascular ferroptosis in diabetic cardiomyopathy: Role of the mitochondria-localized AMPK-Parkin-ACSL4 signaling pathway. Pharmacol Res. (2024) 200:107057. doi: 10.1016/j.phrs.2024.107057
13. Wang Z, Wu C, Yin D, Dou K. Ferroptosis: mechanism and role in diabetes-related cardiovascular diseases. Cardiovasc Diabetol. (2025) 24:60. doi: 10.1186/s12933-025-02614-x
14. Wen J, Li L, Yang Y, Ou D, Yang J, Xie J, et al. Phytochemicals targeting ferroptosis in cardiovascular diseases: Recent advances and therapeutic perspectives. Phytother Res. (2024) 38:4386–405. doi: 10.1002/ptr.v38.9
15. Jin EJ, Jo Y, Wei S, Rizzo M, Ryu D, Gariani K. Ferroptosis and iron metabolism in diabetes: Pathogenesis, associated complications, and therapeutic implications. Front Endocrinol (Lausanne). (2024) 15:1447148. doi: 10.3389/fendo.2024.1447148
16. Wang H, Liu C, Zhao Y, Gao G. Mitochondria regulation in ferroptosis. Eur J Cell Biol. (2020) 99:151058. doi: 10.1016/j.ejcb.2019.151058
17. Capelletti MM, Manceau H, Puy H, Peoc’h K. Ferroptosis in liver diseases: an overview. Int J Mol Sci. (2020) 21(14):4908. doi: 10.3390/ijms21144908
18. Teschke R, Eickhoff A. Wilson disease: copper-mediated cuproptosis, iron-related ferroptosis, and clinical highlights, with comprehensive and critical analysis update. Int J Mol Sci. (2024) 25(9):35. doi: 10.3390/ijms25094753
19. Liu Q, Liu CQ, Yi WZ, Ouyang PW, Yang BF, Liu Q, et al. Ferroptosis contributes to microvascular dysfunction in diabetic retinopathy. Am J Pathol. (2024) 194:1078–89. doi: 10.1016/j.ajpath.2024.01.019
20. Catapano A, Cimmino F, Petrella L, Pizzella A, D’Angelo M, Ambrosio K, et al. Iron metabolism and ferroptosis in health and diseases: the crucial role of mitochondria in meta-bolically active tissues. J Nutr Biochem. (2025) 140:109888. doi: 10.1016/j.jnutbio.2025.109888
21. Guo Y, Lu C, Hu K, Cai C, Wang W. Ferroptosis in cardiovascular diseases: current status, challenges, and future perspectives. Biomolecules. (2022) 12(3). doi: 10.3390/biom12030390
22. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. (2021) 22:266–82. doi: 10.1038/s41580-020-00324-8
23. Sha W, Hu F, Xi Y, Chu Y, Bu S. Mechanism of ferroptosis and its role in type 2 diabetes mellitus. J Diabetes Res. (2021) 2021:9999612. doi: 10.1155/2021/9999612
24. Maiorino M, Conrad M, Ursini F. GPx4, lipid peroxidation, and cell death: discoveries, rediscoveries, and open issues. Antioxid Redox Signal. (2018) 29:61–74. doi: 10.1089/ars.2017.7115
25. Wang M, Tang J, Zhang S, Pang K, Zhao Y, Liu N, et al. Exogenous H(2)S initiating Nrf2/GPx4/GSH pathway through promoting Syvn1-Keap1 interaction in diabetic hearts. Cell Death Discov. (2023) 9:394. doi: 10.1038/s41420-023-01690-w
26. Liu Y, Wan Y, Jiang Y, Zhang L, Cheng W. GPX4: The hub of lipid oxidation, ferroptosis, disease and treatment. Biochim Biophys Acta Rev Cancer. (2023) 1878:188890. doi: 10.1016/j.bbcan.2023.188890
27. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. (2017) 13:91–8. doi: 10.1038/nchembio.2239
28. Ding K, Liu C, Li L, Yang M, Jiang N, Luo S, et al. Acyl-CoA synthase ACSL4: an essential target in ferroptosis and fatty acid metabolism. Chin Med J (Engl). (2023) 136:2521–37. doi: 10.1097/CM9.0000000000002533
29. He S, Li R, Peng Y, Wang Z, Huang J, Meng H, et al. ACSL4 contributes to ferroptosis-mediated rhabdomyolysis in exertional heat stroke. J Cachexia Sarcopenia Muscle. (2022) 13:1717–30. doi: 10.1002/jcsm.12953
30. Cui J, Chen Y, Yang Q, Zhao P, Yang M, Wang X, et al. Protosappanin A protects DOX-induced myocardial injury and cardiac dysfunction by targeting ACSL4/FTH1 axis-dependent ferroptosis. Adv Sci (Weinh). (2024) 11:e2310227. doi: 10.1002/advs.202310227
31. Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radical Biol Med. (2020) 152:175–85. doi: 10.1016/j.freeradbiomed.2020.02.027
32. Hu X, Bao Y, Li M, Zhang W, Chen C. The role of ferroptosis and its mechanism in ischemic stroke. Exp Neurol. (2024) 372:114630. doi: 10.1016/j.expneurol.2023.114630
33. Lin Y, Shen X, Ke Y, Lan C, Chen X, Liang B, et al. Activation of osteoblast ferroptosis via the METTL3/ASK1-p38 signaling pathway in high glucose and high fat (HGHF)-induced diabetic bone loss. FASEB J. (2022) 36:e22147. doi: 10.1096/fj.202101610R
34. Park TJ, Park JH, Lee GS, Lee JY, Shin JH, Kim MW, et al. Quantitative proteomic analyses reveal that GPX4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death Dis. (2019) 10:835. doi: 10.1038/s41419-019-2061-8
35. Gao X, Hu W, Qian D, Bai X, He H, Li L, et al. The mechanisms of ferroptosis under hypoxia. Cell Mol Neurobiol. (2023) 43:3329–41. doi: 10.1007/s10571-023-01388-8
36. Wang B, Wang Y, Zhang J, Hu C, Jiang J, Li Y, et al. ROS-induced lipid peroxidation modulates cell death outcome: mechanisms behind apoptosis, autophagy, and ferroptosis. Arch Toxicol. (2023) 97:1439–51. doi: 10.1007/s00204-023-03476-6
37. Xu X, Xu X-D, Ma M-Q, Liang Y, Cai Y-B, Zhu Z-X, et al. The mechanisms of ferroptosis and its role in atherosclerosis. Biomed Pharmacother. (2024) 171:116112. doi: 10.1016/j.biopha.2023.116112
38. Song Z, Wang J, Zhang L. Ferroptosis: A new mechanism in diabetic cardiomyopathy. Int J Med Sci. (2024) 21:612–22. doi: 10.7150/ijms.88476
39. Ryabov VV, Maslov LN, Vyshlov EV, Mukhomedzyanov AV, Kilin M, Gusakova SV, et al. Ferroptosis, a regulated form of cell death, as a target for the development of novel drugs preventing ischemia/reperfusion of cardiac injury, cardiomyopathy and stress-induced cardiac injury. Int J Mol Sci. (2024) 25:897. doi: 10.3390/ijms25020897
40. Chen Y, Meng Z, Li Y, Liu S, Hu P, Luo E. Advanced glycation end products and reactive oxygen species: uncovering the potential role of ferroptosis in diabetic complications. Mol Med. (2024) 30:141. doi: 10.1186/s10020-024-00905-9
41. Dai Y, Zhou S, Qiao L, Peng Z, Zhao J, Xu D, et al. Non-apoptotic programmed cell deaths in diabetic pulmonary dysfunction: the new side of advanced glycation end products. Front Endocrinol. (2023) 14:1126661. doi: 10.3389/fendo.2023.1126661
42. Stockwell BR. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell. (2022) 185:2401–21. doi: 10.1016/j.cell.2022.06.003
43. Chen X, Li J, Kang R, Klionsky DJ, Tang D. Ferroptosis: machinery and regulation. Autophagy. (2021) 17:2054–81. doi: 10.1080/15548627.2020.1810918
44. Saedi S, Tan Y, Watson SE, Wintergerst KA, Cai L. Potential pathogenic roles of ferroptosis and cuproptosis in cadmium-induced or exacerbated cardiovascular complications in individuals with diabetes. Front Endocrinol. (2024) 15:1461171. doi: 10.3389/fendo.2024.1461171
45. Tan M, Yin Y, Ma X, Zhang J, Pan W, Tan M, et al. Glutathione system enhancement for cardiac protection: pharmacological options against oxidative stress and ferroptosis. Cell Death Dis. (2023) 14:131. doi: 10.1038/s41419-023-05645-y
46. Frangogiannis NG. Cardiac fibrosis. Cardiovasc Res. (2021) 117:1450–88. doi: 10.1093/cvr/cvaa324
47. Li N, Jiang W, Wang W, Xiong R, Wu X, Geng Q. Ferroptosis and its emerging roles in cardiovascular diseases. Pharmacol Res. (2021) 166:105466. doi: 10.1016/j.phrs.2021.105466
48. Chen G-H, Song C-C, Pantopoulos K, Wei X-L, Zheng H, Luo Z. Mitochondrial oxidative stress mediated Fe-induced ferroptosis via the NRF2-ARE pathway. Free Radical Biol Med. (2022) 180:95–107. doi: 10.1016/j.freeradbiomed.2022.01.012
49. Xu R, Wang W, Zhang W. Ferroptosis and the bidirectional regulatory factor p53. Cell Death Discov. (2023) 9:197. doi: 10.1038/s41420-023-01517-8
50. Miao R, Fang X, Zhang Y, Wei J, Zhang Y, Tian J. Iron metabolism and ferroptosis in type 2 diabetes mellitus and complications: mechanisms and therapeutic opportunities. Cell Death Dis. (2023) 14:186. doi: 10.1038/s41419-023-05708-0
51. Gwon HJ, Cho W, Choi SW, Lim DS, Tanriverdi EÇ, Abd El-Aty A, et al. Donepezil improves skeletal muscle insulin resistance in obese mice via the AMPK/FGF21-mediated suppression of inflammation and ferroptosis. Arch Pharmacal Res. (2024) 47(12):940–53. doi: 10.1007/s12272-024-01518-w
52. Galy B, Conrad M, Muckenthaler M. Mechanisms controlling cellular and systemic iron homeostasis. Nat Rev Mol Cell Biol. (2024) 25:133–55. doi: 10.1038/s41580-023-00648-1
53. Meng H, Yu Y, Xie E, Wu Q, Yin X, Zhao B, et al. Hepatic HDAC3 regulates systemic iron homeostasis and ferroptosis via the hippo signaling pathway. Res (Wash D C). (2023) 6:0281. doi: 10.34133/research.0281
54. Feng H, Schorpp K, Jin J, Yozwiak CE, Hoffstrom BG, Decker AM, et al. Transferrin receptor is a specific ferroptosis marker. Cell Rep. (2020) 30:3411–3423. e7. doi: 10.1016/j.celrep.2020.02.049
55. He J, Li Z, Xia P, Shi A, FuChen X, Zhang J, et al. Ferroptosis and ferritinophagy in diabetes complications. Mol Metab. (2022) 60:101470. doi: 10.1016/j.molmet.2022.101470
56. Chen L, Ma Y, Ma X, Liu L, Jv X, Li A, et al. TFEB regulates cellular labile iron and prevents ferroptosis in a TfR1-dependent manner. Free Radic Biol Med. (2023) 208:445–57. doi: 10.1016/j.freeradbiomed.2023.09.004
57. Li L, Yu XJ, Gao L, Cheng L, Sun B, Wang G. Diabetic ferroptosis and pancreatic cancer: foe or friend? Antioxid Redox Signal. (2022) 37:1206–21. doi: 10.1089/ars.2022.0101
58. Zhao Y, Zhao J, Ma H, Han Y, Xu W, Wang J, et al. High hepcidin levels promote abnormal iron metabolism and ferroptosis in chronic atrophic gastritis. Biomedicines. (2023) 11(9):2338. doi: 10.3390/biomedicines11092338
59. Bao X, Luo X, Bai X, Lv Y, Weng X, Zhang S, et al. Cigarette tar mediates macrophage ferroptosis in atherosclerosis through the hepcidin/FPN/SLC7A11 signaling pathway. Free Radic Biol Med. (2023) 201:76–88. doi: 10.1016/j.freeradbiomed.2023.03.006
60. Tsuji Y, Ninomiya-Tsuji J, Shen MY, DiFrancesco BR. Modulation of iron metabolism by new chemicals interacting with the iron regulatory system. Redox Biol. (2025) 79:103444. doi: 10.1016/j.redox.2024.103444
61. Liu J, Kang R, Tang D. Signaling pathways and defense mechanisms of ferroptosis. FEBS J. (2022) 289:7038–50. doi: 10.1111/febs.v289.22
62. Dong B, Jiang Y, Shi B, Zhang Z, Zhang Z. Selenomethionine alleviates decabromodiphenyl ether-induced oxidative stress and ferroptosis via the NRF2/GPX4 pathway in the chicken brain. J Hazard Mater. (2024) 465:133307. doi: 10.1016/j.jhazmat.2023.133307
63. Qiao O, Zhang L, Han L, Wang X, Li Z, Bao F, et al. Rosmarinic acid plus deferasirox inhibits ferroptosis to alleviate crush syndrome-related AKI via Nrf2/Keap1 pathway. Phytomedicine. (2024) 129:155700. doi: 10.1016/j.phymed.2024.155700
64. Zhao Y, Pan B, Lv X, Chen C, Li K, Wang Y, et al. Ferroptosis: roles and molecular mechanisms in diabetic cardiomyopathy. Front Endocrinol (Lausanne). (2023) 14:1140644. doi: 10.3389/fendo.2023.1140644
65. Wang L, Liu Y, Du T, Yang H, Lei L, Guo M, et al. ATF3 promotes erastin-induced ferroptosis by suppressing system Xc(). Cell Death Differ. (2020) 27:662–75. doi: 10.1038/s41418-019-0380-z
66. Guo M, Zhu Y, Shi Y, Meng X, Dong X, Zhang H, et al. Inhibition of ferroptosis promotes retina ganglion cell survival in experimental optic neuropathies. Redox Biol. (2022) 58:102541. doi: 10.1016/j.redox.2022.102541
67. Wu X, Li Y, Zhang S, Zhou X. Ferroptosis as a novel therapeutic target for cardiovascular disease. Theranostics. (2021) 11:3052. doi: 10.7150/thno.54113
68. Xu S, Wu B, Zhong B, Lin L, Ding Y, Jin X, et al. Naringenin alleviates myocardial ischemia/reperfusion injury by regulating the nuclear factor-erythroid factor 2-related factor 2 (Nrf2)/System xc-/glutathione peroxidase 4 (GPX4) axis to inhibit ferroptosis. Bioengineered. (2021) 12:10924–34. doi: 10.1080/21655979.2021.1995994
69. Yu Y, Yan Y, Niu F, Wang Y, Chen X, Su G, et al. Ferroptosis: a cell death connecting oxidative stress, inflammation and cardiovascular diseases. Cell Death Discov. (2021) 7:193. doi: 10.1038/s41420-021-00579-w
70. Zhou Q, Meng Y, Li D, Yao L, Le J, Liu Y, et al. Ferroptosis in cancer: From molecular mechanisms to therapeutic strategies. Signal Transduct Target Ther. (2024) 9:55. doi: 10.1038/s41392-024-01769-5
71. Bellezza I, Giambanco I, Minelli A, Donato R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim Biophys Acta Mol Cell Res. (2018) 1865:721–33. doi: 10.1016/j.bbamcr.2018.02.010
72. Sun J, Wei N, Yu C, Li C, Li W, Sun X, et al. Natural polysaccharides: The potential biomacromolecules for treating diabetes and its complications via AGEs-RAGE-oxidative stress axis. Int Immunopharmacol. (2024) 143:113426. doi: 10.1016/j.intimp.2024.113426
73. Moldogazieva NT, Mokhosoev IM, Mel’nikova TI, Porozov YB, Terentiev AA. Oxidative stress and advanced lipoxidation and glycation end products (ALEs and AGEs) in aging and age-related diseases. Oxid Med Cell Longev. (2019) 2019:3085756. doi: 10.1155/2019/3085756
74. Ouyang J, Zhou L, Wang Q. Spotlight on iron and ferroptosis: research progress in diabetic retinopathy. Front Endocrinol (Lausanne). (2023) 14:1234824. doi: 10.3389/fendo.2023.1234824
75. Liu Y, Zhang Z, Yang J, Wang J, Wu Y, Zhu R, et al. lncRNA ZFAS1 Positively Facilitates Endothelial Ferroptosis via miR-7-5p/ACSL4 Axis in Diabetic Retinopathy. Oxid Med Cell Longev. (2022) 2022:9004738. doi: 10.1155/2022/9004738
76. He W, Chang L, Li X, Mei Y. Research progress on the mechanism of ferroptosis and its role in diabetic retinopathy. Front Endocrinol (Lausanne). (2023) 14:1155296. doi: 10.3389/fendo.2023.1155296
77. Park MW, Cha HW, Kim J, Kim JH, Yang H, Yoon S, et al. NOX4 promotes ferroptosis of astrocytes by oxidative stress-induced lipid peroxidation via the impairment of mitochondrial metabolism in Alzheimer’s diseases. Redox Biol. (2021) 41:101947. doi: 10.1016/j.redox.2021.101947
78. Lu Y, Sun J, Yang M, Xing Y, Zhu W, Zhu J, et al. Myricetin induces ferroptosis and inhibits gastric cancer progression by targeting NOX4. J Agric Food Chem. (2024) 72:6178–88. doi: 10.1021/acs.jafc.3c05243
79. Li J, Jia Y-c, Ding Y-x, Bai J, Cao F, Li F. The crosstalk between ferroptosis and mitochondrial dynamic regulatory networks. Int J Biol Sci. (2023) 19:2756. doi: 10.7150/ijbs.83348
80. Borén J, Öörni K, Catapano AL. The link between diabetes and cardiovascular disease. Atherosclerosis. (2024) 394:117607. doi: 10.1016/j.atherosclerosis.2024.117607
81. Yan M, Li Y, Luo Q, Zeng W, Shao X, Li L, et al. Mitochondrial damage and activation of the cytosolic DNA sensor cGAS-STING pathway lead to cardiac pyroptosis and hypertrophy in diabetic cardiomyopathy mice. Cell Death Discov. (2022) 8:258. doi: 10.1038/s41420-022-01046-w
82. Masenga SK, Kabwe LS, Chakulya M, Kirabo A. Mechanisms of oxidative stress in metabolic syndrome. Int J Mol Sci. (2023) 24(8026):E8. doi: 10.3390/ijms24097898
83. Silveira Rossi JL, Barbalho SM, Reverete de Araujo R, Bechara MD, Sloan KP, Sloan LA. Metabolic syndrome and cardiovascular diseases: Going beyond traditional risk factors. Diabetes Metab Res Rev. (2022) 38:e3502. doi: 10.1002/dmrr.3502
84. Rodríguez-Nuevo A, Torres-Sanchez A, Duran JM, De Guirior C, Martínez-Zamora MA, Böke E. Oocytes maintain ROS-free mitochondrial metabolism by suppressing complex I. Nature. (2022) 607:756–61. doi: 10.1038/s41586-022-04979-5
85. Nolfi-Donegan D, Braganza A, Shiva S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. (2020) 37:101674. doi: 10.1016/j.redox.2020.101674
86. Okoye CN, Koren SA, Wojtovich AP. Mitochondrial complex I ROS production and redox signaling in hypoxia. Redox Biol. (2023) 67:102926. doi: 10.1016/j.redox.2023.102926
87. Da Dalt L, Cabodevilla AG, Goldberg IJ, Norata GD. Cardiac lipid metabolism, mitochondrial function, and heart failure. Cardiovasc Res. (2023) 119:1905–14. doi: 10.1093/cvr/cvad100
88. Chang X, Li Y, Cai C, Wu F, He J, Zhang Y, et al. Mitochondrial quality control mechanisms as molecular targets in diabetic heart. Metabolism. (2022) 137:155313. doi: 10.1016/j.metabol.2022.155313
89. Steenberge LH, Rogers S, Sung AY, Fan J, Pagliarini DJ. Coenzyme Q(4) is a functional substitute for coenzyme Q(10) and can be targeted to the mitochondria. J Biol Chem. (2024) 300:107269. doi: 10.1016/j.jbc.2024.107269
90. Peng Z, Ding YN, Yang ZM, Li XJ, Zhuang Z, Lu Y, et al. Neuron-targeted liposomal coenzyme Q10 attenuates neuronal ferroptosis after subarachnoid hemorrhage by activating the ferroptosis suppressor protein 1/coenzyme Q10 system. Acta Biomater. (2024) 179:325–39. doi: 10.1016/j.actbio.2024.03.023
91. Guile MD, Jain A, Anderson KA, Clarke CF. New insights on the uptake and trafficking of coenzyme Q. Antioxid (Basel). (2023) 12(7):1391. doi: 10.3390/antiox12071391
92. Li W, Liang L, Liu S, Yi H, Zhou Y. FSP1: a key regulator of ferroptosis. Trends Mol Med. (2023) 29:753–64. doi: 10.1016/j.molmed.2023.05.013
93. Nakamura T, Hipp C, Santos Dias Mourão A, Borggräfe J, Aldrovandi M, Henkelmann B, et al. Phase separation of FSP1 promotes ferroptosis. Nature. (2023) 619:371–7. doi: 10.1038/s41586-023-06255-6
94. Kagan VE, Straub AC, Tyurina YY, Kapralov AA, Hall R, Wenzel SE, et al. Vitamin E/coenzyme Q-dependent “Free radical reductases”: redox regulators in ferroptosis. Antioxid Redox Signal. (2024) 40:317–28. doi: 10.1089/ars.2022.0154
95. Ito J, Omiya S, Rusu M-C, Ueda H, Murakawa T, Tanada Y, et al. Iron derived from autophagy-mediated ferritin degradation induces cardiomyocyte death and heart failure in mice. Elife. (2021) 10:e62174. doi: 10.7554/eLife.62174
96. Chen C, Wang J, Zhang S, Zhu X, Hu J, Liu C, et al. Epigenetic regulation of diverse regulated cell death modalities in cardiovascular disease: Insights into necroptosis, pyroptosis, ferroptosis, and cuproptosis. Redox Biol. (2024) 76:103321. doi: 10.1016/j.redox.2024.103321
97. Gan B. Mitochondrial regulation of ferroptosis. J Cell Biol. (2021) 220:e202105043. doi: 10.1083/jcb.202105043
98. Yan F, Li K, Xing W, Dong M, Yi M, Zhang H. Role of iron-related oxidative stress and mitochondrial dysfunction in cardiovascular diseases. Oxid Med Cell Longev. (2022) 2022:5124553. doi: 10.1155/2022/5124553
99. Cao S, Wei Y, Yue Y, Chen Y, Qian J, Wang D, et al. Rosiglitazone retards the progression of iron overload-induced osteoarthritis by impeding chondrocyte ferroptosis. Iscience. (2024) 27(9):110526. doi: 10.1016/j.isci.2024.110526
100. Zhang M, Liu Q, Meng H, Duan H, Liu X, Wu J, et al. Ischemia-reperfusion injury: molecular mechanisms and therapeutic targets. Signal Transduct Target Ther. (2024) 9:12. doi: 10.1038/s41392-023-01688-x
101. Chen Y, Hua Y, Li X, Arslan IM, Zhang W, Meng G. Distinct types of cell death and the implication in diabetic cardiomyopathy. Front Pharmacol. (2020) 11:42. doi: 10.3389/fphar.2020.00042
102. Yuan Y, Wang Y, Yan Y, Kim E, Bai J, Zhao Y, et al. FBLN1 regulates ferroptosis in acute respiratory distress syndrome by reducing free ferrous iron by inhibiting the TGF-β/Smad pathway. PloS One. (2024) 19:e0314750. doi: 10.1371/journal.pone.0314750
103. Deng L, He S, Guo N, Tian W, Zhang W, Luo L. Molecular mechanisms of ferroptosis and relevance to inflammation. Inflammation Res. (2023) 72:281–99. doi: 10.1007/s00011-022-01672-1
104. Mohr ME, Li S, Trouten AM, Stairley RA, Roddy PL, Liu C, et al. Cardiomyocyte-fibroblast interaction regulates ferroptosis and fibrosis after myocardial injury. iScience. (2024) 27:109219. doi: 10.1016/j.isci.2024.109219
105. Tadokoro T, Ikeda M, Ide T, Deguchi H, Ikeda S, Okabe K, et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight. (2020) 5(9). doi: 10.1172/jci.insight.132747
106. Ma X, Mei S, Wuyun Q, Zhou L, Sun D, Yan J. Epigenetics in diabetic cardiomyopathy. Clin Epigenet. (2024) 16:52. doi: 10.1186/s13148-024-01667-1
107. Huang K, Luo X, Liao B, Li G, Feng J. Insights into SGLT2 inhibitor treatment of diabetic cardiomyopathy: focus on the mechanisms. Cardiovasc Diabetol. (2023) 22:86. doi: 10.1186/s12933-023-01816-5
108. Preda A, Montecucco F, Carbone F, Camici GG, Lüscher TF, Kraler S, et al. SGLT2 inhibitors: from glucose-lowering to cardiovascular benefits. Cardiovasc Res. (2024) 120:443–60. doi: 10.1093/cvr/cvae047
109. Du S, Shi H, Xiong L, Wang P, Shi Y. Canagliflozin mitigates ferroptosis and improves myocardial oxidative stress in mice with diabetic cardiomyopathy. Front Endocrinol (Lausanne). (2022) 13:1011669. doi: 10.3389/fendo.2022.1011669
110. Beghini A, Sammartino AM, Papp Z, von Haehling S, Biegus J, Ponikowski P, et al. 2024 update in heart failure. ESC Heart Fail. (2024) 12(1):8–42. doi: 10.1002/ehf2.14857
111. Gupta P, Ekbbal R. Liraglutide improves diabetic cardiomyopathy by downregulation of cardiac inflammatory and apoptosis markers. Curr Drug Res Rev. (2024) 16:289–99. doi: 10.2174/0125899775243787231103075804
112. Subrahmanyan NA, Koshy RM, Jacob K, Pappachan JM. Efficacy and cardiovascular safety of DPP-4 inhibitors. Curr Drug Saf. (2021) 16:154–64. doi: 10.2174/22123911MTA5pMzA20
113. Sawami K, Tanaka A, Node K. Updated evidence on cardiovascular and renal effects of GLP-1 receptor agonists and combination therapy with SGLT2 inhibitors and finerenone: a narrative review and perspectives. Cardiovasc Diabetol. (2024) 23:410. doi: 10.1186/s12933-024-02500-y
114. Wang R, Ye H, Zhao Y, Wei J, Wang Y, Zhang X, et al. Effect of sacubitril/valsartan and ACEI/ARB on glycaemia and the development of diabetes: a systematic review and meta-analysis of randomised controlled trials. BMC Med. (2022) 20:487. doi: 10.1186/s12916-022-02682-w
115. Abushamat LA, Schauer IE, Low Wang CC, Mitchell S, Herlache L, Bridenstine M, et al. Rosiglitazone improves insulin resistance but does not improve exercise capacity in individuals with impaired glucose tolerance: A randomized clinical study. J Invest Med. (2024) 72:294–304. doi: 10.1177/10815589231225183
116. Zhou Y, Huang Y, Ji X, Wang X, Shen L, Wang Y. Pioglitazone for the primary and secondary prevention of cardiovascular and renal outcomes in patients with or at high risk of type 2 diabetes mellitus: a meta-analysis. J Clin Endocrinol Metab. (2020) 105:1670–81. doi: 10.1210/clinem/dgz252
117. Zou R, Nie C, Pan S, Wang B, Hong X, Xi S, et al. Co-administration of hydrogen and metformin exerts cardioprotective effects by inhibiting pyroptosis and fibrosis in diabetic cardiomyopathy. Free Radical Biol Med. (2022) 183:35–50. doi: 10.1016/j.freeradbiomed.2022.03.010
118. Rubin S, Tomaszewski M. Prediction and prevention of ACE-inhibitor-induced angioedema-an unmet clinical need in management of hypertension. Hypertens Res. (2024) 47:257–60. doi: 10.1038/s41440-023-01491-9
119. Zhao J, Tostivint I, Xu L, Huang J, Gambotti L, Boffa J-J, et al. Efficacy of combined abelmoschus manihot and irbesartan for reduction of albuminuria in patients with type 2 diabetes and diabetic kidney disease: a multicenter randomized double-blind parallel controlled clinical trial. Diabetes Care. (2022) 45:e113–5. doi: 10.2337/dc22-0607
120. Singhal S, Patil VM, Verma S, Masand N. Recent advances and structure-activity relationship studies of DPP-4 inhibitors as anti-diabetic agents. Bioorg Chem. (2024) 146:107277. doi: 10.1016/j.bioorg.2024.107277
121. Xie J, Wang Z, Wang J, Feng W, Shan T, Jing S, et al. Intakes of omega-3 fatty acids and risks of all-cause and cause-specific mortality in people with diabetes: a cohort study based on NHANES 1999–2014. Acta Diabetol. (2023) 60:353–62. doi: 10.1007/s00592-022-02003-w
122. Aguilar D, Davis BR. Aspirin dosing in cardiovascular disease and diabetes: insights from ADAPTABLE. Diabetes Care. (2024) 47:52–3. doi: 10.2337/dci23-0078
123. Singh P, Sharma S, Sharma PK, Alam A. Topical anti-ulcerogenic effect of the beta-adrenergic blockers on diabetic foot ulcers: recent advances and future prospectives. Curr Diabetes Rev. (2024) 20:23–37. doi: 10.2174/0115733998249061231009093006
124. Sun Y, Guo LQ, Wang DG, Xing YJ, Bai YP, Zhang T, et al. Metformin alleviates glucolipotoxicity-induced pancreatic β cell ferroptosis through regulation of the GPX4/ACSL4 axis. Eur J Pharmacol. (2023) 956:175967. doi: 10.1016/j.ejphar.2023.175967
125. Xu L, Li W, Chen Y, Liu S, Liu G, Luo W, et al. Metformin regulates cardiac ferroptosis to reduce metabolic syndrome-induced cardiac dysfunction. Appl Biochem Biotechnol. (2025) 197:179–93. doi: 10.1007/s12010-024-05038-7
126. Wu Z, Bai Y, Qi Y, Chang C, Jiao Y, Bai Y, et al. Metformin ameliorates ferroptosis in cardiac ischemia and reperfusion by reducing NOX4 expression via promoting AMPKα. Pharm Biol. (2023) 61:886–96. doi: 10.1080/13880209.2023.2212700
127. Liao HH, Ding W, Zhang N, Zhou ZY, Ling Z, Li WJ, et al. Activation of AMPKα2 attenuated doxorubicin-induced cardiotoxicity via inhibiting lipid peroxidation associated ferroptosis. Free Radic Biol Med. (2023) 205:275–90. doi: 10.1016/j.freeradbiomed.2023.06.004
128. Mohammadi S, Al-Harrasi A. Macrophage modulation with dipeptidyl peptidase-4 inhibitors: A new frontier for treating diabetic cardiomyopathy? World J Diabetes. (2024) 15:1847–52. doi: 10.4239/wjd.v15.i9.1847
129. Al-Rasheed NM, Al-Rasheed NM, Hasan IH, Al-Amin MA, Al-Ajmi HN, Mahmoud AM. Sitagliptin attenuates cardiomyopathy by modulating the JAK/STAT signaling pathway in experimental diabetic rats. Drug Des Devel Ther. (2016) 10:2095–107. doi: 10.2147/DDDT.S109287
130. Pham TK, Nguyen THT, Yi JM, Kim GS, Yun HR, Kim HK, et al. Evogliptin, a DPP-4 inhibitor, prevents diabetic cardiomyopathy by alleviating cardiac lipotoxicity in db/db mice. Exp Mol Med. (2023) 55:767–78. doi: 10.1038/s12276-023-00958-6
131. Jin L, Geng L, Ying L, Shu L, Ye K, Yang R, et al. FGF21-sirtuin 3 axis confers the protective effects of exercise against diabetic cardiomyopathy by governing mitochondrial integrity. Circulation. (2022) 146:1537–57. doi: 10.1161/CIRCULATIONAHA.122.059631
132. Ding M, Liu C, Shi R, Yu M, Zeng K, Kang J, et al. Mitochondrial fusion promoter restores mitochondrial dynamics balance and ameliorates diabetic cardiomyopathy in an optic atrophy 1-dependent way. Acta Physiol. (2020) 229:e13428. doi: 10.1111/apha.v229.1
133. Tadic M, Sala C, Saeed S, Grassi G, Mancia G, Rottbauer W, et al. New antidiabetic therapy and HFpEF: light at the end of tunnel? Heart Failure Rev. (2021) 27:1137–46. doi: 10.1007/s10741-021-10106-9
134. Sciacqua A, Succurro E, Armentaro G, Miceli S, Pastori D, Rengo G, et al. Pharmacological treatment of type 2 diabetes in elderly patients with heart failure: randomized trials and beyond. Heart Fail Rev. (2021) 28(3):15. doi: 10.1007/s10741-021-10182-x
135. Damgaard MV, Treebak JT. What is really known about the effects of nicotinamide riboside supplementation in humans. Sci Adv. (2023) 9:eadi4862. doi: 10.1126/sciadv.adi4862
136. Mushtaq I, Bashir Z, Sarwar M, Arshad M, Ishtiaq A, Khan W, et al. N-acetyl cysteine, selenium, and ascorbic acid rescue diabetic cardiac hypertrophy via mitochondrial-associated redox regulators. Molecules. (2021) 26:7285. doi: 10.3390/molecules26237285
137. Yang J, Zhang Y, Na X, Zhao A. β-Carotene supplementation and risk of cardiovascular disease: a systematic review and meta-analysis of randomized controlled trials. Nutrients. (2022) 14:1284. doi: 10.3390/nu14061284
138. Jiang C, Li D, Chen L, Liu Y, Zhao Y, Mei G, et al. Quercetin ameliorated cardiac injury via reducing inflammatory actions and the glycerophospholipid metabolism dysregulation in a diabetic cardiomyopathy mouse model. Food Funct. (2022) 13:7847–56. doi: 10.1039/D2FO00912A
139. Chudzińska M, Rogowicz D, Wołowiec Ł, Banach J, Sielski S, Bujak R, et al. Resveratrol and cardiovascular system-the unfulfilled hopes. Ir J Med Sci. (2021) 190:981–6. doi: 10.1007/s11845-020-02441-x
140. Jia M, Tan X, Yuan Z, Zhu W, Yan P. Nanoliposomes encapsulated rapamycin/resveratrol to induce apoptosis and ferroptosis for enhanced colorectal cancer therapy. J Pharm Sci. (2024) 113(8):2565–74. doi: 10.1016/j.xphs.2024.05.015
141. Perris A, Bhattacharya S, Jawed JJ, Hoda M. Oncotherapeutic application of resveratrol-based inorganic nanoparticles. Pharm Nanotechnol. (2021) 9:271–80. doi: 10.2174/2211738509666210906164727
142. Mas-Capdevila A, Teichenne J, Domenech-Coca C, Caimari A, Del Bas JM, Escoté X, et al. Effect of hesperidin on cardiovascular disease risk factors: the role of intestinal microbiota on hesperidin bioavailability. Nutrients. (2020) 12:1488. doi: 10.3390/nu12051488
143. Kabil MF, Nasr M. Deferasirox: A comprehensive drug profile. Profiles Drug Subst Excip Relat Method. (2024) 49:1–18.
144. Qiu Y, Cao Y, Cao W, Jia Y, Lu N. The application of ferroptosis in diseases. Pharmacol Res. (2020) 159:104919. doi: 10.1016/j.phrs.2020.104919
145. Yan H-f, Zou T, Tuo Q-z, Xu S, Li H, Belaidi AA, et al. Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther. (2021) 6:49. doi: 10.1038/s41392-020-00428-9
146. Nemeth E, Ganz T. Hepcidin and iron in health and disease. Annu Rev Med. (2023) 74:261–77. doi: 10.1146/annurev-med-043021-032816
147. Chen Y, Shi S, Dai Y. Research progress of therapeutic drugs for doxorubicin-induced cardiomyopathy. Biomed Pharmacother. (2022) 156:113903.
148. Chen W, Yuan X, Li Z, Lu Z, Kong S, Jiang H, et al. CN128: A new orally active hydroxypyridinone iron chelator. J Med Chem. (2020) 63:4215–26. doi: 10.1021/acs.jmedchem.0c00137
149. Kontoghiorghes GJ. The importance and essentiality of natural and synthetic chelators in medicine: increased prospects for the effective treatment of iron overload and iron deficiency. Int J Mol Sci. (2024) 25(9):4654. doi: 10.3390/ijms25094654
150. Shen T, Huang S. Repositioning the old fungicide ciclopirox for new medical uses. Curr Pharm Des. (2016) 22:4443–50.
151. Berndt C, Alborzinia H, Amen VS, Ayton S, Barayeu U, Bartelt A, et al. Ferroptosis in health and disease. Redox Biol. (2024) 75:103211. doi: 10.1016/j.redox.2024.103211
152. Scarpellini C, Klejborowska G, Lanthier C, Hassannia B, Vanden Berghe T, Augustyns K. Beyond ferrostatin-1: a comprehensive review of ferroptosis inhibitors. Trends Pharmacol Sci. (2023) 44:902–16. doi: 10.1016/j.tips.2023.08.012
153. He X, Xiong Y, Liu Y, Li Y, Zhou H, Wu K. Ferrostatin-1 inhibits ferroptosis of vascular smooth muscle cells and alleviates abdominal aortic aneurysm formation through activating the SLC7A11/GPX4 axis. FASEB J. (2024) 38:e23401. doi: 10.1096/fj.202300198RRR
154. Du B, Deng Z, Chen K, Yang Z, Wei J, Zhou L, et al. Iron promotes both ferroptosis and necroptosis in the early stage of reperfusion in ischemic stroke. Genes Dis. (2024) 11:101262. doi: 10.1016/j.gendis.2024.101262
155. Zhang W, Liu Y, Liao Y, Zhu C, Zou Z. GPX4, ferroptosis, and diseases. Biomed Pharmacother. (2024) 174:116512. doi: 10.1016/j.biopha.2024.116512
156. Liu C, Sun W, Zhu T, Shi S, Zhang J, Wang J, et al. Glia maturation factor-β induces ferroptosis by impairing chaperone-mediated autophagic degradation of ACSL4 in early diabetic retinopathy. Redox Biol. (2022) 52:102292. doi: 10.1016/j.redox.2022.102292
157. Wang L, Qiu S, Li X, Zhang Y, Huo M, Shi J. Myocardial-targeting tannic cerium nanocatalyst attenuates ischemia/reperfusion injury. Angew Chem Int Ed Engl. (2023) 62:e202305576. doi: 10.1002/anie.202305576
158. Moghaddam SYZ, Biazar E, Esmaeili J, Kheilnezhad B, Goleij F, Heidari S. Tannic acid as a green cross-linker for biomaterial applications. Mini Rev Med Chem. (2023) 23:1320–40. doi: 10.2174/1389557522666220622112959
159. Gao Y, Wang B, Qin G, Liang S, Yin J, Jiang H, et al. Therapeutic potentials of allicin in cardiovascular disease: advances and future directions. Chin Med. (2024) 19:93. doi: 10.1186/s13020-024-00936-8
160. Sánchez-Gloria JL, Arellano-Buendía AS, Juárez-Rojas JG, García-Arroyo FE, Argüello-García R, Sánchez-Muñoz F, et al. Cellular mechanisms underlying the cardioprotective role of allicin on cardiovascular diseases. Int J Mol Sci. (2022) 23:9082. doi: 10.3390/ijms23169082
161. Aribi N, Denis B, Kilani-Morakchi S, Joly D. Azadirachtin, a natural pesticide with multiple effects. Med Sci: M/S. (2020) 36:44–9. doi: 10.1051/medsci/2019268
162. Liang NN, Guo YY, Zhang XY, Ren YH, He YZ, Liu ZB, et al. Mitochondrial dysfunction-evoked DHODH acetylation is involved in renal cell ferroptosis during cisplatin-induced acute kidney injury. Adv Sci (Weinh). (2024) 11:e2404753. doi: 10.1002/advs.202404753
163. Broome SC, Whitfield J, Karagounis LG, Hawley JA. Mitochondria as nutritional targets to maintain muscle health and physical function during ageing. Sports Med. (2024) 54:2291–309. doi: 10.1007/s40279-024-02072-7
164. Sun Q, Liu D, Cui W, Cheng H, Huang L, Zhang R. Cholesterol mediated ferroptosis suppression reveals essential roles of Coenzyme Q and squalene. Commun Biol. (2023) 6:1108. doi: 10.1038/s42003-023-05477-8
165. Li S, Ren Q-J, Xie C-H, Cui Y, Xu L-T, Wang Y-D, et al. Taurine attenuates activation of hepatic stellate cells by inhibiting autophagy and inducing ferroptosis. World J Gastroenterol. (2024) 30:2143. doi: 10.3748/wjg.v30.i15.2143
166. Alanazi AZ, Alqinyah M, Alhamed AS, Mohammed H, Raish M, Aljerian K, et al. Cardioprotective effects of liposomal resveratrol in diabetic rats: unveiling antioxidant and anti-inflammatory benefits. Redox Rep. (2024) 29:2416835. doi: 10.1080/13510002.2024.2416835
167. Ma L, Chen C, Zhao C, Li T, Ma L, Jiang J, et al. Targeting carnitine palmitoyl transferase 1A (CPT1A) induces ferroptosis and synergizes with immunotherapy in lung cancer. Signal Transduct Target Ther. (2024) 9:64. doi: 10.1038/s41392-024-01772-w
168. Guo Q, Ma M, Yu H, Han Y, Zhang D. Dexmedetomidine enables copper homeostasis in cerebral ischemia/reperfusion via ferredoxin 1. Ann Med. (2023) 55:2209735. doi: 10.1080/07853890.2023.2209735
169. Huang J, Zhao Y, Luo X, Luo Y, Ji J, Li J, et al. Dexmedetomidine inhibits ferroptosis and attenuates sepsis-induced acute kidney injury via activating the Nrf2/SLC7A11/FSP1/CoQ10 pathway. Redox Rep. (2024) 29:2430929. doi: 10.1080/13510002.2024.2430929
170. Lu JS, Wang JH, Han K, Li N. Nicorandil regulates ferroptosis and mitigates septic cardiomyopathy via TLR4/SLC7A11 signaling pathway. Inflammation. (2024) 47:975–88. doi: 10.1007/s10753-023-01954-8
171. Kar S, Shahshahan H, Mishra PK. Hydrogen sulfide protects the heart against ferroptotic cell death in diabetic cardiomyopathy. Circ Res. (2020) 127:A501–1. doi: 10.1161/res.127.suppl_1.501
172. Peng S, Wang M, Zhang S, Liu N, Li Q, Kang J, et al. Hydrogen sulfide regulates SERCA2a SUMOylation by S-Sulfhydration of SENP1 to ameliorate cardiac systole-diastole function in diabetic cardiomyopathy. Biomed Pharmacother. (2023) 160:114200. doi: 10.1016/j.biopha.2022.114200
173. Peng S, Zhao D, Li Q, Wang M, Zhang S, Pang K, et al. Hydrogen sulfide regulates SERCA2a ubiquitylation via muscle RING finger-1 S-sulfhydration to affect cardiac contractility in db/db mice. Cells. (2022) 11:3465. doi: 10.3390/cells11213465
174. Wu S, Zhu J, Wu G, Hu Z, Ying P, Bao Z, et al. 6-gingerol alleviates ferroptosis and inflammation of diabetic cardiomyopathy via the nrf2/HO-1 pathway. Oxid Med Cell Longev. (2022) 2022:3027514. doi: 10.1155/2022/3027514
175. Wu P-Y, Lai S-Y, Su Y-T, Yang K-C, Chau Y-P, Don M-J, et al. β-Lapachone, an NQO1 activator, alleviates diabetic cardiomyopathy by regulating antioxidant ability and mitochondrial function. Phytomedicine. (2022) 104:154255. doi: 10.1016/j.phymed.2022.154255
176. Zhang L, Luo YL, Xiang Y, Bai XY, Qiang RR, Zhang X, et al. Ferroptosis inhibitors: past, present and future. Front Pharmacol. (2024) 15:1407335. doi: 10.3389/fphar.2024.1407335
177. Zilka O, Shah R, Li B, Friedmann Angeli JP, Griesser M, Conrad M, et al. On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent Sci. (2017) 3:232–43. doi: 10.1021/acscentsci.7b00028
178. Li S, Lu S, Wang L, Liu S, Zhang L, Du J, et al. Effects of amygdalin on ferroptosis and oxidative stress in diabetic retinopathy progression via the NRF2/ARE signaling pathway. Exp Eye Res. (2023) 234:109569. doi: 10.1016/j.exer.2023.109569
179. Kolijn D, Pabel S, Tian Y, Lódi M, Herwig M, Carrizzo A, et al. Empagliflozin improves endothelial and cardiomyocyte function in human heart failure with preserved ejection fraction via reduced pro-inflammatory-oxidative pathways and protein kinase Gα oxidation. Cardiovasc Res. (2021) 117:495–507. doi: 10.1093/cvr/cvaa123
180. Wang X, Chen X, Zhou W, Men H, Bao T, Sun Y, et al. Ferroptosis is essential for diabetic cardiomyopathy and is prevented by sulforaphane via AMPK/NRF2 pathways. Acta Pharm Sin B. (2022) 12:708–22. doi: 10.1016/j.apsb.2021.10.005
181. Sun Y, Pham AN, Hider RC, Zheng H, Waite TD. Effectiveness of the iron chelator CN128 in mitigating the formation of dopamine oxidation products associated with the progression of parkinson’s disease. ACS Chem Neurosci. (2020) 11:3646–57. doi: 10.1021/acschemneuro.0c00557
182. Liu Y, Zhang Q, Wang L, Wen Y, Jia X. Resveratrol inhibited nanoparticles stromal interaction molecule 2 in regulating miR-20b-5p signaling pathway to improve mitochondrial function during myocardial ischemia-reperfusion injury. J Biomed Nanotechnol. (2023) 19:2099–107. doi: 10.1166/jbn.2023.3720
183. Fang W, Xie S, Deng W. Ferroptosis mechanisms and regulations in cardiovascular diseases in the past, present, and future. Cell Biol Toxicol. (2024) 40:17. doi: 10.1007/s10565-024-09853-w
184. Zhang X-D, Liu Z-Y, Wang M-S, Guo Y-X, Wang X-K, Luo K, et al. Mechanisms and regulations of ferroptosis. Front Immunol. (2023) 14:1269451. doi: 10.3389/fimmu.2023.1269451
185. Wang Y, Hu J, Wu S, Fleishman JS, Li Y, Xu Y, et al. Targeting epigenetic and posttranslational modifications regulating ferroptosis for the treatment of diseases. Signal Transduct Target Ther. (2023) 8:449. doi: 10.1038/s41392-023-01720-0
186. Huang M, Teng Q, Cao F, Huang J, Pang J. Ferroptosis and ferroptosis-inducing nanomedicine as a promising weapon in combination therapy of prostate cancer. Biomater Sci. (2024) 12:1617–29. doi: 10.1039/D3BM01894F
187. Feng Y, Li X, Yang B, Li M, Du Y, Wang J, et al. The role of ferroptosis in radiotherapy and combination therapy for head and neck squamous cell carcinoma. Oncol Rep. (2024) 51:1–10. doi: 10.3892/or.2024.8738
188. Stockwell BR, Jiang X, Gu W. Emerging mechanisms and disease relevance of ferroptosis. Trends Cell Biol. (2020) 30:478–90. doi: 10.1016/j.tcb.2020.02.009
189. Qi X, Zhao Y, Qi Z, Hou S, Chen J. Machine learning empowering drug discovery: applications, opportunities and challenges. Molecules. (2024) 29:903. doi: 10.3390/molecules29040903
190. Cook DA, Stephenson CR, Wilkinson JM, Maloney S, Foo J. Cost-effectiveness and economic benefit of continuous professional development for drug prescribing: a systematic review. JAMA Netw Open. (2022) 5:e2144973–e2144973. doi: 10.1001/jamanetworkopen.2021.44973
191. Muteeb G, Rehman MT, Shahwan M, Aatif M. Origin of antibiotics and antibiotic resistance, and their impacts on drug development: A narrative review. Pharmaceuticals. (2023) 16:1615. doi: 10.3390/ph16111615
192. Kumfu S, Chattipakorn SC, Chattipakorn N. Iron overload cardiomyopathy: using the latest evidence to inform future applications. Exp Biol Med. (2022) 247:574–83. doi: 10.1177/15353702221076397
193. Pérez A. GLP-1 receptor agonists in clinical practice. Med Clin (Barc). (2024) 163:242–4. doi: 10.1016/j.medcli.2024.02.018
194. Feingold KR. Oral and injectable (non-insulin) pharmacological agents for the treatment of type 2 diabetes. Endotext. (2024). Available online at: https://MDText.com, Inc.
195. Abdulrahman SJ, Abdulhadi MA, Turki Jalil A, Falah D, Merza MS, Almulla AF, et al. Conjugated linoleic acid and glucosamine supplements may prevent bone loss in aging by regulating the RANKL/RANK/OPG pathway. Mol Biol Rep. (2023) 50:10579–88. doi: 10.1007/s11033-023-08839-x
196. Sun H, Chen D, Xin W, Ren L, Li Q, Han X. Targeting ferroptosis as a promising therapeutic strategy to treat cardiomyopathy. Front Pharmacol. (2023) 14:1146651. doi: 10.3389/fphar.2023.1146651
197. Borcherding N, Brestoff JR. The power and potential of mitochondria transfer. Nature. (2023) 623:283–91. doi: 10.1038/s41586-023-06537-z
198. Suomalainen A, Nunnari J. Mitochondria at the crossroads of health and disease. Cell. (2024) 187:2601–27. doi: 10.1016/j.cell.2024.04.037
199. Yao Y, Zhang T, Tang M. Toxicity mechanism of engineered nanomaterials: focus on mitochondria. Environ Pollut. (2024) 343:123231. doi: 10.1016/j.envpol.2023.123231
200. Boulet J, Sridhar VS, Bouabdallaoui N, Tardif J-C, White M. Inflammation in heart failure: pathophysiology and therapeutic strategies. Inflammation Res. (2024) 73:709–23. doi: 10.1007/s00011-023-01845-6
201. Lei G, Mao C, Yan Y, Zhuang L, Gan B. Ferroptosis, radiotherapy, and combination therapeutic strategies. Protein Cell. (2021) 12:836–57. doi: 10.1007/s13238-021-00841-y
202. Zhang X, Li X, Xia R, Zhang H-S. Ferroptosis resistance in cancer: recent advances and future perspectives. Biochem Pharmacol. (2024) 219:115933. doi: 10.1016/j.bcp.2023.115933
203. Baldwin JG, Heuser-Loy C, Saha T, Schelker RC, Slavkovic-Lukic D, Strieder N, et al. Intercellular nanotube-mediated mitochondrial transfer enhances T cell metabolic fitness and antitumor efficacy. Cell. (2024) 187:6614–6630.e21. doi: 10.1016/j.cell.2024.08.029
204. Paratz ED, Mundisugih J, Rowe SJ, Kizana E, Semsarian C. Gene therapy in cardiology: is a cure for hypertrophic cardiomyopathy on the horizon? Can J Cardiol. (2024) 40:777–88. doi: 10.1016/j.cjca.2023.11.024
205. Dhar A, Venkadakrishnan J, Roy U, Vedam S, Lalwani N, Ramos KS, et al. A comprehensive review of the novel therapeutic targets for the treatment of diabetic cardiomyopathy. Ther Adv Cardiovasc Dis. (2023) 17:17539447231210170. doi: 10.1177/17539447231210170
206. Mullowney MW, Duncan KR, Elsayed SS, Garg N, van der Hooft JJJ, Martin NI, et al. Artificial intelligence for natural product drug discovery. Nat Rev Drug Discov. (2023) 22:895–916. doi: 10.1038/s41573-023-00774-7
207. Packer M. Potential interactions when prescribing SGLT2 inhibitors and intravenous iron in combination in heart failure. JACC Heart Fail. (2023) 11:106–14. doi: 10.1016/j.jchf.2022.10.004
208. Han JX, Luo LL, Wang YC, Miyagishi M, Kasim V, Wu SR. SGLT2 inhibitor empagliflozin promotes revascularization in diabetic mouse hindlimb ischemia by inhibiting ferroptosis. Acta Pharmacol Sin. (2023) 44:1161–74. doi: 10.1038/s41401-022-01031-0
209. Shen R, Qin S, Lv Y, Liu D, Ke Q, Shi C, et al. GLP-1 receptor agonist attenuates tubular cell ferroptosis in diabetes via enhancing AMPK-fatty acid metabolism pathway through macropinocytosis. Biochim Biophys Acta Mol Basis Dis. (2024) 1870:167060. doi: 10.1016/j.bbadis.2024.167060
Keywords: diabetic cardiomyopathy, ferroptosis, mitochondrial damage, molecular mechanisms, targeted therapy, combination therapy, clinical applications
Citation: Liu B, Jin Q, Sun YK, Yang ZM, Meng P, Zhang X, Chen Q, Gan P, Zhao T, He JJ, He GP and Xue Q (2025) From bench to bedside: targeting ferroptosis and mitochondrial damage in the treatment of diabetic cardiomyopathy. Front. Endocrinol. 16:1563362. doi: 10.3389/fendo.2025.1563362
Received: 19 January 2025; Accepted: 31 March 2025;
Published: 25 April 2025.
Edited by:
Mirjana Macvanin, University of Belgrade, SerbiaReviewed by:
Kulvinder Kochar Kaur, Kulvinder Kaur Centre For Human Reproduction, IndiaArchita Venugopal Menon, Beam Therapeutics, United States
Copyright © 2025 Liu, Jin, Sun, Yang, Meng, Zhang, Chen, Gan, Zhao, He, He and Xue. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bin Liu, a21oeGlucWluZ0BzaW5hLmNvbQ==; Qiang Xue, eHVlcWlhbmczNTEzQDEyNi5jb20=