 Rupangi C. Vasavada
Rupangi C. Vasavada Sangeeta Dhawan
Sangeeta Dhawan- Department of Translational Research and Cellular Therapeutics, Arthur Riggs Diabetes and Metabolism Research Institute, City of Hope, Duarte, CA, United States
Diminished functional beta-cell mass is a key pathogenic mechanism underlying both type 1 and type 2 diabetes (T1D and T2D), precipitated by the progressive impairment of insulin secretion, loss of cellular identity, and ultimately, beta-cell death. The replenishment of beta-cell deficit through the transplantation of pancreatic islets from cadaveric donors or beta-cells derived from human embryonic stem cells has shown transformative therapeutic potential. However, the regeneration of functional beta-cell mass in vivo remains an important therapeutic goal, as a more physiological and scalable approach. Effective beta-cell replenishment must address the underlying causes of beta-cell loss, such as cellular stress and autoimmunity, while simultaneously promoting beta-cell regeneration, function, and survival. Advances in the mechanistic underpinnings of beta-cell differentiation, growth, and survival, coupled with cutting-edge high-throughput screening methods have accelerated the discovery of novel therapeutic targets and small-molecule interventions. Current strategies for in vivo beta-cell expansion include modulating the cell-cycle to promote replication, reprogramming non-beta-cell lineages into beta-cells, and enhancing beta-cell survival. However, the limited regenerative capacity and inherently high stress sensitivity of beta-cells pose significant barriers to their in vivo expansion, further complicated by the fundamental conflict between replication and functional maintenance, and the high vulnerability of replicating cells in a metabolically stressed environment. There has been tremendous progress in developing approaches that simultaneously promote beta-cell expansion and function. In this review, we discuss the recent advances in beta-cell expansion, along with remaining challenges and emerging opportunities to address them.
1 Introduction
Strategies that promote the regeneration of functional beta-cells in vivo by stimulating their proliferation represent an attractive and physiological therapeutic approach that can benefit patients with T2D as well as T1D, given that residual beta-cells persist in established T1D. Human beta-cell expansion poses a unique therapeutic challenge due to their limited replicative capacity, heightened vulnerability to stress during proliferation, and remodeling of beta-cell heterogeneity throughout diabetes progression. Here, we examine advances in stimulating beta-cell expansion, focusing on how recent mechanistic insights into beta-cell biology are informing the development of therapies that can simultaneously enhance proliferation, function, and survival in diabetes.
2 Targeting beta-cell proliferation for regeneration of beta-cell mass
Targeting the pathways involved in beta-cell proliferation has emerged as a therapeutically promising approach for beta-cell expansion in vivo, leveraging one of the body’s natural mechanisms for growth in early life as well as subsequent adaptive beta-cell expansion in response to increased insulin demand. Many of the pathways regulating beta-cell proliferation are amenable to small molecule therapeutics, making this approach particularly attractive for clinical translation. Consequently, significant efforts have focused on identifying the molecular control of beta-cell replication.
2.1 Harnessing physiological control of beta-cell replication for regeneration
Replication is the primary mechanism of postnatal beta-cell growth and crucial for adaptation to metabolic challenges (1–3) (Figure 1A). Understanding the physiological regulators of replication can reveal potential targets for therapeutic beta-cell expansion in diabetes. Beta-cells undergo substantial replication during early postnatal stages to establish beta-cell mass (4–6). Proliferation gradually declines as beta-cells mature functionally, revealing an inverse relationship between function and replication (7–9). The mTOR/PI3K/Akt pathway integrates nutrient cues and growth signals to promote early postnatal beta-cell expansion, while increased AMPK (AMP-activated protein kinase) activity subsequently enhances functional maturation while restraining proliferation through cell-cycle regulation (10–12). The calcineurin/NFAT pathway is another critical regulator of beta-cell growth and functional maturation, with DYRK1A kinase inhibiting the pro-proliferative activity of NFATs and serving as a cell-cycle brake (13–15).
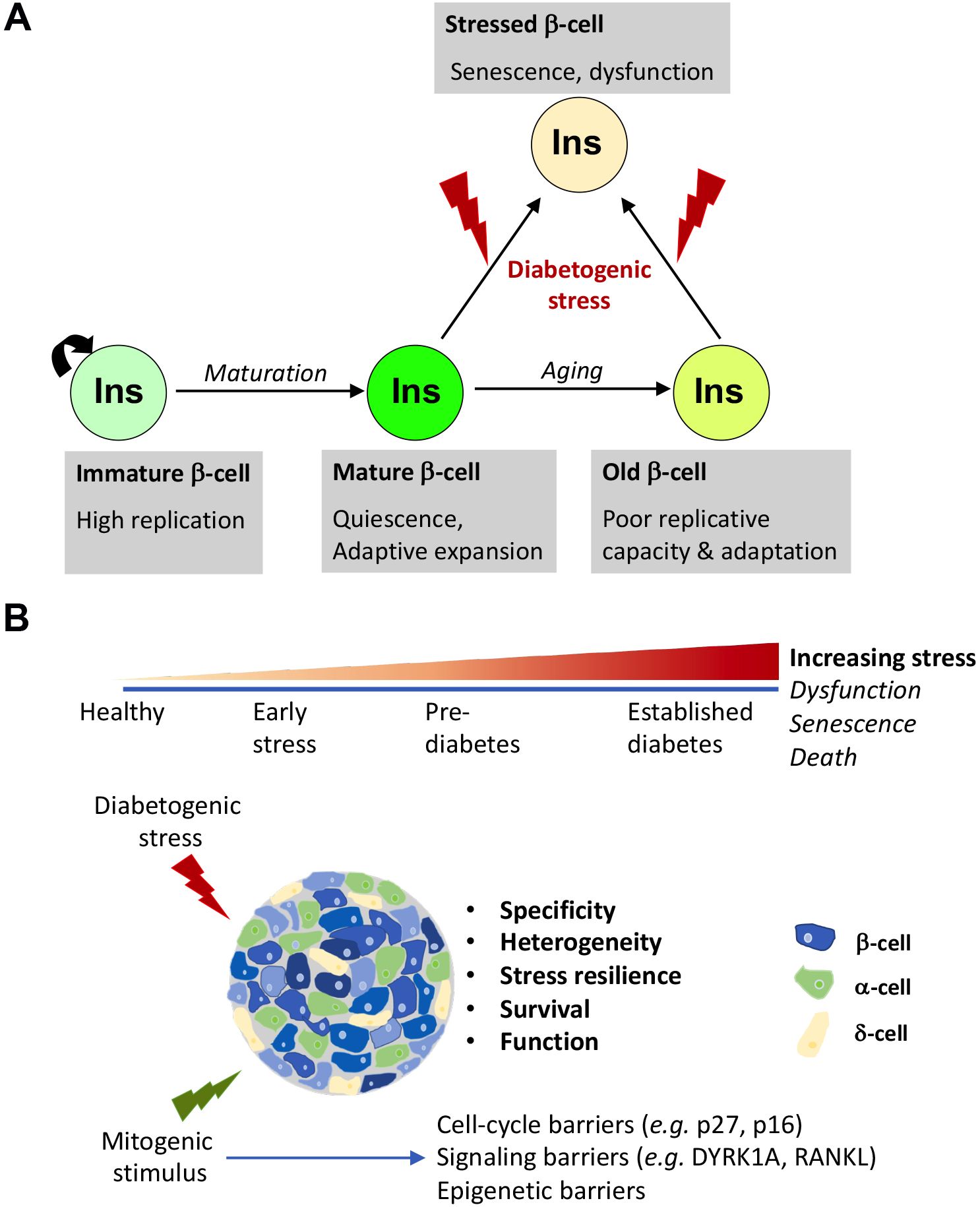
Figure 1. Beta-cell proliferation: natural history and therapeutic considerations. (A) Physiological control of beta-cell expansion in health and disease. The beta-cell proliferation landscape changes substantially throughout life. Early in postnatal life, beta-cells rapidly expand to accommodate growth and establish beta-cell mass. As growth gradually tapers out, beta-cells exit cell-cycle and assume a functionally mature and quiescent state, which retains the capacity to expand on demand, in response to physiological challenges for increased insulin needs, such as pregnancy, obesity, islet injury etc. This capacity of adaptive replication, however, declines with aging due to the onset of p16 accumulation dependent replicative senescence. The replicative response of beta-cells not only depends on age but also varies depending on the nature of the metabolic demand. Continuously high metabolic demand and/or exposure to inflammation can trigger maladaptation and result in dysfunction and a state of permanent cell-cycle exit marked by a pro-inflammatory phenotype: stress-induced senescence. If the stress persists, beta-cells can eventually succumb and undergo cell-death. Ultimately, this can result in diabetes. (B) Factors influencing the efficacy of mitogenic agents on beta-cell expansion in diabetes. The beta-cell phenotype evolves through the course of the initiation and progression of diabetes, amounting to significant heterogeneity of disease. To promote beta-cell expansion in these conditions, the therapeutic agents must not only overcome barriers to beta-cell replication that are especially stringent in human beta-cells, but must also promote resilience to stress and survival, support optimal insulin secretion post-replication, and ensure a healthy milieu by combating inflammation and/or promoting optimal islet niche. This can be achieved either by using mitogenic agents that inherently support beta-cell health or by combining them with therapeutic agents that resolve stress and boost function. Ideally, an optimal therapeutic agent must also selectively target beta-cells to avoid off-target effect. Finally, there is considerable heterogeneity in beta-cell phenotypes, which is further remodeled in disease. Identifying which beta-cell subset maybe most responsive for expansion remains an ongoing line of enquiry that will benefit approaches that involve replication to boost beta-cell mass in diabetes.
The regulation of postnatal beta-cell replication depends on the balance between mitogen-induced cyclin D2-Cdk4 complex formation and inhibitory actions of cyclin kinase inhibitors (CKIs) on the cyclinE-Cdk2 complexes (1, 16–18). As growth progresses, accumulation of the CKI p27 drives cell-cycle exit and a quiescent beta-cell state (19). Beta-cell mass is largely established following the postnatal proliferative phase, with subsequent maintenance occurring primarily through limited replication and survival (6, 20, 21). Multiple mechanisms reinforce beta-cell quiescence: p27 degradation control via SCF ubiquitin ligase and Menin-mediated epigenetic regulation, which maintains CKI expression and co-represses transcription of cell-cycle genes driven by TEAD1, an effector of the Hippo pathway which controls tissue growth (22–25). p27 degradation is essential for proliferation of mature beta-cells in response to increased insulin demand, such as in obesity and pregnancy. The metabolic state of beta-cells is a key determinant of proliferative capacity (26). The postnatal maturation transforms beta-cell proliferative response from growth-driven mass expansion to selective compensatory proliferation upon increased insulin demand (27). However, this capacity diminishes with age due to replicative senescence marked by accumulation of CKI p16 (28–32). This age-related decline involves altered epigenetic control, with young beta-cells repressing p16 through Polycomb proteins (Ezh2, Bmi1) (33, 34). Aging reduces Ezh2 levels and increases Trithorax complex (containing Mll1 and JmjD3) activity to induce p16 accumulation (35). DNA methylation is another epigenetic mechanism that modulates the transcriptional programs involved in beta-cell maturation and proliferation (36). Age-dependent shifts in growth factor signaling drive beta-cell senescence: declining PDGF signaling reduces Ezh2 expression, while increased TGFβ signaling activates Trithorax-dependent p16 accumulation, promoting replicative senescence (37, 38).
Our understanding of pathways that modulate physiological beta-cell expansion has revealed species-, age-, and physiological state-specific differences in replicative potential and identified several therapeutic avenues. For instance, PDGF can stimulate β-cell proliferation only in juvenile human islets, while more downstream manipulation of the PDGF/Ezh2 pathway using beta-cell specific overexpression of Ezh2 can overcome the age-related replicative barrier in mice (35, 37). Similarly, TGFβ inhibitors can successfully drive the expansion of both adult murine and human beta-cells (38). Along these lines, antibody arrays on serum of young mice identified Wnt-1 inducible signaling protein 1 (Wisp1) as a circulating factor that can induce rodent and human beta-cell replication (39). Compensatory beta-cell expansion during insulin resistance (IR) involves critical growth factors produced by liver (40). Proteomic analysis of liver secretome under conditions of IR has identified the hepatocyte-secreted protease inhibitor SerpinB1 as a key factor promoting beta-cell replication through elastase inhibition—an effect that can be mimicked by small molecule elastase inhibitors. In agreement with this, SerpinB1 deficient mice display poor beta-cell compensation in response to IR (41). Pregnancy hormones have also been explored as therapeutic agents for beta-cell expansion. Prolactin (PRL), placental lactogen (PL), estrogen, and other factors such as serotonin—naturally upregulated during pregnancy—induce beta-cell replication, providing another physiology-based approach to beta-cell regeneration (42–45). PRL-dependent beta-cell replication in vivo requires the protein osteoprotegerin (OPG), as PRL fails to induce beta-cell expansion in whole-body OPG knockout mice (46). Notably, while PRL can only enhance beta-cell replication in rodents but not in human islets due to the lack of PRL-receptors (47), OPG can independently induce both rodent and human beta-cell replication in vitro and in vivo (46). Glucagon-like peptide 1 (GLP1) is another circulating factor that promotes beta-cell replication in mice, with GLP1 receptor agonists (GLP1RAs) emerging as potential therapeutic agents when used in combination with other proliferative agents capable of enhancing human beta-cell replication (48–50). Extracellular-vesicles (EVs) have emerged as critical modulators of cellular function in both health and disease, including diabetes (51–53). Recent work suggests that EVs derived from sources that naturally stimulate beta-cell replication and regeneration—such as stem and progenitor cells, and serum from young or pregnant individuals— may hold promise for beta-cell expansion (54).
2.2 Barriers to human beta-cell proliferation: lessons from islet pathologies
Much of our mechanistic understanding of beta-cell replication comes from rodent models, which exhibit higher baseline proliferation and mitogen responsiveness, limiting translational relevance. Mature human beta-cells display remarkable resistance to proliferation, presenting a significant challenge for regenerative approaches (55–57). The cell-cycle machinery differs substantially between species—humans have abundant CDK6 but minimal Cyclin D2, while mice show the reverse pattern (58). While replication is the primary mechanism of postnatal beta-cell growth in both mice and humans, its role in human beta-cell adaptive expansion has been debated (1–3, 59). Pathological conditions exhibiting abnormal beta-cell expansion have provided valuable insights into the molecular barriers that normally restrict human beta-cell replication. Comprehensive genomic, epigenomic, and transcriptional profiling of human insulinomas—rare beta-cell tumors characterized by insulin overproduction—has provided crucial insight into human beta-cell proliferation. Most insulinomas exhibit concurrent mutations in multiple chromatin regulators, particularly in the Polycomb and Trithorax Group genes (such as EZH2, YY1, RING1, BMI1, MEN1, KMT2C and KDM6A). EZH2 overexpression appears in most insulinomas, likely driving hyperproliferation and altered gene expression (60). All insulinomas share an aberrant DNA methylation signature within the 11p15.5-p15.4 sub-region containing critical imprinting control regions for the IGF2/H19 and KCNQ1/CDKN1C loci that regulate body growth (61). Mutations in this region cause Beckwith-Wiedemann syndrome, another condition with beta-cell hyperproliferation (62). Comparison of normal beta-cell and insulinoma transcriptomic profiles has revealed the DREAM (dimerization partner, retinoblastoma-like, E2F and MuvB) repressor complex as a key transcriptional modulator of human beta-cell proliferation. DREAM complex assembly occurs in response to DYRK1A activation to establish and maintain quiescence. Loss of DYRK1A results in the reorganization of DREAM components into a pro-proliferative complex called MMB (MuvB complex with MYBL2) which promotes entry to the S phase (63). Indeed, DYRK1A inhibition has emerged as a major target for inducing human beta-cell proliferation.
2.3 Unbiased approaches for beta-cell expansion
Increasing efforts are focused on unbiased identification of molecules that promote beta-cell expansion with higher efficacy and cell specificity, mitigating off-target effects. These efforts include high-throughput screens (HTS) utilizing human islet cells to screen chemical and RNAi libraries (64–68). Among the most notable advances from these efforts is the identification of the dual-specificity tyrosine-regulated kinase-1a (DYRK1A) as a central negative regulator of beta-cell proliferation. DYRK1A intersects with multiple arms of beta-cell replication machinery, including NFATs and DREAM complex (13, 63). Multiple independent screens converged on DYRK1A, leading to the discovery of Harmine and INDY, small molecules that induce beta-cell replication through DYRK1A inhibition (69). Parallel screens identified aminopyrazine analogs and the adenosine kinase inhibitor 5-iodotubercidin (5-IT) as additional compounds that enhance human beta-cell replication through DYRK1A/NFAT-dependent and -independent mechanisms (70, 71). Another screen leveraged a VGF promoter-reporter to screen for NKX6.1 pathway activators, based on findings that NKX6.1 enhances beta- but not alpha-cell replication and induces VGF expression, discovering a compound that selectively promotes human beta-cell proliferation via a mechanism distinct from DYRK1A inhibition (72).
Complementing these in vitro approaches, zebrafish has emerged as a scalable in vivo screening model system, offering real-time assessment of islet expansion and function. A luminescence-based ubiquitination reporter screen in zebrafish identified an inhibitor of salt-inducible kinases (SIKs) as a potent cross-species stimulator of beta-cell proliferation via unfolded protein response (UPR) activation (73). The relevance of SIKs was independently validated in an RNAi screen of the human G protein-coupled receptor (GPCR) family on human islets, which uncovered GPR3 as a negative regulator of human beta-cell proliferation via modulation of SIK2 activity (74). Similarly, a small molecule screening in zebrafish identified the non-canonical IκB kinase TANK-binding kinase 1 (TBK1) as a negative regulator of beta-cell replication and shown to directly modulate rodent and human beta-cell regeneration (75).
An unbiased search using microarrays identified OPG as a downstream effector of lactogens in the beta-cell. OPG induces both rodent and human beta-cell replication by inhibiting the Receptor Activator of NF-κB (RANK)/RANK ligand (RANKL) pathway (46, 76). This discovery led us to investigate Denosumab, a monoclonal antibody against human RANKL and an FDA-approved osteoporosis drug. Like OPG, Denosumab enhances human beta-cell replication in vitro and in vivo in human islets transplanted into immunodeficient mice (46, 76). Notably, Denosumab is currently in a phase1/2 multi-center clinical trial (NCT06524960) to assess safety and efficacy in improving beta-cell function in early T1D. More recently, we identified Leucine-rich repeat-containing G-protein coupled receptor 4 (LGR4) as a novel physiological inhibitor of the RANK pathway (77). The soluble extracellular domain of LGR4 (LGR4-ECD) has emerged as a potential new therapeutic for osteoporosis, revealing another promising target for enhancing beta-cell mass in diabetes (78).
3 Considerations for therapeutic expansion of beta-cell mass in diabetes
Beta-cell dysfunction and loss characterize both T1D and T2D, making beta-cell expansion an attractive therapeutic goal. However, effective translation of such candidates to clinical application requires careful consideration of other disease relevant aspects. First, the ideal therapeutic must not only enhance beta-cell replication but also improve beta-cell function and resilience. The heterogeneous nature of diabetes pathogenesis coupled with intrinsic beta-cell heterogeneity poses additional challenges, as therapeutic response could vary based on disease stage and corresponding changes in beta-cell phenotype. Additionally, specific targeting to beta-cells is essential to prevent off-target effects and minimize risks of cellular transformation. Successful beta-cell regenerative therapies must overcome these gaps.
3.1 Non-replicative mechanisms in beta-cell expansion: clues for combination therapy
Replication inherently creates cellular vulnerability, especially under diabetic stress conditions (79–81). Replication is also mutually incompatible with glucose-stimulated insulin secretion, as these processes compete for cellular resources and involve different metabolic states (7). Effective beta-cell expansion strategies in diabetes must therefore concurrently promote stress resilience and survival, while ensuring that cells can regain their mature phenotype and functional capacity following replication. Emerging evidence from pre-clinical studies shows that many therapeutic agents can simultaneously enhance human beta-cell replication while improving survival, function and/or maturity. Examples include inhibitors of DYRK1A and the RANK pathway that simultaneously promote beta-cell survival alongside expansion (49, 76, 82). These agents offer a distinct advantage for clinical applications, as they confer multiple beneficial effects on beta-cell health under disease-relevant stressors. Modulation of these pathways likely exerts temporally distinct effects on beta-cells, initially boosting survival and resilience, followed by successful expansion and restoration of function. Recent work on the TMEM219-IGFBP3 axis illustrates this perfectly; the ligand IGFBP3, markedly elevated in sera from both T1D and T2D patients, triggers beta-cell apoptosis by activating its cognate death receptor TMEM219. Pharmacological disruption of this axis using the extracellular domain of TMEM219 (ecto-TMEM219) provides temporally distinct benefits in preclinical diabetes models by initially enhancing beta-cell survival and preventing diabetes onset and promoting beta-cell expansion in the long-term (83).
Recent studies point to an even more complex mechanistic landscape, suggesting that some beta-cell proliferative agents may also induce trans-differentiation of non-beta-cells to beta-cells alongside replication. Single-cell transcriptomic analysis of human islets reveals that treatment with DYRK1A inhibitors targets cycling alpha cells which may transdifferentiate into beta-cells (84). This process could be particularly relevant in T1D, which is characterized by severe beta-cell depletion and widespread senescence among residual beta-cells, thus significantly limiting the pool of replication-competent beta-cells. The islet microenvironment also impacts beta-cell response to proliferative agents, with some molecules showing anti-inflammatory and immunomodulatory effects that create a more permissive niche for regeneration (76, 82).
Given this complexity, approaches that strategically combine agents that target complementary pathways could offer superior outcomes. Combining agents that enhance survival, mitigate ER-stress, or reduce inflammation with mitogenic stimuli can create synergistic effects for sustainable beta-cell expansion. This approach has been demonstrated using DYRK1A inhibitors with GLP-1RAs and TGF-β inhibitors, and combinations of sitagliptin with melatonin (49, 82, 85, 86). GLP-1RAs, widely used in T2D, offer multiple benefits including beta-cell regeneration (87, 88). Selective elimination of senescent beta-cells prevents immune-mediated destruction in T1D models and protects against T2D, with potential to transform a stress-prone environment into one conducive to both replication and trans-differentiation (89, 90). Similarly, pharmaceutical agents targeting the integrated stress response (ISR) have shown promise for enhancing beta-cell survival and function (91–94). Combining agents that target senescence or ISR with those directed at beta-cell proliferation may therefore be an effective way to removing barriers to regeneration in the setting of diabetes while preserving the regenerative potential of remaining healthy cells. Notably, IGFBP3 is a component of beta-cell senescence associated secretory phenotype (SASP) in T1D (89). Thus, targeting of IGFBP3/TMEM219 axis might offer additional protective effects against senescence (83).
Beyond these considerations, achieving cellular specificity remains a critical hurdle for clinical translation. Several innovative approaches are being developed to address this challenge, including leveraging beta-cell-enriched receptors such as GLP1R for drug targeting, engineering sophisticated nanoparticle and recombinant adeno-associated virus (rAAV) delivery systems, and repurposing existing FDA-approved medications with established safety profiles (46, 76, 82, 95–97). For instance, Denosumab has now advanced to early clinical trials (NCT06524960) following promising pre-clinical evidence of beta-cell regeneration. Similarly, a recent phase-1 safety study demonstrated that orally administered Harmine at doses below 2.7 mg/kg produced minimal to no adverse events in healthy volunteers, representing an encouraging step toward clinical application (98). Therapeutic targeting of pathways that are largely beta-cell specific, e.g. the IGFBP3/TMEM219 axis (83), would be crucial to improve specificity. These advances in targeting specificity, combined with approaches addressing overall beta-cell health, provide a comprehensive framework for developing therapeutically viable beta-cell regeneration strategies with acceptable safety profiles.
3.2 The proliferative beta-cell: what, when, where?
A fundamental question in developing therapeutic beta-cell expansion strategies concerns how regeneration may proceed across the heterogeneous pancreatic landscape in diabetes (99–104). For instance, would regeneration in T1D primarily occur in regions with residual beta-cells, in areas completely devoid of beta-cells, or both? Immune infiltration adds additional complexity—regions with active autoimmunity may not be permissive for regeneration despite having residual beta cells, while areas where inflammation has resolved might permit replication even with fewer remaining cells. There is growing recognition of beta-cell heterogeneity, with distinct subpopulations emerging throughout development, aging, and across islet regions, exhibiting variations in molecular characteristics, functional properties, and replicative potential (105–116). For instance, in mice, cells expressing Flattop (Fltp) display a functionally mature, post-mitotic phenotype, while Fltp- cells represent the proliferative beta-cell subset (117). Similarly, CyTOF mass cytometry revealed a small subpopulation of Ki67+ beta-cells in human pancreas (118), suggesting that replicative heterogeneity marks both rodent and human beta-cells. This is particularly relevant for diabetes therapies, where identifying target beta-cell subpopulations with high functionality and replicative potential is crucial.
Beta-cell heterogeneity undergoes significant temporal remodeling in response to metabolic stress during diabetes progression. Transition to a replication-permissive state represents one of the earliest responses to acute beta-cell stress, along with functional compensation and activation of stress-response and pro-survival pathways (119–122). Select beta-cell subpopulations in diabetes recapitulate molecular features of the more proliferative and immature neonatal beta-cells (116, 123–126), suggesting these subsets could be optimal targets for expansion. Emerging evidence shows that modest endoplasmic reticulum stress (ER stress) drives beta-cell replication in response to increased insulin demand, with beta-cells exhibiting an active unfolded protein response (UPR) showing more proliferative capacity (127). However, unresolved stress can eventually prevent cell-cycle progression, instead inducing a pro-inflammatory senescent state (99, 128).
Multiple studies have demonstrated the accumulation of senescent beta-cell subpopulations in both T1D and T2D, highlighting heterogeneous beta-cell proliferative capacity in diabetes (89, 90, 99, 105, 129, 130). Unrelenting stress in diabetes can cause irreversible cellular damage and cause cell-death. Agents that successfully promote beta-cell replication in diabetes must therefore simultaneously be able to mitigate stress, creating optimal milieu for expansion. Emerging data suggests that T2D islets exhibit enhanced responsiveness to proliferative agents. Interestingly, Wang et al. demonstrated that the rare Ki67+ beta-cells detectable in non-diabetic human pancreas were absent in T2D (118), supporting the hypothesis that diabetic conditions impair the ability of beta-cells to progress through cell-cycle. This apparent contradiction may reflect different phases of the disease, with early stress responses enhancing proliferative potential while chronic stress ultimately blocks cell-cycle progression.
Heterogeneity of beta-cell replicative potential and the subtype shifts observed in diabetes raise several questions (Figure 1B). First, which beta-cell subpopulations represent optimal targets for therapeutic expansion? Second, as most therapeutic candidates are tested in human islets from non-diabetic donors, question remains whether these interventions would be effective in diabetic patients. Finally, what impact does ongoing beta-cell expansion have on islet function? Addressing these questions requires understanding how beta-cell states and their replicative potential are dynamically modulated in response to stress. Defining temporal evolution of these populations can determine the optimal disease stage for therapeutic intervention. Advances in whole-pancreas visualization permit comprehensive temporal assessment of beta-cell expansion following drug treatment in pre-clinical models (82, 131). Using single-cell and spatial transcriptomic and proteomic approaches to map molecular trajectories of beta-cell functional and proliferative states at different disease stages will help decipher the optimal therapeutic window and target beta-cell population(s) to boost beta-cell expansion (106, 118, 132–136).
4 Next-Generation approaches for beta-cell regeneration
The future of beta-cell expansion approaches is rapidly evolving toward combinatorial strategies and precision therapies, warranting platforms that enhance target discovery and therapeutic specificity. Gene-editing platforms such as CRISPR have emerged as transformative tools enabling targeted modulation of gene expression, high-throughput screens, and disease modeling (137, 138). Recent proof-of-principle work has leveraged gene-editing tools to induce human beta-cell expansion by altering the epigenetic state and expression of specific cell-cycle modulators (139, 140). Recent work has also demonstrated the feasibility of using CRISPR to target regulatory genomic regions in primary human islets to modulate gene expression and islet function, an important advancement considering the challenges of delivering CRISPR components into quiescent, post-mitotic cells like human beta-cells (141, 142). Genome-wide CRISPR screens have also been used to identify critical regulators of beta-cell function and immune vulnerability (143, 144). Translating these findings to clinical applications requires careful validation in human beta-cells. Advances in long-term culturing of live pancreatic tissue slices now allow study of human beta-cells within their native microenvironment (145). This platform enables in situ lineage tracing to monitor beta-cell formation in real-time, assess therapeutic candidates in disease context, and evaluate off-target effects alongside islet function (132, 146). These technologies offer unprecedented opportunities to develop personalized regenerative approaches for diabetes, potentially transforming treatment paradigms.
Author contributions
SD: Conceptualization, Writing – review & editing, Funding acquisition, Writing – original draft. RCV: Conceptualization, Writing – review & editing, Funding acquisition, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Work in Dhawan laboratory is supported by grants from the National Institutes of Health (R01DK120523), the Wanek Family Project to Cure Type 1 Diabetes, and the Arthur Riggs Diabetes and Metabolism Research Institute Pilot Award Program at City of Hope. Work in Vasavada laboratory is supported by grants from the National Institutes of Health (R01DK102893, R01DK125856 and R01DK133885), Breakthrough T1D (formerly JDRF) (17-2012-37, SRA-2023-1421-M-B), the Wanek Family Project to Cure Type 1 Diabetes, the Arthur Riggs Diabetes and Metabolism Research Institute Pilot Award Program at City of Hope, George and Irina Schaeffer Foundation, and anonymous SS donor.
Acknowledgments
We thank members of the Dhawan and Vasavada labs, Dr. Adolfo Garcia-Ocaña (City of Hope), and Dr. Julia Panzer (City of Hope) for helpful discussions.
Conflict of interest
RCV is a named inventor on two U.S. utility patents, no. 9333239, issued on 10 May 2016, and no. 9724386, issued on 08 August 2017, for “Use of Osteoprotegerin OPG to increase human pancreatic beta cell survival and proliferation”.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Georgia S and Bhushan A. Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J Clin Invest. (2004) 114:963–8. doi: 10.1172/JCI22098
2. Dor Y, Brown J, Martinez OI, and Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. (2004) 429:41–6. doi: 10.1038/nature02520nature
3. Meier JJ, Butler AE, Saisho Y, Monchamp T, Galasso R, Bhushan A, et al. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes. (2008) 57:1584–94. doi: 10.2337/db07-1369
4. Dhawan S, Georgia S, and Bhushan A. Formation and regeneration of the endocrine pancreas. Curr Opin Cell Biol. (2007) 19:634–45. doi: 10.1016/j.ceb.2007.09.015
5. Bonner-Weir S. Life and death of the pancreatic beta cells. Trends Endocrinol Metab. (2000) 11:375–8. doi: 10.1016/s1043-2760(00)00305-2
6. Finegood DT, Scaglia L, and Bonner-Weir S. Dynamics of beta-cell mass in the growing rat pancreas. Estimation with a simple mathematical model. Diabetes. (1995) 44:249–56. doi: 10.2337/diab.44.3.249
7. Puri S, Roy N, Russ HA, Leonhardt L, French EK, Roy R, et al. Replication confers beta cell immaturity. Nat Commun. (2018) 9:485. doi: 10.1038/s41467-018-02939-0
8. Blum B, Hrvatin S, Schuetz C, Bonal C, Rezania A, and Melton DA. Functional beta-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3. Nat Biotechnol. (2012) 30:261–4. doi: 10.1038/nbt.2141
9. Wortham M and Sander M. Transcriptional mechanisms of pancreatic beta-cell maturation and functional adaptation. Trends Endocrinol Metab. (2021) 32:474–87. doi: 10.1016/j.tem.2021.04.011
10. Barsby T and Otonkoski T. Maturation of beta cells: lessons from in vivo and in vitro models. Diabetologia. (2022) 65:917–30. doi: 10.1007/s00125-022-05672-y
11. Jaafar R, Tran S, Shah AN, Sun G, Valdearcos M, Marchetti P, et al. mTORC1 to AMPK switching underlies beta-cell metabolic plasticity during maturation and diabetes. J Clin Invest. (2019) 129:4124–37. doi: 10.1172/JCI127021
12. Helman A, Cangelosi AL, Davis JC, Pham Q, Rothman A, Faust AL, et al. A nutrient-sensing transition at birth triggers glucose-responsive insulin secretion. Cell Metab. (2020) 31:1004–1016 e1005. doi: 10.1016/j.cmet.2020.04.004
13. Heit JJ, Apelqvist AA, Gu X, Winslow MM, Neilson JR, Crabtree GR, et al. Calcineurin/NFAT signalling regulates pancreatic beta-cell growth and function. Nature. (2006) 443:345–9. doi: 10.1038/nature05097
14. Kalwat MA, Scheuner D, Rodrigues-Dos-Santos K, Eizirik DL, and Cobb MH. The pancreatic ss-cell response to secretory demands and adaption to stress. Endocrinology. (2021) 162. doi: 10.1210/endocr/bqab173
15. Goodyer WR, Gu X, Liu Y, Bottino R, Crabtree GR, and Kim SK. Neonatal beta cell development in mice and humans is regulated by calcineurin/NFAT. Dev Cell. (2012) 23:21–34. doi: 10.1016/j.devcel.2012.05.014
16. Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, Reddy EP, et al. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat Genet. (1999) 22:44–52. doi: 10.1038/8751
17. He LM, Sartori DJ, Teta M, Opare-Addo LM, Rankin MM, Long SY, et al. Cyclin D2 protein stability is regulated in pancreatic beta-cells. Mol Endocrinol. (2009) 23:1865–75. doi: 10.1210/me.2009-0057
18. Kushner JA, Ciemerych MA, Sicinska E, Wartschow LM, Teta M, Long SY, et al. Cyclins D2 and D1 are essential for postnatal pancreatic beta-cell growth. Mol Cell Biol. (2005) 25:3752–62. doi: 10.1128/MCB.25.9.3752-3762.2005
19. Georgia S and Bhushan A. p27 Regulates the transition of beta-cells from quiescence to proliferation. Diabetes. (2006) 55:2950–6. doi: 10.2337/db06-0249
20. Scaglia L, Cahill CJ, Finegood DT, and Bonner-Weir S. Apoptosis participates in the remodeling of the endocrine pancreas in the neonatal rat. Endocrinology. (1997) 138:1736–41. doi: 10.1210/endo.138.4.5069
21. Wortham M and Sander M. Mechanisms of beta-cell functional adaptation to changes in workload. Diabetes Obes Metab. (2016) 18 Suppl 1:78–86. doi: 10.1111/dom.12729
22. Zhong L, Georgia S, Tschen SI, Nakayama K, and Bhushan A. Essential role of Skp2-mediated p27 degradation in growth and adaptive expansion of pancreatic beta cells. J Clin Invest. (2007) 117:2869–76. doi: 10.1172/JCI32198
23. Karnik SK, Chen H, McLean GW, Heit JJ, Gu X, Zhang AY, et al. Menin controls growth of pancreatic beta-cells in pregnant mice and promotes gestational diabetes mellitus. Science. (2007) 318:806–9. doi: 10.1126/science.1146812
24. Karnik SK, Hughes CM, Gu X, Rozenblatt-Rosen O, McLean GW, Xiong Y, et al. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc Natl Acad Sci U S A. (2005) 102:14659–64. doi: 10.1073/pnas.0503484102
25. Li F, Liu R, Negi V, Yang P, Lee J, Jagannathan R, et al. VGLL4 and MENIN function as TEAD1 corepressors to block pancreatic beta cell proliferation. Cell Rep. (2023) 42:111904. doi: 10.1016/j.celrep.2022.111904
26. Porat S, Weinberg-Corem N, Tornovsky-Babaey S, Schyr-Ben-Haroush R, Hija A, Stolovich-Rain M, et al. Control of pancreatic beta cell regeneration by glucose metabolism. Cell Metab. (2011) 13:440–9. doi: 10.1016/j.cmet.2011.02.012
27. Stolovich-Rain M, Enk J, Vikesa J, Nielsen FC, Saada A, Glaser B, et al. Weaning triggers a maturation step of pancreatic beta cells. Dev Cell. (2015) 32:535–45. doi: 10.1016/j.devcel.2015.01.002
28. Tschen SI, Dhawan S, Gurlo T, and Bhushan A. Age-dependent decline in beta-cell proliferation restricts the capacity of beta-cell regeneration in mice. Diabetes. (2009) 58:1312–20. doi: 10.2337/db08-1651
29. Kushner JA. The role of aging upon beta cell turnover. J Clin Invest. (2013) 123:990–5. doi: 10.1172/JCI64095
30. Rankin MM and Kushner JA. Adaptive beta-cell proliferation is severely restricted with advanced age. Diabetes. (2009) 58:1365–72. doi: 10.2337/db08-1198
31. Teta M, Long SY, Wartschow LM, Rankin MM, and Kushner JA. Very slow turnover of beta-cells in aged adult mice. Diabetes. (2005) 54:2557–67. doi: 10.2337/diabetes.54.9.2557
32. Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, et al. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. (2006) 443:453–7. doi: 10.1038/nature05092
33. Dhawan S, Tschen SI, and Bhushan A. Bmi-1 regulates the Ink4a/Arf locus to control pancreatic beta-cell proliferation. Genes Dev. (2009) 23:906–11. doi: 10.1101/gad.1742609
34. Chen H, Gu X, Su IH, Bottino R, Contreras JL, Tarakhovsky A, et al. Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev. (2009) 23:975–85. doi: 10.1101/gad.1742509
35. Zhou JX, Dhawan S, Fu H, Snyder E, Bottino R, Kundu S, et al. Combined modulation of polycomb and trithorax genes rejuvenates beta cell replication. J Clin Invest. (2013) 123:4849–58. doi: 10.1172/JCI69468
36. Avrahami D, Li C, Zhang J, Schug J, Avrahami R, Rao S, et al. Aging-Dependent demethylation of regulatory elements correlates with chromatin state and improved beta cell function. Cell Metab. (2015) 22:619–32. doi: 10.1016/j.cmet.2015.07.025
37. Chen H, Gu X, Liu Y, Wang J, Wirt SE, Bottino R, et al. PDGF signalling controls age-dependent proliferation in pancreatic beta-cells. Nature. (2011) 478:349–55. doi: 10.1038/nature10502
38. Dhawan S, Dirice E, Kulkarni RN, and Bhushan A. Inhibition of TGF-beta signaling promotes human pancreatic beta cell replication. Diabetes. (2016) 65(5):1208–18. doi: 10.2337/db15-1331
39. Fernandez-Ruiz R, Garcia-Alaman A, Esteban Y, Mir-Coll J, Serra-Navarro B, Fontcuberta-PiSunyer M, et al. Wisp1 is a circulating factor that stimulates proliferation of adult mouse and human beta cells. Nat Commun. (2020) 11:5982. doi: 10.1038/s41467-020-19657-1
40. El Ouaamari A, Kawamori D, Dirice E, Liew CW, Shadrach JL, Hu J, et al. Liver-derived systemic factors drive beta cell hyperplasia in insulin-resistant states. Cell Rep. (2013) 3:401–10. doi: 10.1016/j.celrep.2013.01.007
41. El Ouaamari A, Dirice E, Gedeon N, Hu J, Zhou JY, Shirakawa J, et al. SerpinB1 promotes pancreatic beta cell proliferation. Cell Metab. (2016) 23:194–205. doi: 10.1016/j.cmet.2015.12.001
42. Vasavada RC, Garcia-Ocana A, Zawalich WS, Sorenson RL, Dann P, Syed M, et al. Targeted expression of placental lactogen in the beta cells of transgenic mice results in beta cell proliferation, islet mass augmentation, and hypoglycemia. J Biol Chem. (2000) 275:15399–406. doi: 10.1074/jbc.275.20.15399
43. Vasavada RC, Gonzalez-Pertusa JA, Fujinaka Y, Fiaschi-Taesch N, Cozar-Castellano I, and Garcia-Ocana A. Growth factors and beta cell replication. Int J Biochem Cell Biol. (2006) 38:931–50. doi: 10.1016/j.biocel.2005.08.003
44. Millette K, Rodriguez K, Sheng X, Finley SD, and Georgia S. Exogenous lactogenic signaling stimulates beta cell replication in vivo and in vitro. Biomolecules. (2022) 12. doi: 10.3390/biom12020215
45. Pretorius M and Huang C. Beta-cell adaptation to pregnancy - role of calcium dynamics. Front Endocrinol (Lausanne). (2022) 13:853876. doi: 10.3389/fendo.2022.853876
46. Kondegowda NG, Fenutria R, Pollack IR, Orthofer M, Garcia-Ocana A, Penninger JM, et al. Osteoprotegerin and denosumab stimulate human beta cell proliferation through inhibition of the receptor activator of NF-kappaB ligand pathway. Cell Metab. (2015) 22:77–85. doi: 10.1016/j.cmet.2015.05.021
47. Chen H, Kleinberger JW, Takane KK, Salim F, Fiaschi-Taesch N, Pappas K, et al. Augmented stat5 signaling bypasses multiple impediments to lactogen-mediated proliferation in human beta cells. Diabetes. (2015) 64(11):3784–97. doi: 10.2337/db15-0083
48. Brubaker PL and Drucker DJ. Minireview: Glucagon-like peptides regulate cell proliferation and apoptosis in the pancreas, gut, and central nervous system. Endocrinology. (2004) 145:2653–9. doi: 10.1210/en.2004-0015
49. Ackeifi C, Wang P, Karakose E, Manning Fox JE, Gonzalez BJ, Liu H, et al. GLP-1 receptor agonists synergize with DYRK1A inhibitors to potentiate functional human beta cell regeneration. Sci Transl Med. (2020) 12. doi: 10.1126/scitranslmed.aaw9996
50. Dai C, Hang Y, Shostak A, Poffenberger G, Hart N, Prasad N, et al. Age-dependent human beta cell proliferation induced by glucagon-like peptide 1 and calcineurin signaling. J Clin Invest. (2017) 127:3835–44. doi: 10.1172/JCI91761
51. Hooten N and Evans MK. Extracellular vesicles as signaling mediators in type 2 diabetes mellitus. Am J Physiol Cell Physiol. (2020) 318:C1189–99. doi: 10.1152/ajpcell.00536.2019
52. Liu J, Zhang Y, Tian Y, Huang W, Tong N, and Fu. Integrative biology of extracellular vesicles in diabetes mellitus and diabetic complications. Theranostics. (2022) 12:1342–72. doi: 10.7150/thno.65778
53. Aguirre RS, Kulkarni A, Becker MW, Lei X, Sarkar S, Ramanadham S, et al. Extracellular vesicles in beta cell biology: Role of lipids in vesicle biogenesis, cargo, and intercellular signaling. Mol Metab. (2022) 63:101545. doi: 10.1016/j.molmet.2022.101545
54. Sylvester-Armstrong KR, Reeder CF, Powell A, Becker MW, Hagan DW, Chen J, et al. Serum from pregnant donors induces human beta cell proliferation. Islets. (2024) 16:2334044. doi: 10.1080/19382014.2024.2334044
55. Kulkarni RN, Mizrachi EB, Ocana AG, and Stewart AF. Human beta-cell proliferation and intracellular signaling: driving in the dark without a road map. Diabetes. (2012) 61:2205–13. doi: 10.2337/db12-0018
56. Stewart AF, Hussain MA, Garcia-Ocana A, Vasavada RC, Bhushan A, Bernal-Mizrachi E, et al. Human beta-Cell proliferation and intracellular signaling: part 3. Diabetes. (2015) 64:1872–85. doi: 10.2337/db14-184364/6/1872
57. Bernal-Mizrachi E, Kulkarni RN, Scott DK, Mauvais-Jarvis F, Stewart AF, et al. Human beta-cell proliferation and intracellular signaling part 2: still driving in the dark without a road map. Diabetes. (2014) 63:819–31. doi: 10.2337/db13-1146
58. Fiaschi-Taesch N, Bigatel TA, Sicari B, Takane KK, Salim F, Velazquez-Garcia S, et al. Survey of the human pancreatic beta-cell G1/S proteome reveals a potential therapeutic role for cdk-6 and cyclin D1 in enhancing human beta-cell replication and function in vivo. Diabetes. (2009) 58:882–93. doi: 10.2337/db08-0631
59. Butler AE, Cao-Minh L, Galasso R, Rizza RA, Corradin A, Cobelli C, et al. Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia. (2010) 53:2167–76. doi: 10.1007/s00125-010-1809-6
60. Wang H, Bender A, Wang P, Karakose E, Inabnet WB, Libutti SK, et al. Insights into beta cell regeneration for diabetes via integration of molecular landscapes in human insulinomas. Nat Commun. (2017) 8:767. doi: 10.1038/s41467-017-00992-9
61. Karakose E, Wang H, Inabnet W, Thakker RV, Libutti S, Fernandez-Ranvier G, et al. Aberrant methylation underlies insulin gene expression in human insulinoma. Nat Commun. (2020) 11:5210. doi: 10.1038/s41467-020-18839-1
62. Eggermann T, Algar E, Lapunzina P, Mackay D, Maher ER, Mannens M, et al. Clinical utility gene card for: Beckwith-Wiedemann Syndrome. Eur J Hum Genet. (2014) 22:435. doi: 10.1038/ejhg.2013.132
63. Wang P, Karakose E, Argmann C, Wang H, Balev M, Brody RI, et al. Disrupting the DREAM complex enables proliferation of adult human pancreatic beta cells. J Clin Invest. (2022) 132. doi: 10.1172/JCI157086
64. Walpita D, Hasaka T, Spoonamore J, Vetere A, Takane KK, Fomina-Yadlin D, et al. A human islet cell culture system for high-throughput screening. J Biomol Screen. (2012) 17:509–18. doi: 10.1177/1087057111430253
65. Walpita D and Wagner BK. Evaluation of compounds in primary human islet cell culture. Curr Protoc Chem Biol. (2014) 6:157–68. doi: 10.1002/9780470559277.ch140088
66. Robitaille K, Rourke JL, McBane JE, Fu A, Baird S, Du Q, et al. High-throughput functional genomics identifies regulators of primary human beta cell proliferation. J Biol Chem. (2016) 291:4614–25. doi: 10.1074/jbc.M115.683912
67. Aamodt KI, Aramandla R, Brown JJ, Fiaschi-Taesch N, Wang P, Stewart AF, et al. Development of a reliable automated screening system to identify small molecules and biologics that promote human beta-cell regeneration. Am J Physiol Endocrinol Metab. (2016) 311:E859–68. doi: 10.1152/ajpendo.00515.2015
68. Abdolazimi Y, Zhao Z, Lee S, Xu H, Allegretti P, Horton TM, et al. CC-401 Promotes beta-Cell Replication via Pleiotropic Consequences of DYRK1A/B Inhibition. Endocrinology. (2018) 159:3143–57. doi: 10.1210/en.2018-00083
69. Wang P, Alvarez-Perez JC, Felsenfeld DP, Liu H, Sivendran S, Bender A, et al. A high-throughput chemical screen reveals that harmine-mediated inhibition of DYRK1A increases human pancreatic beta cell replication. Nat Med. (2015) 21:383–8. doi: 10.1038/nm.3820
70. Shen W, Taylor B, Jin Q, Nguyen-Tran V, Meeusen S, Zhang YQ, et al. Inhibition of DYRK1A and GSK3B induces human beta-cell proliferation. Nat Commun. (2015) 6:8372. doi: 10.1038/ncomms9372
71. Dirice E, Walpita D, Vetere A, Meier BC, Kahraman S, Hu J, et al. Inhibition of DYRK1A stimulates human beta-Cell proliferation. Diabetes. (2016) 65:1660–71. doi: 10.2337/db15-1127
72. Hohmeier HE, Zhang L, Taylor B, Stephens S, Lu D, McNamara P, et al. Identification of a small molecule that stimulates human beta-cell proliferation and insulin secretion, and protects against cytotoxic stress in rat insulinoma cells. PloS One. (2020) 15:e0224344. doi: 10.1371/journal.pone.0224344
73. Charbord J, Ren L, Sharma RB, Johansson A, Agren R, Chu L, et al. In vivo screen identifies a SIK inhibitor that induces beta cell proliferation through a transient UPR. Nat Metab. (2021) 3:682–700. doi: 10.1038/s42255-021-00391-x
74. Iorio C, Rourke JL, Wells L, Sakamaki JI, Moon E, Hu Q, et al. Silencing the G-protein coupled receptor 3-salt inducible kinase 2 pathway promotes human beta cell proliferation. Commun Biol. (2021) 4:907. doi: 10.1038/s42003-021-02433-2
75. Jia YF, Jeeva S, Xu J, Heppelmann CJ, Jang JS, Slama MQ, et al. TBK1 regulates regeneration of pancreatic beta-cells. Sci Rep. (2020) 10:19374. doi: 10.1038/s41598-020-76600-6
76. Kondegowda NG, Filipowska J, Do JS, Leon-Rivera N, Li R, Hampton R, et al. RANKL/RANK is required for cytokine-induced beta cell death; osteoprotegerin, a RANKL inhibitor, reverses rodent type 1 diabetes. Sci Adv. (2023) 9:eadf5238. doi: 10.1126/sciadv.adf5238
77. Filipowska J, Cisneros Z, Varghese SS, Leon-Rivera N, Wang P, Kang R, et al. LGR4 is essential for maintaining beta-cell homeostasis through suppression of RANK. Mol Metab. (2025) 92:102097. doi: 10.1016/j.molmet.2025.102097
78. Filipowska J, Kondegowda NG, Leon-Rivera N, Dhawan S, and Vasavada RC. LGR4, a G protein-coupled receptor with a systemic role: from development to metabolic regulation. Front Endocrinol (Lausanne). (2022) 13:867001. doi: 10.3389/fendo.2022.867001
79. Varghese SS, Hernandez-De La Pena AG, and Dhawan S. Safeguarding genomic integrity in beta-cells: implications for beta-cell differentiation, growth, and dysfunction. Biochem Soc Trans. (2024) 52:2133–44. doi: 10.1042/BST20231519
80. Meier JJ, Ritzel RA, Maedler K, Gurlo T, and Butler PC. Increased vulnerability of newly forming beta cells to cytokine-induced cell death. Diabetologia. (2006) 49:83–9. doi: 10.1007/s00125-005-0069-3
81. Ritzel RA and Butler PC. Replication increases beta-cell vulnerability to human islet amyloid polypeptide-induced apoptosis. Diabetes. (2003) 52:1701–8. doi: 10.2337/diabetes.52.7.1701
82. Rosselot C, Li Y, Wang P, Alvarsson A, Beliard K, Lu G, et al. Harmine and exendin-4 combination therapy safely expands human beta cell mass in vivo in a mouse xenograft system. Sci Transl Med. (2024) 16:eadg3456. doi: 10.1126/scitranslmed.adg3456
83. D’Addio F, Maestroni A, Assi E, Ben Nasr M, Amabile G, Usuelli V, et al. The IGFBP3/TMEM219 pathway regulates beta cell homeostasis. Nat Commun. (2022) 13:684. doi: 10.1038/s41467-022-28360-2
84. Karakose E, Wang X, Wang P, Carcamo S, Demircioglu D, Lambertini L, et al. Cycling alpha cells in regenerative drug-treated human pancreatic islets may serve as key beta cell progenitors. Cell Rep Med. (2024) 5:101832. doi: 10.1016/j.xcrm.2024.101832
85. Wang P, Karakose E, Liu H, Swartz E, Ackeifi C, Zlatanic V, et al. Combined inhibition of DYRK1A, SMAD, and trithorax pathways synergizes to induce robust replication in adult human beta cells. Cell Metab. (2019) 29:638–52:e635. doi: 10.1016/j.cmet.2018.12.005
86. Patel R, Parmar N, Rathwa N, Palit SP, Li Y, Garcia-Ocana A, et al. A novel therapeutic combination of sitagliptin and melatonin regenerates pancreatic beta-cells in mouse and human islets. Biochim Biophys Acta Mol Cell Res. (2022) 1869:119263. doi: 10.1016/j.bbamcr.2022.119263
87. Wei T, Cui X, Jiang Y, Wang K, Wang D, Li F, et al. Glucagon Acting at the GLP-1 Receptor Contributes to beta-Cell Regeneration Induced by Glucagon Receptor Antagonism in Diabetic Mice. Diabetes. (2023) 72:599–610. doi: 10.2337/db22-0784
88. Jiang Y, Yang J, Xia L, Wei T, Cui X, Wang D, et al. Gut microbiota-Tryptophan metabolism-GLP-1 axis participates in beta-Cell regeneration induced by dapagliflozin. Diabetes. (2024) 73:926–40. doi: 10.2337/db23-0553
89. Thompson PJ, Shah A, Ntranos V, Van Gool F, Atkinson M, and Bhushan A. Targeted elimination of senescent beta cells prevents type 1 diabetes. Cell Metab. (2019) 29:1045–1060 e1010. doi: 10.1016/j.cmet.2019.01.021
90. Aguayo-Mazzucato C, Andle J, Lee TB Jr., Midha A, Talemal L, Chipashvili V, et al. Acceleration of beta cell aging determines diabetes and senolysis improves disease outcomes. Cell Metab. (2019) 30:129–142 e124. doi: 10.1016/j.cmet.2019.05.006
91. Thielen LA, Chen J, Jing G, Moukha-Chafiq O, Xu G, Jo S, et al. Identification of an anti-diabetic, orally available small molecule that regulates TXNIP expression and glucagon action. Cell Metab. (2020) 32:353–365 e358. doi: 10.1016/j.cmet.2020.07.002
92. Muralidharan C, Huang F, Enriquez JR, Wang JE, Nelson JB, Nargis T, et al. Inhibition of the eukaryotic initiation factor-2alpha kinase PERK decreases risk of autoimmune diabetes in mice. J Clin Invest. (2024) 134. doi: 10.1172/JCI176136
93. Ovalle F, Grimes T, Xu G, Patel AJ, Grayson TB, Thielen LA, et al. Verapamil and beta cell function in adults with recent-onset type 1 diabetes. Nat Med. (2018) 24:1108–12. doi: 10.1038/s41591-018-0089-4
94. Forlenza GP, McVean J, Beck RW, Bauza C, Bailey R, Buckingham B, et al. Effect of verapamil on pancreatic beta cell function in newly diagnosed pediatric type 1 diabetes: A randomized clinical trial. JAMA. (2023) 329:990–9. doi: 10.1001/jama.2023.2064
95. Chai S, Kim Y, Galivo F, Dorrell C, Wakefield L, Posey J, et al. Development of a beta cell-Specific expression control element for recombinant adeno-Associated virus. Hum Gene Ther. (2022) 33:789–800. doi: 10.1089/hum.2021.219
96. Cristelo C, Nunes R, Pinto S, Marques JM, Gama FM, and Sarmento B. Targeting beta cells with cathelicidin nanomedicines improves insulin function and pancreas regeneration in type 1 diabetic rats. ACS Pharmacol Transl Sci. (2023) 6:1544–60. doi: 10.1021/acsptsci.3c00218
97. Behzadifar S, Barras A, Plaisance V, Pawlowski V, Szunerits S, Abderrahmani A, et al. Polymer-based nanostructures for pancreatic beta-cell imaging and non-invasive treatment of diabetes. Pharmaceutics. (2023) 15. doi: 10.3390/pharmaceutics15041215
98. Ables JL, Israel L, Wood O, Govindarajulu U, Fremont RT, Banerjee R, et al. A Phase 1 single ascending dose study of pure oral harmine in healthy volunteers. J Psychopharmacol. (2024) 38:911–23. doi: 10.1177/02698811241273772
99. Cha J, Aguayo-Mazzucato C, and Thompson PJ. Pancreatic beta-cell senescence in diabetes: mechanisms, markers and therapies. Front Endocrinol (Lausanne). (2023) 14:1212716. doi: 10.3389/fendo.2023.1212716
100. den Hollander NHM and Roep BO. From disease and patient heterogeneity to precision medicine in type 1 diabetes. Front Med (Lausanne). (2022) 9:932086. doi: 10.3389/fmed.2022.932086
101. Eizirik DL, Colli ML, and Ortis F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol. (2009) 5:219–26. doi: 10.1038/nrendo.2009.21
102. Garyu JW, Meffre E, Cotsapas C, and Herold KC. Progress and challenges for treating Type 1 diabetes. J Autoimmun. (2016) 71:1–9. doi: 10.1016/j.jaut.2016.04.004
103. Redondo MJ and Morgan NG. Heterogeneity and endotypes in type 1 diabetes mellitus. Nat Rev Endocrinol. (2023) 19:542–54. doi: 10.1038/s41574-023-00853-0
104. Roep BO. The need and benefit of immune monitoring to define patient and disease heterogeneity, mechanisms of therapeutic action and efficacy of intervention therapy for precision medicine in type 1 diabetes. Front Immunol. (2023) 14:1112858. doi: 10.3389/fimmu.2023.1112858
105. Aguayo-Mazzucato C, van Haaren M, Mruk M, Lee TB Jr., Crawford C, Hollister-Lock J, et al. beta Cell Aging Markers Have Heterogeneous Distribution and Are Induced by Insulin Resistance. Cell Metab. (2017) 25:898–910 e895. doi: 10.1016/j.cmet.2017.03.015
106. Baron M, Veres A, Wolock SL, Faust AL, Gaujoux R, Vetere A, et al. A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra-cell population structure. Cell Syst. (2016) 3:346–360 e344. doi: 10.1016/j.cels.2016.08.011
107. Benninger RKP and Hodson DJ. New understanding of beta-cell heterogeneity and in situ islet function. Diabetes. (2018) 67:537–47. doi: 10.2337/dbi17-0040
108. Benninger RKP and Kravets V. The physiological role of beta-cell heterogeneity in pancreatic islet function. Nat Rev Endocrinol. (2021) 18:9–22. doi: 10.1038/s41574-021-00568-0
109. Da Silva Xavier G and Rutter GA. Metabolic and functional heterogeneity in pancreatic beta cells. J Mol Biol. (2020) 432:1395–406. doi: 10.1016/j.jmb.2019.08.005
110. Dorrell C, Schug J, Canaday PS, Russ HA, Tarlow BD, Grompe MT, et al. Human islets contain four distinct subtypes of beta cells. Nat Commun. (2016) 7:11756. doi: 10.1038/ncomms11756
111. Fang Z, Weng C, Li H, Tao R, Mai W, Liu X, et al. Single-Cell heterogeneity analysis and CRISPR screen identify key beta-Cell-Specific disease genes. Cell Rep. (2019) 26:3132–3144 e3137. doi: 10.1016/j.celrep.2019.02.043
112. Gutierrez GD, Gromada J, and Sussel L. Heterogeneity of the pancreatic beta cell. Front Genet. (2017) 8:22. doi: 10.3389/fgene.2017.00022
113. Pipeleers DG. Heterogeneity in pancreatic beta-cell population. Diabetes. (1992) 41:777–81. doi: 10.2337/diab.41.7.777
114. Parveen N, Wang JK, Bhattacharya S, Cuala J, Rajkumar MS, Butler AE, et al. DNA methylation-dependent restriction of tyrosine hydroxylase contributes to pancreatic beta-cell heterogeneity. Diabetes. (2023) 72:575–89. doi: 10.2337/db22-0506
115. Yu V, Yong F, Marta A, Khadayate S, Osakwe A, Bhattacharya S, et al. Differential CpG methylation at Nnat in the early establishment of beta cell heterogeneity. Diabetologia. (2024) 67(6):1079–94. doi: 10.1007/s00125-024-06123-6
116. Rodnoi P, Rajkumar M, Moin ASM, Georgia SK, Butler AE, and Dhawan S. Neuropeptide Y expression marks partially differentiated beta cells in mice and humans. JCI Insight. (2017) 2. doi: 10.1172/jci.insight.94005
117. Bader E, Migliorini A, Gegg M, Moruzzi N, Gerdes J, Roscioni SS, et al. Identification of proliferative and mature beta-cells in the islets of Langerhans. Nature. (2016) 535:430–4. doi: 10.1038/nature18624
118. Wang YJ, Golson ML, Schug J, Traum D, Liu C, Vivek K, et al. Single-Cell mass cytometry analysis of the human endocrine pancreas. Cell Metab. (2016) 24:616–26. doi: 10.1016/j.cmet.2016.09.007
119. Kulkarni A, Muralidharan C, May SC, Tersey SA, and Mirmira RG. Inside the beta cell: molecular stress response pathways in diabetes pathogenesis. Endocrinology. (2022) 164. doi: 10.1210/endocr/bqac184
120. Weir GC and Bonner-Weir S. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes. (2004) 53 Suppl 3:S16–21. doi: 10.2337/diabetes.53.suppl_3.s16
121. Weir GC, Laybutt DR, Kaneto H, Bonner-Weir S, and Sharma A. Beta-cell adaptation and decompensation during the progression of diabetes. Diabetes. (2001) 50 Suppl 1:S154–159. doi: 10.2337/diabetes.50.2007.s154
122. Ma K and Dhawan S. Yo-yo dieting: mixed messages for beta-cell plasticity. Diabetes. (2022) 71:2253–5. doi: 10.2337/dbi22-0024
123. Talchai C, Xuan S, Lin HV, Sussel L, and Accili D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell. (2012) 150:1223–34. doi: 10.1016/j.cell.2012.07.029
124. Dahan T, Ziv O, Horwitz E, Zemmour H, Lavi J, Swisa A, et al. Pancreatic beta-Cells express the fetal islet hormone gastrin in rodent and human diabetes. Diabetes. (2017) 66:426–36. doi: 10.2337/db16-0641
125. Hudish LI, Reusch JE, and Sussel L. beta Cell dysfunction during progression of metabolic syndrome to type 2 diabetes. J Clin Invest. (2019) 129:4001–8. doi: 10.1172/JCI129188
126. Hunter CS and Stein RW. Evidence for loss in identity, de-differentiation, and trans-differentiation of islet beta-cells in type 2 diabetes. Front Genet. (2017) 8:35. doi: 10.3389/fgene.2017.00035
127. Sharma RB, O’Donnell AC, Stamateris RE, Ha B, McCloskey KM, Reynolds PR, et al. Insulin demand regulates beta cell number via the unfolded protein response. J Clin Invest. (2015) 125:3831–46. doi: 10.1172/JCI79264
128. Varghese SS and Dhawan S. Senescence: a double-edged sword in beta-cell health and failure? Front Endocrinol (Lausanne). (2023) 14:1196460. doi: 10.3389/fendo.2023.1196460
129. Brawerman G, Pipella J, and Thompson PJ. DNA damage to beta cells in culture recapitulates features of senescent beta cells that accumulate in type 1 diabetes. Mol Metab. (2022) 62:101524. doi: 10.1016/j.molmet.2022.101524
130. Aguayo-Mazzucato C. Functional changes in beta cells during ageing and senescence. Diabetologia. (2020) 63:2022–9. doi: 10.1007/s00125-020-05185-6
131. Reissaus CA, Pineros AR, Twigg AN, Orr KS, Conteh AM, Martinez MM, et al. A versatile, portable intravital microscopy platform for studying beta-cell biology in vivo. Sci Rep. (2019) 9:8449. doi: 10.1038/s41598-019-44777-0
132. Doke M, Alvarez-Cubela S, Klein D, Altilio I, Schulz J, Mateus Goncalves L, et al. Dynamic scRNA-seq of live human pancreatic slices reveals functional endocrine cell neogenesis through an intermediate ducto-acinar stage. Cell Metab. (2023) 35:1944–1960 e1947. doi: 10.1016/j.cmet.2023.10.001
133. Olaniru Oe, Kadolsky U, Kannambath S, Vaikkinen H, Fung K, Dhami P, et al. Single-cell transcriptomic and spatial landscapes of the developing human pancreas. Cell Metab. (2023) 35:184–199 e185. doi: 10.1016/j.cmet.2022.11.009
134. Bosi E, Marchetti P, Rutter GA, and Eizirik DL. Human alpha cell transcriptomic signatures of types 1 and 2 diabetes highlight disease-specific dysfunction pathways. iScience. (2022) 25:105056. doi: 10.1016/j.isci.2022.105056
135. Camunas-Soler J, Dai XQ, Hang Y, Bautista A, Lyon J, Suzuki K, et al. Patch-Seq links single-Cell transcriptomes to human islet dysfunction in diabetes. Cell Metab. (2020) 31:1017–1031 e1014. doi: 10.1016/j.cmet.2020.04.005
136. Enge M, Arda HE, Mignardi M, Beausang J, Bottino R, Kim SK, et al. Single-cell analysis of human pancreas reveals transcriptional signatures of aging and somatic mutation patterns. Cell. (2017) 171:321–330 e314. doi: 10.1016/j.cell.2017.09.004
137. Pacesa M, Pelea O, and Jinek M. Past, present, and future of CRISPR genome editing technologies. Cell. (2024) 187:1076–100. doi: 10.1016/j.cell.2024.01.042
138. Wang JY and Doudna JA. CRISPR technology: A decade of genome editing is only the beginning. Science. (2023) 379:eadd8643. doi: 10.1126/science.add8643
139. Ou K, Yu M, Moss NG, Wang YJ, Wang AW, Nguyen SC, et al. Targeted demethylation at the CDKN1C/p57 locus induces human beta cell replication. J Clin Invest. (2019) 129:209–14. doi: 10.1172/JCI99170
140. Bernstein DL, Le Lay JE, Ruano EG, and Kaestner KH. TALE-mediated epigenetic suppression of CDKN2A increases replication in human fibroblasts. J Clin Invest. (2015) 125:1998–2006. doi: 10.1172/JCI77321
141. Bevacqua RJ, Dai X, Lam JY, Gu X, Friedlander MSH, Tellez K, et al. CRISPR-based genome editing in primary human pancreatic islet cells. Nat Commun. (2021) 12:2397. doi: 10.1038/s41467-021-22651-w
142. Bevacqua RJ, Zhao W, Merheb E, Kim SH, Marson A, Gloyn AL, et al. Multiplexed CRISPR gene editing in primary human islet cells with Cas9 ribonucleoprotein. iScience. (2024) 27:108693. doi: 10.1016/j.isci.2023.108693
143. Cai EP, Ishikawa Y, Zhang W, Leite NC, Li J, Hou S, et al. Genome-scale in vivo CRISPR screen identifies RNLS as a target for beta cell protection in type 1 diabetes. Nat Metab. (2020) 2:934–45. doi: 10.1038/s42255-020-0254-1
144. Lim D, Sreekanth V, Cox KJ, Law BK, Wagner BK, Karp JM, et al. Engineering designer beta cells with a CRISPR-Cas9 conjugation platform. Nat Commun. (2020) 11:4043. doi: 10.1038/s41467-020-17725-0
145. Panzer JK, Hiller H, Cohrs CM, Almaca J, Enos SJ, Beery M, et al. Pancreas tissue slices from organ donors enable in situ analysis of type 1 diabetes pathogenesis. JCI Insight. (2020) 5. doi: 10.1172/jci.insight.134525
Keywords: beta cells, replication, proliferation, regeneration, therapeutics, diabetes
Citation: Vasavada RC and Dhawan S (2025) Harnessing beta-cell replication: advancing molecular insights to regenerative therapies in diabetes. Front. Endocrinol. 16:1612576. doi: 10.3389/fendo.2025.1612576
Received: 15 April 2025; Accepted: 04 June 2025;
Published: 19 June 2025.
Edited by:
George K Gittes, University of Pittsburgh, United StatesReviewed by:
Emma Assi, University of Milan, ItalyCopyright © 2025 Vasavada and Dhawan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sangeeta Dhawan, c2RoYXdhbkBjb2gub3Jn; Rupangi C. Vasavada, cnZhc2F2YWRhQGNvaC5vcmc=