
 Mattias Enoksson
Mattias Enoksson
- Clinical Immunology and Allergy, Department of Medicine, Karolinska Institutet, Stockholm, Sweden
Mast cells have been attributed several functions in both health and disease. Mast cell activation and release of inflammatory mediators are associated with the pathogenesis of several diseases, in particular that of allergic diseases. While the notion of mast cells as important, protective sentinel cells is old, this feature of the cell is not well recognized outside the mast cell field. The mast cell is a unique, multifunctional cell of our defense system, with characteristics such as wide-spread tissue distribution, expression of receptors capable of recognizing both endogenous and exogenous agents, and a capability to rapidly respond to triggering factors by selective mediator release. In this review, we discuss the function of mast cells as sentinel cells in the context of cell injury, where mast cells respond by initiating an inflammatory response. In this setting, IL-33 has turned out to be of particular interest. IL-33 is released by necrotic structural cells and is recognized by mast cells via the IL-33 receptor ST2. IL-33 and mast cells probably constitute one important link between cell injury and an inflammatory response that can lead to restoration of tissue function and homeostasis, but might under other circumstances contribute to a vicious circle driving chronic inflammation.
Introduction
Mast cells are peculiar cells with many fascinating properties and functions. They are distributed in almost all tissues, endowed with a broad spectra of receptors that gives them the capacity to respond to a variety of endogenous and exogenous agents, leading to release of pre-formed mediators stored in granules, and/or rapid release of lipid mediators and in addition initiation of protein synthesis and secretion of cytokines, chemokines, interferons, and growth factors. Depending on the stimuli and the tissue environment, the mast cell responds with a differential release of mediators (Theoharides et al., 2007), leading to a specific but at the same time multifunctional effect on the tissue. For instance, IgE-receptor activation leads to degranulation, secretion of eicosanoids, and de novo synthesis of cytokines; while treatment with CD30 induces degranulation independent secretion of chemokines, without any secretion of leukotrienes (Fischer et al., 2006). Because of this, the roles and functions of mast cells in health and disease are diverse and complex, and have been increasingly appreciated (Leslie, 2007; Metz and Maurer, 2007; Kalesnikoff and Galli, 2008; Abraham and St. John, 2010).
In Hans Selye’s book entitled The Mast Cell, published 1965, he reviewed current literature on mast cell research and listed more than 30 theories concerning mast cell functions in, e.g., stress, inflammation, edema formation, arteriosclerosis, temperature regulation, hair growth, sweat secretion, and defensive phenomena (Selye, 1965). Many of these theories were based on the morphological appearance of the highly granulated mast cell, and on its content of histamine, serotonin, and heparin. Some of the theories have been explored further and we have today, thanks to technical developments and animal models, a more in-depth mechanistic understanding of mast cell function, and thereby explanations for many of the different hypotheses. Other early hypotheses have remained more or less unanswered. West (1958) suggested that mast cells act as part of the defense system where they sense and react quickly to any kind of cell injury. The function of mast cells as sentinel cells has been extensively investigated during the last 10–15 years, mainly in the context of pathogen recognition (Galli et al., 1999; Abraham and St. John, 2010). However, how mast cells recognize and react to cell injury has not until recently been deciphered in any detail.
Cell injury can be caused by mechanical trauma, infection, UV-light exposure, radiation, toxins, ischemia, but also by endogenous cytotoxic mediators and be part of a chronic inflammation (Kono and Rock, 2008). In this context, necrotic cells release their intracellular contents containing what is usually referred to as damage-associated molecular patterns (DAMPs), or alarmins, that are recognized by immune cells, which then initiate inflammatory responses (Bianchi, 2007). While there are many well-characterized alarmins, such as high mobility group box 1 (HMGB1; Scaffidi et al., 2002), heat shock proteins (Basu et al., 2000), uric acid (Shi et al., 2003), IL-1α (Eigenbrod et al., 2008), and IL-33 (Moussion et al., 2008), surprisingly few of these have been investigated in the context of mast cell activation, and initiation of inflammatory responses. Recently it was described that necrotic cells specifically activate mast cells through the release of IL-33, with no apparent effect of HMGB1 or uric acid (Enoksson et al., 2011). Thus, IL-33 constitutes the alarmin released by necrotic cells that is recognized by adjacent mast cells. These mast cells then react by releasing pro-inflammatory mediators that subsequently initiate an inflammatory response.
IL-33 belongs to the IL-1 family of cytokines, and was recently identified as a ligand to the previously orphan ST2 receptor. IL-33 is expressed by several different cell types; most notably by epithelial and endothelial cells (Liew et al., 2010) and is a potent inducer of Th2 responses (Schmitz et al., 2005). Unlike other cytokines of the IL-1 family such as IL-1β and IL-18, the full-length form of IL-33 does not require proteolytical cleavage for activation (Cayrol and Girard, 2009). Thus, IL-33 released from necrotic cells is biologically active, whereas IL-33 released from apoptotic cells is inactivated by caspases (Luthi et al., 2009). By signaling through the IL-33 receptor ST2, IL-33 activates several cell types, such as Th2 cells, innate lymphoid cells, mast cells, basophils, and eosinophils. Even though the role of IL-33 in the immune system is not yet fully understood, this cytokine has been implicated, for good or for bad, in several diseases. Since the mast cell is located in all tissues, they are positioned to rapidly recognize IL-33 released from damaged cells and initiate an inflammatory response. Since this is a newly described pathway, this review will focus on IL-33 and mast cells, and their interrelation in acute and chronic inflammations.
Mast Cell Recognition of Cell Injury through IL-33 Released by Necrotic Cells
In a recent study we examined the role of mast cells as sensors of cellular injury (Enoksson et al., 2011). Debris-free supernatant made from freeze–thaw necrotized cells induced mouse mast cells to produce and release leukotrienes and cytokines such as IL-6 and TNF, without any preceding degranulation. Through the use of mast cells derived from receptor deficient mice, the activation was pinpointed to be mediated through a TLR-independent, but totally MyD88- and ST2-dependent route. Silencing of the IL-33 expression in fibroblasts further defined IL-33 and ST2 to be responsible for the necrosis-induced activation of mast cells. The cellular origin of the necrotic cells also turned out to be of importance, and only necrotic structural cells (such as keratinocytes, neuronal cells, and smooth muscle cells) contained IL-33 and therefore possessed the ability to activate mast cells, whereas necrotic hematopoietic cells (bone marrow-derived mouse mast cells, spleen cells, and monocytes) lacked this capacity. Moreover, other endogenous danger signals, such as HMGB1, uric acid, and adenosine, known to activate other immune cells (Scaffidi et al., 2002; Shi et al., 2003; Bours et al., 2006; Bianchi, 2007), did not contribute to the mast cell alarming activity within the necrotic cell supernatant. Thus, IL-33 appears to be the sole danger signal responsible for alarming mast cells of cell injury.
IL-33 Biology
IL-33 was initially described as NF-high endothelial venules (HEV), a nuclear factor strongly expressed in HEVs (Baekkevold et al., 2003). NF-HEV was later identified to be identical to IL-33, and shown to be preferentially expressed in endothelial cells (Carriere et al., 2007). While IL-33 has been shown to be expressed in several tissues and cell types at the mRNA level (Schmitz et al., 2005), IL-33 protein is predominantly expressed in the nuclei of structural cell types such as endothelial cells, fibroblasts, keratinocytes (Moussion et al., 2008) epithelial cells (Prefontaine et al., 2010), and airway smooth muscle cells (Prefontaine et al., 2009). The nuclear role of IL-33 is to date not fully understood, but the recent generation of IL-33−/− mice (Oboki et al., 2010) will likely facilitate future studies aiming at understanding nuclear actions of IL-33. It has been shown that IL-33 associates with heterochromatin in the nucleus, and that IL-33 can regulate transcription by repressing transcriptional activity (Carriere et al., 2007). Interestingly, is has also been suggested that IL-33 interacts with NFκB to reduce NFκB-triggered gene expression (Ali et al., 2011). It is thus clear that IL-33 is a cytokine of dual functions, where nuclear IL-33 functions as a repressor of NFκB, while released IL-33 mainly is involved in pro-inflammatory processes, acting through ST2 signaling activating NFκB.
The subjects of IL-33 activity, processing, and release/active secretion have been matters of some controversy. Full-length IL-33 was originally proposed to be cleaved by caspase-1 (Schmitz et al., 2005), similarly to proIL-1β and proIL-18, two other members of the IL-1 cytokine family. In contrast, several later studies have reported that IL-33 is not at all a target for caspase-1 (Luthi et al., 2009; Talabot-Ayer et al., 2009; Ali et al., 2010), and it has even been reported that caspase-1 treatment of IL-33 results in inactivation (Cayrol and Girard, 2009). Furthermore, it has been demonstrated that caspase-3 and caspase-7 inactivates IL-33 during apoptosis (Luthi et al., 2009). Another study exploring IL-33 processing in vivo implicated calpain as an important player in IL-33 maturation (Hayakawa et al., 2009). This is in contrast to a study by Ohno et al. (2009) in which IL-33 release could be observed in macrophages treated with calpain- and caspase-8-inhibitors as well as in macrophages from caspase-1−/− mice. Several studies have reported that the full-length IL-33 is biologically active (Cayrol and Girard, 2009; Luthi et al., 2009; Talabot-Ayer et al., 2009; Ali et al., 2010), a characteristic that fits well with its proposed role as an alarmin (Moussion et al., 2008; Cayrol and Girard, 2009; Enoksson et al., 2011). As stated above, IL-33 is inactivated during apoptosis but has been shown to be readily released upon induction of necrosis (Schmitz et al., 2005), for instance after endothelial cell damage or mechanical injury (Cayrol and Girard, 2009). To date, necrosis is believed to be the principal way in which IL-33 is released from cells. However, a recent study observed IL-33 secretion from epithelial cells exposed to the fungus Alternaria alternata (Kouzaki et al., 2011), providing evidence for that necrosis is not the only way in which IL-33 is released.
IL-33 signals through a receptor complex composed of ST2 and IL-1R accessory protein (IL-1RAcP; Chackerian et al., 2007). ST2 was for a long time an orphan receptor of the IL-1R family (Tominaga, 1989), until 2005 when IL-33 was revealed as a ST2 ligand by Schmitz et al. (2005). ST2 was shown by Xu et al. (1998) to be a stable surface marker expressed on Th2 but not on Th1 cells. In the same year it was described that ST2 is expressed also on mast cells (Moritz et al., 1998). Three isoforms are encoded by the ST2 gene; a transmembrane form responsible for the ST2/IL-33 signaling on most cells (ST2L), another transmembrane form which is mainly expressed on cells in the gastrointestinal organs (ST2V) and a secreted soluble form (sST2) with a decoy function preventing IL-33 to bind ST2 (Yanagisawa et al., 1993; Tago et al., 2001; Trajkovic et al., 2004). The association of IL-1RAcP with ST2 during IL-33 binding is essential for functional signaling (Ali et al., 2007; Chackerian et al., 2007; Palmer et al., 2008). For instance, IL-1RAcP is required for IL-33-induced effects in vivo (Chackerian et al., 2007), and IL-6 secretion has been shown to be impaired in IL-1RAcP−/− mast cells treated with IL-33 (Palmer et al., 2008). Likewise, mast cell responses to IL-33 could be disrupted by using a neutralizing IL-1RAcP antibody (Ali et al., 2007). Upon binding of IL-33 to its receptor ST2, MyD88, IRAK, IRAK4, and TRAF6 are recruited, resulting in both NFκB phosphorylation and activation of the MAP-kinases Erk1/2 and p38 (Schmitz et al., 2005). This signaling pathway has subsequently been studied in greater detail, revealing that the tyrosine kinase JAK2 is involved in IL-33-induced IκBα-degradation and subsequent NFκB activation (Funakoshi-Tago et al., 2011). In addition, TRAF6 has been demonstrated to be of vital importance, as NFκB, p38, and JNK activation induced by IL-33 is entirely inhibited in TRAF6−/− fibroblasts (Funakoshi-Tago et al., 2008).
The finding that the ST2 receptor is stably expressed on Th2 but not Th1 cells is also well reflected in terms of cell activation. In an experiment performed by Schmitz et al. (2005), it was revealed that Th2 but not Th1 cells from C57BL/6 mice respond to IL-33 treatment with secretion of IL-5 and IL-13. Since then, the capability of IL-33 to activate several other cell types has been described. In particular, IL-33 has emerged as a potent activator of both murine and human mast cells, a subject discussed in detail below and summarized in Figure 1. Aside from mast cells, IL-33 also activates mouse (Kondo et al., 2008; Kroeger et al., 2009; Schneider et al., 2009) and human basophils (Suzukawa et al., 2008a; Pecaric-Petkovic et al., 2009) to release IL-4 and IL-13. In addition, IL-33 enhances eosinophil survival, adhesion, and CD11b expression (Cherry et al., 2008; Suzukawa et al., 2008b). Neutrophils express ST2 and in an experimental sepsis model IL-33 likely acts on neutrophil recruitment directly without involvement of Th2 cytokines by counteracting downregulating effects of TLR ligands on the chemokine receptor CXCR2 (Alves-Filho et al., 2010). In dendritic cells, IL-33, induces TNF, IL-1β, IL-6, and CCL17 production (Besnard et al., 2011), and importantly, it has been shown that IL-33-activated dendritic cells can prime naïve T cells to produce IL-5 and IL-13 (Rank et al., 2009; Besnard et al., 2011). In addition, it has also been demonstrated that both iNKT cells and NK cells produce IFN-γ in the presence of IL-33 (Bourgeois et al., 2009), and iNKT cells treated with IL-33 in the presence of α-galactosylceramide produce both IFN-γ and IL-4 (Smithgall et al., 2008), thus demonstrating that IL-33 is a multi-facetted cytokine involved in several different types of immune responses. Interestingly, several populations of cells in the recently described class of innate lymphoid cells type 2 (ILC2) also express ST2 and both expand in vivo and produce Th2 cytokines such as IL-5 and IL-13 in response to IL-25 and IL-33 (Neill et al., 2010).
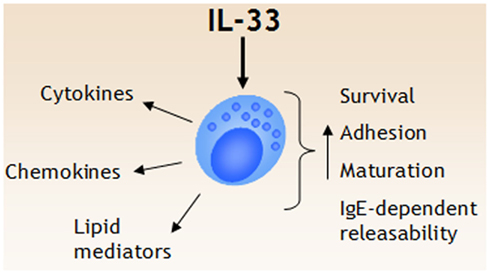
Figure 1. IL-33 is a potent regulator of mast cell functions. IL-33 triggers several events in mast cells, including release of cytokines, chemokines, and lipid mediators. In addition, IL-33 has been shown to promote mast cell survival, adhesion, and maturation. While IL-33 does not seem to induce degranulation in naïve mast cells, IL-33 augment IgE-receptor mediated degranulation and cytokine release.
IL-33 Induces Multifunctional Mast Cell Response
ST2 is expressed already on early mouse mast cell-committed progenitors residing in the bone marrow (Chen et al., 2005). Human CD34+ progenitors also express ST2, and activation through the receptor by IL-33 both accelerates their maturation into tryptase-containing cells and induces release of pro-inflammatory cytokines and chemokines (Allakhverdi et al., 2007b). In vitro-differentiation of mouse mast cells is promoted by co-culture with fibroblasts which provide stem cell factor (Levi-Schaffer et al., 1985; Fujita et al., 1988; Nocka et al., 1990). Besides stem cell factor, which is the main mast cell growth factor, it was recently described that IL-33 in the co-culture system promotes granule accumulation in the developing mast cells (Kaieda et al., 2010). IL-33 also has the capability to enhance the survival of mature human in vitro differentiated mast cells (Iikura et al., 2007), even in the absence of SCF. Furthermore, IL-33 also increases human mast cell adhesion to fibronectin, which might have implications for their tissue distribution (Iikura et al., 2007).
The traditional view of mast cells includes activation of the cells through cross-linking of the IgE-receptor, which typically results in degranulation and release of pre-formed mediators. During the years this view has been modified and today it is obvious that mast cells can be activated by a variety of stimuli and exert different actions upon stimulation, with or without degranulation (Theoharides et al., 2007). IL-33 does not generally induce degranulation of mouse and human mast cells (Allakhverdi et al., 2007b; Ho et al., 2007; Moulin et al., 2007; Silver et al., 2009). The effect of IL-33 on formation of eicosanoids appears to be different between species, where human mast cells do not produce either prostaglandin (PGD2) or leukotrienes (Allakhverdi et al., 2007b), whereas mouse mast cells produce both (Moulin et al., 2007; Enoksson et al., 2011). One important IL-33-induced mast cell-mediated effect is to initiate an orchestration of the inflammatory response with production of pro-inflammatory cytokines (IL-6, TNF), Th2 cytokines (IL-5, IL-10, IL-13, GM-CSF) and chemokines which direct the recruitment of inflammatory cells (CXCL8 and CCL1) (Allakhverdi et al., 2007b; Ho et al., 2007; Iikura et al., 2007; Moulin et al., 2007; Silver et al., 2009). IL-6 and IL-13 belongs to the cytokines produced by mouse mast cells upon IL-33 stimulation (Ho et al., 2007).
One important function of IL-33 is when it works in concert with other stimuli, such as activation of the high-affinity IgE-receptor, FcεRI. In this case the combination of IgE-receptor activation with IL-33 induces a more robust activation of the cells with degranulation and associated release of leukotrienes and prostaglandins together with a synergistically higher production of the cytokines IL-6, IL-13, TNF, and chemokines than IgE-receptor cross-linking alone (Ho et al., 2007; Iikura et al., 2007; Silver et al., 2009). Another example is the effect of thymic stromal lymphopoietin (TSLP) on human mast cells. TSLP requires presence of IL-1 to be able to induce activation of mast cells (Allakhverdi et al., 2007a). With TSLP as catalyzer, human mast cells produce higher amounts of IL-5 and IL-13 upon IL-33 stimulation than is produced with IL-33 as the only stimuli (Allakhverdi et al., 2007b). Other identified synergizers are nerve growth factor, the adenosine receptor agonist NECA and C5a (Silver et al., 2009).
Both human and mouse mast cells have been described to express IL-33 mRNA upon IgE-receptor cross-linking and treatment with ionomycin, but so far no extracellular release has been detected (Ohno et al., 2009; Hsu et al., 2010). In contrast to some other cell types, IL-33 expression in mast cells is not induced upon treatment with bacterial LPS (Ohno et al., 2009).
The Role of IL-33 in Inflammatory Diseases
Although being a recently identified cytokine, the tissue expression of IL-33 and its biological functions have already been investigated in a number of clinical settings and experimental models. IL-33 and its receptor ST2 have been attributed both protective functions, in, e.g., atherosclerosis, cardiac remodeling, and helminth infections; as well as damaging effects in diseases such as autoimmune disorders and asthma (Liew et al., 2010). Both positive and negative effects of IL-33 can be related, at least in part, to its Th2 promoting capacities. A Th2 response is characterized by infiltration of CD4 positive T-lymphocytes, release of IL-4 and IL-5, IgE-synthesis, and activation of eosinophils, basophils, and mast cells.
Asthma
Asthma is usually regarded as a Th2 driven disease, with strong contribution in the pathogenesis of T-lymphocytes, eosinophils, and mast cells. Besides airway hyperresponsiveness, inflammation, and tissue remodeling, there is also a damage of the airway epithelial cell layer, which might lead to release of DAMPs; e.g., IL-33. In a genome-wide association study of more than 10,000 subjects with asthma, a single-nucleotide polymorphism in the IL-33 gene turned out to be one of few genes that significantly associated with the disease (Moffatt et al., 2010). Similar findings were reported recently where the study had been performed on other subjects (Torgerson et al., 2011). These findings strengthens the hypothesis that IL-33 released from structural lung cells, e.g., damaged epithelial cells, is a link to the immune system that activates airway inflammation. It has previously been demonstrated that expression of IL-33 is increased in the airways of subjects with severe asthma. IL-33 protein and mRNA was described to be increased in airway smooth muscle cells (Prefontaine et al., 2009), and thus these cells are one of the sources of IL-33 that can be detected in bronchial alveolar lavage fluid (Prefontaine et al., 2010). Another source of IL-33 is the airway epithelial cells that demonstrate strong nuclear staining (Prefontaine et al., 2010; Kouzaki et al., 2011). Of interest for pulmonary inflammation is the finding that extracts of Alternaria or cockroach can induce secretion of IL-33 from human bronchial airway epithelial cells (Kouzaki et al., 2011). The release is mediated via increase of extracellular ATP accumulation that acts on P2 purinergic receptors leading to IL-33 release. This is of particular interest since increased airborne exposure to especially Alternaria allergens has been associated with acute asthma exacerbations (Bush and Prochnau, 2004).
The role of IL-33 and ST2 in allergic airway inflammation has been studied in several mouse models, including mice deficient in either ST2 or IL-33. Administration of IL-33 through intra-nasal installation generates an allergic inflammation with increased levels of IL-5, IL-13, and eotaxin; enhanced mucus production and cellular infiltration of mainly eosinophils and T-lymphocytes (Kondo et al., 2008; Kurowska-Stolarska et al., 2008; Louten et al., 2011). IL-33 deficient mice sensitized and challenged with ovalbumin exhibit an attenuated airway inflammation and airway hypersensitivity to methacholine compared to wild type mice (Oboki et al., 2010; Louten et al., 2011). Similar findings have been obtained when ST2 deficient mice were used, or where an anti-ST2 blocking antibody was applied to the model. Deficient ST2 activity results in an attenuated airway inflammation and IL-5 production (Kurowska-Stolarska et al., 2008). Of particular interest is the finding that administration of an anti-ST2 antibody during the post challenge period reduces the AHR to background levels, as well as reduction in mucus production and lymphocytic lung infiltration (Kearley et al., 2009). These in vivo studies suggest that the IL-33–ST2 pathway are not only important for the development of allergic airway inflammations, but also for its maintenance.
Although mast cells have a central role in the pathogenesis of asthma (Bradding et al., 2006), it still remains to decipher the impact of the IL-33 – mast cell axis in asthma. With the current knowledge one might foresee that mast cells are activated in the asthmatic lung by released IL-33, which thereby contributes to the airway inflammation.
Rheumatoid Arthritis
Mast cells have an impact on the development and disease severity of different inflammatory and autoimmune conditions such as rheumatoid arthritis (RA), multiple sclerosis, and type 1 diabetes (Walker et al., 2012), which is suggested to be mainly through release of TNF and/or recruitment of neutrophils as a common mechanism (Walker et al., 2012). Other common denominators for these conditions are involvement of IL-33 (Matsuyama et al., 2010; Miller, 2011) and necrosis (Merrill and Scolding, 1999; Kim et al., 2007; Goh and Midwood, 2011), which likely could be the underlying explanation for the involvement of mast cells in these diseases.
The levels of IL-33 in sera and synovial fluid significantly correlates with disease specificity, and in some cases also disease severity; with RA patients being positive for IL-33 whereas patients with osteoarthritis, psoriatic arthritis, or infectious disease and healthy controls do not have IL-33 present in their sera or synovial fluid (Matsuyama et al., 2010; Mu et al., 2010; Talabot-Ayer et al., 2012). However, not all patients with RA do have high levels of IL-33 in their sera, whereas most of them have increased levels in their synovial fluid (Matsuyama et al., 2010).
If disease models can be transferred to mast cell deficient mice the involvement of mast cells can be estimated. Depending on the experimental model of arthritis, mast cells, and IL-33 have been claimed to be important or of no importance (Xu et al., 2008, 2010; Verri et al., 2010). In one of the models (collagen-induced arthritis), IL-33 augments arthritis through mast cell production of cytokines inducing Th17 cells (Xu et al., 2008), and in another model (methylated BSA-induced arthritis) IL-33 functions as a chemoattractant for neutrophils, either directly or indirectly by first activating synoviocytes and macrophages independently of mast cells (Verri et al., 2010). In a third model (autoantibody-induced arthritis), IL-33 amplifies autoantibody-induced arthritis by promoting mast cell degranulation (Xu et al., 2010).
Studies of experimental arthritis in mice have demonstrated that blocking of IL-33, either targeting ST2 signaling by using anti-ST2 antibodies (Palmer et al., 2009), soluble ST2 (Leung et al., 2004) or indirectly by anti-TNF therapy (Verri et al., 2010), diminishes the severity, and even protects against the progression of arthritis. In addition, clinical studies also point in the same direction; RA patients receiving etanercept (TNF blocker) treatment showed lowered levels of IL-33 in serum as well as reduced disease activity after 3 and 6 months of treatment (Kageyama et al., 2011). Treatment with infliximab (anti-TNF) also decreased the serum IL-33 levels (Mu et al., 2010). However, 30–40% of the patients do not respond to treatment targeting TNF and these non-responders have high serum levels of IL-33 (Matsuyama et al., 2011). In experiments using fibroblast-like synoviocytes, IL-1β as well as TNF could induce expression of IL-33 (Kaieda et al., 2010; Matsuyama et al., 2011), suggesting that in the absence of TNF, IL-1β could still potentially induce IL-33 expression in non-responders (Matsuyama et al., 2011).
Skin Inflammations
Mast cells are strategically located in the upper dermis of normal skin, where they are exposed to external insults such as antigens and microbes. During certain challenges of the skin homeostasis such as wounding (Weller et al., 2006), microbial infection (Di Nardo et al., 2003; Marshall, 2004), inflammation in atopic eczema, psoriasis, and skin sclerosis (Harvima and Nilsson, 2011; Ribatti and Crivellato, 2011), the number and/or reactivity of mast cells increase in order to regain or rather create new homeostasis. IL-33 is constitutively expressed in skin (Schmitz et al., 2005; Moussion et al., 2008), and coincidentally there is also a increased expression of IL-33 in many of these conditions (Pushparaj et al., 2009; Alves-Filho et al., 2010; Manetti et al., 2010; Theoharides et al., 2010; Hueber et al., 2011; Yanaba et al., 2011). The exact trigger(s) for IL-33 expression in skin is to a large part unknown (Hueber et al., 2011). However, physical cellular injury induced by wounding, tape stripping, scratching in atopic eczema or UVB radiation, as well as biological challenge such as bacterial infection are stimuli that potentially induce IL-33 production (Ohno et al., 2009; Dickel et al., 2010; Byrne et al., 2011).
Some of the actions of IL-33 through mast cells are augmented with IgE as a companion (Ho et al., 2007; Iikura et al., 2007; Moulin et al., 2007; Pushparaj et al., 2009; Silver et al., 2009). Atopic eczema is a chronic inflammatory skin disease associated with elevated serum IgE levels and involvement of mast cells (Scheynius et al., 2002; Harvima and Nilsson, 2011). An elevated expression of IL-33 can be demonstrated in skin biopsies of atopic eczema patients with more expression in lesional than in non-inflamed skin (Pushparaj et al., 2009). For IL-33 to induce degranulation and increased activation of mast cells, combined action with IgE sensitization or IgE-receptor cross-linking is desirable (Ho et al., 2007; Iikura et al., 2007; Moulin et al., 2007; Pushparaj et al., 2009; Silver et al., 2009). Furthermore, IgE sensitization alone induces increased ST2 expression by mast cells (Ho et al., 2007; Pushparaj et al., 2009). Moreover, there is a significant genetic association between a single-nucleotide polymorphism in the distal ST2 promoter and the risk of developing atopic eczema (Shimizu et al., 2005). With a predisposition for elevated ST2 expression, very low levels of IL-33 might have an impact on the induction of atopic eczema.
Physiologically relevant doses of sun light radiation (consisting of both UVA and UVB) induce IL-33 mRNA and protein expression in the nucleus and cytoplasm of epidermal keratinocytes and dermal fibroblasts (Byrne et al., 2011). Neutrophils are rapidly recruited, possibly through IL-8/CXCL8 from activated mast cells, epidermal keratinocytes (Strickland et al., 1997), or Langerhans cells (Nakagawa et al., 1999), to the radiated skin where they are found adjacent to mast cells and the IL-33 expressing cells (Byrne et al., 2011).
Collectively, IL-33 induces skin inflammation, where at least some of the reactions are mast cell dependent (Theoharides et al., 2010; Byrne et al., 2011; Hueber et al., 2011).
Protective Roles of IL-33
While many studies have demonstrated that IL-33 can exacerbate or in other ways play detrimental roles in certain diseases, there is also a growing body of evidence that IL-33 in other diseases can have a protective role. For instance, it has been shown that IL-33 has important roles in helminth infections, thus providing evidence of functionality for the much studied fact that IL-33 induces Th2 responses, which are essential for parasite expulsion. In a parasite infection model in mice, Humphreys et al. (2008) showed that infected mice could expulse the parasite if treated with IL-33. Here, IL-33 treatment prevented a Th1 polarized response, and instead induced IL-4, IL-9, and IL-13. In a recent study, IL-33 was also demonstrated to have a beneficial role in sepsis. By utilizing an experimental model of sepsis in mice by cecal ligation and puncture, it was demonstrated that mice treated with IL-33 displayed reduced mortality compared to PBS treated mice, following cecal ligation and puncture (Alves-Filho et al., 2010). IL-33-treated mice recruited more neutrophils into the peritoneum, and could thus more effectively combat infection. Interestingly, it was also shown that patients not recovering from sepsis had increased levels of soluble ST2 compared to surviving patients. Increased levels of sST2 in sera leads to suppression of the IL-33 function with lowered recruitment of neutrophils. Furthermore, IL-33 has also been shown to have a cardioprotective role. By inducing cardiac pressure overload trough transverse aortic constriction in mice, Sanada et al. (2007) demonstrated that IL-33 treatment can reduce hypertrophy and fibrosis and thus improve survival. In addition, Miller et al. (2008) have reported that IL-33 treatment can reduce atherosclerosis development in mice.
Summary
IL-33 has emerged as an important alarmin that alerts the sentinel cells of the body to initiate an inflammatory response. Animal models also suggest that IL-33 plays a role in perpetuation of chronic inflammations by activating inflammatory cells of both the innate and adaptive immune system. In this context mast cells recognize IL-33 through the expression of ST2 which mediates a signal to produce inflammatory cytokines, and possibly eicosanoids. These inflammatory mediators recruit and activate other cells like neutrophils and dendritic cells to the site (Figure 2). The IL-33-induced mast cell activation is probably an underlying mechanism in acute inflammations induced by cell injury, but also in chronic inflammatory diseases where IL-33 is released either from necrotic cells or by active secretion. Future studies, in human and animal models, will clarify in more detail how important the IL-33 – mast cell – inflammatory axis is, and if this can be a possible target for new therapies of, e.g., asthma and other inflammatory diseases.
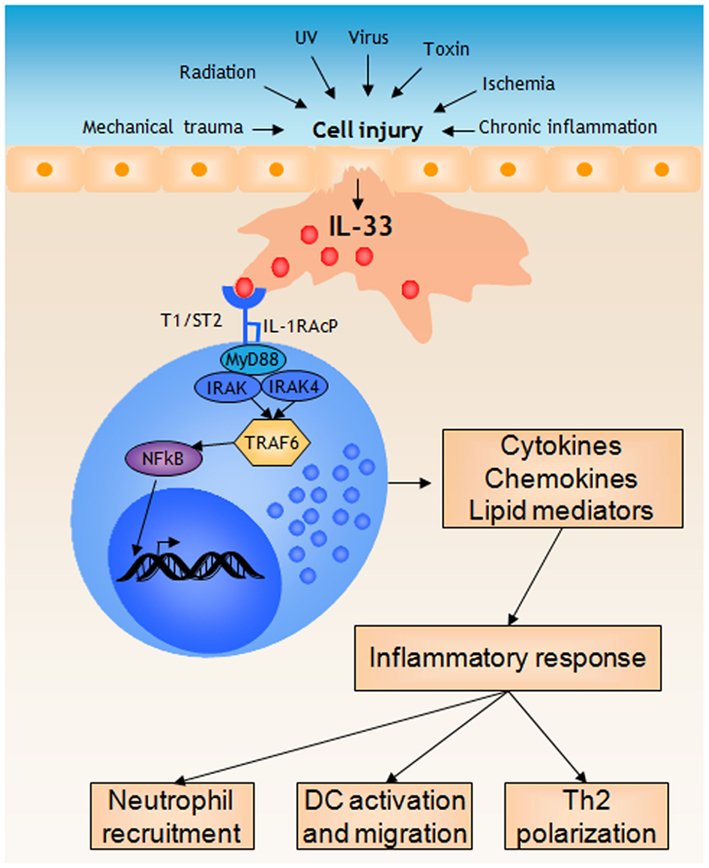
Figure 2. Mast cells respond to IL-33 released during cell injury by initiating a pro-inflammatory response. During cell injury, for instance induced by mechanical trauma, structural cell types such as epithelial cells release IL-33. Mast cells recognize IL-33 through the T1/ST2 receptor, which initiates MyD88-dependent signaling mechanisms eventually resulting in NFkB activation and the subsequent transcription of several pro-inflammatory genes. Release of cytokines, chemokines, and lipid mediators together initiates a inflammatory response, for instance resulting in neutrophil recruitment, activation and migration of dendritic cells, and polarization of T cells.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abraham, S. N., and St. John, A. L. (2010). Mast cell-orchestrated immunity to pathogens. Nat. Rev. Immunol. 10, 440–452.
Ali, S., Huber, M., Kollewe, C., Bischoff, S. C., Falk, W., and Martin, M. U. (2007). IL-1 receptor accessory protein is essential for IL-33-induced activation of T lymphocytes and mast cells. Proc. Natl. Acad. Sci. U.S.A. 104, 18660–18665.
Ali, S., Mohs, A., Thomas, M., Klare, J., Ross, R., Schmitz, M. L., and Martin, M. U. (2011). The dual function cytokine IL-33 interacts with the transcription factor NF-{kappa}B To dampen NF-{kappa}B-stimulated gene transcription. J Immunol. 187, 1609–1616.
Ali, S., Nguyen, D. Q., Falk, W., and Martin, M. U. (2010). Caspase 3 inactivates biologically active full length interleukin-33 as a classical cytokine but does not prohibit nuclear translocation. Biochem. Biophys. Res. Commun. 391, 1512–1516.
Allakhverdi, Z., Comeau, M. R., Jessup, H. K., Yoon, B. R., Brewer, A., Chartier, S., Paquette, N., Ziegler, S. F., Sarfati, M., and Delespesse, G. (2007a). Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potently activates mast cells. J. Exp. Med. 204, 253–258.
Allakhverdi, Z., Smith, D. E., Comeau, M. R., and Delespesse, G. (2007b). Cutting edge: The ST2 ligand IL-33 potently activates and drives maturation of human mast cells. J. Immunol. 179, 2051–2054.
Alves-Filho, J. C., Sonego, F., Souto, F. O., Freitas, A., Verri, W. A. Jr., Auxiliadora-Martins, M., Basile-Filho, A., McKenzie, A. N., Xu, D., Cunha, F. Q., and Liew, F. Y. (2010). Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat. Med. 16, 708–712.
Baekkevold, E. S., Roussigne, M., Yamanaka, T., Johansen, F. E., Jahnsen, F. L., Amalric, F., Brandtzaeg, P., Erard, M., Haraldsen, G., and Girard, J. P. (2003). Molecular characterization of NF-HEV, a nuclear factor preferentially expressed in human high endothelial venules. Am. J. Pathol. 163, 69–79.
Basu, S., Binder, R. J., Suto, R., Anderson, K. M., and Srivastava, P. K. (2000). Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int. Immunol. 12, 1539–1546.
Besnard, A. G., Togbe, D., Guillou, N., Erard, F., Quesniaux, V., and Ryffel, B. (2011). IL-33-activated dendritic cells are critical for allergic airway inflammation. Eur. J. Immunol. 41, 1675–1686.
Bianchi, M. E. (2007). DAMPs, PAMPs and alarmins: all we need to know about danger. J. Leukoc. Biol. 81, 1–5.
Bourgeois, E., Van, L. P., Samson, M., Diem, S., Barra, A., Roga, S., Gombert, J. M., Schneider, E., Dy, M., Gourdy, P., Girard, J. P., and Herbelin, A. (2009). The pro-Th2 cytokine IL-33 directly interacts with invariant NKT and NK cells to induce IFN-gamma production. Eur. J. Immunol. 39, 1046–1055.
Bours, M. J., Swennen, E. L., Di Virgilio, F., Cronstein, B. N., and Dagnelie, P. C. (2006). Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol. Ther. 112, 358–404.
Bradding, P., Walls, A. F., and Holgate, S. T. (2006). The role of the mast cell in the pathophysiology of asthma. J. Allergy Clin. Immunol. 117, 1277–1284.
Bush, R. K., and Prochnau, J. J. (2004). Alternaria-induced asthma. J. Allergy Clin. Immunol. 113, 227–234.
Byrne, S. N., Beaugie, C., O’Ssullivan, C., Leighton, S., and Halliday, G. M. (2011). The immune-modulating cytokine and endogenous alarmin interleukin-33 is upregulated in skin exposed to inflammatory UVB radiation. Am. J. Pathol. 179, 211–222.
Carriere, V., Roussel, L., Ortega, N., Lacorre, D. A., Americh, L., Aguilar, L., Bouche, G., and Girard, J. P. (2007). IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc. Natl. Acad. Sci. U.S.A. 104, 282–287.
Cayrol, C., and Girard, J. P. (2009). The IL-1-like cytokine IL-33 is inactivated after maturation by caspase-1. Proc. Natl. Acad. Sci. U.S.A. 106, 9021–9026.
Chackerian, A. A., Oldham, E. R., Murphy, E. E., Schmitz, J., Pflanz, S., and Kastelein, R. A. (2007). IL-1 receptor accessory protein and ST2 comprise the IL-33 receptor complex. J. Immunol. 179, 2551–2555.
Chen, C. C., Grimbaldeston, M. A., Tsai, M., Weissman, I. L., and Galli, S. J. (2005). Identification of mast cell progenitors in adult mice. Proc. Natl. Acad. Sci. U.S.A. 102, 11408–11413.
Cherry, W. B., Yoon, J., Bartemes, K. R., Iijima, K., and Kita, H. (2008). A novel IL-1 family cytokine, IL-33, potently activates human eosinophils. J. Allergy Clin. Immunol. 121, 1484–1490.
Di Nardo, A., Vitiello, A., and Gallo, R. L. (2003). Cutting edge: mast cell antimicrobial activity is mediated by expression of cathelicidin antimicrobial peptide. J. Immunol. 170, 2274–2278.
Dickel, H., Gambichler, T., Kamphowe, J., Altmeyer, P., and Skrygan, M. (2010). Standardized tape stripping prior to patch testing induces upregulation of Hsp90, Hsp70, IL-33, TNF-alpha and IL-8/CXCL8 mRNA: new insights into the involvement of “alarmins.” Contact Derm. 63, 215–222.
Eigenbrod, T., Park, J. H., Harder, J., Iwakura, Y., and Nunez, G. (2008). Cutting edge: critical role for mesothelial cells in necrosis-induced inflammation through the recognition of IL-1 alpha released from dying cells. J. Immunol. 181, 8194–8198.
Enoksson, M., Lyberg, K., Moller-Westerberg, C., Fallon, P. G., Nilsson, G., and Lunderius-Andersson, C. (2011). Mast cells as sensors of cell injury through IL-33 recognition. J. Immunol. 186, 2523–2528.
Fischer, M., Harvima, I. T., Carvalho, R. F. S., Moller, C., Naukkarinen, A., Enblad, G., and Nilsson, G. (2006). Mast cell CD30 ligand is upregulated in cutaneous inflammation and mediates degranulation-independent chemokine secretion. J. Clin. Invest. 116, 2748–2756.
Fujita, J., Nakayama, H., Onoue, H., Kanakura, Y., Nakano, T., Asai, H., Takeda, S., Honjo, T., and Kitamura, Y. (1988). Fibroblast-dependent growth of mouse mast cells in vitro: duplication of mast cell depletion in mutant mice of W/Wv genotype. J. Cell. Physiol. 134, 78–84.
Funakoshi-Tago, M., Tago, K., Hayakawa, M., Tominaga, S., Ohshio, T., Sonoda, Y., and Kasahara, T. (2008). TRAF6 is a critical signal transducer in IL-33 signaling pathway. Cell. Signal. 20, 1679–1686.
Funakoshi-Tago, M., Tago, K., Sato, Y., Tominaga, S., and Kasahara, T. (2011). JAK2 is an important signal transducer in IL-33-induced NF-kappaB activation. Cell. Signal. 23, 363–370.
Galli, S. J., Maurer, M., and Lantz, C. S. (1999). Mast cells as sentinels of innate immunity. Curr. Opin. Immunol. 11, 53–59.
Goh, F. G., and Midwood, K. S. (2011). Intrinsic danger: activation of Toll-like receptors in rheumatoid arthritis. Rheumatol. (Oxf.). 51, 7–23.
Harvima, I. T., and Nilsson, G. (2011). Mast cells as regulators of skin inflammation and immunity. Acta Derm. Venereol. 91, 644–650.
Hayakawa, M., Hayakawa, H., Matsuyama, Y., Tamemoto, H., Okazaki, H., and Tominaga, S. (2009). Mature interleukin-33 is produced by calpain-mediated cleavage in vivo. Biochem. Biophys. Res. Commun. 387, 218–222.
Ho, L. H., Ohno, T., Oboki, K., Kajiwara, N., Suto, H., Iikura, M., Okayama, Y., Akira, S., Saito, H., Galli, S. J., and Nakae, S. (2007). IL-33 induces IL-13 production by mouse mast cells independently of IgE-FcepsilonRI signals. J. Leukoc. Biol. 82, 1481–1490.
Hsu, C. L., Neilsen, C. V., and Bryce, P. J. (2010). IL-33 is produced by mast cells and regulates IgE-dependent inflammation. PLoS ONE 5, e11944. doi:10.1371/journal.pone.0011944
Hueber, A. J., Alves-Filho, J. C., Asquith, D. L., Michels, C., Millar, N. L., Reilly, J. H., Graham, G. J., Liew, F. Y., Miller, A. M., and McInnes, I. B. (2011). IL-33 induces skin inflammation with mast cell and neutrophil activation. Eur. J. Immunol. 41, 2229–2237.
Humphreys, N. E., Xu, D., Hepworth, M. R., Liew, F. Y., and Grencis, R. K. (2008). IL-33, a potent inducer of adaptive immunity to intestinal nematodes. J. Immunol. 180, 2443–2449.
Iikura, M., Suto, H., Kajiwara, N., Oboki, K., Ohno, T., Okayama, Y., Saito, H., Galli, S. J., and Nakae, S. (2007). IL-33 can promote survival, adhesion and cytokine production in human mast cells. Lab. Invest. 87, 971–978.
Kageyama, Y., Torikai, E., Tsujimura, K., and Kobayashi, M. (2011). Involvement of IL-33 in the pathogenesis of rheumatoid arthritis: the effect of etanercept on the serum levels of IL-33. Mod Rheumatol. 22, 89–93.
Kaieda, S., Shin, K., Nigrovic, P. A., Seki, K., Lee, R. T., Stevens, R. L., and Lee, D. M. (2010). Synovial fibroblasts promote the expression and granule accumulation of tryptase via interleukin-33 and its receptor ST-2 (IL1RL1). J. Biol. Chem. 285, 21478–21486.
Kalesnikoff, J., and Galli, S. J. (2008). New developments in mast cell biology. Nat. Immunol. 9, 1215–1223.
Kearley, J., Buckland, K. F., Mathie, S. A., and Lloyd, C. M. (2009). Resolution of allergic inflammation and airway hyperreactivity is dependent upon disruption of the T1/ST2-IL-33 pathway. Am. J. Respir. Crit. Care Med. 179, 772–781.
Kim, H. S., Han, M. S., Chung, K. W., Kim, S., Kim, E., Kim, M. J., Jang, E., Lee, H. A., Youn, J., Akira, S., and Lee, M. S. (2007). Toll-like receptor 2 senses beta-cell death and contributes to the initiation of autoimmune diabetes. Immunity 27, 321–333.
Kondo, Y., Yoshimoto, T., Yasuda, K., Futatsugi-Yumikura, S., Morimoto, M., Hayashi, N., Hoshino, T., Fujimoto, J., and Nakanishi, K. (2008). Administration of IL-33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lungs in the absence of adaptive immune system. Int. Immunol. 20, 791–800.
Kono, H., and Rock, K. L. (2008). How dying cells alert the immune system to danger. Nat. Rev. Immunol. 8, 279–289.
Kouzaki, H., Iijima, K., Kobayashi, T., O’Grady, S. M., and Kita, H. (2011). The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL-33 release and innate Th2-type responses. J. Immunol. 186, 4375–4387.
Kroeger, K. M., Sullivan, B. M., and Locksley, R. M. (2009). IL-18 and IL-33 elicit Th2 cytokines from basophils via a MyD88- and p38{alpha}-dependent pathway. J Leukoc. Biol. 86, 769–778.
Kurowska-Stolarska, M., Kewin, P., Murphy, G., Russo, R. C., Stolarski, B., Garcia, C. C., Komai-Koma, M., Pitman, N., Li, Y., Niedbala, W., McKenzie, A. N., Teixeira, M. M., Liew, F. Y., and Xu, D. (2008). IL-33 induces antigen-specific IL-5+ T cells and promotes allergic-induced airway inflammation independent of IL-4. J. Immunol. 181, 4780–4790.
Leung, B. P., Xu, D., Culshaw, S., McInnes, I. B., and Liew, F. Y. (2004). A novel therapy of murine collagen-induced arthritis with soluble T1/ST2. J. Immunol. 173, 145–150.
Levi-Schaffer, F., Austen, K. F., Caulfield, J. P., Hein, A., Bloes, W. F., and Stevens, R. L. (1985). Fibroblasts maintain the phenotype and viability of the rat heparin-containing mast cell in vitro. J. Immunol. 135, 3454–3462.
Liew, F. Y., Pitman, N. I., and McInnes, I. B. (2010). Disease-associated functions of IL-33: the new kid in the IL-1 family. Nat. Rev. Immunol. 10, 103–110.
Louten, J., Rankin, A. L., Li, Y., Murphy, E. E., Beaumont, M., Moon, C., Bourne, P., McClanahan, T. K., Pflanz, S., and De Waal Malefyt, R. (2011). Endogenous IL-33 enhances Th2 cytokine production and T-cell responses during allergic airway inflammation. Int. Immunol. 23, 307–315.
Luthi, A. U., Cullen, S. P., Mcneela, E. A., Duriez, P. J., Afonina, I. S., Sheridan, C., Brumatti, G., Taylor, R. C., Kersse, K., Vandenabeele, P., Lavelle, E. C., and Martin, S. J. (2009). Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity 31, 84–98.
Manetti, M., Ibba-Manneschi, L., Liakouli, V., Guiducci, S., Milia, A. F., Benelli, G., Marrelli, A., Conforti, M. L., Romano, E., Giacomelli, R., Matucci-Cerinic, M., and Cipriani, P. (2010). The IL1-like cytokine IL33 and its receptor ST2 are abnormally expressed in the affected skin and visceral organs of patients with systemic sclerosis. Ann. Rheum. Dis. 69, 598–605.
Matsuyama, Y., Okazaki, H., Hoshino, M., Onishi, S., Kamata, Y., Nagatani, K., Nagashima, T., Iwamoto, M., Yoshio, T., Ohto-Ozaki, H., Tamemoto, H., Komine, M., Sekiya, H., Tominaga, S. I., and Minota, S. (2011). Sustained elevation of interleukin-33 in sera and synovial fluids from patients with rheumatoid arthritis non-responsive to anti-tumor necrosis factor: possible association with persistent IL-1beta signaling and a poor clinical response. Rheumatol. Int. doi: 10.1007/s00296-011-1854-6. [Epub ahead of print].
Matsuyama, Y., Okazaki, H., Tamemoto, H., Kimura, H., Kamata, Y., Nagatani, K., Nagashima, T., Hayakawa, M., Iwamoto, M., Yoshio, T., Tominaga, S., and Minota, S. (2010). Increased levels of interleukin 33 in sera and synovial fluid from patients with active rheumatoid arthritis. J. Rheumatol. 37, 18–25.
Merrill, J. E., and Scolding, N. J. (1999). Mechanisms of damage to myelin and oligodendrocytes and their relevance to disease. Neuropathol. Appl. Neurobiol. 25, 435–458.
Metz, M., and Maurer, M. (2007). Mast cells – key effector cells in immune responses. Trends Immunol. 28, 234–241.
Miller, A. M., Xu, D., Asquith, D. L., Denby, L., Li, Y., Sattar, N., Baker, A. H., McInnes, I. B., and Liew, F. Y. (2008). IL-33 reduces the development of atherosclerosis. J. Exp. Med. 205, 339–346.
Moffatt, M. F., Gut, I. G., Demenais, F., Strachan, D. P., Bouzigon, E., Heath, S., Von Mutius, E., Farrall, M., Lathrop, M., and Cookson, W. O. (2010). A large-scale, consortium-based genomewide association study of asthma. N. Engl. J. Med. 363, 1211–1221.
Moritz, D. R., Rodewald, H. R., Gheyselinck, J., and Klemenz, R. (1998). The IL-1 receptor-related T1 antigen is expressed on immature and mature mast cells and on fetal blood mast cell progenitors. J. Immunol. 161, 4866–4874.
Moulin, D., Donze, O., Talabot-Ayer, D., Mezin, F., Palmer, G., and Gabay, C. (2007). Interleukin (IL)-33 induces the release of pro-inflammatory mediators by mast cells. Cytokine 40, 216–225.
Moussion, C., Ortega, N., and Girard, J. P. (2008). The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel “alarmin?” PLoS ONE 3, e3331. doi:10.1371/journal.pone.0003331
Mu, R., Huang, H. Q., Li, Y. H., Li, C., Ye, H., and Li, Z. G. (2010). Elevated serum interleukin 33 is associated with autoantibody production in patients with rheumatoid arthritis. J. Rheumatol. 37, 2006–2013.
Nakagawa, S., Koomen, C. W., Bos, J. D., and Teunissen, M. B. (1999). Differential modulation of human epidermal Langerhans cell maturation by ultraviolet B radiation. J. Immunol. 163, 5192–5200.
Neill, D. R., Wong, S. H., Bellosi, A., Flynn, R. J., Daly, M., Langford, T. K., Bucks, C., Kane, C. M., Fallon, P. G., Pannell, R., Jolin, H. E., and McKenzie, A. N. (2010). Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 464, 1367–1370.
Nocka, K., Buck, J., Levi, E., and Besmer, P. (1990). Candidate ligand for the c-kit transmembrane kinase receptor: KL, a fibroblast derived growth factor stimulates mast cells and erythroid progenitors. EMBO J. 9, 3287–3294.
Oboki, K., Ohno, T., Kajiwara, N., Arae, K., Morita, H., Ishii, A., Nambu, A., Abe, T., Kiyonari, H., Matsumoto, K., Sudo, K., Okumura, K., Saito, H., and Nakae, S. (2010). IL-33 is a crucial amplifier of innate rather than acquired immunity. Proc. Natl. Acad. Sci. U.S.A. 107, 18581–18586.
Ohno, T., Oboki, K., Kajiwara, N., Morii, E., Aozasa, K., Flavell, R. A., Okumura, K., Saito, H., and Nakae, S. (2009). Caspase-1, caspase-8, and calpain are dispensable for IL-33 release by macrophages. J. Immunol. 183, 7890–7897.
Palmer, G., Lipsky, B. P., Smithgall, M. D., Meininger, D., Siu, S., Talabot-Ayer, D., Gabay, C., and Smith, D. E. (2008). The IL-1 receptor accessory protein (AcP) is required for IL-33 signaling and soluble AcP enhances the ability of soluble ST2 to inhibit IL-33. Cytokine 42, 358–364.
Palmer, G., Talabot-Ayer, D., Lamacchia, C., Toy, D., Seemayer, C. A., Viatte, S., Finckh, A., Smith, D. E., and Gabay, C. (2009). Inhibition of interleukin-33 signaling attenuates the severity of experimental arthritis. Arthritis Rheum. 60, 738–749.
Pecaric-Petkovic, T., Didichenko, S. A., Kaempfer, S., Spiegl, N., and Dahinden, C. A. (2009). Human basophils and eosinophils are the direct target leukocytes of the novel IL-1 family member IL-33. Blood 113, 1526–1534.
Prefontaine, D., Lajoie-Kadoch, S., Foley, S., Audusseau, S., Olivenstein, R., Halayko, A. J., Lemiere, C., Martin, J. G., and Hamid, Q. (2009). Increased expression of IL-33 in severe asthma: evidence of expression by airway smooth muscle cells. J. Immunol. 183, 5094–5103.
Prefontaine, D., Nadigel, J., Chouiali, F., Audusseau, S., Semlali, A., Chakir, J., Martin, J. G., and Hamid, Q. (2010). Increased IL-33 expression by epithelial cells in bronchial asthma. J. Allergy Clin. Immunol. 125, 752–754.
Pushparaj, P. N., Tay, H. K., H’ng S, C., Pitman, N., Xu, D., McKenzie, A., Liew, F. Y., and Melendez, A. J. (2009). The cytokine interleukin-33 mediates anaphylactic shock. Proc. Natl. Acad. Sci. U.S.A. 106, 9773–9778.
Rank, M. A., Kobayashi, T., Kozaki, H., Bartemes, K. R., Squillace, D. L., and Kita, H. (2009). IL-33-activated dendritic cells induce an atypical TH2-type response. J. Allergy Clin. Immunol. 123, 1047–1054.
Ribatti, D., and Crivellato, E. (2011). Mast cells, angiogenesis and cancer. Adv. Exp. Med. Biol. 716, 270–288.
Sanada, S., Hakuno, D., Higgins, L. J., Schreiter, E. R., McKenzie, A. N., and Lee, R. T. (2007). IL-33 and ST2 comprise a critical biomechanically induced and cardioprotective signaling system. J. Clin. Invest. 117, 1538–1549.
Scaffidi, P., Misteli, T., and Bianchi, M. E. (2002). Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418, 191–195.
Scheynius, A., Johansson, C., Buentke, E., Zargari, A., and Linder, M. T. (2002). Atopic eczema/dermatitis syndrome and Malassezia. Int. Arch. Allergy Immunol. 127, 161–169.
Schmitz, J., Owyang, A., Oldham, E., Song, Y., Murphy, E., McClanahan, T. K., Zurawski, G., Moshrefi, M., Qin, J., Li, X., Gorman, D. M., Bazan, J. F., and Kastelein, R. A. (2005). IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 23, 479–490.
Schneider, E., Petit-Bertron, A.F., Bricard, R., Levasseur, M., Ramadan, A., Girard, J.P., Herbelin, A., and Dy, M. (2009). IL-33 activates unprimed murine basophils directly in vitro and induces their in vivo expansion indirectly by promoting hematopoietic growth factor production. J. Immunol. 183, 3591–3597.
Shi, Y., Evans, J. E., and Rock, K. L. (2003). Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 425, 516–521.
Shimizu, M., Matsuda, A., Yanagisawa, K., Hirota, T., Akahoshi, M., Inomata, N., Ebe, K., Tanaka, K., Sugiura, H., Nakashima, K., Tamari, M., Takahashi, N., Obara, K., Enomoto, T., Okayama, Y., Gao, P. S., Huang, S. K., Tominaga, S., Ikezawa, Z., and Shirakawa, T. (2005). Functional SNPs in the distal promoter of the ST2 gene are associated with atopic dermatitis. Hum. Mol. Genet. 14, 2919–2927.
Silver, M. R., Margulis, A., Wood, N., Goldman, S. J., Kasaian, M., and Chaudhary, D. (2009). IL-33 synergizes with IgE-dependent and IgE-independent agents to promote mast cell and basophil activation. Inflamm. Res. 59, 207–218.
Smithgall, M. D., Comeau, M. R., Yoon, B. R., Kaufman, D., Armitage, R., and Smith, D. E. (2008). IL-33 amplifies both Th1- and Th2-type responses through its activity on human basophils, allergen-reactive Th2 cells, iNKT and NK cells. Int. Immunol. 20, 1019–1030.
Strickland, I., Rhodes, L. E., Flanagan, B. F., and Friedmann, P. S. (1997). TNF-alpha and IL-8 are upregulated in the epidermis of normal human skin after UVB exposure: correlation with neutrophil accumulation and E-selectin expression. J. Invest. Dermatol. 108, 763–768.
Suzukawa, M., Iikura, M., Koketsu, R., Nagase, H., Tamura, C., Komiya, A., Nakae, S., Matsushima, K., Ohta, K., Yamamoto, K., and Yamaguchi, M. (2008a). An IL-1 cytokine member, IL-33, induces human basophil activation via its ST2 receptor. J. Immunol. 181, 5981–5989.
Suzukawa, M., Koketsu, R., Iikura, M., Nakae, S., Matsumoto, K., Nagase, H., Saito, H., Matsushima, K., Ohta, K., Yamamoto, K., and Yamaguchi, M. (2008b). Interleukin-33 enhances adhesion, CD11b expression and survival in human eosinophils. Lab. Invest. 88, 1245–1253.
Tago, K., Noda, T., Hayakawa, M., Iwahana, H., Yanagisawa, K., Yashiro, T., and Tominaga, S. (2001). Tissue distribution and subcellular localization of a variant form of the human ST2 gene product, ST2V. Biochem. Biophys. Res. Commun. 285, 1377–1383.
Talabot-Ayer, D., Lamacchia, C., Gabay, C., and Palmer, G. (2009). Interleukin-33 is biologically active independently of caspase-1 cleavage. J Biol Chem. 284, 19420–19426.
Talabot-Ayer, D., Mckee, T., Gindre, P., Bas, S., Baeten, D. L., Gabay, C., and Palmer, G. (2012). Distinct serum and synovial fluid interleukin (IL)-33 levels in rheumatoid arthritis, psoriatic arthritis and osteoarthritis. Joint Bone Spine 79, 32–37.
Theoharides, T. C., Kempuraj, D., Tagen, M., Conti, P., and Kalogeromitros, D. (2007). Differential release of mast cell mediators and the pathogenesis of inflammation. Immunol. Rev. 217, 65–78.
Theoharides, T. C., Zhang, B., Kempuraj, D., Tagen, M., Vasiadi, M., Angelidou, A., Alysandratos, K. D., Kalogeromitros, D., Asadi, S., Stavrianeas, N., Peterson, E., Leeman, S., and Conti, P. (2010). IL-33 augments substance P-induced VEGF secretion from human mast cells and is increased in psoriatic skin. Proc. Natl. Acad. Sci. U.S.A. 107, 4448–4453.
Tominaga, S. (1989). A putative protein of a growth specific cDNA from BALB/c-3T3 cells is highly similar to the extracellular portion of mouse interleukin 1 receptor. FEBS Lett. 258, 301–304.
Torgerson, D. G., Ampleford, E. J., Chiu, G. Y., Gauderman, W. J., Gignoux, C. R., Graves, P. E., Himes, B. E., Levin, A. M., Mathias, R. A., Hancock, D. B., Baurley, J. W., Eng, C., Stern, D. A., Celedon, J. C., Rafaels, N., Capurso, D., Conti, D. V., Roth, L. A., Soto-Quiros, M., Togias, A., Li, X., Myers, R. A., Romieu, I., Van Den Berg, D. J., Hu, D., Hansel, N. N., Hernandez, R. D., Israel, E., Salam, M. T., Galanter, J., Avila, P. C., Avila, L., Rodriquez-Santana, J. R., Chapela, R., Rodriguez-Cintron, W., Diette, G. B., Adkinson, N. F., Abel, R. A., Ross, K. D., Shi, M., Faruque, M. U., Dunston, G. M., Watson, H. R., Mantese, V. J., Ezurum, S. C., Liang, L., Ruczinski, I., Ford, J. G., Huntsman, S., Chung, K. F., Vora, H., Li, X., Calhoun, W. J., Castro, M., Sienra-Monge, J. J., Del Rio-Navarro, B., Deichmann, K. A., Heinzmann, A., Wenzel, S. E., Busse, W. W., Gern, J. E., Lemanske, R. F. Jr., Beaty, T. H., Bleecker, E. R., Raby, B. A., Meyers, D. A., London, S. J., Gilliland, F. D., Burchard, E. G., Martinez, F. D., Weiss, S. T., Williams, L. K., Barnes, K. C., Ober, C., and Nicolae, D. L. (2011). Meta-analysis of genome-wide association studies of asthma in ethnically diverse North American populations. Nat. Genet. 43, 887–892.
Trajkovic, V., Sweet, M. J., and Xu, D. (2004). T1/ST2 – an IL-1 receptor-like modulator of immune responses. Cytokine Growth Factor Rev. 15, 87–95.
Verri, W. A. Jr., Souto, F. O., Vieira, S. M., Almeida, S. C., Fukada, S. Y., Xu, D., Alves-Filho, J. C., Cunha, T. M., Guerrero, A. T., Mattos-Guimaraes, R. B., Oliveira, F. R., Teixeira, M. M., Silva, J. S., McInnes, I. B., Ferreira, S. H., Louzada-Junior, P., Liew, F. Y., and Cunha, F. Q. (2010). IL-33 induces neutrophil migration in rheumatoid arthritis and is a target of anti-TNF therapy. Ann. Rheum. Dis. 69, 1697–1703.
Walker, M. E., Hatfield, J. K., and Brown, M. A. (2012). New insights into the role of mast cells in autoimmunity: evidence for a common mechanism of action? Biochim. Biophys. Acta 1822, 57–65.
Weller, K., Foitzik, K., Paus, R., Syska, W., and Maurer, M. (2006). Mast cells are required for normal healing of skin wounds in mice. FASEB J. 20, 2366–2368.
Xu, D., Chan, W. L., Leung, B. P., Huang, F., Wheeler, R., Piedrafita, D., Robinson, J. H., and Liew, F. Y. (1998). Selective expression of a stable cell surface molecule on type 2 but not type 1 helper T cells. J. Exp. Med. 187, 787–794.
Xu, D., Jiang, H. R., Kewin, P., Li, Y., Mu, R., Fraser, A. R., Pitman, N., Kurowska-Stolarska, M., McKenzie, A. N., McInnes, I. B., and Liew, F. Y. (2008). IL-33 exacerbates antigen-induced arthritis by activating mast cells. Proc. Natl. Acad. Sci. U.S.A. 105, 10913–10918.
Xu, D., Jiang, H. R., Li, Y., Pushparaj, P. N., Kurowska-Stolarska, M., Leung, B. P., Mu, R., Tay, H. K., McKenzie, A. N., McInnes, I. B., Melendez, A. J., and Liew, F. Y. (2010). IL-33 exacerbates autoantibody-induced arthritis. J. Immunol. 184, 2620–2626.
Yanaba, K., Yoshizaki, A., Asano, Y., Kadono, T., and Sato, S. (2011). Serum IL-33 levels are raised in patients with systemic sclerosis: association with extent of skin sclerosis and severity of pulmonary fibrosis. Clin. Rheumatol. 30, 825–830.
Keywords: mast cell, injury, inflammation, IL-33, IL-6, migration
Citation: Lunderius-Andersson C, Enoksson M and Nilsson G (2012) Mast cells respond to cell injury through the recognition of IL-33. Front. Immun. 3:82. doi: 10.3389/fimmu.2012.00082
Received: 06 December 2011; Accepted: 03 April 2012;
Published online: 19 April 2012.
Edited by:
Toshiaki Kawakami, La Jolla Institute for Allergy and Immunology, USAReviewed by:
Miriam Wittmann, University of Leeds, UKMasato Kubo, Tokyo University of Science, Japan
Tomohiro Yoshimoto, Hyogo College of Medicine, Jordan
Hirohito Kita, Mayo Clinic, USA
Copyright: © 2012 Lunderius-Andersson, Enoksson and Nilsson. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Gunnar Nilsson, Clinical Immunology and Allergy, Department of Medicine, Karolinska Institutet, Stockholm, Sweden. e-mail:Z3VubmFyLnAubmlsc3NvbkBraS5zZQ==