Abstract
Glucose-dependent insulinotropic polypeptide (GIP) is an intestinal hormone that potentiates glucose-induced insulin secretion in the postprandial state. GIP exerts a broad range of other physiological actions e.g. in the pancreas, bone tissue, and vasculature. In more than 20 years, GIP receptor antagonism has contributed to the discoveries of the role of GIP within both human and animal physiology. In 1986, a fragment of the biological active bovine GIP(1-42), was discovered and characterized as the first GIP receptor antagonist. Several different molecules have been identified, including peptides, vaccines against GIP, GIP antibodies, and antibodies against the GIP receptor. Today, GIP receptor antagonists are not only used as scientific tools but due to significant metabolic effects, they also have a therapeutic purpose. The beneficial clinical effects of GIP receptor antagonism are supported by comparable phenotypic traits of individuals with loss-of-function genetic receptor variants. Novel insights into GIP receptor targeting treatment reveal that both GIP receptor antagonists and agonists, when combined with glucagon like peptide 1 (GLP-1) receptor activation, are associated with improved glycemic control and weight loss. This paradoxical scenario highlights the complexity of GIP receptor pharmacology. Moreover, the long-term effects of therapeutic GIP receptor antagonism in humans are not fully elucidated and are thought to depend on the specific drug molecule, receptor functions, and the extent of GLP-1 receptor activation. With this review, we provide an overview of the preclinical and clinical evidence of GIP receptor antagonism from the central early findings to the current therapeutics in clinical development. Finally, the current therapeutic developments and the further therapeutic potential within GIP receptor antagonism are discussed.
1 Introduction
1.1 Physiological actions of GIP
Glucose-dependent insulinotropic polypeptide (GIP) is a 42 amino acid-peptide and gut-derived hormone. GIP is produced by enteroendocrine K cells (1) found in the proximal part of the small intestine but also located throughout the gastrointestinal tract to a lower extent (2). In the fasting state, the plasma levels of GIP are relatively low (3), but measurable concentrations of GIP are present throughout the entire day and night (4). Food consumption leads to a rapid rise in GIP secretion, and several nutrients have been confirmed as GIP secretagogues: glucose, amino acids, short- and long-chain fatty acids, and bile acids (5–8).
The biologically active GIP(1-42) is short-lived (half-life of 7 minutes) (9). In the circulation and in the liver and kidneys, GIP(1-42) is inactivated by the enzyme dipeptidyl peptidase 4 (DPP-4) through cleavage of the two N-terminal amino acids (Figure 1). The degradation product is the inactive GIP(3-42), which constitutes the majority of the circulating GIP variants (10). Another variant of GIP is the C-terminally truncated form, GIP(1-30)NH2. GIP(1-30)NH2 is derived from the cleavage of the precursor protein pro-GIP by prohormone convertase 2 (11), and circulates in low picomolar concentrations (12). GIP(1-30)NH2 activates the GIP receptor with equivalent potency and efficacy as GIP(1-42) and elicits the same physiological effects on glucose- and bone metabolism in humans (13). Due to the identical amino acid sequence in the first 30 amino acids, GIP(1-30)NH2 also acts as a substrate for DPP-4 (Figure 1). The cleavage of GIP(1-30)NH2 subsequently results in GIP(3-30)NH2, a fragment that binds to the human GIP receptor with high affinity but does not have any intrinsic activation of the human GIP receptor in physiological concentrations (14).
Figure 1

Peptide sequence and bioactive regions of GIP. The N-terminus is essential for GIP receptor activation, whereas the mid-region is crucial for binding to the receptor. Therefore, GIP receptor agonist peptides based on the natural sequence includes the N-terminus. If removed or modified by amino acid substitutions e.g. at position 3, the peptide can be a partial agonist. If the N-terminus is removed and the mid-region retained, the peptide has antagonistic properties. DPP-4, di-peptidyl-peptidase 4; GIP, glucose-dependent insulinotropic polypeptide.
The human GIP receptor can be found in multiple organs and tissues, reflecting its various physiological functions (15). GIP is an incretin hormone that, along with glucagon-like peptide 1 (GLP-1), postprandially potentiates glucose-stimulated insulin secretion from pancreatic beta cells (16). Additionally, GIP is suggested to regulate glucose homeostasis by stimulation of pancreatic alpha cells to secrete glucagon during euglycemia and hypoglycemia (17). A key physiological difference between the two incretin hormones, GIP and GLP-1, is therefore, that GIP is able to stimulate glucagon secretion, whereas GLP-1 suppresses glucagon secretion (18).
Beyond the pancreatic islet cells, the GIP receptor is expressed within the vascular system, including the heart and various endothelial cells. Here, GIP receptor activation may contribute to the increase in heart rate and reduction in arterial blood pressure observed after GIP administration (19, 20). Additionally, GIP is suggested to increase blood circulation in adipose tissue (21, 22) and the gastrointestinal tract (23, 24).
In bone, exogenous and endogenous GIP is found to decrease bone resorption by reducing osteoclast activity and improving osteoblast survival (25–27). In addition, GIP administration increases markers of bone formation (20, 28, 29). Hereby, GIP appears to uncouple the otherwise parallel process of bone remodeling, ultimately resulting in a beneficial impact of GIP on bone metabolism (20, 26).
Finally, infusion of GIP increases the deposition of triacylglycerol in adipose tissue (22). This is thought to be due to the GIP-induced increase in blood flow to subcutaneous adipose tissue and enhanced triglyceride clearance (22). However, although primarily assessed in animal models, the direct effects of GIP in adipocytes and adipose tissue fibroblasts are also possible (30–32). GIP receptor expression has been detected in both human and mouse adipocytes (33, 34), and GIP receptor activation in adipose tissue could protect mice from obesity and induce weight loss (32).
1.2 GIP receptor signaling pathways
The actions of GIP are mediated by the GIP receptor, a G protein-coupled receptor (GPCR) in the class B1 receptor family (35). Activation of the GIP receptor involves ligand binding, leading to conformational changes in the receptor that subsequently initiates intracellular signaling cascades (36). Upon ligand binding that leads to GIP receptor activation, the receptor primarily couples to the Gαs subfamily. When the G protein is coupled to the GIP receptor adenylyl cyclase is activated to increase the intracellular concentrations of the secondary messenger, cyclic adenosine monophosphate (cAMP) (Figure 2A). An increased concentration of cAMP leads to the activation of protein kinase A (PKA), which activates tissue-specific downstream signaling pathways (37). Adding more complexity to the interpretation of GIP receptor pharmacology, the conformational shift in the receptor also facilitates the recruitment of beta arrestin. The beta arrestin recruitment is necessary for GIP receptor function (30, 38) as it desensitizes the receptor by hindering binding to its corresponding G protein and hereby hinders the intracellular signaling cascade. Moreover, beta arrestin recruitment is essential for GIP receptor internalization where the GIP receptors are removed from the cell surface (30). Subsequently, the receptor is taken up by endosomes, where it can either continue its signaling intracellularly, be recycled to the cell surface (resensitization) or undergo lysosomal degradation. This indicates that receptor signaling is tightly regulated by beta arrestins (39). In addition, with sustained stimulation, there will be fewer receptors available on the cell surface due to receptor desensitization and internalization. Hence, with fewer receptors available for targeting chronic stimulation with agonists, is proposed to act as functional antagonism of the GIP receptor (40–42) (Figure 2C).
Figure 2

GIP receptor signaling. (A) The GIP receptor is activated upon binding of GIP, which leads to the coupling of Gαs, and subsequent activation of adenylyl cyclase, that in return increases intracellular cAMP concentrations and initiate further downstream signaling pathways. The recruitment of beta arrestin inhibits Gαs coupling and promotes receptor internalization, leading to intracellular signaling from endosomes, receptor recycling or degradation; (B) A GIP receptor antagonist will block these pathways; (C) Long-term/chronic GIP receptor signaling could enhance receptor internalization and desensitization, leading to a reduced number of receptors available on the cell surface thereby introducing functional antagonism as a response to GIP agonist administration. GIP, glucose-dependent insulinotropic polypeptide; AC, adenylyl cyclase; cAMP, cyclic adenosine monophosphate.
1.3 GIP receptor antagonism
The physiology of GIP has been explored extensively by exogenous GIP infusions resulting in both physiological and supraphysiological concentrations (43–47). However, these findings may not fully reflect the role of endogenous GIP. To understand the physiological effects of endogenous GIP receptor activation, the development of GIP receptor antagonists has been essential. The first GIP receptor antagonist was introduced in 1986 (Figure 3), and in recent years, the truncated and deactivated form of GIP, GIP(3-30)NH2, has proven to be a potent and specific GIP receptor antagonist in humans in high concentrations, and suitable for studies of endogenous GIP physiology and pathophysiology (14) (Table 1). In parallel with the human studies, preclinical and in vitro pharmacological work has played a crucial role in understanding of the physiological and pathophysiological role of endogenous GIP and the potential of GIP receptor antagonism as a therapeutic concept. Various methods to inhibit the GIP system in rodents result in reduced body weight and improve insulin sensitivity (52). Combined with the theory of GIP being an obesogenic hormone (53), these prior findings have highlighted the potential of GIP receptor antagonism, and today the development of GIP receptor antagonists has also emerged as an attractive therapeutic approach for addressing obesity and cardiovascular diseases (54, 55). In the following, we review the preclinical and clinical evidence of GIP receptor antagonism.
Figure 3

Timeline of major discoveries of GIP receptor antagonists. GIP, glucose-dependent insulinotropic polypeptide; VLP, virus-like peptide.
Table 1
| GIP receptor antagonists | Inhibition of GIP(1-42)-induced receptor activation Potency/IC50 (nmol/l) | GIP receptor binding Affinity/Kd or Ki (nmol/l) |
|---|---|---|
| GIP(3-42) (14, 48) | 138 | 22 |
| SKL-14959 (49) | 2,900 | 55 |
| GIP(3-30)NH2 (14) | 2.3 | 3.4 |
| GIP(5-30)NH2 (14) | 5.9 | 5.9 |
| AMG133 (50) | 136 | Not Available |
| AT-7687 (51) | 9.5 | 2.0 |
In vitro results for the most potent and widely used GIP receptor antagonists.
The results were not performed in parallel and originate from various in vitro assays. GIP, glucose-dependent insulinotropic polypeptide; IC50, half-maximal inhibitory concentration; Kd, dissociation constant; Ki, inhibition constant.
2 Development of GIP receptor antagonists
GIP was identified in the 1970s (56) and first described as an enterogastrone that inhibited gastric acid secretion in dogs, and, thus, initially named gastric inhibitory peptide (57). A few years later, GIP was characterized as an insulinotropic gut-derived peptide hormone, and the name was changed to glucose-dependent insulinotropic polypeptide (56). In 1979, intravenous infusions of a GIP antiserum in rats were able to inhibit the action of GIP on glucose-induced insulin release, hereby, showing the first inhibition of GIP activity (58) (Figure 3).
In 1986, the biological activity of bovine GIP(1-42) and several of its fragments was characterized according to their ability to promote insulin secretion from an isolated perfused rat pancreas. The fragment bovine GIP(4-42) was a partial agonist of the rat GIP receptor, concluding that N-terminal truncation of GIP alters its pharmacological properties from full agonist to partial agonist (59). This called for further studies investigating modification of the GIP C-terminus on interaction with the GIP receptor.
Ten years later, in 1996, various peptide fragments of GIP were studied to investigate the potential for identifying a GIP receptor-specific antagonist. GIP(7-30)NH2 was found to be a GIP-specific receptor antagonist that inhibits GIP-stimulated cAMP production and insulin release in a concentration-dependent manner. Due to the compelling findings of antagonistic properties, GIP(7-30)NH2 was named ANTGIP (60). In 1999, infusions of GIP(7-30)NH2 in rats, reduced insulin release after an intragastric glucose meal. This demonstrated the antagonistic effect while also providing evidence for the crucial role of GIP receptor signaling in potentiating the insulin response to oral glucose (61).
Alongside studies of GIP(7-30)NH2, other truncated forms: GIP(10-30), GIP(6-30)NH2 and GIP(7-30) were characterised as GIP receptor antagonists with the ability to block GIP’s activation of the GIP receptor in vitro. Of the three, GIP(6-30)NH2 was the most potent antagonist followed by GIP(7-30) and finally GIP(10-30), ranked by their potencies as inhibitors of GIP-stimulated cAMP production (62).
In 2001, several synthetic peptide GIP fragments were tested to identify the molecule’s bioactive domain. Here, the GIP fragments GIP(15-42), GIP(15-30), GIP(16-30) and GIP(17-30) were characterized as weak antagonists at the human GIP receptor in vitro, and the amino acids number 6-30 are required for effective GIP receptor antagonism as a high-affinity binding domain (63) (Figure 1). The following year, the important role of the N-terminus in GIP’s full biological activity was again demonstrated. In vitro, GIP(17-30) and GIP(4-42) did not increase cellular cAMP production and had only weak insulin releasing activity compared with the native GIP (64).
Simultaneously, the most abundant circulating GIP fragment GIP(3-42) was identified as a GIP receptor antagonist with the ability to moderate GIP-induced cAMP production and insulin secretion in vitro (65) (Table 1). However, in 2006 it became clear that GIP(3-42) does not function as a physiological antagonist in vivo as its maximal circulating levels are insufficient to elicit antagonistic effects (48).
At the same time, in 2002, Gipr knockout mice were found to be protected against both obesity and insulin resistance induced by high-fat feeding (53). These findings led to a suggested role for GIP in the development of obesity, highlighting the therapeutic opportunity of GIP receptor antagonism, and subsequently leading to great interest and research activity within the field (53).
Also in 2002, a GIP analogue with an N-terminal substitution of glutamic acid in position 3 with proline (Pro3), was made as an attempt to prolong the GIP actions by protecting from DPP-4 degradation (66) (Figure 1). In animal models, (Pro3)GIP was found to be a potent enzyme-resistant GIP receptor antagonist by its ability to block GIP’s effects in vitro and in vivo (66, 67). However, years later in 2016, a pharmacological analysis of this ligand demonstrated interspecies differences between the rodent and human GIP system and also led to the conclusion that the human Pro3(GIP) was a potent partial agonist with the ability to activate the human GIP receptor with a maximal efficiency (Emax) of 90% compared to human GIP (68). This finding annulled its clinical potential as a GIP receptor antagonist.
In 2008, an immunization approach, led to the development of an active vaccine with GIP peptides covalently attached to virus-like particles (VLP-GIP). Vaccination of mice with VLP-GIP produced high titers of specific antibodies, that bound to GIP and prevented activation of the GIP receptor. As seen for the GIP receptor knockout mice, VLP-GIP efficiently reduced body weight gain in animals on a high-fat diet (69), supporting that the GIP receptor plays a role in developing obesity.
Two years later in 2010, GIP(4-42), GIP(5-42), GIP(7-42) and GIP(8-42) also proved to have functionally interesting antagonistic effects at the GIP receptor in vitro (70). However, only GIP(8-42) exhibited antagonistic properties in vivo in mice reducing the effects of the native agonist, GIP(1-42).
In 2011, a small molecule, low-molecular GIP receptor antagonist, SKL-14959 (49), was studied both in vitro, and in vivo by injections in mice. The small molecule has a strong affinity for the GIP receptor and inhibition of GIP-stimulated cAMP production in vitro (Table 1). Furthermore, SKL-14959 suppresses insulin secretion and reduces lipolytic effects in vivo (71).
In 2013, Gipg013, a GIP receptor immunoglobin G antibody, was presented. Gipg013 is a specific competitive antagonist with equally high potency across mouse, rat, dog and human GIP receptors (72). In vivo, Gipg013 dose-dependently reduces GIP-induced insulin secretion and was one of the first GIP receptor antagonizing antibodies published (72). The following year, antagonism of the GIP receptor by palmitoylation of GIP analogues with N- and C-terminal modifications reduced body weight and improved metabolic control in high-fat-fed mice. Consequently, GIP receptor antagonism was proposed as a potential strategy for treating obesity-related diabetes (73).
Until 2015, a common limitation of the identified GIP receptor antagonists was the absence of human data, raising concerns about their potential clinical application. In 2015, N-terminally truncated variants of the C-terminally truncated and fully bioactive GIP(1-30)NH2 were studied. Here, GIP(3-30)NH2 and GIP(5-30)NH2 were characterized as potent, competitive and high-affinity antagonists of the human GIP receptor (14) (Table 1). In 2017, GIP(3-30)NH2 was confirmed to be highly selective for the human GIP receptor without any agonistic properties. To complement human studies, mouse GIP(3-30)NH2 was also confirmed as a GIP receptor antagonist with the ability to impair the glucose-lowering effects of human GIP(1-42) in mice (74). In 2017, the first human study with GIP(3-30)NH2 was performed. GIP(3-30)NH2 demonstrated an inhibition of the GIP-potentiated glucose-stimulated insulin secretion by 82%. Importantly, the infusions were well tolerated and did not trigger any adverse effects (30). That same year, human infusions of GIP(3-30)NH2 were also used to block the previously observed effects of exogenous GIP on adipose tissue perfusion (22). In conclusion, GIP(3-30)NH2 was defined as a new scientific tool to investigate GIP physiology (75). Over the following years, in several human studies, GIP(3-30)NH2 was administered intravenously to study the effects of endogenous GIP by inhibiting the GIP receptor and comparing the results with placebo infusions (76). GIP(3-30)NH2 has been infused in both healthy individuals (16, 22, 24, 75), patients with type 1 diabetes (77), type 2 diabetes (78, p. 2) with or without obesity (79), acromegaly (80), before and after gastric bypass surgeries (79), and in totally pancreatectomized patients (81). Species variants of GIP(3-30)NH2 (for especially mice and rats) are also selective and efficient tools (30, 82, 83), and with 15 completed human studies and several ongoing, GIP(3-30)NH2 has become the most widely used tool for studying the role of the GIP system in humans.
In 2018, another non-peptide candidate for the inhibition of the GIP receptor was presented using an antibody-based approach. In mice, treatment with anti-murine GIP receptor antibody (muGIPR-Ab) protected against body weight gain (54). This finding was replicated in nonhuman primates using another antibody, now against the human GIP receptor, supportive of an antagonistic approach for obesity treatment (54).
In 2021, AMG133, a bispecific molecule that combines GIP receptor antagonism and GLP-1 receptor agonism was presented. The molecule is a human monoclonal anti-human GIP receptor antibody conjugated with two GLP-1 receptor agonist peptides. In preclinical efficacy studies in diet-induced obese (DIO) mice and obese monkeys, treatment with AMG133 reduces body weight and improves metabolic parameters to a larger extent than a GLP-1 receptor agonist alone (50). Additionally, AMG133 has an extended pharmacokinetic profile with a half-life of up to 9.1 days in obese monkeys, suitable for less frequent dosing regimens (50). Phase 1 and phase 2 studies of the clinical trial development program for AMG133 are completed with results from phase 1 publicly available (84), while, at this time, phase 2 data have only been disclosed through a company announcement with headline results (85). In 2022, a GIP(5-31) analogue was discovered and proved to function as a potent GIP receptor antagonist (86). In vitro, a GIP(5-31) analogue palmitoylated at a lysin in position, GIP(5-31)-K11, inhibited GIP-induced cAMP production in both human and rodent systems. In vivo, the GIP(5-31) analogue inhibited GIP-stimulated insulin secretion in human islets. Additionally, co-administration of this antagonist with a GLP-1 receptor agonist in DIO mice potentiated weight loss compared to either monotherapy (86).
Latest in 2024, AT-7687, an optimized peptide analogue of the naturally occurring GIP(3-30)NH2 was characterized as a potent and selective GIP receptor antagonist (Table 1) with a circulatory half-life of 27.4 hours (51) (Figure 3). The in vivo effects of AT-7687 were evaluated in obese cynomolgus monkeys, and when combined with the GLP-1 analogue liraglutide, AT-7687 enhances weight reduction induced by liraglutide and improves glycemic control and lipid metabolism (51).
3 Results of reduced GIP receptor activation in humans
Some GIP receptor antagonists have contributed to the elucidation of the physiological properties of endogenous GIP. Moreover, several GIP receptor antagonists have also been used to evaluate the therapeutic potential of inhibiting endogenous GIP actions. Among the described antagonists, GIP(3-30)NH2 has been particularly instrumental in advancing the understanding of the human physiology of the GIP system, whereas modified peptides and antibodies have contributed with therapeutic perspectives. Another relevant contribution to the elucidation of GIP biology is studies of genetic loss-of-function variants of the human GIP receptor. These studies have provided valuable insights into the potential long-term effects of GIP receptor antagonism, and offered a clearer understanding of how impairing GIP receptor activity may impact physiological traits related to GIP’s biological effects. In the following, we describe and discuss the effects of reduced or abolished GIP receptor activation within different organs and tissues in humans (Figure 4).
Figure 4

Effects of GIP receptor inhibition or genetic loss-of function receptor variants in humans. Reduced GIP receptor activity leading to reduced splanchnic blood flow, reduced insulin secretion, increased blood glucose, increased bone resorption and reduced body weight. GIP, glucose-dependent insulinotropic polypeptide.
3.1 Glucose metabolism
GIP(3-30)NH2 administration has yielded significant outcomes within glucose metabolism across human studies. In healthy individuals, GIP(3-30)NH2 infusions inhibit GIP-potentiated glucose-stimulated insulin secretion (75), thereby confirming GIP’s role as a potent incretin hormone (16, 87). In addition, infusions of GIP(3-30)NH2 induce higher postprandial plasma glucose levels (16, 87).
In individuals with type 2 diabetes, GIP(3-30)NH2 infusions decrease insulin secretion, with little effect on postprandial glycemia (78). These findings reveal an insulinotropic effect of endogenous GIP in individuals with type 2 diabetes. However, this contrasts to findings from earlier infusions of exogenous GIP(1-42), which failed to enhance insulin secretion in individuals with type 2 diabetes (88, 89). The lack of insulinotropic response to GIP has long been claimed as the reason for not targeting the GIP receptor in this patient group. Although later, endogenous GIP was revealed to have significant insulinotropic properties. Furthermore, several novel therapeutic compounds (GIP receptor antagonists and agonists including multiple-targeting compounds) have also shown beneficial glucometabolic and body weight lowering effects in individuals with type 2 diabetes and obesity (40, 41). Highlighting the complexity of the GIP system in humans, conclusions based on short-term GIP(1-42) infusions failed to demonstrate its therapeutic potential.
We recently assessed the role of endogenous GIP in individuals with obesity during fasting using GIP(3-30)NH2. Here, we found infusions of GIP(3-30)NH2 to cause higher plasma glucose levels, indicating a glucose-lowering effect of endogenous GIP. However, GIP(3-30)NH2 did not affect insulin secretion in the fasting state (90). A possible explanation for these findings is that endogenous GIP may play a role in the regulation of insulin sensitivity in humans, similar to observations in mice (91).
In gastric bypass-operated individuals with obesity and no history of diabetes, GIP(3-30)NH2 does not affect insulin secretion nor plasma glucose levels (79). These results differ from the effects of GIP(3-30)NH2 in non-operated individuals, and points to a less important role of GIP in glucose metabolism after gastric bypass surgery (79, 92). However, the effect of GIP(3-30)NH2 still appears to differ depending on the type of gastric surgery. Following gastric sleeve, infusions of GIP(3-30)NH2 induce a slight increase in plasma glucose levels, whereas no changes are found after Roux-en-Y gastric bypass. Therefore, endogenous GIP seems to retain a role in postprandial glucose regulation following gastric sleeve but when the first part of the small intestine is bypassed (Roux-en-Y gastric bypass) endogenous GIP does not contribute to the postprandial glucose regulation (79). In totally pancreatectomized individuals, GIP(3-30)NH2 infusions do not affect postprandial plasma glucose levels highlighting the importance of the pancreas in GIP’s glucose-regulating effects (81).
Infusions with GIP(3-30)NH2 in healthy individuals during a mixed meal test or an oral glucose tolerance test as well as in persons with obesity in the fasting state do not affect glucagon levels (16, 87, 90). However, in individuals with type 2 diabetes, infusions of GIP(3-30)NH2 reduced glucagon concentrations (78), illustrating the potential of GIP receptor antagonism to limit the paradoxical postprandial hyperglucagonemia in individuals with type 2 diabetes (93).
Treatment with AMG133 in individuals with obesity decreases HbA1c levels and reduces fasting glucose levels in a dose-responsive manner (84). In DIO mice and obese monkeys, AMG133 decreases fasting insulin levels, but this effect was not seen in the clinical trial of AMG133 (84). Furthermore, AMG133, in higher doses, induces lower fasting glucagon levels compared to placebo (84). The results of the phase 1 studies of AMG133 could provide insights into the role of GIP receptor antagonism within glucose metabolism, however, determining the exact effect of the GIP receptor antagonism remains challenging due to an anticipated influence of GLP-1 receptor agonism.
3.2 Adipose tissue
In humans, infusions of GIP(3-30)NH2 during a hyperinsulinemic and hyperglycemic clamp reduce GIP-induced blood flow to subcutaneous adipose tissue and inhibit the increase in triacylglycerol clearance observed with GIP administration (22). Furthermore, the administration of GIP(3-30)NH2 increased free fatty acid (FFA) output and a higher FFA/glycerol-ratio, suggesting a reduction in in-situ re-esterification of FFA and lipolytic activity (22). However, these findings are based on the inhibition of administered GIP, and, therefore, they do not describe the clinical effect of GIP receptor antagonism and inhibition of endogenous GIP in adipose tissue.
Nevertheless, GIP receptor antagonism is thought to exert beneficial metabolic effects due to the expression of GIP receptors on adipocytes (94), along with elevated circulating GIP levels observed in both individuals with obesity (95) and individuals given a high-fat-diet (96). Therefore, reducing fat deposition through GIP receptor antagonism still hold promising therapeutic potential supported by animal models and the fact that individuals with loss-of-function GIPR genetic variants have a lower body mass index (BMI) (39, 97–99).
3.3 Bone metabolism
Addressing the acute changes in the postprandial state, GIP(3-30)NH2 infusions diminished GIP-induced suppression of the bone resorption marker carboxy-terminal type 1 collagen crosslinks (CTX) in both healthy individuals (26) and individuals with type 2 diabetes (78). Furthermore, GIP(3-30)NH2 has been used to demonstrate an uncoupling of the otherwise coupled processes of bone resorption and formation assessed by a decrease in CTX and a temporary increase in the bone formation marker procollagen type 1 amino terminal propertied (P1NP) in healthy individuals (100). Characterizations of individuals with genetically impaired GIP receptor function corresponds with the findings following GIP receptor agonist or antagonist administrations. Three amino acid-altering GIP receptor variants (R190Q, E288G, and E354Q) are all associated with lower bone mineral density, and in postmenopausal women followed for 10 years, who were homozygous carriers of the E354Q variant, showed a higher risk of non-vertebral fractures (99, 101). However, a recent large meta-analysis showed no association between these three variants and low bone mineral density or fracture prevalence (102).
3.4 Appetite regulation and weight management
Similar to animal studies with deletion of the GIPR gene that exhibit protection toward obesity (53), early genome-wide association studies (GWAS) in humans have revealed the GIPR gene to be associated with BMI (103). Specifically, carriers of GIPR variants that are pharmacologically classified as loss-of-function variants with reduced cell surface expression, cAMP production, beta arrestin 2 recruitment, internalization, and endosomal signaling have consistently been associated with lower adiposity-related traits, including BMI (103) and nominally lower prevalence of obesity (39, 97–99). However, studies with the available GIP receptor antagonists have not shown an acute effect on appetite regulation in humans (16) assessed by an ad libitum meal test and visual analogue scale questionaries (16, 87). When tested in obese cynomolgus monkeys, treatment with AT-7687 in combination with liraglutide results in lower total energy intake and significant body weight loss compared to placebo but no differences when compared to liraglutide alone. Thus, treatment with AT-7687 and liraglutide reduced appetite but, in this first preclinical study, the effect of the GIP receptor antagonist could not be separated from the GLP-1 receptor agonist alone (51). Recently, in a phase 1 clinical study, AMG133 demonstrated a pronounced dose-dependent weight loss in participants with obesity. Participants receiving multiple ascending doses of AMG133 experienced sustained weight loss for up to 150 days after the last dose (84). It is worth noting that GLP-1-induced appetite suppression is a well-established mechanism in the treatment of obesity. However, no clear mechanism behind the potential of weight loss induced by GIP receptor antagonism has been identified. While co-receptor targeting, as seen with AMG133, has shown promising results in weight reduction, it remains unclear whether this is an additive or synergistic effect, or if the main mode of action behind appetite suppression is based on GLP-1 receptor activation (104).
3.5 Nausea
Recent technological advancements have detected the GIP receptor in the brain of mice, specifically in inhibitory gamma-aminobutyric acid (GABA) neurons located within the area postrema, which function as a chemoreceptor trigger zone for vomiting (105). GIP receptor agonists have shown antiemetic effects and reduces nausea-like behavior in animal models (105). However, in the human brain, the presence of the GIP receptor is still elusive. Recently, administration of a long-acting GIP receptor agonist in humans, reduced GLP-1 analogue related gastrointestinal side effects, including nausea and vomiting (106). Although no findings have yet been made with GIP receptor antagonists in this field, the findings suggests that the GIP system is involved in the regulation of central nervous system-mediated anorexigenic and emetic behaviors (106).
3.6 Memory and other cognitive functions
As GIP receptor expression have been demonstrated in different brain regions of animals (105, 107), GIP could play a direct role in modulation aspects of brain functions. Although, there are no human data describing the effect of GIP activity on memory and other cognitive functions, several animal studies have shown that activation of the GIP receptor has a profound effect on neuronal plasticity (108). As an example, in mice, prolonged GIP receptor activation improves cognitive function by an increased recognition index (109). Furthermore, it improves hippocampal synaptic plasticity (109, 110). While the effects of GIP receptor antagonism on these outcomes remain unknow, GIPR knockout mice have been shown to exhibit impaired learning and memory, as well as reduced synaptic plasticity (111).
3.7 Cardiovascular effects
In a GWAS, the genetic GIP receptor variant, E354Q, and the risk of cardiovascular diseases are nominally associated (112), and also, carriers of E354Q have been associated with lower risk of coronary artery disease (113). The carriers of the loss-of-function GIP receptor variants, R190Q and E288G, display higher blood pressure and an increased tendency toward hypertension (99) but a comprehensive study of larger number of loss-of-function GIP receptor variants did not reveal similar effects on cardiovascular traits (39). The direct mechanism of the potential effects of long-term reduced GIP receptor activity in the cardiovascular system is not yet confirmed (55) but recently, infusions of GIP(3-30)NH2, have highlighted the crucial role of endogenous GIP in the vascular system during meal digestion. During oral glucose ingestion, GIP(3-30)NH2 infusion reduces postprandial splanchnic blood flow in healthy men (24). The reduced blood flow in the major gastrointestinal vessels, the superior mesenteric artery and portal vein, demonstrate that endogenous GIP contributes to postprandial hyperemia (24). However, since no measurements of splanchnic hemodynamic changes after more long-term exposure to GIP receptor antagonism have been conducted, in either animal models or humans, the therapeutic implications of this effect are uncertain.
3.8 Adverse effects of GIP receptor antagonism
So far, no adverse effects have been reported with the administration of GIP receptor antagonists in humans or animals. Given the established physiological effects of GIP receptor activation, GIP receptor antagonism could potentially lead to undesirable increases in blood glucose levels due to reduced insulin secretion. Additionally, alterations in lipid distribution could cause concerns about where lipids would be stored and how it would be metabolized (114). Longer treatment with GIP receptor antagonists could also reduce bone mineral density (100), and hereby increase the risk of fractures. Moreover, the reduced splanchnic blood flow could affect nutrient uptake or affect digestion (23). Finally, the expected involvement of GIP in the central nervous system (107) leads to uncertainty regarding effects on memory and cognitive functions.
When other receptor antagonists are administered, a compensatory increase in the circulating hormone levels is seen (115). However, GIP receptor antagonism has not, so far, resulted in higher or lower circulating levels of GIP (46, 75, 87). Importantly, to evaluate adverse or compensatory effects, a GIP receptor antagonist must be administered longer than those presented so far. Moreover, current studies with long-term exposure to GIP receptor antagonism have only been conducted during co-stimulation of the GLP-1 receptor which induce a drug class-specific adverse effect profile, including nausea and vomiting (84, 116). Therefore, further research is needed to determine whether prolonged GIP receptor antagonism can lead to any adverse effects.
When administered in humans, GIP receptor antagonists conjugated to GLP-1 analogues have led to mild gastrointestinal-related adverse events, including nausea and vomiting (50). Considering this, the reported side effects may be GLP-1 receptor activation-related, but the exact mechanism is still unknown.
4 Combinations of GIP receptor targeting treatments and GLP-1 receptor agonism
In recent years, GIP receptor agonists and antagonists have gained attention for their therapeutic potential in treating obesity and type 2 diabetes. Remarkably, combining GIP receptor agonism with GLP-1 receptor agonism, or GIP receptor antagonism with GLP-1 receptor agonism has led to substantial weight loss, improved glycemic control, and better cardiovascular risk profiles in both animal models and clinical trials (50, 51, 54, 84, 86, 117, 118) (Figure 5). However, the mechanism of action of GIP in this paradox and the extent of its role in these outcomes remains unclear. Therefore, growing interest and evidence support both stimulation and inhibition of GIP receptor signaling.
Figure 5
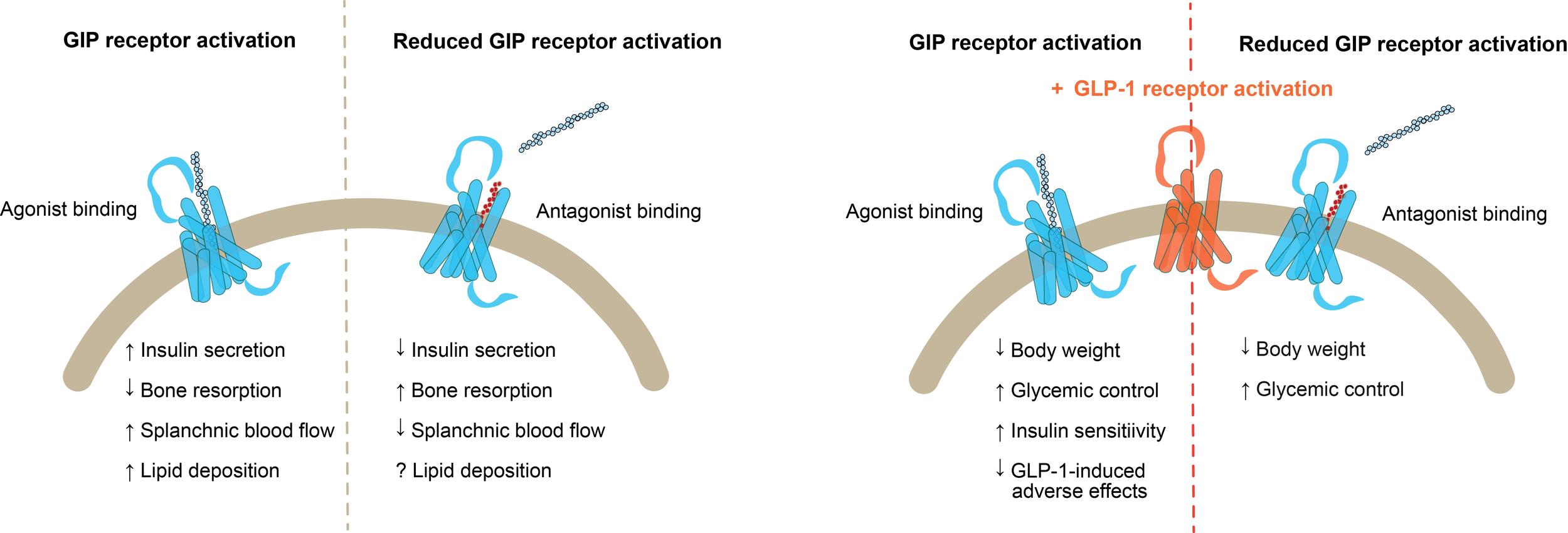
Combinations of GIP receptor targeting treatments and GLP-1 receptor agonism. Left) Outcomes of GIP receptor activation by agonist binding versus reduced GIP receptor activation by antagonist binding; Right) Outcomes of GLP-1 receptor activation in combination with GIP receptor agonist binding versus GIP receptor antagonist binding. GIP, glucose-dependent insulinotropic polypeptide; GLP-1, glucagon like peptide 1.
Understanding the balance between GIP receptor signaling, intracellular recycling, and desensitization is crucial for interpreting the physiological consequences of a given compound. Therefore, the traditional pharmacological terms like agonist and antagonist could be insufficient for characterizing compounds that target the GIP receptor. Today, the majority of the emerging GIP receptor targeting compounds are claimed to be agonists, and to increase insulin secretion (17, 20), to improve insulin sensitivity (91), and also, suppress appetite (119). Most of the GIP receptor targeting compounds are evaluated in vitro as stimulators of a single cellular pathway, which is the Gαs-mediated cAMP production (14, 30, 37). However, assessing only cAMP production may be insufficient, as e.g. beta arrestin recruitment to the GIP receptor is a key regulator of receptor signaling, particularly in post-activation trafficking, including receptor internalization, continued signaling from intracellular compartments like endosomes and recycling of the receptor back to the cell surface. Since the different ‘agonists’/compounds, all of which stimulate Gαs recruitment to the GIP receptor, have distinct pharmacology, they may affect interaction of the various effector molecules with the GIP receptor differently (39, 42) (Figure 2). Hence, a more thorough pharmacological characterization is necessary for each developed compound.
The GIP receptor-GLP-1 receptor dual agonist, tirzepatide, ranks among the most effective therapeutic agents within treatment of obesity and diabetes to date (118). It has demonstrated the ability to reduce body weight by more than 20% in individuals with obesity (118), and to improve glycemic control to a degree that 87-91% of enrolled individuals with type 2 diabetes reach HbA1c levels below the type 2 diabetes diagnostic threshold after 40 weeks of treatment (120). Interestingly, while tirzepatide acts as an agonist of the GLP-1 receptor, it may effectively act as a GIP receptor antagonist. Its binding to the GIP receptor induces receptor internalization which, with continuous treatment, could lead to receptor downregulation and reduced GIP signaling (121). However, clinical evidence of GIP receptor engagement is still lacking and due to similar actions of GIP and GLP-1 receptor activation, the distinct mode of action for this dual-acting compound is complex to address (41).
The only GIP receptor antagonist administered in humans for a longer period, is the aforementioned AMG133, which is an antagonistic GIP receptor antibody conjugated to two agonistic GLP-1 receptor peptide analogues (84). The mode of action for AMG133 is believed to involve this bispecific molecule simultaneously binding to both the GIP- and GLP-1 receptor, where it is demonstrated to increase cAMP production and induce internalization of co-localized GIP and GLP-1 receptors. Notably, higher levels of endosomal markers were observed in cells expressing GLP-1 receptor stimulated with AMG133, suggesting an increased localization of the receptor complex within the endosomes. This enhanced receptor internalization is, therefore, thought to amplify the effects of GLP-1 receptor agonism (50).
Besides the complex pharmacological interplay between the GIP receptor targeting ligands and the receptor addressed above, another aspect of GIP biology is the tissue-specific actions, which might affect the clinical results of GIP targeting treatments. When therapeutically targeting the GIP system, effects on other tissues than the pancreas could partly explain the paradox observed for the weight-reducing effects of GIP receptor agonism and antagonism. GIP’s action to augment splanchnic blood flow, potentially affecting nutrient uptake and its involvement in adipose tissue metabolism and suggested central affects in the regulation of appetite as seen in animal models, all remain to be further explored in humans.
5 Current clinical trials
In a recent press release, the results from the phase 2 study of AMG133 (maridebart cafraglutide or MariTide) are revealed to induce a substantial weight loss following 52 weeks treatment. The phase 2 study showed that participants with overweight or obesity, without type 2 diabetes had an average weight loss of up to 20% and without the occurrence of a weight loss plateau. Participants with type 2 diabetes with obesity or overweight had an average weight loss of up to 17% also without reaching a weight loss plateau. Moreover, they obtained a decrease in average HbA1c of up to 2.2 percentage points. In addition, cardiometabolic parameters were improved, including blood pressure, triglyceride levels, and high-sensitivity C-reactive protein. AMG133 was administered in longer treatment intervals with either monthly or less frequent dosing. The most common adverse effects were mild and transient gastrointestinal symptoms including nausea, vomiting and constipation. Notably, the adverse effects were mainly associated with the first dose. Due to these results, Amgen expects to initiate a phase 3 development program (85).
6 Conclusions
The discoveries of GIP receptor antagonists as scientific tools have elucidated the role of GIP in human physiology. The GIP receptor antagonist GIP(3-30)NH2 has especially contributed to a greater understanding of the GIP system in both human and rodent studies. Importantly, genetic variations within GIP receptors across different species, make the GIP system complex compared to the more conserved GLP-1 system. Thus, translation of GIP physiology and pharmacology across species can be challenging (122). Within the past years, development of GIP receptor antagonists has been an attractive therapeutic approach for combating obesity and treating type 2 diabetes. Today, more compounds that target the GIP receptor are emerging as a valuable complement to GLP-1 based treatments and in combination with e.g. glucagon or GLP-2 receptor agonistic properties as well (123). Recently, administration of AMG133, has shown promising results in promoting weight loss in initial phase 1 and 2 studies. Paradoxically, these findings indicate that similar results can be obtained by combining GLP-1 receptor activation with either GIP receptor agonism or GIP receptor antagonism. These findings have led to greater interest in understanding the underlying mechanisms of targeting the GIP receptor. Although less than a decade has passed since the first GIP(3-30)NH2 injection in humans, substantial research has already contributed to the confirmation of GIP signaling pathways, genetic variations, as well as preclinical and clinical outcomes of GIP receptor targeting compounds (124). Despite the promising therapeutic potential of GIP receptor antagonism, uncertainties remain regarding associated adverse effects. Given that both GIP receptor agonism and antagonism have only been thoroughly investigated in combination with GLP-1 receptor agonism, long-term clinical studies of GIP receptor antagonism alone are needed to evaluate the adverse effect profile.
Statements
Author contributions
FK: Writing – original draft, Visualization. MH: Writing – review & editing. HK: Visualization, Writing – review & editing. AL: Writing – review & editing. MR: Writing – review & editing. LG: Conceptualization, Project administration, Supervision, Visualization, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AC, Adenylyl cyclase; BMI, Body mass index; cAMP, Cyclic adenosine monophosphate; DIO, Diet-induced obese mice; DPP-4, Dipeptidyl peptidase; CTX, Carboxy-terminal type 1 collagen crosslinks; FFA, Free fatty acid; GABA, Gamma-aminobutyric acid; GIP, Glucose-dependent insulinotropic polypeptide; GLP-1, Glucagon like peptide 1; GLP-2, Glucagon like peptide 2; GPCR, G Protein-coupled receptor; GRKs, G Protein-coupled receptor kinases; GWAS, Genome-wide association studies; IC50, Half-maximal inhibitory concentrations; Kd, Dissociation constant; Ki, Inhibition constant; PKA, Protein kinase A; P1NP, Procollagen type 1 amino terminal propeptid; VLP, Virus-like peptide.
References
1
Buchan AMJ Polak JM Capella C Solcia E Pearse AGE . Electronimmunocytochemical evidence for the K cell localization of gastric inhibitory polypeptide (GIP) im man. Histochemistry. (1978) 56:37–44. doi: 10.1007/BF00492251
2
Jorsal T Rhee NA Pedersen J Wahlgren CD Mortensen B Jepsen SL et al . Enteroendocrine K and L cells in healthy and type 2 diabetic individuals. Diabetologia. (2018) 61:284–94. doi: 10.1007/s00125-017-4450-9
3
Holst JJ Gasbjerg LS Rosenkilde MM . The role of incretins on insulin function and glucose homeostasis. Endocrinology. (2021) 162:bqab065. doi: 10.1210/endocr/bqab065
4
Zilstorff DB Richter MM Hannibal J Jørgensen HL Sennels HP Wewer Albrechtsen NJ et al . Secretion of glucagon, GLP-1 and GIP may be affected by circadian rhythm in healthy males. BMC Endocrine Disord. (2024) 24:38. doi: 10.1186/s12902-024-01566-9
5
Rayner CK Park HS Wishart JM Kong MF Doran SM Horowitz M . Effects of intraduodenal glucose and fructose on antropyloric motility and appetite in healthy humans. Am J Physiology-Regulatory Integr Comp Physiol. (2000) 278:R360–6. doi: 10.1152/ajpregu.2000.278.2.R360
6
Carr RD Larsen MO Sörhede Winzell M Jelic K Lindgren O Deacon CF et al . Incretin and islet hormonal responses to fat and protein ingestion in healthy men. Am J Physiology-Endocrinology Metab. (2008) 295:E779–84. doi: 10.1152/ajpendo.90233.2008
7
Lindgren O Carr RD Deacon CF Holst JJ Pacini G Mari A et al . Incretin hormone and insulin responses to oral versus intravenous lipid administration in humans. J Clin Endocrinol Metab. (2011) 96:2519–24. doi: 10.1210/jc.2011-0266
8
Lindgren O Pacini G Tura A Holst JJ Deacon CF Ahrén B et al . Incretin effect after oral amino acid ingestion in humans. J Clin Endocrinol Metab. (2015) 100:1172–6. doi: 10.1210/jc.2014-3865
9
Deacon CF . Circulation and degradation of GIP and GLP-1. Hormone Metab Res. (2005) 36:761–5. doi: 10.1055/s-2004-826160
10
Deacon CF Nauck MA Meier J Hücking K Holst JJ . Degradation of endogenous and exogenous gastric inhibitory polypeptide in healthy and in type 2 diabetic subjects as revealed using a new assay for the intact peptide1. J Clin Endocrinol Metab. (2000) 85:3575–81. doi: 10.1210/jcem.85.10.6855
11
Fujita Y Yanagimachi T Takeda Y Honjo J Takiyama Y Abiko A et al . Alternative form of glucose-dependent insulinotropic polypepide and its physiology. J Diabetes Invest. (2016) 7:33–7. doi: 10.1111/jdi.12445
12
Takeda Y Fujita Y Yanagimachi T Maruyama N Bessho R Sakagami H et al . Establishment of novel specific assay for short-form glucose-dependent insulinotropic polypeptide and evaluation of its secretion in nondiabetic subjects. Physiol Rep. (2020) 8:2519–24. doi: 10.14814/phy2.14469
13
Krogh LL Henriksen K Stensen S Skov-Jeppesen K Bergmann NC Størling J et al . The naturally occurring GIP(1-30)NH2 is a GIP receptor agonist in humans. Eur J Endocrinol. (2023) 188:48–58. doi: 10.1093/ejendo/lvac015
14
Hansen LS Sparre-Ulrich AH Christensen M Knop FK Hartmann B Holst JJ et al . N-terminally and C-terminally truncated forms of glucose-dependent insulinotropic polypeptide are high-affinity competitive antagonists of the human GIP receptor. Br J Pharmacol. (2016) 173:826–38. doi: 10.1111/bph.13384
15
Usdin TB Mezey E Button DC Brownstein MJ Bonner TI . Gastric inhibitory polypeptide receptor, a member of the secretin-vasoactive intestinal peptide receptor family, is widely distributed in peripheral organs and the brain. Endocrinology. (1993) 133:2861–70. doi: 10.1210/endo.133.6.8243312
16
Gasbjerg LS Helsted MM Hartmann B Jensen MH Gabe MBN Sparre-Ulrich AH et al . Separate and combined glucometabolic effects of endogenous glucose-dependent insulinotropic polypeptide and glucagon-like peptide 1 in healthy individuals. Diabetes. (2019) 68:906–17. doi: 10.2337/db18-1123
17
Christensen M Vedtofte L Holst JJ Vilsbøll T Knop FK . Glucose-dependent insulinotropic polypeptide. Diabetes. (2011) 60:3103–9. doi: 10.2337/db11-0979
18
Ørskov C Holst JJ Nielsen OV . Effect of truncated glucagon-like peptide-1 [Proglucagon-(78–107) amide] on endocrine secretion from pig pancreas, antrum, and nonantral stomach. Endocrinology. (1988) 123:2009–13. doi: 10.1210/endo-123-4-2009
19
Wice BM Reeds DN Tran HD Crimmins DL Patterson BW Dunai J et al . Xenin-25 amplifies GIP-mediated insulin secretion in humans with normal and impaired glucose tolerance but not type 2 diabetes. Diabetes. (2012) 61:1793–800. doi: 10.2337/db11-1451
20
Skov-Jeppesen K Svane MS Martinussen C Gabe MBN Gasbjerg LS Veedfald S et al . GLP-2 and GIP exert separate effects on bone turnover: A randomized, placebo-controlled, crossover study in healthy young men. Bone. (2019) 125:178–85. doi: 10.1016/j.bone.2019.05.014
21
Asmar M Simonsen L Arngrim N Holst JJ Dela F Bülow J . Glucose-dependent insulinotropic polypeptide has impaired effect on abdominal, subcutaneous adipose tissue metabolism in obese subjects. Int J Obes. (2014) 38:259–65. doi: 10.1038/ijo.2013.73
22
Asmar M Asmar A Simonsen L Gasbjerg LS Sparre-Ulrich AH Rosenkilde MM et al . The gluco- and liporegulatory and vasodilatory effects of glucose-dependent insulinotropic polypeptide (GIP) are abolished by an antagonist of the human GIP receptor. Diabetes. (2017) 66:2363–71. doi: 10.2337/db17-0480
23
Koffert J Honka H Teuho J Kauhanen S Hurme S Parkkola R et al . Effects of meal and incretins in the regulation of splanchnic blood flow. Endocrine Connections. (2017) 6:179–87. doi: 10.1530/EC-17-0015
24
Gasbjerg LS Rasmussen RS Langberg LS Østergaard F Vestergaard MB Asmar A et al . The gut hormone GIP contributes to the postprandial gastrointestinal hyperaemia in humans. Diabetologia. (2023) 66:288. doi: 10.1007/s00125-023-05969-6
25
Nissen A Christensen M Knop FK Vilsbøll T Holst JJ Hartmann B . Glucose-dependent insulinotropic polypeptide inhibits bone resorption in humans. J Clin Endocrinol Metab. (2014) 99:E2325–9. doi: 10.1210/jc.2014-2547
26
Helsted MM Gasbjerg LS Lanng AR Bergmann NC Stensen S Hartmann B et al . The role of endogenous GIP and GLP-1 in postprandial bone homeostasis. Bone. (2020) 140:1–7. doi: 10.1016/j.bone.2020.115553
27
Hansen MS Søe K Christensen LL Fernandez-Guerra P Hansen NW Wyatt RA et al . GIP reduces osteoclast activity and improves osteoblast survival in primary human bone cells. Eur J Endocrinol. (2023) 188:144–57. doi: 10.1093/ejendo/lvac004
28
Christensen MB Lund A Calanna S Jørgensen NR Holst JJ Vilsbøll T et al . Glucose-dependent insulinotropic polypeptide (GIP) inhibits bone resorption independently of insulin and glycemia. J Clin Endocrinol Metab. (2018) 103:288–94. doi: 10.1210/jc.2017-01949
29
Bergmann NC Lund A Gasbjerg LS Jørgensen NR Jessen L Hartmann B et al . Separate and combined effects of GIP and GLP-1 infusions on bone metabolism in overweight men without diabetes. J Clin Endocrinol Metab. (2019) 104:2953–60. doi: 10.1210/jc.2019-00008
30
Gabe MBN Sparre-Ulrich AH Pedersen MF Gasbjerg LS Inoue A Bräuner-Osborne H et al . Human GIP(3-30)NH2 inhibits G protein-dependent as well as G protein-independent signaling and is selective for the GIP receptor with high-affinity binding to primate but not rodent GIP receptors. Biochem Pharmacol. (2018) 150:97–107. doi: 10.1016/j.bcp.2018.01.040
31
Campbell JE Beaudry JL Svendsen B Baggio LL Gordon AN Ussher JR et al . GIPR is predominantly localized to nonadipocyte cell types within white adipose tissue. Diabetes. (2022) 71:1115–27. doi: 10.2337/db21-1166
32
Yu X Chen S Funcke JB Straub LG Pirro V Emont MP et al . The GIP receptor activates futile calcium cycling in white adipose tissue to increase energy expenditure and drive weight loss in mice. Cell Metab. (2025) 37:187–204.e7. doi: 10.1016/j.cmet.2024.11.003
33
Weaver RE Donnelly D Wabitsch M Grant PJ Balmforth AJ . Functional expression of glucose-dependent insulinotropic polypeptide receptors is coupled to differentiation in a human adipocyte model. Int J Obes. (2008) 32:1705–11. doi: 10.1038/ijo.2008.148
34
Regmi A Aihara E Christe ME Varga G Beyer TP Ruan X et al . Tirzepatide modulates the regulation of adipocyte nutrient metabolism through long-acting activation of the GIP receptor. Cell Metab. (2024) 36:1898–9. doi: 10.1016/j.cmet.2024.06.012
35
Takeda S Kadowaki S Haga T Takaesu H Mitaku S . Identification of G protein-coupled receptor genes from the human genome sequence. FEBS Lett. (2002) 520:97–101. doi: 10.1016/S0014-5793(02)02775-8
36
Gabe MBN van der Velden WJC Smit FX Gasbjerg LS Rosenkilde MM . Molecular interactions of full-length and truncated GIP peptides with the GIP receptor – A comprehensive review. Peptides. (2020) 125:170224. doi: 10.1016/j.peptides.2019.170224
37
McIntosh CH Widenmaier S Kim S . Glucose-dependent insulinotropic polypeptide signaling in pancreatic β-cells and adipocytes. J Diabetes Invest. (2012) 3:96–106. doi: 10.1111/j.2040-1124.2012.00196.x
38
Møller TC Moo EV Inoue A Pedersen MF Bräuner-Osborne H . Characterization of the real-time internalization of nine GPCRs reveals distinct dependence on arrestins and G proteins. Biochim Biophys Acta (BBA) - Mol Cell Res. (2024) 1871:119584. doi: 10.1016/j.bbamcr.2023.119584
39
Kizilkaya HS Chen M Falsey JR Sivits G Hager T Atangan L et al . Characterization of genetic variants of GIPR reveals a contribution of β-arrestin to metabolic phenotypes. Nat Metab. (2024) 6:1268–81. doi: 10.1038/s42255-024-01061-4
40
Killion EA Chen M Falsey JR Sivits G Hager T Atangan L et al . Chronic glucose-dependent insulinotropic polypeptide receptor (GIPR) agonism desensitizes adipocyte GIPR activity mimicking functional GIPR antagonism. Nat Commun. (2020) 11:4981. doi: 10.1038/s41467-020-18751-8
41
Gasbjerg LS Rosenkilde MM Meier JJ Holst JJ Knop FK . The importance of glucose-dependent insulinotropic polypeptide receptor activation for the effects of tirzepatide. Diabetes Obes Metab. (2023) 25:3079–92. doi: 10.1111/dom.15216
42
Davies I Adriaenssens AE Scott WR Carling D Murphy KG Minnion JS et al . Chronic GIPR agonism results in pancreatic islet GIPR functional desensitisation. Mol Metab. (2025) 92:102094. doi: 10.1016/j.molmet.2025.102094
43
Baggio LL Drucker DJ . Biology of incretins: GLP-1 and GIP. Gastroenterology. (2007) 132:2131–57. doi: 10.1053/j.gastro.2007.03.054
44
Holst JJ Windeløv JA Boer GA Pedersen J Svendsen B Christensen M et al . Searching for the physiological role of glucose-dependent insulinotropic polypeptide. J Diabetes Invest. (2016) 7:8–12. doi: 10.1111/jdi.12488
45
Nauck MA Meier JJ . The incretin effect in healthy individuals and those with type 2 diabetes: physiology, pathophysiology, and response to therapeutic interventions. Lancet Diabetes Endocrinol. (2016) 4:525–36. doi: 10.1016/S2213-8587(15)00482-9
46
Gasbjerg LS Bergmann NC Stensen S Christensen MB Rosenkilde MM Holst JJ et al . Evaluation of the incretin effect in humans using GIP and GLP-1 receptor antagonists. Peptides. (2020) 125:170183. doi: 10.1016/j.peptides.2019.170183
47
Holst JJ Rosenkilde MM . Recent advances of GIP and future horizons. Peptides. (2020) 125:170230. doi: 10.1016/j.peptides.2019.170230
48
Deacon CF Plamboeck A Rosenkilde MM de Heer J Holst JJ . GIP-(3–42) does not antagonize insulinotropic effects of GIP at physiological concentrations. Am J Physiology-Endocrinology Metab. (2006) 291:E468–75. doi: 10.1152/ajpendo.00577.2005
49
Nakamura T Tanimoto H Mizuno Y Okamoto M Takeuchi M Tsubamoto Y et al . Gastric inhibitory polypeptide receptor antagonist, SKL-14959, suppressed body weight gain on diet-induced obesity mice. Obes Sci Pract. (2018) 4:194–203. doi: 10.1002/osp4.164
50
Lu S-C Chen M Atangan L Killion EA Komorowski R Cheng Y et al . GIPR antagonist antibodies conjugated to GLP-1 peptide are bispecific molecules that decrease weight in obese mice and monkeys. Cell Rep Med. (2021) 2:100263. doi: 10.1016/j.xcrm.2021.100263
51
Jensen MH Sanni SJ Riber D Holst JJ Rosenkilde MM Sparre-Ulrich AH . AT-7687, a novel GIPR peptide antagonist, combined with a GLP-1 agonist, leads to enhanced weight loss and metabolic improvements in cynomolgus monkeys. Mol Metab. (2024) 88:102006. doi: 10.1016/j.molmet.2024.102006
52
Nasteska D Harada N Suzuki K Yamane S Hamasaki A Joo E et al . Chronic reduction of GIP secretion alleviates obesity and insulin resistance under high-fat diet conditions. Diabetes. (2014) 63:2332–43. doi: 10.2337/db13-1563
53
Miyawaki K et al . Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat Med. (2002) 8:738–42. doi: 10.1038/nm727
54
Killion EA Jinghong Yie J Shi SDH Bates D Min X et al . Anti-obesity effects of GIPR antagonists alone and in combination with GLP-1R agonists in preclinical models. Sci Trans Med. (2018) 10:eaat3392. doi: 10.1126/scitranslmed.aat3392
55
Heimbürger SM Bergmann NC Augustin R Gasbjerg LS Christensen MB Knop FK . Glucose-dependent insulinotropic polypeptide (GIP) and cardiovascular disease. Peptides. (2020) 125:170174. doi: 10.1016/j.peptides.2019.170174
56
Dupre J Ross SA Watson D Brown JC . Stimulation of insulin secretion by gastric inhibitory polypeptide in man.1. J Clin Endocrinol Metab. (1973) 37:826–8. doi: 10.1210/jcem-37-5-826
57
Brown JC Pederson RA Jorpes E Mutt V . Preparation of highly active enterogastrone. Can J Physiol Pharmacol. (1969) 47:113–4. doi: 10.1139/y69-020
58
Ebert R Illmer K Creutzfeldt W . Release of gastric inhibitory polypeptide (GIP) by intraduodenal acidification in rats and humans and abolishment of the incretin effect of acid by GIP-antiserum in rats. Gastroenterology. (1979) 76:515–23. doi: 10.1016/S0016-5085(79)80219-X
59
Maletti M Carlquist M Portha B Kergoat M Mutt V Rosselin G . Structural requirements for gastric inhibitory polypeptide (GIP) receptor binding and stimulation of insulin release. Peptides. (1986) 7:75–8. doi: 10.1016/0196-9781(86)90167-1
60
Tseng CC Kieffer TJ Jarboe LA Usdin TB Wolfe MM . Postprandial stimulation of insulin release by glucose-dependent insulinotropic polypeptide (GIP). Effect of a specific glucose-dependent insulinotropic polypeptide receptor antagonist in the rat. J Clin Invest. (1996) 98:2440–5. doi: 10.1172/JCI119060
61
Tseng C-C Zhang X-Y Wolfe MM . Effect of GIP and GLP-1 antagonists on insulin release in the rat. Am J Physiology-Endocrinology Metab. (1999) 276:E1049–54. doi: 10.1152/ajpendo.1999.276.6.E1049
62
Gelling RW Coy DH Pederson RA Wheeler MB Hinke S Kwan T et al . GIP6–30amide contains the high affinity binding region of GIP and is a potent inhibitor of GIP1–42action in vitro. Regul Peptides. (1997) 69:151–4. doi: 10.1016/S0167-0115(97)00009-8
63
Hinke SA Manhart S Pamir N Demuth HU Gelling RW Pederson RA et al . Identification of a bioactive domain in the amino-terminus of glucose-dependent insulinotropic polypeptide (GIP). Biochim Biophys Acta (BBA) - Protein Structure Mol Enzymology. (2001) 1547:143–55. doi: 10.1016/S0167-4838(01)00181-9
64
Gault V Harriott P Flatt PR O'Harte FPM . Cyclic AMP production and insulin releasing activity of synthetic fragment peptides of glucose-dependent insulinotropic polypeptide. Bioscience Rep. (2002) 22(5–6)523–8. doi: 10.1023/A:1022073819618
65
Gault V Parker J Harriott P Flatt PR O'Harte FP . Evidence that the major degradation product of glucose-dependent insulinotropic polypeptide, GIP(3-42), is a GIP receptor antagonist in vivo. J Endocrinol. (2002) 175:525–33. doi: 10.1677/joe.0.1750525
66
Gault V O’Harte FPM Harriott P Flatt PR . Characterization of the cellular and metabolic effects of a novel enzyme-resistant antagonist of glucose-dependent insulinotropic polypeptide. Biochem And Biophys Res Commun. (2002) 290. https://www.sciencedirect.com/science/article/abs/pii/S0006291X02963643?via%3Dihub.
67
Gault VA O'Harte FPM Harriott P Mooney MH Green BD Flatt PR et al . Effects of the novel (Pro3)GIP antagonist and exendin(9–39)amide on GIP- and GLP-1-induced cyclic AMP generation, insulin secretion and postprandial insulin release in obese diabetic (ob/ob) mice: evidence that GIP is the major physiological incretin. Diabetologia. (2003) 46:222–30. doi: 10.1007/s00125-002-1028-x
68
Sparre-Ulrich AH Hansen LS Svendsen B Christensen M Knop FK Hartmann B et al . Species-specific action of (Pro3)GIP – a full agonist at human GIP receptors, but a partial agonist and competitive antagonist at rat and mouse GIP receptors. Br J Pharmacol. (2016) 173:27–38. doi: 10.1111/bph.13323
69
Fulurija A Lutz TA Sladko K Osto M Wielinga PY Bachmann MF et al . Vaccination against GIP for the treatment of obesity. PloS One. (2008) 3:e3163. doi: 10.1371/journal.pone.0003163
70
Kerr BD Flatt AJS Flatt PR Gault VA . Characterization and biological actions of N-terminal truncated forms of glucose-dependent insulinotropic polypeptide. Biochem Biophys Res Commun. (2010) 404:870–6. doi: 10.1016/j.bbrc.2010.12.077
71
Nakamura T Tanimoto H Mizuno Y Tsubamoto Y Noda H . Biological and functional characteristics of a novel low–molecular weight antagonist of glucose-dependent insulinotropic polypeptide receptor, SKL-14959, in vitro and in vivo. Diabetes Obes Metab. (2011) 14:511–7. doi: 10.1111/j.1463-1326.2011.01555.x
72
Ravn P Madhurantakam C Kunze S Matthews E Priest C O'Brien S et al . Structural and pharmacological characterization of novel potent and selective monoclonal antibody antagonists of glucose-dependent insulinotropic polypeptide receptor. J Biol Chem. (2013) 288:19760–72. doi: 10.1074/jbc.M112.426288
73
Pathak V Gault VA Flatt PR Irwin N . Antagonism of gastric inhibitory polypeptide (GIP) by palmitoylation of GIP analogues with N- and C-terminal modifications improves obesity and metabolic control in high fat fed mice. Mol Cell Endocrinol. (2015) 401:120–9. doi: 10.1016/j.mce.2014.10.025
74
Perry R Craig SL Ng MT Gault VA Flatt PR Irwin N . Characterisation of glucose-dependent insulinotropic polypeptide receptor antagonists in rodent pancreatic beta cells and mice. Clin Med Insights: Endocrinol Diabetes. (2019) 12:1179551419875453. doi: 10.1177/1179551419875453
75
Gasbjerg LS Christensen MB Christensen MB Hartmann B Lanng AR Sparre-Ulrich AH Gabe MBN et al . GIP(3-30)NH2 is an efficacious GIP receptor antagonist in humans: a randomised, double-blinded, placebo-controlled, crossover study. Diabetologia. (2018) 61:413–23. doi: 10.1007/s00125-017-4447-4
76
Lynggaard MB Gasbjerg LS Christensen MB Knop FK . GIP(3-30)NH2 – a tool for the study of GIP physiology. Curr Opin Pharmacol. (2020) 55:31–40. doi: 10.1016/j.coph.2020.08.011
77
Hoe B Heimbürger SMN Gasbjerg LS Lynggaard MB Hartmann B Holst JJ et al . 89-LB: the effect of GIP on plasma glucose in a setting of prandial insulin overdose and physical activity after meal intake in patients with type 1 diabetes. Diabetes. (2020) 69:89–LB. doi: 10.2337/db20-89-LB
78
Stensen S Gasbjerg LS Krogh LL Skov-Jeppesen K Sparre-Ulrich AH Jensen MH et al . Effects of endogenous GIP in patients with type 2 diabetes. Eur J Endocrinol. (2021) 185:33–45. doi: 10.1530/EJE-21-0135
79
Hindsø M Hedbäck N Svane MS Møller A Martinussen C Jørgensen NB et al . The importance of endogenously secreted GLP-1 and GIP for postprandial glucose tolerance and β-cell function after roux-en-Y gastric bypass and sleeve gastrectomy surgery. Diabetes. (2023) 72:336–47. doi: 10.2337/db22-0568
80
Jensen MH Gasbjerg LS Skov-Jeppesen K Jacobsen JCB Poulsen SS Zhou C et al . GIP receptor antagonism eliminates paradoxical growth hormone secretion in some patients with acromegaly. J Clin Endocrinol Metab. (2024) 10, dgae583. doi: 10.1210/clinem/dgae583
81
Krogh LL Nielsen CK Bergmann NC Holst JJ Lund A Gasbjerg LS et al . Extrapancreatic effects of endogenous incretin hormones evaluated using incretin hormone receptor antagonists in totally pancreatectomised patients. Diabetologia. (2023) 66:1–536. doi: 10.1007/s00125-023-05969-6
82
Sparre-Ulrich AH Sparre-Ulrich AH Gabe MN Gasbjerg LS Christiansen CB Svendsen B Hartmann et al . GIP(3–30)NH2 is a potent competitive antagonist of the GIP receptor and effectively inhibits GIP-mediated insulin, glucagon, and somatostatin release. Biochem Pharmacol. (2017) 131:78–88. doi: 10.1016/j.bcp.2017.02.012
83
Baldassano S Gasbjerg LS Kizilkaya HS Rosenkilde MM Holst JJ Hartmann B et al . Increased body weight and fat mass after subchronic GIP receptor antagonist, but not GLP-2 receptor antagonist, administration in rats. Front Endocrinol. (2019) 10:492. doi: 10.3389/fendo.2019.00492
84
Véniant MM Lu S-C Atangan L Komorowski R Stanislaus S Cheng Y et al . A GIPR antagonist conjugated to GLP-1 analogues promotes weight loss with improved metabolic parameters in preclinical and phase 1 settings. Nat Metab. (2024) 6:290–303. doi: 10.1038/s42255-023-00966-w
85
Amgen . Amgen announces robust weight loss with Maritide in people living with obesity or overweight at 52 weeks in a phase 2 study. Thousand Oaks, Californien: Amgen (2024). Available at: https://www.amgen.com/newsroom/press-releases/2024/11/amgen-announces-robust-weight-loss-with-maritide-in-people-living-with-obesity-or-overweight-at-52-weeks-in-a-phase-2-study.
86
Yang B Gelfanov VM El K Chen A Rohlfs R DuBois B et al . Discovery of a potent GIPR peptide antagonist that is effective in rodent and human systems. Mol Metab. (2022) 66:101638. doi: 10.1016/j.molmet.2022.101638
87
Gasbjerg LS Helsted MM Hartmann B Sparre-Ulrich AH Veedfald S Stensen S et al . GIP and GLP-1 receptor antagonism during a meal in healthy individuals. J Clin Endocrinol Metab. (2020) 105:1–14. doi: 10.1210/clinem/dgz175
88
Nauck MA Heimesaat MM Orskov C Holst JJ Ebert R Creutzfeldt W . Preserved incretin activity of glucagon-like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest. (1993) 91:301–7. doi: 10.1172/JCI116186
89
Vilsbøll T Krarup T Madsbad S Holst J . Defective amplification of the late phase insulin response to glucose by GIP in obese Type II diabetic patients. Diabetologia. (2002) 45:1111–9. doi: 10.1007/s00125-002-0878-6
90
Koefoed-Hansen F et al . Blocking the effects of endogenous GIP indcreases fasting plasma glucose levels in individuals with obesity(2024). Available online at: https://www.easd.org/media-centre/!resources/b-blocking-the-effects-of-endogenous-gip-increases-fasting-plasma-glucose-levels-in-individuals-with-obesity-b-ad10301d-56e2-43dc-a81a-d3a421263cfb (Accessed January 31, 2025).
91
Samms RJ Christe ME Collins KAL Pirro V Droz BA Holland AK et al . GIPR agonism mediates weight-independent insulin sensitization by tirzepatide in obese mice. J Clin Invest. (2021) 131:1–14. doi: 10.1172/JCI146353
92
Svane MS Bojsen-Møller KN Nielsen S Jørgensen NB Dirksen C Bendtsen F et al . Effects of endogenous GLP-1 and GIP on glucose tolerance after Roux-en-Y gastric bypass surgery. Am J Physiology-Endocrinology Metab. (2016) 310:E505–14. doi: 10.1152/ajpendo.00471.2015
93
Lund A Vilsbøll T Bagger JI Holst JJ Knop FK . The separate and combined impact of the intestinal hormones, GIP, GLP-1, and GLP-2, on glucagon secretion in type 2 diabetes. Am J Physiology-Endocrinology Metab. (2011) 300:E1038–46. doi: 10.1152/ajpendo.00665.2010
94
Yip RG-C Boylan MO Kieffer TJ Wolfe MM . Functional GIP receptors are present on adipocytes. Endocrinology. (1998) 139:4004–7. doi: 10.1210/endo.139.9.6288
95
Jones I Owens DR Luzio SD Hayes TM . Obesity is associated with increased post-prandial GIP levels which are not reduced by dietary restriction and weight loss. Diabete Metab. (1989) 15:11–22.
96
Wang F Yoder SM Yang Q Kohan AB Kindel TL et al . Chronic high-fat feeding increases GIP and GLP-1 secretion without altering body weight. Am J Physiology-Gastrointestinal Liver Physiol. (2015) 309:G807–15. doi: 10.1152/ajpgi.00351.2013
97
Meidtner K Milani L Mistry V Mitchell P Mohlke KL Moilanen L et al . Protein-altering variants associated with body mass index implicate pathways that control energy intake and expenditure in obesity. Nat Genet. (2018) 50:26–41. doi: 10.1038/s41588-017-0011-x
98
Akbari P Gilani A Sosina O Kosmicki JA Khrimian L Fang YY et al . Sequencing of 640,000 exomes identifies GPR75 variants associated with protection from obesity. Science. (2021) 373:eabf8683. doi: 10.1126/science.abf8683
99
Kizilkaya HS Sørensen KV Kibsgaard CJ Gasbjerg LS Hauser AS Sparre-Ulrich AH et al . Loss of function glucose-dependent insulinotropic polypeptide receptor variants are associated with alterations in BMI, bone strength and cardiovascular outcomes. Front Cell Dev Biol. (2021) 9:749607. doi: 10.3389/fcell.2021.749607
100
Gasbjerg LS Hartmann B Christensen MB Lanng AR Vilsbøll T Jørgensen NR et al . GIP’s effect on bone metabolism is reduced by the selective GIP receptor antagonist GIP(3–30)NH2. Bone. (2020) 130:1–6. doi: 10.1016/j.bone.2019.115079
101
Torekov SS Harsløf T Rejnmark L Eiken P Jensen JB Herman AP et al . A functional amino acid substitution in the glucose-dependent insulinotropic polypeptide receptor (GIPR) gene is associated with lower bone mineral density and increased fracture risk. J Clin Endocrinol Metab. (2014) 99:E729–33. doi: 10.1210/jc.2013-3766
102
Styrkarsdottir U Tragante V Stefansdottir L Thorleifsson G Oddsson A Sørensen E et al . Obesity variants in the GIPR gene are not associated with risk of fracture or bone mineral density. J Clin Endocrinol Metab. (2024) 109:e1608–15. doi: 10.1210/clinem/dgad734
103
Speliotes EK Willer CJ Berndt SI Monda KL Thorleifsson G Jackson AU et al . Association analyses of 249,796 individuals reveal eighteen new loci associated with body mass index. Nat Genet. (2010) 42:937–48. doi: 10.1038/ng.686
104
Novikoff A Müller TD . Antagonizing GIPR adds fire to the GLP-1R flame. Trends Endocrinol Metab. (2024) 35:566–8. doi: 10.1016/j.tem.2024.04.016
105
Hammoud R Drucker DJ . Beyond the pancreas: contrasting cardiometabolic actions of GIP and GLP1. Nat Rev Endocrinol. (2023) 19:201–16. doi: 10.1038/s41574-022-00783-3
106
Knop FK Urva S Rettiganti M Benson CT Roell WC Mather KJ et al . A long-acting glucose-dependent insulinotropic polypeptide receptor agonist improves the gastrointestinal tolerability of glucagon-like peptide-1 receptor agonist therapy. Diabetes Obes Metab. (2024) 26:5474–8. doi: 10.1111/dom.15875
107
Kaplan AM Vigna SR . Gastric inhibitory polypeptide (GIP) binding sites in rat brain. Peptides. (1994) 15:297–302. doi: 10.1016/0196-9781(94)90016-7
108
Irwin N Gault V Flatt PR . Therapeutic potential of the original incretin hormone glucose-dependent insulinotropic polypeptide: diabetes, obesity, osteoporosis and Alzheimer’s disease? Expert Opin Investigational Drugs. (2010) 19:1039–48. doi: 10.1517/13543784.2010.513381
109
Porter DW Irwin NI Flatt PR Hölscher C Gault VA . Prolonged GIP receptor activation improves cognitive function, hippocampal synaptic plasticity and glucose homeostasis in high-fat fed mice. Eur J Pharmacol. (2011) 650:688–93. doi: 10.1016/j.ejphar.2010.10.059
110
Nyberg J Anderson MF Meister BJ Alborn AM Ström AK Brederlau A et al . Glucose-dependent insulinotropic polypeptide is expressed in adult hippocampus and induces progenitor cell proliferation. J Neurosci. (2005) 25:1816–25. doi: 10.1523/JNEUROSCI.4920-04.2005
111
Faivre E Gault VA Thorens B Hölscher C . Glucose-dependent insulinotropic polypeptide receptor knockout mice are impaired in learning, synaptic plasticity, and neurogenesis. J Neurophysiol. (2011) 105:1574–80. doi: 10.1152/jn.00866.2010
112
Nitz I Fisher E Weikert C Burwinkel B Li Y Möhlig M et al . Association analyses of GIP and GIPR polymorphisms with traits of the metabolic syndrome. Mol Nutr Food Res. (2007) 51:1046–52. doi: 10.1002/mnfr.200700048
113
Bowker N Hansford R Burgess S Foley CN Auyeung VPW Erzurumluoglu AM et al . Genetically predicted glucose-dependent insulinotropic polypeptide (GIP) levels and cardiovascular disease risk are driven by distinct causal variants in the GIPR region. Diabetes. (2021) 70:2706–19. doi: 10.2337/db21-0103
114
Gasbjerg LS Gabe MBN Hartmann B Christensen MB Knop FK Holst JJ et al . Glucose-dependent insulinotropic polypeptide (GIP) receptor antagonists as anti-diabetic agents. Peptides. (2018) 100:173–81. doi: 10.1016/j.peptides.2017.11.021
115
Gasbjerg LS Bari EJ Christensen MK Knop FK . Exendin(9-39) NH 2 : Recommendations for clinical use based on a systematic literature review. Diabetes Obes Metab. (2021) 23:2419–36. doi: 10.1111/dom.14507
116
Wilding JPH Batterham RL Calanna S Davies M Van Gaal LF Lingvay I et al . Once-weekly semaglutide in adults with overweight or obesity. New Engl J Med. (2021) 384:989–1002. doi: 10.1056/NEJMoa2032183
117
Frias JP Nauck MA Van J Benson C Bray R Cui X et al . Efficacy and tolerability of tirzepatide, a dual glucose-dependent insulinotropic peptide and glucagon-like peptide-1 receptor agonist in patients with type 2 diabetes: A 12-week, randomized, double-blind, placebo-controlled study to evaluate different dose-escalation regimens. Diabetes Obes Metab. (2020) 22:938–46. doi: 10.1111/dom.13979
118
Jastreboff AM Aronne LJ Ahmad NN Wharton S Connery L Alves B et al . Tirzepatide once weekly for the treatment of obesity. New Engl J Med. (2022) 387:205–16. doi: 10.1056/NEJMoa2206038
119
Adriaenssens AE Biggs EK Darwish T Tadross J Sukthankar T Girish M et al . Glucose-dependent insulinotropic polypeptide receptor-expressing cells in the hypothalamus regulate food intake. Cell Metab. (2019) 30:987–96.e6. doi: 10.1016/j.cmet.2019.07.013
120
Rosenstock J Wysham C Frías JP Kaneko S Lee CJ Fernández Landó L et al . Efficacy and safety of a novel dual GIP and GLP-1 receptor agonist tirzepatide in patients with type 2 diabetes (SURPASS-1): a double-blind, randomised, phase 3 trial. Lancet. (2021) 398:143–55. doi: 10.1016/S0140-6736(21)01324-6
121
Willard FS Douros JD Gabe MBN Showalter AD Wainscott DB Suter TM et al . Tirzepatide is an imbalanced and biased dual GIP and GLP-1 receptor agonist. JCI Insight. (2020) 5:e140532. doi: 10.1172/jci.insight.140532
122
Gasbjerg LS Rasmussen RS Dragan A Lindquist P Melchiorsen JU Stepniewski TM et al . Altered desensitization and internalization patterns of rodent versus human glucose-dependent insulinotropic polypeptide (GIP) receptors. An important drug discovery challenge. Br J Pharmacol. (2024) 181, bph.16478. doi: 10.1111/bph.16478
123
Kusminski CM Perez-Tilve D Müller TD DiMarchi RD Tschöp MH Schere PE . Transforming obesity: The advancement of multi-receptor drugs. Cell. (2024) 187:3829–53. doi: 10.1016/j.cell.2024.06.003
124
Rosenkilde MM Lindquist P Kizilkaya HS Gasbjerg LS . GIP-derived GIP receptor antagonists – a review of their role in GIP receptor pharmacology. Peptides. (2024) 177:171212. doi: 10.1016/j.peptides.2024.171212
Summary
Keywords
GIP -glucose-dependent insulinotropic polypeptide, antagonism, obesity, diabetes, pharmacology
Citation
Koefoed-Hansen F, Helsted MM, Kizilkaya HS, Lund AB, Rosenkilde MM and Gasbjerg LS (2025) The evolution of the therapeutic concept ‘GIP receptor antagonism’. Front. Endocrinol. 16:1570603. doi: 10.3389/fendo.2025.1570603
Received
03 February 2025
Accepted
29 April 2025
Published
21 May 2025
Volume
16 - 2025
Edited by
Jared Rutter, The University of Utah, United States
Reviewed by
Steven Patterson, Glasgow Caledonian University, United Kingdom
Elena Jimenez Marti, University of Valencia, Spain
Nigel Irwin, Ulster University, United Kingdom
Updates
Copyright
© 2025 Koefoed-Hansen, Helsted, Kizilkaya, Lund, Rosenkilde and Gasbjerg.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lærke Smidt Gasbjerg, lsg@sund.ku.dk
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.