 Xing Ji
Xing Ji Xinhua Hu3
Xinhua Hu3 Taotao Xu
Taotao Xu- 1Department of Pharmacology, School of Medicine, Hangzhou City University, Hangzhou, China
- 2Department of Pharmacology, Zhejiang University School of Medicine, Hangzhou, China
- 3Center of Clinical Pharmacology, Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, Zhejiang, China
- 4Department of Orthopedics Surgery, The First Affiliated Hospital of Zhejiang Chinese Medical University (Zhejiang Provincial Hospital of Chinese Medicine), Hangzhou, China
The musculoskeletal system is not only closely linked anatomically, but muscle-derived myokines also play a crucial role in bone development and metabolism beyond the effects of mechanical force. Myokines are essential in muscle-bone crosstalk, significantly influencing bone remodeling and metabolism. In the context of diabetes, including both type 1 (T1DM) and type 2 (T2DM), changes in myokine expression have a substantial impact on bone metabolism, leading to an increased risk of osteoporosis. This review provides a comprehensive examination of the roles of key myokines in regulating osteoblast lineage cells and osteoclast activity. We highlight how different myokines can either promote or inhibit bone formation and resorption and discuss their altered expression levels under diabetic conditions. A deeper understanding of the multifaceted roles of myokines may open new avenues for treating osteoporosis, particularly in diabetic patients.
1 Introduction
Myokines, a term first introduced in 2003 by Pedersen et al. (1), are cytokines and other peptides produced and released by muscle fibers in response to muscular contractions. These molecules act in an autocrine, paracrine, or endocrine manner, influencing not only muscle function but also other tissues, including bone (2, 3). Myokines play crucial roles in muscle hypertrophy, metabolism, and inflammation regulation (4, 5), and they are increasingly recognized for their role in muscle-bone crosstalk, particularly in the context of diabetes (6).
Osteoporosis is a prevalent and serious condition characterized by decreased bone mass and the structural deterioration of bone tissue, which leads to an increased risk of fractures (7). Recent studies increasingly demonstrate the significant impact of factors secreted by other organs on bone health (8, 9). Our review specifically focuses on the role of muscles, which are anatomically closest to bone, beyond the traditionally acknowledged mechanical effects. Myokines play a central role in regulating osteoblast (OB) and osteoclast (OC) activities, thereby contributing to the development and progression of osteoporosis. In particular, myokines such as irisin, myostatin, and interleukin-6 (IL-6) have been found to influence bone metabolism by modulating the differentiation and functional activities of bone-forming osteoblasts and bone-resorbing osteoclasts.
Diabetes is a chronic metabolic disease characterized by persistent hyperglycemia, resulting from defective insulin secretion or action, leading to impaired glucose, lipid, and protein metabolism. Beyond its impact on glycemic control, diabetes negatively affects bone health, increasing the risk of osteoporosis and fractures due to changes in bone metabolism and microarchitecture (10, 11). Growing evidence highlights a significantly elevated fracture risk in people with diabetes, particularly among those with T1DM (12). In a recent meta-analysis, individuals with type 1 diabetes had a significantly increased risk of both hip and non-vertebral fractures. The relative risk of hip fracture was 4.93 (95% CI: 3.06–7.95), while the risk for non-vertebral fractures was 1.92 (95% CI: 0.92–3.99). Notably, hip fracture risk was more pronounced in younger individuals (13). In T2DM, the increase in fracture risk is more modest but becomes substantial in individuals with low BMI, prolonged disease duration, or insulin therapy, where the risk may rise by more than 20% (14, 15). These findings underscore the importance of individualized fracture risk assessment in the context of diabetes. Notably, myokines are key factors that influence systemic metabolism, prompting us to explore how these myokines change within the context of diabetes and their effects on bone.
We have comprehensively cataloged all known myokines and detailed their specific effects on osteoblasts lineage cells and osteoclasts. Special emphasis is placed on the circulating concentrations of these myokines and their alterations in the context of diabetes. Additionally, we critically examine the impact of these changes on bone metabolism in the context of diabetes, highlighting how variations in myokine levels influence bone formation, resorption, and overall skeletal integrity in diabetes. This review aims to provide a comprehensive and systematic review of the role of myokines in bone metabolism, with a particular focus on their role in diabetic conditions. Through this study, we hope to provide a theoretical basis and identify novel therapeutic targets for the prevention and treatment of osteoporosis associated with diabetes.
2 The musculoskeletal unit in diabetes: traditional and emerging views
2.1 Traditional view of mechanical coupling between muscles and bones
The interaction between bones and skeletal muscles has traditionally been regarded as a mechanically driven relationship, where muscles generate forces that stimulate bone adaptation, while bones provide structural support and serve as attachment sites for muscles (16, 17). Mechanical loading is a key determinant of bone strength, influencing osteoblast activity, bone mineralization, and trabecular architecture. During musculoskeletal development, these mechanical forces ensure proper cell fate determination, structural organization, and functional integration of bones, joints, tendons, and muscles. Hippo signaling (YAP1), Indian hedgehog (IHH), and parathyroid hormone-related protein (PTHrP) are among the key regulators of this process (18). However, in diabetes, the mechanical loading response of bone is impaired due to muscle wasting, neuropathy, and altered mechanotransduction signaling, leading to accelerated bone loss (19). While mechanical forces remain essential for bone adaptation, emerging evidence highlights the role of myokines—muscle-derived cytokines that influence bone metabolism through endocrine signaling.
2.2 Myokines: a new frontier in the treatment of diabetic osteoporosis
Myokines act as key mediators in the muscle-bone crosstalk, regulating the balance between bone formation and resorption. Their secretion is modulated by physical activity, metabolic status, and inflammatory signals, and they are significantly altered in diabetes and its complications. In both T1DM and T2DM, dysregulated myokines affect osteoblasts, osteoclasts, and mesenchymal stem cells (MSCs) and disrupt the balance between bone formation and resorption. These changes contribute to diabetes-induced osteoporosis, leading to impaired bone remodeling, increased fragility, and a higher risk of fractures (20). The mechanisms through which specific myokines influence bone metabolism in the context of diabetes will be discussed in detail in the following sections. Understanding the role of myokines within the musculoskeletal unit offers a novel approach to managing bone complications in the context of diabetes, providing insights into potential targeted therapies that leverage muscle-derived factors to enhance both muscle function and bone health in people with diabetes.
3 Myokines and bone metabolism
3.1 Overview of myokines and their impact on bone resorption and formation
Myokines are muscle-derived signaling molecules that influence bone metabolism through a diverse range of mechanisms, impacting both OB and OC. This section explores the phenotypes observed in mouse models for each myokine, further enhancing our understanding of their impact on both OB and OC (Table 1). Additionally, we summarize their roles in bone formation and resorption, while also examining the molecular pathways they regulate (Figures 1, 2), highlighting their contributions to skeletal homeostasis and potential implications for bone health and disease. Their effects are highly context-dependent, influenced by factors such as concentration, receptor availability, and intracellular signaling crosstalk.
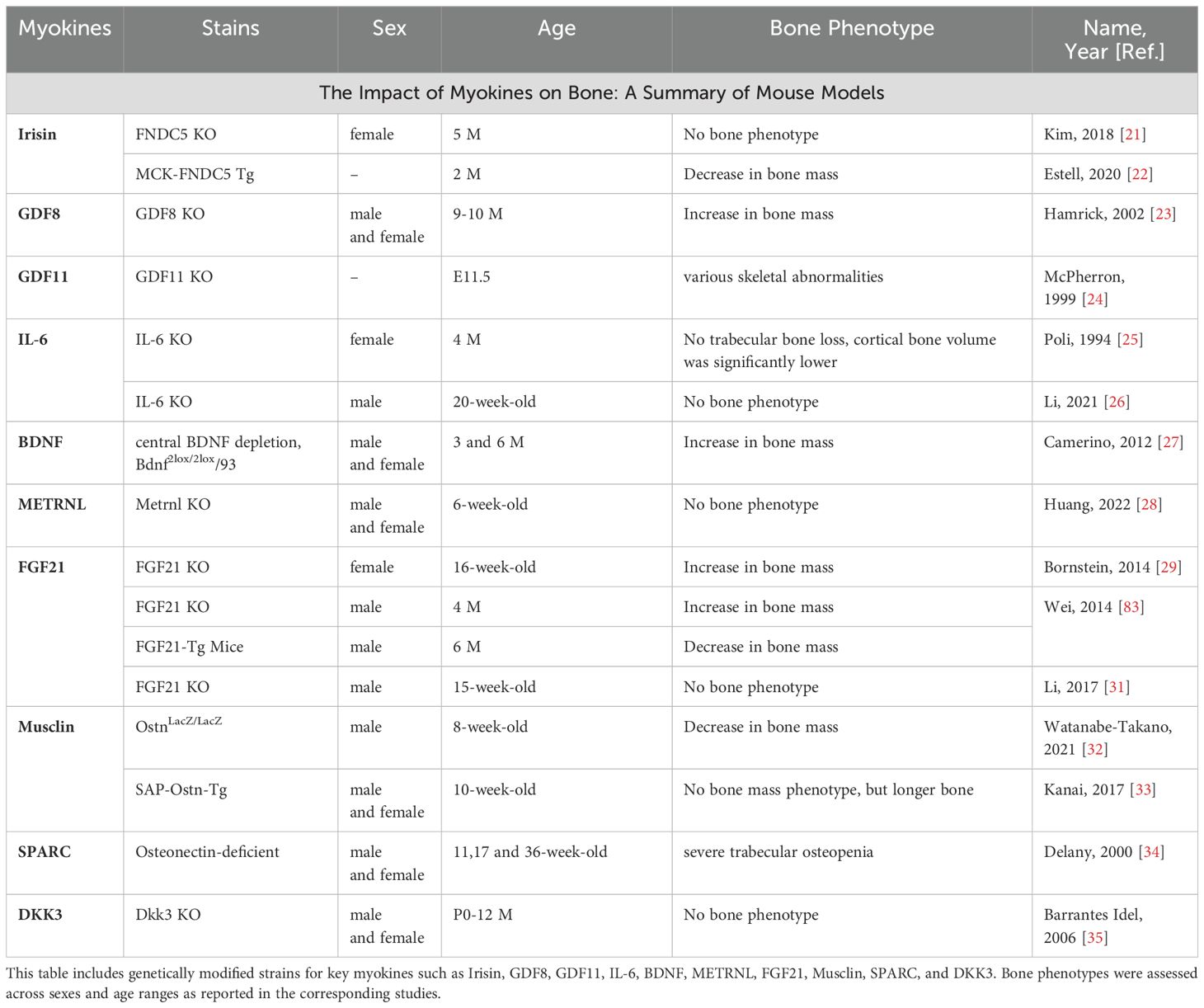
Table 1. Summary of mouse models evaluating the impact of various myokines on bone mass and skeletal phenotype.
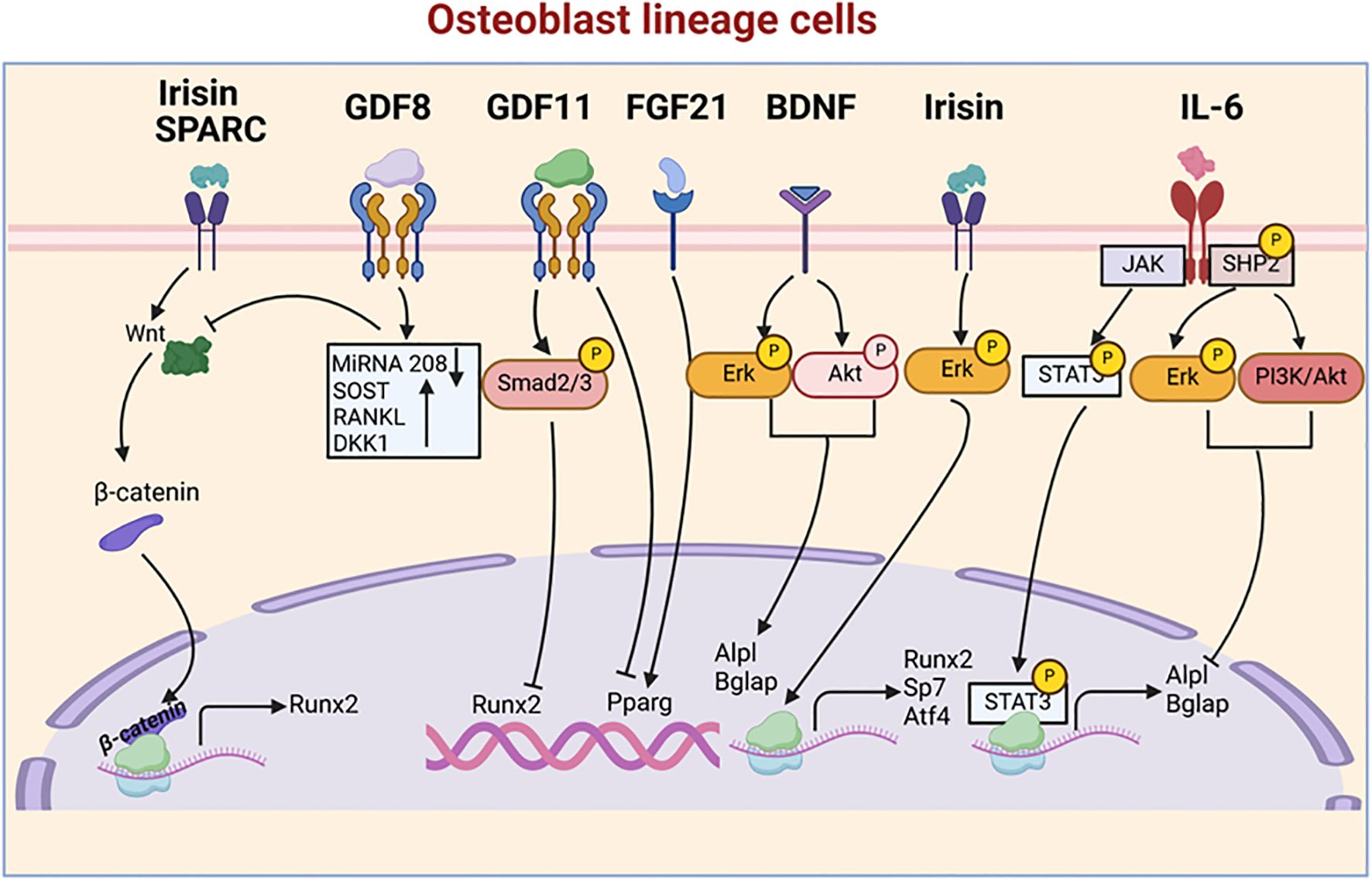
Figure 1. Myokine signaling pathways regulating osteoblastogenesis. This figure illustrates the complex network of myokine signaling pathways that control osteoblast differentiation and function. Key myokines, including Irisin, GDF8, GDF11, FGF21, BDNF, Sparc, and IL-6, activate various signaling cascades, such as Smad, Erk, Akt, and STAT3 pathways. These pathways converge on key transcription factors, including Runx2, Pparg, and Sp7, which regulate the expression of osteoblast-specific genes like Alpl and Bglap.
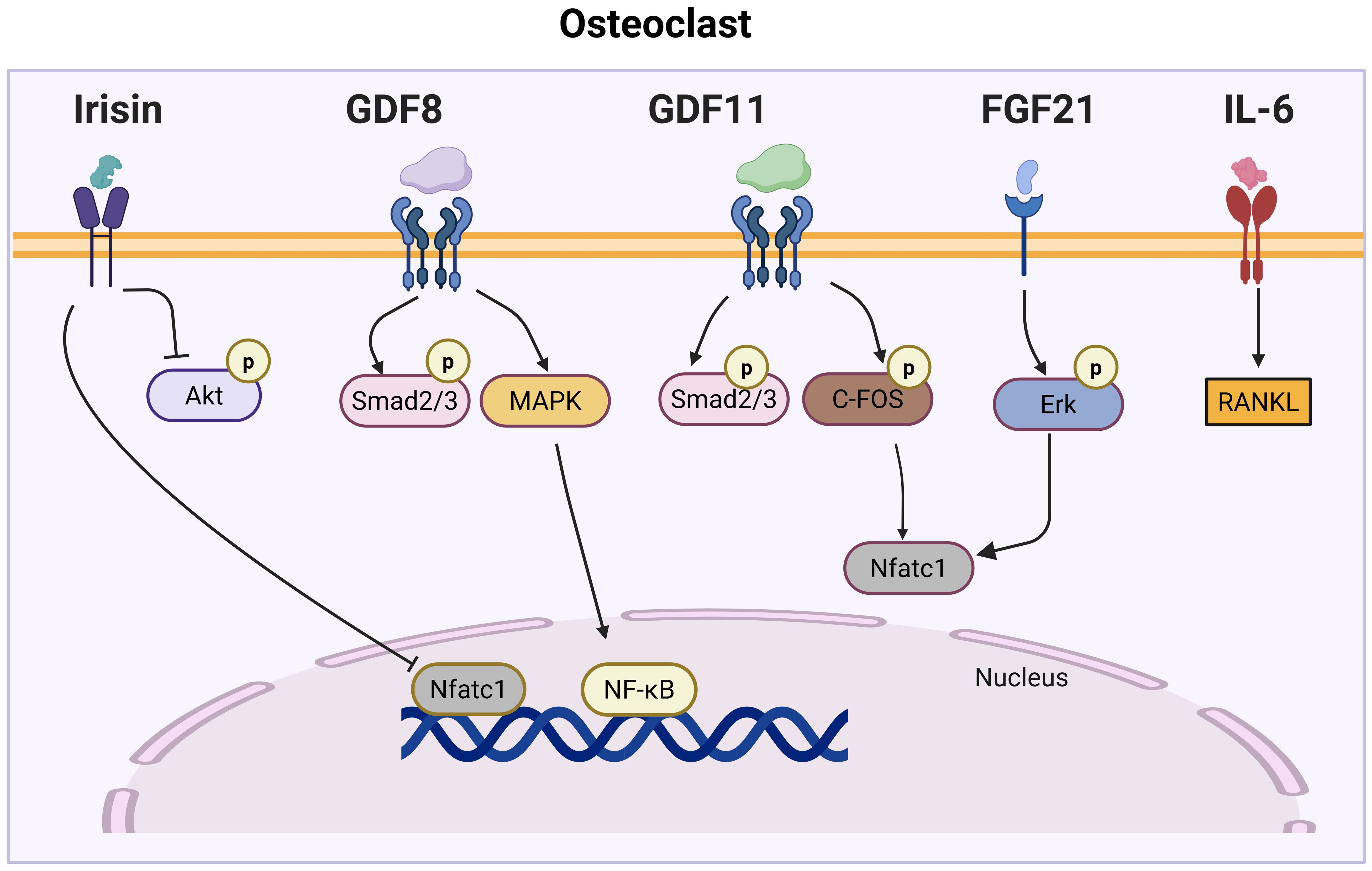
Figure 2. Myokine signaling pathways regulating osteoclast differentiation. This figure illustrates the complex network of myokine signaling pathways that control osteoclast differentiation. Key myokines, including Irisin, GDF8, GDF11, FGF21, and IL-6, activate various signaling cascades, such as Akt, MAPK, Smad2/3, and Erk pathways. These pathways converge on key transcription factors, such as NFATc1 and NF-κB, which regulate the expression of osteoclast-specific genes.
3.2 Specific myokines and their effects on bone cells
3.2.1 Irisin
Irisin, a myokine released during exercise, plays a crucial role in energy metabolism by promoting the conversion of white adipose tissue (WAT) into thermogenic brown fat (BAT)-like cells, enhancing energy expenditure and glucose homeostasis (36, 37). It binds to αV integrins, particularly αV/β5, initiating signaling pathways that influence bone and fat metabolism (21). In bone metabolism, irisin exhibits tissue-specific effects. Irisin is produced by the cleavage of fibronectin type III domain-containing protein 5 (FNDC5), and it is released into the bloodstream. While global FNDC5 knockout (KO) does not significantly affect bone mass, it confers resistance to ovariectomy (OVX)-induced bone loss (21). In contrast, muscle-specific FNDC5 KO reduces bone mass, indicating a regulatory role in skeletal health (22). Intermittent low-dose irisin injections enhance cortical bone mineral density (37), likely through osteoblast stimulation via the Wnt/β-catenin and MAPK pathways, promoting autophagy and osteogenic differentiation (38–40). Additionally, irisin activates AMP-activated protein kinase (AMPK), influencing inflammatory cytokines and promoting the M2 macrophage phenotype, which enhances osteogenesis (41). Irisin’s impact on osteoclasts is dose-dependent. At low concentrations (10 ng/mL), it promotes osteoclast differentiation (22), whereas higher doses (120–480 ng/mL) suppress RANKL-induced osteoclastogenesis by inhibiting Akt phosphorylation and NFATc1 activation (42). Variability in reported circulating irisin levels (3–5 ng/mL vs. commercial kit measurements of ~100 ng/mL) underscores the importance of dosage and detection methods in interpreting its skeletal effects (43, 44). These findings suggest irisin’s role in bone metabolism is complex and context dependent.
3.2.2 GDF8
Myostatin (GDF8), a TGF-β superfamily member, is primarily expressed in skeletal muscle as a negative regulator of muscle growth (45), but it also plays a key role in bone metabolism. It circulates in an inactive form, bound to a propeptide, and is activated by BMP1 or Tolloid metalloproteinases. GDF8 is inhibited by follistatin and FLRG (Follistatin-Related Gene) and signals through the ActRIIB receptor, triggering TGF-β signaling that regulates bone cell activity (46). GDF8 is critically involved in regulating the proliferation and differentiation of mesenchymal stem cells (47, 48). Studies in GDF8 KO mice have demonstrated significant reductions in body fat and notable increases in bone mineral density and strength (23), observed in the appendicular skeleton, vertebral column, and mandible (49, 50). In osteoblasts and osteocytes, GDF8 upregulates SOST, DKK1, and RANKL, while suppressing miR-218, thereby inhibiting osteoblast differentiation (51). It also enhances the mechanosensitivity of bone marrow stromal cells, promoting an osteogenic response to mechanical loading (52, 53). However, GDF8 inhibition alone has limited effects on bone formation, particularly in aged mice, and is less effective than ActRIIB-Fc, which blocks multiple ligands, including BMPs (54). In osteoclasts, GDF8 enhances RANKL-mediated osteoclastogenesis via the Smad2-NFATC1 axis and promotes differentiation through Ccdc50-regulated NF-κB and MAPK signaling (55, 56). Although GDF8-modified osteocytic exosomes can be internalized by osteoclasts, they have minimal impact on osteoclast differentiation, underscoring the multifaceted nature of GDF8’s role in bone metabolism (51).
3.2.3 GDF11
Similar to GDF8, GDF11, another TGF-β superfamily member (57, 58), plays a significant role in skeletal development and adult bone homeostasis. Homozygous Gdf11-null mice (Gdf11-/-) exhibit severe skeletal abnormalities, including anterior homeotic transformations and tail absence, while heterozygous (Gdf11+/-) mice display milder defects (24). In adult mice, daily administration of recombinant GDF11 (rGDF11) reduces trabecular bone volume, increases osteoclast number, and decreases osteoblast activity, leading to lower mineral apposition and bone formation rates (59). At the cellular level, GDF11 inhibits osteoblast differentiation by activating Smad2/3 signaling, which suppresses Runx2 expression (60), thereby impairing osteoblastic differentiation of bone marrow stromal cells (BMSCs). While some studies suggest that GDF11 promotes osteoblastogenesis through PPARγ inhibition (61), others indicate that blocking GDF11 enhances bone formation, highlighting ongoing debate regarding its precise role. In osteoclasts, GDF11 promotes Smad2/3-mediated c-Fos upregulation, leading to increased Nfatc1 expression, which enhances osteoclast differentiation and activity (59). This osteoclastogenic effect aligns with the bone loss observed following rGDF11 administration.
3.2.4 IL-6
Interleukin-6 (IL-6), a pleiotropic cytokine (62, 63), was initially identified in immune regulation but was later recognized as a myokine produced by muscle fibers during contraction (64). IL-6 signaling occurs through three pathways: classic signaling via membrane-bound IL-6R, trans-signaling through soluble IL-6R (sIL-6R), and trans-presentation, where dendritic cells present IL-6 to T cells via IL-6R (65). Studies on IL-6 KO and transgenic mice reveal its complex role in bone metabolism. Female IL-6 KO mice (4 months old) showed preserved trabecular bone despite ovariectomy but had reduced cortical bone volume (25), while male IL-6 KO mice (20 weeks old) showed no significant differences in bone mass (26). In contrast, IL-6 transgenic mice, with chronically elevated IL-6, exhibited smaller skeletons, reduced trabecular and cortical bone, and impaired ossification, likely due to osteoclast overactivity and osteoblast suppression (66). At the cellular level, IL-6 modulates osteoblast activity through different pathways. Since osteoblasts have low membrane-bound IL-6Rα, IL-6 primarily acts through trans-signaling, requiring sIL-6R. IL-6 suppresses osteoblast differentiation via SHP2/MEK2/ERK and SHP2/PI3K/Akt2 pathway s (67), while the gp130-STAT1/3 axis promotes bone formation. Disruption of gp130-STAT1/3 signaling (gp130ΔSTAT/ΔSTAT mice) leads to premature growth plate closure, reduced bone length, and impaired skeletal growth, underscoring its essential role in maintaining bone elongation (68). Conversely, IL-6 strongly promotes osteoclastogenesis by upregulating RANKL in osteoblasts and stromal cells, facilitating osteoclast differentiation. Muscle-derived IL-6, produced during exercise, is required for osteoblast-mediated osteoclast activation and the release of bioactive osteocalcin, which enhances exercise capacity (69). In gp130Y757F/Y757F mice, where the SHP2/Ras/MAPK pathway is impaired, increased osteoclastogenesis and bone resorption lead to osteopenia, highlighting the distinct role of this pathway in bone turnover regulation (68).
3.2.5 Brain-derived neurotrophic factor
Brain-Derived Neurotrophic Factor (BDNF), traditionally known for its role in the central nervous system, has also been identified as a myokine secreted by skeletal muscle during contraction. However, studies suggest that muscle-derived BDNF does not enter systemic circulation, indicating a predominantly local effect within muscle tissue (70). Despite this, BDNF has been increasingly linked to bone development and remodeling. BDNF and its receptor TrkB are co-expressed in chondrocytes of the epiphyseal growth plate, active osteoblasts, and trabecular bone, suggesting a regulatory role in skeletal metabolism (71). Mice with central BDNF depletion exhibit increased bone mass, elongated femurs, and elevated bone mineral density (BMD) and bone mineral content (BMC), highlighting its systemic influence on bone physiology (27). At the cellular level, BDNF enhances osteoblast activity by upregulating osteocalcin, alkaline phosphatase (ALP), bone morphogenetic protein-2 (BMP-2), and osteopontin via TrkB-mediated ERK1/2 and AKT signaling pathways, promoting osteoblast differentiation and migration (72, 73). In osteoclasts, BDNF indirectly regulates osteoclastogenesis by modifying the RANKL/OPG ratio. It increases osteoprotegerin (OPG) expression, which inhibits RANKL-RANK binding, thereby reducing osteoclast differentiation and bone resorption (74). Currently, direct experimental evidence linking muscle-derived BDNF to bone metabolism is lacking. Given its restricted local effects in muscle, further research is needed to clarify its role as a myokine in bone physiology.
3.2.6 Metrnl
Metrnl (Meteorin-like) is a hormone-like protein involved in inflammation, metabolism, and muscle repair, primarily expressed in adipose tissue and skeletal muscle, with detectable levels in bone. It is secreted post-exercise by muscle cells and upon cold exposure by adipose tissue, promoting energy expenditure and glucose homeostasis through beige fat thermogenesis and anti-inflammatory pathways (75). Despite its expression in perichondrium and primary ossification centers during skeletal development, Metrnl-null mice exhibit no significant differences in bone length, bone volume, trabecular thickness, or cortical thickness compared to wild-type mice. Additionally, Metrnl upregulation after bone injury does not affect bone deposition or fracture healing, suggesting that Metrnl is not essential for bone mass maintenance or normal fracture repair in vivo (28). At the cellular level, Metrnl suppresses osteoblast differentiation by inhibiting AP-1 activity, as shown in in vitro studies (76). However, its precise effects on osteoclasts remain unclear.
3.2.7 L-BAIBA
The study identifies β-aminoisobutyric acid (BAIBA) as a small molecule myokine produced by muscle cells in response to exercise (77). A small molecule secreted by skeletal muscle during exercise, exerts a protective effect on osteocytes through the Mas-Related G-Protein Coupled Receptor D (MRGPRD) receptor. The binding of L-BAIBA to MRGPRD activates signaling pathways that prevent ROS-induced mitochondrial breakdown, thereby maintaining osteocyte viability. In vivo experiments demonstrated that L-BAIBA supplementation prevented bone loss in a murine hindlimb unloading model, which is typically used to simulate conditions of disuse osteoporosis (78). The combination of L-BAIBA and sub-optimal mechanical loading significantly increased bone formation markers compared to either treatment alone (79).
3.2.8 FGF21
Fibroblast Growth Factor 21 (FGF21), primarily secreted by the liver, also functions as a myokine, with its expression in skeletal muscle regulated by the PI3K/Akt1 pathway (80). While FGF21 is well known for its role in glucose and lipid metabolism (81, 82), recent studies suggest conflicting effects on bone health. FGF21-deficient mice exhibit a high-bone-mass phenotype, with increased bone formation and reduced bone resorption, whereas transgenic FGF21-overexpressing mice display lower bone mass, reduced trabecular bone volume, and decreased bone thickness and number (83). FGF21 also plays a role in lactation-induced bone remodeling, promoting bone resorption, with knockout studies indicating that FGF21 deficiency preserves bone mass during lactation (29). However, some studies suggest that FGF21 does not significantly impact bone mass or turnover under normal conditions, leaving its precise role in bone metabolism unclear (31). At the cellular level, FGF21 suppresses osteoblast differentiation by upregulating PPARγ, shifting bone marrow stromal cell fate toward adipogenesis rather than osteogenesis, leading to a reduction in bone formation and an increase in marrow adipocytes (83). In osteoclasts, FGF21 indirectly promotes osteoclastogenesis by upregulating IGFBP1 (insulin-like growth factor-binding protein 1) in the liver, which increases circulating IGFBP1 levels. IGFBP1 binds to integrin β1 receptors on osteoclast precursors, activating ERK phosphorylation and NFATc1, thereby enhancing osteoclast differentiation and bone resorption (30). Despite accumulating evidence, the role of FGF21 as a myokine in bone metabolism remains underexplored, with conflicting findings across different studies. Strain, age, and diet variations may contribute to these discrepancies, but they do not fully explain the observed differences in bone phenotype, highlighting the need for further targeted research.
3.2.8 Musclin
Musclin, also known as osteocrin, is a secreted peptide primarily expressed in skeletal muscle and bone (84). As a myokine, Musclin is upregulated in response to exercise, enhancing mitochondrial biogenesis and function in skeletal muscle (85). It also plays a role in insulin sensitivity, glucose metabolism, and fat oxidation. In bone, Musclin regulates skeletal growth and adaptation to mechanical loading. Osteocrin-overexpressing mice (SAP-Ostn-Tg) exhibit significant skeletal overgrowth with elongated limbs and thicker growth plates, while OSTN-deficient mice (OstnLacZ/LacZ) display reduced bone length and trabecular bone volume, indicating that osteocrin is essential for normal bone development and load-induced bone formation (33). In osteocytes, osteocrin is a target gene of Sp7 (Osterix) and is crucial for dendrite formation and osteocyte network integrity. Deleting Sp7 in osteoblasts results in defective osteocyte dendrites, increased cortical porosity, and higher osteocyte apoptosis rates. Overexpression of osteocrin rescues these defects, highlighting its role in maintaining osteocyte function (86). At the cellular level, osteocrin promotes bone elongation and thickness by preventing CNP clearance via NPR-C, thereby amplifying the CNP/GC-B/cGMP signaling pathway (32). Periosteal osteoblasts upregulate osteocrin expression in response to mechanical loading, reinforcing its role in bone adaptation to stress.
3.2.9 SPARC
SPARC (Secreted Protein Acidic and Rich in Cysteine), also known as Osteonectin, is a matricellular glycoprotein that plays a crucial role in bone mineralization, extracellular matrix (ECM) regulation, and cellular processes such as proliferation, migration, and adhesion (87–89). Initially identified as a bone-specific protein, SPARC was later recognized as a myokine, secreted by muscle cells in response to exercise and mechanical stretching (90). While circulating SPARC is rapidly degraded, its degradation products may have biological significance, though direct evidence remains limited. In bone, SPARC interacts with integrin α5β1, activating Wnt/β-catenin signaling, which is central to skeletal homeostasis (91). SPARC-null mice exhibit severe trabecular osteopenia, diminished bone formation, and impaired bone turnover, which worsens with age, leading to reduced bone mass and compromised mechanical integrity (34). At the cellular level, SPARC deficiency reduces both osteoblast and osteoclast number and activity, disrupting normal bone turnover and remodeling. This is accompanied by increased collagen maturity and altered mineral crystallinity, resulting in weakened bone structure and impaired remodeling dynamics (92, 93). Furthermore, osteonectin-null mice exhibit an exaggerated osteoclastic response to parathyroid hormone (PTH) treatment, leading to increased bone resorption instead of the expected anabolic effect, indicating dysregulation of the bone formation-resorption balance (94). Genetic studies have identified a 3’ UTR SNP1599 variant that modulates osteonectin expression via miR-433 binding, influencing bone mass. The SNP1599G variant (haplotype A), more common in osteoporotic patients, introduces a miR-433 binding site, reducing SPARC expression and impairing bone formation. Conversely, the SNP1599C variant (haplotype B), associated with higher SPARC levels, correlates with improved bone health due to the absence of miR-433-mediated repression (95). Despite its established role in bone biology, there is no direct evidence regarding the impact of muscle-secreted SPARC on bone metabolism, highlighting a gap for future research.
3.2.10 DKK3
Dickkopf-3 (DKK3) is a member of the Dickkopf family, which plays a distinct role in bone compared to DKK1, a critical player in the Wnt signaling pathway within bone (96). Moreover, unlike mice deficient in Dkk1 or Dkk2, those with a Dkk3 KO do not exhibit any bone-related phenotypes (35, 97, 98). Recent research has identified DKK3 as a myokine (99), elevated levels of DKK3 have been detected in the bloodstream following exercise in both mice and humans, with in vitro studies showing that cyclic stretching (mimicking muscle contractions) enhances its secretion. Although the direct relationship between DKK3 and diabetes is not well-studied, the protein’s involvement in CKD and CVD, both of which are common diabetes complications, suggests that DKK3 could be an indirect marker for managing diabetes-related complications (100–102). Further research is required to fully understand its role in diabetes, but its potential as a biomarker for conditions closely linked to diabetes is noteworthy.
4 Evidence from animal models and human studies in diabetes
4.1 Overview of myokine research in animal models of diabetes-related bone disorders
Animal models have been instrumental in elucidating the relationship between diabetes, myokines, and bone health. Studies utilizing streptozotocin (STZ)-induced diabetic mice and genetically modified db/db mice have demonstrated how diabetes disrupts myokine expression and its subsequent impact on bone metabolism. In these models, altered myokine profiles are also closely linked to insulin resistance, a key metabolic feature of diabetes. Several myokines, including Irisin (103, 104), GDF8 (105, 106), GDF 11 (107), IL-6 (108, 109), Metrnl (110), and FGF21, influence insulin sensitivity by modulating key components of the insulin signaling pathway such as the insulin receptor, PI3K/Akt, and GLUT4. Despite extensive research on myokine-mediated modulation of insulin signaling in metabolic tissues, their interplay within bone cells remains insufficiently investigated.
4.2 Irisin and diabetic bone loss
Irisin has protective effects on bone health in diabetic models. In T2D mice, irisin prevents bone loss by attenuating inflammasome-associated pyroptosis signaling via the miR-150-FNDC5/Irisin/pyroptosis axis, making it a promising therapeutic target for osteoporosis associated with diabetes (111). Additionally, in T1D models, irisin reduces oxidative stress and prevents periodontal bone destruction, highlighting its broader role in bone protection under hyperglycemic conditions (112).
4.3 GDF8 and bone atrophy in diabetes
GDF8 acts as a negative regulator of muscle and bone mass. In T1D rats, weight-bearing treadmill running counteracts bone atrophy by downregulating GDF8 expression through the Activin A Receptor Type 2B (ActRIIB)/Smad2/3 pathway (113). Additionally, in T2D individuals, increased GDF8 expression in bone tissue has been linked to impaired fracture healing. Blocking myostatin with Follistatin (a natural MSTN inhibitor) enhances bone regeneration, suggesting its potential as a therapeutic approach for improving diabetic bone healing (114). Moreover, treating mice with soluble ActRIIB-Fc, an inhibitor of the activin/myostatin pathway, significantly improves bone mass, strength, and muscle function, underscoring the interplay between muscle-bone crosstalk in diabetic bone loss (115).
4.4 FGF21 and metabolic-bone homeostasis in diabetes
FGF21, a metabolic regulator, plays a complex role in diabetic bone metabolism. In diet-induced obese (DIO) mice, treatment with recombinant human FGF21 (rhFGF21) improved metabolic parameters—including body weight, glucose, insulin, and lipid levels—but did not cause significant bone loss. Parameters such as bone mineral density (BMD), trabecular thickness, and bone volume fraction (BV/TV) remained unchanged, suggesting that FGF21’s metabolic benefits do not necessarily compromise skeletal integrity in diabetes. However, the long-term effects of FGF21 on bone remodeling under chronic hyperglycemia remain unclear (31).
Importantly, insulin is a key regulator of FGF21 expression in skeletal muscle. By activating the PI3K/Akt1 signaling pathway, insulin induces upregulation of FGF21 mRNA and protein levels, as demonstrated in both in vitro and in vivo studies (80, 116). This represents a bidirectional regulatory mechanism, in which insulin not only modulates myokine production but may also amplify the downstream skeletal effects of specific myokines. Given that insulin promotes FGF21 expression in skeletal muscle, it is plausible that the insulin–FGF21 axis contributes to the regulation of bone metabolism, particularly under diabetic conditions.
Research indicates that the expression levels of certain myokines, including irisin and myostain, are significantly altered in these diabetic models. These alterations are associated with changes in bone density and microarchitecture, suggesting that myokines play a contributory role in diabetes-induced bone disorders. Notably, these findings highlight the potential of myokines as therapeutic targets to mitigate bone loss in the context of diabetes. However, translating these animal model findings to human clinical scenarios requires careful consideration, given the differences in myokine expression patterns and bone metabolism between species.
4.4.2 Examination of human studies on myokines, bone density, and fracture
4.4.2.1 Risk in diabetic patients
Clinical studies provide substantial evidence that myokines play a crucial role in bone metabolism in people with diabetes. Significant differences in circulating myokine levels have been observed between individuals with diabetes and non-diabetic controls, as summarized in Table 2. Among these, irisin levels exhibit opposite trends in different forms of diabetes. Patients with T1D show elevated irisin levels (117), whereas those with T2D have significantly reduced levels compared to healthy controls (118, 119). Similarly, GDF8 concentrations in healthy individuals typically range from 1 to 8 ng/mL (120, 121), but in T1D patients, GDF8 levels are significantly elevated, particularly in women, suggesting a stronger inhibitory effect on muscle and bone mass under insulin-deficient conditions (122). In contrast, T2D patients exhibit only a slight elevation in circulating GDF8, yet muscle GDF8 mRNA expression is upregulated (1.4-fold higher), indicating potential dysregulation in skeletal muscle metabolism (123). Serum GDF11 levels average 5.92 ± 2.52 ng/mL and exhibit a negative correlation with bone mineral density (BMD) at the lumbar spine, total hip, and femoral neck (124). Although GDF11 declines with aging, its levels do not appear significantly influenced by obesity or glycemic status in T2D (125). IL-6 plays a complex role in both T1D and T2D pathogenesis, with its concentrations in healthy individuals averaging 5.186 pg/mL but increasing substantially during acute inflammation and muscle activity (126). In T1D, IL-6 levels are elevated, contributing to autoimmune-mediated β-cell destruction (127). In T2D, IL-6 acts as a pro-inflammatory mediator linking adipose tissue inflammation to systemic insulin resistance (128). Elevated IL-6 levels have also been implicated in diabetes-induced bone loss, potentially through increased osteoclastogenesis and altered osteoblast function. Similarly, BDNF concentrations in healthy individuals typically range 32.69 ± 8.33 ng/mL (129), but both T1D and T2D patients exhibit lower circulating BDNF levels, suggesting that BDNF deficiency may contribute to impaired bone remodeling and neuro-metabolic dysfunction in diabetes (130, 131).
In healthy individuals, median circulating FGF21 levels generally fall between 0.1 and 0.2 ng/mL, although significant inter-individual variability exists, with concentrations ranging from 0.021 to 5.3 ng/mL. Serum FGF21 levels are significantly lower in T1DM patients compared to non-diabetic individuals (132), while in T2DM, circulating FGF21 levels are consistently elevated (133, 134). A deficiency in FGF21 disrupts the normal regulatory feedback mechanisms in adipose tissue, leading to impaired systemic lipid and glucose homeostasis (82). In control groups, musclin serum concentrations typically range from 66.35 to 104.33 ng/mL. Elevated musclin levels have been reported in individuals with T2DM (135, 136). Recent research has also suggested that SPARC plays a role in metabolic diseases such as obesity and T2DM, where it may influence both adipose tissue function and insulin sensitivity (137). Interestingly, SPARC blood concentration has been found to increase in T2DM (138). Epidemiological research has also assessed the prognostic potential of myokines as biomarkers for bone health in diabetes. The median plasma DKK3 concentration is 32.8 ng/mL, with an interquartile range (IQR) of 28.0–39.0 ng/mL (139). Urinary DKK3 serves as a kidney-specific biomarker associated with tubular stress and fibrosis, whereas plasma DKK3 levels integrate signals from various tissues, reducing its specificity for chronic kidney disease (CKD) (101, 139). Findings suggest that baseline myokine levels can serve as predictors for future bone loss and fracture risk, underscoring their value as diagnostic and prognostic tools. Nonetheless, the variability in study methodologies, patient demographics, and myokine assay techniques across different studies calls for further validation through large-scale, well-controlled clinical trials. In summary, both animal models and human studies provide compelling evidence that myokines are integral to the pathophysiology of diabetes-related bone disorders. While current findings are encouraging, there is a pressing need for more extensive research to unravel the precise mechanisms through which myokines modulate bone health in the context of diabetes and to evaluate their potential as therapeutic targets.
5 Therapeutic strategies targeting myokines to enhance bone health in diabetic and patients
The recognition of myokines as essential regulators of muscle-bone crosstalk has opened new therapeutic avenues for addressing osteoporosis, particularly in diabetic patients. Given that diabetes can influence bone metabolism in complex ways through altered myokine expression, increased inflammation, and impaired bone remodeling, targeting myokines represents a promising strategy to mitigate osteoporosis and fracture risk in these populations. Multiple therapeutic approaches are currently being explored, including exercise interventions, myokine mimetics, and gene therapies.
5.1 Exercise interventions
Physical activity can stimulate the production of myokines (140, 141). Tailoring individualized exercise programs could also better modulate myokine expression and improve bone health. The specific effects of exercise-induced myokines on bone health in patients with T1DM and T2DM remain areas that require further research.
5.2 Myokine mimetics
Several myokine mimetics have entered clinical trials, offering promising therapeutic options for muscle-related disorders and osteoporosis. These mimetics aim to replicate the actions of naturally occurring myokines. Among the most studied GDF8 inhibitors in clinical research are MYO-029 (Anti-MSTN monoclonal antibody), Domagrozumab (Anti-MSTN monoclonal antibody), LY2495655 (Anti-MSTN monoclonal antibody), REGN1033 (Anti-MSTN monoclonal antibody), SRK-015 (Anti-propeptide monoclonal antibody), Bimagrumab (Anti-receptor monoclonal antibody), ACE-031 (Decoy receptor), and ACE-083 (Follistatin/Fc fusion protein). Broader inhibitors like Bimagrumab and ACE-031 have demonstrated significant effects on muscle mass. However, these inhibitors are also associated with risks of adverse effects, particularly those impacting the vascular system. The specificity of these inhibitors is crucial, as it influences their potential to cause off-target effects (142, 143). Particularly, Bimagrumab emerges as a significant therapeutic agent of interest. In the phase 2 clinical trial, it was demonstrated that Bimagrumab, an inhibitor of activin type II receptors (ActRII), led to a substantial reduction in body fat mass and concurrently enhanced glycemic control among adults afflicted with type 2 diabetes and obesity (144). Although clinical trials are underway, the effects of myokine mimetics on bone still require further investigation, particularly within the complex context of diabetes. More research is needed to fully understand the role of these mimetics in this condition.
5.3 Gene therapy
Direct delivery of myokine genes or gene encoding sequences to muscle tissue might lead to increased local levels and could in turn support bone health. This article focuses on two well-established myokine-based gene therapies: GDF8 and FGF21. The study explores AAV8-mediated delivery of GDF8 propeptide as a therapy in both normal and dystrophic (mdx) mice, demonstrating enhanced muscle growth and improved dystrophic symptoms. AAV8-mediated GDF8 inhibition is identified as a promising strategy for treating muscular dystrophy (145). Additionally, another study investigates the potential of fibroblast growth factor 21 (FGF21) gene therapy using AAV vectors to counteract obesity, insulin resistance, and T2D. Researchers showed that liver-specific AAV-mediated expression of FGF21 had significant and long-lasting effects in high-fat diet (HFD)-fed mice and genetically obese ob/ob mice, and also prevented age-associated metabolic decline in healthy animals (146). While there is ongoing debate about whether FGF21 negatively affects bone mineral density, the effects of these myokine-based gene therapies on bone health, particularly in the context of osteoporosis and diabetes, have yet to be fully studied. Therefore, when considering gene therapy for osteoporosis under physiological and diabetic conditions, it is essential to assess not only the effects of myokines on bone health but also their influence on whole-body metabolism.
6 Current limitations in understanding the role of myokines in bone metabolism in diabetes
6.1 Diversity of myokines and their targets
The range of myokines involved in bone metabolism is extensive. While most animal studies have focused on global myokine knockouts, many findings remain controversial and require further experimental validation. Current research, based on reproducibility of results, supports that GDF8 and GDF11 are well-established as negative regulators of bone metabolism, while Musclin, SPARC, and Irisin have been identified as positive regulators. Nonetheless, additional studies are necessary to confirm and refine these roles. Moreover, research specifically examining muscle-specific myokine knockouts and their effects on bone remains limited. In bone-specific investigations, critical factors such as mouse strain, age, sex, and diet composition must be carefully considered, yet many of these variables are underexplored or poorly understood, particularly in the context of diabetes. This highlights the need for more focused research in this area (Table 2). Furthermore, the full spectrum of muscle-derived factors influencing bone remodeling in diabetic conditions has not been fully characterized. The possibility remains that other yet-to-be-identified myokines may have significant effects on bone homeostasis and pathology in diabetes, emphasizing the importance of comprehensive analyses to further elucidate these complex relationships.
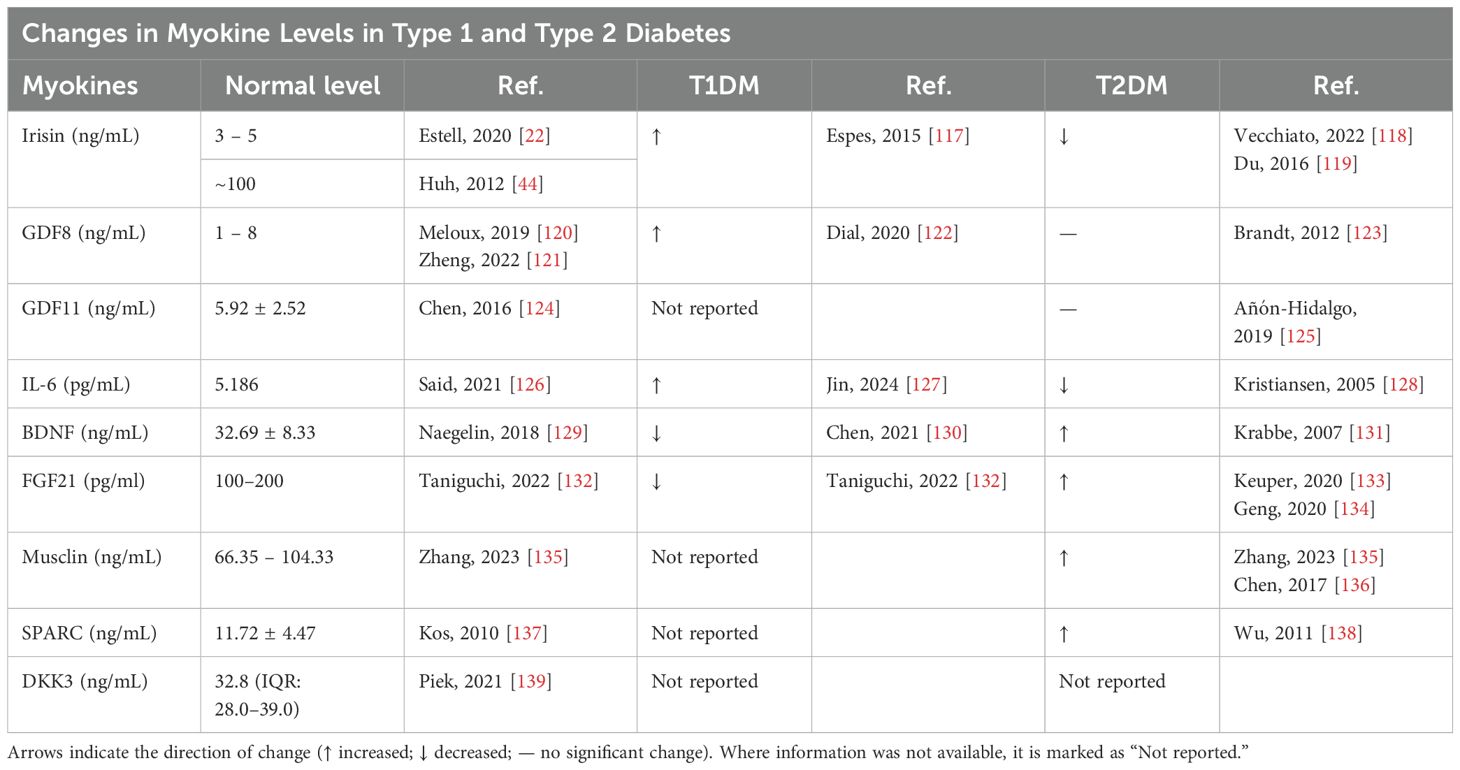
Table 2. Summary of reported levels of selected myokines in individuals with type 1 diabetes mellitus (T1DM) and type 2 diabetes mellitus (T2DM).
6.2 Complexity of diabetes and muscle-bone crosstalk
Diabetes presents a complex pathophysiological challenge, encompassing hyperglycemia, insulin resistance, inflammation, and the accumulation of advanced glycation end products (AGEs). The interplay between these factors and myokines, and their collective impact on bone metabolism, remains poorly understood. Our previous research has shown that bone health is significantly compromised in both T1DM and T2DM (10, 147), particularly due to disruptions in osteoblast glucose metabolism. However, we have observed that focusing solely on bone cell metabolism is insufficient to prevent long-term bone loss. A more comprehensive approach is necessary, one that addresses the broader pathophysiological context of diabetes, including the influence of muscle on bone health. This complexity is further amplified by the bidirectional communication between muscle and bone, which is disrupted under diabetic conditions. This makes it challenging to isolate the specific contributions of myokines from the wider metabolic disturbances associated with diabetes, underscoring the need for a more integrated understanding of these interactions.
6.3 Methodological challenges
One of the key challenges we have faced is the variability in detection methods for myokines, such as Irisin. Different methodologies produce inconsistent concentration measurements, which in turn lead to discrepancies in the experimental concentrations used. As a result, this variability has contributed to contradictory findings in our studies, underscoring the need for standardized detection approaches. Investigating the effects of myokines on bone metabolism in vivo is fraught with methodological difficulties. One major challenge is distinguishing the direct effects of myokines on bone cells from the myriad systemic changes occurring in a diabetic state, such as altered nutrient availability, hormonal imbalances, and inflammatory responses. This makes it difficult to attribute observed changes in bone health solely to myokine activity without the confounding effects of other factors.
7 Future research directions
7.1 Impact of diabetes on myokine production and function
Research should explore how diabetic conditions—characterized by hyperglycemia, insulin resistance, and systemic inflammation—affect the production and function of myokines. Systematically investigating the regulation of myokine expression and secretion under diabetic stress could provide insights into their roles in bone metabolism and highlight potential points for therapeutic intervention.
7.2 Identification of novel myokines
Future research should prioritize the identification and characterization of novel myokines that may influence bone metabolism, particularly under diabetic conditions. This could be achieved through comprehensive proteomic and transcriptomic analyses of muscle tissues from diabetic animal models and human patients, which could reveal new candidates for further investigation. Understanding which specific subpopulations of muscle cells are responsible for secreting myokines remains a pivotal area of investigation for future research. The advent of scRNA-seq technology provides an unparalleled opportunity to delve into this question with great specificity. By applying single-cell sequencing, researchers can distinguish between different muscle cell types, such as satellite cells, myoblasts, and myocytes, and determine their unique roles in myokine production. To further elucidate the function of specific myokines within the context of diabetes, muscle-specific Cre-loxP strategies can be employed to selectively knock out myokine genes in particular muscle cell subtypes. This targeted genetic approach allows researchers to validate the contribution and mechanistic role of these myokines in the pathophysiology of diabetes, including their effects on muscle-bone crosstalk, metabolic regulation, and systemic inflammation. By combining single-cell sequencing with muscle-specific gene editing, it becomes feasible to map out the cellular and molecular landscapes that drive myokine secretion and to better understand how these factors contribute to diabetic complications and overall muscle health.
7.3 Elucidation of myokine signaling pathways
There is a need for detailed studies focused on the specific signaling pathways activated by myokines in osteoblasts lineage cells and osteoclasts. Understanding these pathways could uncover new molecular targets for therapeutic intervention, helping to mitigate the adverse effects of diabetes on bone health.
7.4 Therapeutic potential of myokines
Given the emerging role of myokines in bone metabolism, research should continue to explore their therapeutic potential. Strategies could include developing drugs that enhance the production of beneficial myokines, mimic their effects, or inhibit the actions of detrimental myokines in diabetic patients with bone complications.
7.5 Clinical trials and marker development
Conducting clinical trials to validate the efficacy of myokine-based therapies in diabetic populations is essential. Additionally, the development of markers that reflect the activity of myokines could provide a valuable tool for monitoring treatment responses and predicting long-term outcomes in patients with diabetes-related bone disorders.
8 Conclusion
Myokines are emerging as critical regulators of bone health, particularly in the context of osteoporosis. They mediate the communication between muscle and bone, influencing bone remodeling and mass. As our understanding of these molecules deepens, myokines could become central to new strategies for managing osteoporosis and related disorders. Our review provides a comprehensive and detailed analysis of the bone phenotypes associated with each myokine, as discrepancies in the results may partially stem from these factors. This in-depth examination not only identifies potential sources of inconsistency but also offers valuable insights into the specific roles myokines play in bone physiology. Notably, even under physiological conditions, the bone phenotypes of certain myokines remain a topic of debate. Furthermore, research that isolates the effects of muscle-derived factors on bone is still limited, with the majority of studies relying on global knockout mouse models. In the context of diabetes, particularly T1DM, both animal and clinical studies remain underexplored, highlighting the need for further research in these areas. With the continued advancement of proteomics and single-cell omics, these technologies offer the potential to identify a broader range of myokines and precisely determine the specific cell types responsible for their secretion. Furthermore, they provide an opportunity to systematically elucidate the roles of myokines in bone metabolism within the context of T1DM and T2DM, offering deeper insights into their impact under diabetic conditions. The therapeutic targeting of myokines holds great potential for treating osteoporosis. By modulating the activity of specific myokines, it may be possible to enhance bone density, reduce fracture risk, and improve overall musculoskeletal health, offering a promising avenue for future research and clinical applications.
Author contributions
XJ: Writing – original draft, Writing – review & editing. XH: Writing – original draft. TX: Writing – original draft. WY: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by National Natural Science Foundation of China (No. 82002329,82100934), and Zhejiang Provincial Natural Science Foundation of China (No. LY23H060001, LQ22H070001).
Acknowledgments
We would like to express our gratitude to Chunmei Xiu for her outstanding assistance throughout the course of this review.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. Generative AI is only used for refining grammar and modifying sentences.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ActRIIB, Activin Receptor Type IIB; AGEs, Advanced Glycation End-products; ALP, Alkaline Phosphatase AMPK, AMP-activated Protein Kinase; BAIBA, β-aminoisobutyric acid; BDNF, Brain-Derived Neurotrophic Factor; BMI, Body Mass Index; BMC, Bone Mineral Content; BMD, Bone Mineral Density; BMP, Bone Morphogenetic Protein; BMSCs, Bone Marrow Stromal Cells; BV/TV, Bone Volume/Total Volume; FGF21, Fibroblast Growth Factor 21; FLRG, Follistatin-Related Gene; FNDC5, Fibronectin Type III Domain-Containing Protein 5; GDF11, Growth Differentiation Factor 11; GDF8(MSTN), Growth Differentiation; Factor 8 (Myostatin); HFD, High-Fat Diet; IGFBP1, Insulin-like ; Growth Factor Binding Protein 1; IHH, Indian Hedgehog; IL-6, Interleukin-6; KO, Knockout; MRGPRD, Mas-Related G-Protein Coupled Receptor D; MSCs, Mesenchymal Stem Cells; OC, Osteoclast; OB, Osteoblast; OPG, Osteoprotegerin; OSTN, Osteocrin; OVX, Ovariectomy; PPARγ, Peroxisome Proliferator-Activated Receptor Gamma; PTH, Parathyroid Hormone; PTHrP, Parathyroid ; Hormone-related Protein; scRNA-seq, Single-Cell RNA Sequencing; sIL-6R, Soluble Interleukin-6 Receptor; Sp7, Specificity Protein 7/Osterix; T1D/T1DM, Type 1 Diabetes Mellitus; T2D/T2DM, Type 2 Diabetes Mellitus; WAT/BAT, White/Brown Adipose Tissue.
References
1. Pedersen BK, Steensberg A, Fischer C, Keller C, Keller P, Plomgaard P, et al. Searching for the exercise factor: is IL-6 a candidate? J Muscle Res Cell Moti. (2003) 24(2):113–9. doi: 10.1023/A:1026070911202
2. Severinsen MCK and Pedersen BK. Muscle-Organ Crosstalk: The Emerging Roles of Myokines. Endocr Re. (2020) 41(4):16. doi: 10.1210/endrev/bnaa016
3. Kaji H. Crosstalk between muscle and bone. J Bone Miner Meta. (2023) 8. doi: 10.1007/s00774-023-01472-5
4. Lee JH and Jun HS. Role of myokines in regulating skeletal muscle mass and function. Front Physio. (2019) 10:9. doi: 10.3389/fphys.2019.00042
5. Muñoz-Cánoves P, Scheele C, Pedersen BK, and Serrano AL. Interleukin-6 myokine signaling in skeletal muscle: a double-edged sword? FEBS . (2013) 280(17):4131–48.
6. Shero JA, Lindholm ME, Sandri M, and Stanford KI. Skeletal muscle as a mediator of interorgan crosstalk during exercise: implications for aging and obesity. Circ Re. (2025) 136(11):1407–32. doi: 10.1161/CIRCRESAHA.124.325614
7. Clynes MA, Harvey NC, Curtis EM, Fuggle NR, Dennison EM, and Cooper C. The epidemiology of osteoporosis. Br Med Bul. (2020) 133(1):105–17. doi: 10.1093/bmb/ldaa005
8. Kirk B, Feehan J, Lombardi G, and Duque G. Muscle, bone, and fat crosstalk: the biological role of myokines, osteokines, and adipokines. Curr Osteoporos Re. (2020) 18(4):388–400. doi: 10.1007/s11914-020-00599-y
9. Li Z, Wen X, Li N, Zhong C, Chen L, Zhang F, et al. The roles of hepatokine and osteokine in liver-bone crosstalk: Advance in basic and clinical aspects. Front Endocrinol (Lausanne). (2023) 14:1149233. doi: 10.3389/fendo.2023.1149233
10. Song FF, Lee WD, Marmo T, Ji X, Song C, Liao XY, et al. Osteoblast-intrinsic defect in glucose metabolism impairs bone formation in type II diabetic male mice. eLif. (2023) 12:19. doi: 10.7554/eLife.85714
11. Sellmeyer DE, Civitelli R, Hofbauer LC, Khosla S, Lecka-Czernik B, and Schwartz AV. Skeletal metabolism, fracture risk, and fracture outcomes in type 1 and type 2 diabetes. Diabete. (2016) 65(7):1757–66. doi: 10.2337/db16-0063
12. Hofbauer LC, Busse B, Eastell R, Ferrari S, Frost M, Müller R, et al. Bone fragility in diabetes: novel concepts and clinical implications. Lancet Diabetes Endocrinology. (2022) 10(3):207–20. doi: 10.1016/S2213-8587(21)00347-8
13. Vilaca T, Schini M, Harnan S, Sutton A, Poku E, Allen IE, et al. The risk of hip and non-vertebral fractures in type 1 and type 2 diabetes: A systematic review and meta-analysis update. Bon. (2020) 137:115457. doi: 10.1016/j.bone.2020.115457
14. Schwartz AV, Vittinghoff E, Bauer DC, Hillier TA, Strotmeyer ES, Ensrud KE, et al. Association of BMD and FRAX score with risk of fracture in older adults with type 2 diabetes. Jam. (2011) 305(21):2184–92. doi: 10.1001/jama.2011.715
15. Napoli N, Chandran M, Pierroz DD, Abrahamsen B, Schwartz AV, and Ferrari SL. Mechanisms of diabetes mellitus-induced bone fragility. Nat Rev Endocrino. (2017) 13(4):208–19. doi: 10.1038/nrendo.2016.153
16. Dong YC, Yuan HY, Ma GX, and Cao HL. Bone-muscle crosstalk under physiological and pathological conditions. Cell Mol Life Sc. (2024) 81(1):20. doi: 10.1007/s00018-024-05331-y
17. Brotto M and Bonewald L. Bone and muscle: Interactions beyond mechanical. Bon. (2015) 80:109–14. doi: 10.1016/j.bone.2015.02.010
18. Felsenthal N and Zelzer E. Mechanical regulation of musculoskeletal system development. Developmen. (2017) 144(23):4271–83. doi: 10.1242/dev.151266
19. Walle M, Duseja A, Whittier DE, Vilaca T, Paggiosi M, Eastell R, et al. Bone remodeling and responsiveness to mechanical stimuli in individuals with type 1 diabetes mellitus. J Bone Mineral Re. (2024) 39(2):85–94. doi: 10.1093/jbmr/zjad014
20. Gomarasca M, Banfi G, Lombardi G, and Makowski GS. Myokines: The endocrine coupling of skeletal muscle and bone. Adv Clin Che. (2020) 94:155–218. doi: 10.1016/bs.acc.2019.07.010
21. Kim H, Wrann CD, Jedrychowski M, Vidoni S, Kitase Y, Nagano K, et al. Irisin Mediates Effects on Bone and Fat via αV Integrin Receptors. Cel. (2018) 175(7):1756–68.e17. doi: 10.1016/j.cell.2018.10.025
22. Estell EG, Le PT, Vegting Y, Kim H, Wrann C, Bouxsein ML, et al. Irisin directly stimulates osteoclastogenesis and bone resorption in vitro and in vivo. eLif. (2020) 9:13. doi: 10.7554/eLife.58172
23. Hamrick MW, McPherron AC, and Lovejoy CO. Bone mineral content and density in the humerus of adult myostatin-deficient mice. Calcif Tissue In. (2002) 71(1):63–8. doi: 10.1007/s00223-001-1109-8
24. McPherron AC, Lawler AM, and Lee SJ. Regulation of anterior/posterior patterning of the axial skeleton by growth/differentiation factor 11. Nat Gene. (1999) 22(3):260–4. doi: 10.1038/10320
25. Poli V, Balena R, Fattori E, Markatos A, Yamamoto M, Tanaka H, et al. Interleukin-6 deficient mice are protected from bone loss caused by estrogen depletion. EMBO . (1994) 13(5):1189–96. doi: 10.1002/j.1460-2075.1994.tb06368.x
26. Li Y, Lu L, Xie Y, Chen X, Tian L, Liang Y, et al. Interleukin-6 knockout inhibits senescence of bone mesenchymal stem cells in high-fat diet-induced bone loss. . Front Endocrino. (2021) 11. doi: 10.3389/fendo.2020.622950
27. Camerino C, Zayzafoon M, Rymaszewski M, Heiny J, Rios M, and Hauschka PV. Central depletion of brain-derived neurotrophic factor in mice results in high bone mass and metabolic phenotype. Endocrinolog. (2012) 153(11):5394–405. doi: 10.1210/en.2012-1378
28. Huang R, Balu AR, Molitoris KH, White JP, Robling AG, Ayturk UM, et al. The role of Meteorin-like in skeletal development and bone fracture healing. J Orthop Re. (2022) 40(11):2510–21. doi: 10.1002/jor.v40.11
29. Bornstein S, Brown SA, Le PT, Wang X, DeMambro V, Horowitz MC, et al. FGF-21 and skeletal remodeling during and after lactation in C57BL/6J mice. Endocrinolog. (2014) 155(9):3516–26. doi: 10.1210/en.2014-1083
30. Wang X, Wei W, Krzeszinski JY, Wang Y, and Wan Y. A liver-bone endocrine relay by IGFBP1 promotes osteoclastogenesis and mediates FGF21-induced bone resorption. Cell Meta. (2015) 22(5):811–24. doi: 10.1016/j.cmet.2015.09.010
31. Li X, Stanislaus S, Asuncion F, Niu QT, Chinookoswong N, Villasenor K, et al. FGF21 is not a major mediator for bone homeostasis or metabolic actions of PPARα and PPARγ agonists. J Bone Miner Re. (2017) 32(4):834–45. doi: 10.1002/jbmr.2936
32. Watanabe-Takano H, Ochi H, Chiba A, Matsuo A, Kanai Y, Fukuhara S, et al. Mechanical load regulates bone growth via periosteal Osteocrin. Cell Re. (2021) 36(2):109380. doi: 10.1016/j.celrep.2021.109380
33. Kanai Y, Yasoda A, Mori KP, Watanabe-Takano H, Nagai-Okatani C, Yamashita Y, et al. Circulating osteocrin stimulates bone growth by limiting C-type natriuretic peptide clearance. J Clin Invest. (2017) 127(11):4136–47. doi: 10.1172/JCI94912
34. Delany AM, Amling M, Priemel M, Howe C, Baron R, and Canalis E. Osteopenia and decreased bone formation in osteonectin-deficient mice. J Clin Invest. (2000) 105(7):915–23. doi: 10.1172/JCI7039
35. Barrantes Idel B, Montero-Pedrazuela A, Guadaño-Ferraz A, Obregon MJ, Martinez de Mena R, Gailus-Durner V, et al. Generation and characterization of dickkopf3 mutant mice. Mol Cell Bio. (2006) 26(6):2317–26.
36. Boström P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, et al. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Natur. (2012) 481(7382):463–U72.
37. Colaianni G, Cuscito C, Mongelli T, Pignataro P, Buccoliero C, Liu P, et al. The myokine irisin increases cortical bone mass. Proc Natl Acad Sci U S A. (2015) 112(39):12157–62. doi: 10.1073/pnas.1516622112
38. Luo Y, Ma Y, Qiao X, Zeng R, Cheng R, Nie Y, et al. Irisin ameliorates bone loss in ovariectomized mice. Climacteri. (2020) 23(5):496–504. doi: 10.1080/13697137.2020.1745768
39. Colaianni G, Cuscito C, Mongelli T, Oranger A, Mori G, Brunetti G, et al. Irisin enhances osteoblast differentiation in vitro. Int J Endocrino. (2014) 2014:902186. doi: 10.1155/2014/902186
40. Zhu X, Li X, Wang X, Chen T, Tao F, Liu C, et al. Irisin deficiency disturbs bone metabolism. J Cell Physio. (2021) 236(1):664–76. doi: 10.1002/jcp.v236.1
41. Ye WB, Wang JZ, Lin DS, and Ding ZQ. The immunomodulatory role of irisin on osteogenesis via AMPK-mediated macrophage polarization. Int J Biol Macromol. (2020) 146:25–35. doi: 10.1016/j.ijbiomac.2019.12.028
42. Ma YX, Qiao XY, Zeng RJ, Cheng R, Zhang J, Luo YY, et al. Irisin promotes proliferation but inhibits differentiation in osteoclast precursor cells. FASEB . (2018) 32(11):5813–23. doi: 10.1096/fj.201700983RR
43. Jedrychowski MP, Wrann CD, Paulo JA, Gerber KK, Szpyt J, Robinson MM, et al. Detection and quantitation of circulating human irisin by tandem mass spectrometry. Cell Meta. (2015) 22(4):734–40. doi: 10.1016/j.cmet.2015.08.001
44. Huh JY, Panagiotou G, Mougios V, Brinkoetter M, Vamvini MT, Schneider BE, et al. FNDC5 and irisin in humans: I. Predictors of circulating concentrations in serum and plasma and II. mRNA expression and circulating concentrations in response to weight loss and exercise. Metab-Clin Ex. (2012) 61(12):1725–38.
45. McPherron AC, Lawler AM, and Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Natur. (1997) 387(6628):83–90. doi: 10.1038/387083a0
46. Chiu CS, Peekhaus N, Weber H, Adamski S, Murray EM, Zhang HZ, et al. Increased muscle force production and bone mineral density in ActRIIB-Fc-treated mature rodents. J Gerontol A Biol Sci Med Sc. (2013) 68(10):1181–92. doi: 10.1093/gerona/glt030
47. Lee S-J and McPherron AC. Regulation of myostatin activity and muscle growth. Proc Natl Acad Sc. (2001) 98(16):9306–11. doi: 10.1073/pnas.151270098
48. Rebbapragada A, Benchabane H, Wrana JL, Celeste AJ, and Attisano L. Myostatin signals through a transforming growth factor beta-like signaling pathway to block adipogenesis. Mol Cell Bio. (2003) 23(20):7230–42. doi: 10.1128/MCB.23.20.7230-7242.2003
49. Elkasrawy MN and Hamrick MW. Myostatin (GDF-8) as a key factor linking muscle mass and bone structure. J Musculoskelet Neuronal Interac. (2010) 10(1):56–63.
50. Hamrick MW. Increased bone mineral density in the femora of GDF8 knockout mice. Anat Rec Part . (2003) 272A(1):388–91. doi: 10.1002/ar.a.v272a:1
51. Qin Y, Peng Y, Zhao W, Pan J, Ksiezak-Reding H, Cardozo C, et al. Myostatin inhibits osteoblastic differentiation by suppressing osteocyte-derived exosomal microRNA-218: A novel mechanism in muscle-bone communication. J Biol Che. (2017) 292(26):11021–33. doi: 10.1074/jbc.M116.770941
52. Hamrick MW, Shi X, Zhang W, Pennington C, Thakore H, Haque M, et al. Loss of myostatin (GDF8) function increases osteogenic differentiation of bone marrow-derived mesenchymal stem cells but the osteogenic effect is ablated with unloading. Bon. (2007) 40(6):1544–53. doi: 10.1016/j.bone.2007.02.012
53. Arounleut P, Bialek P, Liang L-F, Upadhyay S, Fulzele S, Johnson M, et al. A myostatin inhibitor (propeptide-Fc) increases muscle mass and muscle fiber size in aged mice but does not increase bone density or bone strength. Exp Gerontology. (2013) 48(9):898–904. doi: 10.1016/j.exger.2013.06.004
54. Bialek P, Parkington J, Li X, Gavin D, Wallace C, Zhang J, et al. A myostatin and activin decoy receptor enhances bone formation in mice. Bon. (2014) 60:162–71. doi: 10.1016/j.bone.2013.12.002
55. Dankbar B, Fennen M, Brunert D, Hayer S, Frank S, Wehmeyer C, et al. Myostatin is a direct regulator of osteoclast differentiation and its inhibition reduces inflammatory joint destruction in mice. Nat Me. (2015) 21(9):1085–90. doi: 10.1038/nm.3917
56. Zhi X, Chen Q, Song S, Gu Z, Wei W, Chen H, et al. Myostatin promotes osteoclastogenesis by regulating Ccdc50 gene expression and RANKL-Induced NF-κB and MAPK Pathways. Front Pharmaco. (2020) 11:565163. doi: 10.3389/fphar.2020.565163
57. Gamer LW, Wolfman NM, Celeste AJ, Hattersley G, Hewick R, and Rosen V. A novel BMP expressed in developing mouse limb, spinal cord, and tail bud is a potent mesoderm inducer in Xenopus embryos. Dev Bio. (1999) 208(1):222–32. doi: 10.1006/dbio.1998.9191
58. Oh SP, Yeo CY, Lee Y, Schrewe H, Whitman M, and Li E. Activin type IIA and IIB receptors mediate Gdf11 signaling in axial vertebral patterning. Genes De. (2002) 16(21):2749–54. doi: 10.1101/gad.1021802
59. Liu W, Zhou L, Zhou C, Zhang S, Jing J, Xie L, et al. GDF11 decreases bone mass by stimulating osteoclastogenesis and inhibiting osteoblast differentiation. Nat Commu. (2016) 7:12794. doi: 10.1038/ncomms12794
60. Lu Q, Tu ML, Li CJ, Zhang L, Jiang TJ, Liu T, et al. GDF11 inhibits bone formation by activating Smad2/3 in bone marrow mesenchymal stem cells. Calcif Tissue In. (2016) 99(5):500–9. doi: 10.1007/s00223-016-0173-z
61. Zhang Y, Shao J, Wang Z, Yang T, Liu S, Liu Y, et al. Growth differentiation factor 11 is a protective factor for osteoblastogenesis by targeting PPARgamma. Gen. (2015) 557(2):209–14. doi: 10.1016/j.gene.2014.12.039
62. Hirano T, Yasukawa K, Harada H, Taga T, Watanabe Y, Matsuda T, et al. Complementary-dna for A novel human interleukin (Bsf-2) that induces lymphocytes-B to produce immunoglobulin. Natur. (1986) 324(6092):73–6. doi: 10.1038/324073a0
63. Kishimoto T. Interleukin-6: discovery of a pleiotropic cytokine. Arthritis Res The. (2006) 8 Suppl 2(Suppl 2):S2. doi: 10.1186/ar1916
64. Pedersen BK and Febbraio MA. Muscle as an endocrine organ: Focus on muscle-derived interleukin-6. Physiol Re. (2008) 88(4):1379–406. doi: 10.1152/physrev.90100.2007
65. Chen Z-T, Weng Z-X, Lin JD, and Meng Z-X. Myokines: metabolic regulation in obesity and type 2 diabetes. Life Meta. (2024) 3(3):loae006. doi: 10.1093/lifemeta/loae006
66. De Benedetti F, Rucci N, Del Fattore A, Peruzzi B, Paro R, Longo M, et al. Impaired skeletal development in interleukin-6-transgenic mice: a model for the impact of chronic inflammation on the growing skeletal system. Arthritis Rheumato. (2006) 54(11):3551–63. doi: 10.1002/art.v54:11
67. Kaneshiro S, Ebina K, Shi K, Higuchi C, Hirao M, Okamoto M, et al. IL-6 negatively regulates osteoblast differentiation through the SHP2/MEK2 and SHP2/Akt2 pathways in vitro. J Bone Miner Meta. (2014) 32(4):378–92. doi: 10.1007/s00774-013-0514-1
68. Sims NA, Jenkins BJ, Quinn JM, Nakamura A, Glatt M, Gillespie MT, et al. Glycoprotein 130 regulates bone turnover and bone size by distinct downstream signaling pathways. J Clin Invest. (2004) 113(3):379–89. doi: 10.1172/JCI19872
69. Chowdhury S, Schulz L, Palmisano B, Singh P, Berger JM, Yadav VK, et al. Muscle-derived interleukin 6 increases exercise capacity by signaling in osteoblasts. J Clin Invest. (2020) 130(6):2888–902. doi: 10.1172/JCI133572
70. Matthews VB, Åström MB, Chan MHS, Bruce CR, Krabbe KS, Prelovsek O, et al. Brain-derived neurotrophic factor is produced by skeletal muscle cells in response to contraction and enhances fat oxidation via activation of AMP-activated protein kinase. Diabetologi. (2009) 52(7):1409–18. doi: 10.1007/s00125-009-1364-1
71. Yamashiro T, Fukunaga T, Yamashita K, Kobashi N, and Takano-Yamamoto T. Gene and protein expression of brain-derived neurotrophic factor and TrkB in bone and cartilage. Bon. (2001) 28(4):404–9. doi: 10.1016/S8756-3282(01)00405-7
72. Zhang ZT, Hu PL, Wang Z, Qiu XS, and Chen YX. BDNF promoted osteoblast migration and fracture healing by up-regulating integrin β1 via TrkB-mediated ERK1/2 and AKT signalling. J Cell Mol Me. (2020) 24(18):10792–802. doi: 10.1111/jcmm.v24.18
73. Xue F, Zhao Z, Gu Y, Han J, Ye K, and Zhang Y. 7,8-Dihydroxyflavone modulates bone formation and resorption and ameliorates ovariectomy-induced osteoporosis. eLif. (2021) 10:e64872. doi: 10.7554/eLife.64872
74. Xiong J, Liao JM, Liu X, Zhang ZH, Adams J, Pacifici R, et al. A TrkB agonist prodrug prevents bone loss via inhibiting asparagine endopeptidase and increasing osteoprotegerin. Nat Commu. (2022) 13(1):16. doi: 10.1038/s41467-022-32435-5
75. Rao RR, Long JZ, White JP, Svensson KJ, Lou J, Lokurkar I, et al. Meteorin-like is a hormone that regulates immune-adipose interactions to increase beige fat thermogenesis. Cel. (2014) 157(6):1279–91. doi: 10.1016/j.cell.2014.03.065
76. Wang S, Wang L, Shi S, Wang X, He C, Yuan L, et al. Inhibition of GDF11 could promote bone healing in the tooth extraction socket and facilitate mesenchymal stem cell osteogenic differentiation in T2DM pigs. J periodontolog. (2020). doi: 10.1002/JPER.20-0011
77. Roberts Lee D, Boström P, O’Sullivan John F, Schinzel Robert T, Lewis Gregory D, Dejam A, et al. β-aminoisobutyric acid induces browning of white fat and hepatic β-oxidation and is inversely correlated with cardiometabolic risk factors. Cell Meta. (2014) 19(1):96–108.
78. Kitase Y, Vallejo JA, Gutheil W, Vemula H, Jähn K, Yi JX, et al. β-aminoisobutyric Acid, L-BAIBA, is a muscle-derived osteocyte survival factor. Cell Re. (2018) 22(6):1531–44. doi: 10.1016/j.celrep.2018.01.041
79. Prideaux M, Smargiassi A, Peng G, Brotto M, and Robling AG/person-group>. L-BAIBA synergizes with sub-optimal mechanical loading to promote new bone formation. . JBMR Plus. (Bonewal2023) 7(6):e10746.
80. Izumiya Y, Bina HA, Ouchi N, Akasaki Y, Kharitonenkov A, and Walsh K. FGF21 is an Akt-regulated myokine. FEBS Let. (2008) 582(27):3805–10. doi: 10.1016/j.febslet.2008.10.021
81. Nishimura T, Nakatake Y, Konishi M, and Itoh N. Identification of a novel FGF, FGF-21, preferentially expressed in the liver. Biochim Biophys Acta-Gene Struct Expression. (2000) 1492(1):203–6. doi: 10.1016/S0167-4781(00)00067-1
82. Kharitonenkov A, Shiyanova TL, Koester A, Ford AM, Micanovic R, Galbreath EJ, et al. FGF-21 as a novel metabolic regulator. J Clin Invest. (2005) 115(6):1627–35. doi: 10.1172/JCI23606
83. Wei W, Dutchak PA, Wang X, Ding X, Wang X, Bookout AL, et al. Fibroblast growth factor 21 promotes bone loss by potentiating the effects of peroxisome proliferator-activated receptor γ. Proc Natl Acad Sci U S A. (2012) 109(8):3143–8. doi: 10.1073/pnas.1200797109
84. Thomas G, Moffatt P, Salois P, Gaumond M-H, Gingras R, Godin É, et al. Osteocrin, a novel bone-specific secreted protein that modulates the osteoblast phenotype*. J Biol Che. (2003) 278(50):50563–71. doi: 10.1074/jbc.M307310200
85. Nishizawa H, Matsuda M, Yamada Y, Kawai K, Suzuki E, Makishima M, et al. Musclin, a novel skeletal muscle-derived secretory factor. J Biol Che. (2004) 279(19):19391–5. doi: 10.1074/jbc.C400066200
86. Wang JS, Kamath T, Mazur CM, Mirzamohammadi F, Rotter D, Hojo H, et al. Control of osteocyte dendrite formation by Sp7 and its target gene osteocrin. Nat Commu. (2021) 12(1):6271. doi: 10.1038/s41467-021-26571-7
87. Termine JD, Kleinman HK, Whitson SW, Conn KM, McGarvey ML, and Martin GR. Osteonectin, a bone-specific protein linking mineral to collagen. Cel. (1981) 26Pt 1):99–105. doi: 10.1016/0092-8674(81)90037-4
88. Rosset EM and Bradshaw AD. SPARC/osteonectin in mineralized tissue. Matrix Bio. (2016) 5-54:78–87. doi: 10.1016/j.matbio.2016.02.001
89. Brekken RA and Sage EH. SPARC, a matricellular protein: at the crossroads of cell-matrix communication. Matrix Bio. (2001) 19(8):816–27. doi: 10.1016/S0945-053X(00)00133-5
90. Aoi W, Naito Y, Takagi T, Tanimura Y, Takanami Y, Kawai Y, et al. A novel myokine, secreted protein acidic and rich in cysteine (SPARC), suppresses colon tumorigenesis via regular exercise. Gu. (2013) 62(6):882–9. doi: 10.1136/gutjnl-2011-300776
91. Nie J, Chang B, Traktuev DO, Sun J, March K, Chan L, et al. IFATS collection: Combinatorial peptides identify alpha5beta1 integrin as a receptor for the matricellular protein SPARC on adipose stromal cells. Stem Cell. (2008) 26(10):2735–45. doi: 10.1634/stemcells.2008-0212
92. Boskey AL, Moore DJ, Amling M, Canalis E, and Delany AM. Infrared analysis of the mineral and matrix in bones of osteonectin-null mice and their wildtype controls. J Bone Miner Re. (2003) 18(6):1005–11. doi: 10.1359/jbmr.2003.18.6.1005
93. Kessler CB and Delany AM. Increased Notch 1 expression and attenuated stimulatory G protein coupling to adenylyl cyclase in osteonectin-null osteoblasts. Endocrinolog. (2007) 148(4):1666–74. doi: 10.1210/en.2006-0443
94. Machado do Reis L, Kessler CB, Adams DJ, Lorenzo J, Jorgetti V, and Delany AM. Accentuated osteoclastic response to parathyroid hormone undermines bone mass acquisition in osteonectin-null mice. Bon. (2008) 43(2):264–73. doi: 10.1016/j.bone.2008.03.024
95. Dole NS, Kapinas K, Kessler CB, Yee SP, Adams DJ, Pereira RC, et al. A single nucleotide polymorphism in osteonectin 3' untranslated region regulates bone volume and is targeted by miR-433. J Bone Miner Re. (2015) 30(4):723–32. doi: 10.1002/jbmr.2378
96. Pinzone JJ, Hall BM, Thudi NK, Vonau M, Qiang YW, Rosol TJ, et al. The role of Dickkopf-1 in bone development, homeostasis, and disease. Bloo. (2009) 113(3):517–25. doi: 10.1182/blood-2008-03-145169
97. Baron R and Rawadi G. Targeting the Wnt/β-Catenin Pathway to Regulate Bone Formation in the Adult Skeleton. Endocrinolog. (2007) 148(6):2635–43. doi: 10.1210/en.2007-0270
98. Mukhopadhyay M, Shtrom S, Rodriguez-Esteban C, Chen L, Tsukui T, Gomer L, et al. Dickkopf1 is required for embryonic head induction and limb morphogenesis in the mouse. Dev Cel. (2001) 1(3):423–34. doi: 10.1016/S1534-5807(01)00041-7
99. Xu J, Li X, Chen W, Zhang Z, Zhou Y, Gou Y, et al. Myofiber Baf60c controls muscle regeneration by modulating Dkk3-mediated paracrine signaling. J Exp Me. (2023) 220(9). doi: 10.1084/jem.20221123
100. Schunk SJ, Floege J, Fliser D, and Speer T. WNT-β-catenin signalling - a versatile player in kidney injury and repair. Nat Rev Nephrol. (2021) 17(3):172–84. doi: 10.1038/s41581-020-00343-w
101. Zewinger S, Rauen T, Rudnicki M, Federico G, Wagner M, Triem S, et al. Dickkopf-3 (DKK3) in urine identifies patients with short-term risk of eGFR loss. J Am Soc Nephrol. (2018) 29(11):2722–33. doi: 10.1681/ASN.2018040405
102. Cheng WL, Yang Y, Zhang XJ, Guo J, Gong J, Gong FH, et al. Dickkopf-3 ablation attenuates the development of atherosclerosis in apoe-deficient mice. J Am Heart Asso. (2017) 6(2). doi: 10.1161/JAHA.116.004690
103. Lee HJ, Lee JO, Kim N, Kim JK, Kim HI, Lee YW, et al. Irisin, a novel myokine, regulates glucose uptake in skeletal muscle cells via AMPK. Mol Endocrino. (2015) 29(6):873–81. doi: 10.1210/me.2014-1353
104. Perakakis N, Triantafyllou GA, Fernández-Real JM, Huh JY, Park KH, Seufert J, et al. Physiology and role of irisin in glucose homeostasis. Nat Rev Endocrino. (2017) 13(6):324–37. doi: 10.1038/nrendo.2016.221
105. Chen Y, Ye J, Cao L, Zhang Y, Xia W, and Zhu D. Myostatin regulates glucose metabolism via the AMP-activated protein kinase pathway in skeletal muscle cells. Int J Biochem Cell Bio. (2010) 42(12):2072–81. doi: 10.1016/j.biocel.2010.09.017
106. Liu XH, Bauman WA, and Cardozo CP. Myostatin inhibits glucose uptake via suppression of insulin-dependent and -independent signaling pathways in myoblasts. Physiol Re. (2018) 6(17):e13837. doi: 10.14814/phy2.13837
107. Lu B, Zhong J, Pan J, Yuan X, Ren M, Jiang L, et al. Gdf11 gene transfer prevents high fat diet-induced obesity and improves metabolic homeostasis in obese and STZ-induced diabetic mice. J Trans Me. (2019) 17(1):422. doi: 10.1186/s12967-019-02166-1
108. Al-Khalili L, Bouzakri K, Glund S, Lönnqvist F, Koistinen HA, and Krook A. Signaling specificity of interleukin-6 action on glucose and lipid metabolism in skeletal muscle. Mol Endocrino. (2006) 20(12):3364–75. doi: 10.1210/me.2005-0490
109. Carey AL, Steinberg GR, Macaulay SL, Thomas WG, Holmes AG, Ramm G, et al. Interleukin-6 increases insulin-stimulated glucose disposal in humans and glucose uptake and fatty acid oxidation in vitro via AMP-activated protein kinase. Diabete. (2006) 55(10):2688–97. doi: 10.2337/db05-1404
110. Lee JO, Byun WS, Kang MJ, Han JA, Moon J, Shin MJ, et al. The myokine meteorin-like (metrnl) improves glucose tolerance in both skeletal muscle cells and mice by targeting AMPKα2. FEBS . (2020) 287(10):2087–104. doi: 10.1111/febs.v287.10
111. Behera J, Ison J, Voor MJ, and Tyagi N. Exercise-linked skeletal irisin ameliorates diabetes associated osteoporosis by inhibiting the oxidative damage-dependent miR-150-FNDC5/Pyroptosis Axis. Diabete. (2022) 71(12):2777–92. doi: 10.2337/db21-0573
112. Li GY, Qin H, Zhou MJ, Zhang TW, Zhang Y, Ding HF, et al. Knockdown of SIRT3 perturbs protective effects of irisin against bone loss in diabetes and periodontitis. Free Radic Biol Me. (2023) 200:11–25. doi: 10.1016/j.freeradbiomed.2023.02.023
113. Yang J, Sun L, Fan X, Yin B, Kang Y, Tang L, et al. Effect of exercise on bone in poorly controlled type 1 diabetes mediated by the ActRIIB/Smad signaling pathway. Exp Ther Me. (2018) 16(4):3686–93. doi: 10.3892/etm.2018.6601
114. Wallner C, Jaurich H, Wagner JM, Becerikli M, Harati K, Dadras M, et al. Inhibition of GDF8 (Myostatin) accelerates bone regeneration in diabetes mellitus type 2. Sci Re. (2017) 7(1):9878. doi: 10.1038/s41598-017-10404-z
115. Puolakkainen T, Ma H, Kainulainen H, Pasternack A, Rantalainen T, Ritvos O, et al. Treatment with soluble activin type IIB-receptor improves bone mass and strength in a mouse model of Duchenne muscular dystrophy. BMC Musculoskeletal Disor. (2017) 18(1):20. doi: 10.1186/s12891-016-1366-3
116. Hojman P, Pedersen M, Nielsen AR, Krogh-Madsen R, Yfanti C, Akerstrom T, et al. Fibroblast growth factor-21 is induced in human skeletal muscles by hyperinsulinemia. Diabete. (2009) 58(12):2797–801. doi: 10.2337/db09-0713
117. Espes D, Lau J, and Carlsson PO. Increased levels of irisin in people with long-standing Type1 diabetes. Diabetic Me. (2015) 32(9):1172–6. doi: 10.1111/dme.2015.32.issue-9
118. Vecchiato M, Zanardo E, Battista F, Quinto G, Bergia C, Palermi S, et al. The effect of exercise training on irisin secretion in patients with type 2 diabetes: a systematic review. J Clin Me. (2022) 12(1). doi: 10.3390/jcm12010062
119. Du XL, Jiang WX, and Lv ZT. Lower circulating irisin level in patients with diabetes mellitus: a systematic review and meta-analysis. Horm Metab Re. (2016) 48(10):644–52. doi: 10.1055/s-0042-108730
120. Meloux A, Rochette L, Maza M, Bichat F, Tribouillard L, Cottin Y, et al. Growth differentiation factor-8 (GDF8)/myostatin is a predictor of troponin i peak and a marker of clinical severity after acute myocardial infarction. . J Clin Me. (2019) 9(1). doi: 10.3390/jcm9010116
121. Zheng X, Zheng Y, Qin D, Yao Y, Zhang X, Zhao Y, et al. Regulatory role and potential importance of gdf-8 in ovarian reproductive activity. Front Endocrinol (Lausanne). (2022) 13:878069. doi: 10.3389/fendo.2022.878069
122. Dial AG, Monaco CMF, Grafham GK, Romanova N, Simpson JA, Tarnopolsky MA, et al. Muscle and serum myostatin expression in type 1 diabetes. Physiol Re. (2020) 8(13):e14500. doi: 10.14814/phy2.14500
123. Brandt C, Nielsen AR, Fischer CP, Hansen J, Pedersen BK, and Plomgaard P. Plasma and muscle myostatin in relation to type 2 diabetes. PloS On. (2012) 7(5):e37236. doi: 10.1371/journal.pone.0037236
124. Chen Y, Guo Q, Zhang M, Song S, Quan T, Zhao T, et al. Relationship of serum GDF11 levels with bone mineral density and bone turnover markers in postmenopausal Chinese women. Bone Re. (2016) 4:16012. doi: 10.1038/boneres.2016.12
125. Añón-Hidalgo J, Catalán V, Rodríguez A, Ramírez B, Silva C, Galofré JC, et al. Circulating GDF11 levels are decreased with age but are unchanged with obesity and type 2 diabetes. Aging (Albany NY). (2019) 11(6):1733–44. doi: 10.18632/aging.101865
126. Said EA, Al-Reesi I, Al-Shizawi N, Jaju S, Al-Balushi MS, Koh CY, et al. Defining IL-6 levels in healthy individuals: A meta-analysis. J Med Viro. (2021) 93(6):3915–24. doi: 10.1002/jmv.26654
127. Jin Z, Zhang Q, Liu K, Wang S, Yan Y, Zhang B, et al. The association between interleukin family and diabetes mellitus and its complications: An overview of systematic reviews and meta-analyses. Diabetes Res Clin Practice. (2024) 210:111615. doi: 10.1016/j.diabres.2024.111615
128. Kristiansen OP and Mandrup-Poulsen T. Interleukin-6 and diabetes: the good, the bad, or the indifferent? Diabet. (2005) 54(suppl_2):S114–S24. doi: 10.2337/diabetes.54.suppl_2.S114
129. Naegelin Y, Dingsdale H, Säuberli K, Schädelin S, Kappos L, and Barde YA. Measuring and validating the levels of brain-derived neurotrophic factor in human serum. eNeur. (2018) 5(2). doi: 10.1523/ENEURO.0419-17.2018
130. Chen H-J, Lee Y-J, Huang C-C, Lin Y-F, and Li S-T. Serum brain-derived neurotrophic factor and neurocognitive function in children with type 1 diabetes. J Formosan Med Asso. 2021120(1, Part 1):157–64.
131. Krabbe KS, Nielsen AR, Krogh-Madsen R, Plomgaard P, Rasmussen P, Erikstrup C, et al. Brain-derived neurotrophic factor (BDNF) and type 2 diabetes. Diabetologi. (2007) 50(2):431–8. doi: 10.1007/s00125-006-0537-4
132. Taniguchi H, Nirengi S, Ishihara K, and Sakane N. Association of serum fibroblast growth factor 21 with diabetic complications and insulin dose in patients with type 1 diabetes mellitus. PloS On. (2022) 17(2):e0263774. doi: 10.1371/journal.pone.0263774
133. Keuper M, Häring HU, and Staiger H. Circulating FGF21 levels in human health and metabolic disease. Exp Clin Endocrinol Diabetes. (2020) 128(11):752–70. doi: 10.1055/a-0879-2968
134. Geng L, Lam KSL, and Xu A. The therapeutic potential of FGF21 in metabolic diseases: from bench to clinic. Nat Rev Endocrino. (2020) 16(11):654–67. doi: 10.1038/s41574-020-0386-0
135. Zhang J, Shi J, Cheng Z, and Hu W. The correlation of serum musclin with diabetic nephropathy. Cytokin. (2023) 167:156211. doi: 10.1016/j.cyto.2023.156211
136. Chen WJ, Liu Y, Sui YB, Zhang B, Zhang XH, and Yin XH. Increased circulating levels of musclin in newly diagnosed type 2 diabetic patients. Diabetes Vasc Dis Re. (2017) 14(2):116–21. doi: 10.1177/1479164116675493
137. Kos K and Wilding JP. SPARC: a key player in the pathologies associated with obesity and diabetes. Nat Rev Endocrino. (2010) 6(4):225–35. doi: 10.1038/nrendo.2010.18
138. Wu D, Li L, Yang M, Liu H, and Yang G. Elevated plasma levels of SPARC in patients with newly diagnosed type 2 diabetes mellitus. Eur J Endocrino. (2011) 165(4):597–601. doi: 10.1530/EJE-11-0131
139. Piek A, Smit L, Suthahar N, Bakker SJL, de Boer RA, and Silljé HHW. The emerging plasma biomarker Dickkopf-3 (DKK3) and its association with renal and cardiovascular disease in the general population. Sci Re. (2021) 11(1):8642. doi: 10.1038/s41598-021-88107-9
140. Pedersen BK, Akerström TC, Nielsen AR, and Fischer CP. Role of myokines in exercise and metabolism. J Appl Physiol (1985). (2007) 103(3):1093–8. doi: 10.1152/japplphysiol.00080.2007
141. Leal LG, Lopes MA, and Batista ML Jr.. Physical exercise-induced myokines and muscle-adipose tissue crosstalk: a review of current knowledge and the implications for health and metabolic diseases. Front Physio. (2018) 9:1307. doi: 10.3389/fphys.2018.01307
142. Suh J and Lee YS. Myostatin inhibitors: panacea or predicament for musculoskeletal disorders? J Bone Meta. (2020) 27(3):151–65. doi: 10.11005/jbm.2020.27.3.151
143. Attie KM, Borgstein NG, Yang Y, Condon CH, Wilson DM, Pearsall AE, et al. A single ascending-dose study of muscle regulator ACE-031 in healthy volunteers. Muscle Nerve. (2013) 47(3):416–23. doi: 10.1002/mus.23539
144. Heymsfield SB, Coleman LA, Miller R, Rooks DS, Laurent D, Petricoul O, et al. Effect of bimagrumab vs placebo on body fat mass among adults with type 2 diabetes and obesity: a phase 2 randomized clinical trial. JAMA Network Ope. (2021) 4(1):e2033457–e. doi: 10.1001/jamanetworkopen.2020.33457
145. Qiao CP, Li JB, Jiang JG, Zhu XD, Wang B, Li J, et al. Myostatin propeptide gene delivery by adeno-associated virus serotype 8 vectors enhances muscle growth and ameliorates dystrophic phenotypes in mdx mice. Hum Gene The. (2008) 19(3):241–54B. doi: 10.1089/hum.2007.159
146. Jimenez V, Jambrina C, Casana E, Sacristan V, Muñoz S, Darriba S, et al. FGF21 gene therapy as treatment for obesity and insulin resistance. EMBO Mol Me. (2018) 10(8). doi: 10.15252/emmm.201708791
Keywords: myokines, diabetes, bone metabolism, osteoporosis, osteoblast lineage cells, osteoclast*, T1DM, T2DM
Citation: Ji X, Hu X, Xu T and Yang W (2025) Roles of myokines in osteoporosis under physiological and diabetic conditions. Front. Endocrinol. 16:1600218. doi: 10.3389/fendo.2025.1600218
Received: 26 March 2025; Accepted: 20 May 2025;
Published: 11 June 2025.
Edited by:
Jannis Bodden, Technical University of Munich, GermanyReviewed by:
Suma Uday, Birmingham Women’s and Children’s Hospital, United KingdomKenda Jawich, Damascus University, Syria
Copyright © 2025 Ji, Hu, Xu and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wanlei Yang, eWFuZ3dhbmxlaUB6Y211LmVkdS5jbg==