 Joanna Hubska1,2*
Joanna Hubska1,2* Zuzanna Roszkowska3
Zuzanna Roszkowska3 Małgorzata Bobrowicz1Sebastian Iwaniuk3
Małgorzata Bobrowicz1Sebastian Iwaniuk3 Beata Rak-Makowska1,4
Beata Rak-Makowska1,4 Urszula Ambroziak1
Urszula Ambroziak1- 1Department of Internal Medicine and Endocrinology, Medical University of Warsaw, Warsaw, Poland
- 2Doctoral School of the Medical University of Warsaw, Warsaw, Poland
- 3Student Scientific Club “Endocrinus” Affiliated to the Department of Internal Medicine and Endocrinology, Medical University of Warsaw, Warsaw, Poland
- 4Laboratory of Experimental Medicine, Medical University of Warsaw, Warsaw, Poland
Congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency (21OHD) is a complex endocrine disorder characterized by impaired cortisol synthesis and androgen excess. Beyond its hormonal and metabolic implications, CAH has been increasingly associated with an elevated risk of cardiovascular complications, including endothelial dysfunction, a critical precursor to atherosclerosis and a risk factor for cardiovascular and metabolic diseases. This review explores the current knowledge on endothelial function in patients with CAH, focusing on the interplay between chronic hormonal imbalance, prolonged glucocorticoid treatment, and associated metabolic disorders. We also discuss in vivo methods for assessing endothelial function alongside the potential utility of novel biomarkers, which may facilitate earlier identification of vascular dysfunction and stratification of cardiovascular risk. By summarizing emerging concepts in this field, we aim to highlight areas for future research and opportunities for improving long-term cardiovascular outcomes in individuals with 21OHD.
1 Introduction
Congenital adrenal hyperplasia (CAH), due to 21-hydroxylase deficiency (21-OHD), is an autosomal recessive condition that is caused by mutations in the gene CYP21A2. It is characterized by impaired cortisol secretion and androgen excess. 21-OHD is the most common cause of CAH, accounting for 95% of cases (1). Based on the residual enzyme activity, CAH shows a spectrum of phenotypes, varying from a severe classic CAH (CCAH), usually diagnosed in newborns, to a non-classic CAH (NCCAH), which is a mild variant often diagnosed late, if ever. CCAH is classified into two forms based on aldosterone deficiency: salt-wasting (SW) and simple virilizing (SV). The primary treatment for CAH, particularly CCAH, involves glucocorticoid and mineralocorticoid replacement to prevent adrenal crises and manage excess androgen production. Achieving a balance between these treatments is essential to avoid both under- and over-treatment, as both extremes can have detrimental effects on long-term metabolic and cardiovascular health. However, even in an era of continuously advancing knowledge about CAH, improved patient care, and the availability of effective treatments, such as those mimicking the circadian rhythm of cortisol, the presence of CAH remains associated with numerous metabolic complications and increased cardiovascular morbidity (2).
Mounting evidence has shown that the dysfunction of endothelial cells in the vasculature is profoundly implicated in the pathogenesis of cardiovascular and metabolic diseases (3). Furthermore, there is a bidirectional relationship between endothelial dysfunction and these disorders. Components of metabolic syndrome, such as abdominal obesity, hypertension, and impaired glycemic control, can contribute to endothelial dysfunction (4). Conversely, structural and functional changes in the endothelium promote the progression of metabolic diseases and atherosclerosis (5). Given the hormonal imbalances and systemic effects of CAH, understanding its potential impact on endothelial function is crucial, as individuals with CAH are at higher risk for these conditions (6, 7).
Despite the growing body of evidence linking CAH to endothelial dysfunction, significant research gaps persist. To date, no studies have systematically assessed the relationship between endothelial dysfunction and glucocorticoid dose, type, or treatment duration. Importantly, the differential impact of various chronic glucocorticoid replacement regimens on endothelial function has not been evaluated in controlled studies (8). Moreover, the progression of endothelial dysfunction over time in individuals with CAH remains insufficiently understood. To date, no randomized controlled trials have been conducted in this area, and no meta-analyses are available to synthesize the existing evidence.
In this review, we provide an overview of current knowledge on endothelial function in individuals with CAH, with a particular focus on the factors that contribute to endothelial damage, methods of endothelial assessment, and novel biomarkers that could help to detect patients at higher risk. We also discuss gaps in knowledge and areas for future research.
2 Endothelial dysfunction and its role in cardiovascular disease
The endothelium, a single-cell layer lining the inner surface of blood vessels, plays a vital role in maintaining vascular homeostasis. The endothelium releases various autocrine, paracrine, and endocrine substances, such as nitric oxide (NO), C-type natriuretic peptide, prostacyclin, and endothelium-derived hyperpolarizing factor (9). These factors collectively inhibit smooth muscle cell proliferation and migration, prevent platelet adhesion and aggregation, and regulate processes that influence thrombogenesis (10).
Endothelial dysfunction involves a shift in endothelial cell behavior, leading to various maladaptive changes in their functional phenotype. This results in disturbances in the regulation of hemostasis, thrombosis, vascular tone, redox balance, and inflammatory processes (11). The underlying pathophysiology is multifaceted, involving several mechanisms.
A key factor in the development of endothelial dysfunction is oxidative stress, which arises from multiple enzymatic sources such as xanthine oxidase, NADPH oxidases, uncoupled endothelial nitric oxide synthase (eNOS), and malfunctioning mitochondria. It occurs when the balance between pro-oxidants and antioxidants is disrupted (12). Elevated reactive oxygen species (ROS) levels can oxidize cellular macromolecules and reduce NO production by promoting the formation of peroxynitrite (12), a toxic compound that degrades the eNOS cofactor tetrahydrobiopterin (13), leading to the “uncoupling” of eNOS and increased oxidative stress. This oxidative imbalance also contributes to impaired endothelial vasodilation and a proinflammatory environment, as well as the upregulation of adhesion molecules such as intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1), along with chemotactic molecules (12) (Figure 1).
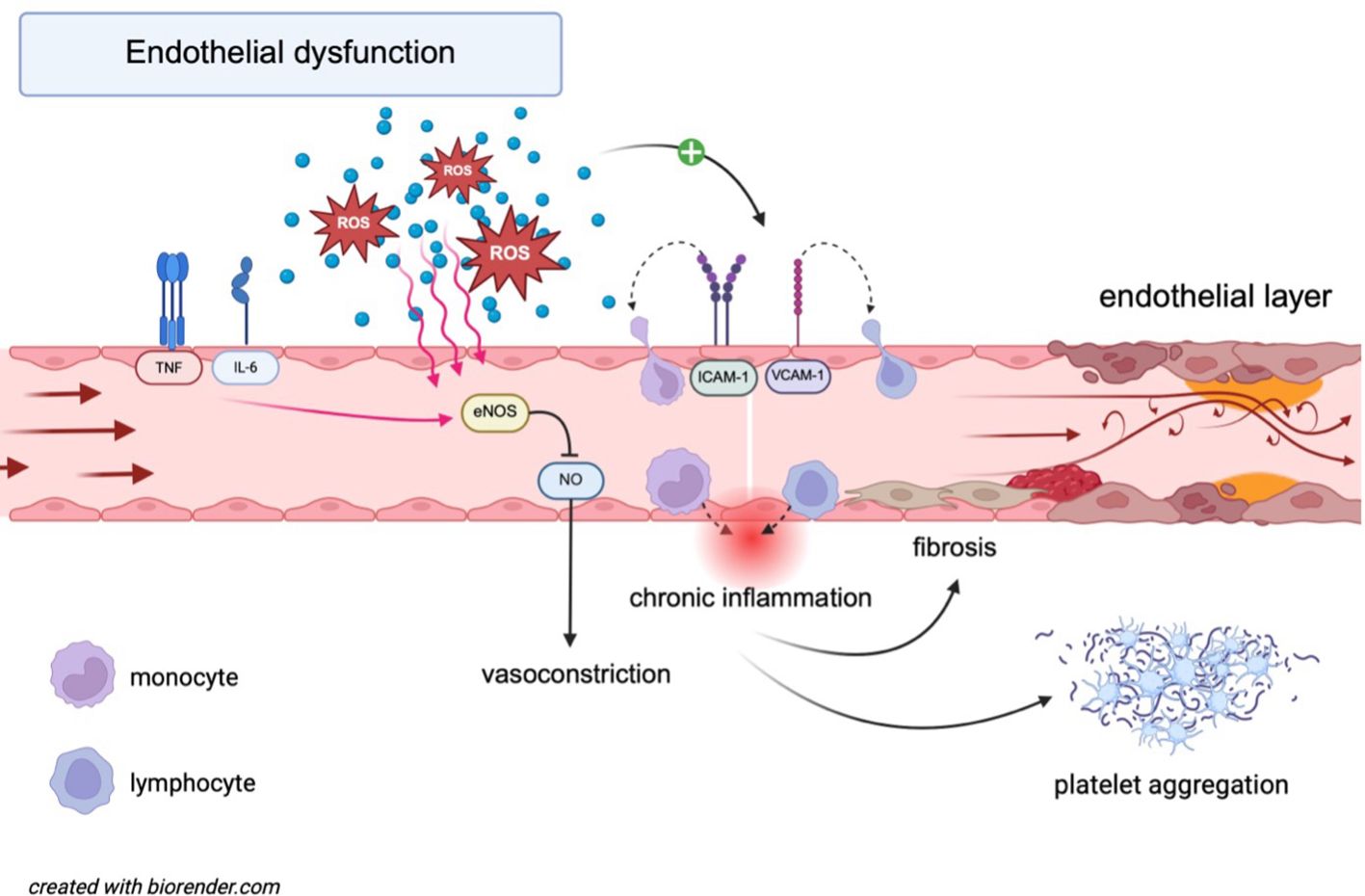
Figure 1. Molecular mechanisms underlying endothelial dysfunction. IL-6 and TNF-α induce eNOS dysfunction, leading to reduced NO bioavailability and vasoconstriction. ROS contribute to oxidative stress, further impairing eNOS activity and promoting chronic inflammation. ROS also upregulate adhesion molecules (ICAM-1, VCAM-1), facilitating monocyte recruitment. This cascade promotes the adhesion and migration of lymphocytes across the endothelial barrier, further activating an inflammatory state. Platelet aggregation is simultaneously enhanced, increasing the risk of thrombosis. Collectively, these processes drive endothelial dysfunction and contribute to the progression of cardiovascular disease. eNOS, endothelial nitric oxide synthase; ICAM-1, intercellular adhesion molecule 1; IL-6, interleukin-6; NO, nitric oxide; ROS, reactive oxygen species; TNF, tumor necrosis factor; VCAM-1, vascular cell adhesion molecule 1.
Inflammation plays a crucial role in the pathogenesis of cardiovascular disease (14). In response to vascular injury, endothelial cells release a variety of inflammatory molecules, including chemokines, interleukin-8, colony-stimulating factors, monocyte chemoattractant protein-1 (MCP-1), adhesion molecules such as ICAM-1 and E-selectin, and growth factors and other inflammatory mediators (15), leading to the attachment of monocytes and their migration into the vessel wall. Monocyte-derived macrophages ingest oxidized low-density lipoprotein (LDL), forming foam cells and fatty streaks, which lead to the development of plaques affecting the coronary arteries, aorta, and carotid arteries, ultimately resulting in atherosclerosis (16). This cascade promotes the adhesion and migration of leukocytes across the endothelial barrier, further activating an inflammatory state (17). In addition, proinflammatory cytokines such as tumor necrosis factor-alpha (TNF-α) and interferon-gamma (IFN-γ) are released by endothelial cells, activating a vicious circle (18).
Endothelial dysfunction is often widespread throughout the body, as individuals with diagnosed atherosclerosis frequently exhibit endothelial dysfunction in peripheral vascular regions that may not yet show overt signs of the disease. It is also observed in those with a family history of early cardiovascular disease despite the absence of other risk factors (19); in individuals with hypertriglyceridemia (20), dyslipidemia (21), nicotine use (22), and insulin resistance (23, 24); first-degree relatives with type 2 diabetes; and elderly patients regardless of the presence of other comorbidities (25). The advancement of endothelial dysfunction is influenced by the severity and duration of established risk factors and the overall risk profile of individual patients (26).
3 The risks for endothelial dysfunction in CAH
The exact mechanisms behind the higher prevalence of cardiometabolic risk elements in patients with CAH remain unclear. Nonetheless, both disease-related and treatment-related elements are being discussed. Importantly, cardiovascular disease is the second most common cause of death in patients with CAH after adrenal crisis (27), and is closely linked to endothelial dysfunction (28).
Individuals with CAH are at an increased risk of developing metabolic syndrome, which is characterized by a cluster of metabolic abnormalities, including central obesity, insulin resistance, hypertension, and atherogenic dyslipidemia (29) (Figure 2). Obesity is the most common component of metabolic syndrome in both children and adults with CAH and acts as a major independent risk factor for cardiovascular diseases (1). In patients with CAH, the prevalence of obesity ranges from 30% to 40% (30–34). It contributes to endothelial dysfunction through associated complications such as hypertension, dyslipidemia, type 2 diabetes, and obstructive sleep apnea (35, 36). The excess fat accumulation in obesity leads to adipocyte dysfunction, triggering oxidative stress and insulin resistance, while also serving as a source of pro-inflammatory cytokines, all of which contribute to endothelial dysfunction (36, 37). Additionally, patients with CAH are more likely to develop increased visceral adipose tissue, a well-established risk factor for cardiovascular diseases (38).
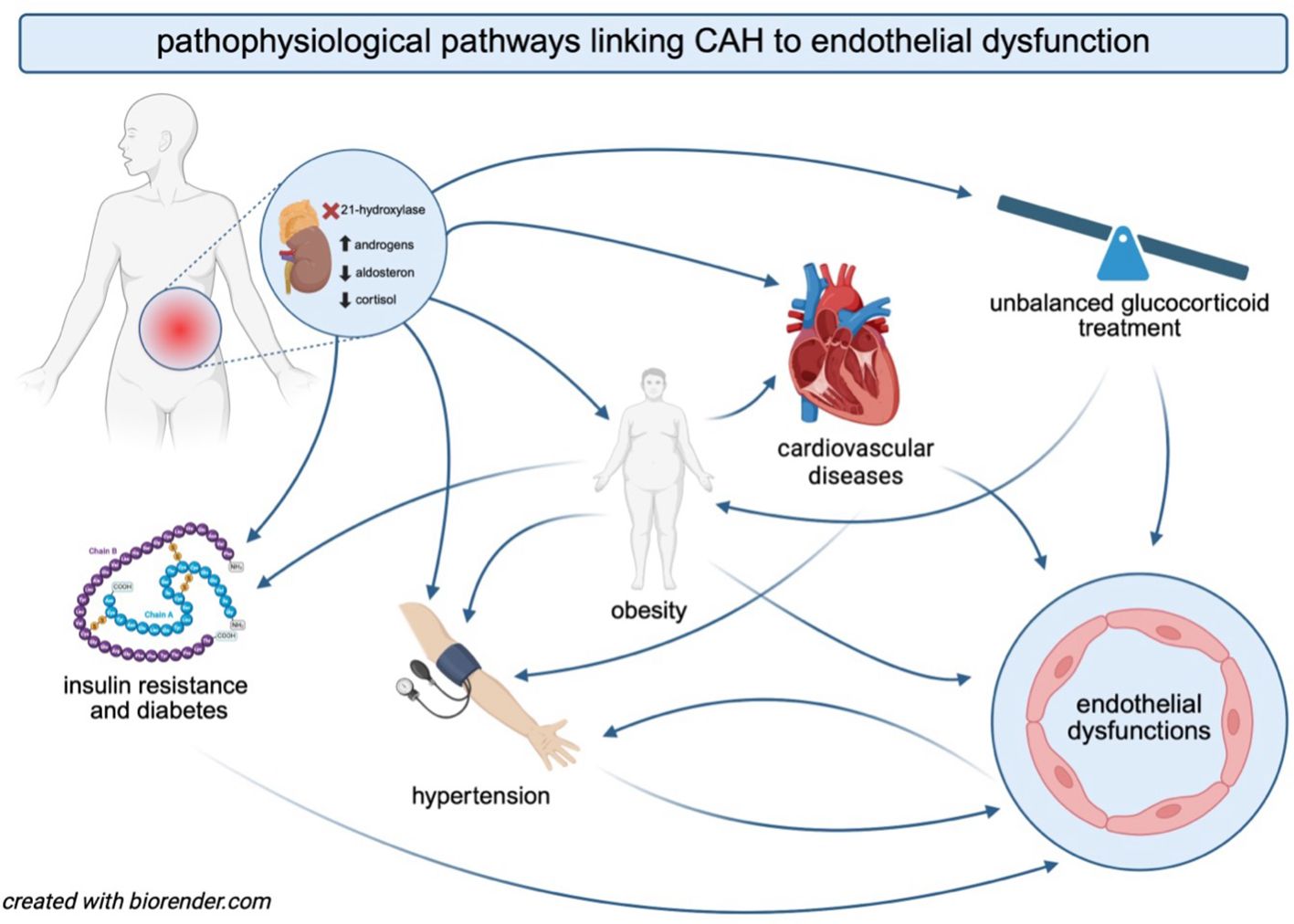
Figure 2. The interplay between CAH, the pathophysiology of comorbidities, and endothelial dysfunction. The figure illustrates the interconnected mechanisms through which CAH contributes to an increased risk of endothelial dysfunction. Hormonal imbalances in CAH promote obesity, insulin resistance, diabetes, and cardiovascular diseases. Glucocorticoid therapy, essential for managing cortisol deficiency in CAH, may further exacerbate endothelial dysfunction and increase cardiovascular risk. Obesity and insulin resistance contribute to the development of hypertension, which, in turn, accelerates endothelial damage and cardiovascular complications. The bidirectional interactions among these factors create a vicious cycle, ultimately predisposing individuals with CAH to an elevated risk of cardiovascular morbidity.
Notably, endothelial dysfunction is a key component of metabolic syndrome, with involvement in both the initiation and propagation of this condition (39). In patients with CAH, these abnormalities arise from cortisol deficiency, excess androgen secretion, and hypercortisolism due to possible glucocorticoid overtreatment. Additionally, dysfunction of the adrenomedullary system, marked by deficient epinephrine secretion, may contribute to reduced lipolysis of triglyceride stores and disruptions in insulin and adipokine regulation (40–42).
Abnormal androgen levels in CAH should be considered among the risk factors for endothelial dysfunction (Figure 2). Both hypoandrogenism in male patients and hyperandrogenism in female patients can contribute to adverse metabolic effects, thereby increasing cardiovascular risk (43–45). Arlt et al. (31) found that the majority of patients with CAH exhibited either elevated or suppressed androgen levels, with only 36% showing normal androstenedione levels. The detrimental effects of androgen excess in CAH were further confirmed in a cohort of women with CCAH SV untreated with glucocorticoids; insulin resistance and unfavorable metabolic markers were notably increased in these patients compared to the control group, and showed a direct correlation to testosterone levels (46). Paizoni et al. (30) also supported the role of androgens in insulin resistance development in women with CCAH, especially in those with poor androgen control. Similarly, there are studies in NCCAH that emphasize the relationship of increased testosterone levels with insulin resistance (47). Moreover, studies in patients with polycystic ovary syndrome (PCOS) have shown a link between hyperandrogenism and impaired endothelial function (48, 49), suggesting that elevated androgen levels may significantly contribute to endothelial dysfunction.
Hypertension is another major risk factor for cardiovascular disease, and in CAH, an increased frequency of hypertension overall is observed across age groups, although more prevalent in children compared to adults (50, 51). Importantly, the relationship between systemic arterial hypertension and endothelial dysfunction is bidirectional, amplifying the severity of both conditions. Endothelial cells influence the development of systemic hypertension through various mediators, while systemic hypertension exacerbates endothelial dysfunction, contributing to a prothrombotic, proinflammatory, and proatherosclerotic state (52). It is well established that excess glucocorticoids can elevate arterial blood pressure mainly through mineralocorticoid mimetic effects, vascular remodeling, and impaired NO signaling (53). However, the role of mineralocorticoid treatment should also be considered; patients with CAH receiving fludrocortisone tend to experience elevated blood pressure more frequently than those who do not (54).
The majority of studies have reported elevated blood pressure values in patients with CAH (32, 55, 56). In youth with CAH, a positive correlation between body mass index (BMI) and blood pressure has been observed (57, 58), highlighting a significant association between hypertension and obesity in this population. Furthermore, even in the absence of clinically overt hypertension (47), individuals with CAH may show a reduced physiological nocturnal dip in blood pressure (30, 59, 60). Gender differences have been examined in a limited number of studies, with most finding a similar prevalence of hypertension between men and women with CAH (32, 61–63). However, two studies suggested that women with CAH may be more affected than men, likely due to excessive androgen exposure (47, 64).
The cardiometabolic status in CAH is strongly affected by the medications used in therapy, as the mainstay of CAH management involves the intake of glucocorticoids and mineralocorticoids (Figure 2). The doses should be substitutive; however, patients frequently fail to adhere to the guidelines. Excessive intake of glucocorticoids and mineralocorticoids can raise cardiovascular risk factors, while inadequate glucocorticoid therapy or poor adherence may result in androgen excess, infertility, and the formation of adrenal rest tumors (65, 66). Therefore, preventing long-term metabolic and cardiovascular complications depends on maintaining an optimal balance between overtreatment and undertreatment, however, it remains a significant challenge in both CCAH and NCCAH (67).
Chronic glucocorticoid therapy has been shown to heighten the risk of developing insulin resistance and, subsequently, type 2 diabetes in patients with CAH (2, 31, 32). Adult patients with CAH exhibit elevated fasting plasma glucose levels (31), reduced insulin sensitivity, and a β-cell response that is unable to compensate for insulin resistance (68). Significantly, insulin resistance in CAH seems to be related not only to the cumulative dose of glucocorticoids but also to the type of glucocorticoid used. Patients on long-term dexamethasone show a higher prevalence of insulin resistance compared to those taking prednisolone or hydrocortisone (8).
The impact of glucocorticoids on vascular function, particularly in the context of treatment adherence, remains poorly understood. Non-adherence to treatment in CAH has been correlated with detrimental effects on health, including greater intima–media thickness (69) and a poorer quality of life (70). Finkielstain et al. (32) emphasized the critical importance of consistent treatment adherence in modulating disease outcomes, further highlighting the urgent need for longitudinal investigations in this population.
Interestingly, the effects of systemic glucocorticoid therapy in CAH can also be influenced at the receptor level. Variations in the glucocorticoid receptor gene (NR3C1) may be associated with either negative or positive metabolic and cardiovascular profiles (71). For example, the A3669G polymorphism is linked to unfavorable lipid profiles in pediatric patients with CAH, while the ER22/23EK haplotype reduces glucocorticoid sensitivity, leading to a more favorable metabolic profile. In contrast, the N363S and BclI restriction fragment length polymorphisms increase glucocorticoid sensitivity, raising the risk of type 2 diabetes, obesity, and cardiovascular diseases (72, 73). BclI heterozygotes with CAH show higher body mass index (BMI), waist circumference, and systolic blood pressure compared to those with the wild-type (74), though this polymorphism is less common in patients with CAH than in the general population (71).
4 Assessment of endothelial function in CAH in vivo
The pivotal role of the endothelium in vascular-related diseases has driven increased scientific attention in examining the endothelial function as a tool for screening, as well as for monitoring the course of the diseases and evaluating treatment outcomes (75). Traditionally, endothelial function is evaluated through endothelium-dependent vasomotion, which can be measured in either the coronary or peripheral circulation. While invasive angiography is still considered the gold standard for measuring coronary endothelial function (76), there is no agreement on the gold standard for the measurement of peripheral endothelial function (77).
Well-established non-invasive techniques for evaluating peripheral endothelial function include strain-gauge venous occlusion forearm plethysmography (78, 79), flow-mediated dilation (FMD) (79, 80), peripheral arterial tonometry (PAT) (79), and laser Doppler flowmetry (79). Less conventional non-invasive techniques include pulse wave velocity (PWV) (81) and indirect endothelial assessment via intima-media thickness (IMT) measurement (82) (Table 1). Unlike invasive techniques, which are associated with risks such as vascular injury, infection, and procedural complications, non-invasive methods are inherently safer, cost-efficient, logistically simpler, and well-suited for implementation in both clinical practice and large-scale epidemiological studies. According to several studies, non-invasive assessment of peripheral vascular function may be useful in identifying patients at risk for cardiac adverse events, including cardiac death, myocardial infarction, revascularization, or hospitalization for cardiac causes (83).
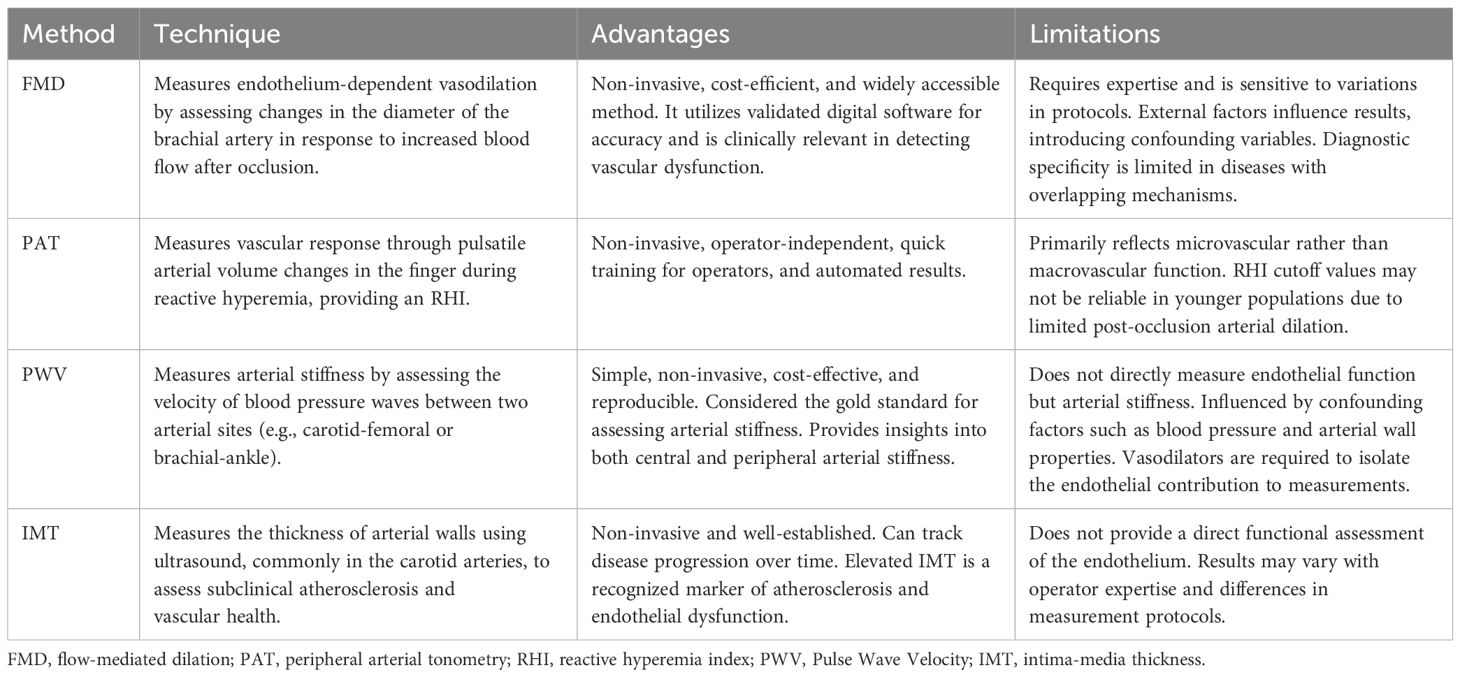
Table 1. Non-invasive methods of assessment of endothelial function.
4.1 Flow-mediated dilatation
One of the most used techniques to study endothelial function in vivo is FMD, which is assessed using ultrasound to measure changes in the diameter of the brachial artery in response to increased blood flow following a period of vascular occlusion created by a blood pressure cuff. This response is highly dependent on the availability of NO. Endothelial dysfunction is indicated by decreased vasodilation, as shown by lower FMD in the brachial artery.
The benefits of the FMD technique encompass cost-efficiency, non-invasiveness, accessibility, and the use of validated digital software for automated analyses. However, achieving optimal examination is technically demanding, and variations in techniques and protocols can impact the consistency and reliability of results. Moreover, factors such as diet, coffee consumption, medication, vitamins, physical activity, tobacco use, air temperature, and the menstrual cycle can influence a patient’s FMD, potentially introducing confounding variables into the study outcomes (28). Nonetheless, the brachial FMD method provides a validated, non-invasive evaluation of endothelial function (76).
Wierzbicka-Chmiel et al. (84) reported that 19 patients with CAH had decreased mean FMD compared with the control group. Farghaly et al. (16) showed that FMD was impaired in 40 patients with CAH and associated with elevated levels of neopterin and high sensitivity C-reactive protein (hs-CRP), the markers of vascular inflammation. According to Harrington et al. (85), impaired FMD was observed in a group of 14 adolescents with CAH, similar to obese control subjects. Given the limited number of studies assessing FMD in CAH, it is noteworthy that impaired FMD is a well-recognized indicator of endothelial dysfunction in other conditions associated with increased cardiovascular risk, such as type 2 diabetes (86–88), PCOS (89–91), obesity (92–94), heart failure (95), and peripheral artery disease (96).
4.2 Peripheral arterial tonometry
The PAT method, which is used in the EndoPAT device (Itamar Medical Inc., Caesarea, Israel), has emerged as a newer than FMD, non-invasive technology for measuring endothelial dysfunction. The device uses non-invasive pneumatic probes placed on both index fingers to measure pulsatile arterial volume changes at rest and during reactive hyperemia, which occurs in response to increased shear stress. A blood pressure cuff is placed over the brachial artery and inflated to occlude blood flow, and the response after deflation is recorded. The pulse wave amplitude (PWA) is measured, and the reactive hyperemia index (RHI) result is automatically calculated. The RHI is calculated as the ratio of the average PWA during the reactive phase to the average amplitude measured during the stabilization period. A suggested RHI threshold for indicating endothelial dysfunction is <1.67 (97–99). To adjust for systemic changes, this ratio is normalized using the concurrent signal from the contralateral finger. The EndoPAT device also assesses the peripheral augmentation index (AI), which measures arterial stiffness, calculated based on PWA.
The main advantages of PAT technology include its easy accessibility, operator independence, automated calculation, and control by the contralateral arm (83). PAT operators can be trained in a relatively short amount of time and do not require specialized certification. A major limitation is the unclear understanding of its pathophysiological basis (100). Unlike FMD, which evaluates macrovascular dilation, PAT assesses microvessel dilation. The endpoint measured by PAT, PWA, is thought to reflect arterial distensibility and venous capacitance in the digital vasculature, suggesting that changes in PWA may indicate vascular function. However, the structure of the digital vasculature is complex, comprising both nutritive vessels and arteriovenous anastomoses. The sympathetic nervous system primarily regulates the resting vascular tone in these vessels, with NO playing a minimal role (101). There are also concerns regarding the cutoff point for RHI, particularly in relation to age. Jujic et al. (102) postulated that an RHI result under 1.67, an early marker of endothelial dysfunction, may not be a suitable measure of endothelial function in individuals under 30 years of age. Their findings suggest that low RHI in young, healthy individuals may not necessarily indicate true endothelial dysfunction, but rather be an artefact of the limited ability of healthy arteries to dilate post-occlusion (102).
Available data on the positive predictive value (PPV) and negative predictive value (NPV) of the EndoPAT test are heterogeneous and vary depending on the studied population and clinical context. For instance, in the evaluation of erectile dysfunction, the PPV was relatively low at 43%, whereas the NPV reached 90%, indicating a greater utility in excluding rather than confirming organic endothelial dysfunction (103). In contrast, when EndoPAT was compared to the acetylcholine provocation test—the gold standard for diagnosing coronary endothelial dysfunction—the sensitivity was reported at 80% and specificity at 85%, supporting its potential role in identifying coronary artery spasm (99). These differences highlight the importance of interpreting EndoPAT results within the appropriate clinical framework.
Despite several limitations, numerous studies in both adult and pediatric literature reveal PAT’s satisfactory reproducibility and reliability (104–106). To date, no study utilizing PAT technology in individuals with CAH has been published. However, reduced RHI values have been reported in patients with other conditions associated with increased cardiovascular risk, including coronary artery disease (107), type 2 diabetes (108, 109), and metabolic syndrome (110). In contrast, the levels of RHI were consistent with preserved endothelial function in both groups of patients with PCOS, whether or not they had non-alcoholic fatty liver disease (NAFLD) (111).
4.3 Pulse wave velocity
PWV is the proposed gold standard for arterial stiffness and an indicator of early atherosclerosis (112), however, this technique can also be applied to studies of endothelial function (81). The predictive value of PWV for the occurrence of cardiovascular diseases has been demonstrated in both the general population and patients with various clinical conditions, including hypertension (113, 114), type 2 diabetes (115), end-stage renal disease (116), stroke (117), and coronary artery disease (118).
Measuring PWV involves the delay in the peak of the peripheral pulse wave, typically in relation to the QRS complex recorded simultaneously using electrocardiography. Increased vessel stiffness results in faster pulse wave propagation. The delay is determined by measurements taken at two different body sites, such as the carotid and femoral arteries (carotid-femoral PWV) or brachial and ankle arteries (brachial-ankle PWV). These two PWVs are most widely used in clinical and research fields. By selecting specific pulse wave recording points, it is possible to assess both central arterial stiffness and peripheral stiffness, independent of aortic condition. In endothelial studies, PWV measurements rely on the assumption that administering vasodilators isolates the contribution of vascular tone (119). PWV measurement is clinically beneficial due to its simplicity, non-invasive nature, cost-effectiveness, and reproducibility (120). Currently, no data are available on the PPV or NPV of PWV measurement in the assessment of endothelial function.
Costa et al. (121) did not observe any significant differences in PWV between 47 women with NCCAH and controls, nor across different therapy groups (dexamethasone vs. contraceptive pills). Similarly, Rosenbaum et al. (122) reported no significant differences in PWV values between 84 patients with CAH (both CCAH and NCCAH) and controls.
However, outside the CAH population, many studies have recently revealed an association between increased PWV and coronary atherosclerosis (123–125). In patients with PCOS, PWV measured at the brachial artery was found to be significantly elevated, although aortic PWV did not differ between the PCOS and control groups (126). Moreover, Wang et al. (127) reported that brachial-ankle PWV was associated with metabolic syndrome and increased progressively with the number of metabolic syndrome components in the general population.
4.4 Intima-media thickness
IMT is a measurement of the thickness of artery walls by ultrasound to detect the presence or track the progression of cardiovascular disease. In clinical practice, the IMT measurement is most commonly performed in the carotid arteries. A common carotid intima-media thickness (cIMT) measurement greater than 0.9 mm has been considered a significant factor influencing cardiovascular prognosis (128). Since increased IMT is a widely recognized sign of endothelial impairment (82) and a recognized indicator of atherosclerosis (129), which is closely linked to endothelial dysfunction, it can be used as an indirect measure of endothelial condition. Several studies have linked endothelial dysfunction with cIMT in patients with established atherosclerosis or coronary artery disease (130, 131). Consistently, endothelial dysfunction, defined as endothelium-dependent vasodilation (EDV) ≤4.5%, has been associated with a sensitivity of 71%, a specificity of 81%, and a PPV of 95% for the presence of coronary artery disease, further supporting the role of early vascular changes as markers of impaired endothelial function (130).
The exploration of IMT in relation to CAH has been the subject of multiple studies (60, 122, 132–138), with results varying significantly. Most of the studies (133–139) have reported notable differences in IMT between patients with CAH and controls, however, some research has shown normal IMT values, similar to those of control groups (60). The increase in cIMT is more pronounced in adults compared to youth (56), however, only three studies have examined adults (122, 133, 140), while the rest have focused on pediatric and adolescent populations (60, 134–140). Rosenbaum et al. (122) observed no significant difference between cIMT values in 84 adult patients with CAH and controls. Kim et al. (140) found that cIMT was associated with elevated androgen levels in 20 adolescents and young adults with CAH, with a loss of sex differences observed in female patients with excess androgen exposure. However, the subjects with CAH did not have significantly different cIMT values compared to controls. They also found that cIMT was significantly greater in obese than in non-obese individuals with CAH (140). Amr et al. (137) reported significantly higher CIMT in 32 children with CAH, without differences between the SV and SW forms of CCAH. Wasniewska et al. (138) observed increased IMT at different sites in 18 adolescents with CAH, with no differences between CCAH and NCCAH. Rodrigues et al. (136) has found remarkably higher values of cIMT in 40 patients with CAH, with no notable differences between those of normal weight and those who were overweight. Özdemir et al. (139) observed that 25 children with CAH exhibited higher IMT and decreased distensibility of the aorta and carotid arteries compared to control subjects, indicating the potential for early subclinical atherosclerosis. Akyürek et al. (134) observed that cIMT was higher in hypertensive compared to normotensive patients with CAH. Metwalley et al. (135) identified a significant correlation between cIMT and markers of disease management, including treatment duration and levels of 17-OHP and testosterone, indicating that elevated androgen levels may contribute to an increased risk of vascular dysfunction. Notably, cIMT showed no correlation with the hydrocortisone dose equivalent (135).
5 Novel biomarkers of endothelial function in CAH
A broad spectrum of potential biomarkers linked to endothelial function has been identified in reviews addressing cardiovascular diseases (11, 141, 142), renal diseases (143), and peripheral vascular diseases (144, 145). The most extensively studied include endothelial progenitor cells (146, 147), endothelial microparticles (148), microRNAs (149, 150), and adhesion molecules such as P-selectin (151), E-selectin (152), ICAM-1 (153), and VCAM-1 (153), along with molecules involved in the coagulation pathway, particularly von Willebrand factor (154). Although the levels of these well-characterized biomarkers have not yet been investigated in individuals with CAH, some data are available on less extensively studied or newly identified biomarkers, including neopterin (155), osteoprotegerin (132, 156), fetuin A (157), homocysteine (158, 159), leptin (42, 57, 160–163), adiponectin (60, 164), C-reactive protein (CRP) (55, 165), hsCRP (38, 57, 135), interleukin 6 (IL-6) (55, 57, 121, 166), and circulating endothelial cells (CECs) (135) (Table 2).
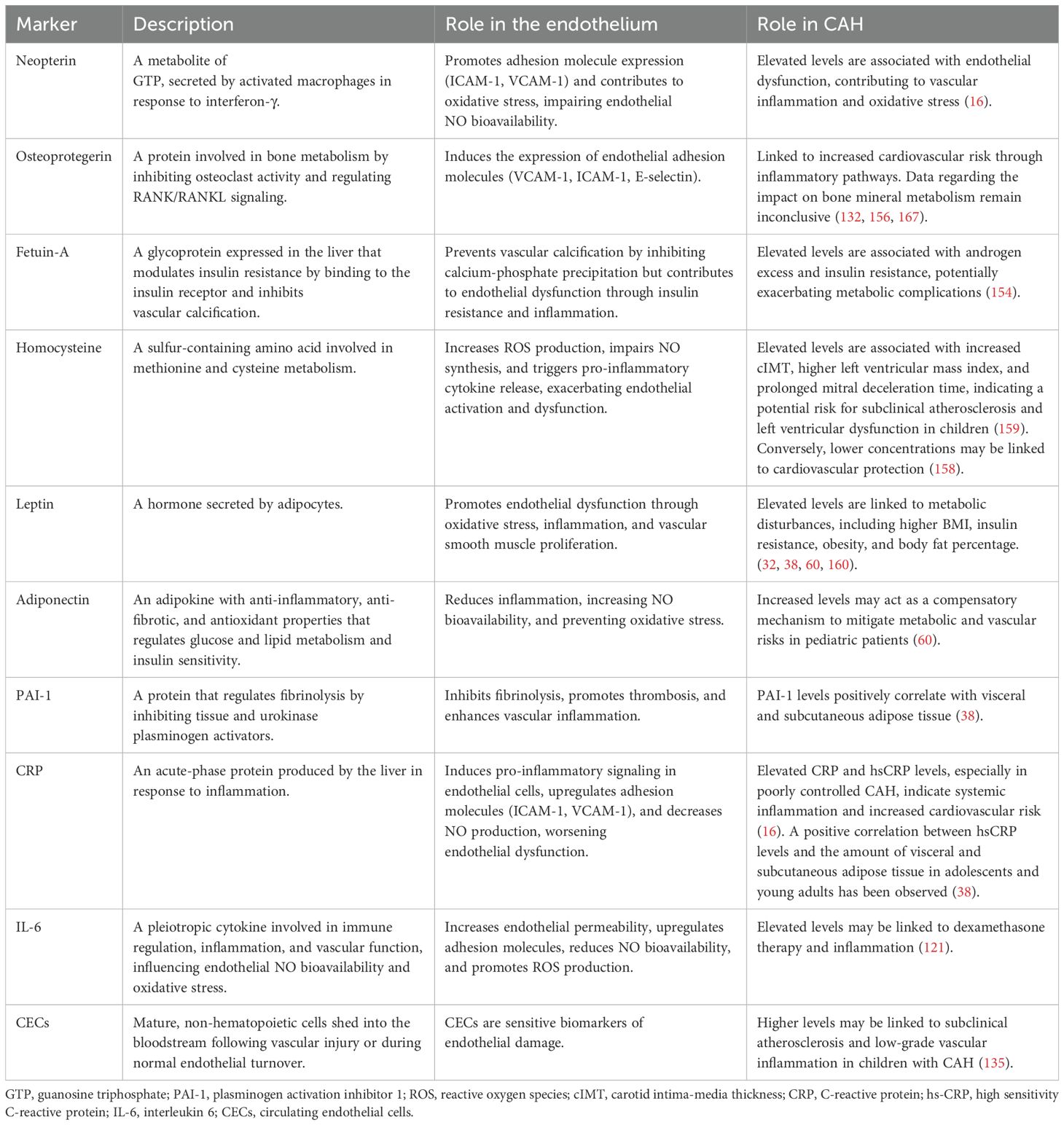
Table 2. Biomarkers that play a significant role in endothelial dysfunction.
Although biomarkers hold significant promise, standardized chemistry tests and protocols for evaluating endothelial damage are not yet available and are currently limited to clinical research applications. It has been suggested that the most effective approach for assessing endothelial function could involve combining tests for circulating endothelial biomarkers with vasomotor response assessments (10). More recently, researchers have been exploring a multibiomarker strategy that integrates both traditional and novel circulating markers (168), offering a potentially more robust tool for cardiovascular risk stratification and therapy monitoring.
5.1 Neopterin
Neopterin, a metabolite of guanosine triphosphate (GTP), is synthesized by activated macrophages in response to stimulation by γ-interferon secreted by activated T-lymphocytes (169). Neopterin promotes atherothrombosis by increasing tissue factor-mRNA transcription and the expression of adhesion molecules ICAM-1 and VCAM-1 (170), however, it also suppresses TNF-α-induced expression of MCP-1, ICAM-1, and VCAM-1, reducing adhesion to endothelial cells, and inhibiting macrophage foam cell formation and smooth muscle cell proliferation (171). Neopterin levels correlate with ROS production and its toxic effects (172), making neopterin an indirect marker of oxidative stress during cell-mediated immune responses. Its vascular role in atherosclerotic processes, either beneficial or deleterious, is still under investigation.
Farghaly et al. (16) reported that patients with CAH had higher neopterin levels compared to healthy controls. These elevated neopterin levels were significantly associated with endothelial dysfunction, as demonstrated by brachial artery FMD measurements (16). Given the limited research on neopterin in CAH, it is relevant to note that increased plasma neopterin levels have also been observed in various other conditions characterized by vascular or systemic inflammation, including atherosclerosis (173, 174), coronary artery disease (175), hypertension (176), and ischemic stroke (177).
5.2 Osteoprotegerin
Osteoprotegerin is a member of the tumor necrosis factor (TNF) receptor superfamily, playing a crucial role in regulating bone metabolism (178). It is a glycoprotein that prevents the differentiation and activity of osteoclasts by binding to the receptor activator of nuclear factor-kappa B ligand (RANKL), thereby inhibiting RANKL from interacting with its receptor, RANK. This blockage of the RANK/RANKL pathway leads to decreased osteoclast formation and reduced survival and activation of mature osteoclasts (179, 180). Through these mechanisms, osteoprotegerin contributes to preserving the balance between bone resorption and formation (179). Additionally, TNF-related apoptosis-inducing ligand (TRAIL), another member of the TNF superfamily, also interacts with osteoprotegerin (181).
Apart from its role as a regulator of bone metabolism, osteoprotegerin has recently emerged as a significant factor in in the pathogenesis of atherosclerosis and cardiovascular diseases (182, 183), amplifying the adverse effects of inflammation and several traditional risk factors such as hyperlipidemia, type 2 diabetes, and hypertension (184). Furthermore, genetic studies have shown associations of osteoprotegerin gene polymorphisms with cardiovascular disease (185, 186). Notably, osteoprotegerin binds directly to RANKL, interfering with its interaction with the RANK receptor on the endothelium and thereby regulating vascular calcification (187, 188). It can act as a receptor for TRAIL, inhibiting the effects of TRAIL on the up-regulation of eNOS and down-regulation of ROS production. In addition, osteoprotegerin also activates the renin-angiotensin system and induces vascular endothelial growth factors, leading to inflammatory and fibrotic processes (189, 190). It has also been demonstrated to induce the expression of VCAM-1, ICAM-1, and E-selectin on endothelial cells, promoting leukocyte adhesion, which is an early step in endothelial cell dysfunction (191).
Although the link between osteoprotegerin concentration and cardiovascular risk in patients with CAH has not been evaluated yet, evidence suggests an association between osteoprotegerin and bone metabolism in people with CAH (132, 156, 167). It was found that osteoprotegerin levels were significantly higher in children with CAH compared to controls, indicating a compensatory mechanism against increased bone resorption in CAH (156). In contrast, another study showed that children with CAH had significantly lower serum osteoprotegerin levels (132), similar to a case report in which a lower serum osteoprotegerin level was found in a child with CAH (167).
Given the limited and inconsistent data on osteoprotegerin in CAH, insights from other conditions characterized by endothelial dysfunction may offer a broader perspective on the potential vascular implications of altered osteoprotegerin levels. Several studies have demonstrated a significant association between osteoprotegerin concentrations and endothelial dysfunction, particularly in patients with hyperuricemia (192), type 2 diabetes mellitus (193), and HIV infection (194). However, studies examining individuals with diabetes (195) or PCOS (196), have reported no correlation between osteoprotegerin levels and alterations in endothelial function.
5.3 Fetuin-A
Fetuin-A is a heterodimer plasma glycoprotein of the cystatin superfamily of protease inhibitors, predominantly expressed in the liver (197). It contributes to insulin resistance by binding to the tandem fibronectin type 3 domains in the extracellular β-subunit of the insulin receptor, inhibiting its activity in peripheral tissues. This binding occurs away from the α-subunits, where insulin binds, with both insulin and fetuin-A targeting the receptor’s ectodomain; however, while insulin activates the receptor’s intrinsic tyrosine kinase activity, fetuin-A deactivates it (198).
Increased fetuin-A levels are associated with various metabolic health factors such as insulin sensitivity (199), glucose tolerance (200), lipid concentrations (201), and inflammatory cytokine levels (202). However, fetuin-A plays an important role in preventing cardiovascular calcification by binding to small calcium-phosphate complexes, preventing their expansion, aggregation, and precipitation (203). It also promotes phagocytosis of extracellular vesicles and apoptotic cells by vascular smooth muscle cells and macrophages, which helps reduce both apoptosis and calcification in conditions with elevated extracellular mineral ion concentration in tissues (204).
Kurnaz et al. (157) observed elevated levels of fetuin-A in 56 patients with CCAH, compared to a control group, along with increased insulin levels and disrupted insulin signaling. Furthermore, the high levels of fetuin-A and insulin substances showed a positive correlation with free and total testosterone. The authors postulated that these findings could be linked to androgen excess and prolonged or high-dose glucocorticoid therapy in people with CAH (157). They suggested that the presence of androgen receptors in liver cells may allow excess androgens to stimulate the overproduction of fetuin-A and insulin, potentially contributing to insulin resistance (157, 205).
The potential involvement of fetuin-A in the metabolic disturbances observed in CAH is further supported by studies in other conditions characterized by insulin resistance and increased cardiometabolic risk. Higher circulating levels of fetuin-A have been linked to obesity (206) and its related conditions, including type 2 diabetes (207), metabolic syndrome (201, 206), PCOS (208) (209), and NAFLD (200). Liu et al. (208) observed that patients with PCOS had higher circulating fetuin-A levels compared to healthy women. Furthermore, higher fetuin-A levels correlated positively with BMI, waist-to-hip ratio, LDL cholesterol and triglyceride concentrations, and other indicators (208). Enli et al. (209) also observed higher fetuin-A levels in patients with PCOS, which correlated positively with insulin, the Homeostasis Model Assessment of Insulin Resistance (HOMA-IR), and the free androgen index.
5.4 Homocysteine
Homocysteine, an amino acid containing a sulfhydryl group, is an intermediate product in the metabolism of methionine and cysteine that affects many cellular biological processes, such as cellular methylation status, cell metabolism, and cell injury (210). Increased concentration of fasting plasma homocysteine (higher than 15 μmol/L) is defined as hyperhomocysteinemia (211), and is associated with various neurodegenerative, metabolic, and cardiovascular disorders (83, 212–218).
Hyperhomocysteinemia contributes to endothelial dysfunction through several mechanisms. First, it stimulates the overproduction of ROS, leading to oxidative stress, which impairs the bioavailability of NO and results in impaired blood vessel relaxation (219). Second, high homocysteine levels induce the release of pro-inflammatory cytokines and other inflammatory mediators, which contribute to the activation of endothelial cells and promote vascular inflammation (220). Lastly, hyperhomocysteine can impair the activity of eNOS (221).
Metwalley et al. (159) observed elevated homocysteine levels in 36 children with CAH, particularly among those with poor disease control. This elevation correlated with increased cIMT, left ventricular mass index, and mitral deceleration time, indicating a potential risk for subclinical atherosclerosis and left ventricular dysfunction (159). Krysiak et al. (222) found that the mean homocysteine concentration was significantly higher in patients with NCCAH compared to the control group. This difference remained significant after adjusting for age and BMI. In contrast, Falhammar et al. (158) reported lower homocysteine levels in male patients with CAH under 30 years of age compared to controls, suggesting a potential association with cardiovascular protection. However, it was proposed that the differing results could be attributed to variations in age, sample size, ethnicity, dietary habits, genetic factors, research methods, and the different steroid treatments (223). Bayraktar et al. found no statistically significant difference in mean homocysteine concentrations between 50 patients with CAH and the control group.
Beyond CAH, hyperhomocysteinemia has been widely studied in other conditions characterized by metabolic dysfunction and increased cardiovascular risk. Elevated serum homocysteine concentrations have been demonstrated in women with PCOS (224–226), in obese and overweight patients with hypertension, and in normotensive obese individuals (227). Furthermore, a study by Vaya et al. (228) reported that homocysteine levels in morbidly obese patients were associated with increased waist circumference and insulin resistance.
5.5 Leptin
Leptin is a pleiotropic hormone secreted by adipocytes that plays a role in various biological processes, such as angiogenesis, vascular function, inflammatory response, bone homeostasis, and reproduction (229). Increased serum leptin levels are directly associated with higher adipose tissue mass and are a significant contributor to obesity and its metabolic complications.
The reports on the role of leptin are contradictory in CAH. A study by Charmandari et al. (42) demonstrated significantly elevated leptin concentrations in 18 young patients (2–12 years old) with CCAH, compared to controls, regardless of BMI and sex. Leptin levels were inversely correlated with epinephrine and free metanephrine concentrations, suggesting a reduced inhibitory effect of catecholamines on leptin secretion (42). Additionally, the group with CAH exhibited a loss of gender dimorphism in leptin concentrations, possibly due to androgen excess in female patients (42). Increased leptin levels and strong correlations between leptin, obesity (32, 38, 60), and HOMA-IR (60) have been observed in studies including both patients with CCAH and NCCAH. Interestingly, Borges et al. (160) demonstrated significantly elevated leptin secretion, along with higher BMI and body fat percentage, exclusively in male patients with CAH compared to controls, while no differences were observed in female patients. In contrast, several studies have reported leptin concentrations in patients with CAH to be comparable to those of controls, despite higher BMI and body fat (57, 161–163). However, Volkl et al. (161) observed significantly lower concentrations of soluble leptin receptors in 51 individuals with CAH compared to the control group, suggesting an increased level of free (unbound) serum leptin.
In addition to findings in CAH, numerous studies have investigated the role of hyperleptinemia in cardiovascular diseases, including congestive heart failure, myocardial infarction, hypertension, and coronary artery disease (230, 231), highlighting its potential contribution to vascular dysfunction. It has been found that elevated plasma leptin levels are associated with coronary artery calcification (232) and with higher serum cholesterol, triglycerides, and CRP levels in patients with coronary artery disease (233). In contrast to these findings, several other studies have suggested that leptin may exert protective effects on blood vessels in obese individuals (234).
5.6 Adiponectin
Adiponectin, an adipokine secreted by adipocytes, is a well-known homeostatic factor that regulates glucose levels, lipid metabolism, and insulin sensitivity through its anti-inflammatory, anti-fibrotic, and antioxidant effects (235). In addition to adipocytes, adiponectin is also expressed in other tissues, including liver parenchyma cells (236), myocytes (237), and epithelial cells (238). The effects of adiponectin are primarily mediated through adiponectin receptors (AdipoR1 and AdipoR2) (239), and also via T-cadherin receptor (240). Adiponectin acts directly on the liver, skeletal muscle (239), and vasculature, including endothelial cells (241), smooth muscle cells (240), and pericytes (242).
Increasing evidence suggests that adiponectin plays a protective role in the cardiovascular system; it was found that adiponectin is inversely correlated with an increased cardiovascular risk, and hypo-adiponectinemia is associated with coronary artery disease and hypertension (243), left ventricular hypertrophy (244), and a greater risk of myocardial infarction (245). Furthermore, adiponectin is linked to the regulation of energy balance, enhanced angiogenesis, anti-inflammatory responses, antiapoptotic effects, and the prevention of interstitial fibrosis (246, 247). It protects the myocardium against oxidative stress and damage from ischemia/reperfusion, and it reduces cardiac remodeling caused by pressure overload or following a myocardial infarction (248, 249).
Adiponectin levels are known to be decreased in conditions such as obesity, insulin resistance, and type 2 diabetes (250), and the administration of glucocorticoids and androgens has been shown to reduce adiponectin levels (251, 252). However, the role of adiponectin in the cardiometabolic risk of patients with CAH remains unclear. Völkl et al. (164) reported higher adiponectin concentrations in 51 individuals with CAH compared to controls, regardless of sex, with no observed changes in serum leptin or the adiponectin/leptin ratio. Adiponectin levels were negatively correlated with BMI, serum dehydroepiandrosterone, and testosterone, but no clear relationship was found with hydrocortisone or fludrocortisone dosage (164). Similarly, Mooij et al. (60) observed a trend towards elevated adiponectin levels in 27 patients with CAH, compared to controls, which may suggest that adiponectin has a protective role in these patients.
5.7 Plasminogen activation inhibitor 1
Plasminogen activation inhibitor 1 (PAI-1) is the primary physiological inhibitor of urokinase plasminogen activator (u-PA) and tissue-plasminogen activator (tPA) and is a member of the serpin superfamily (serine proteinase inhibitors). Its most important function is to regulate plasminogen activator activity, thereby controlling plasmin formation. Consequently, PAI-1 plays a vital role in systemic homeostasis, contributing to the balance of the coagulation and fibrinolytic processes (253).
Data on PAI-1 levels in individuals with CAH and their relationship with cardiovascular and metabolic disorders are limited. Mooij et al. (55) found no significant differences in PAI-1 levels between patients with CAH and controls. Kim et al. (38) observed a positive correlation between PAI-1 levels and the amount of visceral and subcutaneous adipose tissue, both of which were higher in patients with CAH compared to controls. These findings suggest a potential link between PAI-1 and adverse metabolic profiles in CAH, although further research is needed to clarify its role in this population.
Evidence from other conditions shows that elevated PAI-1 levels are linked to the development of cardiovascular and metabolic disturbances, including atherosclerosis (254), type 2 diabetes, obesity, metabolic syndrome (255–257), and PCOS (258, 259). Moreover, increased levels of glucose (260), insulin (261) and its precursors (262), and free fatty acids (263) have been demonstrated to promote PAI-1 expression or to decrease the degradation rate of PAI-1 mRNA (264), further contributing to a prothrombotic and hypofibrinolytic state.
5.8 C-reactive protein
CRP is produced by the liver in response to inflammation and is regulated, in particular, by IL-6 and TNF-α. It is considered a prototypic marker of inflammation. CRP contributes to atherosclerosis by inducing pro-inflammatory signaling in monocytes and increasing cytokine production. In endothelial cells, it upregulates cell adhesion molecules, MCP-1, endothelin-1, and PAI-1, while decreasing prostacyclin release and tPA activity (265). Additionally, CRP has been shown to reduce eNOS activity and lower NO levels (165).
HsCRP is measured using high-sensitivity tests, which enable the detection of very low concentrations, making it valuable for assessing cardiovascular risk in seemingly healthy individuals. A body of evidence supports the utility of this marker in evaluating cardiovascular risk [reviewed by Ridker et al. (266)]. Nevertheless, although CRP has shown consistency as a cardiovascular risk biomarker across large prospective studies, with relative risk ratios approaching those of classical cardiovascular risk factors, its contribution to the current evaluation model of endothelial dysfunction remains minor (267).
Despite the clear impact of inflammation on the development of endothelial dysfunction, data on inflammatory markers in CAH are scarce. In a study of 40 adult patients with CAH, hsCRP levels were shown to be markedly elevated compared to controls, and they were even more pronounced in patients with poorly controlled disease (16). Similar findings have been reported in 32 children (135) and 21 adolescents (57) with CAH. Kim et al. (38) observed a positive correlation between hsCRP levels and the amount of visceral and subcutaneous adipose tissue in 28 adolescents and young adults with CAH. However, a Dutch study involving a cohort of 27 adult patients with CAH showed no differences in the levels of CRP or other inflammatory markers, such as IL-6 and IL-18, compared to healthy controls (55).
5.9 Interleukin 6
IL-6 is a pleiotropic cytokine involved in the regulation of immune responses by recruiting macrophages and lymphocytes to sites of injury or infection, acting as both a pro-inflammatory and anti-inflammatory agent. It also plays a role in a plethora of other functions, ranging from shaping the blood-brain barrier permeability to synovial inflammation, hematopoiesis, embryonic development, and vascular permeability (268–270). As IL-6 is involved in the development of autoimmunity and plays a crucial role in sepsis, targeting the IL-6 axis is an approved pharmacological option in a number of autoimmune disorders. IL-6 is synthesized locally at the inflammation site from where it is transported via the bloodstream and exerts its action on the liver, immune cells, and endothelium [reviewed by Tanaka et al. (271)]. In hepatocytes, its signaling leads to the synthesis of, in particular, CRP, serum amyloid A, antitrypsin, hepcidin, fibrinogen, thrombopoietin, and complement cascade elements (272). In the vessels, IL-6 not only acts directly on endothelial cells to increase the production of cytokines and chemokines, thereby activating the coagulation cascade, but also enhances vascular permeability by inducing VE-cadherin disassembly and promotes clot formation by stimulating tissue factor expression on monocytes (270). Moreover, IL-6 directly affects eNOS activity and increases vascular superoxide, which rapidly inactivates NO, thereby reducing NO bioavailability (273). IL-6 contributes to atherosclerosis by upregulating angiotensin II type 1 receptor expression (274), while increased binding of angiotensin II in turn increases IL-6 signaling (270, 275).
The unique role of IL-6 in shaping cardiovascular risk is demonstrated by experiments in mice with conditional overexpression of IL-6. These experiments demonstrated significantly impaired endothelium-dependent aortic relaxation, ROS formation in the aorta, and vascular dysfunction, all of which led to a markedly decreased survival rate (276). Due to its action on endothelial cells, IL-6 is considered both a marker of cardiovascular risk and a predictor of long-term cardiovascular mortality (277, 278).
According to Ariyawatkul et al. (57), IL-6 was unaltered in the cohort of 21 adolescents with CAH when compared to their healthy peers. Similarly, Delai et al. (166) found no significant differences in IL-6 levels between 31 patients with NCCAH and controls, suggesting that IL-6 may not directly contribute to insulin resistance in this population. Costa et al. (121) reported higher serum IL-6 levels in a group of 28 women with NCCAH treated with dexamethasone compared to 19 women treated with contraceptive pills. However, no statistically significant difference was found in IL-6 serum levels when comparing either group to the control group (121).
5.10 Circulating endothelial cells
CECs are mature, non-hematopoietic cells shed into the bloodstream following vascular injury or during normal endothelial turnover (279). Under physiological conditions, they are present in minimal numbers; however, elevated CEC counts have been documented across a variety of cardiovascular, oncological, and inflammatory diseases, underscoring their potential as sensitive biomarkers of endothelial damage. The enumeration and phenotyping of CECs—primarily via advanced multiparameter flow cytometry protocols—offer a non-invasive “liquid biopsy” approach to assess vascular health (280, 281). Unlike traditional endothelial markers, such as soluble adhesion molecules or von Willebrand factor, CECs directly reflect structural endothelial injury rather than secondary activation processes (282).
In a case-control study by Metwalley et al. (135), 32 children with CAH exhibited significantly higher CEC counts compared to healthy controls, alongside increased CA-IMT and elevated hsCRP levels. These findings support the presence of subclinical atherosclerosis and low-grade vascular inflammation in CAH. Moreover, CEC levels correlated positively with markers of androgen excess (testosterone, 17-OHP) and with measures of cardiac dysfunction, suggesting that chronic hormonal imbalance and insufficient disease control may exacerbate endothelial damage (135).
The observed increase in CECs in CAH is consistent with findings from other clinical contexts, where elevated CEC counts have been established as markers of endothelial injury and disease severity. Increased CEC levels have been reported in cardiovascular conditions such as acute coronary syndromes (283), heart failure (284), and deep vein thrombosis (285). Similarly, increased CEC counts have been documented in patients with solid tumors (e.g., colorectal, breast, small-cell lung cancer) (286–288) and hematological malignancies such as multiple myeloma (289, 290), correlating with tumor progression and response to therapy. Inflammatory and autoimmune diseases, including systemic lupus erythematosus (291, 292) and small-vessel vasculitis (293), are also associated with higher CEC counts, often indicating active disease and worse prognosis.
6 Omics-based tools in understanding endothelial dysfunctions
In parallel with classical biomarkers, omics-based technologies, including genomics, proteomics, and metabolomics, are emerging as powerful tools for the discovery of novel biomarkers of endothelial dysfunction, particularly relevant in CAH. Genomic studies have identified polymorphisms in genes regulating vascular tone, inflammation, and oxidative stress [reviewed by Kim et al. (294)], which may modify individual cardiovascular risk in CAH. Proteomic analyses facilitate the identification of endothelial-derived proteins associated with oxidative stress and vascular remodeling (295, 296), whereas metabolomics provides insights into metabolic alterations affecting endothelial health, including disruptions in arginine metabolism and lipid oxidation (297).
Although omics technologies have demonstrated great promise in various cardiovascular and metabolic diseases, their application to the assessment of endothelial function in patients with CAH has not yet been explored. Despite their advantages, such approaches remain primarily within the domain of scientific research. Their clinical implementation is currently limited by factors such as high costs, technical complexity, and the need for specialized equipment and expertise, which restrict their use to selected research centers. Consequently, omics-derived biomarkers are not routinely used in clinical practice, and further research and validation in well-characterized CAH cohorts are necessary before broader application can be considered.
7 Therapeutic strategies to enhance cardiovascular health in CAH
Current glucocorticoid replacement in CAH remains non-physiological, often leading to androgen excess or glucocorticoid overtreatment. To address these issues, new approaches aiming to replicate circadian cortisol rhythms have been developed, including modified-release hydrocortisone (MR-HC, Efmody/Chronocort), immediate-release tablets with a sustained-release core (SR-HC, Plenadren), and continuous subcutaneous hydrocortisone infusion.
MR-HC improves biochemical control of 17OHP but has shown inconsistent effects on blood pressure, fat mass, and glucose metabolism; early morning cortisol peaks may transiently worsen insulin sensitivity (298–300). Interestingly, a 6-month MR-HC therapy in patients with SW-CAH resulted in a reduction of plasma renin activity and an increase in sodium levels, suggesting more effective mineralocorticoid action, likely due to decreased concentrations of 17OHP, a known mineralocorticoid receptor antagonist (298). SR-HC, although effective in primary and secondary adrenal insufficiency, appears suboptimal in CAH due to insufficient pre-awakening cortisol rise and weak androgen suppression (301, 302). Continuous hydrocortisone infusion more closely mimics physiological cortisol rhythms and enables dose reduction; however, data on its metabolic effects are limited, and its clinical use will likely remain restricted to highly selected patients due to its complexity (303). Emerging therapeutic options, including CRF-1 antagonists (crinecerfont and tildacerfont), have shown potential to reduce adrenocorticotropic hormone (ACTH) and androgen levels, enabling possible glucocorticoid dose reduction, although their long-term impact remains to be established (304–306).
In parallel, adjunctive therapies aiming to counteract androgen excess, such as oral contraceptives or spironolactone, play a role in the management of CAH, although they introduce additional metabolic and cardiovascular concerns requiring careful consideration (307). Combined hormonal contraceptives have been associated with an increased risk of venous thromboembolism and hypertension (308), however, they may assist in regulating menstrual cycles and reducing androgenic symptoms in individuals with CAH. Spironolactone, commonly used in women with androgen excess, improves cardiovascular outcomes in various cardiac conditions. Nevertheless, its cardiometabolic safety in the context of androgen excess remains unestablished, and its use in CAH may be limited due to the frequent coexistence of aldosterone deficiency (309, 310).
Given the elevated cardiometabolic risk in patients with CAH, especially those predisposed to developing diabetes, interventions commonly used in the general population are considered applicable, although evidence specific to CAH remains limited. Intensive lifestyle modifications—encompassing dietary improvements, increased physical activity, smoking cessation, and weight management—are recommended as first-line measures (311). Isolated case reports have demonstrated the beneficial effects of metformin (312, 313), topiramate (314), and bariatric surgery (315, 316) on weight reduction, visceral fat mass, and insulin sensitivity in patients with CAH. Newer antiobesity medications (liraglutide, semaglutide, tirzepatide, and naltrexone/bupropion) have not yet been studied in patients with CAH.
Statin therapy is considered safe and effective for lipid management in CAH, demonstrating significant reductions in total and LDL cholesterol (317, 318) and potential antiandrogenic effects (318). Despite theoretical concerns regarding statin- and glucocorticoid-induced myopathy (319, 320), no such cases have been reported in CAH, likely due to the use of moderate doses.
Management of hypertension in CAH should prioritize angiotensin-converting enzyme (ACE) inhibitors and angiotensin-receptor blockers (ARBs) due to their cardioprotective properties, beneficial effects on endothelial function, and ability to improve insulin sensitivity (321, 322). Thiazide diuretics, β-blockers, and calcium channel blockers can be used as adjuncts, though β-blockers should preferably be third-generation agents (nebivolol, carvedilol) with more favorable metabolic profiles (311, 323, 324).
8 Conclusion
Endothelial dysfunction plays a critical role in the cardiovascular comorbidities observed in individuals with CAH due to 21OHD. The complex interplay of disease-related factors, such as androgen excess, metabolic disturbances, and glucocorticoid therapy, contribute to the heightened cardiovascular and metabolic risk in this population. The pathophysiological mechanisms driving endothelial dysfunction in CAH are multifaceted, involving oxidative stress, inflammation, and altered vascular tone. Although various diagnostic techniques—such as FMD, PAT, PWV, and IMT measurements—offer valuable insights into endothelial function, the optimal in vivo assessment method remains an area of ongoing investigation (Figure 3).

Figure 3. Pathophysiological links between hormonal dysregulation in CAH, endothelial dysfunction, and potential therapeutic targets. CAH, due to 21-hydroxylase deficiency, leads to impaired cortisol synthesis and compensatory ACTH overproduction, resulting in adrenal hyperplasia, androgen excess, and—particularly in salt-wasting forms—aldosterone deficiency. These hormonal disturbances may contribute to endothelial dysfunction through mechanisms such as increased oxidative stress, chronic inflammation, arterial stiffness, and impaired flow-mediated dilation. Therapeutic strategies focus on restoring hormonal balance, providing vascular protection, addressing metabolic dysregulation, and utilizing novel biomarkers and non-invasive vascular assessments to monitor cardiovascular risk. 17-OHP, 17-hydroxyprogesterone; ACE inhibitors, angiotensin-converting enzyme inhibitors; ACTH, adrenocorticotropic hormone; ARBs, angiotensin II receptor blockers; CRP, C-reactive protein; eNOS, endothelial nitric oxide synthase; FMD, flow-mediated dilation; GLP-1, glucagon-like peptide-1; hsCRP, high sensitivity C-reactive protein; ICAM-1, intercellular adhesion molecule 1; IL-6, interleukin-6; IMT, intima-media thickness; NO, nitric oxide; PAI-1, plasminogen activator inhibitor-1; PAT, peripheral arterial tonometry; PWV, pulse wave velocity; ROS, reactive oxygen species; TNF-α, tumor necrosis factor alpha; VCAM-1, vascular cell adhesion molecule 1.
Emerging biomarkers of endothelial dysfunction, including neopterin, osteoprotegerin, fetuin-A, homocysteine, leptin, adiponectin, PAI-1, CRP, IL-6, and CECs, provide additional understanding of the pathophysiology of endothelial damage. Elevated levels of these biomarkers have been documented in populations with cardiovascular risk factors, indicating their potential for early detection and monitoring of endothelial dysfunction in CAH. Despite their promise, several limitations impede their translation into routine clinical practice. The lack of standardized chemical assay protocols for evaluating endothelial damage, high cost, and technical complexity restrict their current application to research settings primarily. Furthermore, many endothelial biomarkers exhibit limited sensitivity and specificity, often overlapping with markers of inflammation, platelet activation, and vascular smooth muscle dysfunction. Small sample sizes in clinical studies further constrain the reliability of these biomarkers, necessitating large-scale, multicenter trials to validate their clinical utility. Some findings remain controversial or inconclusive, highlighting the need for standardized approaches to address discrepancies in the literature. Additionally, the impact of metabolic and cardiovascular comorbidities on endothelial biomarkers remains insufficiently explored, particularly in the context of multiple coexisting conditions, which may have additive or interactive effects on biomarker levels.
Until stronger evidence becomes available, strategies for cardiovascular risk reduction in CAH should follow existing recommendations for high-risk populations, focusing on aggressive management of hypertension, dyslipidemia, disturbances in glucose metabolism, and obesity. Pharmacological strategies to improve cardiovascular health in CAH include optimized glucocorticoid replacement, adjunctive antiandrogen therapies, insulin sensitizers, statins, and renin–angiotensin system inhibitors, although disease-specific evidence remains limited.
Nevertheless, the present analysis is constrained by several important methodological limitations that warrant careful consideration. Most of the studies included in this review are observational, often retrospective in nature, and are limited by small sample sizes. Additionally, various sources of bias, such as sampling bias, recall bias, observation bias, and confounding bias, further weaken the strength of the available evidence and complicate the interpretation of findings related to endothelial dysfunction in CAH. Future research is needed to further investigate novel biomarkers of endothelial dysfunction and to refine diagnostic strategies, potentially integrating endothelial function assessment, to enhance cardiovascular risk evaluation and management. Particularly, well-designed, prospective studies conducted on large cohorts and across different age groups are necessary to better understand biomarker dynamics over time and their predictive value in clinical practice. Longitudinal cohort studies are needed to track changes in endothelial function over time and to evaluate the effects of factors such as age, treatment duration, and adherence to medical regimens. Moreover, the application of omics-based technologies holds promise for the discovery of novel endothelial biomarkers in CAH, potentially enhancing risk stratification and personalized management. Such efforts are crucial to improving long-term cardiovascular outcomes in individuals with CAH. Special attention to vascular health is essential for children with CAH, as atherosclerotic processes typically initiate in childhood and progress more rapidly in high-risk populations (80).
Author contributions
JH: Conceptualization, Formal Analysis, Writing – original draft, Writing – review & editing, Data curation, Methodology, Visualization. ZR: Data curation, Visualization, Writing – review & editing. MB: Supervision, Visualization, Writing – original draft, Writing – review & editing. SI: Data curation, Visualization, Writing – review & editing. BR-M: Conceptualization, Data curation, Formal Analysis, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. UA: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Supervision, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This article was published with financial support from the Fundusz Rozwoju Nauki of the Medical University of Warsaw.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Barbot M, Mazzeo P, Lazzara M, Ceccato F, and Scaroni C. Metabolic syndrome and cardiovascular morbidity in patients with congenital adrenal hyperplasia. Front Endocrinol (Lausanne). (2022) 13:934675. doi: 10.3389/fendo.2022.934675
2. Falhammar H, Frisen L, Hirschberg AL, Norrby C, Almqvist C, Nordenskjold A, et al. Increased Cardiovascular and Metabolic Morbidity in Patients With 21-Hydroxylase Deficiency: A Swedish Population-Based National Cohort Study. J Clin Endocrinol Metab. (2015) 100:3520–8. doi: 10.1210/JC.2015-2093
3. Boulanger CM. Endothelium. Arterioscler Thromb Vasc Biol. (2016) 36:e26–31. doi: 10.1161/ATVBAHA.116.306940
4. Peyter AC, Armengaud JB, Guillot E, and Yzydorczyk C. Endothelial Progenitor Cells Dysfunctions and Cardiometabolic Disorders: From Mechanisms to Therapeutic Approaches. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22136667
5. Toma L, Stancu CS, and Sima AV. Endothelial Dysfunction in Diabetes Is Aggravated by Glycated Lipoproteins; Novel Molecular Therapies. Biomedicines. (2020) 9. doi: 10.3390/biomedicines9010018
6. Torky A, Sinaii N, Jha S, Desai J, El-Maouche D, Mallappa A, et al. Cardiovascular Disease Risk Factors and Metabolic Morbidity in a Longitudinal Study of Congenital Adrenal Hyperplasia. J Clin Endocrinol Metab. (2021) 106:e5247–e57. doi: 10.1210/clinem/dgab133
7. Krysiak R, Claahsen-van der Grinten HL, Reisch N, Touraine P, and Falhammar H. Cardiometabolic Aspects of Congenital Adrenal Hyperplasia. Endocr Rev. (2025) 46:80–148. doi: 10.1210/endrev/bnae026
8. Han TS, Stimson RH, Rees DA, Krone N, Willis DS, Conway GS, et al. Glucocorticoid treatment regimen and health outcomes in adults with congenital adrenal hyperplasia. Clin Endocrinol (Oxf). (2013) 78:197–203. doi: 10.1111/cen.2012.78.issue-2
9. Gallo G, Volpe M, and Savoia C. Endothelial Dysfunction in Hypertension: Current Concepts and Clinical Implications. Front Med (Lausanne). (2021) 8:798958. doi: 10.3389/fmed.2021.798958
10. Deanfield JE, Halcox JP, and Rabelink TJ. Endothelial function and dysfunction: testing and clinical relevance. Circulation. (2007) 115:1285–95. doi: 10.1161/CIRCULATIONAHA.106.652859
11. Gimbrone MA Jr. and García-Cardeña G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ Res. (2016) 118:620–36. doi: 10.1161/CIRCRESAHA.115.306301
12. Endemann DH and Schiffrin EL. Endothelial dysfunction. J Am Soc Nephrol. (2004) 15:1983–92. doi: 10.1097/01.ASN.0000132474.50966.DA
13. Forstermann U and Munzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. (2006) 113:1708–14. doi: 10.1161/CIRCULATIONAHA.105.602532
14. Shaito A, Thuan DTB, Phu HT, Nguyen THD, Hasan H, Halabi S, et al. Herbal Medicine for Cardiovascular Diseases: Efficacy, Mechanisms, and Safety. Front Pharmacol. (2020) 11:422. doi: 10.3389/fphar.2020.00422
15. Xu S, Ilyas I, Little PJ, Li H, Kamato D, Zheng X, et al. Endothelial Dysfunction in Atherosclerotic Cardiovascular Diseases and Beyond: From Mechanism to Pharmacotherapies. Pharmacol Rev. (2021) 73:924–67. doi: 10.1124/pharmrev.120.000096
16. Farghaly HS, Metwalley KA, Raafat DM, Saied GM, Gabri MF, and Algowhary M. Association between vascular endothelial dysfunction and the inflammatory marker neopterin in patients with classic congenital adrenal hyperplasia. Atherosclerosis. (2021) 328:38–43. doi: 10.1016/j.atherosclerosis.2021.05.017
17. Steyers CM 3rd and Miller FJ Jr. Endothelial dysfunction in chronic inflammatory diseases. Int J Mol Sci. (2014) 15:11324–49. doi: 10.3390/ijms150711324
18. Shaito A, Aramouni K, Assaf R, Parenti A, Orekhov A, Yazbi AE, et al. Oxidative Stress-Induced Endothelial Dysfunction in Cardiovascular Diseases. Front Biosci (Landmark Ed). (2022) 27:105. doi: 10.31083/j.fbl2703105
19. Clarkson P, Celermajer DS, Powe AJ, Donald AE, Henry RM, and Deanfield JE. Endothelium-dependent dilatation is impaired in young healthy subjects with a family history of premature coronary disease. Circulation. (1997) 96:3378–83. doi: 10.1161/01.CIR.96.10.3378
20. Lundman P, Eriksson MJ, Stuhlinger M, Cooke JP, Hamsten A, and Tornvall P. Mild-to-moderate hypertriglyceridemia in young men is associated with endothelial dysfunction and increased plasma concentrations of asymmetric dimethylarginine. J Am Coll Cardiol. (2001) 38:111–6. doi: 10.1016/S0735-1097(01)01318-3
21. Ford MA, McConnell JP, Lavi S, Rihal CS, Prasad A, Sandhu GS, et al. Coronary artery endothelial dysfunction is positively correlated with low density lipoprotein and inversely correlated with high density lipoprotein subclass particles measured by nuclear magnetic resonance spectroscopy. Atherosclerosis. (2009) 207:111–5. doi: 10.1016/j.atherosclerosis.2009.04.039
22. Lavi S, Prasad A, Yang EH, Mathew V, Simari RD, Rihal CS, et al. Smoking is associated with epicardial coronary endothelial dysfunction and elevated white blood cell count in patients with chest pain and early coronary artery disease. Circulation. (2007) 115:2621–7. doi: 10.1161/CIRCULATIONAHA.106.641654
23. Shinozaki K, Hirayama A, Nishio Y, Yoshida Y, Ohtani T, Okamura T, et al. Coronary endothelial dysfunction in the insulin-resistant state is linked to abnormal pteridine metabolism and vascular oxidative stress. J Am Coll Cardiol. (2001) 38:1821–8. doi: 10.1016/S0735-1097(01)01659-X
24. Arcaro G, Cretti A, Balzano S, Lechi A, Muggeo M, Bonora E, et al. Insulin causes endothelial dysfunction in humans: sites and mechanisms. Circulation. (2002) 105:576–82. doi: 10.1161/hc0502.103333
25. Celermajer DS, Sorensen KE, Spiegelhalter DJ, Georgakopoulos D, Robinson J, and Deanfield JE. Aging is associated with endothelial dysfunction in healthy men years before the age-related decline in women. J Am Coll Cardiol. (1994) 24:471–6. doi: 10.1016/0735-1097(94)90305-0
26. Poredos P and Kek A. Relation of blunted dilation of the brachial artery in insulin-dependent diabetes mellitus to microalbuminuria. Am J Cardiol. (2000) 86:364–7. doi: 10.1016/S0002-9149(00)00938-3
27. Falhammar H, Frisén L, Norrby C, Hirschberg AL, Almqvist C, Nordenskjöld A, et al. Increased Mortality in Patients With Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency. J Clin Endocrinol Metab. (2014) 99:E2715–E21. doi: 10.1210/jc.2014-2957
28. Drera A, Rodella L, Brangi E, Riccardi M, and Vizzardi E. Endothelial Dysfunction in Heart Failure: What Is Its Role? J Clin Med. (2024) 13. doi: 10.3390/jcm13092534
29. Swarup S, Ahmed I, Grigorova Y, and Zeltser R. Metabolic Syndrome. StatPearls. Treasure Island (FL: StatPearls Publishing Copyright © (2024).
30. Paizoni L, Auer MK, Schmidt H, Hubner A, Bidlingmaier M, and Reisch N. Effect of androgen excess and glucocorticoid exposure on metabolic risk profiles in patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Steroid Biochem Mol Biol. (2020) 197:105540. doi: 10.1016/j.jsbmb.2019.105540
31. Arlt W, Willis DS, Wild SH, Krone N, Doherty EJ, Hahner S, et al. Health status of adults with congenital adrenal hyperplasia: a cohort study of 203 patients. J Clin Endocrinol Metab. (2010) 95:5110–21. doi: 10.1210/jc.2010-0917
32. Finkielstain GP, Kim MS, Sinaii N, Nishitani M, Van Ryzin C, Hill SC, et al. Clinical characteristics of a cohort of 244 patients with congenital adrenal hyperplasia. J Clin Endocrinol Metab. (2012) 97:4429–38. doi: 10.1210/jc.2012-2102
33. Bachelot A, Plu-Bureau G, Thibaud E, Laborde K, Pinto G, Samara D, et al. Long-term outcome of patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Horm Res. (2007) 67:268–76. doi: 10.1159/000098017
34. El-Maouche D, Hannah-Shmouni F, Mallappa A, Hargreaves CJ, Avila NA, and Merke DP. Adrenal morphology and associated comorbidities in congenital adrenal hyperplasia. Clin Endocrinol (Oxf). (2019) 91:247–55. doi: 10.1111/cen.2019.91.issue-2
35. Koenen M, Hill MA, Cohen P, and Sowers JR. Obesity, Adipose Tissue and Vascular Dysfunction. Circ Res. (2021) 128:951–68. doi: 10.1161/CIRCRESAHA.121.318093
36. Kajikawa M and Higashi Y. Obesity and Endothelial Function. Biomedicines. (2022) 10. doi: 10.3390/biomedicines10071745
37. Higashi Y, Noma K, Yoshizumi M, and Kihara Y. Endothelial function and oxidative stress in cardiovascular diseases. Circ J. (2009) 73:411–8. doi: 10.1253/circj.CJ-08-1102
38. Kim MS, Ryabets-Lienhard A, Dao-Tran A, Mittelman SD, Gilsanz V, Schrager SM, et al. Increased Abdominal Adiposity in Adolescents and Young Adults With Classical Congenital Adrenal Hyperplasia due to 21-Hydroxylase Deficiency. J Clin Endocrinol Metab. (2015) 100:E1153–9. doi: 10.1210/jc.2014-4033
39. Deedwania PC. Metabolic syndrome and vascular disease: is nature or nurture leading the new epidemic of cardiovascular disease? Circulation. (2004) 109:2–4. doi: 10.1161/01.CIR.0000110642.73995.BF
40. Kim MS, Ryabets-Lienhard A, Bali B, Lane CJ, Park AH, Hall S, et al. Decreased adrenomedullary function in infants with classical congenital adrenal hyperplasia. J Clin Endocrinol Metab. (2014) 99:E1597–601. doi: 10.1210/jc.2014-1274
41. Merke DP, Chrousos GP, Eisenhofer G, Weise M, Keil MF, Rogol AD, et al. Adrenomedullary dysplasia and hypofunction in patients with classic 21-hydroxylase deficiency. N Engl J Med. (2000) 343:1362–8. doi: 10.1056/NEJM200011093431903
42. Charmandari E, Weise M, Bornstein SR, Eisenhofer G, Keil MF, Chrousos GP, et al. Children with classic congenital adrenal hyperplasia have elevated serum leptin concentrations and insulin resistance: potential clinical implications. J Clin Endocrinol Metab. (2002) 87:2114–20. doi: 10.1210/jcem.87.5.8456
43. Marra AM, Improda N, Capalbo D, Salzano A, Arcopinto M, De Paulis A, et al. Cardiovascular abnormalities and impaired exercise performance in adolescents with congenital adrenal hyperplasia. J Clin Endocrinol Metab. (2015) 100:644–52. doi: 10.1210/jc.2014-1805
44. Rosano GM, Sheiban I, Massaro R, Pagnotta P, Marazzi G, Vitale C, et al. Low testosterone levels are associated with coronary artery disease in male patients with angina. Int J Impot Res. (2007) 19:176–82. doi: 10.1038/sj.ijir.3901504
45. Metwalley KA, Farghaly HS, and Abdelhamid A. Epicardial Fat Thickness in Children with Classic Congenital Adrenal Hyperplasia. J Clin Res Pediatr Endocrinol. (2019) 11:61–9. doi: 10.4274/jcrpe.galenos.2018.2018.0153
46. Zhang HJ, Yang J, Zhang MN, Liu CQ, Xu M, Li XJ, et al. Metabolic disorders in newly diagnosed young adult female patients with simple virilizing 21-hydroxylase deficiency. Endocrine. (2010) 38:260–5. doi: 10.1007/s12020-010-9382-9
47. Janus D, Wojcik M, Tyrawa K, Janeczko M, Bik-Multanowski M, and Starzyk JB. Intima media thickness of common carotids and abdominal aorta in children and adolescents with congenital adrenal hyperplasia due to 21-hydroxylase deficiency in relation to their genotypes. Neuro Endocrinol Lett. (2017) 38:154–62.
48. Berbrier DE, Leone CA, Adler TE, Bender JR, Taylor HS, Stachenfeld NS, et al. Effects of androgen excess and body mass index on endothelial function in women with polycystic ovary syndrome. J Appl Physiol (1985). (2023) 134:868–78. doi: 10.1152/japplphysiol.00583.2022
49. Paradisi G, Steinberg HO, Hempfling A, Cronin J, Hook G, Shepard MK, et al. Polycystic ovary syndrome is associated with endothelial dysfunction. Circulation. (2001) 103:1410–5. doi: 10.1161/01.CIR.103.10.1410
50. Improda N, Barbieri F, Ciccarelli GP, Capalbo D, and Salerno M. Cardiovascular Health in Children and Adolescents With Congenital Adrenal Hyperplasia Due to 21-Hydroxilase Deficiency. Front Endocrinol (Lausanne). (2019) 10:212. doi: 10.3389/fendo.2019.00212
51. Kim MS, Fraga NR, Minaeian N, and Geffner ME. Components of Metabolic Syndrome in Youth With Classical Congenital Adrenal Hyperplasia. Front Endocrinol (Lausanne). (2022) 13:848274. doi: 10.3389/fendo.2022.848274
52. Ambrosino P, Bachetti T, D’Anna SE, Galloway B, Bianco A, D’Agnano V, et al. Mechanisms and Clinical Implications of Endothelial Dysfunction in Arterial Hypertension. J Cardiovasc Dev Dis. (2022) 9. doi: 10.3390/jcdd9050136
53. Barbot M, Ceccato F, and Scaroni C. The Pathophysiology and Treatment of Hypertension in Patients With Cushing’s Syndrome. Front Endocrinol (Lausanne). (2019) 10:321. doi: 10.3389/fendo.2019.00321
54. Maccabee-Ryaboy N, Thomas W, Kyllo J, Lteif A, Petryk A, Gonzalez-Bolanos MT, et al. Hypertension in children with congenital adrenal hyperplasia. Clin Endocrinol (Oxf). (2016) 85:528–34. doi: 10.1111/cen.2016.85.issue-4
55. Mooij CF, Kroese JM, Sweep FC, Hermus AR, and Tack CJ. Adult patients with congenital adrenal hyperplasia have elevated blood pressure but otherwise a normal cardiovascular risk profile. PLoS One. (2011) 6:e24204. doi: 10.1371/journal.pone.0024204
56. Tamhane S, Rodriguez-Gutierrez R, Iqbal AM, Prokop LJ, Bancos I, Speiser PW, et al. Cardiovascular and Metabolic Outcomes in Congenital Adrenal Hyperplasia: A Systematic Review and Meta-Analysis. J Clin Endocrinol Metab. (2018) 103:4097–103. doi: 10.1210/jc.2018-01862
57. Ariyawatkul K, Tepmongkol S, Aroonparkmongkol S, and Sahakitrungruang T. Cardio-metabolic risk factors in youth with classical 21-hydroxylase deficiency. Eur J Pediatr. (2017) 176:537–45. doi: 10.1007/s00431-017-2875-2
58. Vijayan R, Bhavani N, Pavithran PV, Nair V, Menon UV, Menon AS, et al. Metabolic profile, cardiovascular risk factors and health-related quality of life in children, adolescents and young adults with congenital adrenal hyperplasia. J Pediatr Endocrinol Metab. (2019) 32:871–7. doi: 10.1515/jpem-2019-0079
59. Roche EF, Charmandari E, Dattani MT, and Hindmarsh PC. Blood pressure in children and adolescents with congenital adrenal hyperplasia (21-hydroxylase deficiency): a preliminary report. Clin Endocrinol (Oxf). (2003) 58:589–96. doi: 10.1046/j.1365-2265.2003.01757.x
60. Mooij CF, van Herwaarden AE, Sweep F, Roeleveld N, de Korte CL, Kapusta L, et al. Cardiovascular and metabolic risk in pediatric patients with congenital adrenal hyperplasia due to 21 hydroxylase deficiency. J Pediatr Endocrinol Metab. (2017) 30:957–66. doi: 10.1515/jpem-2017-0068
61. Voülkl TMK, Simm D, Doütsch JR, Rascher W, and Doürr HG. Altered 24-Hour Blood Pressure Profiles in Children and Adolescents with Classical Congenital Adrenal Hyperplasia due to 21-Hydroxylase Deficiency. J Clin Endocrinol Metab. (2006) 91:4888–95. doi: 10.1210/jc.2006-1069
62. Hoepffner W, Herrmann A, Willgerodt H, and Keller E. Blood Pressure in Patients with Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency. J Pediatr Endocrinol Metab. (2006) 19:705–12. doi: 10.1515/JPEM.2006.19.5.705
63. Subbarayan A, Dattani MT, Peters CJ, and Hindmarsh PC. Cardiovascular risk factors in children and adolescents with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Clin Endocrinol (Oxf). (2014) 80:471–7. doi: 10.1111/cen.2014.80.issue-4
64. Bonfig W, Roehl FW, Riedl S, Dorr HG, Bettendorf M, Bramswig J, et al. Blood Pressure in a Large Cohort of Children and Adolescents With Classic Adrenal Hyperplasia (CAH) Due to 21-Hydroxylase Deficiency. Am J Hypertens. (2016) 29:266–72. doi: 10.1093/ajh/hpv087
65. El-Maouche D, Arlt W, and Merke DP. Congenital adrenal hyperplasia. Lancet. (2017) 390:2194–210. doi: 10.1016/S0140-6736(17)31431-9
66. Han TS, Walker BR, Arlt W, and Ross RJ. Treatment and health outcomes in adults with congenital adrenal hyperplasia. Nat Rev Endocrinol. (2014) 10:115–24. doi: 10.1038/nrendo.2013.239
67. Speiser PW, Azziz R, Baskin LS, Ghizzoni L, Hensle TW, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. (2010) 95:4133–60. doi: 10.1210/jc.2009-2631
68. de Oliveira DM, Tura A, Vasques ACJ, Camilo DF, Lima MM, de Lemos-Marini SHV, et al. Insulin Resistance in Congenital Adrenal Hyperplasia is Compensated for by Reduced Insulin Clearance. J Clin Endocrinol Metab. (2021) 106:e1574–e85. doi: 10.1210/clinem/dgab010
69. Mnif MF, Kamoun M, Mnif F, Charfi N, Kallel N, Ben Naceur B, et al. Long-term outcome of patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Am J Med Sci. (2012) 344:363–73. doi: 10.1097/MAJ.0b013e31824369e4
70. Ekbom K, Strandqvist A, Lajic S, Hirschberg A, Falhammar H, and Nordenström A. The impact of adherence and therapy regimens on quality of life in patients with congenital adrenal hyperplasia. Clin Endocrinol (Oxf). (2022) 96:666–79. doi: 10.1111/cen.14676
71. Villela TR, Barra CB, Belisario AR, Luizon MR, Simoes ESAC, and Silva IN. Glucocorticoid receptor Gene (NR3C1) Polymorphisms and Haplotypes in patients with congenital adrenal hyperplasia. Mol Cell Endocrinol. (2021) 536:111399. doi: 10.1016/j.mce.2021.111399
72. Lin RC, Wang XL, and Morris BJ. Association of coronary artery disease with glucocorticoid receptor N363S variant. Hypertension. (2003) 41:404–7. doi: 10.1161/01.HYP.0000055342.40301.DC
73. Roussel R, Reis AF, Dubois-Laforgue D, Bellanne-Chantelot C, Timsit J, and Velho G. The N363S polymorphism in the glucocorticoid receptor gene is associated with overweight in subjects with type 2 diabetes mellitus. Clin Endocrinol (Oxf). (2003) 59:237–41. doi: 10.1046/j.1365-2265.2003.01831.x
74. Moreira RP, Bachega TA, Machado MC, Mendonca BB, Bronstein MD, and Villares Fragoso MC. Modulatory effect of BclI GR gene polymorphisms on the obesity phenotype in Brazilian patients with Cushing’s disease. Clinics (Sao Paulo). (2013) 68:579–85. doi: 10.6061/clinics/2013(05)01
75. Musz P, Podhajski P, Grzelakowska K, and Umińska J. Non-invasive assessment of endothelial function — a review of available methods. Med Res J. (2021) 6:53–8. doi: 10.5603/MRJ.a2021.0008
76. Minhas AS, Goerlich E, Corretti MC, Arbab-Zadeh A, Kelle S, Leucker T, et al. Imaging Assessment of Endothelial Function: An Index of Cardiovascular Health. Front Cardiovasc Med. (2022) 9:778762. doi: 10.3389/fcvm.2022.778762
77. Anderson TJ. Arterial stiffness or endothelial dysfunction as a surrogate marker of vascular risk. Can J Cardiol. (2006) 22 Suppl B:72B–80B. doi: 10.1016/S0828-282X(06)70990-4
78. Joannides R, Bellien J, and Thuillez C. Clinical methods for the evaluation of endothelial function– a focus on resistance arteries. Fundam Clin Pharmacol. (2006) 20:311–20. doi: 10.1111/j.1472-8206.2006.00406.x
79. Lekakis J, Abraham P, Balbarini A, Blann A, Boulanger CM, Cockcroft J, et al. Methods for evaluating endothelial function: a position statement from the European Society of Cardiology Working Group on Peripheral Circulation. Eur J Cardiovasc Prev Rehabil. (2011) 18:775–89. doi: 10.1177/1741826711398179
80. Celermajer DS, Sorensen KE, Gooch VM, Spiegelhalter DJ, Miller OI, Sullivan ID, et al. Non-invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosis. Lancet. (1992) 340:1111–5. doi: 10.1016/0140-6736(92)93147-F
81. McEniery CM, Wallace S, Mackenzie IS, McDonnell B, Yasmin, Newby DE, et al. Endothelial function is associated with pulse pressure, pulse wave velocity, and augmentation index in healthy humans. Hypertension. (2006) 48:602–8. doi: 10.1161/01.HYP.0000239206.64270.5f
82. Perrone-Filardi P, Coca A, Galderisi M, Paolillo S, Alpendurada F, de Simone G, et al. Noninvasive cardiovascular imaging for evaluating subclinical target organ damage in hypertensive patients: a consensus article from the European Association of Cardiovascular Imaging, the European Society of Cardiology Council on Hypertension and the European Society of Hypertension. J Hypertens. (2017) 35:1727–41. doi: 10.1097/HJH.0000000000001396
83. Rubinshtein R, Kuvin JT, Soffler M, Lennon RJ, Lavi S, Nelson RE, et al. Assessment of endothelial function by non-invasive peripheral arterial tonometry predicts late cardiovascular adverse events. Eur Heart J. (2010) 31:1142–8. doi: 10.1093/eurheartj/ehq010
84. Wierzbicka-Chmiel J, Chmiel A, Rychlik S, Ogrodowczyk-Bobik M, Marek B, and Kajdaniuk D. Vascular and cardiac function in young adults with classical congenital adrenal hyperplasia. Endokrynol Pol. (2017) 68:505–11. doi: 10.5603/EP.a2017.0046
85. Harrington J, Pena AS, Gent R, Hirte C, and Couper J. Adolescents with congenital adrenal hyperplasia because of 21-hydroxylase deficiency have vascular dysfunction. Clin Endocrinol (Oxf). (2012) 76:837–42. doi: 10.1111/j.1365-2265.2011.04309.x
86. Cutruzzolà A, Parise M, Cacia M, Lucà S, Irace C, and Gnasso A. The relationship between endothelial-dependent flow-mediated dilation and diastolic function in type 2 diabetes. Acta Diabetologica. (2024) 61:1475–82. doi: 10.1007/s00592-024-02313-1
87. Ito H, Nakashima M, Meguro K, Furukawa H, Yamashita H, Takaki A, et al. Flow Mediated Dilatation Is Reduced with the Progressive Stages of Glomerular Filtration Rate and Albuminuria in Type 2 Diabetic Patients without Coronary Heart Disease. J Diabetes Res. (2015) 2015:728127. doi: 10.1155/2015/728127
88. Pillay S, Anderson J, Couper J, Maftei O, Gent R, and Pena AS. Children With Type 1 Diabetes Have Delayed Flow-Mediated Dilation. Can J Diabetes. (2018) 42:276–80. doi: 10.1016/j.jcjd.2017.06.011
89. Soyman Z, Noyan V, Tulmac M, Yucel A, Sagsoz N, Bayrak T, et al. Serum paraoxonase 1 activity, asymmetric dimethylarginine levels, and brachial artery flow-mediated dilatation in women with polycystic ovary syndrome. Fertil Steril. (2011) 95:1067–72. doi: 10.1016/j.fertnstert.2010.12.011
90. Pepene CE. Evidence for visfatin as an independent predictor of endothelial dysfunction in polycystic ovary syndrome. Clin Endocrinol (Oxf). (2012) 76:119–25. doi: 10.1111/j.1365-2265.2011.04171.x
91. Alexandraki K, Protogerou AD, Papaioannou TG, Piperi C, Mastorakos G, Lekakis J, et al. Early microvascular and macrovascular dysfunction is not accompanied by structural arterial injury in polycystic ovary syndrome. Hormones (Athens). (2006) 5:126–36. doi: 10.14310/horm.2002.11176
92. Hussid MF, Jordão CP, Lopes-Vicente WR, Virmondes L, Cepeda F, Katayama K, et al. Flow-Mediated Dilation in Obese Adolescents: Correlation with Waist Circumference and Systolic Blood Pressure. FASEB J. (2018) 32:713.7–.7. doi: 10.1096/fasebj.2018.32.1_supplement.713.7
93. Mori Y. Flow-Mediated Dilatation in Obese Children. Clin Pediatr Endocrinology. (2003) 12:43–8. doi: 10.1297/cpe.12.43
94. Hashimoto M, Akishita M, Eto M, Kozaki K, Ako J, Sugimoto N, et al. The impairment of flow-mediated vasodilatation in obese men with visceral fat accumulation. Int J Obes Relat Metab Disord. (1998) 22:477–84. doi: 10.1038/sj.ijo.0800620
95. Premer C, Blum A, Bellio MA, Schulman IH, Hurwitz BE, Parker M, et al. Allogeneic Mesenchymal Stem Cells Restore Endothelial Function in Heart Failure by Stimulating Endothelial Progenitor Cells. EBioMedicine. (2015) 2:467–75. doi: 10.1016/j.ebiom.2015.03.020
96. Gokce N, Keaney JF Jr., Hunter LM, Watkins MT, Nedeljkovic ZS, Menzoian JO, et al. Predictive value of noninvasively determined endothelial dysfunction for long-term cardiovascular events in patients with peripheral vascular disease. J Am Coll Cardiol. (2003) 41:1769–75. doi: 10.1016/S0735-1097(03)00333-4
97. Heffernan KS, Patvardhan EA, Kapur NK, Karas RH, and Kuvin JT. Peripheral augmentation index as a biomarker of vascular aging: an invasive hemodynamics approach. Eur J Appl Physiol. (2012) 112:2871–9. doi: 10.1007/s00421-011-2255-y
98. Tanaka A, Tomiyama H, Maruhashi T, Matsuzawa Y, Miyoshi T, Kabutoya T, et al. Physiological Diagnostic Criteria for Vascular Failure. Hypertension. (2018) 72:1060–71. doi: 10.1161/HYPERTENSIONAHA.118.11554
99. Bonetti PO, Pumper GM, Higano ST, Holmes DR Jr., Kuvin JT, and Lerman A. Noninvasive identification of patients with early coronary atherosclerosis by assessment of digital reactive hyperemia. J Am Coll Cardiol. (2004) 44:2137–41. doi: 10.1016/j.jacc.2004.08.062
100. Bruno RM, Gori T, and Ghiadoni L. Endothelial function testing and cardiovascular disease: focus on peripheral arterial tonometry. Vasc Health Risk Manage. (2014) 10:577–84. doi: 10.2147/VHRM.S44471
101. Coffman JD. Effects of endothelium-derived nitric oxide on skin and digital blood flow in humans. Am J Physiol. (1994) 267:H2087–90. doi: 10.1152/ajpheart.1994.267.6.H2087
102. Jujic A, Kenneback C, Johansson M, Nilsson PM, and Holm H. The impact of age on endothelial dysfunction measured by peripheral arterial tonometry in a healthy population-based cohort - the Malmo offspring study. Blood Press. (2023) 32:2234059. doi: 10.1080/08037051.2023.2234059
103. Bahouth Z, Zreik R, Graif A, Nativ O, Halachmi S, and Pillar G. Endothelial Function Assessment in Patients with Erectile Dysfunction. Isr Med Assoc J. (2015) 17:682–6.
104. Moerland M, Kales AJ, Schrier L, van Dongen MG, Bradnock D, and Burggraaf J. Evaluation of the EndoPAT as a Tool to Assess Endothelial Function. Int J Vasc Med. (2012) 2012:904141. doi: 10.1155/2012/904141
105. Fatihoglu SG, Jam F, Okutucu S, and Oto A. Noninvasive Investigation of the Presence and Extent of Coronary Artery Disease by the Evaluation of Fingertip-Reactive Hyperemia. Med Princ Pract. (2022) 31:262–8. doi: 10.1159/000522098
106. Selamet Tierney ES, Newburger JW, Gauvreau K, Geva J, Coogan E, Colan SD, et al. Endothelial pulse amplitude testing: feasibility and reproducibility in adolescents. J Pediatr. (2009) 154:901–5. doi: 10.1016/j.jpeds.2008.12.028
107. Heffernan KS, Karas RH, Patvardhan EA, Jafri H, and Kuvin JT. Peripheral arterial tonometry for risk stratification in men with coronary artery disease. Clin Cardiol. (2010) 33:94–8. doi: 10.1002/clc.20705
108. Lal A, Kazi S, Kovoor P, Liew G, Mitchell P, and Thiagalingam A. Assessing endothelial function in type-2-diabetes using a novel method of ECG-gated retinal artery flicker provocation and digital reactive hyperaemia (Endopat&xae);. Heart Lung Circ. (2015) 24:S325. doi: 10.1016/j.hlc.2015.06.500
109. Lal A, Thiagalingam A, Mitchell P, Gibbs O, Barry T, and Kovoor P. PM417 Assessing endothelial function in type-2-diabetes mellitus using ECG-gated retinal artery flicker provocation and digital reactive hyperaemia (Endopat®). Global Heart. (2014) 9:e146. doi: 10.1016/j.gheart.2014.03.1748
110. Zhang H, Sun T, Cheng Y, Zhang J, Zhang H, Krittanawong C, et al. Impact of metabolic syndrome and systemic inflammation on endothelial function in postmenopausal women. Turk Kardiyol Dern Ars. (2022) 50:57–65. doi: 10.5543/tkda.2022.47443
111. Dawson AJ, Sathyapalan T, Smithson JA, Vince RV, Coady AM, Ajjan R, et al. A comparison of cardiovascular risk indices in patients with polycystic ovary syndrome with and without coexisting nonalcoholic fatty liver disease. Clin Endocrinol (Oxf). (2014) 80:843–9. doi: 10.1111/cen.2014.80.issue-6
112. Laurent S, Cockcroft J, Van Bortel L, Boutouyrie P, Giannattasio C, Hayoz D, et al. Expert consensus document on arterial stiffness: methodological issues and clinical applications. Eur Heart J. (2006) 27:2588–605. doi: 10.1093/eurheartj/ehl254
113. Laurent S, Katsahian S, Fassot C, Tropeano AI, Gautier I, Laloux B, et al. Aortic stiffness is an independent predictor of fatal stroke in essential hypertension. Stroke. (2003) 34:1203–6. doi: 10.1161/01.STR.0000065428.03209.64
114. Boutouyrie P, Tropeano AI, Asmar R, Gautier I, Benetos A, Lacolley P, et al. Aortic stiffness is an independent predictor of primary coronary events in hypertensive patients: a longitudinal study. Hypertension. (2002) 39:10–5. doi: 10.1161/hy0102.099031
115. Cruickshank K, Riste L, Anderson SG, Wright JS, Dunn G, and Gosling RG. Aortic pulse-wave velocity and its relationship to mortality in diabetes and glucose intolerance: an integrated index of vascular function? Circulation. (2002) 106:2085–90. doi: 10.1161/01.CIR.0000033824.02722.F7
116. Blacher J, Guerin AP, Pannier B, Marchais SJ, Safar ME, and London GM. Impact of aortic stiffness on survival in end-stage renal disease. Circulation. (1999) 99:2434–9. doi: 10.1161/01.CIR.99.18.2434
117. Gasecki D, Rojek A, Kwarciany M, Kowalczyk K, Boutouyrie P, Nyka W, et al. Pulse wave velocity is associated with early clinical outcome after ischemic stroke. Atherosclerosis. (2012) 225:348–52. doi: 10.1016/j.atherosclerosis.2012.09.024
118. Ki YJ, Choi DH, Lee YM, Lim L, Song H, and Koh YY. Predictive value of brachial-ankle pulse wave velocity for long-term clinical outcomes after percutaneous coronary intervention in a Korean cohort. Int J Cardiol. (2014) 175:554–9. doi: 10.1016/j.ijcard.2014.06.032
119. Musz P, Podhajski P, Grzelakowska K, and Umińska J. Non-invasive assessment of endothelial function – a review of available methods. Med Res J. (2021) 6:53–8. doi: 10.5603/MRJ.a2021.0008
120. Munakata M. Brachial-ankle pulse wave velocity in the measurement of arterial stiffness: recent evidence and clinical applications. Curr Hypertens Rev. (2014) 10:49–57. doi: 10.2174/157340211001141111160957
121. Costa FC, Gomes LG, de Lima TM, Bortolotto LA, Hong V, Verardino R, et al. Cardiovascular risk in women with nonclassical congenital adrenal hyperplasia. J Clin Endocrinol Metab. (2024) 110(1):e72–9. doi: 10.1210/clinem/dgae155
122. Rosenbaum D, Gallo A, Lethielleux G, Bruckert E, Levy BI, Tanguy ML, et al. Early central blood pressure elevation in adult patients with 21-hydroxylase deficiency. J Hypertens. (2019) 37:175–81. doi: 10.1097/HJH.0000000000001850
123. Bechlioulis A, Vakalis K, Naka KK, Bourantas CV, Papamichael ND, Kotsia A, et al. Increased aortic pulse wave velocity is associated with the presence of angiographic coronary artery disease in overweight and obese patients. Am J Hypertens. (2013) 26:265–70. doi: 10.1093/ajh/hps039
124. Duman OO, Goldeli O, Gursul E, Baris N, Ozpelit E, and Simsek MA. The value of aortic pulse wave velocity in predicting coronary artery disease diagnosis and severity. Acta Cardiol. (2015) 70:315–22. doi: 10.1080/ac.70.3.3080636
125. Braber TL, Prakken NH, Mosterd A, Mali WP, Doevendans PA, Bots ML, et al. Identifying Coronary Artery Disease in Asymptomatic Middle-Aged Sportsmen: The Additional Value of Pulse Wave Velocity. PLoS One. (2015) 10:e0131895. doi: 10.1371/journal.pone.0131895
126. Kelly CJ, Speirs A, Gould GW, Petrie JR, Lyall H, and Connell JM. Altered vascular function in young women with polycystic ovary syndrome. J Clin Endocrinol Metab. (2002) 87:742–6. doi: 10.1210/jcem.87.2.8199
127. Wang A, Su Z, Liu X, Yang Y, Chen S, Wang S, et al. Brachial-ankle pulse wave velocity and metabolic syndrome in general population: the APAC study. BMC Cardiovasc Disord. (2016) 16:228. doi: 10.1186/s12872-016-0409-x
128. Mancia G, Fagard R, Narkiewicz K, Redon J, Zanchetti A, Böhm M, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J. (2013) 34(28):2159–219. doi: 10.1093/eurheartj/eht151
129. Naqvi TZ and Lee MS. Carotid intima-media thickness and plaque in cardiovascular risk assessment. JACC Cardiovasc Imaging. (2014) 7:1025–38. doi: 10.1016/j.jcmg.2013.11.014
130. Enderle MD, Schroeder S, Ossen R, Meisner C, Baumbach A, Haering HU, et al. Comparison of peripheral endothelial dysfunction and intimal media thickness in patients with suspected coronary artery disease. Heart. (1998) 80:349–54. doi: 10.1136/hrt.80.4.349
131. Hashimoto M, Eto M, Akishita M, Kozaki K, Ako J, Iijima K, et al. Correlation between flow-mediated vasodilatation of the brachial artery and intima-media thickness in the carotid artery in men. Arterioscler Thromb Vasc Biol. (1999) 19:2795–800. doi: 10.1161/01.ATV.19.11.2795
132. Abd El Dayem SM, Anwar GM, Salama H, Kamel AF, and Emara N. Bone mineral density, bone turnover markers, lean mass, and fat mass in Egyptian children with congenital adrenal hyperplasia. Arch Med Sci. (2010) 6:104–10. doi: 10.5114/aoms.2010.13516
133. Sartorato P, Zulian E, Benedini S, Mariniello B, Schiavi F, Bilora F, et al. Cardiovascular risk factors and ultrasound evaluation of intima-media thickness at common carotids, carotid bulbs, and femoral and abdominal aorta arteries in patients with classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2007) 92:1015–8. doi: 10.1210/jc.2006-1711
134. Akyurek N, Atabek ME, Eklioglu BS, and Alp H. Ambulatory blood pressure and subclinical cardiovascular disease in patients with congenital adrenal hyperplasia: a preliminary report. J Clin Res Pediatr Endocrinol. (2015) 7:13–8. doi: 10.4274/jcrpe.1658
135. Metwalley KA. Left ventricular dysfunction and subclinical atherosclerosis in children with classic congenital adrenal hyperplasia: a single-center study from Upper Egypt. Eur J Pediatr. (2016) 175:415. doi: 10.1007/s00431-015-2678-2
136. Rodrigues TM, Barra CB, Santos JL, Goulart EM, Ferreira AV, and Silva IN. Cardiovascular risk factors and increased carotid intima-media thickness in young patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Arch Endocrinol Metab. (2015) 59:541–7. doi: 10.1590/2359-3997000000119
137. Amr NH, Ahmed AY, and Ibrahim YA. Carotid intima media thickness and other cardiovascular risk factors in children with congenital adrenal hyperplasia. J Endocrinol Invest. (2014) 37:1001–8. doi: 10.1007/s40618-014-0148-8
138. Wasniewska M, Balsamo A, Valenzise M, Manganaro A, Faggioli G, Bombaci S, et al. Increased large artery intima media thickness in adolescents with either classical or non-classical congenital adrenal hyperplasia. J Endocrinol Invest. (2013) 36:12–5. doi: 10.1007/BF03346751
139. Ozdemir R, Korkmaz HA, Kucuk M, Karadeniz C, Mese T, and Ozkan B. Assessment of early atherosclerosis and left ventricular dysfunction in children with 21-hydroxylase deficiency. Clin Endocrinol (Oxf). (2017) 86:473–9. doi: 10.1111/cen.13275
140. Kim MS, Dao-Tran A, Davidowitz E, Tseng T, Gilsanz V, Ryabets-Lienhard A, et al. Carotid Intima-Media Thickness Is Associated with Increased Androgens in Adolescents and Young Adults with Classical Congenital Adrenal Hyperplasia. Horm Res Paediatr. (2016) 85:242–9. doi: 10.1159/000444169
141. Ines D, Ivana J, Ana S, Anita C, Marko S, and Kristina S-R. The Markers of Endothelial Activation. In: Helena L, editor. Endothelial Dysfunction. IntechOpen, Rijeka (2018). p. Ch. 17.
142. Constans J and Conri C. Circulating markers of endothelial function in cardiovascular disease. Clin Chim Acta. (2006) 368:33–47. doi: 10.1016/j.cca.2005.12.030
143. Rabelink TJ, de Boer HC, and van Zonneveld AJ. Endothelial activation and circulating markers of endothelial activation in kidney disease. Nat Rev Nephrol. (2010) 6:404–14. doi: 10.1038/nrneph.2010.65
144. Cooke JP and Wilson AM. Biomarkers of peripheral arterial disease. J Am Coll Cardiol. (2010) 55:2017–23. doi: 10.1016/j.jacc.2009.08.090
145. Zhang J, Defelice AF, Hanig JP, and Colatsky T. Biomarkers of endothelial cell activation serve as potential surrogate markers for drug-induced vascular injury. Toxicol Pathol. (2010) 38:856–71. doi: 10.1177/0192623310378866
146. Qiu Y, Zhang C, Zhang G, and Tao J. Endothelial progenitor cells in cardiovascular diseases. Aging Med (Milton). (2018) 1:204–8. doi: 10.1002/agm2.12041
147. Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, Quyyumi AA, et al. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. (2003) 348:593–600. doi: 10.1056/NEJMoa022287
148. Helbing T, Olivier C, Bode C, Moser M, and Diehl P. Role of microparticles in endothelial dysfunction and arterial hypertension. World J Cardiol. (2014) 6:1135–9. doi: 10.4330/wjc.v6.i11.1135
149. Kumar S, Williams D, Sur S, Wang JY, and Jo H. Role of flow-sensitive microRNAs and long noncoding RNAs in vascular dysfunction and atherosclerosis. Vascul Pharmacol. (2019) 114:76–92. doi: 10.1016/j.vph.2018.10.001
150. Menghini R, Casagrande V, and Federici M. MicroRNAs in endothelial senescence and atherosclerosis. J Cardiovasc Transl Res. (2013) 6:924–30. doi: 10.1007/s12265-013-9487-7
151. Perkins LA, Anderson CJ, and Novelli EM. Targeting P-Selectin Adhesion Molecule in Molecular Imaging: P-Selectin Expression as a Valuable Imaging Biomarker of Inflammation in Cardiovascular Disease. J Nucl Med. (2019) 60:1691–7. doi: 10.2967/jnumed.118.225169
152. Zhang J, Huang S, Zhu Z, Gatt A, and Liu J. E-selectin in vascular pathophysiology. Front Immunol. (2024) 15:1401399. doi: 10.3389/fimmu.2024.1401399
153. Galkina E and Ley K. Vascular adhesion molecules in atherosclerosis. Arterioscler Thromb Vasc Biol. (2007) 27:2292–301. doi: 10.1161/ATVBAHA.107.149179
154. Horvath B, Hegedus D, Szapary L, Marton Z, Alexy T, Koltai K, et al. Measurement of von Willebrand factor as the marker of endothelial dysfunction in vascular diseases. Exp Clin Cardiol. (2004) 9:31–4.
155. Fuchs D, Avanzas P, Arroyo-Espliguero R, Jenny M, Consuegra-Sanchez L, and Kaski JC. The role of neopterin in atherogenesis and cardiovascular risk assessment. Curr Med Chem. (2009) 16:4644–53. doi: 10.2174/092986709789878247
156. B KGEEAYBES. The role of osteoprotegerin and sclerostin in bone metabolism in children with congenital adrenal hyperplasia. In: 25th European Congress of Endocrinology 13 May 2023–16 May 2023. European Society of Endocrinology, Istanbul, Turkey (2023).
157. Kurnaz E, Cetinkaya S, Ozalkak S, Bayramoglu E, Demirci G, Ozturk HS, et al. Serum Fetuin-A and Insulin Levels in Classic Congenital Adrenal Hyperplasia. Horm Metab Res. (2020) 52:654–9. doi: 10.1055/a-1116-2173
158. Falhammar H, Filipsson Nystrom H, Wedell A, and Thoren M. Cardiovascular risk, metabolic profile, and body composition in adult males with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Eur J Endocrinol. (2011) 164:285–93. doi: 10.1530/EJE-10-0877
159. Metwalley KA, Farghaly HS, and Abdelhamid A. Homocysteine Level in Children with Classic Congenital Adrenal Hyperplasia: Relationship to Carotid Intimal Wall Thickness and Left Ventricular Function. Horm Res Paediatr. (2018) 90:228–35. doi: 10.1159/000492900
160. Borges JH, de Oliveira DM, de Lemos-Marini SHV, Geloneze B, Gonçalves EM, and Guerra-Júnior G. Fat Distribution and Lipid Profile of Young Adults with Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Enzyme Deficiency. Lipids. (2021) 56:101–10. doi: 10.1002/lipd.12280
161. Volkl TM, Simm D, Korner A, Rascher W, Kiess W, Kratzsch J, et al. Does an altered leptin axis play a role in obesity among children and adolescents with classical congenital adrenal hyperplasia due to 21-hydroxylase deficiency? Eur J Endocrinol. (2009) 160:239–47. doi: 10.1530/EJE-08-0770
162. Isguven P, Arslanoglu I, Mesutoglu N, Yildiz M, and Erguven M. Bioelectrical impedance analysis of body fatness in childhood congenital adrenal hyperplasia and its metabolic correlates. Eur J Pediatr. (2008) 167:1263–8. doi: 10.1007/s00431-007-0665-y
163. Saygili F, Oge A, and Yilmaz C. Hyperinsulinemia and insulin insensitivity in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency: the relationship between serum leptin levels and chronic hyperinsulinemia. Horm Res. (2005) 63:270–4. doi: 10.1159/000086363
164. Volkl TM, Simm D, Korner A, Kiess W, Kratzsch J, and Dorr HG. Adiponectin levels are high in children with classic congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency. Acta Paediatr. (2009) 98:885–91. doi: 10.1111/j.1651-2227.2009.01231.x
165. Singh U, Devaraj S, Vasquez-Vivar J, and Jialal I. C-reactive protein decreases endothelial nitric oxide synthase activity via uncoupling. J Mol Cell Cardiol. (2007) 43:780–91. doi: 10.1016/j.yjmcc.2007.08.015
166. Delai A, Gomes PM, Foss-Freitas MC, Elias J, Antonini SR, Castro M, et al. Hyperinsulinemic-Euglycemic Clamp Strengthens the Insulin Resistance in Nonclassical Congenital Adrenal Hyperplasia. J Clin Endocrinol Metab. (2022) 107:e1106–e16. doi: 10.1210/clinem/dgab767
167. Angelopoulou MV, Kontogiorgos E, and Emmanouil D. Congenital adrenal hyperplasia: a case report with premature teeth exfoliation and bone resorption. Pediatrics. (2015) 135:e1524–9. doi: 10.1542/peds.2014-3577
168. Walczak M, Suraj J, Kus K, Kij A, Zakrzewska A, and Chlopicki S. Towards a comprehensive endothelial biomarkers profiling and endothelium-guided pharmacotherapy. Pharmacol Rep. (2015) 67:771–7. doi: 10.1016/j.pharep.2015.06.008
169. Gieseg SP, Crone EM, Flavall EA, and Amit Z. Potential to inhibit growth of atherosclerotic plaque development through modulation of macrophage neopterin/7,8-dihydroneopterin synthesis. Br J Pharmacol. (2008) 153:627–35. doi: 10.1038/sj.bjp.0707408
170. Cirillo P, Pacileo M, De Rosa S, CalabrÒ P, Gargiulo A, Angri V, et al. Neopterin induces pro&x2010;atherothrombotic phenotype in human coronary endothelial cells. J Thromb Haemostasis. (2006) 4:2248–55. doi: 10.1111/j.1538-7836.2006.02125.x
171. Shirai R, Sato K, Yamashita T, Yamaguchi M, Okano T, Watanabe-Kominato K, et al. Neopterin Counters Vascular Inflammation and Atherosclerosis. J Am Heart Assoc. (2018) 7. doi: 10.1161/JAHA.117.007359
172. Murr C, Fuith LC, Widner B, Wirleitner B, Baier-Bitterlich G, and Fuchs D. Increased neopterin concentrations in patients with cancer: indicator of oxidative stress? Anticancer Res. (1999) 19:1721–8.
173. Tatzber F, Rabl H, Koriska K, Erhart U, Puhl H, Waeg G, et al. Elevated serum neopterin levels in atherosclerosis. Atherosclerosis. (1991) 89:203–8. doi: 10.1016/0021-9150(91)90061-7
174. Weiss G, Willeit J, Kiechl S, Fuchs D, Jarosch E, Oberhollenzer F, et al. Increased concentrations of neopterin in carotid atherosclerosis. Atherosclerosis. (1994) 106:263–71. doi: 10.1016/0021-9150(94)90131-7
175. Liu Y, Chen F, Zeng Z, Lei C, Chen D, and Zhang X. Neopterin in patients with COPD, asthma, and ACO: association with endothelial and lung functions. Respir Res. (2024) 25:171. doi: 10.1186/s12931-024-02784-4
176. Zhang YY, Tong XZ, Xia WH, Xie WL, Yu BB, Zhang B, et al. Increased plasma neopterin levels are associated with reduced endothelial function and arterial elasticity in hypertension. J Hum Hypertens. (2016) 30:436–41. doi: 10.1038/jhh.2015.72
177. Meng LL and Cao L. Serum neopterin levels and their role in the prognosis of patients with ischemic stroke: A systematic review and meta-analysis. J Clin Neurosci. (2021) 92:55–60. doi: 10.1016/j.jocn.2021.07.033
178. Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Lüthy R, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. (1997) 89:309–19. doi: 10.1016/S0092-8674(00)80209-3
179. Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. (1998) 93:165–76. doi: 10.1016/S0092-8674(00)81569-X
180. Fuller K, Wong B, Fox S, Choi Y, and Chambers TJ. TRANCE is necessary and sufficient for osteoblast-mediated activation of bone resorption in osteoclasts. J Exp Med. (1998) 188:997–1001. doi: 10.1084/jem.188.5.997
181. Emery JG, McDonnell P, Burke MB, Deen KC, Lyn S, Silverman C, et al. Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J Biol Chem. (1998) 273:14363–7. doi: 10.1074/jbc.273.23.14363
182. Tintut Y and Demer L. Role of osteoprotegerin and its ligands and competing receptors in atherosclerotic calcification. J Invest Med. (2006) 54:395–401. doi: 10.2310/6650.2006.06019
183. Ziegler S, Kudlacek S, Luger A, and Minar E. Osteoprotegerin plasma concentrations correlate with severity of peripheral artery disease. Atherosclerosis. (2005) 182:175–80. doi: 10.1016/j.atherosclerosis.2005.01.042
184. Mogelvang R, Pedersen SH, Flyvbjerg A, Bjerre M, Iversen AZ, Galatius S, et al. Comparison of osteoprotegerin to traditional atherosclerotic risk factors and high-sensitivity C-reactive protein for diagnosis of atherosclerosis. Am J Cardiol. (2012) 109:515–20. doi: 10.1016/j.amjcard.2011.09.043
185. Tschiderer L, Willeit J, Schett G, Kiechl S, and Willeit P. Osteoprotegerin concentration and risk of cardiovascular outcomes in nine general population studies: Literature-based meta-analysis involving 26,442 participants. PLoS One. (2017) 12:e0183910. doi: 10.1371/journal.pone.0183910
186. Zhao H, Cao Y, Chen H, Xu W, Sun X, and Pan X. The association between OPG rs3102735 gene polymorphism, microembolic signal and stroke severity in acute ischemic stroke patients. Gene. (2017) 613:25–9. doi: 10.1016/j.gene.2017.02.029
187. Abedin M, Tintut Y, and Demer LL. Vascular calcification: mechanisms and clinical ramifications. Arterioscler Thromb Vasc Biol. (2004) 24:1161–70. doi: 10.1161/01.ATV.0000133194.94939.42
188. Persy V and D’Haese P. Vascular calcification and bone disease: the calcification paradox. Trends Mol Med. (2009) 15:405–16. doi: 10.1016/j.molmed.2009.07.001
189. Rochette L, Meloux A, Rigal E, Zeller M, Cottin Y, and Vergely C. The Role of Osteoprotegerin and Its Ligands in Vascular Function. Int J Mol Sci. (2019) 20. doi: 10.3390/ijms20030705
190. Dutka M, Bobiński R, Wojakowski W, Francuz T, Pająk C, and Zimmer K. Osteoprotegerin and RANKL-RANK-OPG-TRAIL signalling axis in heart failure and other cardiovascular diseases. Heart Fail Rev. (2022) 27:1395–411. doi: 10.1007/s10741-021-10153-2
191. Zauli G, Corallini F, Bossi F, Fischetti F, Durigutto P, Celeghini C, et al. Osteoprotegerin increases leukocyte adhesion to endothelial cells both in vitro and in vivo. Blood. (2007) 110:536–43. doi: 10.1182/blood-2007-01-068395
192. Wang HH and Xiang GD. Changes of plasma concentration of osteoprotegerin and its association with endothelial dysfunction before and after hypouricemic therapy in patients with hyperuricemia. Mod Rheumatol. (2015) 25:123–7. doi: 10.3109/14397595.2014.926852
193. Shin JY, Shin YG, and Chung CH. Elevated serum osteoprotegerin levels are associated with vascular endothelial dysfunction in type 2 diabetes. Diabetes Care. (2006) 29:1664–6. doi: 10.2337/dc06-0631
194. D’Abramo A, Zingaropoli MA, Oliva A, D’Agostino C, Al Moghazi S, De Luca G, et al. Immune activation, immunosenescence, and osteoprotegerin as markers of endothelial dysfunction in subclinical HIV-associated atherosclerosis. Mediators Inflamm. (2014) 2014:192594. doi: 10.1155/2014/192594
195. Gunes M, Temizkan S, Apaydin T, Ilgin C, Haklar G, and Gogas Yavuz D. Serum osteoprotegerin levels, endothelial function and carotid intima-media thickness in type 2 diabetic patients. J Diabetes Complications. (2021) 35:108073. doi: 10.1016/j.jdiacomp.2021.108073
196. Abali R, Tasdemir N, Alpsoy S, Tasdemir UG, Guzel S, Yuksel MA, et al. No relationship between osteoprotegerin concentrations and endothelial dysfunction in non-obese women with and without polycystic ovary syndrome. Arch Gynecol Obstet. (2015) 291:1075–80. doi: 10.1007/s00404-014-3499-7
197. Trepanowski JF, Mey J, and Varady KA. Fetuin-A: a novel link between obesity and related complications. Int J Obesity. (2015) 39:734–41. doi: 10.1038/ijo.2014.203
198. Goustin A-S and Abou-Samra AB. The “thrifty” gene encoding Ahsg/Fetuin-A meets the insulin receptor: Insights into the mechanism of insulin resistance. Cell Signalling. (2011) 23:980–90. doi: 10.1016/j.cellsig.2010.11.003
199. Stefan N, Hennige AM, Staiger H, Machann J, Schick F, Krober SM, et al. Alpha2-Heremans-Schmid glycoprotein/fetuin-A is associated with insulin resistance and fat accumulation in the liver in humans. Diabetes Care. (2006) 29:853–7. doi: 10.2337/diacare.29.04.06.dc05-1938
200. Ou HY, Yang YC, Wu HT, Wu JS, Lu FH, and Chang CJ. Increased fetuin-A concentrations in impaired glucose tolerance with or without nonalcoholic fatty liver disease, but not impaired fasting glucose. J Clin Endocrinol Metab. (2012) 97:4717–23. doi: 10.1210/jc.2012-2414
201. Ix JH, Shlipak MG, Brandenburg VM, Ali S, Ketteler M, and Whooley MA. Association between human fetuin-A and the metabolic syndrome: data from the Heart and Soul Study. Circulation. (2006) 113:1760–7. doi: 10.1161/CIRCULATIONAHA.105.588723
202. Hennige AM, Staiger H, Wicke C, Machicao F, Fritsche A, Haring HU, et al. Fetuin-A induces cytokine expression and suppresses adiponectin production. PLoS One. (2008) 3:e1765. doi: 10.1371/journal.pone.0001765
203. Schafer C, Heiss A, Schwarz A, Westenfeld R, Ketteler M, Floege J, et al. The serum protein alpha 2-Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest. (2003) 112:357–66. doi: 10.1172/JCI17202
204. Jahnen-Dechent W, Heiss A, Schäfer C, and Ketteler M. Fetuin-A regulation of calcified matrix metabolism. Circ Res. (2011) 108:1494–509. doi: 10.1161/CIRCRESAHA.110.234260
205. Hamano T, Matsui I, Mikami S, Tomida K, Fujii N, Imai E, et al. Fetuin-mineral complex reflects extraosseous calcification stress in CKD. J Am Soc Nephrol. (2010) 21:1998–2007. doi: 10.1681/ASN.2009090944
206. Ismail NA, Ragab S, El Dayem SM, Elbaky AA, Salah N, Hamed M, et al. Fetuin-A levels in obesity: differences in relation to metabolic syndrome and correlation with clinical and laboratory variables. Arch Med Sci. (2012) 8:826–33. doi: 10.5114/aoms.2012.31616
207. Ix JH, Wassel CL, Kanaya AM, Vittinghoff E, Johnson KC, Koster A, et al. Fetuin-A and incident diabetes mellitus in older persons. JAMA. (2008) 300:182–8. doi: 10.1001/jama.300.2.182
208. Liu S, Hu W, He Y, Li L, Liu H, Gao L, et al. Serum Fetuin-A levels are increased and associated with insulin resistance in women with polycystic ovary syndrome. BMC Endocr Disord. (2020) 20:67. doi: 10.1186/s12902-020-0538-1
209. Enli Y, Fenkci SM, Fenkci V, and Oztekin O. Serum Fetuin-A levels, insulin resistance and oxidative stress in women with polycystic ovary syndrome. Gynecological Endocrinology. (2013) 29:1036–9. doi: 10.3109/09513590.2013.829442
210. Djuric D, Jakovljevic V, Zivkovic V, and Srejovic I. Homocysteine and homocysteine-related compounds: an overview of the roles in the pathology of the cardiovascular and nervous systems. Can J Physiol Pharmacol. (2018) 96:991–1003. doi: 10.1139/cjpp-2018-0112
211. Paganelli F, Mottola G, Fromonot J, Marlinge M, Deharo P, Guieu R, et al. Hyperhomocysteinemia and Cardiovascular Disease: Is the Adenosinergic System the Missing Link? Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22041690
212. Stehouwer CD, Weijenberg MP, van den Berg M, Jakobs C, Feskens EJ, and Kromhout D. Serum homocysteine and risk of coronary heart disease and cerebrovascular disease in elderly men: a 10-year follow-up. Arterioscler Thromb Vasc Biol. (1998) 18:1895–901. doi: 10.1161/01.ATV.18.12.1895
213. Catena C, Colussi G, Nait F, Capobianco F, and Sechi LA. Elevated Homocysteine Levels Are Associated With the Metabolic Syndrome and Cardiovascular Events in Hypertensive Patients. Am J Hypertens. (2015) 28:943–50. doi: 10.1093/ajh/hpu248
214. Baszczuk A and Kopczynski Z. Hyperhomocysteinemia in patients with cardiovascular disease. Postepy Hig Med Dosw (Online). (2014) 68:579–89. doi: 10.5604/17322693.1102340
215. Shenoy V, Mehendale V, Prabhu K, Shetty R, and Rao P. Correlation of serum homocysteine levels with the severity of coronary artery disease. Indian J Clin Biochem. (2014) 29:339–44. doi: 10.1007/s12291-013-0373-5
216. Jin N, Huang L, Hong J, Zhao X, Chen Y, Hu J, et al. Elevated homocysteine levels in patients with heart failure: A systematic review and meta-analysis. Med (Baltimore). (2021) 100:e26875. doi: 10.1097/MD.0000000000026875
217. Sharma M, Tiwari M, and Tiwari RK. Hyperhomocysteinemia: Impact on Neurodegenerative Diseases. Basic Clin Pharmacol Toxicol. (2015) 117:287–96. doi: 10.1111/bcpt.2015.117.issue-5
218. Skovierova H, Vidomanova E, Mahmood S, Sopkova J, Drgova A, Cervenova T, et al. The Molecular and Cellular Effect of Homocysteine Metabolism Imbalance on Human Health. Int J Mol Sci. (2016) 17. doi: 10.3390/ijms17101733
219. Pushpakumar S, Kundu S, and Sen U. Endothelial dysfunction: the link between homocysteine and hydrogen sulfide. Curr Med Chem. (2014) 21:3662–72. doi: 10.2174/0929867321666140706142335
220. Yuan D, Chu J, Lin H, Zhu G, Qian J, Yu Y, et al. Mechanism of homocysteine-mediated endothelial injury and its consequences for atherosclerosis. Front Cardiovasc Med. (2022) 9:1109445. doi: 10.3389/fcvm.2022.1109445
221. Austin RC, Lentz SR, and Werstuck GH. Role of hyperhomocysteinemia in endothelial dysfunction and atherothrombotic disease. Cell Death Differentiation. (2004) 11:S56–64. doi: 10.1038/sj.cdd.4401451
222. Krysiak R, Kowalcze K, Marek B, and Okopień B. Cardiometabolic risk factors in women with non-classic congenital adrenal hyperplasia. Acta Cardiol. (2020) 75:705–10. doi: 10.1080/00015385.2019.1666964
223. Dierkes J, Jeckel A, Ambrosch A, Westphal S, Luley C, and Boeing H. Factors explaining the difference of total homocysteine between men and women in the European Investigation Into Cancer and Nutrition Potsdam study. Metabolism. (2001) 50:640–5. doi: 10.1053/meta.2001.23286
224. Maleedhu P MV, SBS S, Kodumuri PK, and Devi DV. Status of Homocysteine in Polycystic Ovary Syndrome (PCOS). J Clin Diagn Res. (2014) 8:31–3. doi: 10.7860/JCDR/2014/7070.3999
225. Loverro G, Lorusso F, Mei L, Depalo R, Cormio G, and Selvaggi L. The plasma homocysteine levels are increased in polycystic ovary syndrome. Gynecol Obstet Invest. (2002) 53:157–62. doi: 10.1159/000058367
226. Badawy A, State O, El Gawad SSA, and El Aziz OA. Plasma homocysteine and polycystic ovary syndrome: the missed link. Eur J Obstet Gynecol Reprod Biol. (2007) 131:68–72. doi: 10.1016/j.ejogrb.2006.10.015
227. Karatela RA and Sainani GS. Plasma homocysteine in obese, overweight and normal weight hypertensives and normotensives. Indian Heart J. (2009) 61:156–9.
228. Vayá A, Rivera L, Hernández-Mijares A, de la Fuente M, Solá E, Romagnoli M, et al. Homocysteine levels in morbidly obese patients. Its association with waist circumference and insulin resistance. Clin Hemorheology Microcirculation. (2012) 52:49–56. doi: 10.3233/CH-2012-1544
229. Denver RJ, Bonett RM, and Boorse GC. Evolution of leptin structure and function. Neuroendocrinology. (2011) 94:21–38. doi: 10.1159/000328435
230. Chen M-C, Wang J-H, Lee C-J, and Hsu B-G. Association between hyperleptinemia and cardiovascular outcomes in patients with coronary artery disease. Ther Clin Risk Manage. (2018) 14:1855–62. doi: 10.2147/TCRM.S172231
231. Ren J. Leptin and hyperleptinemia-from friend to foe for cardiovascular function. J Endocrinology. (2004) 181:1–10. doi: 10.1677/joe.0.1810001
232. Reilly MP, Iqbal N, Schutta M, Wolfe ML, Scally M, Localio AR, et al. Plasma leptin levels are associated with coronary atherosclerosis in type 2 diabetes. J Clin Endocrinol Metab. (2004) 89:3872–8. doi: 10.1210/jc.2003-031676
233. Montazerifar F, Bolouri A, Paghalea RS, Mahani MK, and Karajibani M. Obesity, Serum Resistin and Leptin Levels Linked to Coronary Artery Disease. Arq Bras Cardiol. (2016) 107:348–53. doi: 10.5935/abc.20160134
234. Yang H, Guo W, Li J, Cao S, Zhang J, Pan J, et al. Leptin concentration and risk of coronary heart disease and stroke: A systematic review and meta-analysis. PLoS One. (2017) 12:e0166360. doi: 10.1371/journal.pone.0166360
235. Nguyen TMD. Adiponectin: Role in Physiology and Pathophysiology. Int J Prev Med. (2020) 11:136. doi: 10.4103/ijpvm.IJPVM_193_20
236. Yoda-Murakami M, Taniguchi M, Takahashi K, Kawamata S, Saito K, Choi-Miura NH, et al. Change in expression of GBP28/adiponectin in carbon tetrachloride-administrated mouse liver. Biochem Biophys Res Commun. (2001) 285:372–7. doi: 10.1006/bbrc.2001.5134
237. Delaigle AM, Jonas JC, Bauche IB, Cornu O, and Brichard SM. Induction of adiponectin in skeletal muscle by inflammatory cytokines: in vivo and in vitro studies. Endocrinology. (2004) 145:5589–97. doi: 10.1210/en.2004-0503
238. Patel JV, Abraheem A, Dotsenko O, Creamer J, Gunning M, Hughes EA, et al. Circulating serum adiponectin levels in patients with coronary artery disease: relationship to atherosclerotic burden and cardiac function. J Intern Med. (2008) 264:593–8. doi: 10.1111/j.1365-2796.2008.02007.x
239. Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. (2003) 423:762–9. doi: 10.1038/nature01705
240. Tkachuk VA, Bochkov VN, Philippova MP, Stambolsky DV, Kuzmenko ES, Sidorova MV, et al. Identification of an atypical lipoprotein-binding protein from human aortic smooth muscle as T-cadherin. FEBS Lett. (1998) 421:208–12. doi: 10.1016/S0014-5793(97)01562-7
241. Wyder L, Vitaliti A, Schneider H, Hebbard LW, Moritz DR, Wittmer M, et al. Increased expression of H/T-cadherin in tumor-penetrating blood vessels. Cancer Res. (2000) 60:4682–8.
242. Ivanov D, Philippova M, Antropova J, Gubaeva F, Iljinskaya O, Tararak E, et al. Expression of cell adhesion molecule T-cadherin in the human vasculature. Histochem Cell Biol. (2001) 115:231–42. doi: 10.1007/s004180100252
243. Sattar N, Wannamethee G, Sarwar N, Tchernova J, Cherry L, Wallace AM, et al. Adiponectin and coronary heart disease: a prospective study and meta-analysis. Circulation. (2006) 114:623–9. doi: 10.1161/CIRCULATIONAHA.106.618918
244. Iwashima Y, Katsuya T, Ishikawa K, Ouchi N, Ohishi M, Sugimoto K, et al. Hypoadiponectinemia is an independent risk factor for hypertension. Hypertension. (2004) 43:1318–23. doi: 10.1161/01.HYP.0000129281.03801.4b
245. Lam KS and Xu A. Adiponectin: protection of the endothelium. Curr Diabetes Rep. (2005) 5:254–9. doi: 10.1007/s11892-005-0019-y
246. Smekal A and Vaclavik J. Adipokines and cardiovascular disease: A comprehensive review. BioMed Pap Med Fac Univ Palacky Olomouc Czech Repub. (2017) 161:31–40. doi: 10.5507/bp.2017.002
247. Nakamura K, Fuster JJ, and Walsh K. Adipokines: a link between obesity and cardiovascular disease. J Cardiol. (2014) 63:250–9. doi: 10.1016/j.jjcc.2013.11.006
248. Shibata R, Izumiya Y, Sato K, Papanicolaou K, Kihara S, Colucci WS, et al. Adiponectin protects against the development of systolic dysfunction following myocardial infarction. J Mol Cell Cardiol. (2007) 42:1065–74. doi: 10.1016/j.yjmcc.2007.03.808
249. Wang Y, Ma XL, and Lau WB. Cardiovascular Adiponectin Resistance: The Critical Role of Adiponectin Receptor Modification. Trends Endocrinol Metab. (2017) 28:519–30. doi: 10.1016/j.tem.2017.03.004
250. Weyer C, Funahashi T, Tanaka S, Hotta K, Matsuzawa Y, Pratley RE, et al. Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab. (2001) 86:1930–5. doi: 10.1210/jcem.86.5.7463
251. Degawa-Yamauchi M, Moss KA, Bovenkerk JE, Shankar SS, Morrison CL, Lelliott CJ, et al. Regulation of adiponectin expression in human adipocytes: effects of adiposity, glucocorticoids, and tumor necrosis factor alpha. Obes Res. (2005) 13:662–9. doi: 10.1038/oby.2005.74
252. Bottner A, Kratzsch J, Muller G, Kapellen TM, Bluher S, Keller E, et al. Gender differences of adiponectin levels develop during the progression of puberty and are related to serum androgen levels. J Clin Endocrinol Metab. (2004) 89:4053–61. doi: 10.1210/jc.2004-0303
253. Ginsburg D, Zeheb R, Yang AY, Rafferty UM, Andreasen PA, Nielsen L, et al. cDNA cloning of human plasminogen activator-inhibitor from endothelial cells. J Clin Invest. (1986) 78:1673–80. doi: 10.1172/JCI112761
254. Vaughan DE. PAI-1 and atherothrombosis. J Thromb Haemost. (2005) 3:1879–83. doi: 10.1111/j.1538-7836.2005.01420.x
255. Yarmolinsky J, Bordin Barbieri N, Weinmann T, Ziegelmann PK, Duncan BB, and Ines Schmidt M. Plasminogen activator inhibitor-1 and type 2 diabetes: a systematic review and meta-analysis of observational studies. Sci Rep. (2016) 6:17714. doi: 10.1038/srep17714
256. Festa A, D’Agostino R Jr., Tracy RP, and Haffner SM. Insulin Resistance Atherosclerosis S. Elevated levels of acute-phase proteins and plasminogen activator inhibitor-1 predict the development of type 2 diabetes: the insulin resistance atherosclerosis study. Diabetes. (2002) 51:1131–7. doi: 10.2337/diabetes.51.4.1131
257. Alessi MC, Poggi M, and Juhan-Vague I. Plasminogen activator inhibitor-1, adipose tissue and insulin resistance. Curr Opin Lipidol. (2007) 18:240–5. doi: 10.1097/MOL.0b013e32814e6d29
258. Orio F Jr., Palomba S, Cascella T, Tauchmanova L, Nardo LG, Di Biase S, et al. Is plasminogen activator inhibitor-1 a cardiovascular risk factor in young women with polycystic ovary syndrome? Reprod BioMed Online. (2004) 9:505–10. doi: 10.1016/s1472-6483(10)61634-3
259. Tarkun I, Canturk Z, Arslan BC, Turemen E, and Tarkun P. The plasminogen activator system in young and lean women with polycystic ovary syndrome. Endocr J. (2004) 51:467–72. doi: 10.1507/endocrj.51.467
260. Iwasaki H, Okamoto R, Kato S, Konishi K, Mizutani H, Yamada N, et al. High glucose induces plasminogen activator inhibitor-1 expression through Rho/Rho-kinase-mediated NF-kappaB activation in bovine aortic endothelial cells. Atherosclerosis. (2008) 196:22–8. doi: 10.1016/j.atherosclerosis.2006.12.025
261. Schneider DJ and Sobel BE. Synergistic augmentation of expression of plasminogen activator inhibitor type-1 induced by insulin, very-low-density lipoproteins, and fatty acids. Coron Artery Dis. (1996) 7:813–7. doi: 10.1097/00019501-199611000-00004
262. Nordt TK, Schneider DJ, and Sobel BE. Augmentation of the synthesis of plasminogen activator inhibitor type-1 by precursors of insulin. A potential Risk factor Vasc disease. Circulation. (1994) 89:321–30. doi: 10.1161/01.CIR.89.1.321
263. Chen Y, Billadello JJ, and Schneider DJ. Identification and localization of a fatty acid response region in the human plasminogen activator inhibitor-1 gene. Arterioscler Thromb Vasc Biol. (2000) 20:2696–701. doi: 10.1161/01.ATV.20.12.2696
264. Fattal PG, Schneider DJ, Sobel BE, and Billadello JJ. Post-transcriptional regulation of expression of plasminogen activator inhibitor type 1 mRNA by insulin and insulin-like growth factor 1. J Biol Chem. (1992) 267:12412–5. doi: 10.1016/S0021-9258(18)42289-2
265. Verma S, Devaraj S, and Jialal I. Is C-reactive protein an innocent bystander or proatherogenic culprit? C-reactive protein promotes atherothrombosis. Circulation. (2006) 113:2135–50.
266. Ridker PM, Koenig W, Kastelein JJ, Mach F, and Luscher TF. Has the time finally come to measure hsCRP universally in primary and secondary cardiovascular prevention? Eur Heart J. (2018) 39:4109–11. doi: 10.1093/eurheartj/ehy723
267. Emerging Risk Factors C, Kaptoge S, Di Angelantonio E, Pennells L, Wood AM, White IR, et al. C-reactive protein, fibrinogen, and cardiovascular disease prediction. N Engl J Med. (2012) 367:1310–20. doi: 10.1056/NEJMoa1107477
268. Grebenciucova E and VanHaerents S. Interleukin 6: at the interface of human health and disease. Front Immunol. (2023) 14:1255533. doi: 10.3389/fimmu.2023.1255533
269. Kang S and Kishimoto T. Interplay between interleukin-6 signaling and the vascular endothelium in cytokine storms. Exp Mol Med. (2021) 53:1116–23. doi: 10.1038/s12276-021-00649-0
270. Didion SP. Cellular and Oxidative Mechanisms Associated with Interleukin-6 Signaling in the Vasculature. Int J Mol Sci. (2017) 18. doi: 10.3390/ijms18122563
271. Tanaka T, Narazaki M, and Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol. (2014) 6:a016295. doi: 10.1101/cshperspect.a016295
272. Heinrich PC, Castell JV, and Andus T. Interleukin-6 and the acute phase response. Biochem J. (1990) 265:621–36. doi: 10.1042/bj2650621
273. Ali MI, Chen X, and Didion SP. Heterozygous eNOS deficiency is associated with oxidative stress and endothelial dysfunction in diet-induced obesity. Physiol Rep. (2015) 3. doi: 10.14814/phy2.12630
274. Wassmann S, Stumpf M, Strehlow K, Schmid A, Schieffer B, Bohm M, et al. Interleukin-6 induces oxidative stress and endothelial dysfunction by overexpression of the angiotensin II type 1 receptor. Circ Res. (2004) 94:534–41. doi: 10.1161/01.RES.0000115557.25127.8D
275. Han Y, Runge MS, and Brasier AR. Angiotensin II induces interleukin-6 transcription in vascular smooth muscle cells through pleiotropic activation of nuclear factor-kappa B transcription factors. Circ Res. (1999) 84:695–703. doi: 10.1161/01.RES.84.6.695
276. Knopp T, Jung R, Wild J, Bochenek ML, Efentakis P, Lehmann A, et al. Myeloid cell-derived interleukin-6 induces vascular dysfunction and vascular and systemic inflammation. Eur Heart J Open. (2024) 4:oeae046. doi: 10.1093/ehjopen/oeae046
277. Katkenov N, Mukhatayev Z, Kozhakhmetov S, Sailybayeva A, Bekbossynova M, and Kushugulova A. Systematic Review on the Role of IL-6 and IL-1beta in Cardiovascular Diseases. J Cardiovasc Dev Dis. (2024) 11. doi: 10.3390/jcdd11070206
278. Feng Y, Ye D, Wang Z, Pan H, Lu X, Wang M, et al. The Role of Interleukin-6 Family Members in Cardiovascular Diseases. Front Cardiovasc Med. (2022) 9:818890. doi: 10.3389/fcvm.2022.818890
279. Blann AD, Woywodt A, Bertolini F, Bull TM, Buyon JP, Clancy RM, et al. Circulating endothelial cells. Biomarker of vascular disease. Thromb Haemost. (2005) 93:228–35. doi: 10.1160/TH04-09-0578
280. Smadja DM, Mauge L, Sanchez O, Silvestre JS, Guerin C, Godier A, et al. Distinct patterns of circulating endothelial cells in pulmonary hypertension. Eur Respir J. (2010) 36:1284–93. doi: 10.1183/09031936.00130809
281. Calderón-Colmenero J, Massó F, González-Pacheco H, Sandoval J, Guerrero C, Cervantes-Salazar J, et al. Pulmonary arterial hypertension in children with congenital heart disease: a deeper look into the role of endothelial progenitor cells and circulating endothelial cells to assess disease severity. Front Pediatr. (2023) 11:1200395. doi: 10.3389/fped.2023.1200395
282. Sabulski A, Aunins B, Abdullah S, Luebbering N, and Davies SM. Circulating Endothelial Cells As a Marker of Vascular Injury in Pediatric Patients. Biol Blood Marrow Transplant. (2020) 26:S144–S5. doi: 10.1016/j.bbmt.2019.12.675
283. Mutin M, Canavy I, Blann A, Bory M, Sampol J, and Dignat-George F. Direct evidence of endothelial injury in acute myocardial infarction and unstable angina by demonstration of circulating endothelial cells. Blood. (1999) 93:2951–8. doi: 10.1182/blood.V93.9.2951.409k02_2951_2958
284. Martínez-Sales V, Sánchez-Lázaro I, Vila V, Almenar L, Contreras T, and Reganon E. Circulating endothelial cells in patients with heart failure and left ventricular dysfunction. Dis Markers. (2011) 31:75–82. doi: 10.1155/2011/757840
285. Alessio AM, Beltrame MP, Nascimento MC, Vicente CP, de Godoy JA, Silva JC, et al. Circulating progenitor and mature endothelial cells in deep vein thrombosis. Int J Med Sci. (2013) 10:1746–54. doi: 10.7150/ijms.6887
286. Rahbari NN, Schölch S, Bork U, Kahlert C, Schneider M, Rahbari M, et al. Prognostic value of circulating endothelial cells in metastatic colorectal cancer. Oncotarget. (2017) 8:37491–501. doi: 10.18632/oncotarget.16397
287. Goon PK, Lip GY, Stonelake PS, and Blann AD. Circulating endothelial cells and circulating progenitor cells in breast cancer: relationship to endothelial damage/dysfunction/apoptosis, clinicopathologic factors, and the Nottingham Prognostic Index. Neoplasia. (2009) 11:771–9. doi: 10.1593/neo.09490
288. Najjar F, Alammar M, Al-Massarani G, Almalla N, Japawe A, and Ikhtiar A. Circulating endothelial cells and microparticles as diagnostic and prognostic biomarkers in small-cell lung cancer. Lung Cancer. (2018) 124:23–30. doi: 10.1016/j.lungcan.2018.06.033
289. Zhang H, Vakil V, Braunstein M, Smith EL, Maroney J, Chen L, et al. Circulating endothelial progenitor cells in multiple myeloma: implications and significance. Blood. (2005) 105:3286–94. doi: 10.1182/blood-2004-06-2101
290. Szmigielska-Kaplon A, Krawczynska A, Czemerska M, Pluta A, Cebula-Obrzut B, Grzybowska-Izydorczyk O, et al. The kinetics and apoptotic profile of circulating endothelial cells in autologous hematopoietic stem cell transplantation in patients with lymphoproliferative disorders. Ann Hematol. (2013) 92:1255–62. doi: 10.1007/s00277-013-1759-4
291. Attia FM, Maaty A, and Kalil FA. Circulating endothelial cells as a marker of vascular dysfunction in patients with systemic lupus erythematosus by real-time polymerase chain reaction. Arch Pathol Lab Med. (2011) 135:1482–5. doi: 10.5858/arpa.2010-0731-OA
292. Clancy R, Marder G, Martin V, Belmont HM, Abramson SB, and Buyon J. Circulating activated endothelial cells in systemic lupus erythematosus: further evidence for diffuse vasculopathy. Arthritis Rheumatol. (2001) 44:1203–8. doi: 10.1002/1529-0131(200105)44:5<1203::AID-ANR204>3.0.CO;2-C
293. Woywodt A, Streiber F, de Groot K, Regelsberger H, Haller H, and Haubitz M. Circulating endothelial cells as markers for ANCA-associated small-vessel vasculitis. Lancet. (2003) 361:206–10. doi: 10.1016/S0140-6736(03)12269-6
294. Kim SK and Massett MP. Genetic Regulation of Endothelial Vasomotor Function. Front Physiol. (2016) 7:571. doi: 10.3389/fphys.2016.00571
295. Xu S, Chuang CY, Hawkins CL, Hagglund P, and Davies MJ. Quantitative analysis of the proteome and protein oxidative modifications in primary human coronary artery endothelial cells and associated extracellular matrix. Redox Biol. (2025) 81:103524. doi: 10.1016/j.redox.2025.103524
296. Figuer A, Santos FM, Ciordia S, Valera G, Martin-Jouve B, Hernandez-Fonseca JP, et al. Proteomic analysis of endothelial cells and extracellular vesicles in response to indoxyl sulfate: Mechanisms of endothelial dysfunction in chronic kidney disease. Life Sci. (2024) 351:122810. doi: 10.1016/j.lfs.2024.122810
297. Alwahsh M, Alejel R, Hasan A, Abuzaid H, and Al-Qirim T. The Application of Metabolomics in Hyperlipidemia: Insights into Biomarker Discovery and Treatment Efficacy Assessment. Metabolites. (2024) 14. doi: 10.3390/metabo14080438
298. Tschaidse L, Reisch N, Arlt W, Brac de la Perriere A, Linden Hirschberg A, Juul A, et al. Modified-release hydrocortisone is associated with lower plasma renin activity in patients with salt-wasting congenital adrenal hyperplasia. Eur J Endocrinol. (2023) 188:109–17. doi: 10.1093/ejendo/lvac006
299. Merke DP, Mallappa A, Arlt W, Brac de la Perriere A, Lindén Hirschberg A, Juul A, et al. Modified-Release Hydrocortisone in Congenital Adrenal Hyperplasia. J Clin Endocrinol Metab. (2021) 106:e2063–e77. doi: 10.1210/clinem/dgab051
300. Mallappa A, Sinaii N, Kumar P, Whitaker MJ, Daley LA, Digweed D, et al. A phase 2 study of Chronocort, a modified-release formulation of hydrocortisone, in the treatment of adults with classic congenital adrenal hyperplasia. J Clin Endocrinol Metab. (2015) 100:1137–45. doi: 10.1210/jc.2014-3809
301. Arshad MF and Debono M. Current and future treatment options for adrenal insufficiency. Curr Opin Endocrinol Diabetes Obes. (2021) 28:303–11. doi: 10.1097/MED.0000000000000637
302. Mongioì LM, Condorelli RA, Barbagallo F, La Vignera S, and Calogero AE. Dual-release hydrocortisone for treatment of adrenal insufficiency: a systematic review. Endocrine. (2020) 67:507–15. doi: 10.1007/s12020-020-02187-7
303. Mortensen ML, Ornstrup MJ, and Gravholt CH. Patients with Hypocortisolism Treated with Continuous Subcutaneous Hydrocortisone Infusion (CSHI): An Option for Poorly Controlled Patients. Int J Endocrinol. (2023) 2023:5315059. doi: 10.1155/2023/5315059
304. Auchus RJ, Sarafoglou K, Fechner PY, Vogiatzi MG, Imel EA, Davis SM, et al. Crinecerfont Lowers Elevated Hormone Markers in Adults With 21-Hydroxylase Deficiency Congenital Adrenal Hyperplasia. J Clin Endocrinol Metab. (2022) 107:801–12. doi: 10.1210/clinem/dgab749
305. Newfield RS, Sarafoglou K, Fechner PY, Nokoff NJ, Auchus RJ, Vogiatzi MG, et al. Crinecerfont, a CRF1 Receptor Antagonist, Lowers Adrenal Androgens in Adolescents With Congenital Adrenal Hyperplasia. J Clin Endocrinol Metab. (2023) 108:2871–8. doi: 10.1210/clinem/dgad270
306. Sarafoglou K, Barnes CN, Huang M, Imel EA, Madu IJ, Merke DP, et al. Tildacerfont in Adults With Classic Congenital Adrenal Hyperplasia: Results from Two Phase 2 Studies. J Clin Endocrinol Metab. (2021) 106:e4666–e79. doi: 10.1210/clinem/dgab438
307. Maher JY, Gomez-Lobo V, and Merke DP. The management of congenital adrenal hyperplasia during preconception, pregnancy, and postpartum. Rev Endocr Metab Disord. (2023) 24:71–83. doi: 10.1007/s11154-022-09770-5
308. Curtis KM, Tepper NK, Jatlaoui TC, Berry-Bibee E, Horton LG, Zapata LB, et al. U.S. Medical Eligibility Criteria for Contraceptive Use, 2016. MMWR Recomm Rep. (2016) 65:1–103. doi: 10.15585/mmwr.rr6503a1
309. Speiser PW, Agdere L, Ueshiba H, White PC, and New MI. Aldosterone synthesis in salt-wasting congenital adrenal hyperplasia with complete absence of adrenal 21-hydroxylase. N Engl J Med. (1991) 324:145–9. doi: 10.1056/NEJM199101173240302
310. Falhammar H and Nordenström A. Nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency: clinical presentation, diagnosis, treatment, and outcome. Endocrine. (2015) 50:32–50. doi: 10.1007/s12020-015-0656-0
311. Visseren FLJ, Mach F, Smulders YM, Carballo D, Koskinas KC, Bäck M, et al. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice. Eur Heart J. (2021) 42:3227–337. doi: 10.1093/eurheartj/ehab484
312. Mapas-Dimaya AC, Agdere L, Bahtiyar G, Mejia JO, and Sacerdote AS. Metformin-responsive classic salt-losing congenital adrenal hyperplasia due to 21-hydroxylase deficiency: a case report. Endocr Pract. (2008) 14:889–91. doi: 10.4158/EP.14.7.889
313. Krysiak R and Okopien B. The effect of metformin on androgen production in diabetic women with non-classic congenital adrenal hyperplasia. Exp Clin Endocrinol Diabetes. (2014) 122:568–71. doi: 10.1055/s-0034-1382048
314. Seagroves A, Ross HM, Vidmar AP, Geffner ME, Kim WS, Hwang D, et al. Weight Loss During Topiramate Treatment in a Severely Obese Adolescent with Congenital Adrenal Hyperplasia and Migraine. J Clin Res Pediatr Endocrinol. (2023) 15:81–5. doi: 10.4274/jcrpe.galenos.2021.2020.0310
315. Mallappa A, Nella AA, Kumar P, Brooks KM, Perritt AF, Ling A, et al. Alterations in Hydrocortisone Pharmacokinetics in a Patient With Congenital Adrenal Hyperplasia Following Bariatric Surgery. J Endocr Soc. (2017) 1:994–1001. doi: 10.1210/js.2017-00215
316. Kalani A, Thomas N, Sacerdote A, and Bahtiyar G. Roux-en-Y gastric bypass in the treatment of non-classic congenital adrenal hyperplasia due to 11-hydroxylase deficiency. BMJ Case Rep. (2013) 2013. doi: 10.1136/bcr-2012-008416
317. Krysiak R, Kowalcze K, Bednarska-Czerwińska A, and Okopień B. The Effect of Simvastatin on Plasma Steroid Hormone Levels in Metformin-Treated Women with Non-Classic Congenital Adrenal Hyperplasia. Exp Clin Endocrinol Diabetes. (2016) 124:215–9. doi: 10.1055/s-0035-1569375
318. Krysiak R, Kowalcze K, Bednarska-Czerwińska A, and Okopień B. The effect of atorvastatin on cardiometabolic risk factors in women with non-classic congenital adrenal hyperplasia: A pilot study. Pharmacol Rep. (2019) 71:417–21. doi: 10.1016/j.pharep.2019.01.014
319. Selva-O’Callaghan A, Alvarado-Cardenas M, Pinal-Fernández I, Trallero-Araguás E, Milisenda JC, Martínez M, et al. Statin-induced myalgia and myositis: an update on pathogenesis and clinical recommendations. Expert Rev Clin Immunol. (2018) 14:215–24. doi: 10.1080/1744666X.2018.1440206
320. Gupta A and Gupta Y. Glucocorticoid-induced myopathy: Pathophysiology, diagnosis, and treatment. Indian J Endocrinol Metab. (2013) 17:913–6. doi: 10.4103/2230-8210.117215
321. Lévy BI and Mourad JJ. Renin Angiotensin Blockers and Cardiac Protection: From Basis to Clinical Trials. Am J Hypertens. (2022) 35:293–302. doi: 10.1093/ajh/hpab108
322. Nehme A, Zouein FA, Zayeri ZD, and Zibara K. An Update on the Tissue Renin Angiotensin System and Its Role in Physiology and Pathology. J Cardiovasc Dev Dis. (2019) 6. doi: 10.3390/jcdd6020014
323. Katsimardou A, Imprialos K, Stavropoulos K, Sachinidis A, Doumas M, and Athyros V. Hypertension in Metabolic Syndrome: Novel Insights. Curr Hypertens Rev. (2020) 16:12–8. doi: 10.2174/1573402115666190415161813
Keywords: congenital adrenal hyperplasia (CAH), 21-hydroxylase deficiency, endothelial dysfunction, endothelial biomarkers, cardiovascular disease, cardiovascular risk
Citation: Hubska J, Roszkowska Z, Bobrowicz M, Iwaniuk S, Rak-Makowska B and Ambroziak U (2025) Endothelial dysfunction in congenital adrenal hyperplasia due to 21-hydroxylase deficiency: current knowledge and novel biomarkers. Front. Endocrinol. 16:1581681. doi: 10.3389/fendo.2025.1581681
Received: 22 February 2025; Accepted: 12 May 2025;
Published: 03 June 2025.
Edited by:
Ivana Kraljevic, University of Zagreb, CroatiaReviewed by:
Alejandra Tapia-Castillo, Pontificia Universidad Católica de Chile, ChileMirsala Solak, University of Zagreb, Croatia
Sanja Borozan, Clinical Center of Montenegro, Montenegro
Copyright © 2025 Hubska, Roszkowska, Bobrowicz, Iwaniuk, Rak-Makowska and Ambroziak. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joanna Hubska, am9hbm5hLmh1YnNrYUB3dW0uZWR1LnBs