 Bharat Vaidyanathan
Bharat Vaidyanathan Wei-Feng Yen
Wei-Feng Yen Joseph N. Pucella
Joseph N. Pucella Jayanta Chaudhuri
Jayanta Chaudhuri- 1Weill Cornell Graduate School of Medical Sciences, New York, NY, USA
- 2Immunology Program, Memorial Sloan Kettering Cancer Center, Gerstner Sloan Kettering Graduate School, New York, NY, USA
Secondary diversification of the antibody repertoire upon antigenic challenge, in the form of immunoglobulin heavy chain (IgH) class-switch recombination (CSR) endows mature, naïve B cells in peripheral lymphoid organs with a limitless ability to mount an optimal humoral immune response, thus expediting pathogen elimination. CSR replaces the default constant (CH) region exons (Cμ) of IgH with any of the downstream CH exons (Cγ, Cε, or Cα), thereby altering effector functions of the antibody molecule. This process depends on, and is orchestrated by, activation-induced deaminase (AID), a DNA cytidine deaminase that acts on single-stranded DNA exposed during transcription of switch (S) region sequences at the IgH locus. DNA lesions thus generated are processed by components of several general DNA repair pathways to drive CSR. Given that AID can instigate DNA lesions and genomic instability, stringent checks are imposed that constrain and restrict its mutagenic potential. In this review, we will discuss how AID expression and substrate specificity and activity is rigorously enforced at the transcriptional, post-transcriptional, post-translational, and epigenetic levels, and how the DNA-damage response is choreographed with precision to permit targeted activity while limiting bystander catastrophe.
Introduction
B cells are specialized lymphocytes that express Ig receptors (or antibodies) on their cell surface. Antibodies are comprised of immunoglobulin heavy chains (IgH) and light chains (IgL), with the N-termini of IgH and IgL generating the antigen-binding pocket, and the C-terminus of IgH performing effector functions. A salient feature of B-lymphocytes is their ability to recognize an almost infinite array of antigens. This enormous diversity is achieved through V(D)J recombination, a process that assembles the exons encoding the amino-terminal variable regions of IgH and IgL from component variable (V), diversity (D), and joining (J) segments (1). The end product of V(D)J recombination is a mature but naïve IgM+ B cell that exits the bone marrow. In the context of specialized structures called germinal centers in secondary lymphoid organs such as the spleen and lymph nodes, mature B cells interact with antigens and undergo class-switch recombination (CSR) (2, 3).
The mouse IgH locus is comprised of eight constant region (CH) exons, with Cμ most proximal to the variable region segments and Cα being the most distal (Figure 1). CSR exchanges the default Cμ for an alternative set of downstream CH exons, for example, Cγ, Cε, or Cα, so that the B cell switches from expressing IgM to one producing a secondary antibody isotype such as IgG, IgE, or IgA, respectively. CSR occurs between repetitive “switch” (S) DNA elements that precede each set of CH exons. According to the conventional model for CSR, transcription through S regions promotes formation of DNA:RNA hybrid structures, such as R-loops that reveal single-stranded DNA (ssDNA) substrates for activation-induced deaminase (AID)-mediated cytidine deamination (Figure 2). The deaminated residues are processed into DNA double-strand breaks (DSBs) by components of the base-excision repair (BER) and mismatch repair (MMR) pathways (4–6). End-joining of DSBs between two S regions results in the excision of the intervening sequence and juxtaposition of a new set of constant region exons directly downstream of the rearranged V(D)J segment, thereby generating Ig molecules with the same antigen specificity but with new effector functions (2, 3) (Figure 1). Here, we will discuss the intrinsic properties of AID, the factors commissioning its function to produce obligatory DNA DSBs intermediates, and the DNA repair/end-joining pathways that ensure productive recombination.
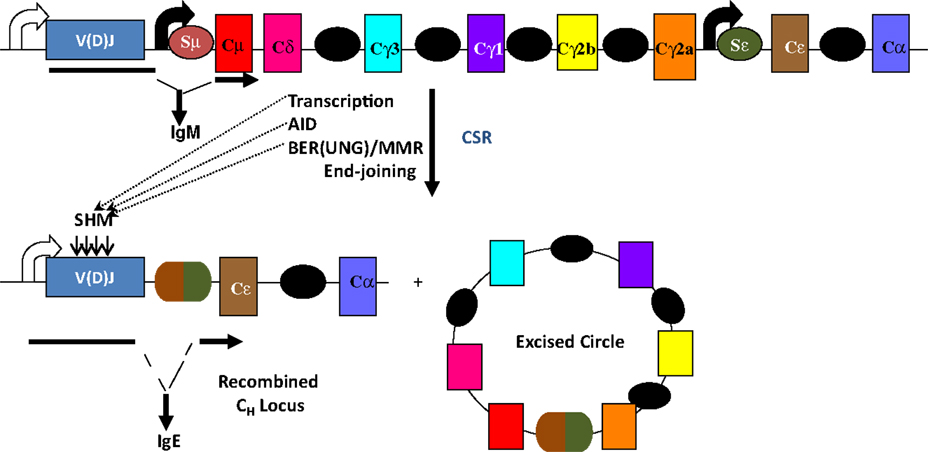
Figure 1. Overview of CSR. CSR is a deletional-recombination reaction between repetitive switch (S) regions (ovals) that precede each set of constant region (CH) exons. Cytokine-induced transcription through S regions drives CSR to specific CH exons. CSR requires AID and components of the base-excision repair (BER) and mismatch repair (MMR) pathways. Somatic hypermutation (SHM) introduces point mutations at a very high rate into variable region exons, V(D)J.
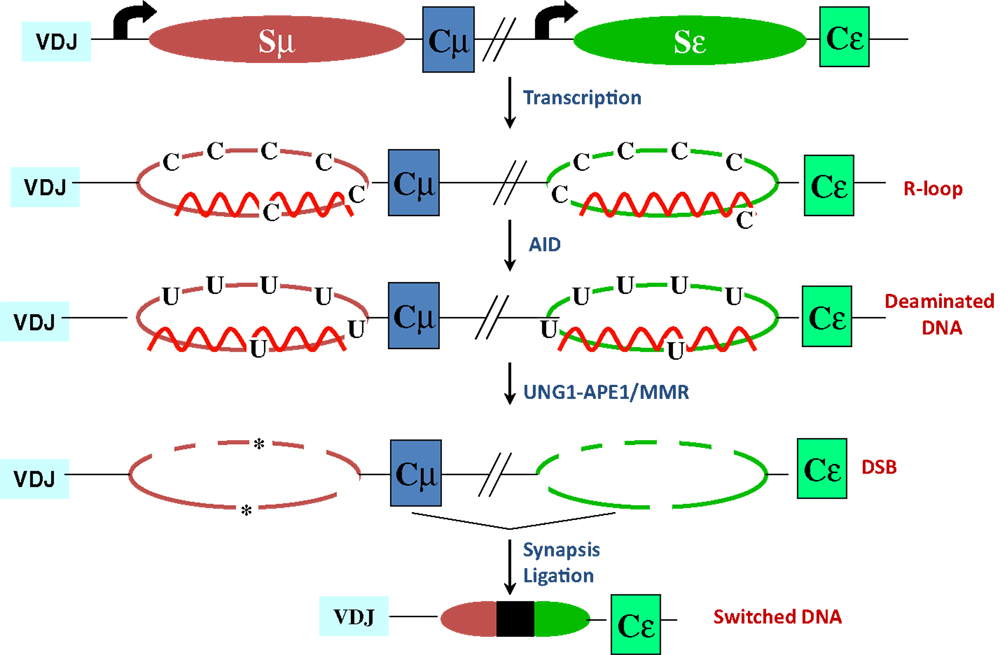
Figure 2. R-loop-based model for CSR. Transcription through S regions generates R-loop structures in which the RNA stably hybridizes to the template strand, displacing the non-template strand as ssDNA. AID activity at R-loops generate uridines in the DNA, which are subsequently processed by BER and MMR proteins to ultimately generate DSBs. End-joining of DSBs completes CSR. Asterisks represent nicks close to each other on opposite strands.
Germinal center B cells also undergo another AID-dependent secondary diversification reaction termed somatic hypermutation (SHM) wherein point mutations, and sometimes insertions and deletions, are introduced at a very high rate (10−2–10−3/bp/generation) into the recombined variable region exons encoding IgH and IgL, so as to select B cells with increased antigen affinity (Figure 1). SHM requires transcription through the variable region exons and occurs primarily, but not exclusively, at RGYW “hot-spot” motifs where R = purine base, Y = pyrimidine base, and W = A or T nucleotide. Details of SHM are outside the scope of this review and have been discussed in multiple excellent reviews [for example, Ref. (7)].
Intrinsic Properties of AID
Following the discovery of AID by subtractive cDNA hybridization from the mouse CH12F3 B lymphoma cells and its proven essentiality for SHM and CSR (8, 9), a huge amount of effort has gone into the characterization of its enzymatic properties. Elegant biochemical work using purified AID from activated splenic B cells, recombinant GST-AID from Sf9 cells, and other epitope-tagged forms, has shed light into the DNA deamination ability of AID in vitro [reviewed in Ref. (2)]. These studies demonstrated unequivocally that AID deaminates deoxycytidines (dCs) in ssDNA, and fails to act on dsDNA, RNA, and DNA:RNA hybrids. Additionally, it was shown that AID could deaminate dCs in the context of transcribed dsDNA (10, 11), suggesting that access to and activity on in vivo substrates might require transcription of the locus. Since the crystal structure of AID has not been determined, the field has faced a bottleneck in explicit elucidation of enzyme biochemistry. Still, based on structural and biochemical insights from bacterial cytidine deaminases and related DNA/RNA deaminases such as APOBECs, the mechanism of Zn2+-dependent catalysis by the active site (H56, E58, C87, C90) residues and preference for RGYW motif (residues 113–123) was cogently demonstrated (12, 13).
In vitro deamination assays using recombinant GST-AID purified from insect cells suggest that AID performs processive catalysis (14), which leads to accumulation of multiple mutations on a single DNA fragment, disfavoring “jumping” onto a second fragment. This finding is in contrast to proposed distributive mode of action based on the high net positive charge (+11 at pH 7.0) of AID that promotes strong binding to nucleic acids (15). Nonetheless, in vitro deamination assays performed on ssDNA substrates revealed that AID-mediated deamination is intrinsically inefficient, haphazard, and a “random bidirectional walk” along DNA, yielding ~3% deamination upon hot-spot encounter (16). Such a mechanism has likely evolved to generate a diverse array of mutations, especially to favor the selection of high affinity antibodies in vivo (16). A note of caution to be borne in mind while interpreting these biochemical analyses is that the bulky tag might affect inherent properties of AID, and GST-AID does not reconstitute CSR in vivo (17), suggesting that in vitro results may not accurately reflect the in vivo scenario. Besides, this form of AID requires the action of RNase A to be active in vitro, which contradicts reports of AID purified from B cells (10), advocating for adventitious properties unique to GST-AID. It is to be noted that AID, based on its homology to the RNA editing enzyme APOBEC1, has been proposed to edit mRNAs and/or micro-RNAs (miRs) required for CSR and SHM, however, there is no experimental evidence yet to support this notion (18). Thus, despite the limitations of AID enzyme biology, strong genetic evidence has propelled DNA deamination as being the generally accepted model, and this review is based on this premise.
Regulation of AID Expression, Localization, and Stability
While the primary and physiological role of AID is to introduce DNA lesions at the Ig loci to drive antibody diversification, AID also poses a threat to genomic integrity. Ectopic expression of AID in non-B cells converts it into a general mutator (19, 20). Even in B cells, mistargeted AID activity is the major underlying cause behind oncogenic translocations that are hallmarks of a large number of B cell malignancies (1, 21). Therefore, regulation of AID expression is fundamental not only for the development of an efficient immune system, but also for the maintenance of genomic integrity inherent to cells expressing a mutator. Thus, it is not surprising that AID comes outfitted with multifaceted transcriptional and post-transcriptional regulatory mechanisms.
Transcriptional Regulation of AID Expression
Activation-induced deaminase is encoded by the Aicda gene, located on chromosome 6 and 12 in mice and humans, respectively. Four highly conserved regulatory regions activate Aicda transcription primarily in activated B cells, and restrict its expression in other cell types (Figure 3). Region 1 is comprised of a TATA-less promoter and enhancer elements that bind HoxC4–Oct1/2 and Sp1/3 [reviewed in Ref. (22)]. This region also contains elements that respond to estrogen and progesterone, hormones that, respectively, activate or repress AID expression (23–25). Region 2 lies within the first Aicda intron and includes binding sites for B-cell-specific Pax5 and E2A proteins (22). This region also harbors silencer elements that could bind repressors E2F and c-Myb in a fashion unrestricted to B cells (22). Deletion of the silencer elements drastically increases AID expression, without inducing transcription in non-B cells, bolstering the notion of extensive checks to AID expression (26). Region 3, approximately 25 kb downstream of Aicda, is necessary to sustain physiological levels of AID expression, likely through a BATF-binding site (22, 27, 28). Region 4 is approximately 8 kb upstream of the Aicda transcriptional initiation site and contains enhancers that bind NF-κB, STAT6, and SMAD3/4, factors that are stimulated by B cell activation (22). Recently, c-Myc was implicated in binding Region 4 to promote robust AID expression (29, 30).
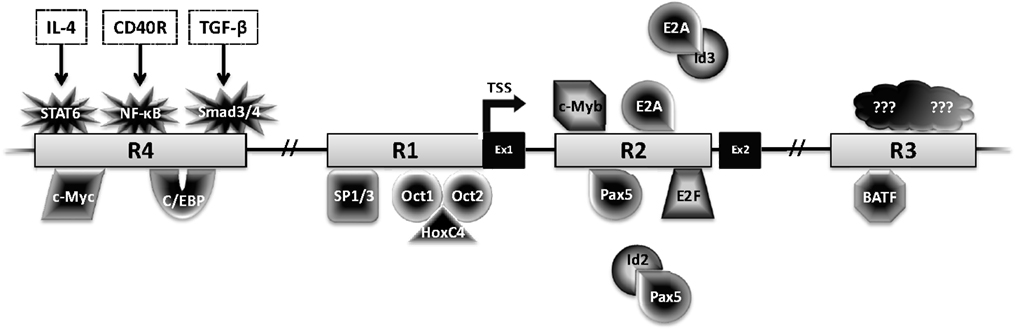
Figure 3. Transcriptional regulation of AID. The Aicda locus contains four conserved regulatory regions (R1–4). The first two exons (Ex1–2) and transcription factors with the potential to bind these regions are shown. Factors with black center and white text are activators, while factors with white center and black text are repressors.
Although physiological AID expression is largely restricted to mature B cells, its expression has also been reported in other settings. AID is expressed in developing B cells in the bone marrow, inducing robust CSR to a subset of isotypes (31, 32). The physiological relevance of CSR in the bone marrow is not clear at present. AID expression has also been observed in intestinal epithelial cells during Helicobacter pylori infection; whether this represents aberrant expression or some uncharacterized response to infection is not known (33, 34). Additionally, AID expression has been observed in prostate cancer cells (35); such aberrant AID expression might be correlative or causal to pathological outcomes.
It is not clear at present whether the non-B-cell-specific expression of AID has any physiological relevance, and the AID fate-mapping mouse does not reveal robust expression pattern in non-lymphoid cells (36). But an intriguing finding is that AID is expressed in primordial germ cells, in embryonic stem (ES) cells, and also in mouse embryonic fibroblasts induced to undergo transcription-factor-mediated reprograming (37, 38). In this regard, AID has been posited to deaminate methylated cytidine, and in concert with DNA BER, promote demethylation of genes required for the maintenance of a pluripotent stem-cell state (37–40). However, AID has extremely weak intrinsic activity on 5mC, and AID-deficient mice do not exhibit any overt phenotype or methylome changes that could be attributed to a failure in active demethylation (41, 42). Thus, in vivo demethylation by AID and the factors regulating AID expression in these settings remain provocative and warrant further research.
Micro-RNA-Mediated Regulation of AID Expression
Another level of regulation exists at the level of stability of Aicda mRNA, enforced by miRs such as miR-155, miR-181b, and miR-361, with miR-155 being the best characterized. miR-155 expression is upregulated upon activation for CSR. The 3′-untranslated region (3′-UTR) of Aicda mRNA has a binding site for miR-155, mutation of which, increased AID expression and doubled the frequency of CSR (43, 44). Surprisingly, although miR-155-deficient B cells upregulate AID expression, they do not undergo increased CSR, perhaps due to dysregulation of other miR-155 targets relevant to CSR (45). The 3′-UTR of Aicda mRNA also contains a binding site for miR-361 (46). Significantly, the transcription factor Bcl6, required for formation of germinal centers, binds and transcriptionally represses both miR-155 and miR-361, in turn relieving repression of AID (46). The role, if any, of miR-361 in the regulation of AID mRNA stability remains to be determined. Finally, ectopic expression of miR-181b in activated murine B cells impaired CSR, likely due to reduced AID mRNA and protein levels (47). Given the emerging significance of canonical and non-canonical miR targeting, it can be conceived that many more miRs affecting AID and CSR are awaiting discovery (48).
Subcellular Localization and Stability
A rational way to constrain AID activity on DNA is by regulating its subcellular localization. AID localization is governed by active nuclear import, cytoplasmic retention, and efficient nuclear export. The majority (greater than 90%) of AID is sequestered in the cytoplasm, possibly through interactions of the C-terminus of AID with eEF1A, chaperone Hsp90, and co-chaperone Hsp40 DnaJa1 (49). Nuclear entry of AID is dependent on importin-3 and a conformational nuclear localization signal (NLS) generated upon folding; a predicted bipartite NLS at the N-terminus of AID might not be functional (49) (Figure 4). In the nucleus, AID was recently found to accumulate in nucleolar structures where it associates with nucleolin and nucleophosmin (50). Mutations that abrogated AID localization to these structures resulted in reduced levels of CSR (50). The nucleoli may serve as a nucleation site for forming complexes, but the precise role of nucleolar AID remains unresolved.
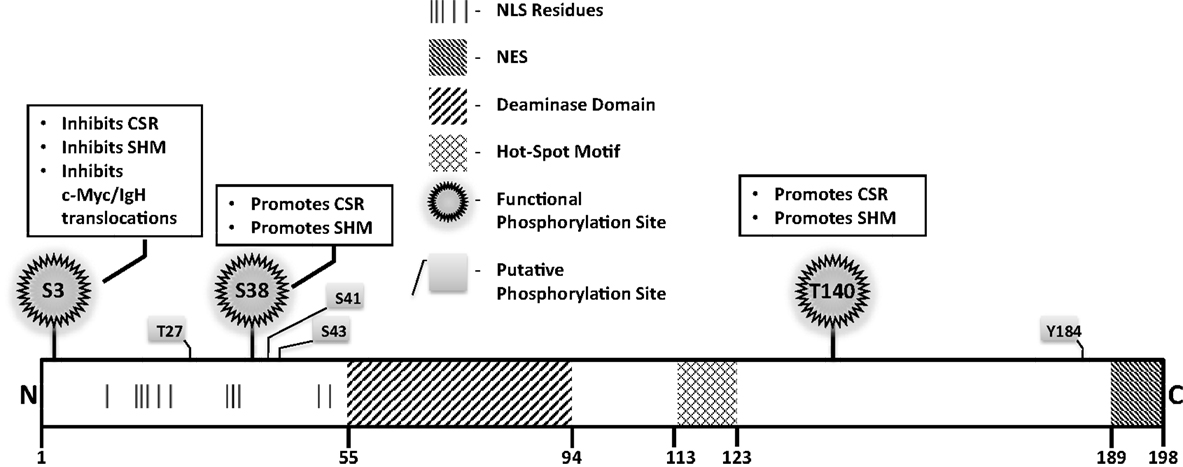
Figure 4. Notable AID domains and residues. Primary structure of AID protein with pertinent domains, motifs, and residues indicated. Panels outlined in black briefly describe functional impact of validated phosphorylation sites.
A mutator protein’s presence in the nucleus must be vigilantly regulated, and a nuclear export signal (NES) within the last 10 amino acids at the C-terminus of AID mediates CRM1-dependent active nuclear export (49) (Figure 4). Mutations in the NES increased levels of nuclear AID, enhanced SHM, but severely impaired CSR, indicating that NES-bearing C-terminus of AID plays a role in CSR beyond export, perhaps in mediating CSR-specific interactions (49). Consistent with this notion, replacement of the C-terminus of AID with a heterologous NES rescued nuclear export, but did not reconstitute CSR (51). Strikingly, the stability of AID was often compromised upon manipulation of the C-terminus of AID, even when nuclear export remained unaffected (51). In this regard, the half-life of nuclear AID is significantly shorter than its cytoplasmic counterpart (~2.5 vs. ~18 h) (52). This is largely due to interactions of AID with the proteasome through ubiquitination or Reg-γ-mediated escort (52, 53). Overall, the involvement of the C-terminus of AID in mediating nuclear export, protein stability, cytoplasmic retention, and CSR-specific interactions render this region one of the most fascinating, yet complicated domain that demands extensive examination.
AID Phosphorylation
Numerous putative phosphorylation sites in AID have been implicated in regulating its ability to effect CSR, SHM, and oncogenic translocations, without affecting stability or deamination potential (Figure 4). Unfortunately, for most, mechanistic insights of functional pertinence remain elusive. Physiologically relevant sites that play critical to modest roles in AID function include serine-3 (S3), threonine-140 (T140), and S38 (54–58). Serine-3 was identified as a site phosphorylated by protein kinase C (PKC) in vitro (55). In contrast to other validated phosphorylation events, phosphorylated S3 inhibits AID function. Mutation of S3 to alanine enhances CSR, SHM, and c-Myc/IgH translocations, despite unperturbed catalytic activity (55); however, the mechanistic underpinnings remain unresolved. PKC can also phosphorylate T140, and T140A mutation perturbs SHM more profoundly than CSR. The mechanism through which phosphorylation at T140 differentially regulates SHM and CSR remains unclear (56). Phosphorylation of AID at serine-38 has been extensively characterized and will be discussed later. Overall, the regulatory mechanisms discussed above, and processes that mediate substrate specificity in vivo as discussed below, impose checkpoints in maintaining physiological functions of AID to facilitate successful and efficient CSR.
Accessibility and Targeting of AID
Since AID is an ssDNA deaminase, mechanisms must exist to generate and reveal such structures at S regions during CSR. Additionally, AID must be actively and specifically recruited to S regions, not only to be productively engaged in CSR, but also to reduce collateral damage associated with expression of a mutator protein. The nature of S regions and their transcription promote AID accessibility to DNA while several proteins have been implicated to specifically recruit AID to the IgH locus during CSR.
S Regions, Transcription, and R-Loops
S regions are 1–12 kb repetitive sequences that are enriched with AID “hot-spot” 5′-RGYW-3′ motifs (59, 60), and are particularly G-rich on the non-template strand. Evidence for the role of S regions came from elegant genetic studies wherein deletion of Sμ dramatically impaired CSR to all isotypes while deletion of Sγ1 abolished CSR to IgG1 (61–63). Recent studies have provocatively suggested that apart from the default donor Sμ, even Sγ1 can serve as a donor and allow sequential switching to IgE (64), an idea that was suggested two decades back when double-isotype expressing B cells were identified (65–68).
The ability of S regions to serve as recombination targets is intricately linked with “germ-line” transcription, an essential prerequisite for CSR (2, 3). Each set of CH exons is an independent transcriptional unit, comprised of an intervening (I)-exon, intronic S region, and the CH exons (Figure 5). The primary transcripts produced constitutively (via μ promoter) or inducibly (for other CH exons), are spliced and polyadenylated but have no protein-coding capacity. These are referred to as germ-line or sterile transcripts. Differential stimulation with distinct sets of activators and cytokines, provided by helper-T cells or through direct interaction with pathogens, induces transcription through different CH exons and promotes CSR to that particular isotype. Significant progress in our understanding of CSR came from ex vivo studies wherein splenic B cells were activated in culture under different conditions. For example, bacterial lipopolysaccharide (LPS) induces germ-line transcription through Cγ2b and Cγ3 and allows CSR to IgG2b and IgG3, while a combination of LPS and interleukin-4 induces Cγ1 and Cε transcription and CSR to IgG1 and IgE. Mutational analyses that altered or deleted the I-exon promoter perturbed CSR dramatically, thus providing experimental evidence for the strong mechanistic link between germ-line transcription and fulfillment of CSR (2, 3).
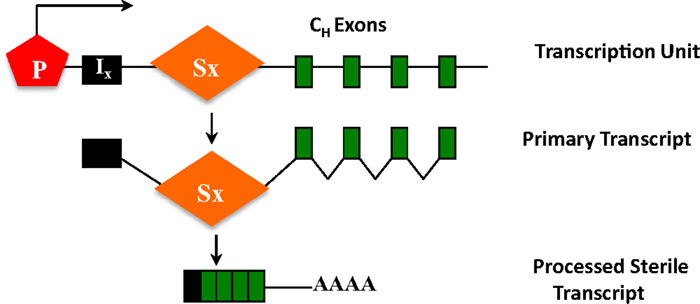
Figure 5. CH exons as transcription units. Each set of CH exons is an independent transcription unit comprised of a promoter (P), an intervening I-exon, an intronic-switch (S) region, and CH exons. The primary transcript is spliced and polyadenylated but does not have protein-coding capacity, and is thus referred to as a sterile transcript.
It is generally believed that transcription through S region sequences promotes formation of R-loops, wherein the template strand stably hybridizes with the G-rich primary transcript (69, 70). This allows the non-template strand to be looped out as ssDNA, providing an ideal substrate for AID (2) (Figure 2). Compelling work in support of the R-loop model came from the observations that a transcribed synthetic DNA fragment with a G-rich non-template strand can support AID deamination in vitro and CSR in B cells, while the inverted sequence (C-rich non-template strand) that does not form R-loops, neither supports AID-mediated deamination in vitro nor CSR in vivo (10, 63). It is to be noted that although the role for germ-line transcription has been well-studied, a possible role of the transcript per se was suggested from the observation that perturbing splicing of primary switch transcripts without affecting transcription impedes CSR (71, 72). However, the neomycin-resistance cassette used in targeting the splice donor site was not removed, leaving open the possibility that the observed CSR defect was due to non-specific effects of this cassette in the IgH locus. Despite this potential caveat, given how non-coding RNAs like HOTAIR and Xist drive PRC2 targeting (73, 74), it would not be surprising if these non-coding switch transcripts play a significant role in AID targeting and activity at S regions.
Factors Promoting Template Strand Deamination
The R-loop model does not account for the mechanism of template strand deamination by AID, a prerequisite for the formation of DSBs. Several models have been put forward to account for deamination of template strand. Anti-sense transcription through the IgH locus has been proposed to facilitate access of AID to the template strand (75); however, anti-sense transcription is not essential for CSR (76). Components of the RNA exosome complex have been shown to interact with AID and mediate accessibility to the template strand by degrading the nascent RNA hybridized to the template strand (77). Recent work cogently elucidated that Nedd4-dependent ubiquitination modulates the fate of AID-associated RNA polymerase II (Pol II), thus generating free 3′-ends that serve as substrates for RNA exosomes (78). RNaseH has also been proposed to facilitate R-loop collapse to ensure template strand deamination (79). However, the kinetics of such R-loop degradation must be stringently regulated in the context of S regions to first allow AID to act on the non-template strand, and elucidation of such intricacies awaits future work.
Targeting AID to DNA
The primary sequence of S regions, transcription, and R-loops set a platform favorable for AID activity. However, for AID to reach this platform inside the nucleus is analogous to finding a needle in a haystack. Although AID-instigated off-target breaks are incurred, the frequency is far less than what is observed for the Ig loci (80–82). The low abundance of AID at the non-Ig genes has led to the debate whether this represents true binding or mere background creeping into the chromatin immunoprecipitation-sequencing (ChIP-seq) analysis used in these experiments (83). While genome-wide occupancy studies suggested that AID associates with accessible chromatin at stalled promoters of transcribed genes (82), reanalysis of the same data set (83) contradicted the notion of genome-wide AID binding. The technicalities and subtleties of data normalization for ChIP-Seq studies seem to be at the heart of such disparate results, and do highlight the need for caution when interrogating chromatin binding of proteins with low nuclear abundance (83). Thus, while both genome-wide (82) and locus-specific ChIP (10, 81, 84) clearly show abundance of AID at the Ig loci, the efficiency of its binding to other genomic sequences needs to be re-evaluated. Nonetheless, AID-induced mutations at non-Ig genes are observed in even normal B cells (80). Thus, it is obvious that the process is stringently orchestrated to prevent bystander damage by AID. Several elements within the Ig loci have been implicated in targeting AID to the variable region exons during SHM (85); however, in this review we will primarily describe the factors that chaperone AID with exquisite precision to the S regions during CSR.
Activation-induced deaminase was shown to be in a complex with Pol II (84), and more specifically with Spt5, a Pol II-associated protein mechanistically linked to transcriptional pausing (81). Genome-wide Spt5 occupancy correlated significantly with stalled Pol II and was predictive of AID-dependent mutations. B cells depleted of Spt5 had a severe defect in CSR, a consequence of decreased AID binding to S regions (81). A comprehensive treatise on the role of RNA pol II pausing at S regions during CSR has been reviewed elsewhere (86). The germinal center-specific GANP protein has also been implicated in mediating AID–Spt5–Pol II interaction (87). However, GANP deficiency does not impair CSR. Thus, in the context of switching B cells, there might be other unidentified players that facilitate AID targeting to stalled Pol II, and recent studies have shown that members of the Pol II-associated factor 1 (PAF1) complex and histone chaperone FACT complex can promote immune diversification by regulating association of AID with Pol II (88).
The 14-3-3 adaptor proteins have been implicated in recruiting AID to DNA through their ability to interact with RGYW sequences (89). It is unclear and somewhat counterintuitive as to what happens to 14-3-3 proteins after they chaperone AID to DNA, and why they do not compete directly with AID for DNA binding. Additionally, recent data suggests that 14-3-3 proteins perform scaffolding function by directly interacting with uracil DNA glycosylase (UNG) and protein kinase A (PKA), two proteins with well-established functions in CSR (90–92). Besides, the data implicate an AID C-terminus-dependent complex formation with 14-3-3 and subsequent targeting, but they fail to reconcile how an AID Δ189–198 mutant that is impaired in 14-3-3 binding, can be targeted to the S regions and generate mutations (93). Future work is warranted to unequivocally establish the role of the 14-3-3 adaptors in AID targeting.
Polypyrimidine-tract-binding protein-2 (PTBP2) was identified as an AID interactor that regulates AID targeting to S regions (94). Originally known to be a splicing regulator in brain (neuronal isoform, nPTB), this protein also interacted with both the sense and anti-sense S region transcripts in primary B cells undergoing CSR. Since splicing might be important for CSR (71), it is tempting to speculate a splicing regulation-associated function of PTBP2 in AID recruitment to S regions. Molecular insights into PTBP2-dependent regulation of AID targeting, and the fate of nuclear AID in the absence of PTBP2 will surely constitute the next phase of investigation.
Role of Chromatin Modifications in Targeting AID
It is becoming increasingly clear that epigenetic marks play crucial roles in mediating S region accessibility (95, 96). Both donor and acceptor S regions are specifically enriched for acetylation and methylation marks at histones H3 and H4, generally associated with “open” chromatin, for example, H3K9/K14ac, H3K27ac, H4K8ac, and H3K4me3. It has been suggested that AID targeting to Sμ is facilitated by the H3K9me3 mark, which tethers AID to the donor S region via the HP1–KAP1 complex (97). Additionally, PTIP, a component of the mixed-lineage leukemia-like complexes that are important regulators of H3K4 methylation, participates in CSR by regulating transcription-coupled chromatin accessibility. PTIP-deficient B cells have a severe defect in CSR due to decreased germ-line transcription of downstream CH exons, and compromised DNA repair (98). Finally, combinatorial H3K9ac and H3S10 phosphorylation (H3K9acS10ph), specifically in the recombining S regions, deposited by GCN5/PCAF in stimulated B cells, leads to 14-3-3 adaptor-dependent AID binding to permit efficient CSR (96). However, it is to be noted that these chromatin marks are not likely to be unique to S regions, and thus cannot be sole determinants of regions permissive to AID activity. Interestingly, it has been shown that R-loops are tightly linked to H3S10ph, a chromatin condensation signature (99). Thus, it can be posited that R-loop formation facilitates H3S10ph chromatin modification, which in combination with H3K9me3 and H3K9ac marks, permits AID-mediated in vivo deamination of S region targets. The precise interplay of chromatin “writers,” “erasers,” and “readers” that regulate these events warrants further investigation, but it is unambiguous that this complex recombination reaction must be impeccably tuned by such epigenetic controls to prevent collateral damage by AID.
Generation of Double-Strand Breaks Downstream of DNA Deamination
All the regulatory mechanisms alluded to above serve to generate AID-instigated dU lesions in S regions. Since CSR proceeds through DSB intermediates, the deaminated S regions need to be processed into DNA nicks, with two closely opposed nicks constituting a DSB (2, 3). This is achieved by components of the BER and MMR pathways.
Role of BER and MMR Pathways
According to the prevailing model for CSR, UNG, a component of the BER pathway, removes the uracil base from deaminated S regions. The abasic site thus generated is converted into a nick by the apurinic/apyrimidinic endonuclease APE1. Two closely spaced nicks on opposite strands constitute a staggered DSB, further processing of which by nucleases or DNA polymerases (fill-in) generates a blunt DSB that can participate in end-joining (4, 5, 100). Consistent with this model, mutations in UNG lead to a severe defect in CSR, likely as a consequence of impaired formation of DSBs in S regions [reviewed in Ref. (2, 3)]. Additionally, APE1± mice and APE1-deficient CH12 cells reflected decreased DSBs in S regions and compromised CSR (101, 102). Components of MMR pathway have also been demonstrated to process DNA during CSR through the ability of Msh2:Msh6 to bind dU:dG mismatches, and subsequently recruit exonuclease 1 (exo1) to potentially process nicks and ssDNA gaps into DSBs. Indeed, mutations in Msh2 and Exo1 alter S region junctions and significantly impair CSR [reviewed in Ref. (2, 3)]. Conversely, deficiency of Pms2 and Mlh1, other members of the MMR machinery, lead to increased microhomology at S region junctions, suggesting that they might act to suppress alternative end-joining (103, 104). However, UNG mutations have a more profound effect on CSR than mutations in MMR proteins, suggesting that CSR is more reliant on the UNG-dependent steps. Whether this reflects an uncharacterized preference for one pathway over the other or supports a proposed non-canonical role for UNG, independent of uracil removal activity during CSR, remains an open question (105).
Generation of High Density of DSBs: Requirement of AID Phosphorylation at Serine-38
The cellular DNA end-joining machinery is highly efficient and it is conceivable that a single DSB at an S region will be repaired before it can synapse with and ligate to a downstream DSB (106). It has therefore been speculated that efficient CSR would require a high density of DSBs at S regions to promote productive long distance synapsis and recombination between acceptor and donor S regions over intra-switch re-ligation (92), a phenomenon commonly observed in B cells that have initiated CSR but failed to complete the process (2). Recent studies have suggested that AID phosphorylated at serine-38 (S38) by PKA interacts with APE1 to actively generate a high density of breaks, a likely prerequisite for CSR (92, 107). In keeping with this notion, mutation of S38 to alanine severely impairs CSR due to a failure to efficiently generate DSBs at S regions (54, 56–58, 92, 107, 108).
Strikingly, AID phosphorylation at S38 was stimulated by DSBs (107). Thus, AID phosphorylation at S38 is both required for, and dependent on DSBs. This suggests the existence of a positive feedback loop wherein a low density of DSBs leads to AID phosphorylation, APE1 binding, and amplification of DSBs that feedback into the loop (Figure 6). It was also demonstrated that ATM, a protein critical for cellular response to DNA-damage, participates in sensing the DSBs at S regions, thereby promoting AID phosphorylation and APE1 interaction (107). Being a master regulator of the DNA-damage response, it is possible to envision ATM as a molecular rheostat that fine tunes DSB formation with efficient repair/recombination and allows safeguarded CSR while minimizing translocations. This is reminiscent of the role of RAG proteins in orchestrating V(D)J recombination by generating DSBs and efficiently channeling them to productive recombination, keeping translocation risks at bay (109–111). Similar to RAG-dependent coordination of break induction and repair, AID phosphorylated at S38 not only facilitates break formation, but also interacts with the ssDNA-binding protein, replication protein A, and likely enforces DNA repair pathways during CSR (112, 113).
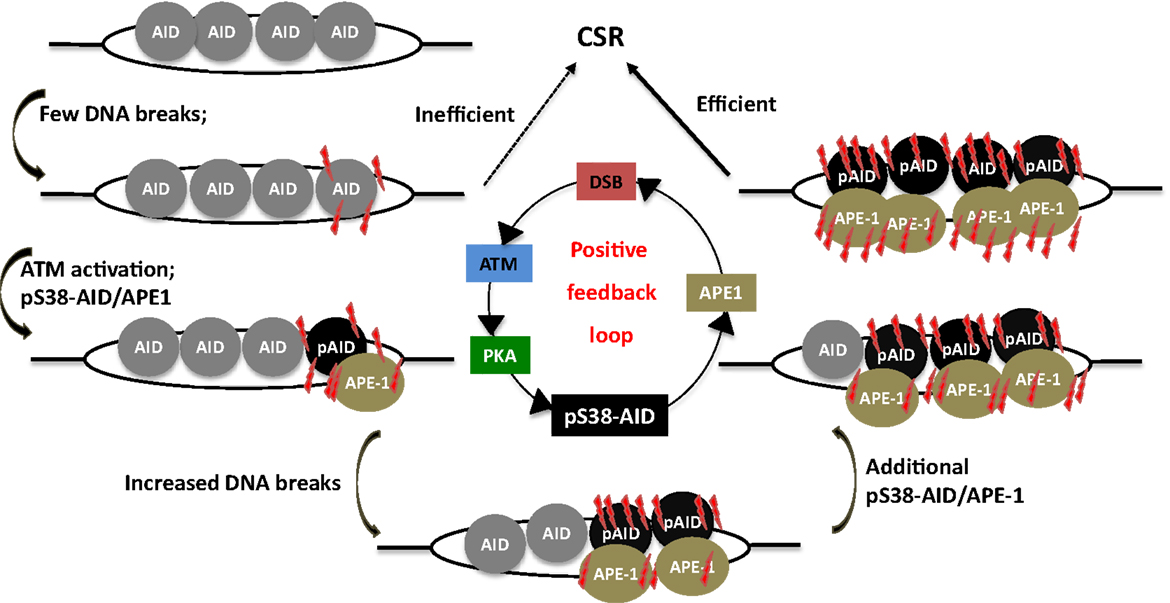
Figure 6. A phosphorylation-dependent positive feedback loop drives CSR. Assembly of AID at S regions induces low-density DSBs and leads to inefficient CSR, but ATM activation. This results in PKA-dependent AID phosphorylation at serine-38, which promotes interaction of AID with APE1. Active recruitment of APE1 to DNA accentuates DSB formation, which in turn induces phosphorylation of additional molecules of AID, thereby perpetuating the DSB amplification loop. This promotes the generation of high density of DSBs at S regions that is required for CSR.
Phosphorylation at S38 actively integrates AID functions into steps downstream of DNA deamination; however, several key questions remain elusive. First, the factors that facilitate APE1 binding to pS38AID need to be identified. Second, the regulatory mechanisms that couple chromatin sensing to DNA-damage signaling remain a mystery. Based on the recent finding of KAT5 (TIP60) tyrosine phosphorylation by DNA-damage to facilitate H3K9me3 binding and subsequent acetylation of ATM (114), it can be conjectured that such a pathway might be involved in the context of S regions and CSR, where the H3K9me3 mark has been shown to play a vital role (97). Finally, the steps between ATM activation and PKA-dependent AID phosphorylation remain a black box. These questions remain an active area of investigation.
Completion of CSR: End-Joining of Switch-Region DSBs
Double-strand breaks generated at two distinct S regions are synapsed and ligated by end-joining during the completion phase of CSR (115). Below, we discuss the DSB response and DNA end-joining pathways that participate in this process.
DSB Response during CSR
During the general DNA-damage response, DSBs are rapidly recognized by the Mre11–Rad50–Nbs1 (MRN) complex (116) (Figure 7). Nbs1 recruits and activates ATM, which phosphorylates H2AX. Phosphorylated H2AX (γH2AX) serves as a docking site for several DNA response proteins and promotes the rapid accumulation of 53BP1, Nbs1, and MDC1 into repair foci near DSBs (116). Deletion or mutation of Nbs1, H2AX, 53BP1, and ATM impaired CSR, indicating that the proteins that participate in sensing and transducing DSBs participate in CSR (116). Additionally, the ATM-dependent DNA-damage response is required for maintenance of genomic integrity and suppression of oncogenic translocations, possibly through enforcing cell-cycle checkpoints (116). Overall, ATM promotes the assembly of macromolecular foci that stabilize DNA ends and facilitate the recruitment of repair factors to ensure productive CSR while preventing oncogenic translocations.
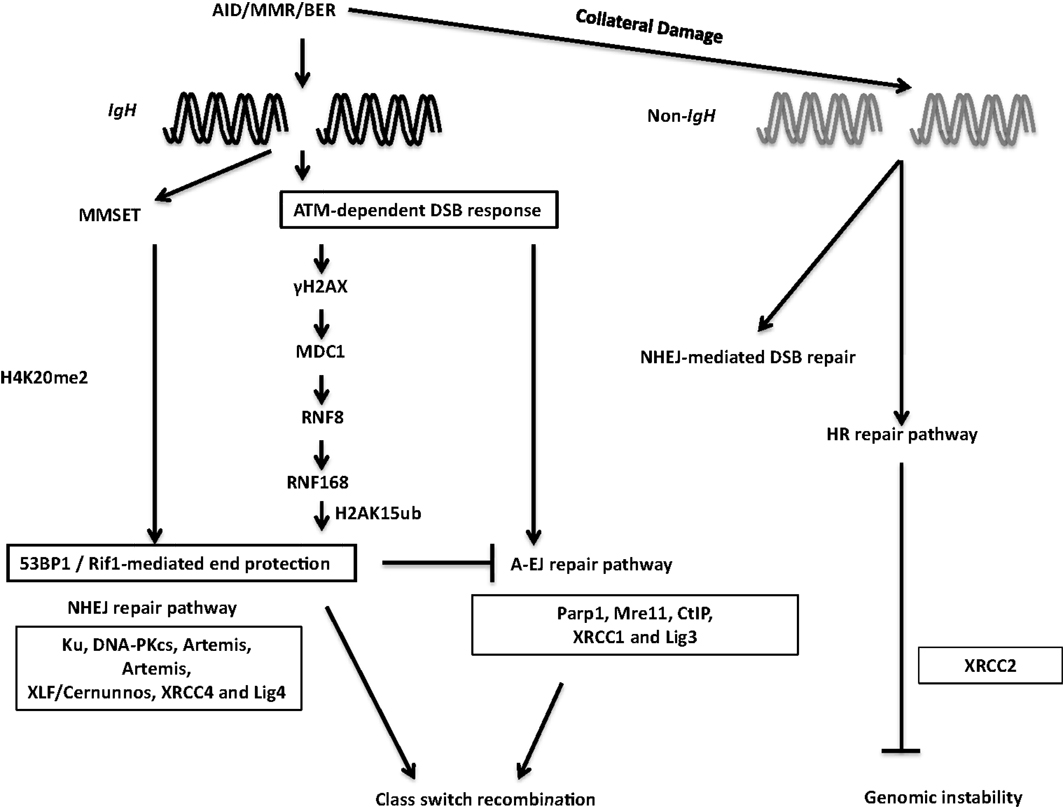
Figure 7. DSB response during CSR. The combination of AID, BER, and MMR proteins generate DSBs physiologically at the IgH locus and aberrantly at non-Ig regions, leading to activation of the cellular DNA-damage response pathways. 53BP1 is recruited to DSBs by directly binding to H4K20me2 or through the γH2AX–RNF8–RNF168 node (see text for details). 53BP1-dependent end protection facilitates ligation of two S regions by NHEJ and/or A-EJ. Collateral damage induced by AID at non-Ig genes is repaired by NHEJ and by XRCC2-dependent homologous recombination.
Among the ATM-activated DSB response factors, 53BP1 deficiency leads to the most pronounced defect in CSR (116). CSR requires “synapsis” or close juxtaposition of donor and acceptor S regions (115, 117) and 53BP1 has been proposed to promote the synapsis of broken S regions during CSR (118). Furthermore, 53BP1 has been shown to associate with Rap1-interacting factor 1 (Rif1) to protect broken DNA ends from resection. Absence of Rif1 in B cells leads to increased DNA end resection, virtually phenocopying 53BP1 deficiency and providing functional significance to the 53BP1–Rif1 interaction during CSR (119–121). A recent study elegantly teased apart differential roles of distinct phosphoprotein interactions of 53BP1, and convincingly illustrated that Rif1 serves as an effector of productive repair, whereas PTIP wards against mutagenic repair. This study clearly demonstrated that 53BP1 is a key player at the crossroads of efficient/aberrant DNA repair pathway choice (122).
The chromatin microenvironment strongly influences the DSB response. Two mechanisms have been proposed to regulate 53BP1 recruitment to DSBs in the context of chromatin. The first relies on its interaction with H4K20me2. Methylation of H4K20 and subsequent 53BP1 recruitment to sites of DNA-damage is regulated by MMSET, a histone methyltransferase (123, 124). MMSET depletion in the CH12F3 B cell line decreases H4K20me2 levels, attenuates 53BP1 accumulation at S regions, and impairs CSR (123). The other process that recruits 53BP1 to DSBs requires the RING finger protein 8 and 168 (RNF8 and RNF168)-dependent histone ubiquitination pathway. RNF8 is recruited to ATM-phosphorylated MDC1 bound to γH2AX at the site of DSBs and catalyzes ubiquitin-dependent recruitment of RNF168 to chromatin flanking the DSBs (125). Recently, 53BP1 has been shown to recognize DNA-damage-induced H2A Lys15 ubiquitination catalyzed by RNF168, revealing the mechanism of RNF8/168-dependent recruitment of 53BP1 at DSBs. RNF8 deficiency compromised recruitment of 53BP1 to S regions in activated B cells and significantly impaired CSR. Additionally, inactivation of RNF168 impaired CSR in mice (126–129). Taken together, these observations suggest that 53BP1 recruitment plays a critical DSB end-protecting role during CSR.
DNA End-Joining
Non-homologous end-joining (NHEJ) is the primary mechanism used for end-joining during CSR (2). The canonical NHEJ machinery includes the Ku70/Ku80 heterodimer (Ku), DNA-dependent protein kinase catalytic subunit (DNA-PKcs), Artemis, XRCC4-like factor (XLF or Cernunnos), XRCC4, and DNA ligase IV (Lig4). Mutations in NHEJ components including Ku70/80, XRCC4, and DNA ligase IV severely compromise CSR. The role of NHEJ proteins in CSR is also evident from the observations that mutations in DNA-PKcs, artemis, and XLF lead to high levels of chromosomal IgH breaks and translocations, even in instances where CSR frequency is not severely impacted (116). Our current knowledge however, does not uncover how the initial recognition of DSBs by the MRN complex leads to the binding of Ku and DNA-PKcs to the broken DNA.
Non-homologous end-joining-deficiency does not abolish CSR, and S junctions in NHEJ-deficient B cells reveal extended microhomology, leading to the proposal that an alternative end-joining process (A-EJ) ligates DSBs during CSR (116). No factors unique to A-EJ have yet been characterized; several proteins involved in other DNA repair pathways have been implicated to function in A-EJ, including Mre11 and CtIP. Mre11 and CtIP have been implicated to trim broken DNA ends to uncover microhomology regions, generating short stretches of complementary nucleotides at DNA breaks, thereby promoting A-EJ during CSR (116). In CH12F3 cells, CtIP depletion impaired CSR to IgA and reduced the overall length of microhomology at the S junctions (130, 131). Notably, CtIP-deficient B cells undergo normal CSR to IgG1 (132). Therefore, elucidation of the role of CtIP in CSR requires further investigation. A major open question relates to the interplay between NHEJ and A-EJ: does A-EJ operate in presence of intact NHEJ and does it have a physiological role other than being a mere backup to NHEJ?
Role of Homology-Dependent Repair in Resolving AID-Induced Breaks during CSR
It has been reported that AID instigates formation of widespread DSBs throughout the genome in activated B cells, albeit at significantly lower levels than that at the IgH locus (133–136). Such off-target DSBs at non-IgH loci are the major underlying lesions contributing to translocations between IgH and non-IgH loci (such as c-Myc) in B cells and are largely responsible for the ontogeny of a large number of B cell lymphomas in humans (21). In addition to aberrant translocations, AID can also induce somatic mutations at numerous loci linked to B cell tumorigenesis (80). While AID-initiated DSBs are observed in the G1 phase of the cell cycle (137), and CSR is likely completed before the cells transit into the S phase, it has been suggested that homologous recombination (HR)-dependent repair has a major role in providing resistance to AID-induced off-target DNA-damage. This is based on the observation that B cells deficient in the HR protein XRCC2 have significantly enhanced AID-dependent genome-wide DSBs (138) (Figure 7). Notably, the interplay between AID-mediated DNA breaks and HR repair pathway has been used in clinically relevant studies wherein AID-expressing human chronic lymphocytic leukemia cells were shown to be hypersensitive to HR inhibitors, possibly due to AID-dependent synthetic cytotoxicity (139). Further studies, in clinical settings, should be an interesting and possibly efficacious way to turn the mutator activity of AID into a therapy for B cell malignancies.
Perspective
The discovery of AID was a watershed event in the field of B cell biology and in deciphering the underlying cause behind the ontogeny of a large number of B cell lymphomas. We now have a working model of how non-coding transcription and DNA deamination initiate CSR and how general DNA repair proteins that function in distinct pathways contribute to the process. Still, a large number of unknowns plague the field. These include the mechanisms that specifically recruit AID to the Ig loci, leaving the rest of the genome largely untouched. We are yet to understand the processes that subvert normal DNA repair machineries and instead wield components of these pathways to promote recombination of DSBs that could be over 100 kb apart. The molecular basis underlying the balance between normal and aberrant repair requires further elucidation. Such basic knowledge can be exploited to shift the fulcrum of repair judiciously in clinical settings of patients with immunodeficiency or lymphoid malignancies to reap translational benefit. Finally, DSB response occurs in, and is strongly influenced by the chromatin microenvironment. The dynamics of chromatin compaction and relaxation at DSBs are just beginning to unravel (140), but clearly much more remains to be unearthed as to how the dynamics of histone and DNA modifications impact and regulate a programed DSB response that ensues during AID-orchestrated CSR. Addressing these exciting issues will be at the forefront of research in the coming years.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank members of the Chaudhuri lab for helpful comments and discussion. This work was supported by grants from the National Institutes of Health (1RO1AI072194) and the Starr Cancer Research Foundation to Jayanta Chaudhuri.
References
1. Alt FW, Zhang Y, Meng FL, Guo C, Schwer B. Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell (2013) 152(3):417–29. doi:10.1016/j.cell.2013.01.007
2. Chaudhuri J, Basu U, Zarrin A, Yan C, Franco S, Perlot T, et al. Evolution of the immunoglobulin heavy chain class switch recombination mechanism. Adv Immunol (2007) 94:157–214. doi:10.1016/S0065-2776(06)94006-1
3. Stavnezer J, Guikema JE, Schrader CE. Mechanism and regulation of class switch recombination. Annu Rev Immunol (2008) 26:261–92. doi:10.1146/annurev.immunol.26.021607.090248
4. Maul RW, Saribasak H, Martomo SA, McClure RL, Yang W, Vaisman A, et al. Uracil residues dependent on the deaminase AID in immunoglobulin gene variable and switch regions. Nat Immunol (2011) 12(1):70–6. doi:10.1038/ni.1970
5. Petersen-Mahrt SK, Harris RS, Neuberger MS. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature (2002) 418(6893):99–103. doi:10.1038/nature00862
6. Rada C, Di Noia JM, Neuberger MS. Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Mol Cell (2004) 16(2):163–71. doi:10.1016/j.molcel.2004.10.011
7. Di Noia JM, Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Annu Rev Biochem (2007) 76:1–22. doi:10.1146/annurev.biochem.76.061705.090740
8. Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell (2000) 102(5):553–63. doi:10.1016/S0092-8674(00)00078-7
9. Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell (2000) 102(5):565–75. doi:10.1016/S0092-8674(00)00079-9
10. Chaudhuri J, Tian M, Khuong C, Chua K, Pinaud E, Alt FW. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature (2003) 422(6933):726–30. doi:10.1038/nature01574
11. Ramiro AR, Stavropoulos P, Jankovic M, Nussenzweig MC. Transcription enhances AID-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nat Immunol (2003) 4(5):452–6. doi:10.1038/ni920
12. Kohli RM, Abrams SR, Gajula KS, Maul RW, Gearhart PJ, Stivers JT. A portable hot spot recognition loop transfers sequence preferences from APOBEC family members to activation-induced cytidine deaminase. J Biol Chem (2009) 284(34):22898–904. doi:10.1074/jbc.M109.025536
13. Vasudevan AA, Smits SH, Hoppner A, Haussinger D, Koenig BW, Munk C. Structural features of antiviral DNA cytidine deaminases. Biol Chem (2013) 394(11):1357–70. doi:10.1515/hsz-2013-0165
14. Pham P, Bransteitter R, Petruska J, Goodman MF. Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature (2003) 424(6944):103–7. doi:10.1038/nature01760
15. Coker HA, Petersen-Mahrt SK. The nuclear DNA deaminase AID functions distributively whereas cytoplasmic APOBEC3G has a processive mode of action. DNA Repair (Amst) (2007) 6(2):235–43. doi:10.1016/j.dnarep.2006.10.001
16. Mak CH, Pham P, Afif SA, Goodman MF. A mathematical model for scanning and catalysis on single-stranded DNA, illustrated with activation-induced deoxycytidine deaminase. J Biol Chem (2013) 288(41):29786–95. doi:10.1074/jbc.M113.506550
17. Dickerson SK, Market E, Besmer E, Papavasiliou FN. AID mediates hypermutation by deaminating single stranded DNA. J Exp Med (2003) 197(10):1291–6. doi:10.1084/jem.20030481
18. Kobayashi M, Aida M, Nagaoka H, Begum NA, Kitawaki Y, Nakata M, et al. AID-induced decrease in topoisomerase 1 induces DNA structural alteration and DNA cleavage for class switch recombination. Proc Natl Acad Sci U S A (2009) 106(52):22375–80. doi:10.1073/pnas.0911879106
19. Martin A, Scharff MD. Somatic hypermutation of the AID transgene in B and non-B cells. Proc Natl Acad Sci U S A (2002) 99(19):12304–8. doi:10.1073/pnas.192442899
20. Yoshikawa K, Okazaki IM, Eto T, Kinoshita K, Muramatsu M, Nagaoka H, et al. AID enzyme-induced hypermutation in an actively transcribed gene in fibroblasts. Science (2002) 296(5575):2033–6. doi:10.1126/science.1071556
21. Nussenzweig A, Nussenzweig MC. Origin of chromosomal translocations in lymphoid cancer. Cell (2010) 141(1):27–38. doi:10.1016/j.cell.2010.03.016
22. Lee-Theilen M, Chaudhuri J. Walking the AID tightrope. Nat Immunol (2010) 11(2):107–9. doi:10.1038/ni0210-107
23. Mai T, Zan H, Zhang J, Hawkins JS, Xu Z, Casali P. Estrogen receptors bind to and activate the HOXC4/HoxC4 promoter to potentiate HoxC4-mediated activation-induced cytosine deaminase induction, immunoglobulin class switch DNA recombination, and somatic hypermutation. J Biol Chem (2010) 285(48):37797–810. doi:10.1074/jbc.M110.169086
24. Pauklin S, Petersen-Mahrt SK. Progesterone inhibits activation-induced deaminase by binding to the promoter. J Immunol (2009) 183(2):1238–44. doi:10.4049/jimmunol.0803915
25. Pauklin S, Sernandez IV, Bachmann G, Ramiro AR, Petersen-Mahrt SK. Estrogen directly activates AID transcription and function. J Exp Med (2009) 206(1):99–111. doi:10.1084/jem.20080521
26. Huong le T, Kobayashi M, Nakata M, Shioi G, Miyachi H, Honjo T, et al. In vivo analysis of Aicda gene regulation: a critical balance between upstream enhancers and intronic silencers governs appropriate expression. PLoS One (2013) 8(4):e61433. doi:10.1371/journal.pone.0061433
27. Ise W, Kohyama M, Schraml BU, Zhang T, Schwer B, Basu U, et al. The transcription factor BATF controls the global regulators of class-switch recombination in both B cells and T cells. Nat Immunol (2011) 12(6):536–43. doi:10.1038/ni.2037
28. Betz BC, Jordan-Williams KL, Wang C, Kang SG, Liao J, Logan MR, et al. Batf coordinates multiple aspects of B and T cell function required for normal antibody responses. J Exp Med (2010) 207(5):933–42. doi:10.1084/jem.20091548
29. Dominguez-Sola D, Victora GD, Ying CY, Phan RT, Saito M, Nussenzweig MC, et al. The proto-oncogene MYC is required for selection in the germinal center and cyclic reentry. Nat Immunol (2012) 13(11):1083–91. doi:10.1038/ni.2428
30. Fernandez D, Ortiz M, Rodriguez L, Garcia A, Martinez D, Moreno de Alboran I. The proto-oncogene c-myc regulates antibody secretion and Ig class switch recombination. J Immunol (2013) 190(12):6135–44. doi:10.4049/jimmunol.1300712
31. Han JH, Akira S, Calame K, Beutler B, Selsing E, Imanishi-Kari T. Class switch recombination and somatic hypermutation in early mouse B cells are mediated by B cell and Toll-like receptors. Immunity (2007) 27(1):64–75. doi:10.1016/j.immuni.2007.05.018
32. Kumar S, Wuerffel R, Achour I, Lajoie B, Sen R, Dekker J, et al. Flexible ordering of antibody class switch and V(D)J joining during B-cell ontogeny. Genes Dev (2013) 27(22):2439–44. doi:10.1101/gad.227165.113
33. Endo Y, Marusawa H, Kinoshita K, Morisawa T, Sakurai T, Okazaki IM, et al. Expression of activation-induced cytidine deaminase in human hepatocytes via NF-kappaB signaling. Oncogene (2007) 26(38):5587–95. doi:10.1038/sj.onc.1210344
34. Matsumoto Y, Marusawa H, Kinoshita K, Endo Y, Kou T, Morisawa T, et al. Helicobacter pylori infection triggers aberrant expression of activation-induced cytidine deaminase in gastric epithelium. Nat Med (2007) 13(4):470–6. doi:10.1038/nm1566
35. Lin C, Yang L, Tanasa B, Hutt K, Ju BG, Ohgi K, et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell (2009) 139(6):1069–83. doi:10.1016/j.cell.2009.11.030
36. Rommel PC, Bosque D, Gitlin AD, Croft GF, Heintz N, Casellas R, et al. Fate mapping for activation-induced cytidine deaminase (AID) marks non-lymphoid cells during mouse development. PLoS One (2013) 8(7):e69208. doi:10.1371/journal.pone.0069208
37. Kumar R, DiMenna L, Schrode N, Liu T-C, Franck P, Munoz-Descalzo S, et al. AID stabilizes stem-cell phenotype by removing epigenetic memory of pluripotency genes. Nature (2013) 500(7460):89–92. doi:10.1038/nature12299
38. Morgan HD, Dean W, Coker HA, Reik W, Petersen-Mahrt SK. Activation-induced cytidine deaminase deaminates 5-methylcytosine in DNA and is expressed in pluripotent tissues: implications for epigenetic reprogramming. J Biol Chem (2004) 279(50):52353–60. doi:10.1074/jbc.M407695200
39. Bhutani N, Brady JJ, Damian M, Sacco A, Corbel SY, Blau HM. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature (2010) 463(7284):1042–7. doi:10.1038/nature08752
40. Popp C, Dean W, Feng S, Cokus SJ, Andrews S, Pellegrini M, et al. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature (2010) 463(7284):1101–5. doi:10.1038/nature08829
41. Nabel CS, Jia H, Ye Y, Shen L, Goldschmidt HL, Stivers JT, et al. AID/APOBEC deaminases disfavor modified cytosines implicated in DNA demethylation. Nat Chem Biol (2012) 8(9):751–8. doi:10.1038/nchembio.1042
42. Fritz EL, Rosenberg BR, Lay K, Mihailovic A, Tuschl T, Papavasiliou FN. A comprehensive analysis of the effects of the deaminase AID on the transcriptome and methylome of activated B cells. Nat Immunol (2013) 14(7):749–55. doi:10.1038/ni.2616
43. Dorsett Y, McBride KM, Jankovic M, Gazumyan A, Thai TH, Robbiani DF, et al. MicroRNA-155 suppresses activation-induced cytidine deaminase-mediated Myc-Igh translocation. Immunity (2008) 28(5):630–8. doi:10.1016/j.immuni.2008.04.002
44. Teng G, Hakimpour P, Landgraf P, Rice A, Tuschl T, Casellas R, et al. MicroRNA-155 is a negative regulator of activation-induced cytidine deaminase. Immunity (2008) 28(5):621–9. doi:10.1016/j.immuni.2008.03.015
45. Vigorito E, Perks KL, Abreu-Goodger C, Bunting S, Xiang Z, Kohlhaas S, et al. microRNA-155 regulates the generation of immunoglobulin class-switched plasma cells. Immunity (2007) 27(6):847–59. doi:10.1016/j.immuni.2007.10.009
46. Basso K, Schneider C, Shen Q, Holmes AB, Setty M, Leslie C, et al. BCL6 positively regulates AID and germinal center gene expression via repression of miR-155. J Exp Med (2012) 209(13):2455–65. doi:10.1084/jem.20121387
47. de Yebenes VG, Belver L, Pisano DG, Gonzalez S, Villasante A, Croce C, et al. miR-181b negatively regulates activation-induced cytidine deaminase in B cells. J Exp Med (2008) 205(10):2199–206. doi:10.1084/jem.20080579
48. Loeb GB, Khan AA, Canner D, Hiatt JB, Shendure J, Darnell RB, et al. Transcriptome-wide miR-155 binding map reveals widespread noncanonical microRNA targeting. Mol Cell (2012) 48(5):760–70. doi:10.1016/j.molcel.2012.10.002
49. Orthwein A, Di Noia JM. Activation induced deaminase: how much and where? Semin Immunol (2012) 24(4):246–54. doi:10.1016/j.smim.2012.05.001
50. Hu Y, Ericsson I, Torseth K, Methot SP, Sundheim O, Liabakk NB, et al. A combined nuclear and nucleolar localization motif in activation-induced cytidine deaminase (AID) controls immunoglobulin class switching. J Mol Biol (2013) 425(2):424–43. doi:10.1016/j.jmb.2012.11.026
51. Geisberger R, Rada C, Neuberger MS. The stability of AID and its function in class-switching are critically sensitive to the identity of its nuclear-export sequence. Proc Natl Acad Sci U S A (2009) 106(16):6736–41. doi:10.1073/pnas.0810808106
52. Aoufouchi S, Faili A, Zober C, D’Orlando O, Weller S, Weill JC, et al. Proteasomal degradation restricts the nuclear lifespan of AID. J Exp Med (2008) 205(6):1357–68. doi:10.1084/jem.20070950
53. Uchimura Y, Barton LF, Rada C, Neuberger MS. REG-gamma associates with and modulates the abundance of nuclear activation-induced deaminase. J Exp Med (2011) 208(12):2385–91. doi:10.1084/jem.20110856
54. Basu U, Chaudhuri J, Alpert C, Dutt S, Ranganath S, Li G, et al. The AID antibody diversification enzyme is regulated by protein kinase A phosphorylation. Nature (2005) 438(7067):508–11. doi:10.1038/nature04255
55. Gazumyan A, Timachova K, Yuen G, Siden E, Di Virgilio M, Woo EM, et al. Amino-terminal phosphorylation of activation-induced cytidine deaminase suppresses c-myc/IgH translocation. Mol Cell Biol (2011) 31(3):442–9. doi:10.1128/MCB.00349-10
56. McBride KM, Gazumyan A, Woo EM, Barreto VM, Robbiani DF, Chait BT, et al. Regulation of hypermutation by activation-induced cytidine deaminase phosphorylation. Proc Natl Acad Sci U S A (2006) 103(23):8798–803. doi:10.1073/pnas.0603272103
57. McBride KM, Gazumyan A, Woo EM, Schwickert TA, Chait BT, Nussenzweig MC. Regulation of class switch recombination and somatic mutation by AID phosphorylation. J Exp Med (2008) 205(11):2585–94. doi:10.1084/jem.20081319
58. Pasqualucci L, Kitaura Y, Gu H, Dalla-Favera R. PKA-mediated phosphorylation regulates the function of activation-induced deaminase (AID) in B cells. Proc Natl Acad Sci U S A (2006) 103(2):395–400. doi:10.1073/pnas.0509969103
59. Rogozin IB, Diaz M. Cutting edge: DGYW/WRCH is a better predictor of mutability at G:C bases in Ig hypermutation than the widely accepted RGYW/WRCY motif and probably reflects a two-step activation-induced cytidine deaminase-triggered process. J Immunol (2004) 172(6):3382–4.
60. Zarrin AA, Alt FW, Chaudhuri J, Stokes N, Kaushal D, Du Pasquier L, et al. An evolutionarily conserved target motif for immunoglobulin class-switch recombination. Nat Immunol (2004) 5(12):1275–81. doi:10.1038/ni1137
61. Khamlichi AA, Glaudet F, Oruc Z, Denis V, Le Bert M, Cogne M. Immunoglobulin class-switch recombination in mice devoid of any S mu tandem repeat. Blood (2004) 103(10):3828–36. doi:10.1182/blood-2003-10-3470
62. Luby TM, Schrader CE, Stavnezer J, Selsing E. The mu switch region tandem repeats are important, but not required, for antibody class switch recombination. J Exp Med (2001) 193(2):159–68. doi:10.1084/jem.193.2.159
63. Shinkura R, Tian M, Smith M, Chua K, Fujiwara Y, Alt FW. The influence of transcriptional orientation on endogenous switch region function. Nat Immunol (2003) 4(5):435–41. doi:10.1038/ni918
64. Zhang T, Franklin A, Boboila C, McQuay A, Gallagher MP, Manis JP, et al. Downstream class switching leads to IgE antibody production by B lymphocytes lacking IgM switch regions. Proc Natl Acad Sci U S A (2010) 107(7):3040–5. doi:10.1073/pnas.0915072107
65. Jabara HH, Loh R, Ramesh N, Vercelli D, Geha RS. Sequential switching from mu to epsilon via gamma 4 in human B cells stimulated with IL-4 and hydrocortisone. J Immunol (1993) 151(9):4528–33.
66. Mills FC, Thyphronitis G, Finkelman FD, Max EE. Ig mu-epsilon isotype switch in IL-4-treated human B lymphoblastoid cells. Evidence for a sequential switch. J Immunol (1992) 149(3):1075–85.
67. Yoshida K, Matsuoka M, Usuda S, Mori A, Ishizaka K, Sakano H. Immunoglobulin switch circular DNA in the mouse infected with Nippostrongylus brasiliensis: evidence for successive class switching from mu to epsilon via gamma 1. Proc Natl Acad Sci U S A (1990) 87(20):7829–33. doi:10.1073/pnas.87.20.7829
68. Kunimoto DY, Sneller MC, Claflin L, Mushinski JF, Strober W. Molecular analysis of double isotype expression in IgA switching. J Immunol (1993) 150(4):1338–47.
69. Tian M, Alt FW. Transcription-induced cleavage of immunoglobulin switch regions by nucleotide excision repair nucleases in vitro. J Biol Chem (2000) 275(31):24163–72. doi:10.1074/jbc.M003343200
70. Yu K, Chedin F, Hsieh CL, Wilson TE, Lieber MR. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat Immunol (2003) 4(5):442–51. doi:10.1038/ni919
71. Lorenz M, Jung S, Radbruch A. Switch transcripts in immunoglobulin class switching. Science (1995) 267(5205):1825–8. doi:10.1126/science.7892607
72. Hein K, Lorenz MG, Siebenkotten G, Petry K, Christine R, Radbruch A. Processing of switch transcripts is required for targeting of antibody class switch recombination. J Exp Med (1998) 188(12):2369–74. doi:10.1084/jem.188.12.2369
73. Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science (2008) 322(5902):750–6. doi:10.1126/science.1163045
74. Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature (2010) 464(7291):1071–6. doi:10.1038/nature08975
75. Perlot T, Li G, Alt FW. Antisense transcripts from immunoglobulin heavy-chain locus V(D)J and switch regions. Proc Natl Acad Sci U S A (2008) 105(10):3843–8. doi:10.1073/pnas.0712291105
76. Haddad D, Dougier HL, Laviolette N, Puget N, Khamlichi AA. Replacement of Imu-Cmu intron by NeoR gene alters Imu germ-line expression but has no effect on V(D)J recombination. Mol Immunol (2010) 47(5):961–71. doi:10.1016/j.molimm.2009.11.024
77. Basu U, Meng FL, Keim C, Grinstein V, Pefanis E, Eccleston J, et al. The RNA exosome targets the AID cytidine deaminase to both strands of transcribed duplex DNA substrates. Cell (2011) 144(3):353–63. doi:10.1016/j.cell.2011.01.001
78. Sun J, Keim CD, Wang J, Kazadi D, Oliver PM, Rabadan R, et al. E3-ubiquitin ligase Nedd4 determines the fate of AID-associated RNA polymerase II in B cells. Genes Dev (2013) 27(16):1821–33. doi:10.1101/gad.210211.112
79. Yu K, Lieber MR. Nucleic acid structures and enzymes in the immunoglobulin class switch recombination mechanism. DNA Repair (Amst) (2003) 2(11):1163–74. doi:10.1016/j.dnarep.2003.08.010
80. Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, et al. Two levels of protection for the B cell genome during somatic hypermutation. Nature (2008) 451(7180):841–5. doi:10.1038/nature06547
81. Pavri R, Gazumyan A, Jankovic M, Di Virgilio M, Klein I, Ansarah-Sobrinho C, et al. Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell (2010) 143(1):122–33. doi:10.1016/j.cell.2010.09.017
82. Yamane A, Resch W, Kuo N, Kuchen S, Li Z, Sun HW, et al. Deep-sequencing identification of the genomic targets of the cytidine deaminase AID and its cofactor RPA in B lymphocytes. Nat Immunol (2011) 12(1):62–9. doi:10.1038/ni.1964
83. Hogenbirk MA, Velds A, Kerkhoven RM, Jacobs H. Reassessing genomic targeting of AID. Nat Immunol (2012) 13(9):797–8; author reply 798–800. doi:10.1038/ni.2367
84. Nambu Y, Sugai M, Gonda H, Lee CG, Katakai T, Agata Y, et al. Transcription-coupled events associating with immunoglobulin switch region chromatin. Science (2003) 302(5653):2137–40. doi:10.1126/science.1092481
85. Kothapalli NR, Fugmann SD. Targeting of AID-mediated sequence diversification to immunoglobulin genes. Curr Opin Immunol (2011) 23(2):184–9. doi:10.1016/j.coi.2010.12.009
86. Kenter AL. AID targeting is dependent on RNA polymerase II pausing. Semin Immunol (2012) 24(4):281–6. doi:10.1016/j.smim.2012.06.001
87. Singh SK, Maeda K, Eid MM, Almofty SA, Ono M, Pham P, et al. GANP regulates recruitment of AID to immunoglobulin variable regions by modulating transcription and nucleosome occupancy. Nat Commun (2013) 4:1830. doi:10.1038/ncomms2823
88. Willmann KL, Milosevic S, Pauklin S, Schmitz KM, Rangam G, Simon MT, et al. A role for the RNA pol II-associated PAF complex in AID-induced immune diversification. J Exp Med (2012) 209(11):2099–111. doi:10.1084/jem.20112145
89. Xu Z, Fulop Z, Wu G, Pone EJ, Zhang J, Mai T, et al. 14-3-3 adaptor proteins recruit AID to 5′-AGCT-3′-rich switch regions for class switch recombination. Nat Struct Mol Biol (2010) 17(9):1124–35. doi:10.1038/nsmb.1884
90. Lam T, Thomas LM, White CA, Li G, Pone EJ, Xu Z, et al. Scaffold functions of 14-3-3 adaptors in B cell immunoglobulin class switch DNA recombination. PLoS One (2013) 8(11):e80414. doi:10.1371/journal.pone.0080414
91. Rada C, Williams GT, Nilsen H, Barnes DE, Lindahl T, Neuberger MS. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr Biol (2002) 12(20):1748–55. doi:10.1016/S0960-9822(02)01215-0
92. Vuong BQ, Lee M, Kabir S, Irimia C, Macchiarulo S, McKnight GS, et al. Specific recruitment of protein kinase A to the immunoglobulin locus regulates class-switch recombination. Nat Immunol (2009) 10(4):420–6. doi:10.1038/ni.1708
93. Barreto V, Reina-San-Martin B, Ramiro AR, McBride KM, Nussenzweig MC. C-terminal deletion of AID uncouples class switch recombination from somatic hypermutation and gene conversion. Mol Cell (2003) 12(2):501–8. doi:10.1016/S1097-2765(03)00309-5
94. Nowak U, Matthews AJ, Zheng S, Chaudhuri J. The splicing regulator PTBP2 interacts with the cytidine deaminase AID and promotes binding of AID to switch-region DNA. Nat Immunol (2011) 12(2):160–6. doi:10.1038/ni.1977
95. Wang L, Wuerffel R, Feldman S, Khamlichi AA, Kenter AL. S region sequence, RNA polymerase II, and histone modifications create chromatin accessibility during class switch recombination. J Exp Med (2009) 206(8):1817–30. doi:10.1084/jem.20081678
96. Li G, White CA, Lam T, Pone EJ, Tran DC, Hayama KL, et al. Combinatorial H3K9acS10ph histone modification in IgH locus S regions targets 14-3-3 adaptors and AID to specify antibody class-switch DNA recombination. Cell Rep (2013) 5(3):702–14. doi:10.1016/j.celrep.2013.09.031
97. Jeevan-Raj BP, Robert I, Heyer V, Page A, Wang JH, Cammas F, et al. Epigenetic tethering of AID to the donor switch region during immunoglobulin class switch recombination. J Exp Med (2011) 208(8):1649–60. doi:10.1084/jem.20110118
98. Daniel JA, Santos MA, Wang Z, Zang C, Schwab KR, Jankovic M, et al. PTIP promotes chromatin changes critical for immunoglobulin class switch recombination. Science (2010) 329(5994):917–23. doi:10.1126/science.1187942
99. Castellano-Pozo M, Santos-Pereira JM, Rondon AG, Barroso S, Andujar E, Perez-Alegre M, et al. R loops are linked to histone h3 s10 phosphorylation and chromatin condensation. Mol Cell (2013) 52(4):583–90. doi:10.1016/j.molcel.2013.10.006
100. Chaudhuri J, Alt FW. Class-switch recombination: interplay of transcription, DNA deamination and DNA repair. Nat Rev Immunol (2004) 4(7):541–52. doi:10.1038/nri1425
101. Masani S, Han L, Yu K. Apurinic/apyrimidinic endonuclease 1 is the essential nuclease during immunoglobulin class switch recombination. Mol Cell Biol (2013) 33(7):1468–73. doi:10.1128/MCB.00026-13
102. Schrader CE, Guikema JE, Wu X, Stavnezer J. The roles of APE1, APE2, DNA polymerase beta and mismatch repair in creating S region DNA breaks during antibody class switch. Philos Trans R Soc Lond B Biol Sci (2009) 364(1517):645–52. doi:10.1098/rstb.2008.0200
103. Ehrenstein MR, Rada C, Jones AM, Milstein C, Neuberger MS. Switch junction sequences in PMS2-deficient mice reveal a microhomology-mediated mechanism of Ig class switch recombination. Proc Natl Acad Sci U S A (2001) 98(25):14553–8. doi:10.1073/pnas.241525998
104. Schrader CE, Vardo J, Stavnezer J. Role for mismatch repair proteins Msh2, Mlh1, and Pms2 in immunoglobulin class switching shown by sequence analysis of recombination junctions. J Exp Med (2002) 195(3):367–73. doi:10.1084/jem.20011877
105. Begum NA, Stanlie A, Doi T, Sasaki Y, Jin HW, Kim YS, et al. Further evidence for involvement of a noncanonical function of uracil DNA glycosylase in class switch recombination. Proc Natl Acad Sci U S A (2009) 106(8):2752–7. doi:10.1073/pnas.0813252106
106. Rush JS, Fugmann SD, Schatz DG. Staggered AID-dependent DNA double strand breaks are the predominant DNA lesions targeted to Sm in immunoglobulin class switch recombination. Int Immunol (2004) 16:549–57. doi:10.1093/intimm/dxh057
107. Vuong BQ, Herrick-Reynolds K, Vaidyanathan B, Pucella JN, Ucher AJ, Donghia NM, et al. A DNA break- and phosphorylation-dependent positive feedback loop promotes immunoglobulin class-switch recombination. Nat Immunol (2013) 14(11):1183–9. doi:10.1038/ni.2732
108. Cheng HL, Vuong BQ, Basu U, Franklin A, Schwer B, Astarita J, et al. Integrity of the AID serine-38 phosphorylation site is critical for class switch recombination and somatic hypermutation in mice. Proc Natl Acad Sci U S A (2009) 106(8):2717–22. doi:10.1073/pnas.0812304106
109. Lee GS, Neiditch MB, Salus SS, Roth DB. RAG proteins shepherd double-strand breaks to a specific pathway, suppressing error-prone repair, but RAG nicking initiates homologous recombination. Cell (2004) 117(2):171–84. doi:10.1016/S0092-8674(04)00301-0
110. Chaumeil J, Micsinai M, Ntziachristos P, Roth DB, Aifantis I, Kluger Y, et al. The RAG2 C-terminus and ATM protect genome integrity by controlling antigen receptor gene cleavage. Nat Commun (2013) 4:2231. doi:10.1038/ncomms3231
111. Deriano L, Chaumeil J, Coussens M, Multani A, Chou Y, Alekseyenko AV, et al. The RAG2 C terminus suppresses genomic instability and lymphomagenesis. Nature (2011) 471(7336):119–23. doi:10.1038/nature09755
112. Chaudhuri J, Khuong C, Alt FW. Replication protein A interacts with AID to promote deamination of somatic hypermutation targets. Nature (2004) 430(7003):992–8. doi:10.1038/nature02821
113. Yamane A, Robbiani DF, Resch W, Bothmer A, Nakahashi H, Oliveira T, et al. RPA accumulation during class switch recombination represents 5′-3′ DNA-end resection during the S-G2/M phase of the cell cycle. Cell Rep (2013) 3(1):138–47. doi:10.1016/j.celrep.2012.12.006
114. Kaidi A, Jackson SP. KAT5 tyrosine phosphorylation couples chromatin sensing to ATM signalling. Nature (2013) 498(7452):70–4. doi:10.1038/nature12201
115. Wuerffel R, Wang L, Grigera F, Manis J, Selsing E, Perlot T, et al. S-S synapsis during class switch recombination is promoted by distantly located transcriptional elements and activation-induced deaminase. Immunity (2007) 27(5):711–22. doi:10.1016/j.immuni.2007.09.007
116. Boboila C, Alt FW, Schwer B. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv Immunol (2012) 116:1–49. doi:10.1016/B978-0-12-394300-2.00001-6
117. Sellars M, Reina-San-Martin B, Kastner P, Chan S. Ikaros controls isotype selection during immunoglobulin class switch recombination. J Exp Med (2009) 206(5):1073–87. doi:10.1084/jem.20082311
118. Reina-San-Martin B, Difilippantonio S, Hanitsch L, Masilamani RF, Nussenzweig A, Nussenzweig MC. H2AX is required for recombination between immunoglobulin switch regions but not for intra-switch region recombination or somatic hypermutation. J Exp Med (2003) 197(12):1767–78. doi:10.1084/jem.20030569
119. Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkac J, et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell (2013) 49(5):872–83. doi:10.1016/j.molcel.2013.01.001
120. Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas-Loba A, et al. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell (2013) 49(5):858–71. doi:10.1016/j.molcel.2013.01.002
121. Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T. 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science (2013) 339(6120):700–4. doi:10.1126/science.1231573
122. Callen E, Di Virgilio M, Kruhlak MJ, Nieto-Soler M, Wong N, Chen HT, et al. 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell (2013) 153(6):1266–80. doi:10.1016/j.cell.2013.05.023
123. Pei H, Zhang L, Luo K, Qin Y, Chesi M, Fei F, et al. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature (2011) 470(7332):124–8. doi:10.1038/nature09658
124. Hajdu I, Ciccia A, Lewis SM, Elledge SJ. Wolf-Hirschhorn syndrome candidate 1 is involved in the cellular response to DNA damage. Proc Natl Acad Sci U S A (2011) 108(32):13130–4. doi:10.1073/pnas.1110081108
125. Stewart GS. Solving the RIDDLE of 53BP1 recruitment to sites of damage. Cell Cycle (2009) 8(10):1532–8. doi:10.4161/cc.8.10.8351
126. Li L, Halaby MJ, Hakem A, Cardoso R, El Ghamrasni S, Harding S, et al. Rnf8 deficiency impairs class switch recombination, spermatogenesis, and genomic integrity and predisposes for cancer. J Exp Med (2010) 207(5):983–97. doi:10.1084/jem.20092437
127. Ramachandran S, Chahwan R, Nepal RM, Frieder D, Panier S, Roa S, et al. The RNF8/RNF168 ubiquitin ligase cascade facilitates class switch recombination. Proc Natl Acad Sci U S A (2010) 107(2):809–14. doi:10.1073/pnas.0913790107
128. Santos MA, Huen MS, Jankovic M, Chen HT, Lopez-Contreras AJ, Klein IA, et al. Class switching and meiotic defects in mice lacking the E3 ubiquitin ligase RNF8. J Exp Med (2010) 207(5):973–81. doi:10.1084/jem.20092308
129. Bohgaki T, Bohgaki M, Cardoso R, Panier S, Zeegers D, Li L, et al. Genomic instability, defective spermatogenesis, immunodeficiency, and cancer in a mouse model of the RIDDLE syndrome. PLoS Genet (2011) 7(4):e1001381. doi:10.1371/journal.pgen.1001381
130. Lee-Theilen M, Matthews AJ, Kelly D, Zheng S, Chaudhuri J. CtIP promotes microhomology-mediated alternative end joining during class-switch recombination. Nat Struct Mol Biol (2011) 18(1):75–9. doi:10.1038/nsmb.1942
131. Cortizas EM, Zahn A, Hajjar ME, Patenaude AM, Di Noia JM, Verdun RE. Alternative end-joining and classical nonhomologous end-joining pathways repair different types of double-strand breaks during class-switch recombination. J Immunol (2013) 191(11):5751–63. doi:10.4049/jimmunol.1301300
132. Bothmer A, Rommel PC, Gazumyan A, Polato F, Reczek CR, Muellenbeck MF, et al. Mechanism of DNA resection during intrachromosomal recombination and immunoglobulin class switching. J Exp Med (2013) 210(1):115–23. doi:10.1084/jem.20121975
133. Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, Ho YJ, et al. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell (2011) 147(1):107–19. doi:10.1016/j.cell.2011.07.049
134. Hasham MG, Donghia NM, Coffey E, Maynard J, Snow KJ, Ames J, et al. Widespread genomic breaks generated by activation-induced cytidine deaminase are prevented by homologous recombination. Nat Immunol (2010) 11(9):820–6. doi:10.1038/ni.1909
135. Klein IA, Resch W, Jankovic M, Oliveira T, Yamane A, Nakahashi H, et al. Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell (2011) 147(1):95–106. doi:10.1016/j.cell.2011.07.048
136. Staszewski O, Baker RE, Ucher AJ, Martier R, Stavnezer J, Guikema JE. Activation-induced cytidine deaminase induces reproducible DNA breaks at many non-Ig Loci in activated B cells. Mol Cell (2011) 41(2):232–42. doi:10.1016/j.molcel.2011.01.007
137. Petersen S, Casellas R, Reina-San-Martin B, Chen HT, Difilippantonio MJ, Wilson PC, et al. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature (2001) 414(6864):660–5. doi:10.1038/414660a
138. Hasham MG, Snow KJ, Donghia NM, Branca JA, Lessard MD, Stavnezer J, et al. Activation-induced cytidine deaminase-initiated off-target DNA breaks are detected and resolved during S phase. J Immunol (2012) 189(5):2374–82. doi:10.4049/jimmunol.1200414
139. Lamont KR, Hasham MG, Donghia NM, Branca J, Chavaree M, Chase B, et al. Attenuating homologous recombination stimulates an AID-induced antileukemic effect. J Exp Med (2013) 210(5):1021–33. doi:10.1084/jem.20121258
Keywords: cytidine deamination, DNA recombination, DNA repair, class-switching, R-loops
Citation: Vaidyanathan B, Yen W-F, Pucella JN and Chaudhuri J (2014) AIDing chromatin and transcription-coupled orchestration of immunoglobulin class-switch recombination. Front. Immunol. 5:120. doi: 10.3389/fimmu.2014.00120
Received: 10 January 2014; Accepted: 07 March 2014;
Published online: 28 March 2014.
Edited by:
Ananda L. Roy, Tufts University School of Medicine, USAReviewed by:
Amy L. Kenter, University of Illinois College of Medicine, USASebastian Dudo Fugmann, Chang Gung University, Taiwan
Copyright: © 2014 Vaidyanathan, Yen, Pucella and Chaudhuri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jayanta Chaudhuri, Immunology Program, Memorial Sloan Kettering Cancer Center, Gerstner Sloan Kettering Graduate School, New York, NY 10065, USA e-mail:Y2hhdWRodWpAbXNrY2Mub3Jn
†Bharat Vaidyanathan, Wei-Feng Yen and Joseph N. Pucella have contributed equally to this work.