 Paola Giordano1
Paola Giordano1 Giuseppina Leonetti1Vanja Granberg1Rosa Maria Pia Casolino1
Giuseppina Leonetti1Vanja Granberg1Rosa Maria Pia Casolino1 Giuseppe Lassandro1
Giuseppe Lassandro1 Maurizio Delvecchio2*
Maurizio Delvecchio2* Giovanna Linguiti1
Giovanna Linguiti1- 1Department of Interdisciplinary Medicine, Pediatric Unit “B. Trambusti”, Cystic Fibrosis Regional Reference Center, University of Bari “Aldo Moro”, Bari, Italy
- 2Department of Biotechnological and Applied Clinical Sciences, University of L’Aquila, L’Aquila, Italy
Introduction: Cystic fibrosis (CF) is an autosomal recessive disorder caused by mutations in the CFTR gene, leading to impaired chloride transport, thickened mucus, and multiorgan dysfunction. Among its complications, cystic fibrosis-related diabetes (CFRD) is a major concern, characterized by progressive b-cell dysfunction and insulin deficiency. The advent of CFTR modulators, including ivacaftor, lumacaftor/ivacaftor, and elexacaftor/tezacaftor/ivacaftor (ETI), has revolutionized CF management by improving pulmonary function, nutritional status, and overall survival. However, their effects on glucose metabolism remain under investigation.
Methods: This systematic review (systematic review registration: PROSPERO 2025 CRD420251021499) analyzes recent evidence on the impact of CFTR modulators on CFRD in children and young adults. Results: Ivacaftor demonstrates potential benefits in glucose regulation, enhancing insulin secretion and glucagon control, particularly in patients with gating mutations. Conversely, lumacaftor/ivacaftor exhibits inconsistent effects, with some studies indicating glucose tolerance improvements while others report insulin sensitivity decline.
Discussion: ETI therapy shows modest but generally positive effects on glycemic control, with reductions in HbA1c and fasting glucose, though without significant changes in insulin secretion. While CFTR modulators improve systemic health, their role in directly preventing or reversing CFRD remains unclear. Further longitudinal studies are needed to optimize therapeutic strategies and elucidate the long-term metabolic effects of CFTR modulation in CF patients.
Systematic review registration: https://www.crd.york.ac.uk/PROSPERO/, identifier CRD420251021499.
1 Introduction
Cystic fibrosis (CF) is the most prevalent life-limiting autosomal recessive disorder among Caucasian populations, with an incidence of approximately 1 in 2,500–3,500 live births in Europe and North America (1). The disease results from mutations in the CFTR (Cystic Fibrosis Transmembrane Conductance Regulator) gene, located on chromosome 7q31.2, which encodes a chloride channel crucial for ion and fluid homeostasis across epithelial surfaces (2, 3). Dysfunctional CFTR activity disrupts ion transport, leading to the production of thick, dehydrated mucus that obstructs multiple organ systems, primarily affecting the respiratory, gastrointestinal, and reproductive tracts. CF manifests as a multisystemic disorder with highly variable phenotypic expression (4). The predominant clinical features involve the respiratory, gastrointestinal, endocrine, and reproductive systems. Respiratory complications include persistent cough, bronchiectasis, recurrent pulmonary infections (particularly with Pseudomonas aeruginosa) progressive decline in lung function, and eventual respiratory failure (5). Gastrointestinal involvement is characterized by pancreatic exocrine insufficiency, malabsorption, meconium ileus, distal intestinal obstruction syndrome (DIOS) (6), and hepatobiliary disease. Moreover, male patients frequently present with infertility due to congenital bilateral absence of the vas deferens (7). The pancreas is one of the earliest organs affected, with mucus accumulation leading to exocrine pancreatic insufficiency (EPI) due to progressive ductal obstruction, chronic inflammation, and acinar cell destruction. This results in fat malabsorption and deficiencies in fat-soluble vitamins (8), necessitating enzyme replacement therapy and dietary supplementation. In addition to exocrine dysfunction, pancreatic endocrine impairment progresses gradually, culminating in defective insulin secretion. Endocrine dysfunction commonly manifests as cystic fibrosis-related diabetes (CFRD) (7–9). CFRD exhibits characteristics of both type 1 diabetes (β-cell dysfunction) and type 2 diabetes (insulin resistance, particularly during pulmonary exacerbations) (9, 10) (Table 1). However, CFRD is marked by a progressive decline in insulin secretion with an often-subtle onset, underscoring the importance of early glucose monitoring.
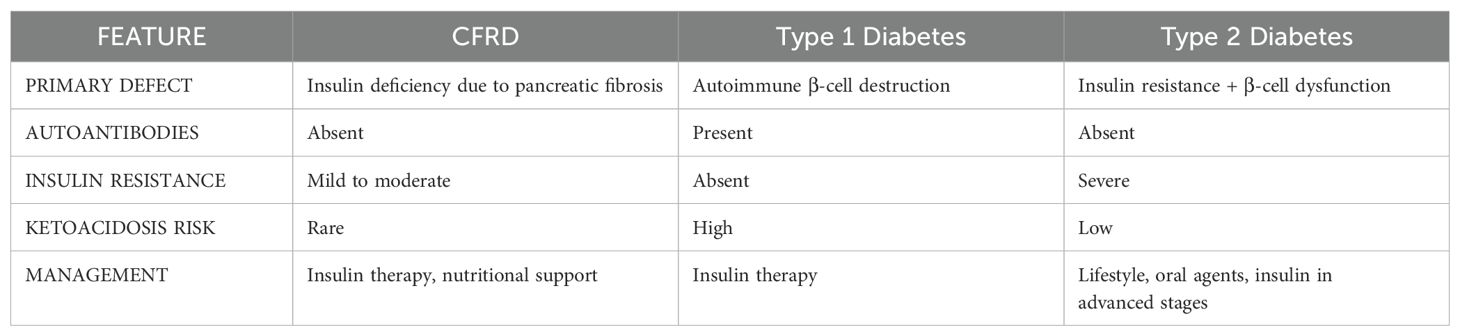
Table 1. Main characteristics of the three types of diabetes.
CFRD primarily arises from insulin deficiency due to progressive destruction of pancreatic islets within a fibrotic and inflamed pancreas (11, 12). Over time, the inflammatory environment, compounded by fibrosis and ischemia, leads to islet damage, reducing β-cell mass and impairing insulin production (13). Even in preclinical stages, patients with cystic fibrosis frequently exhibit delayed and diminished insulin secretion, particularly in response to oral glucose intake. Although insulin deficiency is the principal mechanism underlying CFRD (14), insulin resistance also contributes (15), particularly during pulmonary exacerbations. Chronic inflammation in cystic fibrosis leads to elevated cytokine levels, such as TNF-α and IL-6, which interfere with insulin signaling (16). Additionally, frequent infections precipitate an increase in cortisol and catecholamines, further antagonizing insulin action. Nutritional status also modulates insulin sensitivity, as malnutrition reduces muscle mass (17) (a primary site for insulin-mediated glucose uptake) while corticosteroid use during exacerbations induces transient hyperglycemia.
Glucagon dysregulation and impaired incretin responses further contribute to CFRD pathophysiology. Some individuals exhibit abnormal glucagon secretion, exacerbating postprandial hyperglycemia. Additionally, deficiencies in incretin hormones, such as GLP-1 and GIP, further compromise insulin secretion and glucose homeostasis (18).
The prevalence of CFRD increases with age, affecting over 50% of patients older than 30 years (19, 20). Its clinical impact is profound, being associated with accelerated pulmonary decline, heightened infection susceptibility, nutritional deterioration, and increased mortality.A customized and individualized insulin therapy remains the recommended treatment for children and adolescents with CFRD (21), whereas there is only limited evidence for use of oral hypoglycemic agents (22). Beyond glycemic regulation, insulin therapy plays a pivotal role in improving nutritional status and pulmonary function. Nutritional support is crucial, with dietary recommendations emphasizing a high-calorie, high-fat intake (22), supplemented with pancreatic enzyme replacement therapy (PERT) (23) and fat-soluble vitamins to optimize nutrient absorption. Regular physical activity further enhances metabolic stability by improving insulin sensitivity, increasing muscle mass (24), and promoting respiratory function.
The advent of CFTR modulators represents a landmark advancement in cystic fibrosis therapy. They are administered orally, typically twice daily with fat-containing meals to optimize absorption. Treatment initiation necessitates genotypic confirmation of CFTR mutations and vigilant monitoring for potential adverse effects, including hepatotoxicity and drug interactions. Given their profound impact on disease progression, CFTR modulators are now considered standard therapy for eligible patients. Unlike conventional treatments aimed at symptomatic relief, these agents directly target the underlying molecular defect caused by CFTR gene mutations.
CFTR modulators are categorized into 1. potentiators and 2. correctors. The most advanced combination, elexacaftor/tezacaftor/ivacaftor (ETI, marketed as Trikafta/Kaftrio), has demonstrated substantial benefits, including improved lung function, reduced exacerbation frequency, and enhanced nutritional status in patients harboring at least one F508del allele (25–28). Their introduction has significantly enhanced pulmonary function, nutritional status, and life expectancy in eligible patients (29). Notably, CFTR modulators have been associated with improvements in pancreatic function, leading to enhanced fat and vitamin absorption and a reduced reliance on dietary supplementation (30). However, these benefits are not universally experienced.
This literature review synthesizes recent findings on the efficacy of CFTR modulators on metabolic control, nutritional outcomes and the treatment of Children and Young Adults with CFRD.
2 Materials and methods
2.1 Search strategy
We searched electronic databases (Pubmed and Web of Science) for studies published between 1st January 2010 (the year the first study was published) and 31th December 2024. Search terms, or “MESH” (MEdical Subject Headings) for this systematic review included various combinations: “hyperglycemia” or “diabetes” or “glucose disorder” AND “cystic fibrosis” AND “elexacaftor” OR “Ivacaftor” OR “Tezacaftor”. To avoid missing any relevant studies, we also screened the reference list of eligible studies. We formulated 5 questions related to the efficacy of the CFTR modulators and for each one we established the outcomes listed below. The protocol was registered with the International Prospective Register of Systematic Reviews database (PROSPERO https://www.crd.york.ac.uk/PROSPERO, number CRD420251021499).
2.2 Criteria for study selection
We conducted a systematic search of the literature based on the PICOS model (Population, Intervention, Comparison, Outcomes, Study design) (Table 2).

Table 2. PICOS model.
Inclusion criteria were as follows: i) Study population: Children (birth to 18 years) and young adults (19 to 24 years) with hyperglycemia or cystic fibrosis-related diabetes (CFRD) who received CFTR modulator therapy, with available follow-up data; ii) Study design: Observational studies (cohort, case-control), exploratory studies, and experimental research; iii) Review articles were excluded, but their reference lists were screened for potentially eligible studies; iv) Only full-text papers were included; abstracts only were excluded; v) Data on therapy efficacy: Information on the age at the start of therapy, duration of treatment, and outcomes; vi) Publication date: Studies published in the last 15 years (2010–2024).
Exclusion criteria included: i) Studies with data available without baseline status (cross-sectional studies); ii) Studies with only baseline data, without follow-up; iii) Animal studies; iv) Full-text not available; v) studies not yet published; vi) Studies not reporting the selected outcomes; vii) Studies on CFRD without data on CFTR modulator therapy; viii) Studies in which data concerning the population of interest cannot be extracted; ix) Studies published in languages other than English were not excluded a priori.
2.3 Data extraction and management
Two independent investigators (VG, GL) screened for inclusion the title and abstract of all the studies identified using the search strategy. Any discrepancies between them were resolved by consensus. After abstract selection, the same two investigators conducted the full paper analysis.
The following characteristics were evaluated for each study in the full paper: i) reference details: authorship(s); published or unpublished; year of publication; period in which the study was conducted; other relevant cited papers; ii) study characteristics: study design, topic, treatment period, follow up duration, region; iii) population characteristics: number of participants, age and demographic data; comparator characteristics; iv) methodology: measures to assess the outcomes; v) main results: outcome measures.
2.4 Characteristics of included studies
After removal of duplicate, the titles and abstract of 503 unique references were evaluated for eligibility. A total of 486 were excluded, and 17 full-text articles were extracted and screened for eligibility. Of these, 9 (31–39) original studies met eligibility criteria (Figure 1). Of these studies, 4 were conducted in the United States of America, 1 in Italy, 2 in Israel, 1 in France and 1 in Germany (Table 3).
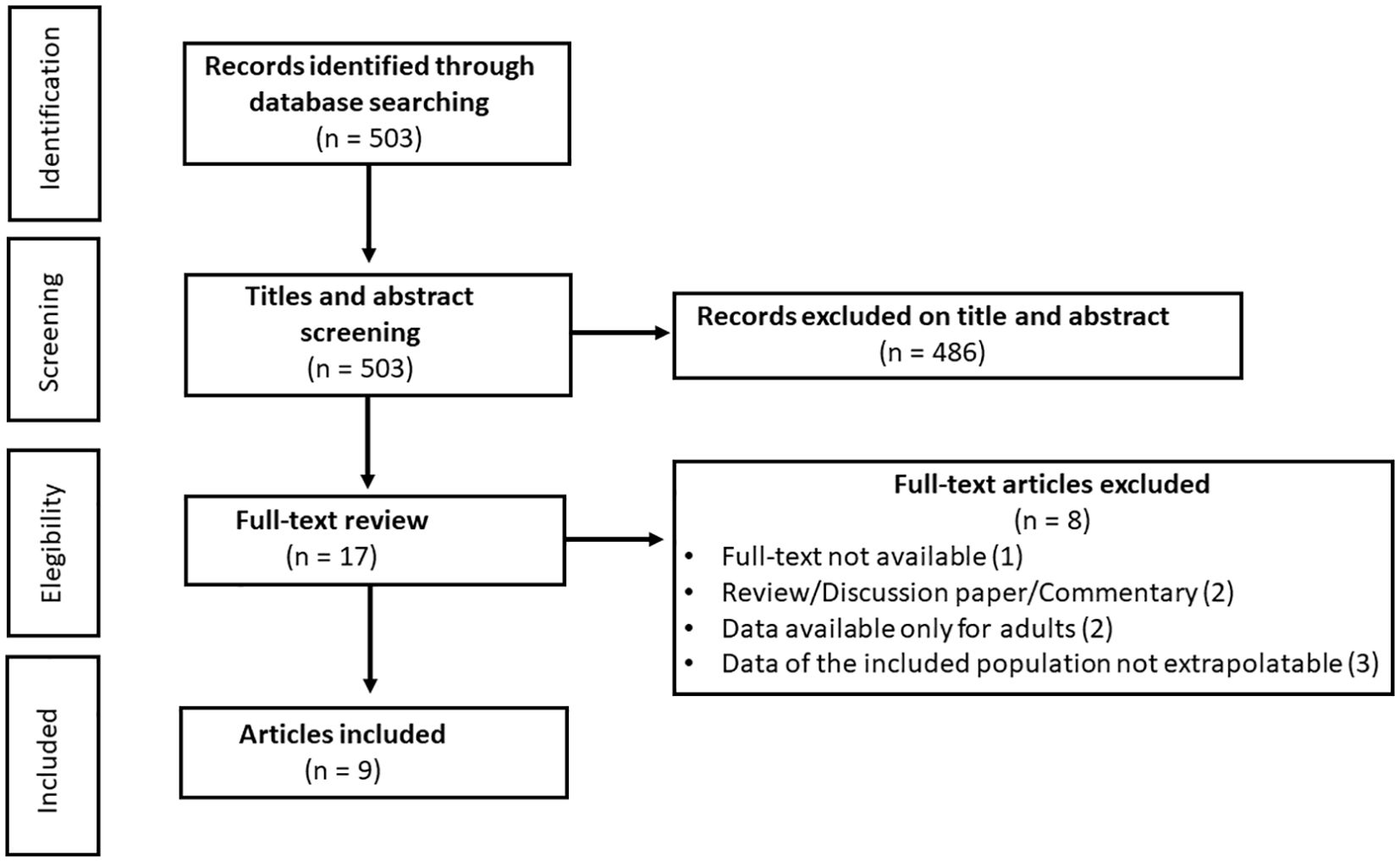
Figure 1. Flow chart of study inclusion in systematic review.
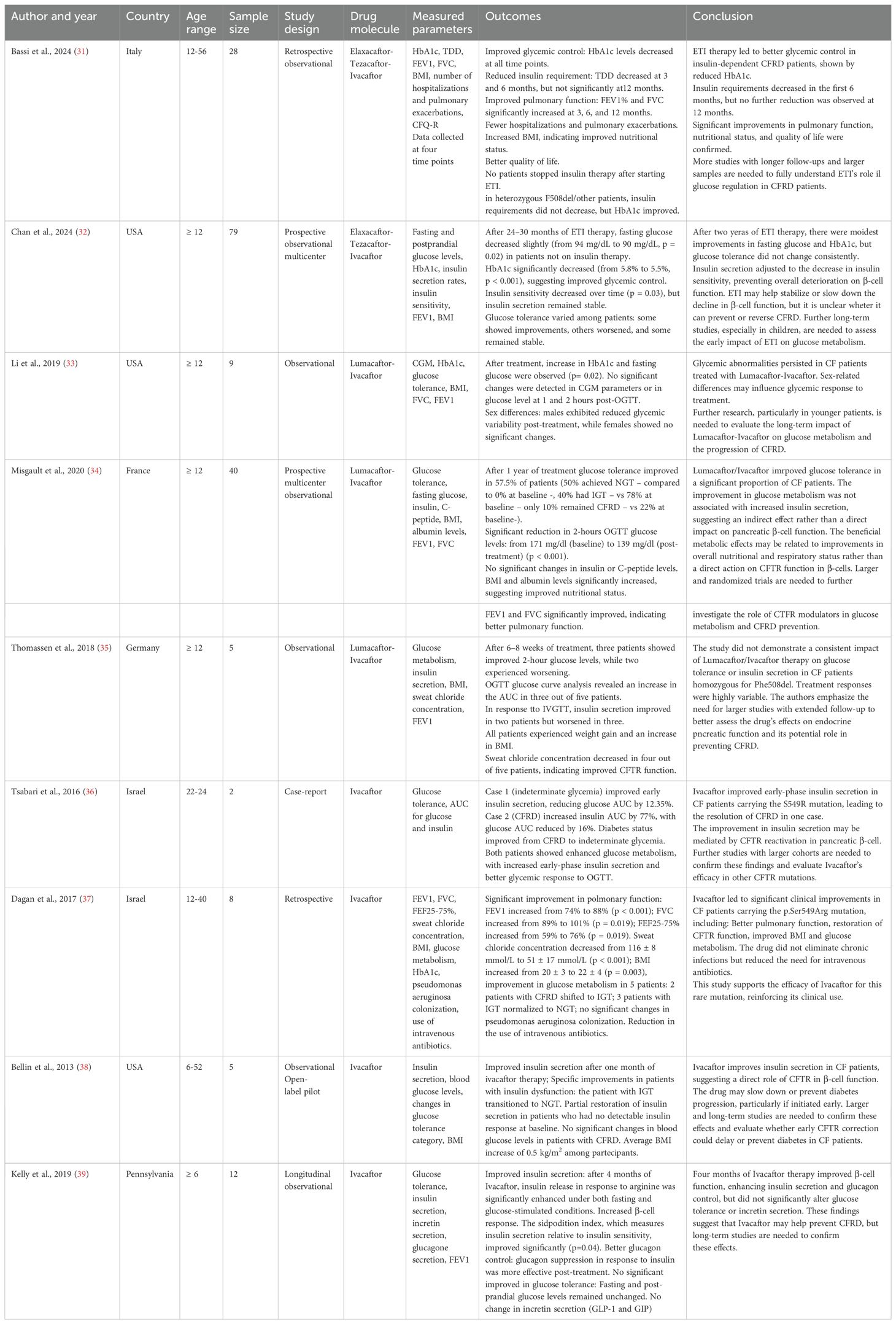
Table 3. Characteristic table.
3 Results
3.1 Ivacafactor
Ivacaftor appears to exert a direct effect on insulin secretion. More than 10 years ago, Bellin et al. (2013) (38) reported the first data about this drug showing that it may improve insulin secretion in a one-month study. Notably, one patient with impaired glucose tolerance achieved normoglycemia, while the others exhibited partial restoration of insulin secretion. Unfortunately, in patients with established CFRD, no significant changes in blood glucose levels were observed. This study suggested for the first time that Ivacaftor may be effective in preventing diabetes progression if initiated early, but not in reversing CFRD once it has developed.
Further, Tsabari et al. (2016) (36) investigated its effects in two CF patients with glucose abnormalities and found that both demonstrated improved insulin secretion, with one patient transitioning from CFRD to an intermediate glycemic status. These findings confirmed that Ivacaftor could contribute to β-cell function restoration, suggesting that even in patients with CFRF blood glucose can improce.
Dagan et al. (2017) (37) demonstrated that some patients carrying the p.Ser549Arg mutation treated with Ivacaftor experienced enhanced glucose metabolism, with transitions from CFRD to impaired glucose tolerance and from impaired glucose tolerance to normoglycemia.
Finally, Kelly et al. (2019) (39) confirmed that Ivacaftor enhances β-cell function and glucagon regulation without significantly altering glucose tolerance or incretin secretion. These findings indicate that Ivacaftor may support pancreatic function but does not fully correct the metabolic abnormalities associated with CF.
3.2 Lumacaftor/ivacaftor
The effects of Lumacaftor/Ivacaftor on glucose metabolism have been explored in multiple studies, with inconsistent findings. On one hand, Li et al. (2019) (33) observed an increase in HbA1c and fasting glucose levels following treatment, with no significant changes in continuous glucose monitoring (CGM) parameters. Moreover, sex differences were identified, with male patients exhibiting reduced glycemic variability, while female patients showed no significant changes. This study suggests that Lumacaftor/Ivacaftor does not effectively correct glycemic abnormalities in CF patients and that individual factors may influence metabolic responses to treatment.
In contrast, Misgault et al. (2020) (34) reported that 57.5% of CF patients treated with Lumacaftor/Ivacaftor experienced an improvement in glucose tolerance after one year of therapy. Specifically, a greater proportion of patients achieved normal glucose tolerance, while fewer persisted with CFRD. However, this improvement was not associated with increased insulin secretion, suggesting that the observed metabolic benefits were likely secondary to enhancements in nutritional and respiratory status rather than a direct effect on pancreatic β-cell function.
Thomassen et al. (2018) (35) did not observe significant improvements in glucose regulation following Lumacaftor/Ivacaftor treatment. However, some patients experienced weight gain and an increase in BMI, suggesting a potential nutritional benefit.
3.3 Elexacaftor-tezacaftor-ivacaftor
Conversely, Bassi et al. (2024) (31) reported that treatment with ETI led to improved glycemic control, as evidenced by a reduction in HbA1c levels across all study time points. Additionally, a decrease in insulin requirements was observed during the first six months of therapy, although no further reduction was noted at 12 months. Patients receiving ETI also exhibited significant improvements in pulmonary function, a reduction in hospitalizations and pulmonary exacerbations, increased BMI, and an overall enhancement in quality of life. However, no patient was able to discontinue insulin therapy, suggesting that while ETI improves glycemic control, it is not sufficient to normalize glucose metabolism in CFRD patients.
Chan et al. (2024) (32) investigated the effects of ETI over a longer period (24–30 months). Their findings revealed a modest decrease in fasting glucose levels and a significant reduction in HbA1c, indicating improved glycemic control. However, insulin sensitivity declined over time, and glucose tolerance exhibited variable responses among patients—some showed improvement, others worsened, and some remained stable. These data suggest that ETI may help stabilize or slow the decline of β-cell function, but whether it can prevent or reverse CFRD remains uncertain.
4 Discussion
CFTR modulators have revolutionized the treatment landscape for cystic fibrosis CF by targeting specific genetic mutations and restoring defective CFTR protein function. While these therapies have significantly improved pulmonary function and nutritional status, their effects on glucose metabolism and CFRD remain an area of active investigation.
Ivacaftor has demonstrated significant metabolic benefits in patients with gating mutations such as G551D and S549N (Table 4). Clinical studies indicate improved glucose metabolism, with increased insulin secretion and reduced postprandial glucose levels (36–39). These effects likely stem from enhanced pancreatic duct function, reducing exocrine pancreatic stress and creating a more favorable endocrine environment. Ivacaftor has also been associated with improved glucose tolerance, particularly in patients treated early in disease progression (36–39). The direct role of CFTR in pancreatic β-cell function has been suggested, given that Ivacaftor increases early-phase insulin secretion and enhances glucagon suppression (38, 39), which may slow CFRD progression.
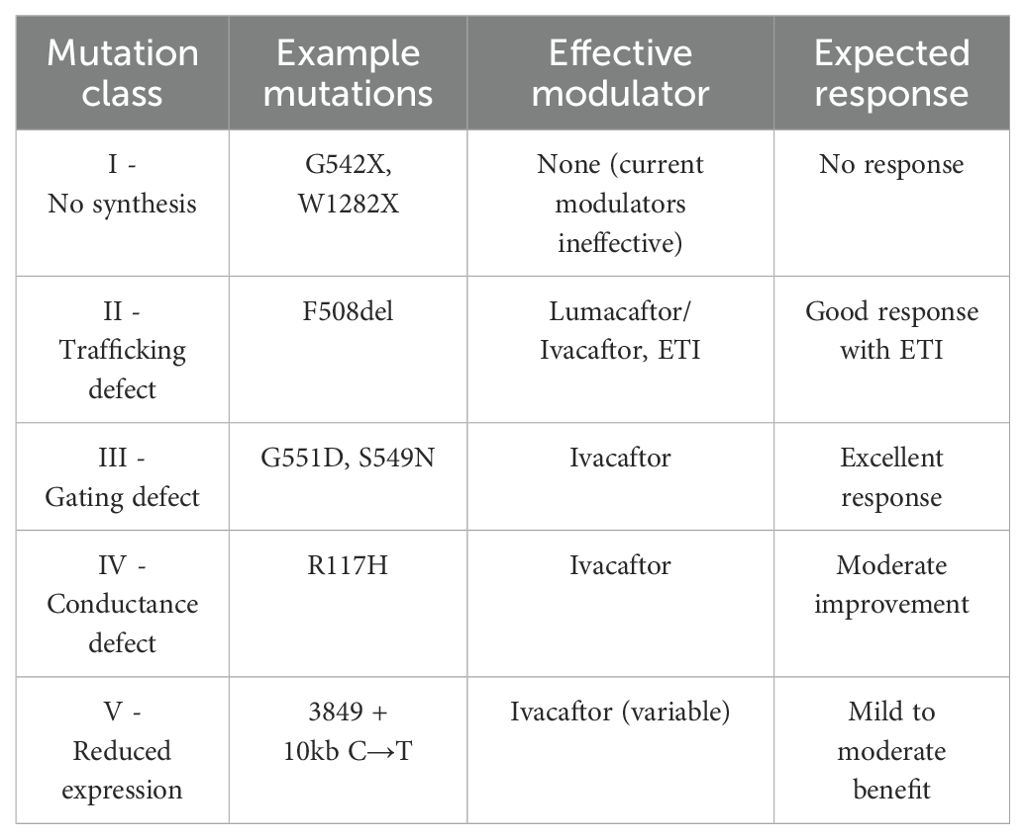
Table 4. Relationship between CFTR mutations and modulator efficacy.
In addition to metabolic benefits, Ivacaftor significantly enhances pulmonary function, with FEV1 improvements exceeding those observed with other modulators (37). FVC and BMI also increase, likely due to improved nutrient absorption and overall better health status. However, its efficacy is largely limited to patients with Class III (gating) and some Class IV (conductance) mutations, with minimal to no effect in Class I and II mutations, which involve defective CFTR synthesis or trafficking (40). Lumacaftor/Ivacaftor has produced mixed results regarding glucose metabolism. Some studies suggest improvements in glucose tolerance (34), while others indicate no significant changes or even worsening glycemic control (33, 41, 42). Insulin secretion remains largely unchanged, suggesting that any metabolic improvements are indirect, likely resulting from reduced systemic inflammation and improved overall health rather than direct β-cell restoration. Contrary to expectations, Piona et al. demonstrated that insulin sensitivity worsened in CF patients treated with lumacaftor/ivacaftor (43).
Pulmonary function benefits are moderate but less pronounced than those observed with Ivacaftor or ETI. FEV1 increases are modest, and FVC shows slight improvements. BMI also exhibits a mild to moderate increase, likely secondary to improved pulmonary status. However, the drug’s tolerability issues and its limited efficacy in heterozygous F508del patients have restricted its widespread adoption. ETI therapy has significantly expanded treatment options for CF patients carrying at least one F508del allele. Its effects on glucose metabolism appear modest but generally positive, with reductions in HbA1c and slight decreases in fasting glucose (31, 32). While insulin secretion shows mild increases in some patients, overall β-cell function remains largely unchanged. Notably, glucose tolerance does not consistently improve, and CFRD progression does not appear to be significantly altered. Beyond glucose metabolism, ETI produces the most substantial improvements in pulmonary function among all CFTR modulators, with FEV1 increases of 10–15% and a reduction in pulmonary exacerbations. FVC improves, particularly in patients with advanced respiratory impairment. BMI increases considerably, reflecting enhanced nutrient absorption and improved pulmonary status. According to Grancini et al. (44), initiation of ETI therapy was linked to better glycaemic control in insulin-treated CFRD patients. Similarly, Bassi et al. demonstrated an improvement in glycaemic control accompanied by a significant reduction in insulin requirements in a cohort of both pediatric and adult patients with CFRD (31). Given its efficacy in patients with at least one F508del allele, ETI represents the most versatile and broadly effective CFTR modulator currently available. The effectiveness of CFTR modulators is closely linked to the type of CFTR mutation present. Ivacaftor is highly effective in Class III (gating) mutations, where it enhances CFTR channel opening, while it has minimal impact on Class I and II mutations. Lumacaftor/Ivacaftor provides moderate benefits to homozygous F508del patients by improving CFTR protein trafficking, although its metabolic effects remain inconsistent. ETI, by contrast, offers substantial clinical benefits to a broader CF population, including those with at least one F508del allele (Table 4).
While CFTR modulators have revolutionized CF care, their metabolic effects, particularly in the context of CFRD, remain an area requiring further research. Ivacaftor demonstrates the most direct benefits on β-cell function, whereas Lumacaftor/Ivacaftor and ETI appear to exert indirect metabolic effects, likely mediated through systemic improvements in health. Future research should focus on addressing treatment gaps, particularly for Class I mutations, through emerging therapies such as gene editing and read-through compounds.
CFTR modulators have transformed CF treatment, but their impact on glucose metabolism and CFRD progression remains an area of active investigation. While Ivacaftor appears to have a direct positive impact on insulin secretion, Lumacaftor/Ivacaftor and ETI seem to exert more indirect metabolic effects, likely mediated by improvements in nutritional and respiratory status. The findings remain heterogeneous, highlighting the need for further research to fully elucidate the role of CFTR modulators in the prevention and management of CFRD.
A deeper understanding of the interplay between CFTR function and pancreatic endocrine regulation will be essential in developing targeted therapies that not only improve pulmonary health but also address metabolic dysfunction in CF patients.
The variability in metabolic responses highlights the complexity of CFRD pathophysiology and the need for personalized treatment approaches. Future research should focus on early intervention strategies, particularly in young patients, to assess if CFTR correction can delay β-cell deterioration. Next-generation CFTR modulators and emerging genetic therapies, such as gene editing and RNA-based approaches, may offer new avenues for more effective CFRD management.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author/s.
Author contributions
PG: Writing – review & editing, Supervision, Writing – original draft, Conceptualization. GLe: Writing – review & editing. VG: Writing – review & editing, Writing – original draft, Data curation, Methodology. CP: Methodology, Writing – review & editing. GLa: Writing – review & editing. MD: Methodology, Conceptualization, Supervision, Writing – review & editing. GLi: Writing – review & editing, Methodology, Conceptualization, Writing – original draft.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
We acknowledge the use of ChatGPT Team (version GPT-4) to improve the English grammar, syntax, and flow of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript to improve the English grammar, syntax, and flow of the manuscript [ChatGPT Team (version GPT-4)].
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ioannou L, McClaren BJ, Massie J, Lewis S, Metcalfe SA, Forrest L, et al. Population-based carrier screening for cystic fibrosis: a systematic review of 23 years of research. Genet Med. (2014) 16:207–16. doi: 10.1038/gim.2013.125
2. Hanssens LS, Duchateau J, and Casimir GJ. CFTR protein: not just a chloride channel? Cells. (2021) 10:2844. doi: 10.3390/cells10112844
3. Fenker DE, McDaniel CT, Panmanee W, Panos RJ, Sorscher EJ, Sabusap C, et al. A comparison between two pathophysiologically different yet microbiologically similar lung diseases: cystic fibrosis and chronic obstructive pulmonary disease. Int J Respir Pulm Med. (2018) 5:098. doi: 10.23937/2378-3516/1410098668
4. Diab Cáceres L and Zamarrón De Lucas E. Fibrosis quística: epidemiología, clínica, diagnóstico y tratamiento. Med Clínica. (2023) 161:389–96.
5. Rajan S. Pulmonary infections in patients with cystic fibrosis. Semin Respir Infect. (2002) 17:47–56. doi: 10.1053/srin.2002.31690
6. Carroll W, Green J, and Gilchrist FJ. Interventions for preventing distal intestinal obstruction syndrome (DIOS) in cystic fibrosis. Cochrane Database Syst Rev. (2021) 12:CD012619. doi: 10.1002/14651858.CD012619.pub2
7. Chillón M, Casals T, Mercier B, Bassas L, Lissens W, Silber S, et al. Mutations in the cystic fibrosis gene in patients with congenital absence of the vas deferens. N Engl J Med. (1995) 332:1475–80. doi: 10.1056/NEJM199506013322204
8. McDonald CM, Reid EK, Pohl JF, Yuzyuk TK, Padula LM, Vavrina K, et al. Cystic fibrosis and fat malabsorption: Pathophysiology of the cystic fibrosis gastrointestinal tract and the impact of highly effective CFTR modulator therapy. Nutr Clin Pract. (2024) 39:S57–S77. doi: 10.1002/ncp.11122
9. Moran A, Hardin D, Rodman D, Allen HF, Beall RJ, Borowitz D, et al. Diagnosis, screening and management of cystic fibrosis related diabetes mellitus. Diabetes Res Clin Pract. (1999) 45:61–73. doi: 10.1016/S0168-8227(99)00058-3
10. Nathan BM, Laguna T, and Moran A. Recent trends in cystic fibrosis-related diabetes. Curr Opin Endocrinol Diabetes Obes. (2010) 17:335–41. doi: 10.1097/MED.0b013e32833a780d
11. Iannucci A, Mukai K, Johnson D, and Burke B. Endocrine pancreas in cystic fibrosis: An immunohistochemical study. Hum Pathol. (1984) 15:278–84. doi: 10.1016/S0046-8177(84)80191-4
12. Löhr M, Goertchen P, Nizze H, Gould NS, Gould VE, Oberholzer M, et al. Cystic fibrosis associated islet changes may provide a basis for diabetes: An immunocytochemical and morphometrical study. Virchows Arch A Pathol Anat Histopathol. (1989) 414:179–85. doi: 10.1007/BF00718598
13. Gibson-Corley KN, Meyerholz DK, and Engelhardt JF. Pancreatic pathophysiology in cystic fibrosis. J Pathol. (2016) 238:311–20. doi: 10.1002/path.4634
14. Lombardo F, De Luca F, Rosano M, Sferlazzas C, Lucanto C, Arrigo T, et al. Natural history of glucose tolerance, beta-cell function and peripheral insulin sensitivity in cystic fibrosis patients with fasting euglycemia. Eur J Endocrinol. (2003) 149:53–9. doi: 10.1530/eje.0.1490053
15. Colomba J, Boudreau V, Lehoux-Dubois C, Desjardins K, Coriati A, Tremblay F, et al. The main mechanism associated with progression of glucose intolerance in older patients with cystic fibrosis is insulin resistance and not reduced insulin secretion capacity. J Cyst Fibros. (2019) 18:551–6. doi: 10.1016/j.jcf.2019.01.009
16. Krogh-Madsen R, Plomgaard P, Møller K, Mittendorfer B, and Pedersen BK. Influence of TNF-α and IL-6 infusions on insulin sensitivity and expression of IL-18 in humans. Am J Physiol-Endocrinol Metab. (2006) 291:E108–14. doi: 10.1152/ajpendo.00471.2005
17. Prado CM, Landi F, Chew STH, Atherton PJ, Molinger J, Ruck T, et al. Advances in muscle health and nutrition: A toolkit for healthcare professionals. Clin Nutr. (2022) 41:2244–63. doi: 10.1016/j.clnu.2022.07.041
18. Sun X, Yi Y, Liang B, Yang Y, He N, Ode KL, et al. Incretin dysfunction and hyperglycemia in cystic fibrosis: Role of acyl-ghrelin. J Cyst Fibros Off J Eur Cyst Fibros Soc. (2019) 18:557–65. doi: 10.1016/j.jcf.2019.01.010
19. Moran A, Dunitz J, Nathan B, Saeed A, Holme B, and Thomas W. Cystic fibrosis–related diabetes: current trends in prevalence, incidence, and mortality. Diabetes Care. (2009) 32:1626–31. doi: 10.2337/dc09-0586
20. Barrio R. MANAGEMENT OF ENDOCRINE DISEASE: Cystic fibrosis-related diabetes: novel pathogenic insights opening new therapeutic avenues. Eur J Endocrinol. (2015) 172:R131–41. doi: 10.1530/EJE-14-0644
21. Schiaffini R and Pampanini V. Diabetes and prediabetes in children with cystic fibrosis. Curr Opin Pediatr. (2023) 35:481–5. doi: 10.1097/MOP.0000000000001259
22. Turck D, Braegger CP, Colombo C, Declercq D, Morton A, Pancheva R, et al. ESPEN-ESPGHAN-ECFS guidelines on nutrition care for infants, children, and adults with cystic fibrosis. Clin Nutr. (2016) 35:557–77. doi: 10.1016/j.clnu.2016.03.004
23. Ng C, Major G, and Smyth AR. A systematic Cochrane Review of the timing of pancreatic enzyme replacement therapy (PERT) in cystic fibrosis. Paediatr Respir Rev. (2021) 40:44–5. doi: 10.1016/j.prrv.2021.09.002
24. Małkowska P. Positive effects of physical activity on insulin signaling. Curr Issues Mol Biol. (2024) 46:5467–87. doi: 10.3390/cimb46060327
25. Gramegna A, Contarini M, Aliberti S, Casciaro R, Blasi F, and Castellani C. From ivacaftor to triple combination: A systematic review of efficacy and safety of CFTR modulators in people with cystic fibrosis. Int J Mol Sci. (2020) 21:5882. doi: 10.3390/ijms21165882
26. Middleton PG, Mall MA, Dřevínek P, Lands LC, McKone EF, Polineni D, et al. Elexacaftor–tezacaftor–ivacaftor for cystic fibrosis with a single phe508del allele. N Engl J Med. (2019) 381:1809–19. doi: 10.1056/NEJMoa1908639
27. Keating D, Marigowda G, Burr L, Daines C, Mall MA, McKone EF, et al. VX-445–tezacaftor–ivacaftor in patients with cystic fibrosis and one or two phe508del alleles. N Engl J Med. (2018) 379:1612–20. doi: 10.1056/NEJMoa1807120
28. Fiedorczuk K and Chen J. Molecular structures reveal synergistic rescue of Δ508 CFTR by Trikafta modulators. Science. (2022) 378:284–90. doi: 10.1126/science.ade2216
29. McGarry ME and McColley SA. Cystic fibrosis patients of minority race and ethnicity less likely eligible for CFTR modulators based on CFTR genotype. Pediatr Pulmonol. (2021) 56:1496–503. doi: 10.1002/ppul.25285
30. Ramsey ML, Li SS, Lara LF, Gokun Y, Akshintala VS, Conwell DL, et al. Cystic fibrosis transmembrane conductance regulator modulators and the exocrine pancreas: A scoping review. J Cyst Fibros. (2023) 22:193–200. doi: 10.1016/j.jcf.2022.08.008
31. Bassi M, Strati MF, Spiandorello G, Scalas M, Cresta F, Calevo MG, et al. One-year effect of elexacaftor/tezacaftor/ivacaftor therapy on hbA1c levels and insulin requirement in patients with insulin-dependent cystic fibrosis-related diabetes: A retrospective observational study. Life. (2024) 14:1309. doi: 10.3390/life14101309
32. Chan CL, Shirley Bezerra M, Stefanovski D, Gallop RJ, Walega R, Donaldson SH, et al. Glycemia and insulin secretion in cystic fibrosis 2 years after elexacaftor/tezacaftor/ivacaftor: PROMISE-ENDO. J Clin Endocrinol Metab. (2024), dgae857. doi: 10.1210/clinem/dgae857
33. Li A, Vigers T, Pyle L, Zemanick E, Nadeau K, Sagel SD, et al. Continuous glucose monitoring in youth with cystic fibrosis treated with lumacaftor-ivacaftor. J Cyst Fibros. (2019) 18:144–9. doi: 10.1016/j.jcf.2018.07.010
34. Misgault B, Chatron E, Reynaud Q, Touzet S, Abely M, Melly L, et al. Effect of one-year lumacaftor–ivacaftor treatment on glucose tolerance abnormalities in cystic fibrosis patients. J Cyst Fibros. (2020) 19:712–6. doi: 10.1016/j.jcf.2020.03.002
35. Thomassen JC, Mueller MI, Alejandre Alcazar MA, Rietschel E, and Van Koningsbruggen-Rietschel S. Effect of Lumacaftor/Ivacaftor on glucose metabolism and insulin secretion in Phe508del homozygous cystic fibrosis patients. J Cyst Fibros. (2018) 17:271–5. doi: 10.1016/j.jcf.2017.11.016
36. Tsabari R, Elyashar HI, Cymberknowh MC, Breuer O, Armoni S, Livnat G, et al. CFTR potentiator therapy ameliorates impaired insulin secretion in CF patients with a gating mutation. J Cyst Fibros. (2016) 15:e25–7. doi: 10.1016/j.jcf.2015.10.012
37. Dagan A, Cohen-Cymberknoh M, Shteinberg M, Levine H, Vilozni D, Bezalel Y, et al. Ivacaftor for the p.Ser549Arg (S549R) gating mutation – The Israeli experience. Respir Med. (2017) 131:225–8. doi: 10.1016/j.rmed.2017.08.026
38. Bellin MD, Laguna T, Leschyshyn J, Regelmann W, Dunitz J, Billings J, et al. Insulin secretion improves in cystic fibrosis following ivacaftor correction of CFTR: a small pilot study: Insulin secretion following CFTR correction. Pediatr Diabetes. (2013) 14:417–21. doi: 10.1111/pedi.12026
39. Kelly A, De Leon DD, Sheikh S, Camburn D, Kubrak C, Peleckis AJ, et al. Islet hormone and incretin secretion in cystic fibrosis after four months of ivacaftor therapy. Am J Respir Crit Care Med. (2019) 199:342–51. doi: 10.1164/rccm.201806-1018OC
40. Ramananda Y, Naren AP, and Arora K. Functional consequences of CFTR interactions in cystic fibrosis. Int J Mol Sci. (2024) 25:3384. doi: 10.3390/ijms25063384
41. Moheet A, Beisang D, Zhang L, Sagel SD, VanDalfsen JM, Heltshe SL, et al. Lumacaftor/ivacaftor therapy fails to increase insulin secretion in F508del/F508del CF patients. J Cyst Fibros. (2021) 20:333–8. doi: 10.1016/j.jcf.2020.09.001
42. Colombo C, Foppiani A, Bisogno A, Gambazza S, Daccò V, Nazzari E, et al. Lumacaftor/ivacaftor in cystic fibrosis: effects on glucose metabolism and insulin secretion. J Endocrinol Invest. (2021) 44:2213–8. doi: 10.1007/s40618-021-01525-4
43. Piona C, Mozzillo E, Tosco A, Volpi S, Rosanio FM, Cimbalo C, et al. Impact of CFTR modulators on beta-cell function in children and young adults with cystic fibrosis. J Clin Med. (2022) 11:4149. doi: 10.3390/jcm11144149
44. Grancini V, Gramegna A, Zazzeron L, Alicandro G, Porcaro LL, Piedepalumbo F, et al. Effects of elexacaftor/tezacaftor/ivacaftor triple combination therapy on glycaemic control and body composition in patients with cystic fibrosis-related diabetes. Diabetes Metab. (2023) 49:101466. doi: 10.1016/j.diabet.2023.101466
Keywords: cystic fibrosis, CFTR modulators, CFRD, glucose metabolism, lumacaftor/ivacaftor, elexacaftor-ivacaftor-tezacaftor
Citation: Giordano P, Leonetti G, Granberg V, Casolino RMP, Lassandro G, Delvecchio M and Linguiti G (2025) Effect of CFTR modulators on glucose homeostasis in children and young adults with cystic fibrosis-related diabetes: a systematic review. Front. Endocrinol. 16:1623654. doi: 10.3389/fendo.2025.1623654
Received: 06 May 2025; Accepted: 18 July 2025;
Published: 06 August 2025.
Edited by:
Fabrizio Barbetti, University of Rome Tor Vergata, ItalyReviewed by:
Enza Mozzillo, University of Naples Federico II, ItalyValeria Grancini, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Italy
Copyright © 2025 Giordano, Leonetti, Granberg, Casolino, Lassandro, Delvecchio and Linguiti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maurizio Delvecchio, bWF1cml6aW8uZGVsdmVjY2hpbzFAdW5pdmFxLml0