 Xinyi Lv1,2
Xinyi Lv1,2 Shengguang Ding1*
Shengguang Ding1*- 1Department of Thoracic Surgery, Affiliated Hospital 2 of Nantong University, Nantong First People’s Hospital, Nantong, China
- 2School of Medicine, Nantong University, Nantong, China
Cancer cachexia is a complex, multifactorial syndrome characterized by severe weight loss, muscle wasting, and systemic inflammation, significantly contributing to cancer-related morbidity and mortality. Signal transducer and activator of transcription 3 (STAT3) has emerged as a central mediator in the pathogenesis of this multifactorial condition. STAT3 regulates a broad range of cellular processes including inflammation, proteolysis, and mitochondrial dysfunction across multiple tissues, particularly skeletal muscle and adipose tissue. Persistent activation of STAT3 in response to tumor-derived and host-derived cytokines drives catabolic signaling cascades, disrupts anabolic pathways, and impairs energy homeostasis. Recent studies have illuminated the cross-talk between STAT3 and other signaling pathways that exacerbate cachexia-related metabolic imbalances. These findings position STAT3 not only as a critical mediator of cachexia progression but also as a promising therapeutic target. Pharmacological inhibition of STAT3 signaling has demonstrated efficacy in preclinical models, offering potential avenues for clinical intervention. This review provides a comprehensive overview of the molecular mechanisms by which STAT3 contributes to cancer cachexia and discusses emerging therapeutic strategies aimed at modulating STAT3 activity to mitigate the progression of this debilitating syndrome.
Introduction
Cancer cachexia is a multifactorial syndrome characterized by severe body weight loss, muscle atrophy, anorexia, and fatigue, which cannot be fully reversed through conventional nutritional support or pharmacological interventions (1, 2). This condition arises from a complex interplay of metabolic dysfunction and inflammatory mediators, including elevated levels of pro-inflammatory cytokines such as interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α), increased energy expenditure, and tumor-derived factors that accelerate muscle and fat wasting (3, 4). Affecting up to 80% of patients in advanced cancer stages, cachexia significantly contributes to morbidity and mortality by impairing physical function, reducing treatment tolerance, and altering drug metabolism due to progressive muscle depletion and overall physiological decline (5–7). The associated symptoms-weight loss, muscle wasting, appetite loss, fatigue, and diminished quality of life-underscore the urgent need for effective management strategies. A multidisciplinary approach integrating nutritional support, pharmacological therapies such as anti-inflammatory agents and appetite stimulants, and structured physical exercise is essential to mitigating muscle loss and improving functional outcomes (8). Given the profound impact of cancer cachexia on patient survival and treatment efficacy, advancing the understanding of its complex etiology remains critical for developing more targeted and effective therapeutic interventions.
The mammalian Signal Transducer and Activator of Transcription (STAT) family comprises STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6, all of which mediate critical intracellular signaling pathways (9). Among these, STAT3 serves as a key transcription factor regulating diverse cellular processes, including cell growth (10, 11), apoptosis (12), and immune response modulation (13, 14). Notably, STAT3 is integral to both inflammation and cancer progression, as its persistent activation is frequently observed in various malignancies, where it drives tumorigenesis by promoting cell proliferation, inhibiting apoptosis, and facilitating angiogenesis (15, 16). Beyond oncogenesis, STAT3 plays a central role in immune regulation, particularly through the modulation of pro-inflammatory cytokines and the differentiation of Th17 cells (17–19), further linking it to chronic inflammation and autoimmune disorders (20, 21). Clinically, aberrant STAT3 signaling is implicated in multiple diseases, with constitutive activation often correlating with poor prognosis in cancer due to its involvement in sustaining tumor growth and survival (17, 21). Given its broad impact, STAT3 has emerged as a prominent therapeutic target, prompting extensive research into the development of STAT3 inhibitors and RNA interference strategies aimed at mitigating its pathological activity (22–24). These ongoing efforts underscore the significance of STAT3 in both physiological and disease contexts, highlighting its potential as a target for novel therapeutic interventions.
STAT3’s involvement in cancer cachexia is primarily driven by its role in mediating inflammatory responses (25) and its contribution to muscle wasting and dysregulated fat metabolism (26, 27). Elevated levels of pro-inflammatory cytokines, such as IL-6 and TNF-α, which are commonly observed in cancer cachexia, trigger STAT3 activation (27, 28), leading to the transcription of genes that exacerbate systemic inflammation, accelerate muscle protein degradation, and disrupt lipid metabolism (1, 29, 30). Furthermore, STAT3 facilitates the interaction between cancer cells and the host immune system, amplifying cachexia’s systemic effects by fostering immunosuppression and promoting tumor progression (17, 31). This interplay between chronic inflammation, metabolic dysfunction, and immune dysregulation underscores STAT3’s pivotal role in cancer cachexia pathogenesis. Consequently, a deeper understanding of STAT3’s function in this condition is essential for developing targeted therapeutic strategies aimed at mitigating its debilitating effects, improving patient outcomes, and enhancing quality of life.
Overview of STAT3 activation pathways
STAT3 is a pivotal transcription factor that regulates diverse cellular functions, including proliferation, differentiation, survival, inflammation, and immune response (32, 33). STAT3 activation occurs primarily through cytokine and growth factor signaling, with the canonical pathway involving tyrosine phosphorylation (33). In this mechanism, cytokines from the interleukin-6 (IL-6) family (e.g., IL-6, IL-11, IL-31, leukemia inhibitory factor [LIF], oncostatin M [OSM], ciliary neurotrophic factor [CNTF], and cardiotrophin-1 [CT-1]), IL-10 family cytokines (IL-10, IL-19, IL-20, IL-22, IL-24, and IL-26), growth factors such as epidermal growth factor (EGF) and platelet-derived growth factor (PDGF), and interferons (IFNs) (9, 32, 34–36) bind to their respective receptors, triggering intracellular kinase activation. Janus kinases (JAK1, JAK2, TYK2) and receptor tyrosine kinases (EGFR, PDGFR, FGFR) phosphorylate STAT3 at Tyr705, inducing dimerization via SH2-domain interactions (32, 37, 38). The activated STAT3 dimer then translocates to the nucleus, where it binds to specific STAT-binding elements (SBEs) in gene promoters, regulating genes associated with survival (Bcl-xL), proliferation (Cyclin D1, c-Myc), inflammation (IL-6, COX2), metastasis (MMPs), angiogenesis (VEGF), and immune evasion (14, 39–43). This tightly regulated signaling cascade underscores STAT3’s pivotal role in cellular homeostasis and disease pathology.
Beyond the canonical pathway, STAT3 activation also occurs through non-canonical mechanisms involving alternative post-translational modifications and extranuclear functions (44, 45). One such mechanism is serine phosphorylation, in which kinases like cyclin-dependent kinase 5 (CDK5) and EGFR phosphorylate STAT3 at Ser727, thereby enhancing its transcriptional activity and modulating mitochondrial function (45, 46). Additionally, a mitochondrial variant of STAT3 (mitoSTAT3) localizes to mitochondria, interacting with electron transport chain (ETC) complexes I and II to regulate ATP production and reactive oxygen species (ROS) generation (45). This function facilitates metabolic adaptation and promotes cancer cell survival (44, 47, 48). Moreover, non-phosphorylated STAT3 (npSTAT3) is implicated in cytoplasmic processes such as microtubule stabilization and protein degradation, demonstrating its diverse functional repertoire beyond transcriptional regulation (45, 49), as detailed in Figure 1A.
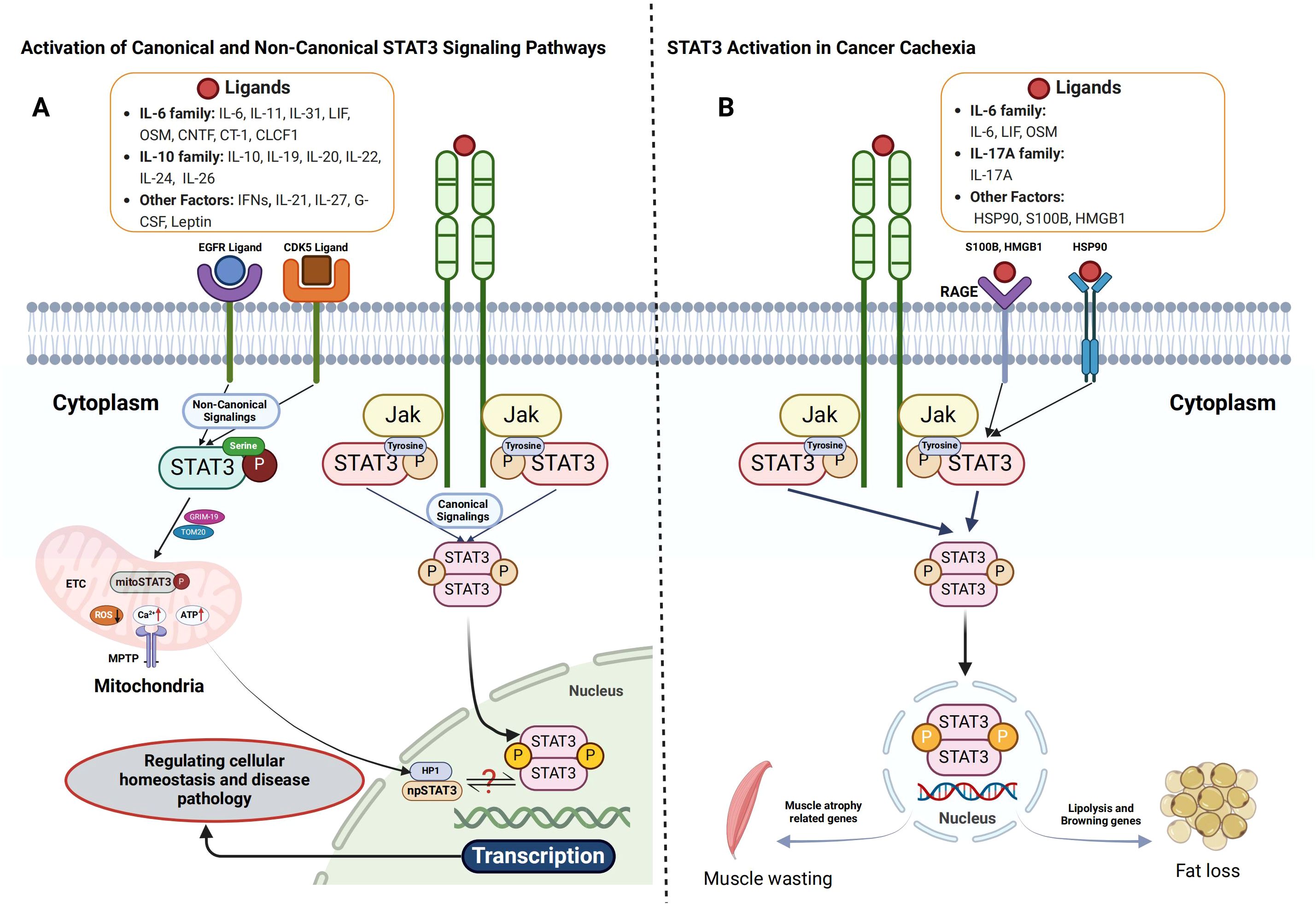
Figure 1. Activation of STAT3 signaling pathways and their role in cancer cachexia. (A) Both canonical and non-canonical STAT3 signaling pathways are crucial for cellular signaling. In the canonical pathway, cytokines such as IL-6, LIF, and OSM bind to their receptors, inducing receptor dimerization and the recruitment of JAKs. This interaction results in receptor phosphorylation at specific tyrosine residues, creating docking sites for STAT3. STAT3 is then phosphorylated by JAKs, dissociates from the receptor, forms homodimers or heterodimers, and translocates to the nucleus to regulate gene transcription. In contrast, the non-canonical STAT3 pathway involves mitochondrial STAT3 (mtSTAT3), unphosphorylated STAT3, and serine 727-phosphorylated STAT3 (p-STAT3 Ser727), alone or in combination with tyrosine 705-phosphorylated STAT3 (p-STAT3 Tyr705). These variants play a role in mitochondrial function, emphasizing STAT3’s involvement beyond transcriptional regulation. (B) In cancer cachexia, STAT3 activation occurs through both receptor- and non-receptor-mediated mechanisms. Pro-inflammatory cytokines or growth factors bind to cell surface receptors, leading to tyrosine phosphorylation, which facilitates the recruitment of JAKs or direct binding of STAT3 via its Src homology 2 (SH2) domain. Phosphorylated STAT3 dimerizes and enters the nucleus, promoting the transcription of genes involved in catabolic processes, driving tissue degradation and metabolic imbalance.
STAT3 activation in cancer cachexia
Cancer cachexia, a debilitating syndrome characterized by muscle and adipose tissue wasting, is driven by elevated levels of pro-inflammatory cytokines, including IL-6-type cytokines (IL-6, LIF, OSM) (15, 25, 50), IL-17A (51), tumor necrosis factor-alpha (TNF-α), and interleukin-1 beta (IL-1β) (52, 53). Among these, IL-6 plays a central role in activating the JAK/STAT3 signaling pathway, a key mediator of systemic inflammation (54–56). Upon IL-6 binding to its receptor, JAK kinases phosphorylate STAT3, leading to dimerization and nuclear translocation (43). Within the nucleus, phosphorylated STAT3 regulates genes associated with inflammation, muscle protein degradation, and adipose tissue loss, exacerbating the imbalance between muscle synthesis and degradation that characterizes cachexia (54, 56–58). Correspondingly, cytokines such as OSM (50), IL-17A (51), and LIF (59) exploit STAT3 signaling to mediate muscle atrophy in cachectic models.
In addition to cytokine-driven activation, tumor-derived cachexia-inducing factors [such as IL-6, LIF, and G-CSF (59, 60)] and immune cells within the tumor microenvironment secrete active molecules that further enhance STAT3 activation, thereby promoting muscle atrophy. Among these factors, heat shock protein 90 (HSP90), receptor for advanced glycation end-products (RAGE) ligands, and S100B have emerged as novel contributors to muscle degradation. These factors induce muscle wasting through the p38 mitogen-activated protein kinase (MAPK)/myogenin axis (61) and STAT3 signaling (61, 62). Furthermore, receptor tyrosine kinases (RTKs), such as EGFR (63, 64), and metabolic regulators like leptin (65, 66) activate STAT3 via their respective homologous receptors, further implicating STAT3 in cachexia pathology. This intricate signaling network highlights the potential of STAT3 as a therapeutic target in cachexia management (Figure 1B). Future research focusing on STAT3 modulation could provide novel insights into intervention strategies, ultimately improving patient outcomes and quality of life.
Mechanistic insights into STAT3 in cancer cachexia
As previously mentioned, pro-inflammatory cytokines such as IL-6 activate STAT3, which subsequently induces the expression of atrogenes, including atrogin-1 and MuRF1. These atrogenes play a key role in promoting muscle proteolysis and impeding muscle regeneration by inhibiting satellite cell differentiation. In addition to its effects on muscle tissue, STAT3 also contributes to adipose tissue loss by upregulating lipolysis and suppressing lipogenesis. Furthermore, STAT3 facilitates the browning of white adipose tissue, leading to increased energy expenditure. This multifaceted role of STAT3 encompasses the regulation of protein and lipid metabolism, appetite control, and tumor immune responses. In the following sections, we will explore the key functions of STAT3 (Figure 2) and its cross-talk with other signaling pathways involved in these processes (Figure 3).
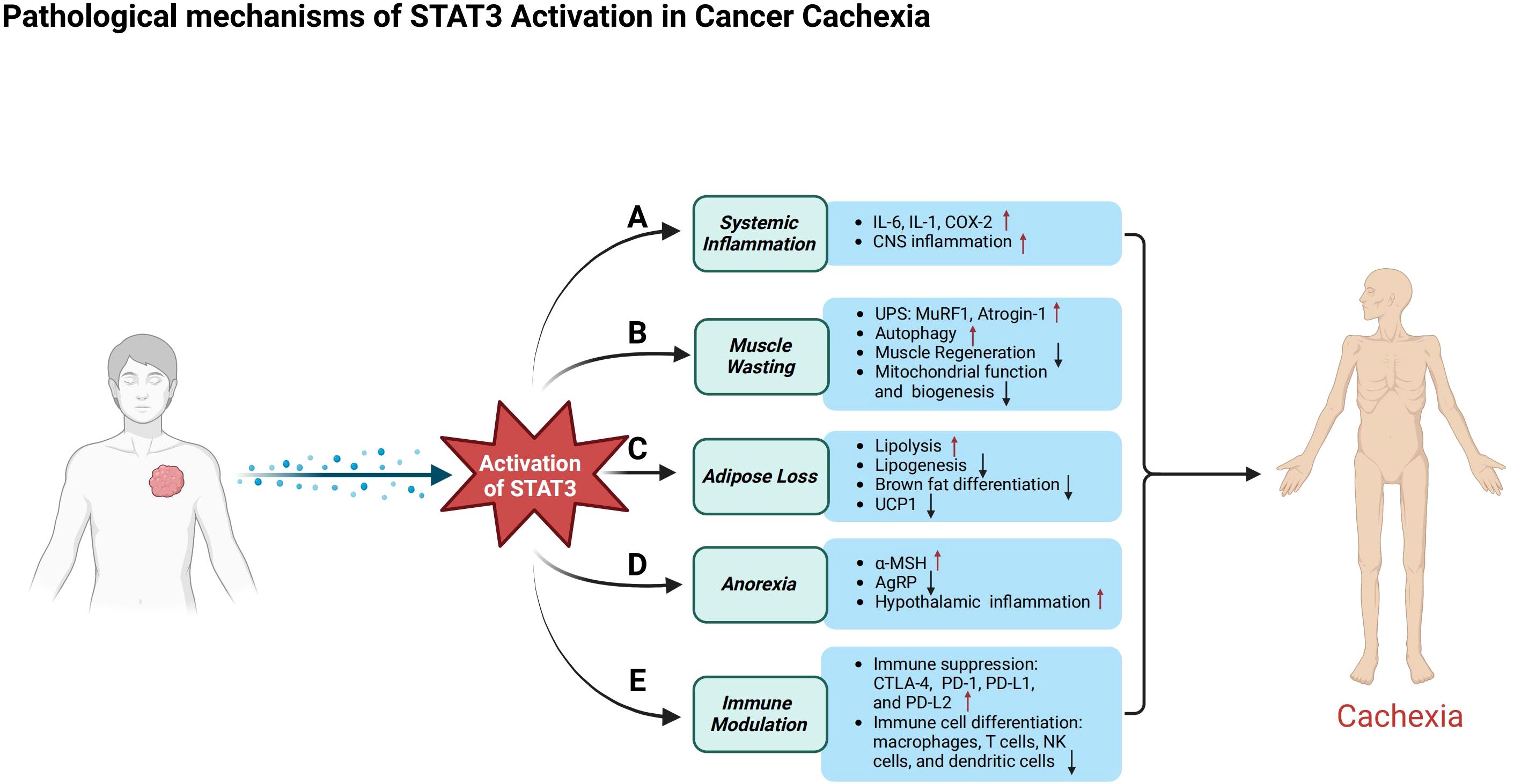
Figure 2. Pathological mechanisms of STAT3 in cancer cachexia. This figure illustrates the diverse roles of STAT3 activation in cancer patients, emphasizing its contribution to systemic inflammation, muscle atrophy, metabolic dysfunction, appetite regulation, and immune suppression. (A) In systemic inflammation, activated STAT3 triggers the release of pro-inflammatory cytokines such as IL-6, IL-1, and COX-2, exacerbates inflammation in the central nervous system, and contributes to anorexia. (B) In skeletal muscle, STAT3 activation induces atrophy by stimulating the ubiquitin–proteasome system and autophagy-related pathways, while impairing muscle regeneration by disrupting the differentiation, proliferation, and self-renewal of muscle satellite cells. Additionally, it compromises mitochondrial function. (C) In adipose tissue, activated STAT3 promotes lipolysis and metabolic dysregulation, inhibits lipogenesis, and suppresses brown adipose tissue differentiation and the expression of uncoupling protein 1 (UCP1). (D) In appetite regulation, STAT3 enhances the activity of pro-opiomelanocortin (POMC) neurons, increasing α-melanocyte-stimulating hormone (α-MSH) production to promote satiety, while simultaneously suppressing agouti-related peptide (AgRP) neurons that normally stimulate hunger, leading to reduced food intake and body weight loss. (E) STAT3 contributes to immune suppression by upregulating immune checkpoint molecules such as CTLA-4, PD-1, PD-L1, and PD-L2. Its activation alters the differentiation and function of immune cells, including macrophages, T cells, natural killer (NK) cells, and dendritic cells, reshaping the immune microenvironment and accelerating tumor progression.
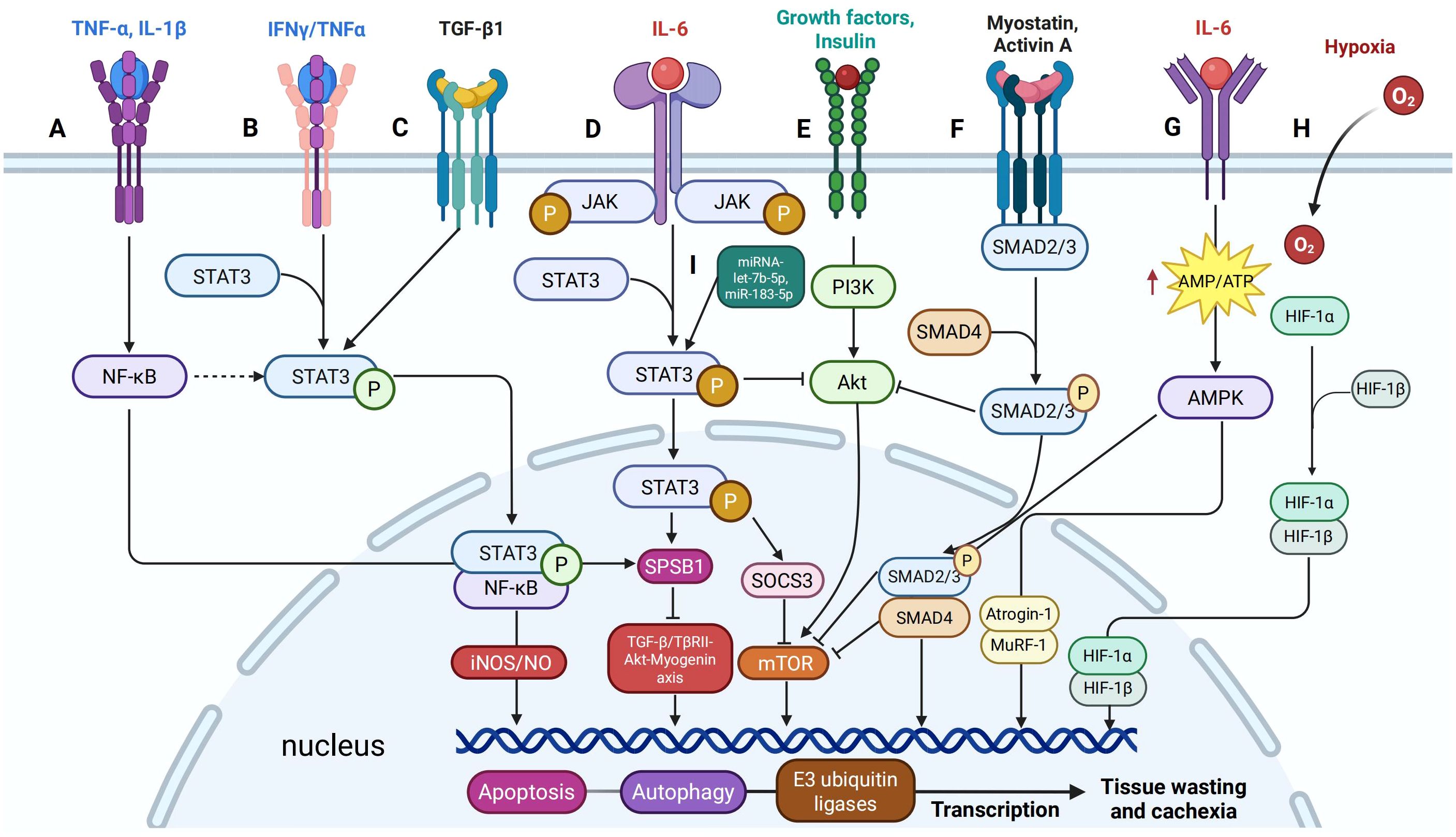
Figure 3. Signaling cross-talk between STAT3 and other pathways in cancer cachexia. (A) IFNγ/TNFα signaling induces phosphorylation of STAT3 at Y705, promoting its interaction with NF-κB to form a nuclear complex that activates the iNOS/NO pathway, a critical mediator of muscle loss. (B) TNF-α and IL-1β upregulate SOCS-box protein 1 (SPSB1) expression through NF-κB signaling. (C) IL-6 enhances SPSB1 expression via the glycoprotein 130/JAK2/STAT3 pathway, while TGF-β activates STAT3 in a SMAD-dependent manner. (D) TGFβ1 induces Tyr705 phosphorylation of STAT3 in C2C12 cells. (E) STAT3 signaling interacts with the PI3K/Akt/mTOR pathway by suppressing p-Akt activity. (F) Myostatin and Activin A activate SMAD2/3 signaling similarly and inhibit the insulin/IGF-1/Akt/mTOR pathway, reducing muscle mass and function. (G) IL-6 increases AMPK activity in C2C12 cells and mouse cancer cachexia models, and AMPK activation enhances myofibrillar protein degradation. (H) HIF-1α shifts muscle metabolism by upregulating glycolysis and downregulating oxidative phosphorylation. (I) The role of miRNAs in regulating STAT3 activation and enhancing its cachectic effects.
STAT3 and systemic inflammation
Chronic inflammation, sustained by elevated levels of IL-6 and other pro-inflammatory cytokines, perpetuates persistent activation of STAT3, establishing a self-reinforcing cycle that drives the progression of cancer cachexia (14, 15, 29, 30). STAT3 activation enhances the transcription of inflammatory mediators such as IL-6, IL-1β, and COX-2, further amplifying cytokine production and systemic inflammation (14, 17, 30, 67). This pro-inflammatory feedback loop is compounded by multiple cytokines, including IL-6, leukemia inhibitory factor (LIF), and oncostatin M (OSM) (50, 56, 58, 68), as well as IFN-γ, TNF-α (69), and IL-17A (51), which converge on the STAT3 pathway, ensuring its sustained activation and contributing to metabolic dysregulation. While acute exposure to IL-6 and LIF can transiently promote protein synthesis via STAT3 and the Akt-mTORC1 pathway, it also induces SOCS3 expression to limit cytokine signaling (70). However, in chronic inflammatory states, prolonged STAT3 activity overrides this regulatory feedback, promoting muscle catabolism and systemic metabolic imbalance (68). Importantly, the influence of STAT3 extends beyond peripheral tissues. Neuroinflammatory signaling impacts the hypothalamus, disrupting appetite regulation by impairing orexigenic pathways and exacerbating anorexia, one of the cardinal features of cancer cachexia (25, 71, 72). This highlights the multifactorial nature of STAT3’s role, linking peripheral inflammation to central behavioral and metabolic dysfunction.
STAT3 also operates in concert with other inflammatory signaling cascades that further disrupt metabolic homeostasis. For instance, IFN-γ and TNF-α promote STAT3 phosphorylation at Y705, facilitating its interaction with NF-κB and forming a nuclear complex that induces the iNOS/NO pathway, a key contributor to inflammation-induced muscle wasting (69) (Figure 3A). Simultaneously, TNF-α and IL-1β activate NF-κB-dependent transcription of SOCS-box protein 1 (SPSB1) (Figure 3B), while IL-6 induces SPSB1 via the gp130/JAK2/STAT3 axis, collectively impairing myogenic differentiation and enhancing proteolytic activity through the ubiquitin–proteasome system (73, 74) (Figure 3D). Although NF-κB can inhibit apoptosis in some contexts, its chronic activation sustains oxidative stress and promotes muscle degradation (75). Furthermore, STAT3 impairs anabolic signaling by upregulating SOCS3, which suppresses the PI3K/Akt/mTOR pathway (Figure 3E). This inhibition reduces protein synthesis and exacerbates proteolysis, intensifying muscle atrophy (30, 76). These interactions position STAT3 as a central mediator linking inflammatory signaling to disrupted muscle metabolism.
In parallel, circulating cytokines influence AMP-activated protein kinase (AMPK), a key regulator of energy homeostasis. IL-6 increases AMPK activity in C2C12 cells and murine cachexia models, contributing to catabolic signaling (77, 78) (Figure 3G). In contrast, exercise training during early cachexia attenuates AMPK activation, restoring mTOR signaling and anabolic balance (78). Conversely, TNF-α suppresses AMPK via TNFR1 engagement, exacerbating metabolic dysfunction (79). Notably, chronic AMPK activation enhances protein degradation through both the autophagy–lysosome pathway and the ubiquitin–proteasome system (80–82). Collectively, these mechanisms form an intricate inflammatory-metabolic axis that drives the relentless tissue wasting characteristic of cancer cachexia.
STAT3 in muscle wasting
Muscle wasting, a hallmark of cancer cachexia, is closely associated with aberrant activation of the STAT3 signaling pathway, which exerts multifaceted effects on muscle cell physiology and pathology (83, 84). STAT3 promotes muscle atrophy by mediating crosstalk between C/EBPδ and myostatin pathways, thereby repressing muscle growth and enhancing catabolic signaling (83). This leads to the upregulation of muscle-specific E3 ubiquitin ligases, including MuRF1 and Atrogin-1, which are central components of the ubiquitin–proteasome system responsible for proteolytic degradation of myofibrillar proteins (76, 85). In addition to proteasomal degradation, STAT3 contributes to autophagy-mediated muscle catabolism. Both nuclear and cytoplasmic STAT3 modulate autophagy-related processes, and its inhibition has been shown to restore autophagic flux by upregulating autophagy-related genes and altering eukaryotic initiation factor 2α (eIF2α) phosphorylation (86). This suggests that targeting STAT3 may be a viable strategy to rebalance protein turnover in cachectic muscle. Moreover, excessive STAT3 activity disrupts skeletal muscle regeneration by impairing satellite cell differentiation, proliferation, and self-renewal, ultimately leading to regenerative failure, premature differentiation, and age-associated muscle decline (86–88). Collectively, these mechanisms highlight STAT3’s central role in promoting muscle atrophy through enhanced proteolysis, dysregulated autophagy, and impaired regenerative capacity, key drivers of cancer cachexia progression.
Mitochondrial dysfunction further amplifies muscle degeneration and is increasingly recognized as a critical feature of cachexia-associated muscle wasting (89). STAT3 impairs mitochondrial biogenesis and function, leading to energetic deficits and contributing to muscle fatigue and weakness (88). Pharmacological inhibition of the JAK/STAT3 pathway has been shown to reverse myotube atrophy and restore mitochondrial protein levels in murine models of colorectal cancer cachexia (90) Additionally, IL-6 signaling through the gp130 receptor regulates mitochondrial quality control in skeletal muscle. During Lewis lung carcinoma (LLC)-induced cachexia, gp130-STAT3 signaling activates p38 MAPK, which in turn stimulates FOXO3 and Atrogin-1 expression, promoting muscle degradation (91, 92). STAT3 also exhibits immunomodulatory properties by engaging the PI3K/Akt axis, indicating potential crosstalk between inflammatory and metabolic pathways in cachectic muscle (93). Together, these findings highlight STAT3’s critical role in muscle wasting by integrating mitochondrial dysfunction, protein degradation, and metabolic disturbances, thereby reinforcing its potential as a therapeutic target for mitigating cancer cachexia.
The interplay between STAT3 and other muscle-wasting signaling pathways further exacerbates muscle atrophy. STAT3 upregulates myostatin, a potent inhibitor of muscle growth, which activates Smad2/3 signaling to suppress the Akt/mTOR axis and reinforce proteasomal degradation via MuRF1 and Atrogin-1 (26, 76, 83, 84, 94) (Figure 3F). TGF-β, another activator of Smad2/3, synergizes with STAT3 to intensify muscle wasting and fibrosis, in part through IL-6 amplification (95, 96). Notably, STAT3 activity correlates with cachexia severity in mouse models overexpressing TGF-β1 in skeletal muscle, further emphasizing this pathological axis (97) (Figure 3C). Synchronously, hypoxia-inducible factor 1α (HIF-1α), which is elevated in tumor-bearing conditions (98, 99), collaborates with STAT3 to shift muscle metabolism from oxidative phosphorylation to glycolysis (Figure 3H). This metabolic reprogramming impairs mitochondrial ATP production and contributes to muscle fatigue. Intriguingly, tumor-derived exosomal miR-183-5p activates both HIF-1α and STAT3, establishing a link between hypoxic signaling and enhanced proteolysis (100). The integration of these inflammatory, catabolic, and metabolic pathways underscores STAT3’s pivotal role in the rapid loss of muscle mass and function characteristic of advanced cancer cachexia.
STAT3 in adipose loss
Cancer cachexia is marked not only by progressive skeletal muscle wasting but also by profound adipose tissue atrophy. Emerging evidence identifies STAT3 as a central mediator of these metabolic disturbances. In the C26 mouse model of cancer cachexia, elevated levels of STAT3 and its activated form (pY705-STAT3) have been observed in white adipose tissue, implicating this pathway in adipocyte dysfunction and fat loss (101). STAT3 activation drives lipolysis and systemic metabolic imbalance by promoting the breakdown of triglycerides and impairing lipid storage mechanisms, thereby exacerbating energy depletion characteristic of cachexia. IL-6 family cytokines, particularly leukemia inhibitory factor (LIF), initiate adipocyte lipolysis through a STAT3-dependent mechanism. Specifically, LIF-induced STAT3 activation upregulates adipose triglyceride lipase (ATGL) and its coactivator CGI-58, facilitating triglyceride hydrolysis (57). Concurrently, STAT3 suppresses peroxisome proliferator-activated receptor alpha (PPARα), a key transcription factor involved in lipid uptake and storage, thereby reducing lipogenesis and worsening adipose tissue dysfunction (59). Inhibiting the JAK/STAT3 pathway in murine models significantly attenuates lipolysis and prevents adipose tissue wasting, highlighting the therapeutic potential of STAT3 blockade in cancer cachexia (57). Moreover, STAT3 activation correlates with elevated lactate dehydrogenase (LDH) levels and diminished adiponectin production in adipocytes, further contributing to systemic metabolic disruption (102). Beyond white adipose tissue, STAT3 also regulates brown adipose tissue (BAT) homeostasis. Constitutive activation of STAT3 enhances brown fat thermogenesis and energy expenditure, as demonstrated by the reversal of obesity in TYK2 knockout mice through increased BAT differentiation (103). Conversely, STAT3 inhibition boosts the expression of uncoupling protein 1 (UCP1) and improves mitochondrial function in brown adipocytes, suggesting its dual role in modulating both lipolytic activity and thermogenic capacity (104).
STAT3’s regulatory role in adipose tissue wasting is further compounded by its interaction with the AMPK and Wnt/β-catenin pathways. While AMPK activation generally supports energy homeostasis by promoting fatty acid oxidation, its chronic stimulation in cachexia leads to destabilization of the AMPK complex in adipocytes, aggravating lipid depletion and energy imbalance (105, 106). Notably, systemic delivery of an AMPK-stabilizing peptide in tumor-bearing mice preserved adipose tissue mass and mitigated body weight loss without affecting tumor growth, underscoring the therapeutic relevance of modulating AMPK in concert with STAT3 signaling (106). Simultaneously, Wnt/β-catenin signaling, traditionally known for its role in adipogenesis and adipocyte differentiation, may intersect with STAT3 to regulate lipid turnover and adipocyte browning (107). Although this interaction remains under active investigation, preliminary findings suggest that Wnt/β-catenin-STAT3 crosstalk may influence adipocyte plasticity and energy metabolism, presenting another potential target for intervention. Collectively, these findings underscore STAT3’s multifaceted role in mediating adipose tissue atrophy in cancer cachexia through its regulation of lipolytic enzymes, suppression of lipogenesis, impairment of mitochondrial function, and modulation of key metabolic pathways. Targeting STAT3 and its interacting partners represents a promising strategy to alleviate the systemic metabolic derangements that characterize this debilitating syndrome.
STAT3 and anorexia
Cancer cachexia-associated anorexia represents a critical clinical challenge, affecting up to 60% of patients with advanced malignancies (108). This condition significantly diminishes quality of life, compromises treatment efficacy, and correlates with poor survival outcomes (109, 110). Central to the regulation of appetite in this context is the transcription factor signal transducer and activator of transcription 3 (STAT3), which integrates peripheral metabolic and inflammatory signals in the hypothalamus. Leptin, a hormone secreted by adipose tissue, regulates energy homeostasis by binding to the long isoform of the leptin receptor (LepRb) expressed in the arcuate nucleus (ARC) of the hypothalamus. This interaction activates the JAK2-STAT3 signaling cascade, which modulates feeding behavior by acting on two distinct neuronal populations (111, 112). STAT3 enhances the activity of pro-opiomelanocortin (POMC) neurons, promoting satiety through the production of α-melanocyte-stimulating hormone (α-MSH) (113, 114), while concurrently inhibiting agouti-related peptide (AgRP) neurons that stimulate appetite (115). As part of a negative feedback mechanism, STAT3 upregulates suppressor of cytokine signaling 3 (SOCS3), which inhibits further JAK2-STAT3 signaling and contributes to leptin resistance (116). Functional studies underscore the physiological relevance of this pathway. Mice lacking STAT3 in POMC neurons develop obesity due to impaired satiety signaling, whereas constitutive STAT3 activation in AgRP neurons suppresses food intake (115). These findings illustrate the critical role of STAT3 in maintaining energy balance and suggest that its dysregulation can lead to pathological anorexia or hyperphagia depending on context.
Beyond leptin signaling, STAT3 is also activated by pro-inflammatory cytokines such as TNF-α (117) and CNTF (118), both of which suppress food intake through hypothalamic STAT3-dependent mechanisms (119). Moreover, STAT3 may mediate crosstalk between leptin and insulin signaling in the hypothalamus, though this interaction remains incompletely understood (120). Chronic overexpression of SOCS3, a direct STAT3 target, has been implicated in leptin and insulin resistance (113, 121), highlighting the importance of tightly regulated STAT3 activity for metabolic homeostasis. Leptin resistance is particularly relevant in cancer cachexia, where elevated leptin levels fail to restore appetite due to SOCS3-mediated inhibition of LepRb signaling (122, 123). This dysregulation is exacerbated in obesity-associated cancers, where both systemic inflammation and leptin resistance contribute to anorexia. Concurrently, pro-inflammatory cytokines activate the NF-κB pathway, which acts synergistically with STAT3 to enhance transcription of anorexigenic mediators and promote hypothalamic inflammation (124, 125).
In addition to inflammatory and hormonal signaling, STAT3 modulates nutrient-sensing pathways. It has been shown to inhibit AMP-activated protein kinase (AMPK), a key promoter of hunger, and potentially stimulate mechanistic target of rapamycin (mTOR) signaling, both of which suppress appetite during inflammatory states (126, 127). Moreover, recent studies implicate STAT3 in the anorexigenic GDF15-GFRAL signaling axis. Growth differentiation factor 15 (GDF15), elevated in several cancer types, signals through its receptor GFRAL in the area postrema and nucleus of the solitary tract (NTS), regions involved in nausea and appetite regulation. STAT3 activation may converge with GDF15 signaling in these brainstem nuclei, amplifying the anorectic response (128, 129). Altogether, these findings position STAT3 as a central node in the neuroimmune circuitry underlying cancer cachexia-associated anorexia. Through its integration of leptin, cytokine, and nutrient-sensing pathways, STAT3 contributes to appetite suppression and energy imbalance. Targeting STAT3 or its regulatory partners, such as SOCS3, may offer a promising therapeutic strategy for alleviating anorexia and improving outcomes in cachectic patients.
STAT3 in immune modulation
Dysregulation of immune checkpoints is a well-established mechanism by which many cancers evade immune surveillance, thereby facilitating tumor progression (17). A growing body of evidence implicates STAT3 as a central regulator of this immunosuppressive network. Elevated STAT3 activity, frequently driven by pro-inflammatory cytokines such as IL-6 (18, 130), promotes immune escape through the transcriptional upregulation of key immune checkpoint molecules, including CTLA-4 (17), programmed cell death protein 1 (PD-1) (131), and its ligands PD-L1 and PD-L2 (132, 133). This STAT3-mediated enhancement of immune checkpoint expression contributes to tumor immune evasion and may help explain the limited efficacy of immune checkpoint blockade therapy in end-stage cancer patients, particularly those suffering from cancer cachexia (134, 135). Beyond checkpoint regulation, STAT3 profoundly alters T cell dynamics within the tumor microenvironment (TME). It promotes the differentiation of CD4+ T cells into regulatory T cells (Tregs), which facilitate immune tolerance, while simultaneously impairing the generation and cytotoxic function of CD8+ cytotoxic T lymphocytes (CTLs), the primary effectors of anti-tumor immunity (136). At the same time, STAT3 activation enhances the differentiation, expansion, and immunosuppressive activity of myeloid-derived suppressor cells (MDSCs) (137), further dampening adaptive immune responses and contributing to systemic inflammation. Emerging evidence highlights that STAT3 hyperactivation occurs not only in tumor cells but also in immune cells infiltrating the tumor microenvironment (TME) (138), affecting a broad spectrum of immune populations, including macrophages (139), T cells (140, 141), NK cells (142), and dendritic cells (143). This widespread STAT3 activity reshapes the immune landscape to favor tumor progression and contributes to the development of cancer cachexia.
In cachexia-prone malignancies, the TME is enriched with pro-inflammatory cytokines such as IL-6, TNF-α, IL-1β, and IFN-γ, secreted by both tumor and immune cells including macrophages, MDSCs, and T cells (4, 144). These cytokines promote chronic systemic inflammation and metabolic dysfunction, driving catabolic processes in skeletal muscle and adipose tissue. Within this inflammatory milieu, STAT3 plays a key role in skewing macrophage polarization toward the M2 phenotype (145, 146), which, although traditionally viewed as anti-inflammatory (147), contributes to tumor immune evasion by producing immunosuppressive cytokines such as IL-10 and TGF-β (148, 149). These cytokines, in turn, sustain a suppressive TME and reinforce STAT3 signaling, forming a self-perpetuating loop that fuels both tumor growth and cachexia-associated wasting (150). Importantly, preclinical studies have demonstrated that blockade of the IL-6/STAT3 axis restores anti-tumor immune responses by relieving T cell suppression and enhancing adaptive immunity (151). These findings underscore the pivotal role of STAT3 in orchestrating immune evasion and systemic catabolism, positioning it as a promising therapeutic target for addressing both cancer progression and the immunometabolic dysfunctions characteristic of cancer cachexia.
Regulatory networks and post-transcriptional modulation: role of microRNAs
MicroRNAs (miRNAs) serve as key post-transcriptional regulators of STAT3 and its associated pathways. For instance, miR-203 targets SOCS3, indirectly enhancing STAT3 signaling, while miR-183-5p simultaneously activates Smad3 and STAT3, promoting muscle degradation via upregulation of Atrogin-1 and MuRF1 (100, 152). Additionally, pancreatic cancer-derived exosomal miRNA let-7b-5p activates STAT3/FOXO1 signaling, exacerbating insulin resistance and muscle wasting (153). These findings underscore the critical role of miRNAs in modulating STAT3 activity and amplifying its cachectic effects (Figure 3I).
Therapeutic perspectives
Given its central role in orchestrating inflammation, metabolic dysregulation, and tissue catabolism, STAT3 represents a highly promising but complex therapeutic target in cancer cachexia. This section explores current and emerging strategies aimed at modulating STAT3 signaling through cytokine inhibition, pharmacological agents, physical exercise, nutritional modulation, and integrated multi-targeted approaches (Figure 4).
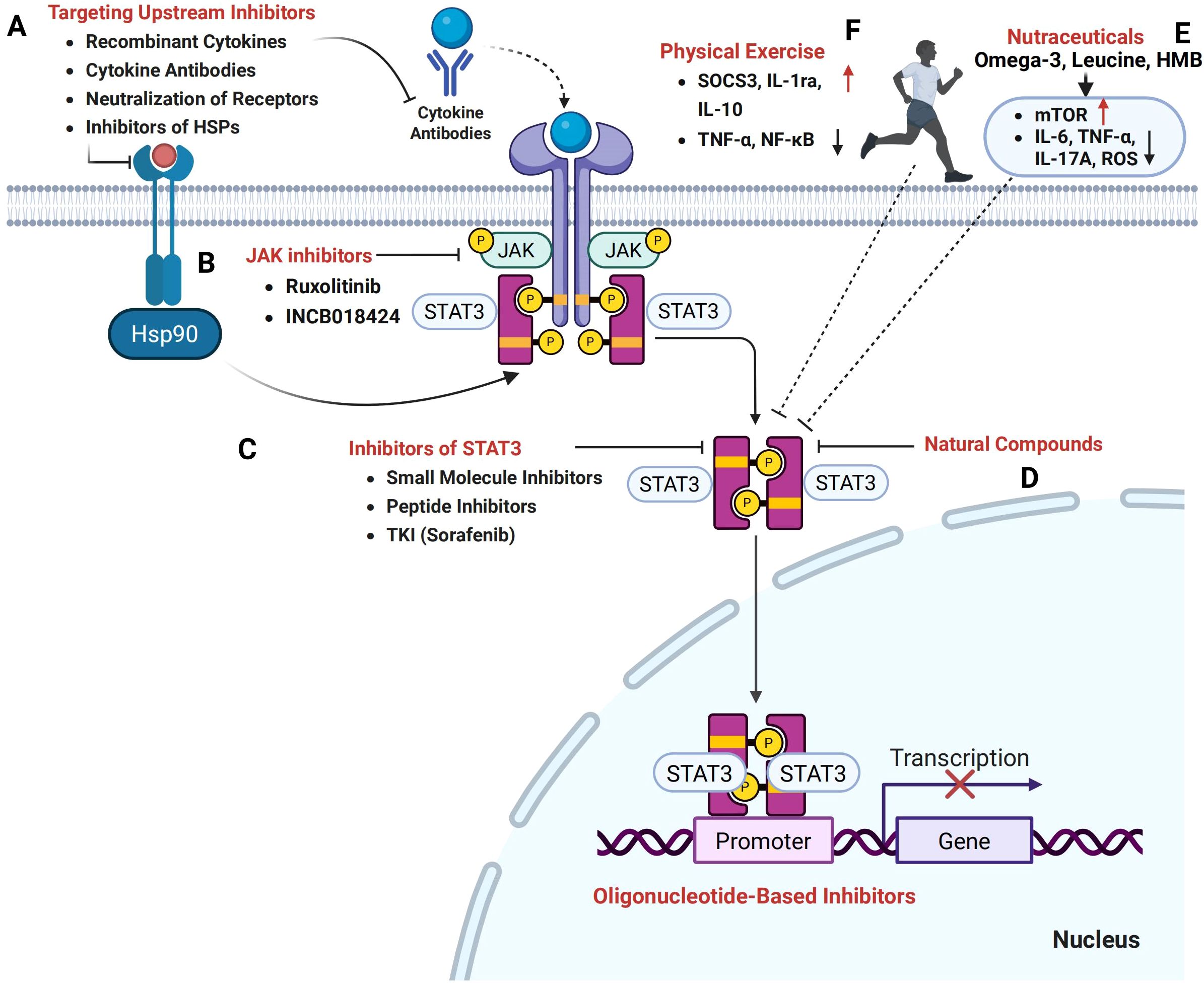
Figure 4. Therapeutic potential of targeting the STAT3 signaling pathway in cancer cachexia. (A) Therapeutic strategies aimed at upstream inhibitors, including recombinant cytokines, cytokine antibodies, receptor neutralization, and inhibitors of heat shock proteins (HSPs), hold promise in modulating STAT3 activation and mitigating cachexia. (B) Janus kinase (JAK) inhibitors, such as Ruxolitinib and INCB018424, effectively inhibit the JAK/STAT3 pathway. (C) Direct STAT3 inhibition can be achieved through small molecule inhibitors, peptide inhibitors, and tyrosine kinase inhibitors (TKIs) such as Sorafenib, which prevent STAT3 dimerization and nuclear translocation. (D) Natural compounds, including plant-derived phytochemicals, also target the STAT3 signaling axis. (E) Nutritional and metabolic modulators, particularly nutraceuticals, have the potential to influence STAT3-mediated pathways and restore the balance between protein synthesis and degradation. (F) Furthermore, exercise interventions targeting STAT3 signaling offer a promising strategy to counteract the muscle wasting and metabolic imbalance characteristic of cancer cachexia.
Upstream cytokine blockade
The JAK-STAT3 pathway, predominantly activated by cytokines such as IL-6, IFNγ, LIF, and OSM (69, 154, 155), plays a central role in cancer cachexia pathogenesis. Among these, IL-6 is a major activator of STAT3, with neutralization strategies showing substantial therapeutic promise. Preclinical studies using anti-IL-6 or anti-IL-6 receptor (IL-6R) antibodies in tumor-bearing mice demonstrate attenuation of muscle atrophy, adipose tissue loss, and systemic inflammation (27, 41, 145–147). Clinically, tocilizumab, a humanized monoclonal antibody targeting IL-6R, has improved symptoms and prognosis in cachectic patients, with Phase II trials in lung cancer confirming its ability to alleviate anorexia and weight loss (149, 156).
Other IL-6 family cytokines, including oncostatin M (OSM) and leukemia inhibitory factor (LIF), also contribute to cachexia through STAT3 activation. OSM induces myotube atrophy, while muscle-specific deletion of the OSM receptor (OSMR) preserves muscle mass in preclinical models (32). LIF promotes hepatic metabolic dysfunction via STAT3, and liver-specific deletion of the LIF receptor ameliorates cachexia-related lipid abnormalities (62). Neutralizing antibodies against OSM and LIF have shown efficacy in preclinical studies, making them viable adjuncts or alternatives to IL-6 blockade (150). Further, inhibition of IL-17A and small-molecule blockade of STAT3 phosphorylation (e.g., AG490) reduce cachexia severity (33), reinforcing the need for multi-cytokine targeting strategies (Figure 4A). Such combinatorial approaches may be particularly relevant for malignancies with high cytokine redundancy, such as pancreatic ductal adenocarcinoma (PDAC).
JAK inhibitors
Inhibiting Janus kinases (JAKs) upstream of STAT3 has emerged as another promising therapeutic strategy. Agents such as ruxolitinib and INCB018424 have shown preclinical efficacy in mitigating cachexia symptoms by suppressing STAT3 phosphorylation, normalizing cytokine/adipokine profiles, and reducing tissue wasting (54, 57, 157, 158). Notably, JAK inhibition also prolongs survival in murine models of cancer cachexia, possibly by dampening systemic inflammation and restoring metabolic homeostasis (159). However, JAK inhibitors may affect multiple downstream targets beyond STAT3, necessitating strategies to enhance tissue specificity and minimize immunosuppressive side effects (Figure 4B).
Pharmacological inhibitors of STAT3
Direct inhibition of STAT3 presents a promising therapeutic strategy, offering greater specificity and efficacy in preclinical models of cancer cachexia. Small-molecule inhibitors such as STATTIC, C188-9, and napabucasin act by disrupting STAT3 dimerization, DNA binding, or phosphorylation (160). Notably, C188–9 significantly reduced phosphorylated STAT3 (pY705-STAT3) levels in skeletal muscle from both chronic kidney disease and C26 tumor-bearing mice, accompanied by preservation of muscle mass, grip strength, and myofiber size (25, 161, 162). These inhibitors also ameliorate muscle wasting by restoring mitochondrial function, reducing proteolysis, and improving systemic metabolic parameters (15, 163–166). In addition to intrinsic STAT3 activation, tumor-associated macrophages (TAMs) exacerbate muscle degeneration by secreting pro-inflammatory cytokines, particularly IL-1α and IL-6, which activate STAT3 signaling in muscle fibers. Pharmacological blockade of macrophage-derived cytokines or direct inhibition of STAT3 in myofibers significantly attenuates muscle atrophy in pancreatic cancer models (139). Furthermore, targeting the HSP90/STAT3/FOXO1 signaling axis using inhibitors such as 17-DMAG and PU-H71 has been shown to suppress atrogene expression and mitigate muscle wasting (42). Moreover, the multi-kinase inhibitor sorafenib has demonstrated the ability to modulate STAT3 activity, preventing the accumulation of atrogin-1 and Pax7 in skeletal muscle, thereby improving functional capacity and reducing fatigue in tumor-bearing animals (167). In total, these findings underscore the therapeutic potential of STAT3-targeted interventions in cancer cachexia (Figure 4C).
Despite advances, several barriers hinder effective STAT3-targeted therapy. To enhance efficacy and minimize systemic toxicity, the development of tissue-specific delivery systems, such as nanoparticles or antisense oligonucleotides, is essential. First, tissue-specific actions of STAT3 necessitate precision in therapeutic targeting to avoid adverse effects (25). Second, off-target toxicity and resistance mechanisms limit the utility of current inhibitors (15, 168, 169). Development of highly selective, delivery-optimized agents (e.g., nanoparticle-based siRNAs or antisense oligonucleotides) is urgently needed. Additionally, identifying reliable biomarkers for STAT3 activation and cachexia progression remains a challenge. Personalized therapeutic approaches based on patient-specific cytokine profiles, tumor types, and metabolic status will be crucial for optimizing outcomes. Future directions include exploring combination therapies and integrating pharmacologic treatments with exercise and nutrition. A comprehensive, multi-targeted approach centered on STAT3 inhibition holds promise to transform cachexia management and improve quality of life in cancer patients.
Natural products targeting STAT3
Several natural compounds have shown promising potential in inhibiting STAT3 activity, and are currently being investigated for their anti-cachexia effects (Figure 4D, Table 1). These compounds exhibit anti-inflammatory, lipid-sparing, and muscle-preserving properties by targeting various mechanisms, including the inhibition of STAT3 activation and the downregulation of pro-inflammatory cytokines, which drive muscle degradation. For instance, alantolactone (28), ursolic acid (170), and brassinin (171) reduce pro-inflammatory cytokines (such as IL-6 and TNF-α), as well as COX-2, and directly inhibit STAT3 signaling, thus alleviating tissue wasting. Additionally, compounds such as atractylenolide I (172), saikosaponin D (173), cucurbitacin IIb (174), imperatorin (175), ginsenoside Rd (176), cryptotanshinone (177), and Z526 (178) have also demonstrated the ability to suppress STAT3 activation and mitigate body weight and muscle loss in cachectic models. Notably, alantolactone (28), atractylenolide I (172), imperatorin (175), ursolic acid (170, 179), and brassinin (171) have further emphasized the therapeutic potential of targeting STAT3 to prevent fat loss in cancer cachexia. While these natural compounds show encouraging preclinical efficacy in targeting STAT3 to alleviate cancer cachexia, further clinical studies are necessary to confirm their therapeutic potential and safety for use in treating this condition.
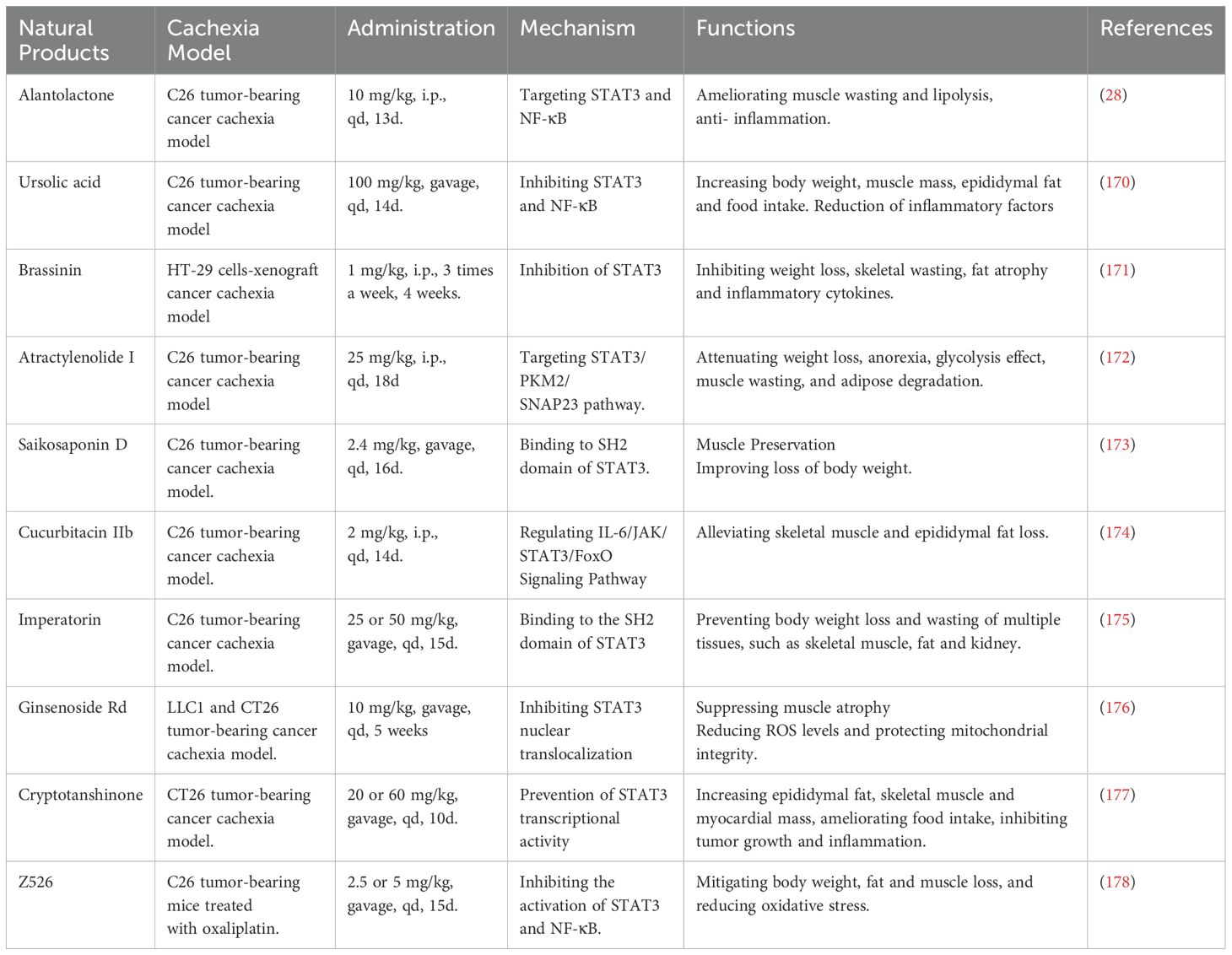
Table 1. Natural compounds in ameliorating cancer cachexia via targeting STAT3 signaling.
Physical exercise targeting STAT3
A growing body of evidence from both preclinical and clinical studies indicates that multimodal exercise interventions can effectively mitigate STAT3-driven muscle atrophy in cancer cachexia by modulating inflammatory signaling and enhancing anabolic pathways. During exercise, skeletal muscle fibers release IL-6 via a TNF-independent mechanism, which promotes the systemic release of anti-inflammatory cytokines such as IL-1 receptor antagonist (IL-1ra) and IL-10, while concurrently suppressing pro-inflammatory mediators including TNF-α and NF-κB activity (180–183). This anti-inflammatory cytokine profile contributes to the attenuation of chronic inflammation that sustains aberrant STAT3 activation in skeletal muscle. Resistance training, in particular, plays a critical role by not only reducing IL-6-induced STAT3 phosphorylation but also by upregulating suppressor of cytokine signaling 3 (SOCS3), a negative feedback regulator that directly inhibits STAT3 signaling (184). Endurance exercise complements these effects by reducing circulating levels of TNF-α and IL-6 and by promoting mitochondrial biogenesis and oxidative metabolism (185), thereby preserving muscle mass and function. Interestingly, exercise-induced IL-6 also exerts context-dependent protective effects, counteracting the catabolic actions of systemic inflammatory cytokines such as TNF-α, likely through autocrine and paracrine mechanisms (186–189). All in all, these findings highlight the therapeutic potential of structured exercise interventions in modulating STAT3 activity, offering a promising, non-pharmacological strategy to ameliorate muscle wasting and improve clinical outcomes in cancer cachexia.
Nutritional and metabolic modulators
Recent therapeutic strategies have increasingly focused on targeting the STAT3 pathway to mitigate the muscle and adipose tissue wasting characteristic of cancer cachexia. Among these strategies, nutritional and metabolic modulators, particularly nutraceuticals, have emerged as promising adjuncts to conventional therapies (Figure 4E). Notably, omega-3 polyunsaturated fatty acids (n-3 PUFAs), especially eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), have demonstrated significant anti-inflammatory properties. These effects are primarily mediated through the downregulation of pro-inflammatory cytokine signaling upstream of STAT3. For instance, n-3 PUFAs have been shown to suppress levels of IL-6 and TNF-α in cancer patients (190), as well as reduce IL-17A-mediated inflammation (191) and IL-11 expression in hepatocytes during acetaminophen-induced hepatotoxicity (192). These cytokines are known activators of STAT3 and play critical roles in sustaining chronic inflammation and tissue catabolism.
Beyond their anti-inflammatory properties, n-3 PUFAs have potential applications in enhancing exercise recovery and preserving skeletal muscle mass and strength (193–195). Mechanistically, supplementation with n-3 PUFAs has been found to activate the mechanistic target of rapamycin complex 1 (mTORC1) pathway, reduce intracellular protein degradation, and promote mitochondrial biogenesis and function (196). Additionally, dietary n-3 PUFAs may protect against muscle mitochondrial oxidative stress and attenuate muscle wasting in chronic heart failure (197), likely by enhancing oxidative phosphorylation efficiency, increasing ATP production, and reducing reactive oxygen species (ROS) accumulation and muscle fatigue.
In tandem, other nutritional agents such as leucine and its metabolite β-hydroxy β-methylbutyrate (HMB) have also demonstrated anti-cachectic potential, partly through modulation of STAT3 activity. Leucine enhances mitochondrial biogenesis and activates mTOR signaling in skeletal muscle while simultaneously decreasing STAT3 phosphorylation and associated inflammatory signaling in tumor-bearing mice (198, 199). Moreover, HMB has been shown to reduce muscle proteolysis and prevent apoptosis of myonuclei by inhibiting both the ubiquitin–proteasome and autophagy–lysosome pathways (197, 200). These effects collectively support its therapeutic value in various forms of cachexia.
Challenges and future directions
Despite significant progress in elucidating the role of STAT3 in cancer cachexia, several key challenges remain that hinder the development of effective therapeutic strategies. One major obstacle is the complexity of STAT3 signaling pathways. STAT3 mediates a broad array of cellular functions, including immune regulation, tumor progression, tumor inflammation, and metabolic reprogramming (15), with tissue-specific responses across various disease contexts (26, 43). Its multifaceted involvement makes it difficult to delineate the exact mechanisms by which STAT3 contributes to cachexia and to develop therapies that selectively disrupt its pathological actions. STAT3’s central role in maintaining immune homeostasis, particularly in anti-tumor immunity, further complicates therapeutic targeting, as systemic inhibition may result in unintended immunosuppressive effects (17, 165).
A second major challenge lies in the tissue-specific and sometimes opposing roles of STAT3. For instance, while it drives skeletal muscle wasting in cancer cachexia, it also regulates lipolysis in adipose tissue and metabolic dysfunction in the liver (25). Dissecting these tissue-specific functions is essential for designing targeted interventions that can suppress STAT3’s deleterious effects in muscle without impairing its roles in other organs. Additionally, the TME is intimately involved in STAT3-driven cachexia, as STAT3 regulates immune cell polarization, cytokine production, and metabolic crosstalk between tumor and host tissues (15, 201). Therapeutic targeting of STAT3 must therefore be carefully balanced to avoid inadvertently promoting tumor progression while alleviating cachexia.
Despite the identification of numerous STAT3 inhibitors, translating these agents into clinical practice remains challenging. Many compounds suffer from poor pharmacokinetics, including low stability, poor oral bioavailability, and limited tissue penetration. Off-target effects and insufficient specificity have limited the efficacy and safety profiles of several candidates in human trials (15, 169, 202–204). Furthermore, because STAT3 is ubiquitously expressed and involved in essential physiological processes, global inhibition carries the risk of side effects such as immunosuppression, hepatotoxicity, and impaired tissue repair (18, 205). These concerns underscore the need for the development of next-generation STAT3 modulators that are highly selective, stable, and capable of achieving tissue-specific delivery (206).
An additional barrier to effective STAT3-targeted therapies is the emergence of resistance mechanisms. Tumors can activate alternative signaling pathways, such as NF-κB (20), or PI3K/AKT (207, 208), to circumvent STAT3 inhibition. This suggests that STAT3 monotherapy may be insufficient in many clinical contexts. Combination strategies involving STAT3 inhibitors with agents such as chemotherapy, radiotherapy, targeted therapy, or immunotherapy are promising approaches to enhance efficacy and mitigate resistance (15, 17). Despite extensive efforts to target STAT3 in cancer therapy, cancer remains a major clinical challenge, even with the advent of novel treatment strategies (168). Another critical unmet need is the lack of reliable biomarkers for early detection and monitoring of STAT3 activation in cancer cachexia. Non-invasive biomarkers, such as circulating cytokines, or phosphorylated STAT3, could facilitate early diagnosis, enable stratification of patients most likely to benefit from STAT3-targeted therapies. Incorporating such biomarkers into clinical trial design will be essential to advance STAT3 inhibitors from bench to bedside (209).
Given the complexity of STAT3’s biological roles and its diverse effects depending on tumor type and disease stage, future therapeutic strategies should adopt a personalized medicine approach. Integration of genomic, transcriptomic, and single-cell data can help identify context-specific STAT3 targets, allowing for more precise intervention. Such strategies would also consider the patient’s tumor profile, metabolic state, immune function, and comorbidities. Personalized treatment paradigms are likely to represent the future direction of STAT3-based therapies in cancer cachexia. In summary, successful translation of STAT3-targeted therapies into clinical use for cancer cachexia will require a multifaceted approach: (1) comprehensive characterization of tissue-specific STAT3 mechanisms, (2) development of highly selective and bioavailable inhibitors, (3) rational combination with other therapeutic agents, (4) discovery and application of predictive biomarkers, and (5) implementation of individualized treatment plans. Addressing these challenges will be key to unlocking the full therapeutic potential of STAT3 modulation in managing cancer cachexia.
Conclusion
This review underscores the critical role of STAT3 in the pathophysiology of cancer cachexia, a debilitating syndrome characterized by progressive muscle wasting, systemic inflammation, and metabolic dysregulation. Activation of STAT3 by pro-inflammatory cytokines such as IL-6 and HSPs fosters a catabolic environment that accelerates disease progression. Mechanistically, STAT3 signaling contributes to muscle degradation by upregulating muscle-specific E3 ubiquitin ligases while simultaneously suppressing protein synthesis. Furthermore, it exacerbates fat loss by promoting lipolysis and disrupting adipokine homeostasis in adipose tissue. Given its central role in cachexia pathogenesis, therapeutic targeting of the STAT3 pathway through small molecule inhibitors, monoclonal antibodies, or combination therapies presents a promising avenue for symptom management and improved patient outcomes. However, the intricate interplay between STAT3 signaling, tumor biology, and host metabolism remains incompletely understood, necessitating further research to refine therapeutic strategies and validate their clinical efficacy and safety. The heterogeneity of cancer cachexia, influenced by tumor type, disease stage, and individual metabolic profiles, further complicates treatment approaches, highlighting the need for personalized interventions. Additionally, the potential risks associated with prolonged STAT3 inhibition, including immune suppression and impaired tissue regeneration, must be carefully balanced against its therapeutic benefits. Advancing our understanding of STAT3-targeted interventions and translating these findings into clinical practice could significantly enhance quality of life and survival outcomes for cancer cachexia patients. Future research should prioritize the identification of predictive biomarkers, the optimization of combination therapies addressing both muscle wasting and metabolic dysfunction, and the integration of adjunctive strategies such as exercise and nutritional support. By bridging the gap between molecular research and clinical application, STAT3-targeted therapies hold the potential to revolutionize cachexia management, offering a more comprehensive and effective approach to improving patient care.
Author contributions
XL: Writing – original draft, Writing – review & editing, Investigation. SD: Funding acquisition, Writing – review & editing, Writing – original draft, Supervision, Visualization.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Natural Science Foundation of China (Grant No. 82103021 to S. Ding) and the China Postdoctoral Science Foundation (Grant No. 2022M721725 to S. Ding). Additional support was provided by the “14th Five-Year Plan” Nantong Clinical Medical Center (Innovation Platform) and the Youth Medical Expert Training Program, both funded by the Nantong Municipal Health Commission, as well as the 2024 School-Level Graduate Research and Practice Innovation Program of Medical School, Nantong University (Grant No. YJS2004006 to X Lv).
Acknowledgments
We acknowledge BioRender (BioRender.com) for enabling the creation of all figures in this work.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Setiawan T, Sari IN, Wijaya YT, Julianto NM, Muhammad JA, Lee H, et al. Cancer cachexia: molecular mechanisms and treatment strategies. J Hematol Oncol. (2023) 16:54. doi: 10.1186/s13045-023-01454-0
2. Baracos VE, Martin L, Korc M, Guttridge DC, and Fearon KCH. Cancer-associated cachexia. Nat Rev Dis Primers. (2018) 4:17105. doi: 10.1038/nrdp.2017.105
3. Wu Q, Li B, Li J, Sun S, Yuan J, and Sun S. Cancer-associated adipocytes as immunomodulators in cancer. biomark Res. (2021) 9:2. doi: 10.1186/s40364-020-00257-6
4. Wu Q, Liu Z, Li B, Liu YE, and Wang P. Immunoregulation in cancer-associated cachexia. J Adv Res. (2024) 58:45–62. doi: 10.1016/j.jare.2023.04.018
5. Nishikawa H, Goto M, Fukunishi S, Asai A, Nishiguchi S, and Higuchi K. Cancer cachexia: its mechanism and clinical significance. Int J Mol Sci. (2021) 22:8491. doi: 10.3390/ijms22168491
6. Wang Y and Ding S. Extracellular vesicles in cancer cachexia: deciphering pathogenic roles and exploring therapeutic horizons. J Transl Med. (2024) 22:506. doi: 10.1186/s12967-024-05266-9
7. Mariean CR, Tiucă OM, Mariean A, and Cotoi OS. Cancer cachexia: new insights and future directions. Cancers (Basel). (2023) 15:5590. doi: 10.3390/cancers15235590
8. Nishie K, Nishie T, Sato S, and Hanaoka M. Update on the treatment of cancer cachexia. Drug Discov Today. (2023) 28:103689. doi: 10.1016/j.drudis.2023.103689
9. Darnell JE Jr., Kerr IM, and Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. (1994) 264:1415–21. doi: 10.1126/science.8197455
10. Zhou J, Qu Z, Yan S, Sun F, Whitsett JA, Shapiro SD, et al. Differential roles of STAT3 in the initiation and growth of lung cancer. Oncogene. (2015) 34:3804–14. doi: 10.1038/onc.2014.318
11. Priester M, Copanaki E, Vafaizadeh V, Hensel S, Bernreuther C, Glatzel M, et al. STAT3 silencing inhibits glioma single cell infiltration and tumor growth. Neuro Oncol. (2013) 15:840–52. doi: 10.1093/neuonc/not025
12. Fathi N, Rashidi G, Khodadadi A, Shahi S, and Sharifi S. STAT3 and apoptosis challenges in cancer. Int J Biol Macromol. (2018) 117:993–1001. doi: 10.1016/j.ijbiomac.2018.05.121
13. Hillmer EJ, Zhang H, Li HS, and Watowich SS. STAT3 signaling in immunity. Cytokine Growth Factor Rev. (2016) 31:1–15. doi: 10.1016/j.cytogfr.2016.05.001
14. Mohan CD, Rangappa S, Preetham HD, Chandra Nayaka S, Gupta VK, Basappa S, et al. Targeting STAT3 signaling pathway in cancer by agents derived from Mother Nature. Semin Cancer Biol. (2022) 80:157–82. doi: 10.1016/j.semcancer.2020.03.016
15. Hu Y, Dong Z, and Liu K. Unraveling the complexity of STAT3 in cancer: molecular understanding and drug discovery. J Exp Clin Cancer Res. (2024) 43:23. doi: 10.1186/s13046-024-02949-5
16. Loh CY, Arya A, Naema AF, Wong WF, Sethi G, and Looi CY. Signal transducer and activator of transcription (STATs) proteins in cancer and inflammation: functions and therapeutic implication. Front Oncol. (2019) 9:48. doi: 10.3389/fonc.2019.00048
17. Zou S, Tong Q, Liu B, Huang W, Tian Y, and Fu X. Targeting STAT3 in cancer immunotherapy. Mol Cancer. (2020) 19:145. doi: 10.1186/s12943-020-01258-7
18. Johnson DE, O’Keefe RA, and Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. (2018) 15:234–48. doi: 10.1038/nrclinonc.2018.8
19. Damasceno LEA, Prado DS, Veras FP, Fonseca MM, Toller-Kawahisa JE, Rosa MH, et al. PKM2 promotes Th17 cell differentiation and autoimmune inflammation by fine-tuning STAT3 activation. J Exp Med. (2020) 217:e20190613. doi: 10.1084/jem.20190613
20. Fan Y, Mao R, and Yang J. NF-κB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell. (2013) 4:176–85. doi: 10.1007/s13238-013-2084-3
21. Gharibi T, Babaloo Z, Hosseini A, Abdollahpour-Alitappeh M, Hashemi V, Marofi F, et al. Targeting STAT3 in cancer and autoimmune diseases. Eur J Pharmacol. (2020) 878:173107. doi: 10.1016/j.ejphar.2020.173107
22. Poria DK, Sheshadri N, Balamurugan K, Sharan S, and Sterneck E. The STAT3 inhibitor Stattic acts independently of STAT3 to decrease histone acetylation and modulate gene expression. J Biol Chem. (2021) 296:100220. doi: 10.1074/jbc.RA120.016645
23. Amara CS, Kami Reddy KR, Yuntao Y, Chan YS, Piyarathna DWB, Dobrolecki LE, et al. The IL6/JAK/STAT3 signaling axis is a therapeutic vulnerability in SMARCB1-deficient bladder cancer. Nat Commun. (2024) 15:1373. doi: 10.1038/s41467-024-45132-2
24. Yang J, Li N, Zhao X, Guo W, Wu Y, Nie C, et al. WP1066, a small molecule inhibitor of STAT3, chemosensitizes paclitaxel-resistant ovarian cancer cells to paclitaxel by simultaneously inhibiting the activity of STAT3 and the interaction of STAT3 with Stathmin. Biochem Pharmacol. (2024) 221:116040. doi: 10.1016/j.bcp.2024.116040
25. Zimmers TA, Fishel ML, and Bonetto A. STAT3 in the systemic inflammation of cancer cachexia. Semin Cell Dev Biol. (2016) 54:28–41. doi: 10.1016/j.semcdb.2016.02.009
26. Martin A, Gallot YS, and Freyssenet D. Molecular mechanisms of cancer cachexia-related loss of skeletal muscle mass: data analysis from preclinical and clinical studies. J Cachexia Sarcopenia Muscle. (2023) 14:1150–67. doi: 10.1002/jcsm.13073
27. Hu W, Ru Z, Zhou Y, Xiao W, Sun R, Zhang S, et al. Lung cancer-derived extracellular vesicles induced myotube atrophy and adipocyte lipolysis via the extracellular IL-6-mediated STAT3 pathway. Biochim Biophys Acta Mol Cell Biol Lipids. (2019) 1864:1091–102. doi: 10.1016/j.bbalip.2019.04.006
28. Shen Q, Kuang JX, Miao CX, Zhang WL, Li YW, Zhang XW, et al. Alantolactone ameliorates cancer cachexia-associated muscle atrophy mainly by inhibiting the STAT3 signaling pathway. Phytomedicine. (2022) 95:153858. doi: 10.1016/j.phymed.2021.153858
29. Rao VK, Das D, and Taneja R. Cancer cachexia: signaling and transcriptional regulation of muscle catabolic genes. Cancers. (2022) 14:4258. doi: 10.3390/cancers14174258
30. Bonetto A, Aydogdu T, Kunzevitzky N, Guttridge DC, Khuri S, Koniaris LG, et al. STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PloS One. (2011) 6:e22538. doi: 10.1371/journal.pone.0022538
31. Jin J, Li Y, Zhao Q, Chen Y, Fu S, and Wu J. Coordinated regulation of immune contexture: crosstalk between STAT3 and immune cells during breast cancer progression. Cell Commun Signal: CCS. (2021) 19:50. doi: 10.1186/s12964-021-00705-2
32. Hu X, Li J, Fu M, Zhao X, and Wang W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther. (2021) 6:402. doi: 10.1038/s41392-021-00791-1
33. Owen KL, Brockwell NK, and Parker BS. JAK-STAT signaling: A double-edged sword of immune regulation and cancer progression. Cancers (Basel). (2019) 11:2002. doi: 10.3390/cancers11122002
34. Zhong Z, Wen Z, and Darnell JE Jr. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. (1994) 264:95–8. doi: 10.1126/science.8140422
35. Ruff-Jamison S, Zhong Z, Wen Z, Chen K, Darnell JE Jr., and Cohen S. Epidermal growth factor and lipopolysaccharide activate Stat3 transcription factor in mouse liver. J Biol Chem. (1994) 269:21933–5. doi: 10.1016/S0021-9258(17)31735-0
36. Heinrich PC, Behrmann I, Müller-Newen G, Schaper F, and Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J. (1998) 334:297–314. doi: 10.1042/bj3340297
37. Berishaj M, Gao SP, Ahmed S, Leslie K, Al-Ahmadie H, Gerald WL, et al. Stat3 is tyrosine-phosphorylated through the interleukin-6/glycoprotein 130/Janus kinase pathway in breast cancer. Breast Cancer Res. (2007) 9:R32. doi: 10.1186/bcr1680
38. Rodig SJ, Meraz MA, White JM, Lampe PA, Riley JK, Arthur CD, et al. Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell. (1998) 93:373–83. doi: 10.1016/s0092-8674(00)81166-6
39. Chen J, Wang J, Lin L, He L, Wu Y, Zhang L, et al. Inhibition of STAT3 signaling pathway by nitidine chloride suppressed the angiogenesis and growth of human gastric cancer. Mol Cancer Ther. (2012) 11:277–87. doi: 10.1158/1535-7163.Mct-11-0648
40. Al Zaid Siddiquee K and Turkson J. STAT3 as a target for inducing apoptosis in solid and hematological tumors. Cell Res. (2008) 18:254–67. doi: 10.1038/cr.2008.18
41. Wang Y, van Boxel-Dezaire AH, Cheon H, Yang J, and Stark GR. STAT3 activation in response to IL-6 is prolonged by the binding of IL-6 receptor to EGF receptor. Proc Natl Acad Sci U S A. (2013) 110:16975–80. doi: 10.1073/pnas.1315862110
42. Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, et al. Stat3 as an oncogene. Cell. (1999) 98:295–303. doi: 10.1016/s0092-8674(00)81959-5
43. Bharadwaj U, Kasembeli MM, Robinson P, and Tweardy DJ. Targeting janus kinases and signal transducer and activator of transcription 3 to treat inflammation, fibrosis, and cancer: rationale, progress, and caution. Pharmacol Rev. (2020) 72:486–526. doi: 10.1124/pr.119.018440
44. Srivastava J and DiGiovanni J. Non-canonical Stat3 signaling in cancer. Mol Carcinog. (2016) 55:1889–98. doi: 10.1002/mc.22438
45. Gai L, Zhu Y, Zhang C, and Meng X. Targeting canonical and non-canonical STAT signaling pathways in renal diseases. Cells. (2021) 10:1610. doi: 10.3390/cells10071610
46. Courapied S, Sellier H, de Carné Trécesson S, Vigneron A, Bernard AC, Gamelin E, et al. The cdk5 kinase regulates the STAT3 transcription factor to prevent DNA damage upon topoisomerase I inhibition. J Biol Chem. (2010) 285:26765–78. doi: 10.1074/jbc.M109.092304
47. Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, and Levy DE. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. (2009) 324:1713–6. doi: 10.1126/science.1171721
48. Avalle L and Poli V. Nucleus, mitochondrion, or reticulum? STAT3 à La carte. Int J Mol Sci. (2018) 19:2820. doi: 10.3390/ijms19092820
49. Mohr A, Chatain N, Domoszlai T, Rinis N, Sommerauer M, Vogt M, et al. Dynamics and non-canonical aspects of JAK/STAT signalling. Eur J Cell Biol. (2012) 91:524–32. doi: 10.1016/j.ejcb.2011.09.005
50. Domaniku-Waraich A, Agca S, Toledo B, Sucuoglu M, Özen SD, Bilgic SN, et al. Oncostatin M signaling drives cancer-associated skeletal muscle wasting. Cell Rep Med. (2024) 5:101498. doi: 10.1016/j.xcrm.2024.101498
51. Ying L, Yao Y, Lv H, Lu G, Zhang Q, Yang Y, et al. IL-17A contributes to skeletal muscle atrophy in lung cancer-induced cachexia via JAK2/STAT3 pathway. Am J Physiol Cell Physiol. (2022) 322:C814–c24. doi: 10.1152/ajpcell.00463.2021
52. Patel HJ and Patel BM. TNF-α and cancer cachexia: Molecular insights and clinical implications. Life Sci. (2017) 170:56–63. doi: 10.1016/j.lfs.2016.11.033
53. Paval DR, Patton R, McDonald J, Skipworth RJE, Gallagher IJ, and Laird BJ. A systematic review examining the relationship between cytokines and cachexia in incurable cancer. J Cachexia Sarcopenia Muscle. (2022) 13:824–38. doi: 10.1002/jcsm.12912
54. Bonetto A, Aydogdu T, Jin X, Zhang Z, Zhan R, Puzis L, et al. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. Am J Physiol Endocrinol Metab. (2012) 303:E410–21. doi: 10.1152/ajpendo.00039.2012
55. Narsale AA and Carson JA. Role of interleukin-6 in cachexia: therapeutic implications. Curr Opin Support Palliat Care. (2014) 8:321–7. doi: 10.1097/spc.0000000000000091
56. Eskiler GG, Bezdegumeli E, Ozman Z, Ozkan AD, Bilir C, Kucukakca BN, et al. IL-6 mediated JAK/STAT3 signaling pathway in cancer patients with cachexia. Bratisl Lek Listy. (2019) 66:819–26. doi: 10.4149/bll_2019_136
57. Gandhi AY, Yu J, Gupta A, Guo T, Iyengar P, and Infante RE. Cytokine-mediated STAT3 transcription supports ATGL/CGI-58-dependent adipocyte lipolysis in cancer cachexia. Front Oncol. (2022) 12:841758. doi: 10.3389/fonc.2022.841758
58. Miller A, McLeod L, Alhayyani S, Szczepny A, Watkins DN, Chen W, et al. Blockade of the IL-6 trans-signalling/STAT3 axis suppresses cachexia in Kras-induced lung adenocarcinoma. Oncogene. (2017) 36:3059–66. doi: 10.1038/onc.2016.437
59. Yang X, Wang J, Chang CY, Zhou F, Liu J, Xu H, et al. Leukemia inhibitory factor suppresses hepatic de novo lipogenesis and induces cachexia in mice. Nat Commun. (2024) 15:627. doi: 10.1038/s41467-024-44924-w
60. Kandarian SC, Nosacka RL, Delitto AE, Judge AR, Judge SM, Ganey JD, et al. Tumour-derived leukaemia inhibitory factor is a major driver of cancer cachexia and morbidity in C26 tumour-bearing mice. J Cachexia Sarcopenia Muscle. (2018) 9:1109–20. doi: 10.1002/jcsm.12346
61. Chiappalupi S, Sorci G, Vukasinovic A, Salvadori L, Sagheddu R, Coletti D, et al. Targeting RAGE prevents muscle wasting and prolongs survival in cancer cachexia. J Cachexia Sarcopenia Muscle. (2020) 11:929–46. doi: 10.1002/jcsm.12561
62. Niu M, Song S, Su Z, Wei L, Li L, Pu W, et al. Inhibition of heat shock protein (HSP) 90 reverses signal transducer and activator of transcription (STAT) 3-mediated muscle wasting in cancer cachexia mice. Br J Pharmacol. (2021) 178:4485–500. doi: 10.1111/bph.15625
63. Bi J, Wu Z, Zhang X, Zeng T, Dai W, Qiu N, et al. TMEM25 inhibits monomeric EGFR-mediated STAT3 activation in basal state to suppress triple-negative breast cancer progression. Nat Commun. (2023) 14:2342. doi: 10.1038/s41467-023-38115-2
64. Zhao C, Yang L, Zhou F, Yu Y, Du X, Xiang Y, et al. Feedback activation of EGFR is the main cause for STAT3 inhibition-irresponsiveness in pancreatic cancer cells. Oncogene. (2020) 39:3997–4013. doi: 10.1038/s41388-020-1271-y
65. Shen L, Zhang C, Cui K, Liang X, Zhu G, and Hong L. Leptin secreted by adipocytes promotes EMT transition and endometrial cancer progression via the JAK2/STAT3 signalling pathway. Adipocyte. (2024) 13:2293273. doi: 10.1080/21623945.2023.2293273
66. Vaisse C, Halaas JL, Horvath CM, Darnell JE Jr., Stoffel M, and Friedman JM. Leptin activation of Stat3 in the hypothalamus of wild-type and ob/ob mice but not db/db mice. Nat Genet. (1996) 14:95–7. doi: 10.1038/ng0996-95
67. Chang Q, Bournazou E, Sansone P, Berishaj M, Gao SP, Daly L, et al. The IL-6/JAK/Stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia. (2013) 15:848–62. doi: 10.1593/neo.13706
68. Agca S and Kir S. The role of interleukin-6 family cytokines in cancer cachexia. FEBS J. (2024) 291:4009–23. doi: 10.1111/febs.17224
69. Ma JF, Sanchez BJ, Hall DT, Tremblay AK, Di Marco S, and Gallouzi IE. STAT3 promotes IFNγ/TNFα-induced muscle wasting in an NF-κB-dependent and IL-6-independent manner. EMBO Mol Med. (2017) 9:622–37. doi: 10.15252/emmm.201607052
70. Gao S, Durstine JL, Koh HJ, Carver WE, Frizzell N, and Carson JA. Acute myotube protein synthesis regulation by IL-6-related cytokines. Am J Physiol Cell Physiol. (2017) 313:C487–c500. doi: 10.1152/ajpcell.00112.2017
71. Burfeind KG, Michaelis KA, and Marks DL. The central role of hypothalamic inflammation in the acute illness response and cachexia. Semin Cell Dev Biol. (2016) 54:42–52. doi: 10.1016/j.semcdb.2015.10.038
72. Mori H, Hanada R, Hanada T, Aki D, Mashima R, Nishinakamura H, et al. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat Med. (2004) 10:739–43. doi: 10.1038/nm1071
73. Li Y, Dörmann N, Brinschwitz B, Kny M, Martin E, Bartels K, et al. SPSB1-mediated inhibition of TGF-β receptor-II impairs myogenesis in inflammation. J Cachexia Sarcopenia Muscle. (2023) 14:1721–36. doi: 10.1002/jcsm.13252
74. Carson JA and Baltgalvis KA. Interleukin 6 as a key regulator of muscle mass during cachexia. Exerc Sport Sci Rev. (2010) 38:168–76. doi: 10.1097/JES.0b013e3181f44f11
75. Argilés JM, Busquets S, Stemmler B, and López-Soriano FJ. Cancer cachexia: understanding the molecular basis. Nat Rev Cancer. (2014) 14:754–62. doi: 10.1038/nrc3829
76. Silva KA, Dong J, Dong Y, Dong Y, Schor N, Tweardy DJ, et al. Inhibition of Stat3 activation suppresses caspase-3 and the ubiquitin-proteasome system, leading to preservation of muscle mass in cancer cachexia. J Biol Chem. (2015) 290:11177–87. doi: 10.1074/jbc.M115.641514
77. Fearon KC, Glass DJ, and Guttridge DC. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab. (2012) 16:153–66. doi: 10.1016/j.cmet.2012.06.011
78. White JP, Puppa MJ, Gao S, Sato S, Welle SL, and Carson JA. Muscle mTORC1 suppression by IL-6 during cancer cachexia: a role for AMPK. Am J Physiol Endocrinol Metab. (2013) 304:E1042–52. doi: 10.1152/ajpendo.00410.2012
79. Steinberg GR, Michell BJ, van Denderen BJ, Watt MJ, Carey AL, Fam BC, et al. Tumor necrosis factor alpha-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab. (2006) 4:465–74. doi: 10.1016/j.cmet.2006.11.005
80. Guo Y, Meng J, Tang Y, Wang T, Wei B, Feng R, et al. AMP-activated kinase α2 deficiency protects mice from denervation-induced skeletal muscle atrophy. Arch Biochem Biophys. (2016) 600:56–60. doi: 10.1016/j.abb.2016.04.015
81. Zungu M, Schisler JC, Essop MF, McCudden C, Patterson C, and Willis MS. Regulation of AMPK by the ubiquitin proteasome system. Am J Pathol. (2011) 178:4–11. doi: 10.1016/j.ajpath.2010.11.030
82. Nakashima K and Yakabe Y. AMPK activation stimulates myofibrillar protein degradation and expression of atrophy-related ubiquitin ligases by increasing FOXO transcription factors in C2C12 myotubes. Biosci Biotechnol Biochem. (2007) 71:1650–6. doi: 10.1271/bbb.70057
83. Zhang L, Pan J, Dong Y, Tweardy DJ, Dong Y, Garibotto G, et al. Stat3 activation links a C/EBPδ to myostatin pathway to stimulate loss of muscle mass. Cell Metab. (2013) 18:368–79. doi: 10.1016/j.cmet.2013.07.012
84. Tierney MT, Aydogdu T, Sala D, Malecova B, Gatto S, Puri PL, et al. STAT3 signaling controls satellite cell expansion and skeletal muscle repair. Nat Med. (2014) 20:1182–6. doi: 10.1038/nm.3656
85. Pang X, Zhang P, Chen X, and Liu W. Ubiquitin-proteasome pathway in skeletal muscle atrophy. Front Physiol. (2023) 14:1289537. doi: 10.3389/fphys.2023.1289537
86. Catarinella G, Bracaglia A, Skafida E, Procopio P, Ruggieri V, Parisi C, et al. STAT3 inhibition recovers regeneration of aged muscles by restoring autophagy in muscle stem cells. Life Sci Alliance. (2024) 7:e202302503. doi: 10.26508/lsa.202302503
87. Zhu H, Xiao F, Wang G, Wei X, Jiang L, Chen Y, et al. STAT3 regulates self-renewal of adult muscle satellite cells during injury-induced muscle regeneration. Cell Rep. (2016) 16:2102–15. doi: 10.1016/j.celrep.2016.07.041
88. Guadagnin E, Mázala D, and Chen YW. STAT3 in skeletal muscle function and disorders. Int J Mol Sci. (2018) 19:2265. doi: 10.3390/ijms19082265
89. Chen X, Ji Y, Liu R, Zhu X, Wang K, Yang X, et al. Mitochondrial dysfunction: roles in skeletal muscle atrophy. J Transl Med. (2023) 21:503. doi: 10.1186/s12967-023-04369-z
90. Huot JR, Novinger LJ, Pin F, and Bonetto A. HCT116 colorectal liver metastases exacerbate muscle wasting in a mouse model for the study of colorectal cancer cachexia. Dis Model Mech. (2020) 13:dmm043166. doi: 10.1242/dmm.043166
91. Fix DK, Hardee JP, Gao S, VanderVeen BN, Velázquez KT, and Carson JA. Role of gp130 in basal and exercise-trained skeletal muscle mitochondrial quality control. J Appl Physiol. (2018) 124:1456–70. doi: 10.1152/japplphysiol.01063.2017
92. Puppa MJ, Gao S, Narsale AA, and Carson JA. Skeletal muscle glycoprotein 130’s role in Lewis lung carcinoma-induced cachexia. FASEB J. (2014) 28:998–1009. doi: 10.1096/fj.13-240580
93. Salminen A, Kaarniranta K, and Kauppinen A. Insulin/IGF-1 signaling promotes immunosuppression via the STAT3 pathway: impact on the aging process and age-related diseases. Inflammation Res. (2021) 70:1043–61. doi: 10.1007/s00011-021-01498-3
94. Miyamoto Y, Hanna DL, Zhang W, Baba H, and Lenz HJ. Molecular pathways: cachexia signaling-A targeted approach to cancer treatment. Clin Cancer Res. (2016) 22:3999–4004. doi: 10.1158/1078-0432.Ccr-16-0495
95. Chen JL, Walton KL, Hagg A, Colgan TD, Johnson K, Qian H, et al. Specific targeting of TGF-β family ligands demonstrates distinct roles in the regulation of muscle mass in health and disease. Proc Natl Acad Sci U S A. (2017) 114:E5266–e75. doi: 10.1073/pnas.1620013114
96. Gallardo FS, Cruz-Soca M, Bock-Pereda A, Faundez-Contreras J, Gutiérrez-Rojas C, Gandin A, et al. Role of TGF-β/SMAD/YAP/TAZ signaling in skeletal muscle fibrosis. Am J Physiol Cell Physiol. (2025) 328:C1015–28. doi: 10.1152/ajpcell.00541.2024
97. Guadagnin E, Narola J, Bönnemann CG, and Chen YW. Tyrosine 705 phosphorylation of STAT3 is associated with phenotype severity in TGFβ1 transgenic mice. BioMed Res Int. (2015) 2015:843743. doi: 10.1155/2015/843743
98. Muthamil S, Kim HY, Jang HJ, Lyu JH, Shin UC, Go Y, et al. Understanding the relationship between cancer associated cachexia and hypoxia-inducible factor-1. BioMed Pharmacother. (2023) 163:114802. doi: 10.1016/j.biopha.2023.114802
99. Devine RD, Bicer S, Reiser PJ, and Wold LE. Increased hypoxia-inducible factor-1α in striated muscle of tumor-bearing mice. Am J Physiol Heart Circ Physiol. (2017) 312:H1154–h62. doi: 10.1152/ajpheart.00090.2016
100. Kuang JX, Shen Q, Zhang RQ, Fang QY, Deng X, Fan M, et al. Carnosol attenuated atrophy of C2C12 myotubes induced by tumour-derived exosomal miR-183-5p through inhibiting Smad3 pathway activation and keeping mitochondrial respiration. Basic Clin Pharmacol Toxicol. (2022) 131:500–13. doi: 10.1111/bcpt.13795
101. Tsoli M, Schweiger M, Vanniasinghe AS, Painter A, Zechner R, Clarke S, et al. Depletion of white adipose tissue in cancer cachexia syndrome is associated with inflammatory signaling and disrupted circadian regulation. PloS One. (2014) 9:e92966. doi: 10.1371/journal.pone.0092966
102. Mannelli M, Bartoloni B, Cantini G, Nencioni E, Magherini F, Luconi M, et al. STAT3 signalling drives LDH up-regulation and adiponectin down-regulation in cachectic adipocytes. Int J Mol Sci. (2023) 24:16343. doi: 10.3390/ijms242216343
103. Derecka M, Gornicka A, Koralov SB, Szczepanek K, Morgan M, Raje V, et al. Tyk2 and Stat3 regulate brown adipose tissue differentiation and obesity. Cell Metab. (2012) 16:814–24. doi: 10.1016/j.cmet.2012.11.005
104. Song L, Cao X, Ji W, Zhao L, Yang W, Lu M, et al. Inhibition of STAT3 enhances UCP1 expression and mitochondrial function in brown adipocytes. Eur J Pharmacol. (2022) 926:175040. doi: 10.1016/j.ejphar.2022.175040
105. Langer HT, Rohm M, Goncalves MD, and Sylow L. AMPK as a mediator of tissue preservation: time for a shift in dogma? Nat Rev Endocrinol. (2024) 20:526–40. doi: 10.1038/s41574-024-00992-y
106. Ji H, Englmaier F, Morigny P, Giroud M, Gräsle P, Brings S, et al. Development of a peptide drug restoring AMPK and adipose tissue functionality in cancer cachexia. Mol Ther. (2023) 31:2408–21. doi: 10.1016/j.ymthe.2023.06.020
107. Cantwell MT, Farrar JS, Lownik JC, Meier JA, Hyun M, Raje V, et al. STAT3 suppresses Wnt/β-catenin signaling during the induction phase of primary Myf5+ brown adipogenesis. Cytokine. (2018) 111:434–44. doi: 10.1016/j.cyto.2018.05.023
108. Van Lancker A, Velghe A, Van Hecke A, Verbrugghe M, Van Den Noortgate N, Grypdonck M, et al. Prevalence of symptoms in older cancer patients receiving palliative care: a systematic review and meta-analysis. J Pain Symptom Manage. (2014) 47:90–104. doi: 10.1016/j.jpainsymman.2013.02.016
109. Yavuzsen T, Davis MP, Walsh D, LeGrand S, and Lagman R. Systematic review of the treatment of cancer-associated anorexia and weight loss. J Clin Oncol. (2005) 23:8500–11. doi: 10.1200/jco.2005.01.8010
110. Tarricone R, Ricca G, Nyanzi-Wakholi B, and Medina-Lara A. Impact of cancer anorexia-cachexia syndrome on health-related quality of life and resource utilisation: A systematic review. Crit Rev Oncol Hematol. (2016) 99:49–62. doi: 10.1016/j.critrevonc.2015.12.008
111. Saxton RA, Caveney NA, Moya-Garzon MD, Householder KD, Rodriguez GE, Burdsall KA, et al. Structural insights into the mechanism of leptin receptor activation. Nat Commun. (2023) 14:1797. doi: 10.1038/s41467-023-37169-6
112. Targa G, Mottarlini F, Rizzi B, Taddini S, Parolaro S, Fumagalli F, et al. Anorexia-induced hypoleptinemia drives adaptations in the JAK2/STAT3 pathway in the ventral and dorsal hippocampus of female rats. Nutrients. (2024) 16:1171. doi: 10.3390/nu16081171
113. Münzberg H, Huo L, Nillni EA, Hollenberg AN, and Bjørbaek C. Role of signal transducer and activator of transcription 3 in regulation of hypothalamic proopiomelanocortin gene expression by leptin. Endocrinology. (2003) 144:2121–31. doi: 10.1210/en.2002-221037
114. Gao Q, Wolfgang MJ, Neschen S, Morino K, Horvath TL, Shulman GI, et al. Disruption of neural signal transducer and activator of transcription 3 causes obesity, diabetes, infertility, and thermal dysregulation. Proc Natl Acad Sci U S A. (2004) 101:4661–6. doi: 10.1073/pnas.0303992101
115. Heldsinger A, Grabauskas G, Wu X, Zhou S, Lu Y, Song I, et al. Ghrelin induces leptin resistance by activation of suppressor of cytokine signaling 3 expression in male rats: implications in satiety regulation. Endocrinology. (2014) 155:3956–69. doi: 10.1210/en.2013-2095
116. Ladyman SR and Grattan DR. JAK-STAT and feeding. Jakstat. (2013) 2:e23675. doi: 10.4161/jkst.23675
117. Rizk NM, Stammsen D, Preibisch G, and Eckel J. Leptin and tumor necrosis factor-alpha induce the tyrosine phosphorylation of signal transducer and activator of transcription proteins in the hypothalamus of normal rats in vivo. Endocrinology. (2001) 142:3027–32. doi: 10.1210/endo.142.7.8225
118. Lambert PD, Anderson KD, Sleeman MW, Wong V, Tan J, Hijarunguru A, et al. Ciliary neurotrophic factor activates leptin-like pathways and reduces body fat, without cachexia or rebound weight gain, even in leptin-resistant obesity. Proc Natl Acad Sci U S A. (2001) 98:4652–7. doi: 10.1073/pnas.061034298
119. Buchanan JB and Johnson RW. Regulation of food intake by inflammatory cytokines in the brain. Neuroendocrinology. (2007) 86:183–90. doi: 10.1159/000108280
120. Sahu M, Anamthathmakula P, and Sahu A. Hypothalamic phosphodiesterase 3B pathway mediates anorectic and body weight-reducing effects of insulin in male mice. Neuroendocrinology. (2017) 104:145–56. doi: 10.1159/000445523
121. Hübschle T, Thom E, Watson A, Roth J, Klaus S, and Meyerhof W. Leptin-induced nuclear translocation of STAT3 immunoreactivity in hypothalamic nuclei involved in body weight regulation. J Neurosci. (2001) 21:2413–24. doi: 10.1523/jneurosci.21-07-02413.2001
122. Myers MG Jr. Leptin receptor signaling and the regulation of mammalian physiology. Recent Prog Horm Res. (2004) 59:287–304. doi: 10.1210/rp.59.1.287
123. Bjørbaek C, El-Haschimi K, Frantz JD, and Flier JS. The role of SOCS-3 in leptin signaling and leptin resistance. J Biol Chem. (1999) 274:30059–65. doi: 10.1074/jbc.274.42.30059
124. Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med. (2005) 11:183–90. doi: 10.1038/nm1166
125. Zhang X, Zhang G, Zhang H, Karin M, Bai H, and Cai D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell. (2008) 135:61–73. doi: 10.1016/j.cell.2008.07.043
126. Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature. (2004) 428:569–74. doi: 10.1038/nature02440
127. Dagon Y, Hur E, Zheng B, Wellenstein K, Cantley LC, and Kahn BB. p70S6 kinase phosphorylates AMPK on serine 491 to mediate leptin’s effect on food intake. Cell Metab. (2012) 16:104–12. doi: 10.1016/j.cmet.2012.05.010
128. Hsu JY, Crawley S, Chen M, Ayupova DA, Lindhout DA, Higbee J, et al. Non-homeostatic body weight regulation through a brainstem-restricted receptor for GDF15. Nature. (2017) 550:255–9. doi: 10.1038/nature24042
129. Yang L, Chang CC, Sun Z, Madsen D, Zhu H, Padkjær SB, et al. GFRAL is the receptor for GDF15 and is required for the anti-obesity effects of the ligand. Nat Med. (2017) 23:1158–66. doi: 10.1038/nm.4394
130. Taniguchi K and Karin M. NF-κB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol. (2018) 18:309–24. doi: 10.1038/nri.2017.142
131. Yoyen-Ermis D, Tunali G, Tavukcuoglu E, Horzum U, Ozkazanc D, Sutlu T, et al. Myeloid maturation potentiates STAT3-mediated atypical IFN-γ signaling and upregulation of PD-1 ligands in AML and MDS. Sci Rep. (2019) 9:11697. doi: 10.1038/s41598-019-48256-4
132. Atsaves V, Tsesmetzis N, Chioureas D, Kis L, Leventaki V, Drakos E, et al. PD-L1 is commonly expressed and transcriptionally regulated by STAT3 and MYC in ALK-negative anaplastic large-cell lymphoma. Leukemia. (2017) 31:1633–7. doi: 10.1038/leu.2017.103
133. Song TL, Nairismägi ML, Laurensia Y, Lim JQ, Tan J, Li ZM, et al. Oncogenic activation of the STAT3 pathway drives PD-L1 expression in natural killer/T-cell lymphoma. Blood. (2018) 132:1146–58. doi: 10.1182/blood-2018-01-829424
134. Yu Y, Yan L, Huang T, Wu Z, and Liu J. Cancer cachexia reduces the efficacy of immune checkpoint inhibitors in cancer patients. Aging (Albany NY). (2024) 16:5354–69. doi: 10.18632/aging.205652
135. Miyawaki T, Naito T, Kodama A, Nishioka N, Miyawaki E, Mamesaya N, et al. Desensitizing effect of cancer cachexia on immune checkpoint inhibitors in patients with advanced NSCLC. JTO Clin Res Rep. (2020) 1:100020. doi: 10.1016/j.jtocrr.2020.100020
136. Brayer J, Cheng F, Wang H, Horna P, Vicente-Suarez I, Pinilla-Ibarz J, et al. Enhanced CD8 T cell cross-presentation by macrophages with targeted disruption of STAT3. Immunol Lett. (2010) 131:126–30. doi: 10.1016/j.imlet.2010.03.004
137. Zhang K, Zakeri A, Alban T, Dong J, Ta HM, Zalavadia AH, et al. VISTA promotes the metabolism and differentiation of myeloid-derived suppressor cells by STAT3 and polyamine-dependent mechanisms. Cell Rep. (2024) 43:113661. doi: 10.1016/j.celrep.2023.113661
138. Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. (2005) 11:1314–21. doi: 10.1038/nm1325
139. Shukla SK, Markov SD, Attri KS, Vernucci E, King RJ, Dasgupta A, et al. Macrophages potentiate STAT3 signaling in skeletal muscles and regulate pancreatic cancer cachexia. Cancer Lett. (2020) 484:29–39. doi: 10.1016/j.canlet.2020.04.017
140. Herrmann A, Kortylewski M, Kujawski M, Zhang C, Reckamp K, Armstrong B, et al. Targeting Stat3 in the myeloid compartment drastically improves the in vivo antitumor functions of adoptively transferred T cells. Cancer Res. (2010) 70:7455–64. doi: 10.1158/0008-5472.Can-10-0736
141. Siegel AM, Heimall J, Freeman AF, Hsu AP, Brittain E, Brenchley JM, et al. A critical role for STAT3 transcription factor signaling in the development and maintenance of human T cell memory. Immunity. (2011) 35:806–18. doi: 10.1016/j.immuni.2011.09.016
142. Gotthardt D, Putz EM, Straka E, Kudweis P, Biaggio M, Poli V, et al. Loss of STAT3 in murine NK cells enhances NK cell-dependent tumor surveillance. Blood. (2014) 124:2370–9. doi: 10.1182/blood-2014-03-564450
143. Iwata-Kajihara T, Sumimoto H, Kawamura N, Ueda R, Takahashi T, Mizuguchi H, et al. Enhanced cancer immunotherapy using STAT3-depleted dendritic cells with high Th1-inducing ability and resistance to cancer cell-derived inhibitory factors. J Immunol. (2011) 187:27–36. doi: 10.4049/jimmunol.1002067
144. Argilés JM, Busquets S, Stemmler B, and López-Soriano FJ. Cachexia and sarcopenia: mechanisms and potential targets for intervention. Curr Opin Pharmacol. (2015) 22:100–6. doi: 10.1016/j.coph.2015.04.003
145. Yu H, Kortylewski M, and Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. (2007) 7:41–51. doi: 10.1038/nri1995
146. Chen S, Wang M, Lu T, Liu Y, Hong W, He X, et al. JMJD6 in tumor-associated macrophage regulates macrophage polarization and cancer progression via STAT3/IL-10 axis. Oncogene. (2023) 42:2737–50. doi: 10.1038/s41388-023-02781-9
147. Padoan A, Plebani M, and Basso D. Inflammation and pancreatic cancer: focus on metabolism, cytokines, and immunity. Int J Mol Sci. (2019) 20:676. doi: 10.3390/ijms20030676
148. Liu Q, Yang C, Wang S, Shi D, Wei C, Song J, et al. Wnt5a-induced M2 polarization of tumor-associated macrophages via IL-10 promotes colorectal cancer progression. Cell Commun Signal. (2020) 18:51. doi: 10.1186/s12964-020-00557-2
149. Zhu X, Liang R, Lan T, Ding D, Huang S, Shao J, et al. Tumor-associated macrophage-specific CD155 contributes to M2-phenotype transition, immunosuppression, and tumor progression in colorectal cancer. J Immunother Cancer. (2022) 10:e004219. doi: 10.1136/jitc-2021-004219
150. Huang L, Zhao Y, Shan M, Wang S, Chen J, Liu Z, et al. Targeting crosstalk of STAT3 between tumor-associated M2 macrophages and Tregs in colorectal cancer. Cancer Biol Ther. (2023) 24:2226418. doi: 10.1080/15384047.2023.2226418
151. Neo SY, Tong L, Chong J, Liu Y, Jing X, Oliveira MMS, et al. Tumor-associated NK cells drive MDSC-mediated tumor immune tolerance through the IL-6/STAT3 axis. Sci Transl Med. (2024) 16:eadi2952. doi: 10.1126/scitranslmed.adi2952
152. Xu Y, Ji Y, Lan X, Gao X, Chen HD, and Geng L. miR−203 contributes to IL−17−induced VEGF secretion by targeting SOCS3 in keratinocytes. Mol Med Rep. (2017) 16:8989–96. doi: 10.3892/mmr.2017.7759
153. Wang L, Li X, Wu J, and Tang Q. Pancreatic cancer-derived exosomal miR-let-7b-5p stimulates insulin resistance in skeletal muscle cells through RNF20/STAT3/FOXO1 axis regulation. Diabetes Metab Syndr Obes. (2023) 16:3133–45. doi: 10.2147/dmso.S430443
154. Schindler C, Levy DE, and Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem. (2007) 282:20059–63. doi: 10.1074/jbc.R700016200
155. Cramer Z, Sadek J, Vazquez GG, Di Marco S, Pause A, Pelletier J, et al. eIF4A inhibition prevents the onset of cytokine-induced muscle wasting by blocking the STAT3 and iNOS pathways. Sci Rep. (2018) 8:8414. doi: 10.1038/s41598-018-26625-9
156. Bayliss TJ, Smith JT, Schuster M, Dragnev KH, and Rigas JR. A humanized anti-IL-6 antibody (ALD518) in non-small cell lung cancer. Expert Opin Biol Ther. (2011) 11:1663–8. doi: 10.1517/14712598.2011.627850
157. Gilabert M, Calvo E, Airoldi A, Hamidi T, Moutardier V, Turrini O, et al. Pancreatic cancer-induced cachexia is Jak2-dependent in mice. J Cell Physiol. (2014) 229:1437–43. doi: 10.1002/jcp.24580
158. Jatoi A. Anti-inflammatory therapy: exploring exercise, serum-derived bovine immunoglobulin/protein isolates, and ruxolitinib for the cancer-associated weight loss syndrome. Curr Opin Support Palliat Care. (2013) 7:339–41. doi: 10.1097/spc.0000000000000014
159. Arora G, Gupta A, Guo T, Gandhi A, Laine A, Williams D, et al. JAK inhibitors suppress cancer cachexia-associated anorexia and adipose wasting in mice. JCSM Rapid Commun. (2020) 3:115–28. doi: 10.1002/rco2.24
160. Zhang L, Wang Y, Dong Y, Chen Z, Eckols TK, Kasembeli MM, et al. Pharmacokinetics and pharmacodynamics of TTI-101, a STAT3 inhibitor that blocks muscle proteolysis in rats with chronic kidney disease. Am J Physiol Renal Physiol. (2020) 319:F84–f92. doi: 10.1152/ajprenal.00603.2019
161. Tang Y, Luo Y, Jiang Z, Ma Y, Lin CJ, Kim C, et al. Jak/Stat3 signaling promotes somatic cell reprogramming by epigenetic regulation. Stem Cells. (2012) 30:2645–56. doi: 10.1002/stem.1225
162. Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. (2010) 363:1117–27. doi: 10.1056/NEJMoa1002028
163. Zanders L, Kny M, Hahn A, Schmidt S, Wundersitz S, Todiras M, et al. Sepsis induces interleukin 6, gp130/JAK2/STAT3, and muscle wasting. J Cachexia Sarcopenia Muscle. (2022) 13:713–27. doi: 10.1002/jcsm.12867
164. Madaro L, Passafaro M, Sala D, Etxaniz U, Lugarini F, Proietti D, et al. Denervation-activated STAT3-IL-6 signalling in fibro-adipogenic progenitors promotes myofibres atrophy and fibrosis. Nat Cell Biol. (2018) 20:917–27. doi: 10.1038/s41556-018-0151-y
165. Yu H, Pardoll D, and Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. (2009) 9:798–809. doi: 10.1038/nrc2734
166. Ono Y, Saito M, Sakamoto K, Maejima Y, Misaka S, Shimomura K, et al. C188-9, a specific inhibitor of STAT3 signaling, prevents thermal burn-induced skeletal muscle wasting in mice. Front Pharmacol. (2022) 13:1031906. doi: 10.3389/fphar.2022.1031906
167. Toledo M, Penna F, Busquets S, López-Soriano FJ, and Argilés JM. Distinct behaviour of sorafenib in experimental cachexia-inducing tumours: the role of STAT3. PloS One. (2014) 9:e113931. doi: 10.1371/journal.pone.0113931
168. Dong J, Cheng XD, Zhang WD, and Qin JJ. Recent update on development of small-molecule STAT3 inhibitors for cancer therapy: from phosphorylation inhibition to protein degradation. J Med Chem. (2021) 64:8884–915. doi: 10.1021/acs.jmedchem.1c00629
169. Thilakasiri PS, Dmello RS, Nero TL, Parker MW, Ernst M, and Chand AL. Repurposing of drugs as STAT3 inhibitors for cancer therapy. Semin Cancer Biol. (2021) 68:31–46. doi: 10.1016/j.semcancer.2019.09.022
170. Chen L, Chen Y, Wang M, Lai L, Zheng L, and Lu H. Ursolic acid alleviates cancer cachexia by inhibiting STAT3 signaling pathways in C2C12 myotube and CT26 tumor-bearing mouse model. Eur J Pharmacol. (2024) 969:176429. doi: 10.1016/j.ejphar.2024.176429
171. Yang MH, Jung YY, Um JY, Sethi G, and Ahn KS. Brassinin alleviates cancer cachexia by suppressing diverse inflammatory mechanisms in mice. MedComm. (2024) 5:e558. doi: 10.1002/mco2.558
172. Fan M, Gu X, Zhang W, Shen Q, Zhang R, Fang Q, et al. Atractylenolide I ameliorates cancer cachexia through inhibiting biogenesis of IL-6 and tumour-derived extracellular vesicles. J Cachexia Sarcopenia Muscle. (2022) 13:2724–39. doi: 10.1002/jcsm.13079
173. Chen LL, Xia LY, Zhang JP, Wang Y, Chen JY, Guo C, et al. Saikosaponin D alleviates cancer cachexia by directly inhibiting STAT3. Phytother Res. (2023) 37:809–19. doi: 10.1002/ptr.7676
174. Wang Y, Sun X, Yang Q, and Guo C. Cucurbitacin IIb attenuates cancer cachexia induced skeletal muscle atrophy by regulating the IL-6/STAT3/FoxO signaling pathway. Phytother Res. (2023) 37:3380–93. doi: 10.1002/ptr.7811
175. Chen L, Xu W, Yang Q, Zhang H, Wan L, Xin B, et al. Imperatorin alleviates cancer cachexia and prevents muscle wasting via directly inhibiting STAT3. Pharmacol Res. (2020) 158:104871. doi: 10.1016/j.phrs.2020.104871
176. Wijaya YT, Setiawan T, Sari IN, Park K, Lee CH, Cho KW, et al. Ginsenoside Rd ameliorates muscle wasting by suppressing the signal transducer and activator of transcription 3 pathway. J Cachexia Sarcopenia Muscle. (2022) 13:3149–62. doi: 10.1002/jcsm.13084
177. Chen L, Yang Q, Zhang H, Wan L, Xin B, Cao Y, et al. Cryptotanshinone prevents muscle wasting in CT26-induced cancer cachexia through inhibiting STAT3 signaling pathway. J Ethnopharmacol. (2020) 260:113066. doi: 10.1016/j.jep.2020.113066
178. Gu X, Lu S, Fan M, Xu S, Lin G, Zhao Y, et al. Compound Z526 alleviates chemotherapy-induced cachectic muscle loss by ameliorating oxidative stress-driven protein metabolic imbalance and apoptosis. Eur J Pharmacol. (2024) 974:176538. doi: 10.1016/j.ejphar.2024.176538
179. Tao W, Ouyang Z, Liao Z, Li L, Zhang Y, Gao J, et al. Ursolic acid alleviates cancer cachexia and prevents muscle wasting via activating SIRT1. Cancers (Basel). (2023) 15:2378. doi: 10.3390/cancers15082378
180. Petersen AM and Pedersen BK. The anti-inflammatory effect of exercise. J Appl Physiol. (2005) 98:1154–62. doi: 10.1152/japplphysiol.00164.2004
181. Daou HN. Exercise as an anti-inflammatory therapy for cancer cachexia: a focus on interleukin-6 regulation. Am J Physiol Regul Integr Comp Physiol. (2020) 318:R296–r310. doi: 10.1152/ajpregu.00147.2019
182. Drenth JP, Van Uum SH, Van Deuren M, Pesman GJ, van der Ven-Jongekrijg J, and van der Meer JW. Endurance run increases circulating IL-6 and IL-1ra but downregulates ex vivo TNF-alpha and IL-1 beta production. J Appl Physiol. (1995) 79:1497–503. doi: 10.1152/jappl.1995.79.5.1497
183. Castellani L, Perry CG, Macpherson RE, Root-McCaig J, Huber JS, Arkell AM, et al. Exercise-mediated IL-6 signaling occurs independent of inflammation and is amplified by training in mouse adipose tissue. J Appl Physiol. (2015) 119:1347–54. doi: 10.1152/japplphysiol.00551.2015
184. Yao B, Li L, Guan X, Zhu J, Liu Q, Qu B, et al. Endurance training inhibits the JAK2/STAT3 pathway to alleviate sarcopenia. Physiol Res. (2024) 73:295–304. doi: 10.33549/physiolres.935234
185. Testa MTJ, Cella PS, Marinello PC, Frajacomo FTT, Padilha CS, Perandini PC, et al. Resistance training attenuates activation of STAT3 and muscle atrophy in tumor-bearing mice. Front Oncol. (2022) 12:880787. doi: 10.3389/fonc.2022.880787
186. Chow BC, Li S, Zhu X, Jiao J, Quach B, Baker JS, et al. Effects of descending or ascending stair exercise on body composition, insulin sensitivity, and inflammatory markers in young Chinese women with obesity: A randomized controlled trial. J Sports Sci. (2021) 39:496–502. doi: 10.1080/02640414.2020.1829362
187. Mavropalias G, Sim M, Taaffe DR, Galvão DA, Spry N, Kraemer WJ, et al. Exercise medicine for cancer cachexia: targeted exercise to counteract mechanisms and treatment side effects. J Cancer Res Clin Oncol. (2022) 148:1389–406. doi: 10.1007/s00432-022-03927-0
188. Huang Q, Wu M, Wu X, Zhang Y, and Xia Y. Muscle-to-tumor crosstalk: The effect of exercise-induced myokine on cancer progression. Biochim Biophys Acta Rev Cancer. (2022) 1877:188761. doi: 10.1016/j.bbcan.2022.188761
189. Pedersen BK, Steensberg A, and Schjerling P. Muscle-derived interleukin-6: possible biological effects. J Physiol. (2001) 536:329–37. doi: 10.1111/j.1469-7793.2001.0329c.xd
190. Guo Y, Ma B, Li X, Hui H, Zhou Y, Li N, et al. n-3 PUFA can reduce IL-6 and TNF levels in patients with cancer. Br J Nutr. (2023) 129:54–65. doi: 10.1017/s0007114522000575
191. Chehimi M, Ward R, Pestel J, Robert M, Pesenti S, Bendridi N, et al. Omega-3 polyunsaturated fatty acids inhibit IL-17A secretion through decreased ICAM-1 expression in T cells co-cultured with adipose-derived stem cells harvested from adipose tissues of obese subjects. Mol Nutr Food Res. (2019) 63:e1801148. doi: 10.1002/mnfr.201801148
192. Liu Y, Lin J, Chen Y, Li Z, Zhou J, Lu X, et al. Omega−3 polyunsaturated fatty acids inhibit IL−11/STAT3 signaling in hepatocytes during acetaminophen hepatotoxicity. Int J Mol Med. (2021) 48:190. doi: 10.3892/ijmm.2021.5023
193. Fernández-Lázaro D, Arribalzaga S, Gutiérrez-Abejón E, Azarbayjani MA, Mielgo-Ayuso J, and Roche E. Omega-3 fatty acid supplementation on post-exercise inflammation, muscle damage, oxidative response, and sports performance in physically healthy adults-A systematic review of randomized controlled trials. Nutrients. (2024) 16:2044. doi: 10.3390/nu16132044
194. Kyriakidou Y, Wood C, Ferrier C, Dolci A, and Elliott B. The effect of Omega-3 polyunsaturated fatty acid supplementation on exercise-induced muscle damage. J Int Soc Sports Nutr. (2021) 18:9. doi: 10.1186/s12970-020-00405-1
195. Smith GI, Julliand S, Reeds DN, Sinacore DR, Klein S, and Mittendorfer B. Fish oil-derived n-3 PUFA therapy increases muscle mass and function in healthy older adults. Am J Clin Nutr. (2015) 102:115–22. doi: 10.3945/ajcn.114.105833
196. Therdyothin A, Phiphopthatsanee N, and Isanejad M. The effect of omega-3 fatty acids on sarcopenia: mechanism of action and potential efficacy. Mar Drugs. (2023) 21:399. doi: 10.3390/md21070399
197. Gortan Cappellari G, Aleksova A, Dal Ferro M, Cannatà A, Semolic A, Guarnaccia A, et al. n-3 PUFA-enriched diet preserves skeletal muscle mitochondrial function and redox state and prevents muscle mass loss in mice with chronic heart failure. Nutrients. (2023) 15:3108. doi: 10.3390/nu15143108
198. Lee HW, Baker E, Lee KM, Persinger AM, Hawkins W, and Puppa M. Effects of low-dose leucine supplementation on gastrocnemius muscle mitochondrial content and protein turnover in tumor-bearing mice. Appl Physiol Nutr Metab. (2019) 44:997–1004. doi: 10.1139/apnm-2018-0765
199. Cruz B, Oliveira A, Ventrucci G, and Gomes-Marcondes MCC. A leucine-rich diet modulates the mTOR cell signalling pathway in the gastrocnemius muscle under different Walker-256 tumour growth conditions. BMC Cancer. (2019) 19:349. doi: 10.1186/s12885-019-5448-0
200. Holeček M. Beta-hydroxy-beta-methylbutyrate supplementation and skeletal muscle in healthy and muscle-wasting conditions. J Cachexia Sarcopenia Muscle. (2017) 8:529–41. doi: 10.1002/jcsm.12208
201. Li YJ, Zhang C, Martincuks A, Herrmann A, and Yu H. STAT proteins in cancer: orchestration of metabolism. Nat Rev Cancer. (2023) 23:115–34. doi: 10.1038/s41568-022-00537-3
202. Yue P and Turkson J. Targeting STAT3 in cancer: how successful are we? Expert Opin Investig Drugs. (2009) 18:45–56. doi: 10.1517/13543780802565791
203. Jung JE, Kim HS, Lee CS, Shin YJ, Kim YN, Kang GH, et al. STAT3 inhibits the degradation of HIF-1alpha by pVHL-mediated ubiquitination. Exp Mol Med. (2008) 40:479–85. doi: 10.3858/emm.2008.40.5.479
204. Siveen KS, Sikka S, Surana R, Dai X, Zhang J, Kumar AP, et al. Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors. Biochim Biophys Acta. (2014) 1845:136–54. doi: 10.1016/j.bbcan.2013.12.005
205. Heinrich PC, Behrmann I, Haan S, Hermanns HM, Müller-Newen G, and Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. (2003) 374:1–20. doi: 10.1042/bj20030407
206. Song J, Wang J, Tian S, and Li H. Discovery of STAT3 inhibitors: recent advances and future perspectives. Curr Med Chem. (2023) 30:1824–47. doi: 10.2174/0929867329666220819093117
207. Sun Z, Jiang Q, Gao B, Zhang X, Bu L, Wang L, et al. AKT blocks SIK1-mediated repression of STAT3 to promote breast tumorigenesis. Cancer Res. (2023) 83:1264–79. doi: 10.1158/0008-5472.Can-22-3407
208. Liu C, Feng H, Song L, Li S, Wu Y, and Yang L. Synergistic effects of thalidomide and cisplatin are mediated via the PI3K/AKT and JAK1/STAT3 signaling pathways in cervical cancer. Oncol Rep. (2022) 48:169. doi: 10.3892/or.2022.8384
Keywords: cancer cachexia, STAT3, systemic inflammation, muscle wasting, pathogenic mechanisms, therapeutic strategies
Citation: Lv X and Ding S (2025) Unraveling the role of STAT3 in Cancer Cachexia: pathogenic mechanisms and therapeutic opportunities. Front. Endocrinol. 16:1608612. doi: 10.3389/fendo.2025.1608612
Received: 12 April 2025; Accepted: 11 June 2025;
Published: 09 July 2025.
Edited by:
Claudia Lagranha, Federal University of Pernambuco, BrazilReviewed by:
Phablo Abreu, University of Minnesota Twin Cities, United StatesRuoxin Fang, Huazhong University of Science and Technology, China
Copyright © 2025 Lv and Ding. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shengguang Ding, c2hlbmdndWFuZ2RpbmdAbnR1LmVkdS5jbg==