 Busayo Oladun
Busayo Oladun Smita Mall
Smita Mall Min-Hyun Kim
Min-Hyun Kim- College of Health Solutions, Arizona State University, Phoenix, AZ, United States
The hypothalamus plays a central role in regulating metabolism by integrating neuropeptide signaling with environmental cues to maintain energy homeostasis. Adverse environmental factors, such as obesogenic diet, undernutrition, stress, and sedentary lifestyles, can disrupt the normal regulation of key hypothalamic neuropeptides and metabolic hormone receptors through epigenetic mechanisms, including DNA methylation, histone modifications, and microRNA regulation. These epigenetic alterations are not merely transient; they can be heritable and may influence metabolic health across generations, highlighting the critical need to understand the underlying epigenetic mechanisms. In this review, we provide a comprehensive overview of how environmental factors shape the epigenetic landscape of hypothalamic neuropeptides (pre-opiomelanocortin, neuropeptide Y, and agouti-related peptide) and metabolic hormone receptors (leptin receptor and insulin receptor), thereby modulating their expression and contributing to long-term metabolic outcomes. A better understanding of environment-epigenome interactions holds promise for the development of innovative therapeutic strategies to combat obesity and metabolic disorders.
1 Introduction
The brain is a critical organ in regulating metabolism, with several anatomical regions pivotal to maintaining energy homeostasis (1, 2). Among these, the hypothalamus emerges as a central regulator, coordinating many physiological processes in the body, including the control of appetite, energy metabolism, stress response, thermoregulation, circadian rhythms, the autonomic nervous system, the regulation of body fluid, control of endocrine systems, and emotional and behavioral responses (3). Key hypothalamic nuclei involved in energy regulation include the arcuate nucleus (ARC), ventromedial hypothalamus (VMH), dorsomedial hypothalamus (DMH), and paraventricular nucleus (PVN) (4–10). These structures work synergistically to integrate internal and external cues, maintaining energy balance through neuropeptides and metabolic receptors.
Hypothalamic neuropeptides such as neuropeptide Y (NPY), agouti-related peptide (AgRP), pro-opiomelanocortin (POMC), and cocaine- and amphetamine-regulated transcript (CART) play contrasting roles in energy homeostasis. NPY and AGRP stimulate appetite, promote food intake, and reduce energy expenditure (11, 12), whereas POMC and CART inhibit food intake, promote satiety, and increase energy expenditure (13). Additionally, receptors for metabolic hormones like leptin and insulin contribute to metabolic function in the hypothalamus. Dysregulation of leptin and insulin signaling has been linked to the development of metabolic disorders (14, 15).
Environmental factors, including diet, stress, and physical activity, modulate hypothalamic neuropeptides and metabolic hormone receptors expression, and dysfunction in their expression has been linked to metabolic disorders (16–19). Overnutrition, particularly energy-dense diets, and chronic stress have been implicated in epigenetic modifications leading to the development and progression of metabolic diseases, particularly obesity (20). Such changes are often prolonged, persisting across an individual’s lifespan and potentially being transmitted across generations (21, 22). This has led to a growing interest in understanding the role of epigenetics as a key regulator of gene-environment interactions in the development of metabolic disorders. Thus, exploring the epigenetic underpinnings of these structures and their activities will improve our understanding of the anomalies associated with metabolic health and provide potential remedies.
Epigenetic mechanisms, including DNA methylation, histone modifications, and non-coding RNAs such as microRNAs (miRNAs), have been identified as key regulators of hypothalamic neuropeptides and metabolic hormone receptors (23). These mechanisms influence cellular transcriptional potentials, thereby modulating phenotypes associated with energy regulation (24). For instance, stress and diet-induced epigenetic changes have been implicated in altered expression of neuropeptides like POMC, AgRP, and NPY, and receptors such as the leptin receptor (LepRb) and the insulin receptor (InsR) (25–28). Additionally, the CART and endocannabinoid system (ECS) represent other epigenetically modulated pathways with implications on energy homeostasis. Recent advancements in understanding the complex interactions between epigenetic modifications and neuropeptide gene expression have increased the potential for therapeutic interventions. This review considers an overview of the current understanding of the epigenetic regulation of hypothalamic neuropeptides and some metabolic hormone receptors and their functional roles, followed by an in-depth analysis of how environmental factors influence their responses or expression. This will deepen our understanding and hold promise to advance personalized therapeutic strategies in preventing and treating metabolic diseases.
1.1 DNA methylation
DNA methylation is a key epigenetic mechanism involving the addition of a methyl group to the fifth carbon of cytosine (one of the four nitrogen bases in DNA, the others being adenine, thymine, and guanine) to form 5-methylcytosine (29). This modification primarily occurs at cytosines adjacent to guanine nucleotides, known as CpG sites, although evidence suggests methylation can also occur at non-CpG sites (30). CpG islands, regions with a high density of CpG sites, are often located near gene promoters, especially the housekeeping genes (31). These regions are crucial in gene regulation, as they regulate chromatin structure and transcription binding factors. Methylation of the CpG islands typically results in gene silencing, whereas unmethylation or demethylation promotes gene expression (32). Environmental factors, such as diet and stress, have been implicated in altering DNA methylation patterns, linking this mechanism to metabolic disorders like diabetes and obesity (33). Given that the thymus and the brain are the two most highly methylated human tissues (34), the hypothalamus, being a critical regulator of energy homeostasis, emerges as an important area for exploring the crosstalk between DNA methylation and metabolic regulation.
1.2 Histone modifications
The DNA is condensed in a complex structure known as chromatin, which is made up of histones H2A, H2B, H3, and H4 (35, 36). The degree of chromatin compaction determines the accessibility of DNA to transcription, repair, and replication machinery. Opening up the chromatin structure facilitates gene activation, while more tightly packed chromatin suppresses gene expression. Histone modifications, such as acetylation, methylation, phosphorylation, sumoylation, deamination, and proline isomerization, play a crucial role in gene expression by influencing the access of transcription factors to DNA (37). Acetylation and phosphorylation promote transcription by loosening the chromatin and enhancing transcription factor access (38), whereas sumoylation, deamination, and proline isomerization generally lead to gene silencing. Depending on the specific lysine residues involved, methylation and ubiquitination exhibit dual roles, either activating or repressing genes (39, 40). Two primary mechanisms underlie these histone modifications: (a) disrupting nucleosome-nucleosome interactions to open chromatin and (b) recruiting non-histone proteins that further modify chromatin through enzymatic activities (39). These dynamic modifications are critical for understanding the regulation of metabolic pathways, especially in the hypothalamus, which governs energy balance and appetite control.
1.3 MicroRNAs
miRNAs are small non-coding RNA molecules of 21 – 26 nucleotides that regulate gene expression by binding to the 3’ untranslated region (3’UTR) of message-encoding RNAs (41–43). This binding can either degrade the resulting messenger RNA (mRNA) or inhibit its translation, depending on the level of complementarity between the miRNA and its target mRNA. Perfect complementarity results in cleavages and degradation, whereas imperfect complementarity initiates mRNA silencing by specific mechanisms, including translational repression, sequestration in cytoplasmic processing bodies, and/or slicer-dependent mRNA degradation (44). The outcome of miRNA processes is gene repression or silencing, which has been implicated in many diseases, especially metabolic disorders (45–47). Dysregulated miRNAs influence key hypothalamic neuropeptides involved in appetite and energy balance, contributing to conditions like obesity. Thus, we shall review the metabolic effects of hypothalamic neuropeptides and receptors in light of these epigenetic changes.
1.4 Pro-opiomelanocortin
In the brain, POMC, a 30-kDa prohormone, is highly expressed in the arcuate nucleus of the hypothalamus, pituitary gland, and brain stem (48, 49). Depending on the expression levels of prohormone convertases, POMC gives rise to various active peptides and hormones, such as melanocyte-stimulating hormones (MSH) (α-MSH, β-MSH, and γ-MSH), adrenocorticotropic hormone (ACTH), corticotropin-like intermediate lobe peptide (CLIP), Lipotropin (γ-lipotropin and β-lipotropin), and met-enkephalin (50, 51). As an anorexigenic peptide, POMC suppresses appetite and increases energy expenditure, leading to weight loss primarily through the melanocortin signaling pathway, an important mechanism for maintaining energy balance (52). Beyond regulating metabolism, these products also regulate stress response, the immune system, and sexual functions (53–55).
The pivotal role of POMC was illustrated by Krude et al. (56), who described two patients with congenital absence of the Pomc gene and its derived peptides. One patient carried two nonsense mutations in exon 3, which led to losing key Pomc-derived peptides, including ACTH, α-MSH, and β-MSH. The second patient had a homozygous mutation in the 5’-untranslated region of Pomc, impairing the proper translational initiation of the POMC protein. Both patients presented with early-onset obesity due to hyperphagia linked to impaired melanocortin signaling in the hypothalamus, along with hypercortisolemia and distinctive physical traits like pale skin and red hair due to reduced activation of MC1R in melanocytes (56). Further evidence by Farooqi et al. (57), demonstrated that loss of even one allele of Pomc predisposes individuals to obesity, with 11 out of 12 heterozygous patients being either overweight or obese (57). Animal studies also alluded to the role of Pomc in energy balance, as Pomc-deficient mice and zebrafish developed severe obesity, increased food intake, and insulin resistance (58–62). These findings emphasize that hypothalamic POMC is critical for regulating energy balance.
Various factors mediate the epigenetic modifications of Pomc, influencing the phenotypic expression and metabolic outcomes. For instance, a high-fat diet (HFD) has been shown to influence DNA methylation of the Pomc gene promoter, thus affecting its expression. In a study by Cifani et al. (63), rats fed an HFD (45% kcal fat) were classified as diet-induced obese or diet-resistant rats based on their weight gain. While there was no difference in the Pomc expression levels at the 5th week between diet-induced obese and diet-resistant rats, by the 21st week, Pomc levels were elevated in diet-resistant rats. Examination of the Pomc promoter revealed decreased DNA methylation at the CpG sites 1,2,6, and 7 in diet-resistant rats compared to diet-induced obese rats (63). Furthermore, rats fed an HFD (60% kcal fat) from post-weaning to adulthood exhibited DNA hypermethylation in specific regions of the Pomc promoter, leading to reduced Pomc expression and increased body weight when compared to the control (64). These findings indicate that HFD promotes DNA hypermethylation of the Pomc promoter, which decreases Pomc expression. This reduction disrupts energy balance and feeding behavior, potentially contributing to HFD-induced obesity.
Similar results were observed in human studies. However, due to the challenges associated with directly investigating epigenetic changes in the living human hypothalamus, researchers have explored the use of alternative approaches, such as postmortem hypothalamus. In a study using postmortem hypothalamic samples from obese and normal-weight individuals with no history of neurodegenerative disease or cancer, researchers performed a laser microdissection of MSH-positive neurons. They found that the Pomc methylation in these neurons positively correlated with the basal metabolic index of the individual (65). Specifically, obese individuals showed increased methylation at a CpG island at the intersection between Pomc intron and coding exon 3, compared to non-obese individuals, thereby suggesting that the Pomc hypermethylation in the MSH neurons may impair satiety signaling and promote obesity. Additionally, using leukocytes, one study examined the effects of an 8-week hypocaloric weight loss program designed to induce a 30% energy restriction (500 – 600 kcal/day) with a macronutrient composition of 55% carbohydrate, 15% protein, and 30% fat, on Pomc methylation in obese male subjects. The participants were categorized as either “weight regainers” or “non-regainers” based on weight changes observed 32 weeks after stopping dieting. Interestingly, the CpG sites 10 and 11 of Pomc showed higher methylation levels in the regainers than non-regainers at baseline (66). This finding suggests that the methylation status of the Pomc promoter region is critical to regulating weight gain and may provide a useful biomarker for potential early detection and differential diagnosis of the predisposition to regain dietary-induced weight loss.
Another factor that affects the methylation status of Pomc is undernutrition, which is culpable in anorexia nervosa. The peripheral blood mononuclear cells of healthy women, underweight patients, and weight-recovered patients with anorexia nervosa were examined for specific DNA methylation of the Pomc gene expression (67). The researchers found that Pomc expression was higher in underweight patients than in weight-recovered patients or healthy controls. Although there was no significant difference in the overall DNA methylation between the groups, specific associations were found between the DNA methylation of single CpG residues and the expression of Pomc mRNA (67). This suggests that changes in Pomc expression observed in patients with anorexia nervosa may be more closely related to nutritional deficiencies rather than the direct effects of DNA methylation patterns or modifications.
Other factors, like stress and gender, also influence the DNA methylation of Pomc. A study on the early life stress caused by 3 hours of daily separation of pups from their dams over 10 consecutive days reduced DNA methylation, increasing Pomc mRNA in the pituitary gland (68). This suggests that early stress exposure may upregulate Pomc mRNA, potentially influencing long-term energy homeostasis. However, how much stress exposure will be beneficial is still debatable, given that prolonged stress has long-term metabolic consequences. Sex-specific differences in the DNA methylation status of the Pomc gene have been reported in various species, highlighting the role of epigenetic regulation of hypothalamic neuropeptides in metabolism. For instance, a study that determined the sex differences in the DNA methylation patterns across eight sites of the Pomc promoter of a 3-week-old chicken observed different methylation patterns across the promoter. Female chickens displayed higher methylation levels across the Pomc promoter than males (69). This elevated methylation in females was associated with reduced Pomc gene expression, suggesting that sex-specific epigenetic modifications may contribute to differences in metabolic regulation between male and female chickens.
However, demethylating the Pomc promoter does not always produce lean or energy-regulated phenotypes. For example, the cafeteria diet (CAF), which is an experimental rodent diet model that provides up to 4.85 kcal/g, 49% of energy as fat, 7% as protein, and 44% as carbohydrate, has been shown to influence Pomc expression (70). Exposure to the CAF diet was found to reduce methylation at the Pomc promoter in rats after 11 weeks and 20 weeks of feeding; however, this demethylation of the Pomc promoter did not adequately counteract the increased food intake observed, suggesting that an additional orexigenic mechanism is responsible for the increase in body weight observed (71). Similarly, targeted demethylation of the Pomc promoter by using CRISPR-dCas9-TET1 in HFD (60% kcal fat) fed rats did not prevent weight gain. Methylation repression using CRISPR-dCas9-DNMT3a also failed to alter body weight significantly (72), suggesting that Pomc promoter methylation changes may result from weight gain during obesity development rather than being its direct cause.
Further studies explored how adverse environmental factors influence histone modifications at the Pomc enhancer region using chromatin immunoprecipitation (ChIP) analysis. In one study, pups fed a high-carbohydrate diet exhibited decreased histone acetylation of H3K9 (H3K9ac) at the Pomc promoter compared to mother-fed pups, thereby reducing Pomc expression and contributing to diet-induced obesity (73). Adolescent alcohol exposure in rats was shown to increase histone acetylation of H3K9/14 at the Pomc promoter, leading to elevated Pomc mRNA levels that continued into adulthood (74). However, the result is different for offspring born to parents who consumed alcohol. A study also showed that prenatal exposure to ethanol in rats led to the production of offspring with suppressed histone activation marks (H3K4me3) and increased repressive marks (H3K9me2), thereby reducing Pomc mRNA levels (75). The differences might be because prenatal exposure affects the early development of the brain during critical epigenetic reprogramming, whereas key brain structures like the hypothalamus are already developed before adolescent alcohol exposure. Nonetheless, it is clear that environmental factors such as diet and alcohol can influence histone marks and Pomc expression, affecting metabolic outcomes.
miRNAs play critical regulatory roles in gene expression, including modulating hypothalamic Pomc expression, which is pivotal in maintaining energy homeostasis. For example, in leptin-deficient (ob/ob) mice, several miRNAs, such as miR-383, miR-384-3p, and miR-488, were upregulated and have been shown to negatively regulate Pomc mRNA expression (76). The downregulation of Pomc leads to disruptions in energy balance and contributes to the development of obesity. Importantly, with the treatment of leptin in these animals, there was the restoration of Pomc mRNA levels, underscoring the dynamic regulation of Pomc by leptin-induced changes in miRNAs, positioning them as potential therapeutic targets for metabolic disorders (76). Further evidence that shows the involvement of miRNAs in metabolic regulation comes from a study on the effect of prenatal alcohol exposure to mice between postnatal day 2 and postnatal day 6, which is the equivalent of the third trimester in humans due to the similarity in brain development. It was observed that prenatal alcohol exposure upregulated miR-383 and miR-384 while the Pomc gene expression was reduced in the mediobasal hypothalamus at postnatal days 6 and 60, respectively (77). These findings emphasize the influence of environmental factors, such as diet and prenatal exposure, on miRNA-mediated Pomc regulation, revealing a critical pathway by which miRNAs may impact metabolic health and disease risk across the lifespan.
Moreover, miRNA-specific manipulations can influence Pomc expression in the brain, particularly the hypothalamus. For instance, the knockdown of miR-342 in mice increased Pomc expression in response to a high-fat, high-sucrose diet, leading to reduced food intake and reduced body weight, thus implicating miR-342 expression in the development and progression of obesity (78). Additionally, loss of miR-29a in Pomc neurons contributes to hyperphagia, decreased energy expenditure, and obesity in female mice (79). Suffice it to mention that miR-375 has been identified as a negative regulator of Pomc expression in the pituitary gland. Inhibition of miR-375 led to an approximately 40% increase in the Pomc expression levels, while upregulation of miR-375 decreased Pomc expression levels (80). Put together, these findings highlight the intricate regulatory network governed by miRNAs in the hypothalamus, particularly in the Pomc neurons, and targeting these miRNAs could be beneficial in managing obesity and its related comorbidities.
The transgenerational effect of diet and environmental factors on epigenetic regulation of DNA methylation of hypothalamic neuropeptides is gaining attention among researchers. The DNA methylation of the Pomc promoter can occur as early as embryonic development and at different cell states. One study examining the Pomc DNA methylation of human embryonic stem cells (hESCs) observed that naive cells showed decreased DNA methylation compared to primed H1 cells, even after differentiation (81). However, when capacitated cells were used for hypothalamic neuronal differentiation, there was a marked increase in Pomc DNA methylation at the progenitor stage and even in Pomc-expressing neurons. The researchers further identified a negative correlation between Pomc DNA methylation and Pomc gene expression at CpG 1 to 7 during human embryogenesis (81). This early occurrence of DNA methylation of the Pomc gene implies that metabolic disease risk could be programmed at the cellular level before birth. As a result, if the methylation patterns persist or are exacerbated postnatally by either diet or environmental factors, they could predispose individuals to metabolic diseases by disrupting energy balance and promoting obesity.
Neonatal DNA methylation is often associated with maternal gestational age and nutritional patterns during these periods (82). In one study, offspring of mice dams fed with a high-fat, high-sucrose diet during pregnancy exhibited increased Pomc mRNA expression and hypomethylation of Pomc promoter in the hypothalamus, contributing to metabolic dysfunction in adulthood (83). Also, mice dams exposed to ethanol consumption had offspring with increased DNA-methylation enzyme (DNMT1), causing an increase in Pomc gene methylation and a concurrent reduction in Pomc mRNA expression (75). These findings suggest that the parents’ nutritional status has the potential to alter the methylation status of Pomc in the offspring, which may impact the Pomc expression, causing altered regulation of food intake, energy expenditure, and glucose homeostasis in adulthood.
1.5 Agouti-related peptide
AgRP is a potent orexigenic neuropeptide that stimulates appetite and reduces energy expenditure, acting as an antagonist to melanocortin receptors and predominantly present in the arcuate nucleus (84–86). In mice, using designer receptors exclusively activated by designer drugs (DREADD) to stimulate AgRP neurons resulted in increased food intake, reduced energy expenditure, and increased adiposity. On the other hand, inhibition of AgRP neuronal activity reduced food intake, highlighting the critical role of AgRP in regulating feeding and energy balance (87). Also, when rats were exposed to a low-protein diet, they exhibited increased AgRP mRNA expression and enhanced food intake, whereas intracerebroventricular injection of amino acids decreased AgRP mRNA and suppressed food intake (88). When AgRP is knocked out in mice and Siberian hamsters, the animals exhibit a reduced feeding drive and reduced weight gain (89, 90). All of these findings show the pivotal role of AgRP in influencing energy balance.
It is noteworthy that the different parts of the hypothalamus do not respond the same way to factors initiating AgRP activation. For example, a study in rats found an increased AgRP expression in the PVN and VMH, and a reduction in the ARC, following a cafeteria diet (91). When rat dams were fed a restricted protein diet during gestation and lactation, their pups displayed reduced AgRP expression in the PVN, while there was no change in the dorsomedial and lateral hypothalamus among pups fed by (92).
DNA methylation of the AgRp promoter region is less studied because of the lack of CpG sites in the promoter region. Some researchers could not even examine the DNA methylation of the AgRP promoter because it does not contain any CpG sites in the 1200 nucleotides upstream of the transcription sites (91, 92). In one study where rat pups born to caloric-restricted dams showed an increased AgRP expression and a decrease in birth weight, the researchers found that three CpG sites were 75% to 96% methylated (93). Another study on maternal HFD exposure revealed sex-specific differences in AgRP promoter methylation; male offspring displayed increased methylation at four CpG sites associated with a reduction in AgRP expression when compared to female offspring (94). However, all these sites are positioned outside the CpG islands of the AgRP promoter region.
Histone modification of AgRP has been observed to play a crucial role in regulating feeding behavior in the hypothalamus by influencing its expression and involvement in energy homeostasis. In rats that experienced caloric restriction following HFD-induced obesity, reduced repressive histone 3 lysine 9 methylation (H3K9me2) at the promoter of AgRP led to increased AgRP expression and heightened hunger signaling, suggesting anxadaptive response to restore energy homeostasis (95). Additionally, male rat offspring nursed by high-fat fed (60% Kcal fat) dams and weaned to control diet (10% Kcal fat) had elevated AgRP expression at six months with a significant increase in lysine-specific histone demethylase 1 (LSD1) and a decrease in histone deacetylase 1 (HDAC1) highlighting the effects of maternal HFD on the hypothalamic energy regulation of their offspring (96).
Not many studies examined the regulatory roles of miRNAs in AgRP gene expression, likely due to the intricate feedback and redundant signaling mechanisms that control appetite and metabolism. In sheep, brain-derived neurotrophic factor (BDNF) infusion increases AgRP expression in the ARC with a concurrent increase in miRNA-33a-5p, miRNA-33 b-5p, and a reduction in miRNA-377-3p and miRNA-214-3p, suggesting the role of these miRNAs in the regulation of AgRP expression (97). Further research, however, is still required to examine how miRNAs influence AgRP gene expression in the hypothalamus, as this gene is crucial in energy homeostasis.
1.6 Neuropeptide Y
NPY is an orexigenic peptide that is abundantly present in the arcuate nucleus of the hypothalamus (98). It plays a key role in regulating feeding behavior by stimulating appetite and increasing hunger, leading to increased food consumption, fat accumulation, weight gain, and obesity (99). NPY exerts its effects by binding to its G protein-coupled receptors, primarily Y1R, which is a key receptor driving appetite, along with other receptors like Y2R, Y4R, Y5R, and Y6R (100). Npy and AgRP neurons are colocalized in the arcuate nucleus, providing synergistic effects (101). A study reported that mice deficient in Npy exhibited normal food intake and body weight under basal conditions, a phenomenon similarly observed in AgRP knockout mice (102). A knockout of Npy or its receptors rarely yields a marked phenotype because compensatory mechanisms, such as other appetite-regulating neuropeptides like AgRP, adjust to offset the deficiency (102).
Recent research has significantly advanced our understanding of how epigenetic modifications impact Npy mRNA and its promoter under different environmental influences. For example, a study employed the use of restriction enzymes to assess the DNA methylation status of the Npy promoter in rats fed a cafeteria diet (CAF) at different feeding periods (71). They found that there was a reduction in DNA methylation of the Npy promoter at 11 weeks (medium term) and 20 weeks (long term) but not 4 weeks (short term), leading to an increase in Npy mRNA contributing to overeating and weight gain (71). This finding indicates that the duration of environmental exposure significantly influences epigenetic modifications in neuropeptides like Npy, potentially explaining variations in metabolic phenotypes. Another study showed that diet-resistant rats exhibited DNA methylation at the 5th CpG site of the Npy promoter region when compared to diet-induced obese rats (63). The increased methylation in diet-resistant rats corresponded with reduced Npy expression, which correlated with lower food intake and weight gain resistance compared to diet-induced obese rats, thus preventing the rats from gaining weight. Additionally, a clinical study examined the methylation status of the Npy promoter region in obese males undergoing an 8-week hypocaloric diet intervention and observed at 8 and 32 weeks after the intervention, using leukocytes. The weight regainers showed a significant decrease in the methylation levels of Npy CpG sites 4 and 8 compared to non-regainers, even at baseline (66). Apart from the fact that hypomethylation at these sites is associated with decreased Npy expression, this finding also suggests that the methylation status of the Npy promoter region may provide a useful biomarker for potential early detection and differential diagnosis of the predisposition to regain dietary-induced weight loss.
Furthermore, studies have also shown that there is a transgenerational effect of diet-induced epigenetic changes in Npy expression. For example, pups of dams fed a high-carbohydrate diet showed reduced DNA methylation at the Npy promoter, leading to increased Npy expression (73). These epigenetic alterations were associated with hyperphagia, leading to increased weight gain and, subsequently, obesity. This finding highlights that parental epigenetic marks can persist through epigenetic reprogramming during conception, thereby influencing the susceptibility to metabolic diseases in adulthood.
Histone modification at the Npy promoter has also been studied. Rats fed a high-carbohydrate diet showed increased acetylation of H3K9 at the Npy promoter, which was linked to elevated Npy expression, contributing to obesity. The same diet did not significantly affect H3K9 methylation levels, suggesting that histone acetylation may play a more prominent role in diet-induced obesity (73). In another study where sex differences in the mice hypothalamic neurons are explored, the researchers found that H3K27me3 at the Npy promoter was higher in males than in females, indicating a more suppressive effect in males. siRNA-mediated knockdown of Kdm6a, a histone demethylase that specifically targets H3K27, resulted in increased H3K27me3 at the Npy promoter in females but not in males (103). This suggests that different types of histone modifications, such as methylation and acetylation, have distinct roles in regulating the target gene expression of Npy and may be sex-specific.
Besides, stress-related factors have been shown to be associated with epigenetic changes in Npy. For example, traumatic brain injury was associated with a significant reduction in H3K9ac levels at the Npy promoter in the arcuate nucleus, reducing food intake (104). Likewise, phenyl butyric acid, an endoplasmic reticulum stress inhibitor as well as a histone deacetylase inhibitor, has been shown to increase Npy mRNA levels by increasing H3K9/14ac at the promoter of Npy, further implicating a potential role of histone acetylation in the development of obesity (105). These findings suggest that stress or injury-induced changes to histone marks can suppress Npy expression, thereby reducing appetite and potentially altering energy balance. Moreover, pharmacologically targeting histone acetylation can modulate Npy expression and influence feeding behavior and weight gain.
miRNAs are also involved in regulating Npy expression. For example, the downregulation of miR-103/107 in mice is associated with reduced Npy mRNA expression, thereby affecting energy homeostasis (106). Mice lacking miR-342 showed a significant reduction in the total number of activated Npy in response to a high-fat, high-sugar diet in mice (78). In humans, miR-4713 and miR-452 were shown to be associated with increased Npy1r among obese children when assessed by miRwalk2.0, a tool for miRNA target prediction (107). These findings suggest that interventions targeting specific miRNAs might enhance our understanding of metabolic control to combat obesity through the modulation of Npy signaling.
1.7 Leptin receptor
The leptin receptor (LepR) belongs to the class 1 cytokine receptor family (108, 109). It exists in two primary isoforms: short forms and long forms (110). While the short form has limited functionality, the long form (LepRb) plays a crucial role in leptin signaling (111). LepRb is predominantly expressed in the central nervous system, especially in the hypothalamus, a region central to energy homeostasis. Circulating leptin, produced from the adipose tissue, mediates its effect by binding to LepRb in the brain, stimulating downstream signaling to regulate energy homeostasis. Studies have demonstrated that the dysfunction of LepRb or leptin signaling is involved in the development and progression of obesity. For instance, a mutation in the LepRb gene in humans resulted in weight gain and hyperphagia similar to those observed in leptin-deficient transgenic mice (112, 113). Deletion of LepRb in somatotrophs of the hypothalamus results in obesity (114), emphasizing the essential role of hypothalamic leptin receptors in maintaining energy balance.
The epigenetic modification of LepRb within the hypothalamus is critical to metabolism. One study in mice observed that a maternal HFD increased the DNA methylation levels of the LepRb promoter, increasing weight gain and reducing LepRb expression among the pups (115). Although specific CpG sites were not identified, higher global methylation levels were correlated with the decreased expression of LepRb. Several studies have failed to detect significant DNA methylation and found no significant DNA methylation of LepRb in the hypothalamus (116–118), potentially due to the unique epigenetic environment of this brain region. It is likely that leptin signaling may rely more on post-translational modifications or receptor sensitivity rather than DNA methylation changes.
Histone modification of leptin receptors in the hypothalamus has been shown to regulate metabolism. It was found that mice fed a high-fat diet upregulated slug, a transcription factor that recruits EZH2, which represses LepRb expression by increasing H3K27 dimethylation/trimethylation (H3K27me2/3) at the LepRb promoter in the hypothalamus (119), suggesting that HFD can epigenetically impact leptin receptor expression, thereby promoting and developing metabolic disorders.
miRNA also regulates LepRb expression. The inhibition of miR-200 in the hypothalamus was observed to increase the expression of LepRb, resulting in suppressed appetite, reduced food intake, and ultimately leading to reduced body weight (120), suggesting the potential roles of microRNAs in regulating LepRb mRNA. Further research on histone and miRNA modifications is needed to fully elucidate the impact of epigenetic changes on regulating LepRb to combat metabolic diseases.
1.8 Insulin receptor
In the brain, InsR is highly expressed in various brain regions, including the hypothalamus, olfactory bulb, cerebral cortex, cerebellum, and choroid plexus (121–123). InsR has two isoforms known as “A” and “B” (124). The “A” isoform is primarily found in the brain, while the “B” isoform is expressed in the liver, muscle, adipocytes, and kidney (125). Insulin, produced from the beta cells of the pancreas, crosses the blood-brain barrier to activate InsR in the brain. However, InsR’s role extends beyond glucose metabolism because only a fraction of glucose supplied to the neurons is insulin-dependent. In the hypothalamus, InsR, as well as their substrates, are vital for energy balance (126). In a transgenic mouse model where Insr-2 is knocked out, the female mice had an increase in food intake and body weight when compared to the control mice. No changes were observed in male mice (127). When a selective decrease in hypothalamic insulin receptor expression was performed using osmotic pumps to infuse oligodeoxynucleotide antisense, the rats became hyperphagic, had increased fat mass, and glucose intolerance (128, 129). These findings highlight the importance of hypothalamic insulin receptors in energy homeostasis and the need to examine how epigenetic modifications impact their functions.
Overfeeding or overnutrition has been shown to alter the DNA methylation of InsR. In a study that examined the effects of neonatal overfeeding on InsR in the hypothalamus, the researchers observed an increase in methylation of the CpG island of the insulin receptor promoter in over-nourished rats, resulting in rapid weight gain and the development of metabolic syndrome (130). Another study in rats showed that a maternal HFD led to hypermethylation of the hypothalamic InsR in male offspring but not female, leading to hyperleptinemia, hyperinsulinemia, impaired glucose tolerance, increased insulin resistance, and obesity (131). Collectively, these findings suggest early life overnutrition can induce sex -specific epigenetic modifications of hypothalamus InsR, thereby predisposing offspring to long-term metabolic dysregulation and obesity.
miRNAs have been observed to regulate InsR in the hypothalamus. In diabetic rats, miR-194-5p and miR-200a-3p are upregulated, leading to reduced InsR protein levels while Inhibition of these miRNAs restores InsR levels (132). Similarly, inhibition of miR-200 in the hypothalamus of db/db mice increased InsR expression, suppressed appetite, reduced food intake, and decreased body weight (120). These findings highlight the roles of hypothalamic miRNAs in regulating central insulin sensitivity and energy balance.
1.9 CART and endocannabinoid system
In addition to well-characterized neuropeptides such as NPY and POMC, several other hypothalamic regulators, including CART, and components of endocannabinoid systems, play a pivotal role in controlling energy homeostasis, feeding behaviors, and metabolic adaptation. Although their physiological significance is well established, the epigenetic mechanism governing their expression and activity in response to environmental cues remains incompletely understood.
CART is a key neuropeptide in the hypothalamus that plays a crucial role in regulating energy balance and feeding behavior. As an anorexigenic peptide, CART suppresses appetite and reduces food intake (133). Knockout studies have shown that deletion of Cart results in increased body weight and the development of obesity (134, 135), emphasizing its critical role in maintaining energy balance. Hypermethylation of the Cart promoter has been linked to reduced Cart mRNA expression in calorie-restricted rats, contributing to imbalanced energy intake and obesity (136). Although the epigenetic regulation of Cart remains underexplored, it has been suggested that many of its physiological effects may overlap with those of Pomc. This highlights the need for more studies on the epigenetic regulation of Cart, particularly in the context of metabolic adaptations and disease.
The endocannabinoid system, which consists of the cannabinoid receptor 1 (CB1R) and cannabinoid receptor 2 (CB2R), is an important mechanism involved in metabolism. Activation of CB1R in the ARC and VMH stimulates appetite and promotes energy storage, contributing to obesity, inflammation, and insulin resistance (137, 138). Mice lacking CB1R are resistant to diet-induced obesity, demonstrating the regulatory roles of endocannabinoid signaling in energy balance (139). Maternal HFD increases CB1R expression in the offspring and leads to sex-specific epigenetic modifications. For instance, female offspring of rats not affected by maternal HFD exhibited lower methylation at a CpG site within the CB1R intron but higher histone H3 acetylation in the promoter region. In contrast, male offspring exposed to maternal HFD showed increased histone acetylation (140). These findings suggest that diet-induced epigenetic modifications in CB1R may differ by sex and environmental exposures. Although CB2R has also been implicated in body weight control and glucose homeostasis, its epigenetic regulation within the hypothalamus remains unexplored.
2 Conclusion and future direction
The hypothalamus is a complex neuronal network with notable cell populations equipped to maintain energy homeostasis by integrating hormonal, neural, and nutrient-related signals. It regulates both short-term appetite and long-term metabolic adaptation to prevent energy imbalance and the development of metabolic disorders such as obesity (141). Despite increasing evidence linking hypothalamic dysfunction to metabolic disease, the role of epigenetic regulation in this brain region remains underexplored, warranting a comprehensive review to synthesize current knowledge and identify future research gaps. In this review, we have discussed how adverse environmental factors such as malnutrition, alcohol consumption, stress, traumatic brain injury, and sex-specific differences can trigger epigenetic changes in the hypothalamus. Clinical and preclinical studies have demonstrated that epigenetic changes, including DNA methylation, histone modification, and regulation by non-coding RNA (miRNAs), can modify the expression of genes critical for energy balance, such as the neuropeptides (POMC, AgRP, NPY, CART) and metabolic hormone receptors (LepRb, InsR, cannabinoid receptors). Disruption of these signaling pathways within the hypothalamic nuclei (ARC, VMH, DMH, PVN) impairs the brain’s ability to integrate metabolic cues, leading to abnormal appetite regulation and reduced energy expenditure. Over time, these changes promote the development of obesity and related metabolic disorders (Figure 1).
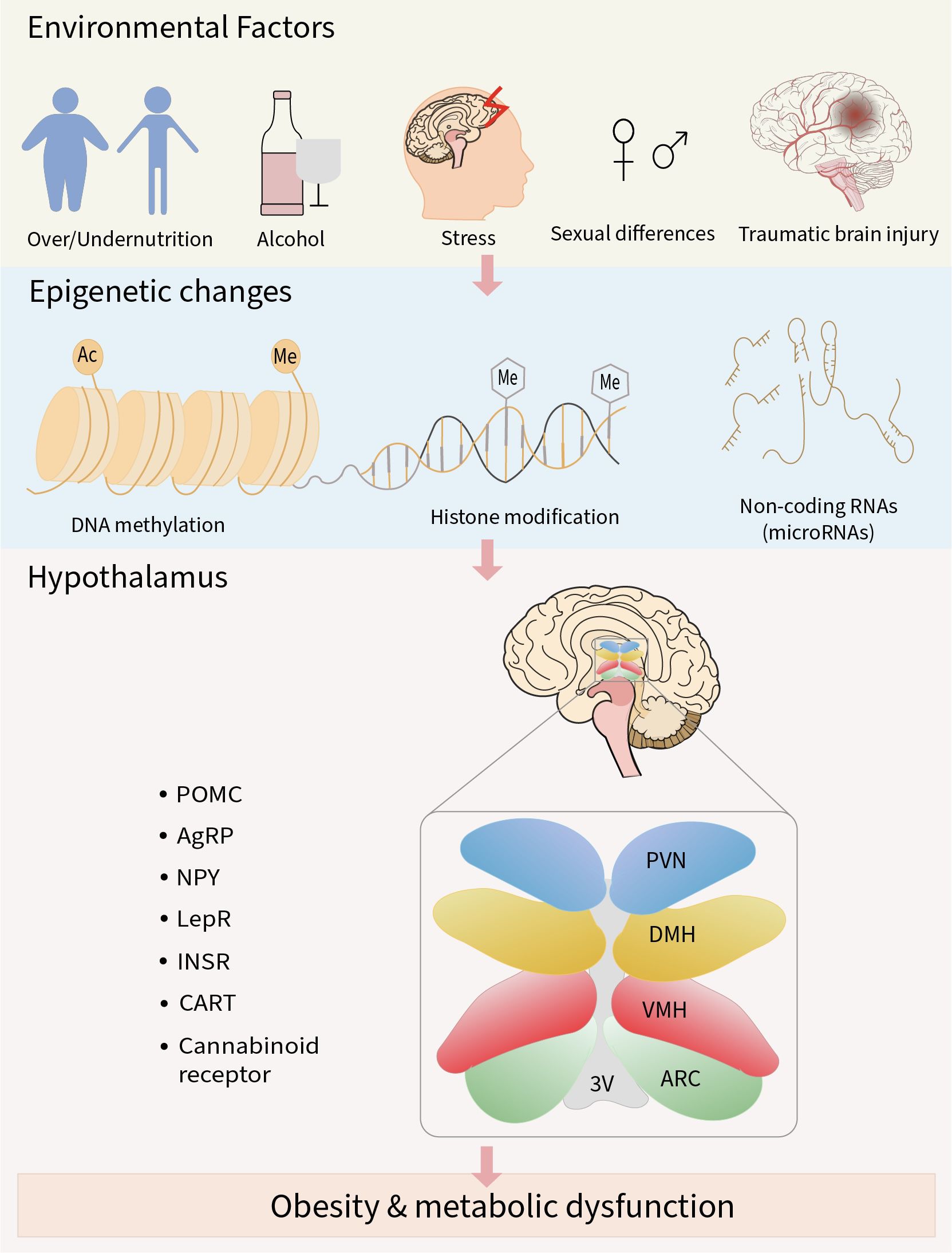
Figure 1. Schematic overview of the review. Adverse environmental factors can result in abnormal epigenetic modifications in neuropeptides and metabolic hormone receptors involved in the regulation of energy metabolism. Sustained exposure to these factors may lead to impaired metabolic signaling, contributing to the development of obesity and related metabolic disorders.
Despite robust evidence from animal models, translating these mechanistic insights to humans remains challenging. Ethical and logistical constraints severely limit access to human hypothalamic tissue, hindering direct validation of animal-based findings. Potential solutions include the use of postmortem hypothalamic tissue repositories, the generation of human induced pluripotent stem cell (iPSC)-derived hypothalamic neurons, and the development of 3D brain organoids to model human-specific epigenetic regulation (142). Furthermore, non-invasive neuroimaging integrated with circulating biomarkers, such as miRNAs, may provide functional proxies for hypothalamic activity in living subjects. Beyond accessibility, the complexity of epigenetic regulation, being cell-type specific, dynamic, and shaped by multiple environmental inputs, requires advanced methodologies. Future research should incorporate single-cell epigenomic profiling, longitudinal study designs, and careful consideration of sex differences to achieve a more precise understanding of hypothalamic epigenetic mechanisms in metabolic diseases.
A deeper grasp of these mechanisms creates a foundation for novel therapeutic strategies targeting hypothalamic epigenetic dysregulation in metabolic disease. One promising strategy is miRNA modulation, such as miR-200 and miR-34a, which influence neuropeptide and hormone receptor expression involved in appetite control and energy homeostasis (120, 143–145). Therapeutic agents like miRNA mimics or antagomirs could normalize dysregulated pathways and restore metabolic balance. Another approach involves reprogramming histone modification states through drugs that activate or inhibit histone deacetylases(HDACs), histone methyltransferases (HMTs), or demethylases. Importantly, some HDAC inhibitors are already in clinical use or undergoing trials for cancer (146–149), offering opportunities for repurposing toward metabolic disorders. Finally, precision epigenetic editing tools, such as CRISPR-Cas9-based epigenetic editors, could allow locus-specific modulation of gene expression, directly correcting pathogenic chromatin states within the hypothalamic neurons. Collectively, these strategies rapidly define an emerging field, where mechanistic insight and technological innovation converge to combat obesity and metabolic syndrome at their epigenetic roots.
Author contributions
BO: Conceptualization, Writing – review & editing, Project administration, Writing – original draft, Methodology, Validation, Investigation, Data curation, Formal analysis. SM: Data curation, Writing – review & editing, Investigation, Writing – original draft, Visualization, Formal analysis. M-HK: Funding acquisition, Validation, Methodology, Writing – review & editing, Formal analysis, Supervision, Project administration, Investigation, Resources, Conceptualization, Writing – original draft.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study was supported by the NIH R01DK139038.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2025.1645474/full#supplementary-material
References
1. Harrold JA, Dovey TM, Blundell JE, and Halford JCG. CNS regulation of appetite. Neuropharmacology. (2012), 3–17. doi: 10.1016/j.neuropharm.2012.01.007
2. Grill HJ, Schwartz MW, Kaplan JM, Foxhall JS, Breininger J, and Baskin DG. Evidence that the caudal brainstem is a target for the inhibitory effect of leptin on food intake. Endocrinology. (2002) 143:239–46. doi: 10.1210/endo.143.1.8589
3. Saper CB and Lowell BB. The hypothalamus. Curr Biol. (2014), R1111–6. doi: 10.1016/j.cub.2014.10.023
4. Sainsbury A and Zhang L. Role of the arcuate nucleus of the hypothalamus in regulation of body weight during energy deficit. Mol Cell Endocrinology. (2010) 316:109–19. doi: 10.1016/j.mce.2009.09.025
5. Sutton AK, Myers MG, and Olson D. The role of PVH circuits in leptin action and energy balance. Annu Rev Physiol. (2016), 207–21. doi: 10.1146/annurev-physiol-021115-105347
6. Yang L, Scott KA, Hyun J, Tamashiro KL, Tray N, Moran TH, et al. Role of dorsomedial hypothalamic neuropeptide Y in modulating food intake and energy balance. J Neurosci. (2009) 29:179–90. doi: 10.1523/JNEUROSCI.4379-08.2009
7. Gavini CK, Jones WC, and Novak CM. Ventromedial hypothalamic melanocortin receptor activation: regulation of activity energy expenditure and skeletal muscle thermogenesis. J Physiol. (2016) 594:5285–301. doi: 10.1113/tjp.2016.594.issue-18
8. Monda M, Amaro IS, and De Luca B. The influence of exercise on energy balance changes induced by ventromedial hypothalamic lesion in the rat. Physiol Behav. (1993) 54(6):1057–61. doi: 10.1016/0031-9384(93)90324-9
9. Rui L. Brain regulation of energy balance and body weight. Rev Endocr Metab Disord. (2013) 14:387–407. doi: 10.1007/s11154-013-9261-9
10. Rossi MA. Control of energy homeostasis by the lateral hypothalamic area. Trends Neurosciences. (2023), 738–49. doi: 10.1016/j.tins.2023.05.010
11. Deem JD, Faber CL, and Morton GJ. AgRP neurons: Regulators of feeding, energy expenditure, and behavior. FEBS J. (2022), 2362–81. doi: 10.1111/febs.v289.8
12. Luo N, Marcelin G, Liu SM, Schwartz G, and Chua S. Neuropeptide Y and Agouti-Related Peptide Mediate Complementary Functions of Hyperphagia and Reduced Energy Expenditure in Leptin Receptor Deficiency. Endocrinology. (2011) 152(3):883–9.
13. Hill JW. Gene expression and the control of food intake by hypothalamic POMC/CART neurons. Open Neuroendocrinol J. (2010) 3:21–7.
14. Könner AC and Brüning JC. Selective insulin and leptin resistance in metabolic disorders. Cell Metab. (2012), 144–52. doi: 10.1016/j.cmet.2012.07.004
15. Hosoi T and Maffei M. Editorial: Leptin resistance in metabolic disorders: Possible mechanisms and treatments. Front Endocrinol. (2017). doi: 10.3389/fendo.2017.00300
16. Carmo-Silva S and Cavadas C. Hypothalamic dysfunction in obesity and metabolic disorders. Adv Neurobiol. (2017), 73–116. doi: 10.1007/978-3-319-63260-5_4
17. de Lima RMS, dos Santos Bento LV, di Marcello Valladão Lugon M, Barauna VG, Bittencourt AS, Dalmaz C, et al. Early life stress and the programming of eating behavior and anxiety: Sex-specific relationships with serotonergic activity and hypothalamic neuropeptides. Behav Brain Res. (2020) 379:112399. doi: 10.1016/j.bbr.2019.112399
18. Benite-Ribeiro SA, Putt DA, and Santos JM. The effect of physical exercise on orexigenic and anorexigenic peptides and its role on long-term feeding control. Med Hypotheses. (2016) 93:30–3. doi: 10.1016/j.mehy.2016.05.005
19. Ziotopoulou M, Mantzoros CS, Hileman SM, and Flier JS. Differential expression of hypothalamic neuropeptides in the early phase of diet-induced obesity in mice. Am J Physiology-Endocrinology Metab. (2000) 279:E838–45. Available online at: http://www.ajpendo.orgE838.
20. Alegría-Torres JA, Baccarelli A, and Bollati V. Epigenetics and lifestyle. Epigenomics. (2011) 3:267–77. doi: 10.2217/epi.11.22
21. Vickers M. Early life nutrition, epigenetics and programming of later life disease. Nutrients. (2014) 6:2165–78. doi: 10.3390/nu6062165
22. Sales VM, Ferguson-Smith AC, and Patti ME. Epigenetic mechanisms of transmission of metabolic disease across generations. Cell Metab. (2017) 25:559–71. doi: 10.1016/j.cmet.2017.02.016
23. Gibney ER and Nolan CM. Epigenetics and gene expression. Heredity. (2010), 4–13. doi: 10.1038/hdy.2010.54
24. Holliday R. Epigenetics: A historical overview. Epigenetics. (2006) 1:76–80. doi: 10.4161/epi.1.2.2762
25. Mariano IR, Yamada LA, Soares Rabassi R, Rissi Sabino VL, Bataglini C, Azevedo SCSF, et al. Differential responses of liver and hypothalamus to the nutritional condition during lactation and adult life. Front Physiol. (2020) 11. doi: 10.3389/fphys.2020.00553
26. Gołyszny M, Obuchowicz E, and Zieliński M. Neuropeptides as regulators of the hypothalamus-pituitary-gonadal (HPG) axis activity and their putative roles in stress-induced fertility disorders. Neuropeptides. (2022). doi: 10.1016/j.npep.2021.102216
27. Marco A, Kisliouk T, Weller A, and Meiri N. High fat diet induces hypermethylation of the hypothalamic Pomc promoter and obesity in post-weaning rats. Psychoneuroendocrinology. (2013) 38:2844–53. doi: 10.1016/j.psyneuen.2013.07.011
28. Plagemann A, Harder T, Brunn M, Harder A, Roepke K, Wittrock-Staar M, et al. Hypothalamic proopiomelanocortin promoter methylation becomes altered by early overfeeding: An epigenetic model of obesity and the metabolic syndrome. J Physiol. (2009) 587:4963–76. doi: 10.1113/tjp.2009.587.issue-20
29. Moore LD, Le T, and Fan G. DNA methylation and its basic function. Neuropsychopharmacology. (2013), 23–38. doi: 10.1038/npp.2012.112
30. Xie W, Barr CL, Kim A, Yue F, Lee AY, Eubanks J, et al. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell. (2012) 148:816–31. doi: 10.1016/j.cell.2011.12.035
31. Saxonov S, Berg P, and Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proceedings of the National Academy of Sciences. (2006) 103(5):1412–7. doi: 10.1073/pnas.0510310103
32. Mohn F, Weber M, Rebhan M, Roloff TC, Richter J, Stadler MB, et al. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol Cell. (2008) 30:755–66. doi: 10.1016/j.molcel.2008.05.007
33. Barres R and Zierath JR. DNA methylation in metabolic disorders. Am J Clin Nutr. (2011) 93(4):897S–900S. doi: 10.3945/ajcn.110.001933
34. Ehrlich M, A-Gama-Sosa M, Huang L-H, Midgett RM, Kuo KC, McCune RA, et al. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues or cells. Nucleic Acids Res. (1982) 10(8):2709–21. doi: 10.1093/nar/10.8.2709
35. Zhang Y, Sun Z, Jia J, Du T, Zhang N, Tang Y, et al. Overview of histone modification. (2021), 1–16. doi: 10.1007/978-981-15-8104-5_1
36. Roth SY, Denu JM, and David Allis C. Histone acetyltransferases. Annu Rev Biochem. (2001) 70(1):81–120. doi: 10.1146/annurev.biochem.70.1.81
37. Zhao Y and Garcia BA. Comprehensive catalog of currently documented histone modifications. Cold Spring Harb Perspect Biol. (2015) 7(9):a025064. doi: 10.1101/cshperspect.a025064
38. Bannister AJ and Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. (2011), 381–95. doi: 10.1038/cr.2011.22
39. Kouzarides T. Chromatin modifications and their function. Cell. (2007), 693–705. doi: 10.1016/j.cell.2007.02.005
40. Jaskelioff M and Peterson CL. Chromatin and transcription: histones continue to make their marks. Nat Cell Biol. (2003) 5:395–9. doi: 10.1038/ncb0503-395
41. Lagos-Quintana M, Rauhut R, Lendeckel W, and Tuschl T. Identification of novel genes coding for small expressed RNAs. Science (1979). (2001) 294(5543):853–8. doi: 10.1126/science.1064921
42. Schneeberger M, Gomez-Valadés AG, Ramirez S, Gomis R, and Claret M. Hypothalamic miRNAs: Emerging roles in energy balance control. Front Neurosci. (2015) 9. doi: 10.3389/fnins.2015.00041
43. Ha M and Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. (2014), 509–24. doi: 10.1038/nrm3838
44. Roberts TC. The microRNA machinery. In MicroRNA: Basic Science: Molecular Biology to Clinical Practice. Cham: Springer International Publishing. (2015), p. 15–30. doi: 10.1007/978-3-319-22380-3_2
45. Li Y and Kowdley KV. MicroRNAs in common human diseases. Genomics Proteomics Bioinf. (2012), 246–53. doi: 10.1016/j.gpb.2012.07.005
46. Fernández-Hernando C, Ramírez CM, Goedeke L, and Suárez Y. MicroRNAs in metabolic disease. Arterioscler Thromb Vasc Biol. (2013) 33:178–85. doi: 10.1161/ATVBAHA.112.300144
47. Rottiers V, Najafi-Shoushtari SH, Kristo F, Gurumurthy S, Zhong L, Li Y, et al. Micrornas in metabolism and metabolic diseases. Cold Spring Harb Symp Quant Biol. (2011) 76:225–33. doi: 10.1101/sqb.2011.76.011049
48. Young JI, Nica Otero V, Cerdá MG, Falzone TSL, Chan EC, Low MJ, et al. Authentic cell-specific and developmentally regulated expression of pro-opiomelanocortin genomic fragments in hypothalamic and hindbrain neurons of transgenic mice. The Journal of Neuroscience. (1998) 18(17):6631–40. doi: 10.1523/JNEUROSCI.18-17-06631.1998
49. Diano S. New aspects of melanocortin signaling: A role for PRCP in α-MSH degradation. Front Neuroendocrinology. (2011), 70–83. doi: 10.1016/j.yfrne.2010.09.001
50. Krieger DT, Yamaguchi H, and Liotta AS. Human plasma ACTH, lipotropin, and endorphin. Adv Biochem Psychopharmacol. (1981) 28:541–56.
51. Toda C, Santoro A, Kim JD, and Diano S. The annual review of physiology. Annu Rev Physiol. (2017) 79(1):209–36. doi: 10.1146/annurev-physiol-022516-034110
52. Millington GWM. The role of proopiomelanocortin (POMC) neurones in feeding behaviour. Nutr Metab. (2007) 4(1):18. doi: 10.1186/1743-7075-4-18
53. Faulkner LD, Dowling AR, Stuart RC, Nillni EA, and Hill JW. Reduced melanocortin production causes sexual dysfunction in male mice with POMC neuronal insulin and leptin insensitivity. Endocrinol (United States). (2015) 156:1372–85. doi: 10.1210/en.2014-1788
54. Wang W, Guo DY, Lin YJ, and Tao YX. Melanocortin regulation of inflammation. Front Endocrinol. (2019) 10. doi: 10.3389/fendo.2019.00683
55. Qu N, He Y, Wang C, Xu P, Yang Y, Cai X, et al. A POMC-originated circuit regulates stress-induced hypophagia, depression, and anhedonia. Mol Psychiatry. (2020) 25:1006–21. doi: 10.1038/s41380-019-0506-1
56. Krude H, Biebermann H, Luck W, Horn R, Brabant G, and Grüters A. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet. (1998) 19:155–7. doi: 10.1038/509
57. Farooqi IS, Drop S, Clements A, Keogh JM, Biernacka J, Lowenbein S, et al. Heterozygosity for a POMC-null mutation and increased obesity risk in humans. Diabetes. (2006) 55:2549–53. doi: 10.2337/db06-0214
58. Yaswen L, Diehl N, Brennan MB, and Hochgeschwender U. Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat Med. (1999) 5:1066–70. doi: 10.1038/12506
59. Chhabra KH, Adams JM, Fagel B, Lam DD, Qi N, Rubinstein M, et al. Hypothalamic POMC deficiency improves glucose tolerance despite insulin resistance by increasing Glycosuria. Diabetes. (2016) 65:660–72. doi: 10.2337/db15-0804
60. Shi C, Lu Y, Zhai G, Huang J, Shang G, Lou Q, et al. Hyperandrogenism in POMCa-deficient zebrafish enhances somatic growth without increasing adiposity. J Mol Cell Biol. (2020) 12:291–304. doi: 10.1093/jmcb/mjz053
61. Yang Z, Wong J, Wang L, Sun F, and Yue GH. pomc knockout increases growth in zebrafish. Aquaculture. (2023), 574. doi: 10.1016/j.aquaculture.2023.739707
62. Fei F, Sun SY, Yao YX, and Wang X. Generation and phenotype analysis of zebrafish mutations of obesity-related genes lepr and mc4r. Sheng Li Xue Bao. (2017) 69:61–9.
63. Cifani C, Micioni Di Bonaventura MV, Pucci M, Giusepponi ME, Romano A, Di Francesco A, et al. Regulation of hypothalamic neuropeptides gene expression in diet induced obesity resistant rats: Possible targets for obesity prediction? Front Neurosci. (2015) 9. doi: 10.3389/fnins.2015.00187
64. Marco A, Kisliouk T, Tabachnik T, Weller A, and Meiri N. DNA CpG methylation (5-methylcytosine) and its derivative (5-hydroxymethylcytosine) alter histone posttranslational modifications at the Pomc promoter, affecting the impact of perinatal diet on leanness and obesity of the offspring. Diabetes. (2016) 65:2258–67. doi: 10.2337/db15-1608
65. Kühnen P, Handke D, Waterland RA, Hennig BJ, Silver M, Fulford AJ, et al. Interindividual variation in DNA methylation at a putative POMC metastable epiallele is associated with obesity. Cell Metab. (2016) 24:502–9. doi: 10.1016/j.cmet.2016.08.001
66. Crujeiras AB, Campion J, Díaz-Lagares A, Milagro FI, Goyenechea E, Abete I, et al. Association of weight regain with specific methylation levels in the NPY and POMC promoters in leukocytes of obese men: A translational study. Regul Pept. (2013) 186:1–6. doi: 10.1016/j.regpep.2013.06.012
67. Ehrlich S, Weiss D, Burghardt R, Infante-Duarte C, Brockhaus S, Muschler MA, et al. Promoter specific DNA methylation and gene expression of POMC in acutely underweight and recovered patients with anorexia nervosa. J Psychiatr Res. (2010) 44:827–33. doi: 10.1016/j.jpsychires.2010.01.011
68. Wu Y, Patchev AV, Daniel G, Almeida OFX, and Spengler D. Early-Life stress reduces dna methylation of the pomc gene in male mice. Endocrinology. (2014) 155:1751–62. doi: 10.1210/en.2013-1868
69. Rancourt RC, Schellong K, Tzschentke B, Henrich W, and Plagemann A. DNA methylation and expression of proopiomelanocortin (POMC) gene in the hypothalamus of three-week-old chickens show sex-specific differences. FEBS Open Bio. (2018) 8:932–9. doi: 10.1002/feb4.2018.8.issue-6
70. Sampey BP, Vanhoose AM, Winfield HM, Freemerman AJ, Muehlbauer MJ, Fueger PT, et al. Cafeteria diet is a robust model of human metabolic syndrome with liver and adipose inflammation: comparison to high-fat diet. Obesity. (2011) 19:1109–17. doi: 10.1038/oby.2011.18
71. Lazzarino GP, Acutain MF, Canesini G, Andreoli MF, and Ramos JG. Cafeteria diet induces progressive changes in hypothalamic mechanisms involved in food intake control at different feeding periods in female rats. Mol Cell Endocrinol. (2019) 498:110542. doi: 10.1016/j.mce.2019.110542
72. McFadden T, Gaito N, Carucci I, Fletchall E, Farrell K, and Jarome TJ. Controlling hypothalamic DNA methylation at the Pomc promoter does not regulate weight gain during the development of obesity. PloS One. (2023) 18(4):e0284286. doi: 10.1371/journal.pone.0284286
73. Mahmood S, Smiraglia DJ, Srinivasan M, and Patel MS. Epigenetic changes in hypothalamic appetite regulatory genes may underlie the developmental programming for obesity in rat neonates subjected to a high-carbohydrate dietary modification. J Dev Orig Health Dis. (2013) 4:479–90. doi: 10.1017/S2040174413000238
74. Kokare DM, Kyzar EJ, Zhang H, Sakharkar AJ, and Pandey SC. Adolescent alcohol exposure-induced changes in alpha-melanocyte stimulating hormone and neuropeptide y pathways via histone acetylation in the brain during adulthood. Int J Neuropsychopharmacol. (2017) 20:758–68. doi: 10.1093/ijnp/pyx041
75. Bekdash RA, Zhang C, and Sarkar DK. Gestational choline supplementation normalized fetal alcohol-induced alterations in histone modifications, DNA methylation, and proopiomelanocortin (POMC) gene expression in β-endorphin-producing POMC neurons of the hypothalamus. Alcohol Clin Exp Res. (2013) 37:1133–42. doi: 10.1111/acer.2013.37.issue-7
76. Derghal A, Djelloul M, Airault C, Pierre C, Dallaporta M, Troadec JD, et al. Leptin is required for hypothalamic regulation of miRNA stargeting POMC 3′UTR. Front Cell Neurosci. (2015) 9. doi: 10.3389/fncel.2015.00172
77. Gangisetty O, Chaudhary S, Tarale P, Cabrera MA, and Sarkar DK. miRNA-383 and miRNA-384 suppress proopiomelanocortin gene expression in the hypothalamus: effects of early life ethanol exposure. Neuroendocrinology. (2023) 113:844–58. doi: 10.1159/000530289
78. Zhang D, Yamaguchi S, Zhang X, Yang B, Kurooka N, Sugawara R, et al. Upregulation of mir342 in diet-induced obesity mouse and the hypothalamic appetite control. Front Endocrinol (Lausanne). (2021) 12. doi: 10.3389/fendo.2021.727915
79. Ma Y, Murgia N, Liu Y, Li Z, Sirakawin C, Konovalov R, et al. Neuronal miR-29a protects from obesity in adult mice. Mol Metab. (2022) 61:101507. doi: 10.1016/j.molmet.2022.101507
80. Zhang N, Lin JK, Chen J, Liu XF, Liu JL, Luo HS, et al. MicroRNA 375 mediates the signaling pathway of corticotropin-releasing factor (CRF) regulating pro-opiomelanocortin (POMC) Expression by targeting Mitogen-activated protein Kinase 8. J Biol Chem. (2013) 288:10361–73. doi: 10.1074/jbc.M112.425504
81. Lechner L, Opitz R, Silver MJ, Krabusch PM, Prentice AM, Field MS, et al. Early-set POMC methylation variability is accompanied by increased risk for obesity and is addressable by MC4R agonist treatment. Sci Transl Med. (2023). doi: 10.1126/scitranslmed.adg1659
82. Schroeder JW, Conneely KN, Cubells JC, Kilaru V, Jeffrey Newport D, Knight BT, et al. Neonatal DNA methylation patterns associate with gestational age. Epigenetics. (2011) 6:1498–504. doi: 10.4161/epi.6.12.18296
83. Zheng J, Xiao X, Zhang Q, Yu M, Xu J, Wang Z, et al. Maternal and post-weaning high-fat, high-sucrose diet modulates glucose homeostasis and hypothalamic POMC promoter methylation in mouse offspring. Metab Brain Dis. (2015) 30:1129–37. doi: 10.1007/s11011-015-9678-9
84. Yang YK, Thompson DA, Dickinson CJ, Wilken J, Barsh GS, Kent SB, et al. Characterization of agouti-related protein binding to melanocortin receptors. Molecular Endocrinology. (1999) 13(1):148–55. doi: 10.1210/mend.13.1.0223
85. Nijenhuis WAJ, Oosterom J, and Adan RAH. AgRP(83 - 132) acts as an inverse agonist on the human-melanocortin-4 receptor. Molecular Endocrinology. (2001) 15(1):164–71. doi: 10.1210/mend.15.1.0578
86. Ming Fong T, Mao C, MacNeil T, Kalyani R, Smith T, Weinberg D, et al. ART (Protein product of agouti-related transcript) as an antagonist of MC - 3 and MC - 4 receptors. (1997) 237:.
87. Krashes MJ, Koda S, Ye CP, Rogan SC, Adams AC, Cusher DS, et al. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J Clin Invest. (2011) 121:1424–8. doi: 10.1172/JCI46229
88. Morrison CD, Xi X, White CL, Ye J, and Martin RJ. Amino acids inhibit Agrp gene expression via an mTOR-dependent mechanism. Am J Physiol Endocrinol Metab. (2007) 293:165–71. doi: 10.1152/ajpendo.00675.2006
89. Barnes MJ, Argyropoulos G, and Bray GA. Preference for a high fat diet, but not hyperphagia following activation of mu opioid receptors is blocked in AgRP knockout mice. Brain Res. (2010) 1317:100–7. doi: 10.1016/j.brainres.2009.12.051
90. Thomas MA, Tran V, Ryu V, Xue B, and Bartness TJ. AgRP knockdown blocks long-term appetitive, but not consummatory, feeding behaviors in Siberian hamsters. Physiol Behav. (2018), 61–70. doi: 10.1016/j.physbeh.2017.10.008
91. Lazzarino GP, Andreoli MF, Rossetti MF, Stoker C, Tschopp MV, Luque EH, et al. Cafeteria diet differentially alters the expression of feeding-related genes through DNA methylation mechanisms in individual hypothalamic nuclei. Mol Cell Endocrinol. (2017) 450:113–25. doi: 10.1016/j.mce.2017.05.005
92. Coupé B, Amarger V, Grit I, Benani A, and Parnet. Nutritional programming affects hypothalamic organization and early response to leptin. Endocrinology. (2010) 151:702–13. doi: 10.1210/en.2009-0893
93. Shin BC, Dai Y, Thamotharan M, Gibson LC, and Devaskar SU. Pre- and postnatal calorie restriction perturbs early hypothalamic neuropeptide and energy balance. J Neurosci Res. (2012) 90:1169–82. doi: 10.1002/jnr.23013
94. Schellong K, Melchior K, Ziska T, Henrich W, Rancourt RC, and Plagemann A. Sex-specific epigenetic alterations of the hypothalamic Agrp-Pomc system do not explain ‘diabesity’ in the offspring of high-fat diet (HFD) overfed maternal rats. J Nutr Biochem. (2020) 75:108257. doi: 10.1016/j.jnutbio.2019.108257
95. Rapps K, Kisliouk T, Marco A, Weller A, and Meiri N. Dieting reverses histone methylation and hypothalamic AgRP regulation in obese rats. Front Endocrinol (Lausanne). (2023) 14. doi: 10.3389/fendo.2023.1121829
96. Desai M, Han G, and Ross MG. Programmed hyperphagia in offspring of obese dams: Altered expression of hypothalamic nutrient sensors, neurogenic factors and epigenetic modulators. Appetite. (2016) 99:193–9. doi: 10.1016/j.appet.2016.01.023
97. Przybył BJ, Szlis M, and Wójcik-Gładysz A. Brain-Derived Neurotrophic Factor Affects mRNA and miRNA Expression of the Appetite Regulating Centre in the Sheep Arcuate Nucleus. Ann Anim Science. (2020) 20:853–69. doi: 10.2478/aoas-2020-0015
98. Lin S, Boey D, and Herzog H. NPY and Y receptors: Lessons from transgenic and knockout models. Neuropeptides. (2004), 189–200. doi: 10.1016/j.npep.2004.05.005
99. Mercer RE, Chee MJS, and Colmers WF. The role of NPY in hypothalamic mediated food intake. Front Neuroendocrinology. (2011), 398–415. doi: 10.1016/j.yfrne.2011.06.001
100. Horsnell H and Baldock PA. Osteoblastic actions of the neuropeptide Y system to regulate bone and energy homeostasis. Curr Osteoporosis Rep. (2016), 26–31. doi: 10.1007/s11914-016-0300-9
101. Engström Ruud L, Pereira MMA, de Solis AJ, Fenselau H, and Brüning JC. NPY mediates the rapid feeding and glucose metabolism regulatory functions of AgRP neurons. Nat Commun. (2020) 11:442. doi: 10.1038/s41467-020-14291-3
102. Erickson JC, Clegg KE, and Palmiter RD. Sensitivity to leptin and susceptibility to seizures of mice lacking neuropeptide Y. Nature. (1996) 381:415–8. doi: 10.1038/381415a0
103. Cabrera Zapata LE, Cambiasso MJ, and Arevalo MA. Epigenetic modifier Kdm6a/Utx controls the specification of hypothalamic neuronal subtypes in a sex-dependent manner. Front Cell Dev Biol. (2022) 10. doi: 10.3389/fcell.2022.937875
104. Balasubramanian N, Sagarkar S, Jadhav M, Shahi N, Sirmaur R, and Sakharkar AJ. Role for histone deacetylation in traumatic brain injury-induced deficits in neuropeptide y in arcuate nucleus: possible implications in feeding behavior. Neuroendocrinology. (2021) 111:1187–200. doi: 10.1159/000513638
105. Krunic A, Loganathan N, Nkechika V, and Belsham DD. Phenylbutyric acid robustly increases Npy mRNA expression in hypothalamic neurons by increasing H3K9/14 acetylation at the Npy promoter. Biochem Biophys Res Commun. (2023) 658:18–26. doi: 10.1016/j.bbrc.2023.03.031
106. Croizier S, Park S, Maillard J, and Bouret SG. Central Dicer-miR-103/107 controls developmental switch of POMC progenitors into NPY neurons and impacts glucose homeostasis. Elife. (2018) 7. doi: 10.7554/eLife.40429
107. Feng X, Ding Y, Zhou M, Song N, and Ding Y. Integrative Analysis of Exosomal miR-452 and miR-4713 Downregulating NPY1R for the Prevention of Childhood Obesity. Dis Markers. (2022) 2022:1–12. doi: 10.1155/2022/2843353
108. Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, et al. Identification and expression cloning of a leptin receptor, OB-R. Cell. (1995). doi: 10.1016/0092-8674(95)90151-5
109. Peelman F, Waelput W, Iserentant H, Lavens D, Eyckerman S, Zabeau L, et al. Leptin: Linking adipocyte metabolism with cardiovascular and autoimmune diseases. Prog Lipid Res. (2004), 283–301. doi: 10.1016/j.plipres.2004.03.001
111. Bates SH and Myers MG. The role of leptin receptor signaling in feeding and neuroendocrine function. Trends Endocrinol Metab. (2003), 447–52. doi: 10.1016/j.tem.2003.10.003
112. Cohen P, Zhao C, Cai X, Montez JM, Rohani SC, Feinstein P, et al. Selective deletion of leptin receptor in neurons leads to obesity. J Clin Invest. (2001) 108:1113–21. doi: 10.1172/JCI200113914
113. Nunziata A, Funcke JB, Borck G, Von Schnurbein J, Brandt S, Lennerz B, et al. Functional and phenotypic characteristics of human leptin receptor mutations. J Endocr Soc. (2019) 3:27–41. doi: 10.1210/js.2018-00123
114. Childs GV, Akhter N, Haney A, Syed M, Odle A, Cozart M, et al. The somatotrope as a metabolic sensor: Deletion of leptin receptors causes obesity. Endocrinology. (2011) 152:69–81. doi: 10.1210/en.2010-0498
115. Zhang Q, Xiao X, Zheng J, Li M, Yu M, Ping F, et al. Maternal inulin alleviates high-fat diet-induced lipid disorder in offspring by epigenetically modulating hypothalamus feeding circuit-related genes. Food Funct. (2023). doi: 10.1039/D3FO02223D
116. Fan C, Liu X, Shen W, Deckelbaum RJ, and Qi K. The regulation of leptin, leptin receptor and pro-opiomelanocortin expression by N - 3 PUFAs in diet-induced obese mice is not related to the methylation of their promoters. Nutr Metab (Lond). (2011) 8(1):31. doi: 10.1186/1743-7075-8-31
117. Batury VL, Walton E, Tam F, Wronski ML, Buchholz V, Frieling H, et al. DNA methylation of ghrelin and leptin receptors in underweight and recovered patients with anorexia nervosa. J Psychiatr Res. (2020) 131:271–8. doi: 10.1016/j.jpsychires.2020.08.026
118. Palou M, Picó C, McKay JA, Sánchez J, Priego T, Mathers JC, et al. Protective effects of leptin during the suckling period against later obesity may be associated with changes in promoter methylation of the hypothalamic pro-opiomelanocortin gene. Br J Nutr. (2011), 769–78. doi: 10.1017/S0007114511000973
119. Kim MH, Li Y, Zheng Q, Jiang L, Myers MG, Wu WS, et al. LepRb+ cell–specific deletion of Slug mitigates obesity and nonalcoholic fatty liver disease in mice. J Clin Invest. (2023) 133(4). doi: 10.1172/JCI156722
120. Crépin D, Benomar Y, Riffault L, Amine H, Gertler A, and Taouis M. The over-expression of miR-200a in the hypothalamus of ob/ob mice is linked to leptin and insulin signaling impairment. Mol Cell Endocrinol. (2014) 384:1–11. doi: 10.1016/j.mce.2013.12.016
121. Schulingkamp RJ, Pagano TC, Hung D, and Raffa RB. Insulin receptors and insulin action in the brain: review and clinical implications. Neurosci Biobehav Rev. (2000) 24(8):855–72. doi: 10.1016/S0149-7634(00)00040-3
122. Werther GA, Hogg A, Oldfield BJ, Mckinley MJ, Figdor R, Allen AM, et al. Localization and characterization of insulin receptors in rat brain and pituitary gland using in vitro autoradiography and computerized densitometry*. Endocrinology. (1987) 121:1562–70. doi: 10.1210/endo-121-4-1562
123. Folli F, Ghidella S, Bonfanti L, Kahn CR, and Merighi A. The early intracellular signaling pathway for the insulin/insulin-like growth factor receptor family in the mammalian central nervous system. Mol Neurobiol. (1996) 13:155–83. doi: 10.1007/BF02740639
124. Mosthaf L, Grako K, Dull TJ, Coussens L, Ullrich A, and McClain DA. Functionally distinct insulin receptors generated by tissue-specific alternative splicing. EMBO J. (1990) 9:2409–13. doi: 10.1002/j.1460-2075.1990.tb07416.x
125. Frasca F, Pandini G, Scalia P, Sciacca L, Mineo R, Costantino A, et al. a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol Cell Biol. (1999) 19:3278–88. doi: 10.1128/MCB.19.5.3278
126. Dodd GT and Tiganis T. Insulin action in the brain: Roles in energy and glucose homeostasis. J Neuroendocrinol. (2017) 29(10). doi: 10.1111/jne.2017.29.issue-10
127. Masaki T, Chiba S, Noguchi H, Yasuda T, Tobe K, Suzuki R, et al. Obesity in insulin receptor substrate-2–deficient mice: disrupted control of arcuate nucleus neuropeptides. Obes Res. (2004) 12:878–85. doi: 10.1038/oby.2004.106
128. Obici S, Feng Z, Karkanias G, Baskin DG, and Rossetti L. Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat Neurosci. (2002) 5:566–72. doi: 10.1038/nn0602-861
129. Paranjape SA, Chan O, Zhu W, Horblitt AM, Grillo CA, Wilson S, et al. Chronic reduction of insulin receptors in the ventromedial hypothalamus produces glucose intolerance and islet dysfunction in the absence of weight gain. Am J Physiol Endocrinol Metab. (2011) 301:978–83. doi: 10.1152/ajpendo.00304.2011
130. Plagemann A, Roepke K, Harder T, Brunn M, Harder A, Wittrock-Staar M, et al. Epigenetic malprogramming of the insulin receptor promoter due to developmental overfeeding. J Perinat Med. (2010) 38:393–400. doi: 10.1515/jpm.2010.051
131. Schellong K, Melchior K, Ziska T, Ott R, Henrich W, Rancourt RC, et al. Hypothalamic insulin receptor expression and DNA promoter methylation are sex-specifically altered in adult offspring of high-fat diet (HFD)-overfed mother rats. J Nutr Biochem. (2019) 67:28–35. doi: 10.1016/j.jnutbio.2019.01.014
132. Pandur E, Szabó I, Hormay E, Pap R, Almási A, Sipos K, et al. Alterations of the expression levels of glucose, inflammation, and iron metabolism related miRNAs and their target genes in the hypothalamus of STZ-induced rat diabetes model. Diabetol Metab Syndr. (2022) 14(1):147. doi: 10.1186/s13098-022-00919-5
133. Menyhért J, Wittmann G, Lechan RM, Keller E, Liposits Z, and Fekete C. Cocaine- and amphetamine-regulated transcript (CART) is colocalized with the orexigenic neuropeptide Y and agouti-related protein and absent from the anorexigenic α-melanocyte-stimulating hormone neurons in the infundibular nucleus of the human hypothalamus. Endocrinology. (2007) 148:4276–81. doi: 10.1210/en.2007-0390
134. Wierup N, Richards WG, Bannon AW, Kuhar MJ, Ahrén B, and Sundler F. CART knock out mice have impaired insulin secretion and glucose intolerance, altered beta cell morphology and increased body weight. Regul Pept. (2005) 129:203–11. doi: 10.1016/j.regpep.2005.02.016
135. Asnicar MA, Smith DP, Yang DD, Heiman ML, Fox N, Chen YF, et al. Absence of cocaine- and amphetamine-regulated transcript results in obesity in mice fed a high caloric diet. Endocrinology. (2001) 142:4394–400. doi: 10.1210/endo.142.10.8416
136. Gibson LC, Shin BC, Dai Y, Freije W, Kositamongkol S, Cho J, et al. Early leptin intervention reverses perturbed energy balance regulating hypothalamic neuropeptides in the pre- and postnatal calorie-restricted female rat offspring. J Neurosci Res. (2015) 93:902–12. doi: 10.1002/jnr.23560
137. Osei-Hyiaman D, Harvey-White J, Bátkai S, and Kunos G. The role of the endocannabinoid system in the control of energy homeostasis. Int J Obes. (2006), S33–8. doi: 10.1038/sj.ijo.0803276
138. Cruz-Martínez AM, Tejas-Juárez JG, Mancilla-Díaz JM, Florán-Garduño B, López-Alonso VE, and Escartín-Pérez RE. CB1 receptors in the paraventricular nucleus of the hypothalamus modulate the release of 5-HT and GABA to stimulate food intake in rats. Eur Neuropsychopharmacol. (2018) 28:1247–59. doi: 10.1016/j.euroneuro.2018.08.002
139. Quarta C, Bellocchio L, Mancini G, Mazza R, Cervino C, Braulke LJ, et al. CB1 signaling in forebrain and sympathetic neurons is a key determinant of endocannabinoid actions on energy balance. Cell Metab. (2010) 11:273–85. doi: 10.1016/j.cmet.2010.02.015
140. Almeida MM, Dias-Rocha CP, Reis-Gomes CF, Wang H, Atella GC, Cordeiro A, et al. Maternal high-fat diet impairs leptin signaling and up-regulates type-1 cannabinoid receptor with sex-specific epigenetic changes in the hypothalamus of newborn rats. Psychoneuroendocrinology. (2019) 103:306–15. doi: 10.1016/j.psyneuen.2019.02.004
141. Basu R and Flak JN. Hypothalamic neural circuits regulating energy expenditure. (2025), 79–124. doi: 10.1016/bs.vh.2024.07.004
142. Sarrafha L, Neavin DR, Parfitt GM, Kruglikov IA, Whitney K, Reyes R, et al. Novel human pluripotent stem cell-derived hypothalamus organoids demonstrate cellular diversity. iScience. (2023) 26(9):107525. doi: 10.1016/j.isci.2023.107525
143. Fu T and Kemper JK. MicroRNA-34a and impaired FGF19/21 signaling in obesity. Vitamins Hormones. Academic Press Inc.; (2016), 175–96. doi: 10.1016/bs.vh.2016.02.002
144. Lavery CA, Kurowska-Stolarska M, Holmes WM, Donnelly I, Caslake M, Collier A, et al. miR-34a–/– mice are susceptible to diet-induced obesity. Obesity. (2016) 24:1741–51. doi: 10.1002/oby.21561
145. Briones-Espinoza MJ, Cortés-García JD, Vega-Cárdenas M, Uresti-Rivera EU, Gómez-Otero A, López-López N, et al. Decreased levels and activity of Sirt1 are modulated by increased miR-34a expression in adipose tissue mononuclear cells from subjects with overweight and obesity: A pilot study. Diabetes Metab Syndrome: Clin Res Rev. (2020) 14:1347–54. doi: 10.1016/j.dsx.2020.07.014
146. Di Bello E, Noce B, Fioravanti R, and Mai A. Current HDAC inhibitors in clinical trials. Chimia (Aarau). (2022) 76:448–53. doi: 10.2533/chimia.2022.448
147. West AC and Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. (2014), 30–9. doi: 10.1172/JCI69738
148. Hontecillas-Prieto L, Flores-Campos R, Silver A, de Álava E, Hajji N, and García-Domínguez DJ. Synergistic enhancement of cancer therapy using HDAC inhibitors: opportunity for clinical trials. Front Genet. (2020) 11. doi: 10.3389/fgene.2020.578011
Keywords: hypothalamus, epigenetics, neuropeptides, metabolism, obesity, metabolic hormone receptors, energy homeostasis
Citation: Oladun B, Mall S and Kim MH (2025) Epigenetic modification of hypothalamic neuropeptides and metabolic hormone receptors in metabolic health. Front. Endocrinol. 16:1645474. doi: 10.3389/fendo.2025.1645474
Received: 11 June 2025; Accepted: 21 August 2025;
Published: 17 September 2025.
Edited by:
Stuart Tobet, Colorado State University, United StatesReviewed by:
Daisuke Kohno, Gunma University, JapanMaria Claudia Gonzalez Deniselle, CONICET Institute of Biology and Experimental Medicine (IBYME), Argentina
Copyright © 2025 Oladun, Mall and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min-Hyun Kim, TWluLUh5dW4uS2ltQGFzdS5lZHU=