 Raunaq Singh Nagi
Raunaq Singh Nagi Ashish Shekhar Bhat
Ashish Shekhar Bhat Himanshu Kumar
Himanshu Kumar- 1Laboratory of Immunology, Department of Biological Sciences, Indian Institute of Science Education and Research, Bhopal, India
- 2Laboratory of Host Defense, WPI, Immunology Frontier Research Center, Osaka University, Osaka, Japan
Introduction
Cancer is a disease of complex etiology and multistep progression, manipulating the regular routes to homeostasis. Any deviation from homeostasis alerts the innate immune system and provokes inflammation. Inflammation is generated by the signaling cascades launched by the pattern recognition receptors (PRRs), the germ-line encoded molecules dedicated to sense pathogen, or danger-associated molecular patterns (PAMPs or DAMPs) in case of pathogen/foreign matter invasion and intrinsic disturbances, respectively (1–3). Through inflammation, PRRs eliminate stress signals and re-establish homeostasis in the body, via drawing the required cellular machinery to the inflammatory sites. However, the same lympho-reticular infiltrate has been linked with incidence of cancer at the site of chronic inflammation, since 1863, by Rudolf Virchow (4). From 1990s vast amount of literature has accumulated associating soluble and cellular factors of innate immune system with prevalence and progression of cancer. Furthermore, in the past decade, several pathogens have been linked with cancer as well [Ref. (5, 6) and references therein].
Fascinatingly, it is remarkable how the tightly regulated sensory system for stress removal and maintenance of homeostasis functions anomalously and promotes occurrence and progression of cancers (7–9).
PRR-Mediated Responses and Cancer Progression
All PRR-dependent pathways activate a particular set of transcription factors to generate appropriate responses. The same factors govern cellular proliferation, apoptosis, tissue remodeling, or angiogenesis, and exhibit a perturbed activity during cancer. One such key protein is nuclear factor κB (NFκB); up-regulation of which leads to production of pro-inflammatory cytokines. Additionally, it induces anti-apoptotic proteins like Bcl2 or inhibitors of apoptotic proteins (IAPs) and angiogenic proteins, such as angiopoietin or vascular endothelial growth factor (VEGF). NFκB also induces nitrous oxide synthase-2 (NOS-2), thus producing nitrous oxide (NO) in the immune cells, which along with reactive oxygen species (ROS) eradicates infected cells by lipid per-oxidation and DNA damage (10–14). Conversely, genomic instability and free radicals thus produced act as DAMPs, leading to sensitization of neighboring PRRs and further immune activation, for instance, the DNA fragments released can activate local DNA sensors, resulting in production of Type I IFN by DAI-TBK1, and activate KRAS pathway of cellular proliferation via TBK1-Sec5 complex, which leads to further activation of NFκB and production of anti-apoptotic proteins (15). That is, detouring regular anti-cancer pathway toward proliferation. Also, RONS induce DNA methylases, which lead to methylation and silencing of tumor suppressor and DNA damage repair genes (2, 16–18).
Another pathway crucial in immunity and cancer is the Janus kinases (JAK)-signal transducers and activators of transcription (STAT) pathway. Triggered primarily by interferons and some other mediators, this pathway stimulates various proliferative genes, such as IL-6-mediated induction of myc and CyclinD1/D2 through JAK; also TNFα-mediated up-regulation of STAT-3 leading to activation of Ras-mitogen activated protein kinase (MAPK) pathway, which leads to the expression of transcription factor activating protein (AP)-1, and epidermal growth factor (EGFs) along with eukaryotic initiation factor (eIF)-4. AP-1 couples with NFκB, inducing matrix metalloproteinase (MMP)-9, a protein involved in tissue remodeling required during angiogenesis (19). Thus, the pro-inflammatory signal culminates in the production of proteins aiding tumor survival, proliferation, and development of tumor-associated vasculature (18).
Furthermore, NFκB is also involved in the expression of NLRP3, which assembles with apoptosis-associated speck-like protein containing a CARD (ASC) caspase-1 to form multi-protein complexes, the inflammasomes, and responds to DAMPs, especially nucleotides released from damaged or necrotic tissue (due to cytotoxicity of free radicals) (20). Likewise, absent in myeloma (AIM)-2 inflammasomes also organize in response to the formation of DNA adducts (DNA and cytosolic protein HMGB-1) from the dying tissue (21). These assemblies lead to activation of IL-1β–IL-1βR pair; a system found commonly over-activated in many cancers (2, 22). Additionally, NFκB also generates cyclo-oxygenase-2 (COX-2) enzyme, which converts arachidonic acid into prostaglandin-E2 (PGE-2), one of the dual (pro-inflammatory and/or anti-inflammatory) mediators of immune response. PGE-2 enhances T-cell activation and represses B-cell activity (23, 24). Another common enzyme, activation-induced deaminase (AID), also induced by NFκB, involved in somatic hypermutation and class switch recombination in B-cells, causes genome instability and releases additional DAMPs into the microenvironment (25). Thus, the immune mediators produced for protection can divert inflammation toward pro-tumor facet (26).
A set of pro-inflammatory cytokines consisting of TNF-α and IL-1 and 6 is essentially tumor directing. TNF-α promotes tumor initiation and DNA damage. It also up-regulates hypoxia-inducible factor (HIF)-1α (attributed to the increasingly low oxygen levels due to multiplying cells) aiding in angiogenesis (27). IL-1β aids in tumor invasiveness and adhesion required during metastasis to new sites. IL-1α, the membrane bound form, induces IL-1 expression, associated with tissue damage, compensatory cell proliferation, and activation of JAK-STAT pathway, as seen in hepatocellular carcinomas and colitis-associated cancers (22, 28).
Cigarette smoking has long been associated with incidence of cancer. Cigarette smoke contains numerous compounds with known cytotoxicity, mutagenicity, and carcinogenicity, most of which are particulate. Stable ROS present in the smoke damage DNA and cause lipid per-oxidation, sensitizing the PRRs present from the buccal cavity to lungs leading to increased IL-8 and TNF-α (11). In addition, both, NFκB and AP-1 are up-regulated exaggerating the pro-inflammatory signal, at the same time homeostatic activity of both is reduced, compensating normal immune response. Such a response coupled with prolonged exposure can spontaneously lead to cellular transformations and their expansion (29, 30).
Role of Cellular Components of Innate Immunity in Cancer Progression
Specialized cells of the immune system are equipped with PRRs, and are responsible for clearance of diseased/damaged cells. Pro-inflammatory cytokines draw these cells toward the inflammatory site and direct them for removal of pathogens, particulates, or immune debris. These populations recede as the signal resolves. Since Virchow proposed their role at the site of chronic inflammation and cancer, a number of cellular populations and their effector responses have been ascertained for the same. In cases of prolonged exposure to PAMPs/DAMPs, infiltrating cells fail to withdraw and differentiate into M2 macrophages, identified as tumor-associated macrophages (TAMs), an integral population programed for tissue remodeling and tumor progression. Upon activation of their PRRs, TAMs promote various properties of cancer by releasing a range of inflammatory and angiogenic bio-chemicals. These cells stimulate proliferation of stromal tissue and macrophages by growth factors such as platelet-derived growth factor (PDGF) and colony stimulating factor (CSF)-1 respectively (31). Moreover, they organize a route to metastasis by digesting the substratum, basal lamina and release inactive growth factors via, MMPs. In addition, they assist in cellular movements by releasing cell adhesion molecules such as intercellular adhesion molecule (ICAM)-1 (32–34).
Another such population, the NK cells meant to carry out cytotoxic clearance of all the cells which do not express human leukocyte antigen (HLA) A/B/C and thus fail to activate the membrane-expressed inhibitory receptors (NKp30/44/46). NK cells are also responsible for killing any cell, irrespective of HLA tag, which presents them with stress/abnormality/tumor-associated antigens, via, activating receptor (NKG2D). In addition, they also participate in antibody-dependent cell-mediated cytotoxicity (ADCC), on cells tagged through FCRγIII (35, 36). A number of cytokines activate NK cells and turn them into lymphokine-activated killers, causing them to display their killing property at the site of recruitment (37). Tumor cells escape NK cells by blocking the activating receptor. Also, even if recruited, NK cells can only bring about killing of the outer cells in a solid tumor (38, 39). Furthermore, a reduction in NK cell cytotoxic function as well as NK cell dependent tumor surveillance is evident as a tumor directing effect of cigarette smoke (29, 30).
Another newly characterized population of cells called myeloid-derived suppressor cells (MDSCs) are recruited by inflammatory mediators, which inhibit the anti-tumor responses and release pro-tumor molecules, like, NOS-2 and TGF-β. Arginase-1 and indolamine-2,3-dioxygenase produced by MDSCs are involved in silencing the anti-tumor immunity by reducing Th1 activization (40, 41).
Soluble and cellular factors work in union. Cellular proliferation at an enhanced rate at tumor sites gives rise to hypoxia, low concentration of oxygen that stabilizes HIF-1α, which activates NFκB to secrete an angiogenic protein, VEGF and also induces expression of IL-12 and TNF-α. These mediators collectively induce STAT-3 production that up-regulates PGE-2 which further recruits NK cells and their cytotoxic function releases DAMPs in vicinity attracting TAMs which aid in shaping neo-vasculature and creating area for growing cell mass. Deregulation of Th1 responses by PGE-2 and MDSC/TAMs creates an imbalance (42–44). HIF-1α which causes glycolytic environment, such conditions may tip the balance into a state when IL-12 production is replaced by IL-23, that is, a shift from anti-tumor to pro-tumor (45, 46). In this manner, the PRR triggered pathways to eliminate PAMPs/DAMPs, are rerouted in a manner that leads to exaggeration of the initial signal causing chronic inflammation; moreover eliciting other pathways concluding in tumor growth and metastasis (1, 47, 48).
Regulatory Mechanisms and Progression of Cancer
To maintain homeostasis in the body, all cellular processes, including PRR generated immune responses are regulated by various mechanisms. This control is exercised at various levels. At transcriptional level certain cytokines can inhibit transcription factors, such as IL-4/13 which hinder NFκB. Alternatively; some cytokines directly inhibit other cytokines at protein level, such as inhibition of TNF-α by IL-10. At post-transcriptional level microRNAs play a crucial role by either resolving inflammation or potentiating pro-tumor effects of cytokines (18). For instance, TNF-α/IL-1β induced mir-146a inhibits IRAK/TRAF6, that is, the downstream signaling of TLR pathway, thus resolving inflammation (23). In contrast IL-6 induced mir-21 targets tumor suppressor genes, such as phosphatase and tensin homolog (PTEN), programed cell death (PDCD)-4, and others, thereby hampering the anti-tumor effects (49–52). Another microRNA found commonly up-regulated in cancer, miR-155, induces NOS-2 and inhibits apoptosis by down-regulating TP53INP1 (a downstream molecule of p53 signaling) (53). Thus, a slight imbalance in the pro-inflammatory and anti-inflammatory signals can shift the equilibrium toward oncogenic transformations (18).
Pattern recognition receptor elicited signaling cascades induce synthesis of several zinc-finger proteins that help in regulating the inflammatory signals. For example, ZC3H12a/c and Zfp36 proteins degrade the cellular mRNA coding for pro-inflammatory cytokines; while ZAP proteins directly degrade the viral RNA, to be precise, the PAMP itself (54, 55). Thus, these proteins degrade the PAMPs or signal generated by them to curb inflammation. In addition, certain PRRs themselves restrain the downstream signaling such as inhibition of IPS-1, adaptor molecule of RLR, by NLRX-1. Mutations within these genes or their regulatory elements or imbalance at cellular level can skew the balance toward tumorigenesis (6, 55) (Figure 1).
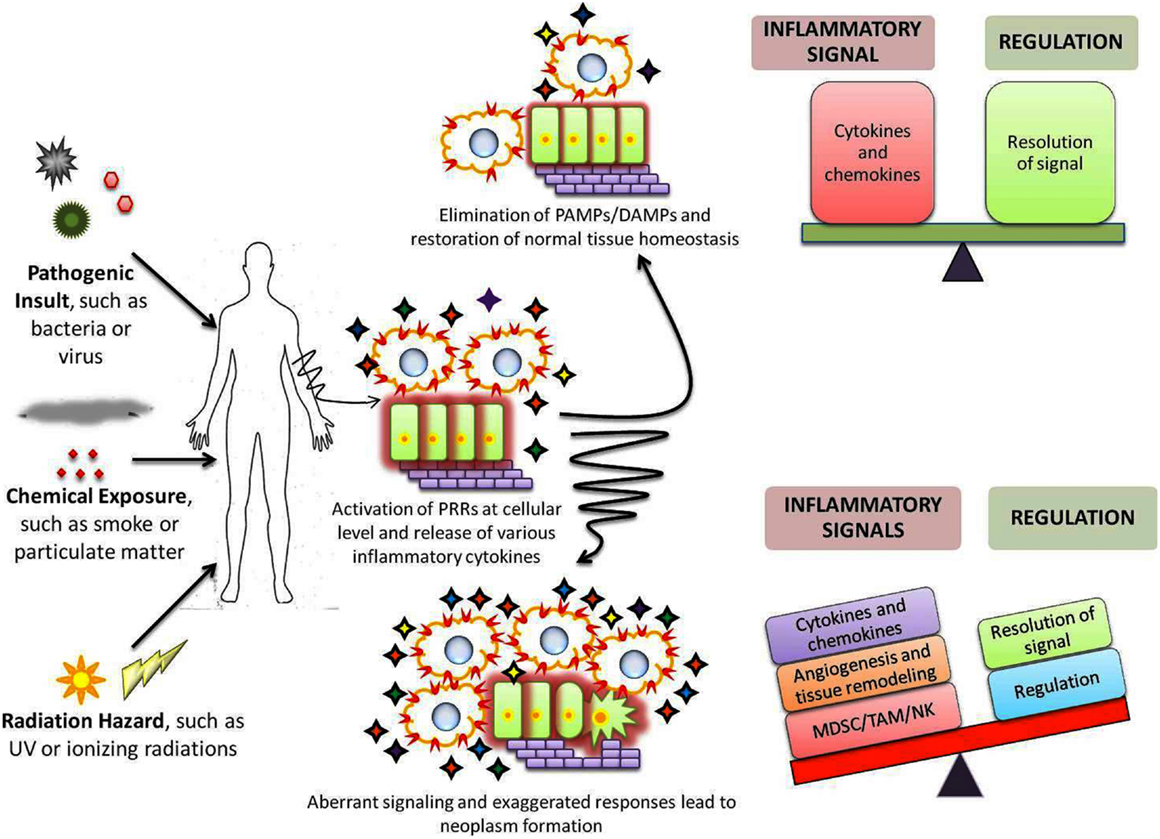
Figure 1. Role of PRRs in maintenance of homeostasis and cancer development. Exposure to any of the various stress stimuli leads to activation of immune system through PRRs ( ) which results in expansion of a variety of cellular populations and production of numerous cytokines (
) which results in expansion of a variety of cellular populations and production of numerous cytokines ( ). A precise sensitive balance at the inflammatory and regulatory front eliminates and reinstates homeostasis within the host. However, under certain circumstances, the precise balance is altered due to aberrant/exaggerated inflammation or failure of regulatory mechanisms to restrict them. Such imbalance results in accumulation of cells and chemicals that deregulate the proliferative pathways; induce angiogenesis and damage the extracellular matrix, thus aiding in origin and/or progression of cancers.
). A precise sensitive balance at the inflammatory and regulatory front eliminates and reinstates homeostasis within the host. However, under certain circumstances, the precise balance is altered due to aberrant/exaggerated inflammation or failure of regulatory mechanisms to restrict them. Such imbalance results in accumulation of cells and chemicals that deregulate the proliferative pathways; induce angiogenesis and damage the extracellular matrix, thus aiding in origin and/or progression of cancers.
New Facet in the Field
A relatively new but noteworthy field in analyzing cancer biology is polymorphisms. Single nucleotide polymorphisms (SNPs) are single base changes in the DNA, which may produce totally drastic effects on the structure and function of the encoded molecules. PRRs and the associated machinery, such as the adaptors or receptors are also susceptible to such polymorphisms and many such SNPs have been reported. SNPs could promote the anti-tumor effect if they prevent the PRR cascade from commencing, or support the pro-tumor effect, if they cause spontaneous induction of signaling without any stimulus. The cytoplasmic domains of the PRRs and cytokine receptors are of prime importance in this context as they form the docking site for progression of inflammatory response. Many polymorphisms have been identified to be associated with cancers of various origins. Mostly CLRs and RLRs have been associated with sensing PAMPs of oncogenic origin, and polymorphisms in their genes have been correlated with cancers of mesoderm, endoderm, and also ectoderm origin. Also several genes and mutations have been correlated with cigarette smoke in association with cancers (56). Still, a rather comprehensive endeavor would be required to establish the integral role of these SNPs at the molecular level to outline their part in incidences and progression of cancers [Ref. (6) and references therein, Ref. (56)].
Conclusion
Recognition of pathogen/stress is one of the essential processes of the host. Tumor cells are in fact, abnormal cells which are steadily eliminated through PRR-mediated pathways. However, hyper or anomalous behavior of same pathways can divert the protective route toward malignancy, by contributing to abnormal proliferation, angiogenesis, or modifying tissue architecture. The origin of anomalous behavior maybe external and internal, such as pathogen/foreign insults tissue damage/necrosis or mutations and polymorphisms in vital signaling components, respectively. Unfolding the root of these irregularities and malfunctions shall help in better understanding of the disease and thus, create new and personalized prospects for treatment.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work is supported by research grants number SR/S2/RJN-55/2009, BT/PR6009/GBD/27/382/2012 from Department of Science and Technology (DST) and Department of Biotechnology (DBT), Government of India to Himanshu Kumar and Intramural Research Grant from IISER, Bhopal, India; Raunaq Singh Nagi would like to thank DST INSPIRE for doctoral fellowship support (IF 130017).
References
1. Lu H, Ouyang W, Huang C. Inflammation, a key event in cancer development. Mol Cancer Res (2006) 4:221–33. doi: 10.1158/1541-7786.MCR-05-0261
2. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140:805–20. doi:10.1016/j.cell.2010.01.022
3. Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol (2011) 30:16–34. doi:10.3109/08830185.2010.529976
4. Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet (2001) 357:539–45. doi:10.1016/S0140-6736(00)04046-0
5. Chen K, Huang J, Gong W, Iribarren P, Dunlop NM, Wang JM. Toll-like receptors in inflammation, infection and cancer. Int Immunopharmacol (2007) 7:1271–85. doi:10.1016/j.intimp.2007.05.016
6. Kutikhin AG, Yuzhalin AE. Inherited variation in pattern recognition receptors and cancer: dangerous liaisons? Cancer Manag Res (2012) 4:31–8. doi:10.2147/CMAR.S28688
7. Cristofori G. New signals from the invasive front. Nature (2006) 441:444–50. doi:10.1038/nature04872
8. Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science (2013) 339:286–91. doi:10.1126/science.1232227
9. de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer (2006) 6:24–37. doi:10.1038/nrc1782
10. Aggarwal BB, Shishodia S, Sandur SK, Pandey MK, Sethi G. Inflammation and cancer: how hot is the link? Biochem Pharmacol (2006) 72:1605–21. doi:10.1016/j.bcp.2006.06.029
11. Closa D, Folch-Puy E. Oxygen free radicals and the systemic inflammatory response. IUBMB Life (2004) 56:185–91. doi:10.1080/15216540410001701642
12. Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature (2004) 431:461–6. doi:10.1038/nature02924
13. Ying L, Hofseth AB, Browning DD, Nagarkatti M, Nagarkatti PS, Hofseth LJ. Nitric oxide inactivates the retinoblastoma pathway in chronic inflammation. Cancer Res (2007) 67:9286–93. doi:10.1158/0008-5472.CAN-06-2149
14. Meira LB, Bugni JM, Green SL, Lee CW, Pang B, Borenshtein D, et al. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J Clin Invest (2008) 118:2516–25. doi:10.1172/JCI35073
15. Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature (2009) 462:108–12. doi:10.1038/nature08460
16. Karin M. Nuclear factor-kappaB in cancer development and progression. Nature (2006) 441:431–6. doi:10.1038/nature04870
17. Sato Y, Goto Y, Narita N, Hoon DS. Cancer cells expressing toll-like receptors and the tumor microenvironment. Cancer Microenviron (2009) 2(Suppl 1):205–14. doi:10.1007/s12307-009-0022-y
18. Schetter AJ, Heegaard NH, Harris CC. Inflammation and cancer: interweaving microRNA, free radical, cytokine and p53 pathways. Carcinogenesis (2010) 31:37–49. doi:10.1093/carcin/bgp272
19. Giraudo M, Inoue M, Hanahan D. An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J Clin Invest (2004) 114:623–33. doi:10.1172/JCI200422087
20. Chen GY, Nunez G. Inflammasomes in intestinal inflammation and cancer. Gastroenterology (2011) 141:1986–99. doi:10.1053/j.gastro.2011.10.002
21. Yanai H, Ban T, Wang Z, Choi MK, Kawamura T, Negishi H, et al. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature (2009) 462:99–103. doi:10.1038/nature08512
22. Song X, Voronov E, Dvorkin T, Fima E, Cagnano E, Benharroch D, et al. Differential effects of IL-1 alpha and IL-1 beta on tumorigenicity patterns and invasiveness. J Immunol (2003) 171:6448–56.
23. Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol (2010) 22:231–7. doi:10.1016/j.coi.2010.01.009
24. Simmons DL, Botting RM, Hla T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev (2004) 56:387–437. doi:10.1124/pr.56.3.3
25. Robbiani DF, Bothmer A, Callen E, Reina-San-Martin B, Dorsett Y, Difilippantonio S, et al. AID is required for the chromosomal breaks in c-myc that lead to c-myc/IgH translocations. Cell (2008) 135:1028–38. doi:10.1016/j.cell.2008.09.062
26. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140:883–99. doi:10.1016/j.cell.2010.01.025
27. Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, et al. Karin: NF-κB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1α. Nature (2008) 453:807–11. doi:10.1038/nature06905
28. Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature (2006) 441:437–43. doi:10.1038/nature04871
29. Lee J, Taneja V, Vassallo R. Cigarette smoking and inflammation: cellular and molecular mechanisms. J Dent Res (2012) 91:142–9. doi:10.1177/0022034511421200
30. Mehta H, Nazzal K, Sadikot RT. Cigarette smoking and innate immunity. Inflamm Res (2008) 57:497–503. doi:10.1007/s00011-008-8078-6
31. Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med (2001) 193:727–40. doi:10.1084/jem.193.6.727
32. Chen JJW, Lin YC, Yao PL, Yuan A, Chen HY, Shun CT, et al. Tumor-associated macrophages: the double-edged sword in cancer progression. J Clin Oncol (2005) 23:953–64. doi:10.1200/JCO.2005.12.172
33. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature (2008) 454:436–44. doi:10.1038/nature07205
34. Quatromoni JG, Eruslanov E. Tumor-associated macrophages: function, phenotype, and link to prognosis in human lung cancer. Am J Transl Res (2012) 4(4):376–89.
35. Nakagomi H, Petersson M, Magnusson I, Juhlin C, Matsuda M, Mellstedt H, et al. Decreased expression of the signal-transducing zeta chains in tumor-infiltrating T-cells and NK cells of patients with colorectal carcinoma. Cancer Res (1993) 53(23):5610–2.
36. Lutz CT, Kurago ZB. Human leukocyte antigen class I expression on squamous cell carcinoma cells regulates natural killer cell activity. Cancer Res (1999) 59:5793–9.
37. Glas R, Franksson L, Une C, Eloranta ML, Ohlen C, Orn A, et al. Recruitment and activation of natural killer (NK) cells in vivo determined by the target cell phenotype. An adaptive component of NK cell-mediated responses. J Exp Med (2000) 191:129–38. doi:10.1084/jem.191.1.129
38. Chouaib S, Thiery J, Gati A, Guerra N, El Behi M, Dorothee G, et al. Tumor escape from killing: role of killer inhibitory receptors and acquisition of tumor resistance to cell death. Tissue Antigens (2002) 60:273–81. doi:10.1034/j.1399-0039.2002.600401.x
39. Jewett A, Tseng HC. Tumor induced inactivation of natural killer cell cytotoxic function; implication in growth, expansion and differentiation of cancer stem cells. J Cancer (2011) 2:443–57. doi:10.7150/jca.2.443
40. Muller AJ, Sharma MD, Chandler PR, Duhadaway JB, Everhart ME, Johnson BA III, et al. Chronic inflammation that facilitates tumor progression creates local immune suppression by inducing indoleamine 2,3 dioxygenase. Proc Natl Acad Sci U S A (2008) 105:17073–8. doi:10.1073/pnas.0806173105
41. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9:162–74. doi:10.1038/nri2506
42. Kusmartsev S, Gabrilovich DI. STAT1 signaling regulates tumor-associated macrophage-mediated T cell deletion. J Immunol (2005) 174:4880–91.
43. Ochoa AC, Zea AH, Hernandez C, Rodriguez PC. Arginase, prostaglandins, and myeloid-derived suppressor cells in renal cell carcinoma. Clin Cancer Res (2007) 13:721s–6s. doi:10.1158/1078-0432.CCR-06-2197
44. Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res (2007) 67:4507–13. doi:10.1158/0008-5472.CAN-06-4174
45. Shime H, Yabu M, Akazawa T, Kodama K, Matsumoto M, Seya T, et al. Tumor-secreted lactic acid promotes IL-23/IL-17 proinflammatory pathway. J Immunol (2008) 180:7175–83.
46. Kortylewski M, Xin H, Kujawski M, Lee H, Liu Y, Harris T, et al. Regulation of the IL-23 and IL-12 balance by STAT3 signaling in the tumor microenvironment. Cancer Cell (2009) 15:114–23. doi:10.1016/j.ccr.2008.12.018
47. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
48. Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol (2011) 31:986–1000. doi:10.1161/ATVBAHA.110.207449
49. Frankel LB, Christoffersen NR, Jacobsen A, Lindow M, Krogh A, Lund AH. Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J Biol Chem (2008) 283:1026–33. doi:10.1074/jbc.M707224200
50. Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology (2007) 133:647–58. doi:10.1053/j.gastro.2007.05.022
51. Löffler D, Brocke-Heidrich K, Pfeifer G, Stocsits C, Hackermüller J, Kretzschmar AK, et al. Interleukin-6 dependent survival of multiple myeloma cells involves the STAT3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood (2007) 110:1330–3. doi:10.1182/blood-2007-03-081133
52. Gironella M, Seux M, Xie MJ, Cano C, Tomasini R, Gommeaux J, et al. Tumor protein 53-induced nuclear protein 1 expression is repressed by miR-155, and its restoration inhibits pancreatic tumor development. Proc Natl Acad Sci U S A (2007) 104:16170–5. doi:10.1073/pnas.0703942104
53. Jones MR, Quinton LJ, Blahna MT, Neilson JR, Fu S, Ivanov AR, et al. Zcchc11-dependent uridylation of microRNA directs cytokine expression. Nat Cell Biol (2009) 11:1157–63. doi:10.1038/ncb1931
54. Matsushita K, Takeuchi O, Standley DM, Kumagai Y, Kawagoe T, Miyake T, et al. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature (2009) 458:1185–90. doi:10.1038/nature07924
55. Kutikhin AG, Yuzhalin AE. C-type lectin receptors and RIG-I-like receptors: new points on the oncogenomics map. Cancer Manag Res (2012) 4:39–53. doi:10.2147/CMAR.S28983
Keywords: cancer, pattern recognition receptors, innate immunity, inflammation, regulation
Citation: Nagi RS, Bhat AS and Kumar H (2014) Cancer: a tale of aberrant PRR response. Front. Immunol. 5:161. doi: 10.3389/fimmu.2014.00161
Received: 26 March 2014; Accepted: 27 March 2014;
Published online: 09 April 2014.
Edited and reviewed by: Anton G. Kutikhin, Research Institute for Complex Issues of Cardiovascular Diseases, Russia
Copyright: © 2014 Nagi, Bhat and Kumar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence:aGt1bWFyQGlpc2VyYi5hYy5pbg==